 Awtum M. Brashear
Awtum M. Brashear Liwang Cui
Liwang Cui- Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
Malaria elimination includes neglected human malaria parasites Plasmodium vivax, Plasmodium ovale spp., and Plasmodium malariae. Biological features such as association with low-density infection and the formation of hypnozoites responsible for relapse make their elimination challenging. Studies on these parasites rely primarily on clinical samples due to the lack of long-term culture techniques. With improved methods to enrich parasite DNA from clinical samples, whole-genome sequencing of the neglected malaria parasites has gained increasing popularity. Population genomics of more than 2200 P. vivax global isolates has improved our knowledge of parasite biology and host-parasite interactions, identified vaccine targets and potential drug resistance markers, and provided a new way to track parasite migration and introduction and monitor the evolutionary response of local populations to elimination efforts. Here, we review advances in population genomics for neglected malaria parasites, discuss how the rich genomic information is being used to understand parasite biology and epidemiology, and explore opportunities for the applications of malaria genomic data in malaria elimination practice.
Introduction
Malaria is caused by infection with protozoan parasites from the genus Plasmodium. Five species are commonly accepted as human malaria parasites: Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale curtisi, and Plasmodium ovale wallikeri. The latter two are recognized as morphologically indistinguishable, sympatric, but non-recombining species only in the last decade (Sutherland et al., 2010) and are frequently combined as P. ovale spp. Additionally, several species infecting non-human primates occasionally cause zoonotic malaria in humans—including at least P. knowlesi and P. cynomolgi in Asia (Antinori et al., 2013; Raja et al., 2020) and P. simium in South America (Brasil et al., 2017). The occurrence of the zoonotic malaria reflects the distribution of their primary hosts. Among the human malaria parasites, P. falciparum is associated with most malaria-related deaths (Gething et al., 2016), whereas P. vivax is the most widespread and dominant species outside of Africa (Howes et al., 2016; Figure 1). It is estimated there were 7.5 million vivax malaria cases globally in 2018, more than half of which occurred in Southeast Asia (World Health Organization [WHO], 2019). In comparison, P. malariae and P. ovale spp. are distributed globally, but the incidence is low. Though generally less virulent than P. falciparum, P. vivax can also cause severe pathology, and recurrent episodes greatly increase morbidity (Anstey et al., 2009; Baird, 2013). P. malariae and P. ovale spp. are sporadically associated with severe clinical manifestations such as anemia, acute respiratory distress, and renal failure (Lau et al., 2013; Langford et al., 2015; Kotepui et al., 2020). Resources for malaria control and research are largely committed to P. falciparum, while the neglected human malaria parasites are understudied, resulting in significant gaps in our understanding of their fundamental biology (Mueller et al., 2009). We refer to these non-falciparum human malaria parasites as “neglected malaria parasites.”
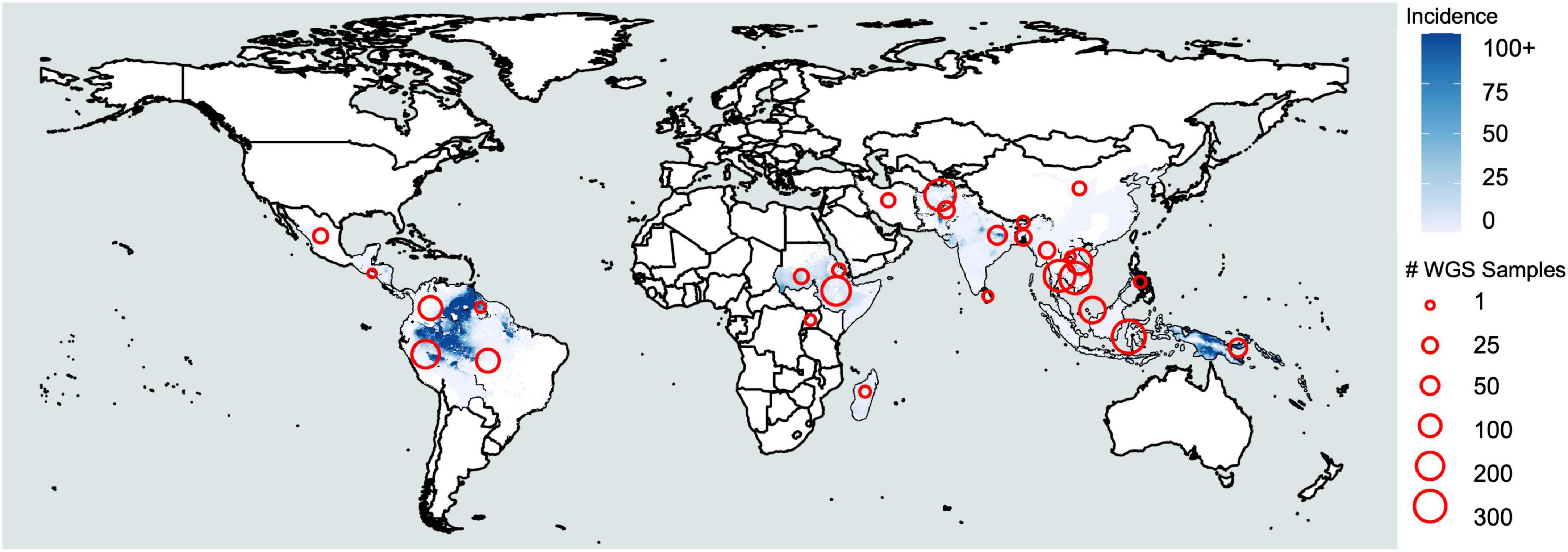
Figure 1. Plasmodium vivax incidence and whole-genome sequencing. Coloration represents incidence per 1000 based on raster data from the Malaria Atlas Project (https://malariaatlas.org/malaria-burden/) in 2017, the most recent year for which data is available. Red circles represent whole-genome sequencing samples collected by country based on Supplementary Table 1.
All malaria parasites share a similar life cycle involving a vertebrate intermediate host such as a human and a definitive host such as an Anopheles mosquito (Figure 2). The neglected malaria parasites have several distinctive features favoring and sustaining transmission. P. vivax and P. ovale spp. invade young reticulocytes (Simpson et al., 1999; Lim et al., 2016), leading to low-density infections. P. malariae is also associated with low-density infections in part due to a preference for older red blood cells (RBCs), production of fewer merozoites per schizont (average of 8), and an extended erythrocytic cycle (72 h) (Collins and Jeffery, 2007). Low-density infections are often missed by routine diagnostic methods such as microscopy and rapid diagnostic tests and may constitute an adaptation of the parasites for sustained transmission (Sutherland, 2016). Gametocytes emerge sooner in P. vivax than in P. falciparum, resulting in more efficient transmission, even among mixed infections (Balasubramanian et al., 2020). Finally, P. vivax and P. ovale spp. form dormant hypnozoites in the liver, which can relapse months or years later (White and Imwong, 2012). These characteristics bestow the neglected malaria parasites the ability to withstand traditional control measures designed for P. falciparum, with their prevalence or proportions often increased in areas of co-endemicity with P. falciparum (Cotter et al., 2013; Geng et al., 2019; Yman et al., 2019). Since neglected malaria parasites must also be eliminated (Baird, 2010; Lover et al., 2018), a better understanding of their biology, epidemiology, and evolution is needed.
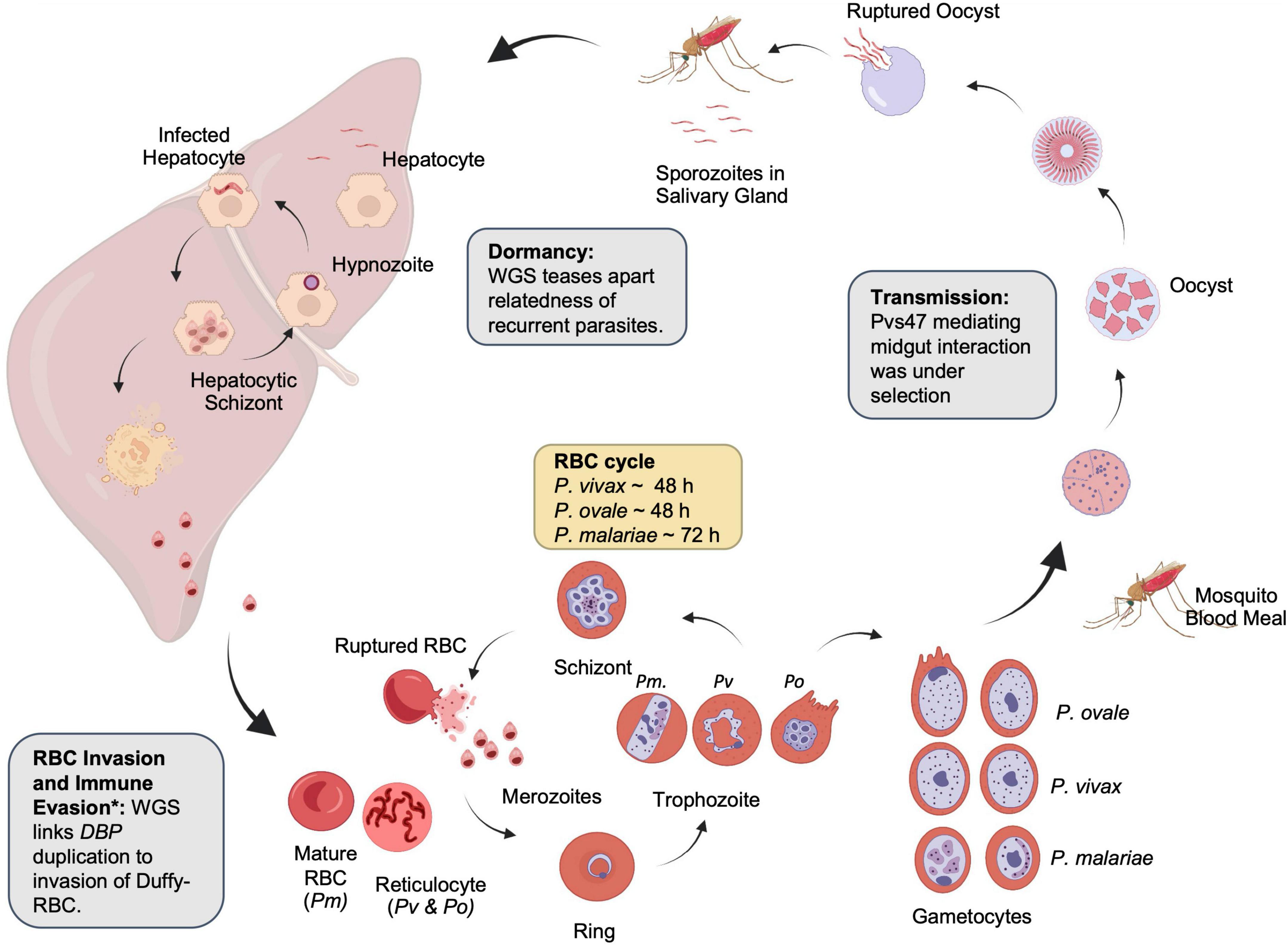
Figure 2. Lifecycle of non-falciparum malaria parasites. Examples of the contribution of whole-genome sequencing to parasite biology are shown in boxes. Created with BioRender.com. *The identification of the relationship between duffy-binding protein duplication and immune evasion was identified with qPCR.
Advances in next-generation sequencing (NGS) technologies have enabled whole-genome sequencing (WGS) of a large number of clinical isolates of malaria parasites, allowing the study of parasite populations at the genomic level – population genomics. However, there have been significant barriers to using the WGS technology within the neglected malaria species. The most notable is the reliance on clinical samples, which often have low parasitemia and contamination with human genomic DNA. Since the human genome (∼3000 Mb) is about 100 times larger than the malaria parasite genome (25–35 Mb), the problem associated with the human genome is obvious. Therefore, leukocyte depletion using filtration methods (e.g., CF-11 columns or commercial filters) is instrumental for obtaining high-quality parasite genomes (Auburn et al., 2011, 2013; Venkatesan et al., 2012; Rutledge et al., 2017b; Brashear et al., 2020b). Additional whole-genome amplification (WGA) may be necessary to further increase the total parasite DNA (Dharia et al., 2010; Brashear et al., 2019). For archived samples (often preserved as dried blood spots on filter paper) or samples collected in endemic areas where on-site leukocyte depletion cannot be performed, parasite DNA needs to be enriched. An in-solution hybridization method called hybrid selection or genome capture (Melnikov et al., 2011) has been used with genome-wide synthetic biotinylated RNA baits (Bright et al., 2012, 2014) or synthetic oligos (Hupalo et al., 2016), but the synthesis of baits can be costly, and this procedure is prone to bias as regions with higher GC content will hybridize more efficiently. Recently, selective WGA (SWGA), which uses specifically-designed small sets of short primers specific to the genome of interest to amplify the target genome preferentially, has become a cost-effective and robust procedure suited to enrich Plasmodium DNA (Leichty and Brisson, 2014). SWGA protocols have been designed for P. falciparum, P. vivax, and P. malariae (Oyola et al., 2016; Cowell et al., 2017; Ibrahim et al., 2020). Another innovative method, combining cell sorting with WGA to obtain highly accurate parasite genome information from single parasite-infected erythrocytes (Nair et al., 2014), is particularly suited for studying within-host variations of malaria infections from hyperendemic regions and teasing out the genetic complexities and relatedness of parasites within single hosts (Nkhoma et al., 2020). Here we review the recent progress in population genomics of neglected parasites focusing on P. vivax due to the breadth of literature available and discuss how the genomic data can be exploited to understand parasite biology and guide malaria elimination efforts.
Establishing reference genomes
In terms of accuracy and completeness, the P. falciparum 3D7 genome is the most complete reference Plasmodium genome (Gardner et al., 2002), and it has been further validated using the PacBio long-read sequencing technology (Otto et al., 2018). However, P. falciparum genomes are not an ideal reference for other medically important malaria parasites. The first draft of the P. vivax Sal-I nuclear genome consisted of over 2500 scaffolds totaling ∼26.8 Mb, of which ∼22.6 Mb were assigned to the 14 chromosomes (Carlton et al., 2008). Shortly after, the NGS technology enabled a multitude of whole-genome assemblies, including four geographically divergent P. vivax strains from North Korea, India, Brazil, and Mauritania (Neafsey et al., 2012). De novo assembly of a Cambodian P. vivax isolate highlighted shortcomings of the Sal-I assembly by identifying nearly 800 unaccounted-for genes (Hester et al., 2013). The 2016 P. vivax PvP01 genome from Papua New Guinea (PNG) dramatically improved assembly quality, reflected in the substantially reduced number of unassigned contigs and better resolved subtelomeric regions where there were nearly four times as many hypervariable pir genes as identified from the Sal-I assembly (Auburn et al., 2016a). The recently assembled new reference genomes of P. vivax strains from the China-Myanmar border region were of comparable quality to the PvP01 genome (Brashear et al., 2020b). With this new information, the P. vivax genome is estimated at ∼30 Mb. The expansion of subtelomeric gene families likely explains the increased genome sizes of the neglected malaria parasites compared to ∼23 Mb of the P. falciparum genome. Genomes for two P. ovale curtisi strains, two P. ovale wallikeri strains, and one P. malariae strain were first reported in 2016 (Ansari et al., 2016). The two P. ovale wallikeri and one P. ovale curtisi isolates were recent clinical samples from travelers who were infected in Gabon and Nigeria, respectively, while the P. malariae (Uganda) and one P. ovale curtisi (Nigeria) were historic samples isolated in 1974 and 1977, respectively, and passaged through chimpanzees. Subsequently, a high-quality genome assembly of a P. malariae strain from Uganda using long-read sequencing technology and draft genomes of P. ovale curtisi (from Ghana) and P. ovale wallikeri (from Cameroon) assembled from P. falciparum co-infections were reported (Rutledge et al., 2017a). Compared with P. vivax and P. falciparum, the genome sizes were much larger for P. ovale spp. (33–38 Mb) and P. malariae (∼34 Mb).
Fragmented reference genome assemblies can pose problems for analyses that rely on highly accurate alignment to reference genomes. Programs that detect insertions or deletions (indels) are especially affected as they depend on discordant read alignment and read depth (Abyzov et al., 2011; Layer et al., 2014; Beghain et al., 2016), both confounded by mis-mapping. Since the parasite genomic DNA of non-falciparum species has to be obtained almost exclusively from clinical samples, genome assemblies for these species have mostly relied on short-read sequencing, which is ineffective at resolving the abundant repetitive regions. The limitations of short-read sequencing may soon be overcome with long-read technologies such as the PacBio and Oxford Nanopore sequencing technologies, as demonstrated with recent successful applications in improving Plasmodium genome assemblies (Vembar et al., 2016; Rutledge et al., 2017a; Otto et al., 2018). Fortunately, incomplete reference genomes should not substantially hinder traditional population genomic studies of the neglected malaria parasites—wherein sequence reads of field isolates are typically aligned to reference genomes for calling single nucleotide polymorphisms (SNPs)—given the high-level synteny and conservation of the core genomes.
Notably, the gene content and quality of annotation are continually improving. Despite P. falciparum being the most well-annotated species, the most recent version of PlasmoDB (Update 57) included updates of 223 P. falciparum genes and less than 60 updated genes for any other human or primate Plasmodium species (PlasmoDB, 2022). Meanwhile, certain annotation resources, such as the Malaria Parasite Metabolic Pathways (MPMP), are exclusively available for P. falciparum (Ginsburg and Abdel-Haleem, 2016). Researchers often use studies performed in orthologs from P. falciparum or a rodent model to annotate the genomes of neglected malaria parasites, although they differ substantially. It was found that P. vivax, P. malariae, P. knowlesi, P. ovale curtisi, and P. ovale wallikeri shared just 3429 ortholog groups, with P. ovale wallikeri having 1036 unique ortholog groups (Ansari et al., 2016). The expansion of subtelomeric genes in non-falciparum malaria parasites is something noted in many genomic assemblies, but very little is known about the purpose of the additional gene family members. Surfin or STP1 genes, for example, have few members in P. vivax and P. falciparum and are lacking in P. knowlesi, but have dozens to hundreds of members in P. malariae and P. ovale spp. (Ansari et al., 2016). Likewise, PfEMP1 and SICAvar proteins are present in P. falciparum and P. knowlesi, respectively (Frech and Chen, 2013). Thus, the neglected parasites deserve special attention in genomics and gene annotations.
Understanding parasite biology using genomics
Population genomics of P. vivax is instrumental for improving our knowledge about its distinct parasite biology, including reticulocyte preference, invasion of Duffy-positive RBCs, and relapse (Figure 2). The rarity of vivax malaria transmission in African populations has been postulated to be due to the absence of Duffy antigen/receptor for chemokines (DARC) gene expression, once considered a requirement for erythrocyte invasion. Confirmed cases of P. vivax parasites in Duffy-negative Africans and the potential spread of such parasites suggest that P. vivax may have evolved to invade Duffy-negative individuals (Menard et al., 2010; Gunalan et al., 2016; Kepple et al., 2021). Duplication of the Duffy binding protein 1 (pvdbp1) gene was common in Malagasy P. vivax strains infecting Duffy-negative individuals (Menard et al., 2013). A very high proportion (>50%) of parasites with amplified pvdbp1 gene was detected in Ethiopia, where ∼35% human population is Duffy negative (Hostetler et al., 2016; Pearson et al., 2016; Auburn et al., 2019; Lo et al., 2019; Ford et al., 2020). However, it is intriguing that parasites with amplified pvdbp1 also reached modest levels (20–38%) in the Greater Mekong subregion (GMS), where nearly all local populations are Duffy positive (Hostetler et al., 2016). WGS analysis provided details of the Southeast Asian pvdbp1 amplification events, revealing two types of duplication, the Cambodian-type and the Malagasy-type, and indicated independent evolution of pvdbp1 duplication on the different genetic backgrounds (Hostetler et al., 2016; Auburn et al., 2019; Lo et al., 2019; Ford et al., 2020). Though the significance of pvdbp1 expansion in the invasion of Duffy-negatives in Africa remains to be tested, pvdbp1 amplification in Cambodian parasites was shown to result in increased pvdbp1 mRNA levels and associated with the ability of the parasites to counteract host anti-PvDBP antibodies (Popovici et al., 2020).
Relapses account for a significant source of blood-stage infections for P. vivax and P. ovale spp. (Robinson et al., 2015). The phenotypes of relapse in P. vivax malaria are well characterized, but the mechanisms of hypnozoite formation and reactivation remain unknown (White and Imwong, 2012). Relapsing parasites often differ from the parasites in the initial acute infection (Chen et al., 2007; Imwong et al., 2007), but it is difficult and sometimes impossible to determine whether a recurrent blood-stage infection arises from recrudescence, relapse, or new infection. Amplicon deep sequencing of polymorphic antigens demonstrated the complexity of the initial blood-stage infections and provided a scheme to assign the probability of a recurrent infection as a relapse (Lin et al., 2015). WGS data offers additional power to dissect the relatedness of individual parasite clones within a blood-stage infection. In a confirmed case of multiple relapses in a patient who acquired the initial infection in Eritrea, WGS revealed that parasites from three relapse episodes were genetically related meiotic siblings (Bright et al., 2014). In one study of Peruvian P. vivax, WGS was used to examine 23 paired clinical samples from the same patients at the times of initial infection and recurrent infection after three primaquine regimens (Cowell et al., 2018). By comparing the degrees of Identity-by-Descent (IBD, Box 1) between the paired samples, these researchers identified 12 cases of homologous relapses (IBD > 98%) and three heterologous relapses with highly related parasites which could be meiotic siblings. In the same study, WGS allowed more accurate assignment of recurrent cases compared to that based on microsatellite genotyping data (Durand et al., 2014). Similarly, a study examining relapse dynamics was conducted in Cambodia in 20 P. vivax patients within 2 months after CQ treatment (Popovici et al., 2018). WGS of five paired samples and SNP typing of others confirmed the multiclonality nature of relapses, and the relapsing parasites showed various degrees of relatedness in IBD to the parasites present in the original infection, consistent with the higher endemicity level of the study area (Popovici et al., 2018, 2019). Environmental factors such as epidemiology impact the nature of recurrence, with more intensive transmission increasing the chance of heterologous relapses (Imwong et al., 2012). Recent models have incorporated microsatellite-based IBD data and time between initial and recurrent infections for estimating recurrent infections as either recrudescence, relapse, or reinfection (Taylor et al., 2019b). Applying this multi-factor principle to genomic data may be incredibly fruitful for classifying infections in the future.
| BOX 1. Identity-by-descent methodologies. | |
| One important technique empowered by WGS is Identity-by-Descent estimation. IBD segments are defined as being inherited from the same ancestor, so longer lengths of IBD segments can suggest a more recent divergence between two individuals (Shetty et al., 2019; Taylor et al., 2019a). Various tools allow researchers to infer relatedness via IBD inference using SNP data. HmmIBD benefits non-falciparum malaria species due to its built-in consideration of SNPs for distance rather than genetic time (Schaffner et al., 2018). This feature removes the necessity of a genetic map, which has not been standardized via hybrid crossing for non-falciparum species. IsoRelate (Henden et al., 2018) does require a genetic map which users may improvise from linkage disequilibrium data, but it can consider infections harboring multiple parasite strains. DEploid-IBD can infer the relatedness of multiple infecting strains (Zhu et al., 2018); however, reliance on a high-confidence reference haplotype panel is problematic for neglected species. It is important to note that while the proportion of two genomes that are in IBD is correlative with estimated relatedness, it is not a perfect indicator, and it can be biased by the chosen SNP sets. Therefore, comparing IBD across whole genome studies is not recommended (Taylor et al., 2019a). | |
Understanding epidemiology from estimates of the complexity of infections
Complexity of infection (COI) defines the degree of multiple infections and the similarity of those infections (Pearson et al., 2016). It is sometimes used to reflect the transmission intensity. However, the relationship between parasite prevalence and COI is often non-linear, likely sensitive to other factors, and in some cases, potentially more spurious in P. vivax than P. falciparum (Fola et al., 2017; Lopez and Koepfli, 2021). As genomics techniques have improved the ability to determine how closely related co-infections are, it also allows us to gleam the likelihood of a superinfection (from more than two separate mosquito bites) or co-transmission (more than two parasite clones from one mosquito bite) (Cowell et al., 2018). COI is frequently used to extrapolate the multiplicity of infection (MOI). Polyclonal samples are frequently omitted in population genomic methods because multiple infections can be problematic for certain profile-based analyses if separate genetic locations are called from two or more parasites. Therefore, inferring COI is typically one of the first analyses done in Plasmodium genomics studies. MOI may be estimated by genotyping at individual loci by amplicon deep sequencing, at a set of loci such as microsatellite markers or SNPs, or more recently, using WGS data (Zhong et al., 2018). When coverage is high enough for effective variant calling across a majority of the genome, WGS offers superior sensitivity in detecting multiclonal infections that are generally missed by genotyping a limited number of markers (Flannery et al., 2015; Ibrahim et al., 2020) and for differentiating closely-related clones such as the meiotic siblings in an infection (Figure 3A; Bright et al., 2014). Single-cell genomics provides the best resolution of the genetic differences of individual parasite clones within very complex infections (Nkhoma et al., 2020), but may be too costly for resolving infections when minor-allele clones are less than 5% or for assessing a large number of infections.
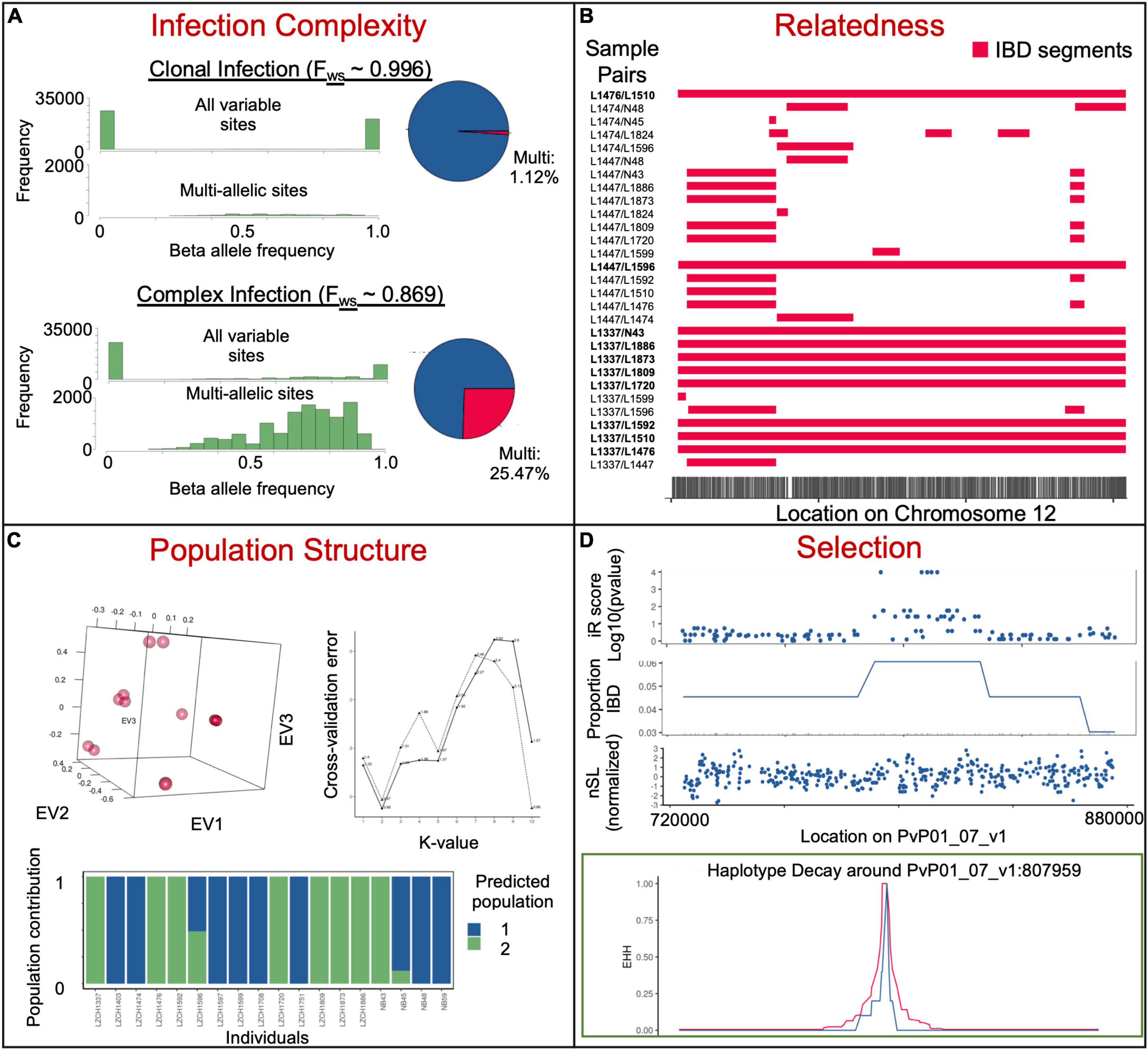
Figure 3. Example insights into parasite populations from whole-genome sequencing data. (A) Different beta allele frequencies for samples previously determined to be clonal (upper) and complex (lower) via Fws analysis. The top histogram shows all sites, while the bottom shows only sites with reads mapping to more than one allele. Pie charts on the right represent the number of sites with more than 5% of mapped reads mapping to each of the two alleles. (B) Identical-by-descent segments on chromosome 12 within selected sample pairs. (C) Population structure of 18 clonal samples from the China–Myanmar border. PCA (top left), admixture K-value cross-validation identifying ideal population numbers based on SNPs (top right) and admixture analysis at the ideal K-value (bottom). (D) Genome scans for selection (iR score, proportion pairs IBD and nSL) on a fragment of chromosome 7 and haplotype decay 100 kb around an SNP at the center of a region based on 11 genetically distinct China-Myanmar border samples. Data is subset from a larger dataset (Brashear et al., 2020a).
Multiple methods exist to estimate COI from whole-genome data. The within-host diversity (FWS) statistic is one popular method (Manske et al., 2012), although it is technically a measurement of inbreeding. FWS compares within-sample, and within-population heterozygosity, and the resulting values are continuous in a range between 0 and 1, with higher values representing samples with higher inbreeding. While high levels of inbreeding are not necessarily due to a lack of clones, FWS ≥ 0.95 is a popular cut-off for clonal infections. In addition to multiple implementations of FWS (Chang et al., 2017), other methods for estimating the MOI have been developed for SNP data based on either minor allele frequency (MAF) distribution (Galinsky et al., 2015) or haplotype structure (Zhu et al., 2018). MAF-based methods include plotting or examining minor allele frequencies at each SNP to look for an unexpected number of heterozygous SNPs (Galinsky et al., 2015; Pearson et al., 2016; Brashear et al., 2019). This approach doesn’t require reference datasets which can be beneficial when looking at individual clones, but can also be biased by data with imperfect mapping. Haplotype structures identify stretches of the genome traveling together and look at the number of each haplotype to predict the number of genetically distinct parasites (Zhu et al., 2018). This can result in higher accuracy at delineating closely related parasites but predicting haplotypes from short-read data is unreliable without strong reference haplotypes. One method to mitigate this concern was employed by the EstMOI program, which looks at strings of SNPs on the same reads or read pairs (default 3 SNPs) as haplotypes and calculates parasite prevalence (Assefa et al., 2014). Importantly, amplification techniques may cause an overestimation or underestimation of minor clones within an infection. Methodological advances in haplotype prediction may bolster future population genomic analyses.
Relapses in P. vivax and P. ovale spp. increase the chance of multi-clonal infections and may result in high MOI even in low-endemicity settings. For example, amplicon sequencing of the pvmsp1 gene detected over 90% of infections from low-endemicity areas of Cambodia as polyclonal; some infections contained as many as 10 parasite clones, and about half of the recurrent infections were attributable to relapses (Lin et al., 2015). Though the changes in MOI and malaria incidence are complex and their relationship is not linear, especially in species known for relapsing, reduced malaria transmission is generally associated with reduced MOI or increased levels of clonality. On a global level, eastern SE Asia and Oceania have more complex P. vivax infections than other parts of the globe, despite P. falciparum having the most complex infections within Africa (MalariaGEN et al., 2022). FWS analysis of P. vivax populations showed Malaysia had a significantly higher proportion (84%) of monoclonal infection (FWS ≥ 0.95) than other endemic areas such as Thailand, Indonesia, and Ethiopia (52–71%), consistent with the decline of vivax malaria incidence in Malaysia followed by unstable transmission and outbreaks (Auburn et al., 2018, 2019). Similarly, P. vivax infections from the China–Myanmar border also displayed a significantly higher level of clonality than in neighboring areas, reflecting lineage expansions during an outbreak (Brashear et al., 2020a).
Characterizing populations with genome-wide single nucleotide polymorphisms
Knowledge about the genetic diversity and population structure of malaria parasites improves understanding of the epidemiology, transmission patterns, population dynamics, parasite distribution and movement, and response to control measures of natural parasite populations, thus providing appraisal and guidance for malaria control and elimination activities (Arnott et al., 2012). The genetic diversity of P. vivax populations has been extensively studied using single polymorphic genes (Sanger sequencing or amplicon deep sequencing), microsatellite markers, and more recently, SNPs (as a subset of loci, such as in barcodes, or genome-wide) (Auburn and Barry, 2017; Escalante and Pacheco, 2019). As NGS costs continue to decline, population genomic studies using genome-wide SNPs have gained popularity. Small-scale WGS of P. vivax field isolates started a decade ago (Chan et al., 2012), which culminated in large-scale genomic analyses of > 400 global P. vivax field isolates in 2016 (Hupalo et al., 2016; Pearson et al., 2016), and more recently > 1500 samples from across the globe (MalariaGEN et al., 2022). Together with other regional P. vivax sequencing efforts including Southeast Asia [Cambodia (Parobek et al., 2016), southern China (Shen et al., 2017), Malaysia (Auburn et al., 2019), Myanmar (Brashear et al., 2020a)], South America [Colombia (Winter et al., 2015), Peru (Flannery et al., 2015), and Brazil (De Oliveira et al., 2017, 2020)], and Africa [Ethiopia (Auburn et al., 2019; Ford et al., 2020)] (Figure 1 and Supplementary Table 1), P. vivax genomic studies delved into the demographic history of this parasite and detected events of parasite lineage expansion, contraction, or introduction. The use of genome-wide SNPs within core genomic regions, often exceeding 200,000, has dramatically increased the capability of population studies, facilitating the fine-grain mapping of population genetic structure, identification of genes under selection by host immunity or drugs, and resolution of the relationships of parasites within a population or even within a single host. Population differentiation can be gauged using grouping-agnostic methods such as principal component analysis (PCA), phylogeny, pairwise IBD, and admixture. It may also be more directly studied by comparing populations using Wright’s fixation index (FST) and shared IBD (Figure 3). IBD is measured between each pair of individuals, while the average pairwise relatedness is population informative. FST is a population differentiation metric derived from allele frequencies within individual populations. Comparing shared IBD is more informative than FST for recent demographic events and datasets where parasites are closely related (Taylor et al., 2017).
Analysis of contemporary global P. vivax populations using genome-wide SNPs firmly established population differentiation among the continents and identified distinct populations from Oceania (PNG), South Asia (India), and Southeast Asia (Hupalo et al., 2016; MalariaGEN et al., 2022), with improved resolutions compared to microsatellites (Koepfli et al., 2015). Within continents, parasite populations generally conform to isolation by distance (MalariaGEN et al., 2022); populations from more distantly separated countries form distinctive clusters and correlate well to countries of origin, but parasites from neighboring countries are sometimes difficult to distinguish, even using genome-wide SNPs (De Oliveira et al., 2017; Brashear et al., 2020a). In South America, for example, population differentiation is evident between Mexico and Brazil but less clear between Peru and Colombia (Hupalo et al., 2016; De Oliveira et al., 2017). Similarly, parasites from Indonesia and Malaysia are well separated from those in the GMS (Auburn et al., 2018), but parasites from Cambodia and Vietnam are not readily differentiated, reflecting physical connectivity and frequent gene flow between the two countries (Pearson et al., 2016; Brashear et al., 2020a). Interestingly, however, the malaria-free central plain of Thailand serves as a gene flow barrier, resulting in substantial population division between parasites from the eastern and western GMS that can be distinguished with as few as four microsatellite markers (Kittichai et al., 2017). Population differences characterized by genomics can be harnessed to gauge and guide malaria control and elimination practice.
Monitoring temporal population changes using genomics
Investigations of temporal changes of parasite populations offer a way to monitor the progress of malaria control and elimination. As elimination efforts increase selection pressure on parasites and reduce gene flow, parasite populations become more fragmented and structured, even though the effective population size may not vary significantly, as demonstrated in microsatellite data from parasites spanning a decade from the South Pacific and GMS (Waltmann et al., 2018; Li et al., 2020). WGS provides higher resolution to detect population contractions, bottlenecks, introductions, and expansions, all of which may occur during malaria elimination. For example, P. vivax subpopulations showing high degrees of IBD-sharing—suggestive of a history of severe bottleneck and/or clonal expansion—were identified in sites approaching malaria elimination, including Sabah, Malaysia (Auburn et al., 2018), and Panama (Buyon et al., 2020). Likewise, WGS of P. vivax parasites from a transmission hotspot in Brazil also detected a fragmented parasite structure, with nearly half of the samples sharing over 50% of their genome in IBD segments, indicating highly inbred parasite populations (De Oliveira et al., 2020). Interestingly, these sympatric lineages observed in Brazil and the predominant lineage identified in Panama are remarkably stable over time, suggesting the existence of persistent reservoirs (Buyon et al., 2020; De Oliveira et al., 2020). In addition to significant IBD sharing, population expansion or reduction may also be reflected in Tajima’s D values – e.g., a negative value may indicate population expansion (De Oliveira et al., 2017). Depending on the local scenarios, rapidly expanded parasite lineages may represent epidemic events such as outbreaks (Brashear et al., 2020a) or evolution adaptation of the parasites to selective pressures such as drug-resistant founder populations.
Tracking parasite movement using genomics
The superb resolution of the parasite genotypes from WGS data makes the accurate tracking of the origins and movement of the parasites possible. With the ability to persist as liver hypnozoites, P. vivax and P. ovale spp. can be introduced as silent infections by migrant workers and travelers (Cao et al., 2016; Spanakos et al., 2018; Zhou et al., 2019). For non-endemic countries, genomic information can approximate where the parasites are imported from (Diez Benavente et al., 2020). In Panama, which is close to elimination, population genomics allowed accurate tracking of the origins of malaria parasites in patients with travel histories (Buyon et al., 2020). For endemic countries, malaria parasites between neighboring countries or different endemic “pockets” within a country have become increasingly divergent with the scale-up of malaria control activities. Thus, information about the sources and sinks of the parasites can be used to identify the routes of parasite movement and block parasite introduction (Buyon et al., 2020). With the shrinking of parasite populations and inbreeding within isolated populations, local parasites may become more closely related. Genetic divergence (e.g., estimated by pairwise FST) or genetic relatedness (e.g., IBD-sharing) can be compared with geographical distances by the Mantel test to identify differentiation beyond what is expected from geographic isolation alone. In one instance, the identification of P. vivax parasites with significant IBD sharing from two sites 700 km apart in the Brazilian Amazon evidenced long-distance migration (De Oliveira et al., 2020), implying the presence of “corridors” for long-distance migration of parasites as suggested from microsatellite analysis of P. vivax parasites from Colombia (Pacheco et al., 2019). For geo-referenced parasite samples across a geographic region, the spatial patterns of gene flow (or migration routes) can be visually presented using Effective Estimation of Migration Surfaces (EEMS), a tool that maps relative migration rates among samples by comparing a matrix of pairwise sample dissimilarity to pairwise distance and identifying regions where dissimilarity is higher or lower than expected (Petkova et al., 2016). In the GMS, EEMS analysis of P. falciparum and P. vivax populations using genome-wide SNP data detected central Thailand and some international borders as common migration barriers for both species (Shetty et al., 2019; Brashear et al., 2020a), although the P. vivax samples were much more spatially sparse. It is noteworthy that both IBD correlations and EEMS assume equal transmission in both directions, requiring additional analysis to detect the migration sources. MIGRATE-N, which uses Bayesian inference to output posterior probability for proposed models, has been used with microsatellite data to infer the migration direction for P. vivax between China and Myanmar (Lo et al., 2017b), within Peru (Delgado-Ratto et al., 2016), and within Ethiopia (Lo et al., 2017a), but is not computationally feasible for the large number of SNPs frequently used in WGS studies. Fine-scaled mapping of asymmetrical migration to identify sources and sinks of migrating parasites, especially on small geographical scales and across political boundaries, may help make actionable plans to prevent parasite introduction during elimination.
Detecting signatures of selection
Compared to P. falciparum, P. vivax shows much higher genetic diversity and genomic plasticity, which may enhance its ability to evade host immunity and develop resistance to chemotherapy (Neafsey et al., 2012; Hupalo et al., 2016; Pearson et al., 2016). Positive natural selection forces, such as antimalarial drugs, can lead to decreased genomic diversity at associated and linked sites, producing a “selection valley” of reduced haplotype heterozygosity. Accordingly, either a single advantageous genotype (hard sweep) or multiple adaptive genotypes (soft sweep) will rise to high frequency in the population. A good example is the hard transnational sweep of P. falciparum strains carrying PfK13 mutations and plasmepsin 2/3 amplification in the GMS, resulting from dihydroartemisinin-piperaquine selection (Imwong et al., 2017). Studies highlighting drug selection as a more recent selective force shaping P. falciparum population genetic structure suggest that this principle should also apply to P. vivax (Miotto et al., 2015). Alternatively, diversifying selection may occur at antigenic sites where host immune responses discourage highly similar antigen sequences within a population. By looking at genome composition at the population level, researchers can understand the selective pressures driving parasite evolution and design more effective strategies guiding malaria elimination.
To detect signatures of natural selection, high-density SNPs from large-scale WGS data are necessary for high-transmission areas, where the level of recombination is high and pairwise linkage disequilibrium is low (Park et al., 2012). High linkage disequilibium or notable population structure may bias many statistics, as will insufficient sample sizes. Without phenotypic data, allele frequency-derived statistics such as nucleotide diversity (π), Tajima’s D, and the ratio of non-synonymous/synonymous mutation (πN/πS) may be used to identify genes under selection. This type of analysis allows the identification of genes showing elevated genetic diversity and positive D (balancing selection, e.g., surface proteins and antigens) or negative D values (negative selection or selection sweep) (Mu et al., 2007; Amambua-Ngwa et al., 2012; Pearson et al., 2016). The ability to gauge homozygosity over long chromosomal distances with the WGS data allows the applications of haplotype-based statistics such as integrative haplotype score (iHS), cross-population extended haplotype homozygosity (XP-EHH), number of segregating sites by length (nSL), and most recently the isorelate Xir to detect selective sweep within parasite populations. Within a population, an exceptionally long IBD region is likely to have undergone fewer recombination events, suggesting a deleterious effect of mutations in that region. With this in mind, IsoRelate creates the isorelate statistic (Xir) to determine the relatedness of individuals and find regions under selection. It was successfully used to detect drug resistance genes in P. falciparum (Henden et al., 2018). However, other statistics, such as iHS and XP-EHH, take advantage of a similar concept called “extended haplotype homozygosity,” which scans for especially long shared haplotypes. nSL differs only slightly from iHS in that it is capable of using segregating sites to estimate genetic distance rather than an explicit genetic map; a study using both nSL and iHS in P. vivax found overlaps in key selection signals (Parobek et al., 2016). XP-EHH and Rsb compare extended haplotypes between two populations and is useful when two populations of interest likely have different selective pressures. In multiple P. vivax populations, signatures of selection have been identified in invasion-related genes (e.g., msp5, msp10, and ama1), AP2 transcription factors, and genes potentially related to drug resistance (Parobek et al., 2016; Auburn et al., 2019; Table 1). As P. vivax populations from different geographic regions reflect different demographic histories, such as adaptation to local hosts and vectors and different drug histories, their comparison may reveal population-specific selection pressure (Hupalo et al., 2016). The divergence between New World and Old World P. vivax populations on the Pvs47 gene, the ortholog of which in P. falciparum has been selected by the New World vectors, may likewise be due to selection by different vectors (Hupalo et al., 2016).
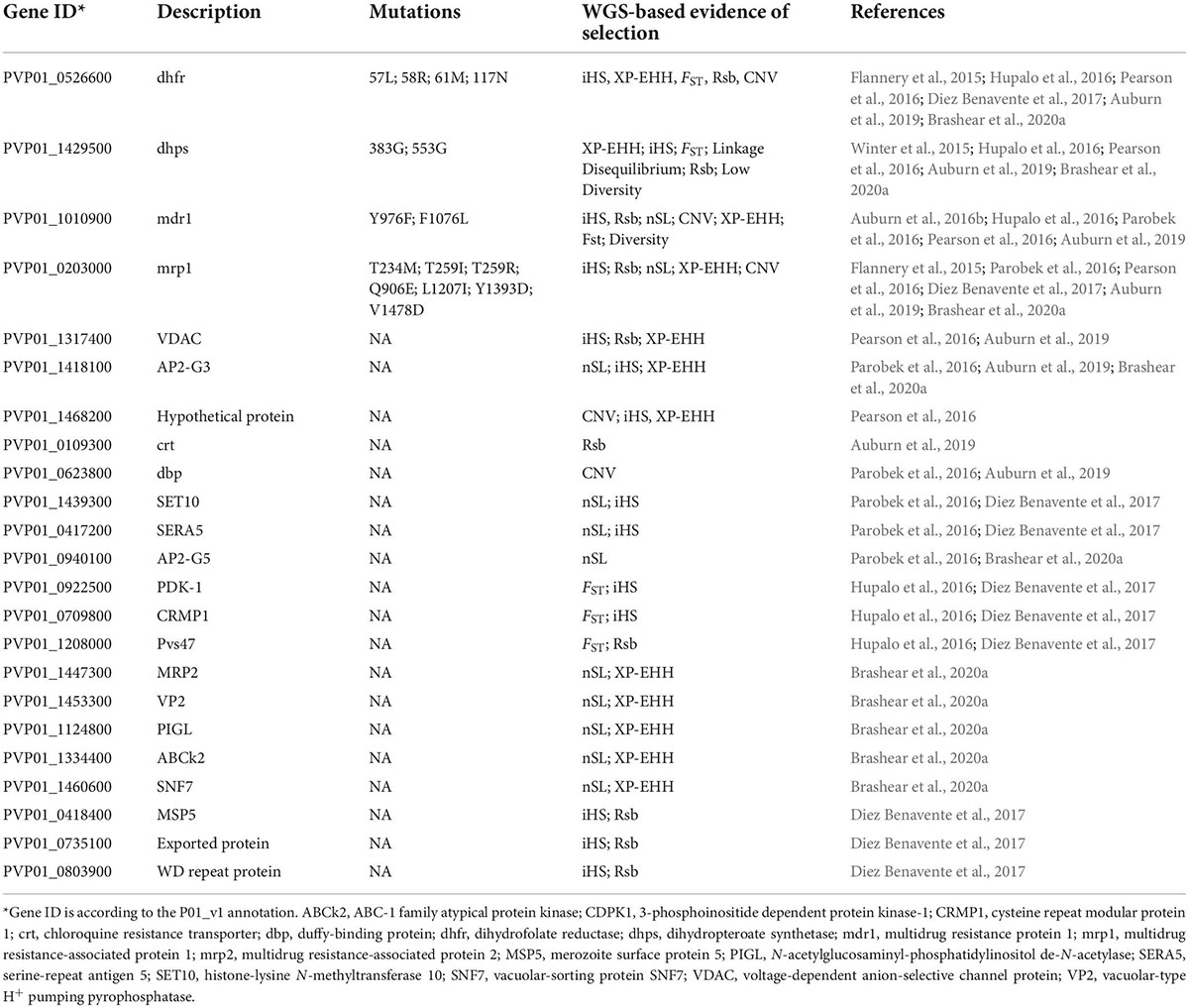
Table 1. Genes implicated as being under directional selection from population genomics studies.
Selection signals from population genomics are especially useful for neglected malaria parasites where resources are scarce and traditional means of genetics are difficult. Genetic linkage mapping using experimental crosses and genome-wide association studies (GWAS) have successfully mapped loci associated with drug resistance in P. falciparum (Su et al., 2007; Volkman et al., 2017). To date, no GWAS has been carried out for the neglected parasites, although drug resistance phenotypes can be obtained from clinical studies or ex vivo drug assays. Clinical phenotypic data are typically more variable in nature, necessitating larger sample sizes to find genuine associations. Ex vivo assays are more controllable but labor intensive, particularly for samples that are not easily cultured. Sá et al. (2019) recently overcame tremendous barriers to perform a genetic cross of two subpopulations of an NIH-1993 line of P. vivax in a splenectomized chimpanzee. By comparing the prevalence of 37 polymorphic markers from chloroquine (CQ)-resistant and –sensitive parental strains before and after CQ administration, the authors calculated an effect e-value for each marker and identified those on chromosome 1 with consistently high e-values across progeny pairs. Targeted sequencing on chromosome 1 and transcriptomic analysis suggested that CQ resistance may be mediated by changes in pvcrt transcription resulting from an upstream motif. While sample sizes for these analyses were small (4–10 linkage group selection pairs), and it is uncertain how mutations in primate-adapted strains may affect the results, they represent an important step forward in phenotypic analyses in P. vivax. In general, however, the lack of a long-term culture system and the need for non-human primates make it less feasible to apply phenotypic assays to genetic mapping in neglected malaria parasites. Therefore, we rely heavily on population genomics to infer underlying resistance mechanisms.
Signatures of selection between populations may reflect different drug histories and provide hints about the mechanisms of drug resistance in this parasite. Strong selective sweeps have been detected in genomic regions harboring Pvdhps and Pvdhfr, mediating resistance to the antifolate drugs sulfadoxine and pyrimethamine (SP), respectively (Winter et al., 2015; Hupalo et al., 2016; Parobek et al., 2016; Pearson et al., 2016; Brashear et al., 2020a). These genes also show significant geographic divergence between the New World and Old World, between PNG and Thailand, and even between geographically adjacent countries, reflecting the varied histories of SP as a treatment for sympatric P. falciparum (Hupalo et al., 2016; Pearson et al., 2016; Earland et al., 2019; Brashear et al., 2020a). Of note, some of the mutations in PvDHFR and PvDHPS are relatively rare in South and Central American P. vivax populations (De Oliveira et al., 2017). CQ resistance (CQR) has been detected in P. vivax populations from many endemic regions, but the resistance mechanism is not understood and appears different from P. falciparum (Price et al., 2014). Unlike the selective sweep found at the pfcrt gene, the major determinant of CQR in P. falciparum, no selection signals were identified at the pvcrt locus (Parobek et al., 2016; Pearson et al., 2016) except an extended haplotype upstream of pvcrt differing between Thai and Ethiopian P. vivax populations (Auburn et al., 2019). This corroborates findings from a genetic cross showing that pvcrt expression levels in the CQ-sensitive and -resistant parasites were correlated with variations in the pvcrt upstream sequence motifs, potential sites for transcription factors (Sá et al., 2019). In this context, positive selection in AP2-domain transcription factors may suggest their involvement in regulating the expression of drug resistance genes (Dharia et al., 2010; Parobek et al., 2016; Auburn et al., 2019). Pvmdr1, which emerged as a potential CQR marker during ex vivo studies (Suwanarusk et al., 2007), has displayed substantial population-level variation at major mutations (Y976F and F1076L). Both mutations were fixed in Indonesia, where high-grade CQR is found, with low to modest prevalence in the GMS (Auburn et al., 2019; Brashear et al., 2020a). In P. vivax from Indonesia, a strong selection signal was also identified downstream of, but not overlapping with, pvmdr1 (Pearson et al., 2016). In addition, population genomic studies of global P. vivax populations have identified signals of selection in other chromosomal regions, including around genes encoding putative transporters such as multidrug resistance-associated protein 1 (pvmrp1) and 2 (pvmrp2) (Dharia et al., 2010; Flannery et al., 2015; Parobek et al., 2016; Pearson et al., 2016; Auburn et al., 2019; Brashear et al., 2020a).
Copy number variations (CNVs) may also differ among populations and potentially lead to drug resistance. CNVs with a marked geographical difference in frequency were identified between P. vivax populations from Oceania and the GMS, some of which harbor genes related to drug resistance (Pearson et al., 2016). Among them, pvmdr1 amplification, previously associated with mefloquine resistance (Suwanarusk et al., 2008), had ∼20% prevalence in Thailand (which has extensively deployed mefloquine to treat P. falciparum) but was absent in Indonesia or Ethiopia (Auburn et al., 2016b,2019; Pearson et al., 2016). A short 3-kb duplication on chromosome 14, including the gene PVX_101445, was found only in Oceania parasites (Pearson et al., 2016). Detecting CNVs from short-read data can be tricky due to mismapping, especially if reference genomes are not complete, while methods incorporating discordant reads and/or read depth have found some success. Genome amplification techniques prior to sequencing (e.g., SWGA) can worsen this technical concern, while long-read sequencing data is considered more reliable for CNV detection.
Population genomic studies have identified various genes under selection, some of which may mediate drug resistance, but the lack of a long-term P. vivax culture or convenient animal models for genetic manipulation precludes direct evaluations of their roles in drug resistance. The expression of these identified P. vivax genes in transgenic P. knowlesi (Verzier et al., 2019), a closely related species that can be cultured in human RBCs, and adaptation of the marker-free CRISPR gene-editing technology in P. knowlesi (Mohring et al., 2020), have opened a new way for downstream functional analysis of P. vivax genes. Incorporating population genomics into the study of neglected malaria parasites may provide insights into the nature of the selective pressure.
Using single nucleotide polymorphism barcodes for high-throughput genotyping
Even with the continuous reduction of WGS costs, it is still prohibitively expensive to perform large-scale population genomic studies of malaria parasites. Yet, the availability of WGS data of parasites from various endemic areas such as those generated from the MalariaGEN Consortium has informed researchers to design ‘genetic barcodes’, which offer a reductionist approach to inferring transmission networks and population structures by using a small set of SNPs (Daniels et al., 2008; Preston et al., 2014; Baniecki et al., 2015). Barcode SNPs are selected as highly differentiated among subpopulations and can be affordably genotyped from small amounts of blood such as dried blood spots using various platforms such as the high resolution melting analysis (Baniecki et al., 2015), molecular inversion probes (Verity et al., 2020), or the Sequenom platform (Omedo et al., 2017). The P. falciparum barcode consisting of 24 SNPs (Daniels et al., 2008; Preston et al., 2014) has proved to be applicable to studying parasite populations in a range of endemic settings of the world (Auburn and Barry, 2017), although a much larger SNP set may be needed for the resolution of global parasite populations and IBD analysis. The barcodes for P. vivax populations appear to require more SNPs, likely due to higher genetic diversity (Neafsey et al., 2012). A set of 42 SNPs was initially designed based on a limited number of P. vivax genomes available at that time (Baniecki et al., 2015). Though the 42-SNP barcode can discriminate parasites from different continents, it was insufficient to differentiate parasite populations from South and Central America or countries of Southeast Asia (De Oliveira et al., 2017; Brashear et al., 2020a). The addition of 37 out of 42 SNPs from the initial barcode to a set of 28 sites selected with machine learning improved its resolution for country-level classification with more flexibility for missing data (Trimarsanto et al., 2019). Recently, as P. vivax WGS data have become available from more geographic regions, a new 71-SNP barcode has been designed, allowing the geographic origins of parasites to be predicted at 91.4% accuracy (Diez Benavente et al., 2020). Though this new barcode remains to be tested, because barcodes are designed for specific purposes based on variability expected in that dataset, barcodes produced from global datasets often underperform in local and regional analyses, prompting additional barcodes for more targeted use in tracking transmission within countries or between neighboring countries. For example, barcodes based on 100 SNPs and 36 SNPs were shown to be able to differentiate parasites in South and Central Americas and the GMS, respectively (De Oliveira et al., 2017; Brashear et al., 2020a), while their performance remains to be evaluated. Establishing the origin of samples is important when determining malaria-free status by the WHO (World Health Organization [WHO], 2021), but it is important to consider the purpose and scope of a barcode in use.
Implications for zoonotic human-infecting malaria parasites
Zoonotic malaria species infecting humans such as P. knowlesi, P. cynomolgi, and P. simium are increasingly recognized as additional targets for malaria elimination. Malaysia has been cleared of human malaria since 2018 but continues to report thousands of human cases of P. knowlesi (World Health Organization [WHO], 2021). As global deforestation continues, the displacement of primate species that carry zoonotic malaria may further increase the risk of transmission to humans (Stark et al., 2019). Genomic studies of the zoonotic species are also challenging, and the parasites infecting humans may be genetically divergent from those infecting their natural non-human primate hosts (Assefa et al., 2015). The adaptation of zoonotic species to in vitro culture in human RBCs has opened new venues to study these parasite species in the lab, which may serve as model systems for functional genomics of their closely related human malaria species, such as P. vivax (Moon et al., 2013; Chua et al., 2019). In 2008, P. knowlesi from a rhesus macaque was sequenced, revealing a 23.5 Mb nuclear genome (Pain et al., 2008). Subsequent long-read sequencing on the PacBio platform with hi-C correction improved the annotation of the P. knowlesi genome of 24.77 Mb to consist of 14 contigs with 25 gaps (Lapp et al., 2018). Recently, the Oxford nanopore long-read sequencing has opened the door to de novo assemblies of clinical field samples (Oresegun et al., 2022). P. cynomolgi was sequenced in 2012, resulting in three draft genomes (Tachibana et al., 2012); the assembly was later improved to have 56 contigs with no gaps, accounting for 6632 genes (Pasini et al., 2017). The P. simium draft genome was only recently sequenced, containing over 2000 scaffolds (Mourier et al., 2021). Improvement in the quality and annotation of these reference genomes will benefit future functional and population genomic studies.
Population genomics techniques have a clear benefit in neglected Plasmodium species due to the ability to amass a large amount of data with few samples and little a priori knowledge. Of the zoonotic species, population genomics studies have been undertaken primarily on P. knowlesi due to the large number of human infections occurring in Malaysia (Divis et al., 2021). Most notably, these studies have provided clear evidence that P. knowlesi populations are structured not just geographically and between clinical and laboratory samples but also by primate hosts (Assefa et al., 2015; Hocking et al., 2020). The original division between two known monkey hosts—the pig-tailed macaque and the long-tailed macaque—was somewhat intuitive (Assefa et al., 2015). A recent finding that peninsular samples actually had three genomic subpopulations in overlapping geographic regions (Hocking et al., 2020) leaves the possibility that there may be additional host reservoirs or additional factors contributing to parasite transmission. High transmission intensity in monkey hosts leads to high genomic and within-host diversity (Lee et al., 2011; Assefa et al., 2015). In P. simium, advancement in population genomics includes compelling evidence that the species diverged from P. vivax in a rare occurrence of reverse zoonosis (De Oliveira et al., 2021; Mourier et al., 2021). Analysis of 11 P. simium samples revealed high genomic similarity to new-world P. vivax samples, but not to old-world P. vivax samples (De Oliveira et al., 2021), supporting the hypothesis that P. vivax spread to the Americas and diverged from its Asian cousins before adapting to local macaques. Finally, very little genomic research has been done with P. cynomolgi field populations. The development of genomic tools and resources for studying zoonotic malaria species may provide crucial findings on the parasite’s host adaptation and the biology of closely related human parasites.
Conclusion
With technical improvements to enrich parasite genomes from very small amounts of DNA, the number of genomes of the neglected malaria parasites sequenced is rapidly growing, providing a new means to address many aspects of parasite biology. High-coverage WGS paves the way to precisely determine the relatedness of recurring parasites with parasites from the initial infection, setting up a platform for studying the biology of relapses (Cowell et al., 2018; Popovici et al., 2018). Population genomics provides a more accurate way to infer the connectivity and ancestry of the parasite populations. Under the malaria elimination scenario, analysis of longitudinal samples enables the detection of population events such as contraction, bottleneck, and expansion, allowing close monitoring of the malaria elimination progress (Auburn et al., 2018; Buyon et al., 2020). The availability of parasite genomes from many endemic regions globally and the genomic resolution of parasite populations allowed the conclusive detection of parasite introduction (Buyon et al., 2020), which is critical for areas approaching and achieving elimination status. Finally, identifying genomic regions and genes under selection provides drug resistance candidate markers for future evaluation (Pearson et al., 2016).
Population genomics of neglected malaria parasites offers a new frontier of malaria research. While the malaria community has recently generated an impressive number of over 2200 whole-genome sequences for P. vivax (Supplementary Table 1), the studies on other neglected malaria parasites have just begun (Ibrahim et al., 2020). In P. ovale spp. and P. malariae, researchers would benefit immensely from large numbers of genomic data collected from various populations to hasten our understanding of their epidemiology and biology. Insights into how they transmit and recur or relapse in different settings will become especially important as P. falciparum decreases and other species stand in the way of malaria elimination. In simian species, including P. knowlesi, P. cynomolgi, and P. simium, population genomics may give insights into their transmission and possible additional hosts, and may even help researchers assess the threat of human transmission. For P. vivax, increased sample acquisition on finer geographical scales remains an important objective, especially for tracking regional parasite movement. We still lack P. vivax genomes from west Africa, where Duffy negativity is most prevalent. Additional locations where there are relatively large numbers of cases but relatively few WGS samples include the areas between Bangladesh and Myanmar, and northeastern South America, specifically around Venezuela (Figure 1 and Supplementary Table 1). Furthermore, with technological breakthroughs in WGS, better genome assemblies from long-read sequencing may be realized soon (Rutledge et al., 2017a). Improved reference genomes will improve population genomic studies in neglected malaria parasites, serving haplotype reference panels for phasing inference and genetic maps for models reliant on genetic distance. We envision that the increasing popularity of population genomics of neglected malaria parasites will translate into a genomic surveillance practice in malaria elimination.
Author contributions
AB and LC conceived, drafted, revised, and gave final approval to the article. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institute for Allergy and Infectious Diseases, The National Institute of Health (NIH) (U19 AI089672).
Acknowledgments
We would like to thank the reviewers of this manuscript who gave constructive feedback that ultimately improved this manuscript in terms of content and clarity.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.984394/full#supplementary-material
Supplementary Table 1 | Whole-genome sequences available for P. vivax grouped by study and location.
References
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. doi: 10.1101/gr.114876.110
Amambua-Ngwa, A., Tetteh, K. K., Manske, M., Gomez-Escobar, N., Stewart, L. B., Deerhake, M. E., et al. (2012). Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PLoS Genet. 8:e1002992. doi: 10.1371/journal.pgen.1002992
Ansari, H. R., Templeton, T. J., Subudhi, A. K., Ramaprasad, A., Tang, J., Lu, F., et al. (2016). Genome-scale comparison of expanded gene families in Plasmodium ovale wallikeri and Plasmodium ovale curtisi with Plasmodium malariae and with other Plasmodium species. Int. J. Parasitol. 46, 685–696. doi: 10.1016/j.ijpara.2016.05.009
Anstey, N. M., Russell, B., Yeo, T. W., and Price, R. N. (2009). The pathophysiology of vivax malaria. Trends Parasitol. 25, 220–227.
Antinori, S., Galimberti, L., Milazzo, L., and Corbellino, M. (2013). Plasmodium knowlesi: The emerging zoonotic malaria parasite. Acta Trop. 125, 191–201. doi: 10.1016/j.actatropica.2012.10.008
Arnott, A., Barry, A. E., and Reeder, J. C. (2012). Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar. J. 11:14. doi: 10.1186/1475-2875-11-14
Assefa, S., Lim, C., Preston, M. D., Duffy, C. W., Nair, M. B., Adroub, S. A., et al. (2015). Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc. Natl. Acad. Sci. U.S.A. 112, 13027–13032. doi: 10.1073/pnas.1509534112
Assefa, S. A., Preston, M. D., Campino, S., Ocholla, H., Sutherland, C. J., and Clark, T. G. (2014). estMOI: Estimating multiplicity of infection using parasite deep sequencing data. Bioinformatics 30, 1292–1294. doi: 10.1093/bioinformatics/btu005
Auburn, S., and Barry, A. E. (2017). Dissecting malaria biology and epidemiology using population genetics and genomics. Int. J. Parasitol. 47, 77–85.
Auburn, S., Benavente, E. D., Miotto, O., Pearson, R. D., Amato, R., Grigg, M. J., et al. (2018). Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics. Nat. Commun. 9:2585. doi: 10.1038/s41467-018-04965-4
Auburn, S., Bohme, U., Steinbiss, S., Trimarsanto, H., Hostetler, J., Sanders, M., et al. (2016a). A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 1:4. doi: 10.12688/wellcomeopenres.9876.1
Auburn, S., Serre, D., Pearson, R. D., Amato, R., Sriprawat, K., To, S., et al. (2016b). Genomic analysis reveals a common breakpoint in amplifications of the Plasmodium vivax multidrug resistance 1 Locus in Thailand. J. Infect. Dis. 214, 1235–1242. doi: 10.1093/infdis/jiw323
Auburn, S., Campino, S., Clark, T. G., Djimde, A. A., Zongo, I., Pinches, R., et al. (2011). An effective method to purify Plasmodium falciparum DNA directly from clinical blood samples for whole genome high-throughput sequencing. PLoS One 6:e22213. doi: 10.1371/journal.pone.0022213
Auburn, S., Getachew, S., Pearson, R. D., Amato, R., Miotto, O., Trimarsanto, H., et al. (2019). Genomic analysis of Plasmodium vivax in southern ethiopia reveals selective pressures in multiple parasite mechanisms. J. Infect. Dis. 220, 1738–1749. doi: 10.1093/infdis/jiz016
Auburn, S., Marfurt, J., Maslen, G., Campino, S., Ruano Rubio, V., Manske, M., et al. (2013). Effective preparation of Plasmodium vivax field isolates for high-throughput whole genome sequencing. PLoS One 8:e53160. doi: 10.1371/journal.pone.0053160
Baird, J. K. (2013). Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin. Microbiol. Rev. 26, 36–57.
Balasubramanian, S., Rahman, R. S., Lon, C., Parobek, C., Ubalee, R., Hathaway, N., et al. (2020). Efficient transmission of mixed Plasmodium falciparum/vivax infections from humans to mosquitoes. J. Infect. Dis. 221, 428–437. doi: 10.1093/infdis/jiz388
Baniecki, M. L., Faust, A. L., Schaffner, S. F., Park, D. J., Galinsky, K., Daniels, R. F., et al. (2015). Development of a single nucleotide polymorphism barcode to genotype Plasmodium vivax infections. PLoS Negl. Trop. Dis. 9:e0003539. doi: 10.1371/journal.pntd.0003539
Beghain, J., Langlois, A. C., Legrand, E., Grange, L., Khim, N., Witkowski, B., et al. (2016). Plasmodium copy number variation scan: Gene copy numbers evaluation in haploid genomes. Malar. J. 15:206. doi: 10.1186/s12936-016-1258-x
Brashear, A. M., Huckaby, A. C., Fan, Q., Dillard, L. J., Hu, Y., Li, Y., et al. (2020b). New Plasmodium vivax genomes from the China-Myanmar border. Front. Microbiol. 11:1930. doi: 10.3389/fmicb.2020.01930
Brashear, A. M., Fan, Q., Hu, Y., Li, Y., Zhao, Y., Wang, Z., et al. (2020a). Population genomics identifies a distinct Plasmodium vivax population on the China-Myanmar border of Southeast Asia. PLoS Negl. Trop. Dis. 14:e0008506. doi: 10.1371/journal.pntd.0008506
Brashear, A. M., Roobsoong, W., Siddiqui, F. A., Nguitragool, W., Sattabongkot, J., Lopez-Uribe, M. M., et al. (2019). A glance of the blood stage transcriptome of a Southeast Asian Plasmodium ovale isolate. PLoS Negl. Trop. Dis. 13:e0007850. doi: 10.1371/journal.pntd.0007850
Brasil, P., Zalis, M. G., De Pina-Costa, A., Siqueira, A. M., Junior, C. B., Silva, S., et al. (2017). Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: A molecular epidemiological investigation. Lancet Glob. Health e1038–e1046. doi: 10.1016/S2214-109X(17)30333-9
Bright, A. T., Manary, M. J., Tewhey, R., Arango, E. M., Wang, T., Schork, N. J., et al. (2014). A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl. Trop. Dis. 8:e2882. doi: 10.1371/journal.pntd.0002882
Bright, A. T., Tewhey, R., Abeles, S., Chuquiyauri, R., Llanos-Cuentas, A., Ferreira, M. U., et al. (2012). Whole genome sequencing analysis of Plasmodium vivax using whole genome capture. BMC Genomics 13:262. doi: 10.1186/1471-2164-13-262
Buyon, L. E., Santamaria, A. M., Early, A. M., Quijada, M., Barahona, I., Lasso, J., et al. (2020). Population genomics of Plasmodium vivax in Panama to assess the risk of case importation on malaria elimination. PLoS Negl. Trop. Dis. 14:e0008962. doi: 10.1371/journal.pntd.0008962
Cao, Y., Wang, W., Liu, Y., Cotter, C., Zhou, H., Zhu, G., et al. (2016). The increasing importance of Plasmodium ovale and Plasmodium malariae in a malaria elimination setting: An observational study of imported cases in Jiangsu Province, China, 2011-2014. Malar. J. 15:459. doi: 10.1186/s12936-016-1504-2
Carlton, J. M., Adams, J. H., Silva, J. C., Bidwell, S. L., Lorenzi, H., Caler, E., et al. (2008). Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763. doi: 10.1038/nature07327
Chan, E. R., Menard, D., David, P. H., Ratsimbasoa, A., Kim, S., Chim, P., et al. (2012). Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl. Trop. Dis. 6:e1811. doi: 10.1371/journal.pntd.0001811
Chang, H. H., Worby, C. J., Yeka, A., Nankabirwa, J., Kamya, M. R., Staedke, S. G., et al. (2017). THE REAL McCOIL: A method for the concurrent estimation of the complexity of infection and SNP allele frequency for malaria parasites. PLoS Comput. Biol. 13:e1005348. doi: 10.1371/journal.pcbi.1005348
Chen, N., Auliff, A., Rieckmann, K., Gatton, M., and Cheng, Q. (2007). Relapses of Plasmodium vivax infection result from clonal hypnozoites activated at predetermined intervals. J. Infect. Dis. 195, 934–941. doi: 10.1086/512242
Chua, A. C. Y., Ong, J. J. Y., Malleret, B., Suwanarusk, R., Kosaisavee, V., Zeeman, A. M., et al. (2019). Robust continuous in vitro culture of the Plasmodium cynomolgi erythrocytic stages. Nat. Commun. 10:3635.
Collins, W. E., and Jeffery, G. M. (2007). Plasmodium malariae: Parasite and disease. Clin. Microbiol. Rev. 20, 579–592.
Cotter, C., Sturrock, H. J., Hsiang, M. S., Liu, J., Phillips, A. A., Hwang, J., et al. (2013). The changing epidemiology of malaria elimination: New strategies for new challenges. Lancet 382, 900–911.
Cowell, A. N., Loy, D. E., Sundararaman, S. A., Valdivia, H., Fisch, K., Lescano, A. G., et al. (2017). Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. MBio 8, e2257–e2216. doi: 10.1128/mBio.02
Cowell, A. N., Valdivia, H. O., Bishop, D. K., and Winzeler, E. A. (2018). Exploration of Plasmodium vivax transmission dynamics and recurrent infections in the Peruvian Amazon using whole genome sequencing. Genome Med. 10:52. doi: 10.1186/s13073-018-0563-0
Daniels, R., Volkman, S. K., Milner, D. A., Mahesh, N., Neafsey, D. E., Park, D. J., et al. (2008). A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar. J. 7, 223–223. doi: 10.1186/1475-2875-7-223
De Oliveira, T. C., Corder, R. M., Early, A., Rodrigues, P. T., Ladeia-Andrade, S., Alves, J. M. P., et al. (2020). Population genomics reveals the expansion of highly inbred Plasmodium vivax lineages in the main malaria hotspot of Brazil. PLoS Negl. Trop. Dis. 14:e0008808. doi: 10.1371/journal.pntd.0008808
De Oliveira, T. C., Rodrigues, P. T., Early, A. M., Duarte, A. M. R. C., Buery, J. C., Bueno, M. G., et al. (2021). Plasmodium simium: Population genomics reveals the origin of a reverse zoonosis. J. Infect. Dis. 224, 1950–1961. doi: 10.1093/infdis/jiab214
De Oliveira, T. C., Rodrigues, P. T., Menezes, M. J., Goncalves-Lopes, R. M., Bastos, M. S., Lima, N. F., et al. (2017). Genome-wide diversity and differentiation in New World populations of the human malaria parasite Plasmodium vivax. PLoS Negl. Trop. Dis. 11:e0005824. doi: 10.1371/journal.pntd.0005824
Delgado-Ratto, C., Gamboa, D., Soto-Calle, V. E., Van Den Eede, P., Torres, E., Sánchez-Martínez, L., et al. (2016). Population genetics of Plasmodium vivax in the Peruvian Amazon. PLoS Negl. Trop. Dis. 10:e0004376. doi: 10.1371/journal.pntd.0004376
Dharia, N. V., Bright, A. T., Westenberger, S. J., Barnes, S. W., Batalov, S., Kuhen, K., et al. (2010). Whole-genome sequencing and microarray analysis of ex vivo Plasmodium vivax reveal selective pressure on putative drug resistance genes. Proc. Natl. Acad. Sci. U.S.A. 107, 20045–20050. doi: 10.1073/pnas.1003776107
Diez Benavente, E., Campos, M., Phelan, J., Nolder, D., Dombrowski, J. G., Marinho, C. R. F., et al. (2020). A molecular barcode to inform the geographical origin and transmission dynamics of Plasmodium vivax malaria. PLoS Genet. 16:e1008576. doi: 10.1371/journal.pgen.1008576
Diez Benavente, E., Ward, Z., Chan, W., Mohareb, F. R., Sutherland, C. J., Roper, C., et al. (2017). Genomic variation in Plasmodium vivax malaria reveals regions under selective pressure. PLoS One 12:e0177134. doi: 10.1371/journal.pone.0177134
Divis, P. C. S., Singh, B., and Conway, D. J. (2021). “Chapter Five – Molecular epidemiology and population genomics of Plasmodium knowlesi,” in Advances in parasitology, ed. C. Drakeley (Cambridge, MA: Academic Press), 191–223.
Durand, S., Cabezas, C., Lescano, A. G., Galvez, M., Gutierrez, S., Arrospide, N., et al. (2014). Efficacy of three different regimens of primaquine for the prevention of relapses of Plasmodium vivax malaria in the Amazon Basin of Peru. Am. J. Trop. Med. Hyg. 91, 18–26. doi: 10.4269/ajtmh.13-0053
Earland, D., Buchwald, A. G., Sixpence, A., Chimenya, M., Damson, M., Seydel, K. B., et al. (2019). Impact of multiplicity of Plasmodium falciparum infection on clinical disease in Malawi. Am. J. Trop. Med. Hyg. 101, 412–415. doi: 10.4269/ajtmh.19-0093
Escalante, A. A., and Pacheco, M. A. (2019). Malaria molecular epidemiology: An evolutionary genetics perspective. Microbiol. Spectr. 7:10.1 128/microbiolsec.AME–0010–2019. doi: 10.1128/microbiolspec.AME-0010-2019
Flannery, E. L., Wang, T., Akbari, A., Corey, V. C., Gunawan, F., Bright, A. T., et al. (2015). Next-generation sequencing of patient samples shows evidence of direct evolution in drug-resistance genes. ACS Infect. Dis. 1, 367–379. doi: 10.1021/acsinfecdis.5b00049
Fola, A. A., Harrison, G. L. A., Hazairin, M. H., Barnadas, C., Hetzel, M. W., Iga, J., et al. (2017). Higher complexity of infection and genetic diversity of Plasmodium vivax Than Plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am. J. Trop. Med. Hyg. 96, 630–641. doi: 10.4269/ajtmh.16-0716
Ford, A., Kepple, D., Abagero, B. R., Connors, J., Pearson, R., Auburn, S., et al. (2020). Whole genome sequencing of Plasmodium vivax isolates reveals frequent sequence and structural polymorphisms in erythrocyte binding genes. PLoS Negl. Trop. Dis. 14:e0008234. doi: 10.1371/journal.pntd.0008234
Frech, C., and Chen, N. (2013). Variant surface antigens of malaria parasites: Functional and evolutionary insights from comparative gene family classification and analysis. BMC Genomics 14:427. doi: 10.1186/1471-2164-14-427
Galinsky, K., Valim, C., Salmier, A., De Thoisy, B., Musset, L., Legrand, E., et al. (2015). COIL: A methodology for evaluating malarial complexity of infection using likelihood from single nucleotide polymorphism data. Malar. J. 14:4. doi: 10.1186/1475-2875-14-4
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R. W., et al. (2002). Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511.
Geng, J., Malla, P., Zhang, J., Xu, S., Li, C., Zhao, Y., et al. (2019). Increasing trends of malaria in a border area of the Greater Mekong Subregion. Malar. J. 18:309. doi: 10.1186/s12936-019-2924-6
Gething, P. W., Casey, D. C., Weiss, D. J., Bisanzio, D., Bhatt, S., Cameron, E., et al. (2016). Mapping plasmodium falciparum mortality in africa between 1990 and 2015. N. Engl. J. Med. 375, 2435–2445.
Ginsburg, H., and Abdel-Haleem, A. M. (2016). Malaria parasite metabolic pathways (MPMP) upgraded with targeted chemical compounds. Trends Parasitol. 32, 7–9. doi: 10.1016/j.pt.2015.10.003
Gunalan, K., Lo, E., Hostetler, J. B., Yewhalaw, D., Mu, J., Neafsey, D. E., et al. (2016). Role of Plasmodium vivax duffy-binding protein 1 in invasion of Duffy-null Africans. Proc. Natl. Acad. Sci. U.S.A. 113, 6271–6276. doi: 10.1073/pnas.1606113113
Henden, L., Lee, S., Mueller, I., Barry, A., and Bahlo, M. (2018). Identity-by-descent analyses for measuring population dynamics and selection in recombining pathogens. PLoS Genet. 14:e1007279. doi: 10.1371/journal.pgen.1007279
Hester, J., Chan, E. R., Menard, D., Mercereau-Puijalon, O., Barnwell, J., Zimmerman, P. A., et al. (2013). De novo assembly of a field isolate genome reveals novel Plasmodium vivax erythrocyte invasion genes. PLoS Negl. Trop. Dis. 7:e2569. doi: 10.1371/journal.pntd.0002569
Hocking, S. E., Divis, P. C. S., Kadir, K. A., Singh, B., and Conway, D. J. (2020). Population genomic structure and recent evolution of Plasmodium knowlesi, Peninsular Malaysia. Emerg. Infect. Dis. 26, 1749–1758. doi: 10.3201/eid2608.190864
Hostetler, J. B., Lo, E., Kanjee, U., Amaratunga, C., Suon, S., Sreng, S., et al. (2016). Independent origin and global distribution of distinct Plasmodium vivax Duffy binding protein gene duplications. PLoS Negl. Trop. Dis. 10:e0005091. doi: 10.1371/journal.pntd.0005091
Howes, R. E., Battle, K. E., Mendis, K. N., Smith, D. L., Cibulskis, R. E., Baird, J. K., et al. (2016). Global epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 95, 15–34.
Hupalo, D. N., Luo, Z., Melnikov, A., Sutton, P. L., Rogov, P., Escalante, A., et al. (2016). Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 48, 953–958. doi: 10.1038/ng.3588
Ibrahim, A., Diez Benavente, E., Nolder, D., Proux, S., Higgins, M., Muwanguzi, J., et al. (2020). Selective whole genome amplification of Plasmodium malariae DNA from clinical samples reveals insights into population structure. Sci. Rep. 10:10832. doi: 10.1038/s41598-020-67568-4
Imwong, M., Boel, M. E., Pagornrat, W., Pimanpanarak, M., Mcgready, R., Day, N. P., et al. (2012). The first Plasmodium vivax relapses of life are usually genetically homologous. J. Infect. Dis. 205, 680–683. doi: 10.1093/infdis/jir806
Imwong, M., Snounou, G., Pukrittayakamee, S., Tanomsing, N., Kim, J. R., Nandy, A., et al. (2007). Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J. Infect. Dis. 195, 927–933. doi: 10.1086/512241
Imwong, M., Suwannasin, K., Kunasol, C., Sutawong, K., Mayxay, M., Rekol, H., et al. (2017). The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: A molecular epidemiology observational study. Lancet Infect. Dis. 17, 491–497. doi: 10.1016/S1473-3099(17)30048-8
Kepple, D., Hubbard, A., Ali, M. M., Abargero, B. R., Lopez, K., Pestana, K., et al. (2021). Plasmodium vivax from Duffy-negative and Duffy-positive individuals shares similar gene pool in east Africa. J. Infect. Dis. 224, 1422–1431. doi: 10.1093/infdis/jiab063
Kittichai, V., Koepfli, C., Nguitragool, W., Sattabongkot, J., and Cui, L. (2017). Substantial population structure of Plasmodium vivax in Thailand facilitates identification of the sources of residual transmission. PLoS Negl. Trop. Dis. 11:e0005930. doi: 10.1371/journal.pntd.0005930
Koepfli, C., Rodrigues, P. T., Antao, T., Orjuela-Sanchez, P., Van Den Eede, P., Gamboa, D., et al. (2015). Plasmodium vivax diversity and population structure across four continents. PLoS Negl. Trop. Dis. 9:e0003872. doi: 10.1371/journal.pntd.0003872
Kotepui, M., Kotepui, K. U., Milanez, G. D., and Masangkay, F. R. (2020). Global prevalence and mortality of severe Plasmodium malariae infection: A systematic review and meta-analysis. Malar. J. 19:274.
Langford, S., Douglas, N. M., Lampah, D. A., Simpson, J. A., Kenangalem, E., Sugiarto, P., et al. (2015). Plasmodium malariae infection associated with a high burden of anemia: A hospital-based surveillance study. PLoS Negl. Trop. Dis. 9:e0004195. doi: 10.1371/journal.pntd.0004195
Lapp, S. A., Geraldo, J. A., Chien, J. T., Ay, F., Pakala, S. B., Batugedara, G., et al. (2018). PacBio assembly of a Plasmodium knowlesi genome sequence with Hi-C correction and manual annotation of the SICAvar gene family. Parasitology 145, 71–84. doi: 10.1017/S0031182017001329
Lau, Y. L., Lee, W. C., Tan, L. H., Kamarulzaman, A., Syed Omar, S. F., Fong, M. Y., et al. (2013). Acute respiratory distress syndrome and acute renal failure from Plasmodium ovale infection with fatal outcome. Malar. J. 12:389. doi: 10.1186/1475-2875-12-389
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 15:R84. doi: 10.1186/gb-2014-15-6-r84
Lee, K. S., Divis, P. C., Zakaria, S. K., Matusop, A., Julin, R. A., Conway, D. J., et al. (2011). Plasmodium knowlesi: Reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 7:e1002015. doi: 10.1371/journal.ppat.1002015
Leichty, A. R., and Brisson, D. (2014). Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics 198, 473–481. doi: 10.1534/genetics.114.165498
Li, Y., Hu, Y., Zhao, Y., Wang, Q., Ngassa Mbenda, H. G., Kittichai, V., et al. (2020). Dynamics of Plasmodium vivax populations in border areas of the Greater Mekong sub-region during malaria elimination. Malar. J. 19:145. doi: 10.1186/s12936-020-03221-9
Lim, C., Pereira, L., Saliba, K. S., Mascarenhas, A., Maki, J. N., Chery, L., et al. (2016). Reticulocyte preference and stage development of Plasmodium vivax isolates. J. Infect. Dis. 214, 1081–1084. doi: 10.1093/infdis/jiw303
Lin, J. T., Hathaway, N. J., Saunders, D. L., Lon, C., Balasubramanian, S., Kharabora, O., et al. (2015). Using amplicon deep sequencing to detect genetic signatures of Plasmodium vivax relapse. J. Infect. Dis. 212, 999–1008. doi: 10.1093/infdis/jiv142
Lo, E., Lam, N., Hemming-Schroeder, E., Nguyen, J., Zhou, G., Lee, M.-C., et al. (2017b). Frequent spread of Plasmodium vivax malaria maintains high genetic diversity at the Myanmar-China Border, without distance and landscape barriers. J. Infecti. Dis. 216, 1254–1263. doi: 10.1093/infdis/jix106
Lo, E., Hemming-Schroeder, E., Yewhalaw, D., Nguyen, J., Kebede, E., Zemene, E., et al. (2017a). Transmission dynamics of co-endemic Plasmodium vivax and P. falciparum in Ethiopia and prevalence of antimalarial resistant genotypes. PLoS Negl. Trop. Dis. 11:e0005806. doi: 10.1371/journal.pntd.0005806
Lo, E., Hostetler, J. B., Yewhalaw, D., Pearson, R. D., Hamid, M. M. A., Gunalan, K., et al. (2019). Frequent expansion of Plasmodium vivax duffy binding protein in Ethiopia and its epidemiological significance. PLoS Negl. Trop. Dis. 13:e0007222. doi: 10.1371/journal.pntd.0007222
Lopez, L., and Koepfli, C. (2021). Systematic review of Plasmodium falciparum and Plasmodium vivax polyclonal infections: Impact of prevalence, study population characteristics, and laboratory procedures. PLoS One 16:e0249382. doi: 10.1371/journal.pone.0249382
Lover, A. A., Baird, J. K., Gosling, R., and Price, R. N. (2018). Malaria elimination: Time to target all species. Am. J. Trop. Med. Hyg. 99, 17–23.
MalariaGEN, Adam, I., Alam, M., Alemu, S., Amaratunga, C., Amato, R., et al. (2022). An open dataset of Plasmodium vivax genome variation in 1,895 worldwide samples [version 1; peer review: 2 approved]. Wellcome Open Res. 7:136.
Manske, M., Miotto, O., Campino, S., Auburn, S., Almagro-Garcia, J., Maslen, G., et al. (2012). Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379. doi: 10.1038/nature11174
Melnikov, A., Galinsky, K., Rogov, P., Fennell, T., Van Tyne, D., Russ, C., et al. (2011). Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 12:R73.
Menard, D., Barnadas, C., Bouchier, C., Henry-Halldin, C., Gray, L. R., Ratsimbasoa, A., et al. (2010). Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. U.S.A. 107, 5967–5971. doi: 10.1073/pnas.0912496107
Menard, D., Chan, E. R., Benedet, C., Ratsimbasoa, A., Kim, S., Chim, P., et al. (2013). Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl. Trop. Dis. 7:e2489. doi: 10.1371/journal.pntd.0002489
Miotto, O., Amato, R., Ashley, E. A., Macinnis, B., Almagro-Garcia, J., Amaratunga, C., et al. (2015). Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234.
Mohring, F., Hart, M. N., Patel, A., Baker, D. A., and Moon, R. W. (2020). CRISPR-Cas9 genome editing of Plasmodium knowlesi. Bio Protoc. 10:e3522.
Moon, R. W., Hall, J., Rangkuti, F., Ho, Y. S., Almond, N., Mitchell, G. H., et al. (2013). Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 110, 531–536. doi: 10.1073/pnas.1216457110
Mourier, T., De Alvarenga, D. A. M., Kaushik, A., De Pina-Costa, A., Douvropoulou, O., Guan, Q., et al. (2021). The genome of the zoonotic malaria parasite Plasmodium simium reveals adaptations to host switching. BMC Biol. 19:219. doi: 10.1186/s12915-021-01139-5
Mu, J., Awadalla, P., Duan, J., Mcgee, K. M., Keebler, J., Seydel, K., et al. (2007). Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat. Genet. 39, 126–130. doi: 10.1038/ng1924
Mueller, I., Galinski, M. R., Baird, J. K., Carlton, J. M., Kochar, D. K., Alonso, P. L., et al. (2009). Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 9, 555–566. doi: 10.1016/S1473-3099(09)70177-X
Nair, S., Nkhoma, S. C., Serre, D., Zimmerman, P. A., Gorena, K., Daniel, B. J., et al. (2014). Single-cell genomics for dissection of complex malaria infections. Genome Res. 24, 1028–1038. doi: 10.1101/gr.168286.113
Neafsey, D. E., Galinsky, K., Jiang, R. H., Young, L., Sykes, S. M., Saif, S., et al. (2012). The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat. Genet. 44, 1046–1050. doi: 10.1038/ng.2373
Nkhoma, S. C., Trevino, S. G., Gorena, K. M., Nair, S., Khoswe, S., Jett, C., et al. (2020). Co-transmission of related malaria parasite lineages shapes within-host parasite diversity. Cell. Host. Microbe 27, 93.e104–103.e104. doi: 10.1016/j.chom.2019.12.001
Omedo, I., Mogeni, P., Rockett, K., Kamau, A., Hubbart, C., Jeffreys, A., et al. (2017). Geographic-genetic analysis of Plasmodium falciparum parasite populations from surveys of primary school children in Western Kenya. Wellcome Open Res. 2:29. doi: 10.12688/wellcomeopenres.11228.2
Oresegun, D. R., Thorpe, P., Benavente, E. D., Campino, S., Muh, F., Moon, R. W., et al. (2022). De novo assembly of Plasmodium knowlesi genomes from clinical samples explains the counterintuitive intrachromosomal organization of variant SICAvar and kir multiple gene family members. Front. Genet. 13:855052. doi: 10.3389/fgene.2022.855052
Otto, T. D., Bohme, U., Sanders, M., Reid, A., Bruske, E. I., Duffy, C. W., et al. (2018). Long read assemblies of geographically dispersed Plasmodium falciparum isolates reveal highly structured subtelomeres. Wellcome Open Res. 3:52. doi: 10.12688/wellcomeopenres.14571.1
Oyola, S. O., Ariani, C. V., Hamilton, W. L., Kekre, M., Amenga-Etego, L. N., Ghansah, A., et al. (2016). Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J. 15:597. doi: 10.1186/s12936-016-1641-7
Pacheco, M. A., Schneider, K. A., Cespedes, N., Herrera, S., Arevalo-Herrera, M., and Escalante, A. A. (2019). Limited differentiation among Plasmodium vivax populations from the northwest and to the south Pacific Coast of Colombia: A malaria corridor? PLoS Negl. Trop. Dis. 13:e0007310. doi: 10.1371/journal.pntd.0007310
Pain, A., Bohme, U., Berry, A. E., Mungall, K., Finn, R. D., Jackson, A. P., et al. (2008). The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455, 799–803. doi: 10.1038/nature07306
Park, D. J., Lukens, A. K., Neafsey, D. E., Schaffner, S. F., Chang, H. H., Valim, C., et al. (2012). Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc. Natl. Acad. Sci. U.S.A. 109, 13052–13057. doi: 10.1073/pnas.1210585109
Parobek, C. M., Lin, J. T., Saunders, D. L., Barnett, E. J., Lon, C., Lanteri, C. A., et al. (2016). Selective sweep suggests transcriptional regulation may underlie Plasmodium vivax resilience to malaria control measures in Cambodia. Proc. Natl. Acad. Sci. U.S.A. 113, E8096–E8105. doi: 10.1073/pnas.1608828113
Pasini, E. M., Böhme, U., Rutledge, G. G., Voorberg-Van Der Wel, A., Sanders, M., Berriman, M., et al. (2017). An improved Plasmodium cynomolgi genome assembly reveals an unexpected methyltransferase gene expansion. Wellcome Open Res. 2:42. doi: 10.12688/wellcomeopenres.11864.1
Pearson, R. D., Amato, R., Auburn, S., Miotto, O., Almagro-Garcia, J., Amaratunga, C., et al. (2016). Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 48, 959–964. doi: 10.1038/ng.3599
Petkova, D., Novembre, J., and Stephens, M. (2016). Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 48, 94–100.
PlasmoDB (2022). PlasmoDB 57 released. Available online at: https://plasmodb.org/plasmo/app/static-content/PlasmoDB/news.html#PlasmoDB57Released (accessed April 21, 2022).
Popovici, J., Friedrich, L. R., Kim, S., Bin, S., Run, V., Lek, D., et al. (2018). Genomic analyses reveal the common occurrence and complexity of Plasmodium vivax relapses in Cambodia. mBio 9, e1888–e1817. doi: 10.1128/mBio.01888-17
Popovici, J., Pierce-Friedrich, L., Kim, S., Bin, S., Run, V., Lek, D., et al. (2019). Recrudescence, reinfection, or relapse? A more rigorous framework to assess chloroquine efficacy for Plasmodium vivax malaria. J. Infect. Dis. 219, 315–322.
Popovici, J., Roesch, C., Carias, L. L., Khim, N., Kim, S., Vantaux, A., et al. (2020). Amplification of Duffy binding protein-encoding gene allows Plasmodium vivax to evade host anti-DBP humoral immunity. Nat. Commun. 11:953. doi: 10.1038/s41467-020-14574-9
Preston, M. D., Campino, S., Assefa, S. A., Echeverry, D. F., Ocholla, H., and Amambua-Ngwa, A. (2014). A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat. Commun. 5:4052. doi: 10.1038/ncomms5052
Price, R. N., Von Seidlein, L., Valecha, N., Nosten, F., Baird, J. K., and White, N. J. (2014). Global extent of chloroquine-resistant Plasmodium vivax: A systematic review and meta-analysis. Lancet Infect. Dis. 14, 982–991.
Raja, T. N., Hu, T. H., Kadir, K. A., Mohamad, D. S. A., Rosli, N., Wong, L. L., et al. (2020). Naturally acquired human Plasmodium cynomolgi and P. knowlesi Infections, Malaysian Borneo. Emerg. Infect. Dis. 26, 1801–1809. doi: 10.3201/eid2608.200343
Robinson, L. J., Wampfler, R., Betuela, I., Karl, S., White, M. T., Li Wai Suen, C. S., et al. (2015). Strategies for understanding and reducing the Plasmodium vivax and Plasmodium ovale hypnozoite reservoir in Papua New Guinean children: A randomised placebo-controlled trial and mathematical model. PLoS Med. 12:e1001891. doi: 10.1371/journal.pmed.1001891
Rutledge, G. G., Marr, I., Huang, G. K. L., Auburn, S., Marfurt, J., Sanders, M., et al. (2017b). Genomic characterization of recrudescent Plasmodium malariae after treatment with artemether/lumefantrine. Emerg. Infect. Dis. 23, 1300–1307. doi: 10.3201/eid2308.161582
Rutledge, G. G., Bohme, U., Sanders, M., Reid, A. J., Cotton, J. A., Maiga-Ascofare, O., et al. (2017a). Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature 542, 101–104.
Sá, J. M., Kaslow, S. R., Moraes Barros, R. R., Brazeau, N. F., Parobek, C. M., Tao, D., et al. (2019). Plasmodium vivax chloroquine resistance links to pvcrt transcription in a genetic cross. Nat. Commun. 10, 4300–4300. doi: 10.1038/s41467-019-12256-9
Schaffner, S. F., Taylor, A. R., Wong, W., Wirth, D. F., and Neafsey, D. E. (2018). hmmIBD: Software to infer pairwise identity by descent between haploid genotypes. Malar. J. 17:196. doi: 10.1186/s12936-018-2349-7
Shen, H. M., Chen, S. B., Wang, Y., Xu, B., Abe, E. M., and Chen, J. H. (2017). Genome-wide scans for the identification of Plasmodium vivax genes under positive selection. Malar. J. 16:238. doi: 10.1186/s12936-017-1882-0
Shetty, A. C., Jacob, C. G., Huang, F., Li, Y., Agrawal, S., Saunders, D. L., et al. (2019). Genomic structure and diversity of Plasmodium falciparum in Southeast Asia reveal recent parasite migration patterns. Nat. Commun. 10:2665. doi: 10.1038/s41467-019-10121-3
Simpson, J. A., Silamut, K., Chotivanich, K., Pukrittayakamee, S., and White, N. J. (1999). Red cell selectivity in malaria: A study of multiple-infected erythrocytes. Trans. R. Soc. Trop. Med. Hyg. 93, 165–168. doi: 10.1016/s0035-9203(99)90295-x
Spanakos, G., Snounou, G., Pervanidou, D., Alifrangis, M., Rosanas-Urgell, A., Baka, A., et al. (2018). Genetic spatiotemporal anatomy of Plasmodium vivax malaria episodes in Greece, 2009-2013. Emerg. Infect. Dis. 24, 541–548. doi: 10.3201/eid2403.170605
Stark, D. J., Fornace, K. M., Brock, P. M., Abidin, T. R., Gilhooly, L., Jalius, C., et al. (2019). Long-tailed macaque response to deforestation in a Plasmodium knowlesi-endemic area. Ecohealth 16, 638–646. doi: 10.1007/s10393-019-01403-9
Su, X., Hayton, K., and Wellems, T. E. (2007). Genetic linkage and association analyses for trait mapping in Plasmodium falciparum. Nat. Rev. Genet. 8, 497–506. doi: 10.1038/nrg2126
Sutherland, C. J. (2016). Persistent parasitism: The adaptive biology of malariae and ovale malaria. Trends Parasitol. 32, 808–819. doi: 10.1016/j.pt.2016.07.001
Sutherland, C. J., Tanomsing, N., Nolder, D., Oguike, M., Jennison, C., Pukrittayakamee, S., et al. (2010). Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J. Infect. Dis. 201, 1544–1550. doi: 10.1086/652240
Suwanarusk, R., Chavchich, M., Russell, B., Jaidee, A., Chalfein, F., Barends, M., et al. (2008). Amplification of pvmdr1 associated with multidrug-resistant Plasmodium vivax. J. Infect. Dis. 198, 1558–1564. doi: 10.1086/592451
Suwanarusk, R., Russell, B., Chavchich, M., Chalfein, F., Kenangalem, E., Kosaisavee, V., et al. (2007). Chloroquine resistant Plasmodium vivax: In vitro characterisation and association with molecular polymorphisms. PLoS One 2:e1089. doi: 10.1371/journal.pone.0001089
Tachibana, S., Sullivan, S. A., Kawai, S., Nakamura, S., Kim, H. R., Goto, N., et al. (2012). Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat. Genet. 44, 1051–1055. doi: 10.1038/ng.2375
Taylor, A. R., Watson, J. A., Chu, C. S., Puaprasert, K., Duanguppama, J., Day, N. P. J., et al. (2019b). Resolving the cause of recurrent Plasmodium vivax malaria probabilistically. Nat. Commun. 10:5595. doi: 10.1038/s41467-019-13412-x
Taylor, A. R., Jacob, P. E., Neafsey, D. E., and Buckee, C. O. (2019a). Estimating relatedness between malaria parasites. Genetics 212, 1337–1351.
Taylor, A. R., Schaffner, S. F., Cerqueira, G. C., Nkhoma, S. C., Anderson, T. J. C., Sriprawat, K., et al. (2017). Quantifying connectivity between local Plasmodium falciparum malaria parasite populations using identity by descent. PLoS Genet. 13:e1007065. doi: 10.1371/journal.pgen.1007065
Trimarsanto, H., Amato, R., Pearson, R. D., Sutanto, E., Noviyanti, R., Trianty, L., et al. (2019). A molecular barcode and online tool to identify and map imported infection with Plasmodium vivax. bioRxiv [Preprint] 776781. doi: 10.1101/776781
Vembar, S. S., Seetin, M., Lambert, C., Nattestad, M., Schatz, M. C., Baybayan, P., et al. (2016). Complete telomere-to-telomere de novo assembly of the Plasmodium falciparum genome through long-read (> 11kb), single molecule, real-time sequencing. DNA Res. 23, 339–351. doi: 10.1093/dnares/dsw022
Venkatesan, M., Amaratunga, C., Campino, S., Auburn, S., Koch, O., Lim, P., et al. (2012). Using CF11 cellulose columns to inexpensively and effectively remove human DNA from Plasmodium falciparum-infected whole blood samples. Malar. J. 11:41. doi: 10.1186/1475-2875-11-41
Verity, R., Aydemir, O., Brazeau, N. F., Watson, O. J., Hathaway, N. J., Mwandagalirwa, M. K., et al. (2020). The impact of antimalarial resistance on the genetic structure of Plasmodium falciparum in the DRC. Nat. Commun. 11:2107. doi: 10.1038/s41467-020-15779-8
Verzier, L. H., Coyle, R., Singh, S., Sanderson, T., and Rayner, J. C. (2019). Plasmodium knowlesi as a model system for characterising Plasmodium vivax drug resistance candidate genes. PLoS Negl. Trop. Dis. 13:e0007470. doi: 10.1371/journal.pntd.0007470
Volkman, S. K., Herman, J., Lukens, A. K., and Hartl, D. L. (2017). Genome-wide association studies of drug-resistance determinants. Trends Parasitol. 33, 214–230.
Waltmann, A., Koepfli, C., Tessier, N., Karl, S., Fola, A., Darcy, A. W., et al. (2018). Increasingly inbred and fragmented populations of Plasmodium vivax associated with the eastward decline in malaria transmission across the Southwest Pacific. PLoS Negl. Trop. Dis. 12:e0006146. doi: 10.1371/journal.pntd.0006146
Winter, D. J., Pacheco, M. A., Vallejo, A. F., Schwartz, R. S., Arevalo-Herrera, M., Herrera, S., et al. (2015). Whole genome sequencing of field isolates reveals extensive genetic diversity in Plasmodium vivax from Colombia. PLoS Negl. Trop. Dis. 9:e0004252. doi: 10.1371/journal.pntd.0004252
World Health Organization [WHO] (2019). World Malaria Report 2019. Geneva: World Health Organization.
World Health Organization [WHO] (2021). World Malaria Report 2021. Geneva: World Health Organization.
Yman, V., Wandell, G., Mutemi, D. D., Miglar, A., Asghar, M., Hammar, U., et al. (2019). Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in eastern Tanzania. PLoS Negl. Trop. Dis. 13:e0007414. doi: 10.1371/journal.pntd.0007414
Zhong, D., Koepfli, C., Cui, L., and Yan, G. (2018). Molecular approaches to determine the multiplicity of Plasmodium infections. Malar. J. 17:172.
Zhou, R., Li, S., Zhao, Y., Yang, C., Liu, Y., Qian, D., et al. (2019). Characterization of Plasmodium ovale spp. imported from Africa to Henan province, China. Sci. Rep. 9:2191. doi: 10.1038/s41598-019-38629-0
Keywords: malaria, neglected, genomics, population genomics, vivax malaria
Citation: Brashear AM and Cui L (2022) Population genomics in neglected malaria parasites. Front. Microbiol. 13:984394. doi: 10.3389/fmicb.2022.984394
Received: 02 July 2022; Accepted: 22 August 2022;
Published: 08 September 2022.
Edited by:
Ashwani Sharma, Paul Scherrer Institut (PSI), SwitzerlandReviewed by:
Sarah Auburn, Charles Darwin University, AustraliaWenn-Chyau Lee, University of Malaya, Malaysia
Copyright © 2022 Brashear and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liwang Cui, bGl3YW5nY3VpQHVzZi5lZHU=