 Nikola Palevich
Nikola Palevich Faith P. Palevich
Faith P. Palevich Amanda Gardner
Amanda Gardner John Mills
John Mills
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 September 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.976152
The diversity of the genus Shewanella and their roles across a variety of ecological niches is largely unknown highlighting the phylogenetic diversity of these bacteria. From a food safety perspective, Shewanella species have been recognized as causative spoilage agents of vacuum-packed meat products. However, the genetic basis and metabolic pathways for the spoilage mechanism are yet to be explored due to the unavailability of relevant Shewanella strains and genomic resources. In this study, whole-genome sequencing of 32 Shewanella strains isolated from vacuum-packaged refrigerated spoiled lamb was performed to examine their roles in meat spoilage. Phylogenomic reconstruction revealed their genomic diversity with 28 Shewanella spp. strains belonging to the same putative novel species, two Shewanella glacialipiscicola strains (SM77 and SM91), Shewanella xiamenensis NZRM825, and Shewanella putrefaciens DSM 50426 (ATCC 8072) isolated from butter. Genome-wide clustering of orthologous gene families revealed functional groupings within the major Shewanella cluster but also considerable plasticity across the different species. Pan-genome analysis revealed conserved occurrence of spoilage genes associated with sulfur and putrescine metabolism, while the complete set of trimethylamine metabolism genes was observed in only Shewanella sp. SM74, S. glacialipiscicola SM77 and SM91 strains. Through comparative genomics, some variations were also identified pertaining to genes associated with adaptation to environmental cues such as temperature, osmotic, salt, oxidative, antimicrobial peptide, and drug resistance stresses. Here we provide a reference collection of draft Shewanella genomes for subsequent species descriptions and future investigations into the molecular spoilage mechanisms for further applications in the meat industry.
The genus Shewanella was first validly published in 1985 (MacDonell and Colwell, 1985) and was included within the family Shewanellaceae in 2004 (Ivanova et al., 2004), having been moved from its previous position within the Alteromonadaceae. A total of 76 species are currently recognized as being validly published with correct names within this taxonomic group (Parte et al., 2020). The genus comprises species that are ubiquitous in aquatic environments, being found in both hot and cold climates, marine and fresh waters, deep-sea sediments, and oil field wastes (Nealson and Scott, 2006). Several Shewanella species have been associated with emerging opportunistic human infections (Holt et al., 2005; Janda and Abbott, 2014; Fang et al., 2017; Martín-Rodríguez and Meier-Kolthoff, 2022), while others, notably Shewanella putrefaciens and Shewanella baltica, are implicated in the spoilage of high pH meat products and seafood (Gill and Newton, 1979; Gram et al., 1987; Vogel et al., 2005).
The pH of beef, lamb, and venison can range from 5.3 to 6.9 depending on the species, muscle type, slaughter season, and other variables; however, meat presenting desirable eating attributes typically has a pH < 5.8, whereas meat with pH > 5.8 (other than lamb shoulder and shank) has inferior qualities such as darker color and shorter shelf life (Reis and Rosenvold, 2014). On high pH meats (pH > 6.0), S. putrefaciens has been shown to cause spoilage both aerobically and anaerobically, utilizing the sulfur-rich amino acids serine and cysteine even when glucose is abundant, producing organic sulfides that contribute to off odors and flavors (Gill, 1986). Anaerobically in vacuum packs, while not fermentative, these sulfides are utilized as alternate terminal electron acceptors rather than oxygen to maintain respiratory metabolism (Gill and Newton, 1979); the abundant hydrogen sulfide (H2S) produced causes green sulfmyoglobin discoloration at the meat surface as well as degradation of the odor and flavor (Devine and Dikeman, 2014). On fish, Shewanella become dominant organisms during storage, producing deleterious odors due to trimethylamine oxide (TMAO) reduction to trimethylamine, and production of H2S (Nealson and Scott, 2006).
Shewanella putrefaciens DSM 50426 (ATCC 8072) isolated from butter and all additional 31 Shewanella spp. strains isolated from vacuum-packaged refrigerated spoiled lamb are psychotropic, facultative anaerobic, Gram-negative gammaproteobacteria that inhabit diverse environments and their phylogenetic relationships are largely uncharacterized (Lemaire et al., 2020). By studying the Shewanella genomes, we aim to determine their biochemical mechanisms and spoilage pathways that enable these species to metabolize sulfur-containing amino acids or proteins to produce H2S resulting in a fishy odor that is characteristic of Shewanella-spoiled meat.
Shewanella putrefaciens DSM 50426 (ATCC 8072) was acquired from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, and Shewanella xiamenensis NZRM825 from the NZRM Culture Collection. The remaining 30 Shewanella spp. strains were isolated from vacuum-packaged lamb that had no pack distension, with meat discoloration (some green spots) and sweaty feet odor for some strains. Meat samples were added into Whirl-Pak bags with 50 ml 0.1% peptone diluent (Fort Richard Laboratories) and stomached (Seward, United Kingdom) for 2 min at full speed. The liquid suspension was transferred to 50 ml falcon tubes and centrifuged at 5,000 × g for 10 min. The supernatant was discarded, and the pellet resuspended in 10 ml 0.1% peptone diluent. The suspensions were diluted 10-fold in 0.1% peptone diluent and the 0.1 ml of the dilutions plated onto Peptone iron agar (Fort Richard Laboratories). Plates were incubated at 25°C for 2 days. Individual black colonies on the Peptone iron agar were streaked onto Columbia Sheep Blood agar (Fort Richard Laboratories) to obtain pure colonies. Isolate identity was checked by PCR using 16S rRNA universal primers pA (5′-AGA GTT TGA TCC TGG CTC AG-3′) and pH (5′-AAG GAG GTG ATC CAG CCG CA-3′; Edwards et al., 1989).
A single colony from each isolate was used to inoculate 10 ml TSB and incubated at 25°C for 2 days. Cells were pelleted by centrifugation at 5,000 × g for 10 min. High-molecular-weight genomic DNA was extracted using a modified phenol-chloroform procedure (5). Bacterial strain identity was verified by 16S rRNA gene amplification and sequencing of genomic DNA was verified by automated Sanger sequencing of the 16S rRNA gene following PCR amplification from genomic DNA. Total DNA concentrations were determined using a NanoDrop® ND-1000 (Thermo Scientific Inc.) and a Qubit Fluorometer dsDNA BR Kit (Invitrogen, United States), in accordance with the manufacturer’s instructions. Genomic DNA integrity was verified by agarose gel electrophoresis and using a 2000 BioAnalyzer (Agilent, United States). DNA libraries were prepared using the Illumina Nextera method chemistry kit v1 according to the manufacturer’s protocol and samples were sequenced to the requested depths on the Illumina MiSeq 500 platform (2 × 250 bp) to generate paired-end reads.
Reads were examined with FastQC v0.11.5 (Andrews, 2010), trimmed with Trimmomatic v0.39 (Bolger et al., 2014), and assembled using the A5-miseq pipeline v20169825 with standard parameters (Coil et al., 2015). The de novo assemblies were annotated using NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v5.2 (Tatusova et al., 2016) and Blast2GO suite (Götz et al., 2008). CheckM (Parks et al., 2015) was used to estimate genome completeness with default software settings and parameters were used throughout unless specified otherwise.
Taxonomic assignments of our strains were explored initially by pairwise comparison of their Average Nucleotide Identity (ANIb) and correlation indexes of their Tetra-nucleotide signatures (TETRA) using JSpeciesWS v3.8.2 (Richter et al., 2016). In addition, whole nucleotide sequences were uploaded to the Type (Strain) Genome Server (TYGS) for whole-genome-based taxonomic analysis (Meier-Kolthoff and Göker, 2019). For the phylogenomic inference, all pairwise comparisons among the set of genomes were conducted using Genome BLAST Distance Phylogeny (GBDP) and accurate intergenomic distances were inferred under the algorithm “trimming” and distance formula d5 (Meier-Kolthoff et al., 2013). One hundred distance replicates were calculated each, and digital DDH values and confidence intervals were calculated using the recommended settings of the Genome-to-Genome Distance Calculator (GGDC)1 v3.01 under recommended settings. Pairwise digital DNA:DNA hybridization values (dDDH) were inferred accordingly with the resulting distance matrix subjected to clustering based on established thresholds for delineating species (DDH > 70%) and subspecies (DDH > 79%), respectively (Meier-Kolthoff et al., 2013). One hundred pseudo-bootstrap replicates were assessed under the same settings each. Finally, a balanced minimum evolution tree was inferred using FastME v2.1.4 with SPR postprocessing (Lefort et al., 2015).
To determine homologs and investigate orthologous gene (OG) family groups, the annotated protein sequences for all Shewanella genomes were analyzed using to OrthoFinder v2.5.2 (Emms and Kelly, 2019) with default settings. The resulting ortho-group counts were imported into R v4.1.12 and the top 20 most common were displayed using UpSetR v1.4.0 (Conway et al., 2017).
The Shewanella spp. genomes assemblies ranged between 32 and 193 contigs, 4.1 to 5.5 Mbp in size, N50 contigs between 66,739 and 625,775 bp, G + C content of 44.1–46.3%, and a mean genome coverage between 45- and 87-fold (Table 1). Overall, the total numbers of annotated protein-coding genes (PCGs) ranged from 3,856 to 5,240, with 108–131 RNA genes consisting of 11–19 rRNAs and 96–112 tRNAs identified. The genome sequence features and data (NCBI SRA, GenBank, and Biosample database accessions) of the Shewanella spp. strains are summarized in Table 1.
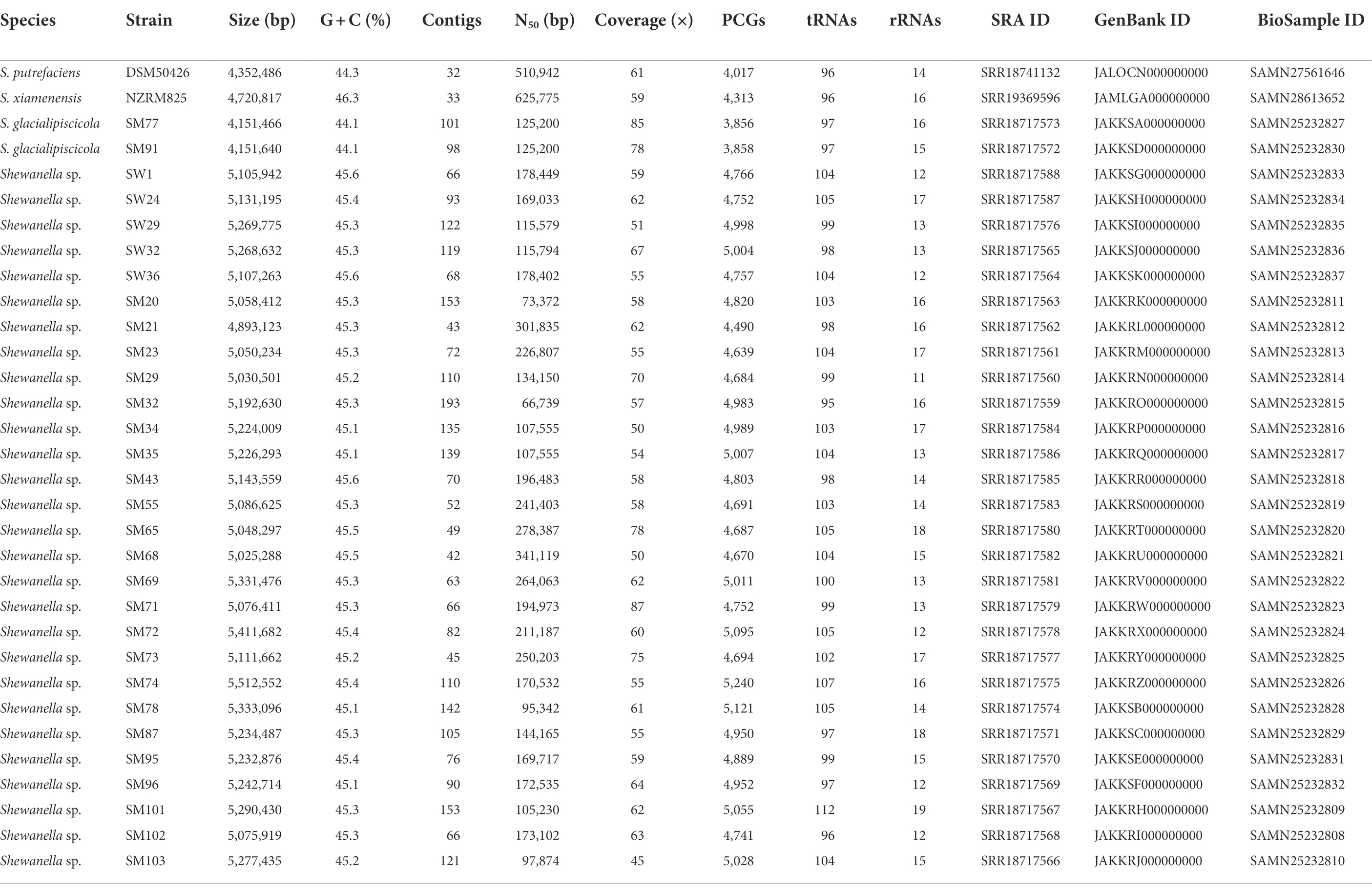
Table 1. General genome features of Shewanella spp. strains.
At both the species and genus levels, the usefulness of complete or partial 16S rRNA gene sequence similarity as a taxonomic marker for Shewanellaceae has been limited (Martín-Rodríguez and Meier-Kolthoff, 2022). Thus, we investigated the taxonomy of our Shewanella genomes, using whole-genome sequence-based pairwise methods including TETRA correlation, average nucleotide identity based on BLAST+ (ANIb), and genome-wide identity indexes using digital DNA:DNA hybridization (dDDH) analyses. Our analyses resulted in the identification of 28 Shewanella spp. strains that represent uncharacterized clades or novel species (with TETRA/ANIb of >0.999/>96.6%), including six putative subspecies that await formal description (Figure 1). From a taxonomic perspective, our phylogenomic reconstruction supported the reassignment of both Shewanella sp. SM77 and SM91 to Shewanella glacialipiscicola due to high sequence similarity (with TETRA/ANIb of ~0.975/>85.0% and dDDH of >90% for both) to the S. glacialipiscicola T147T type strain. The exceptions were S. putrefaciens DSM50426, and S. xiamenensis NZRM825 with a high G + C content of 46.3% and as determined by dDDH and ANIb that will require further experimental investigation. Our genome-wide analyses have highlighted the complexity of the taxonomic relationships within the genus Shewanella with all Shewanella type strains currently sequenced and publicly available from NCBI/TYGS (Martín-Rodríguez and Meier-Kolthoff, 2022). The reported Shewanella spp. genome collection may serve as a valuable resource for prospective work investigating their taxonomic diversity and reference for unravelling their genetic mechanisms associated with spoilage of vacuum packaged meat.
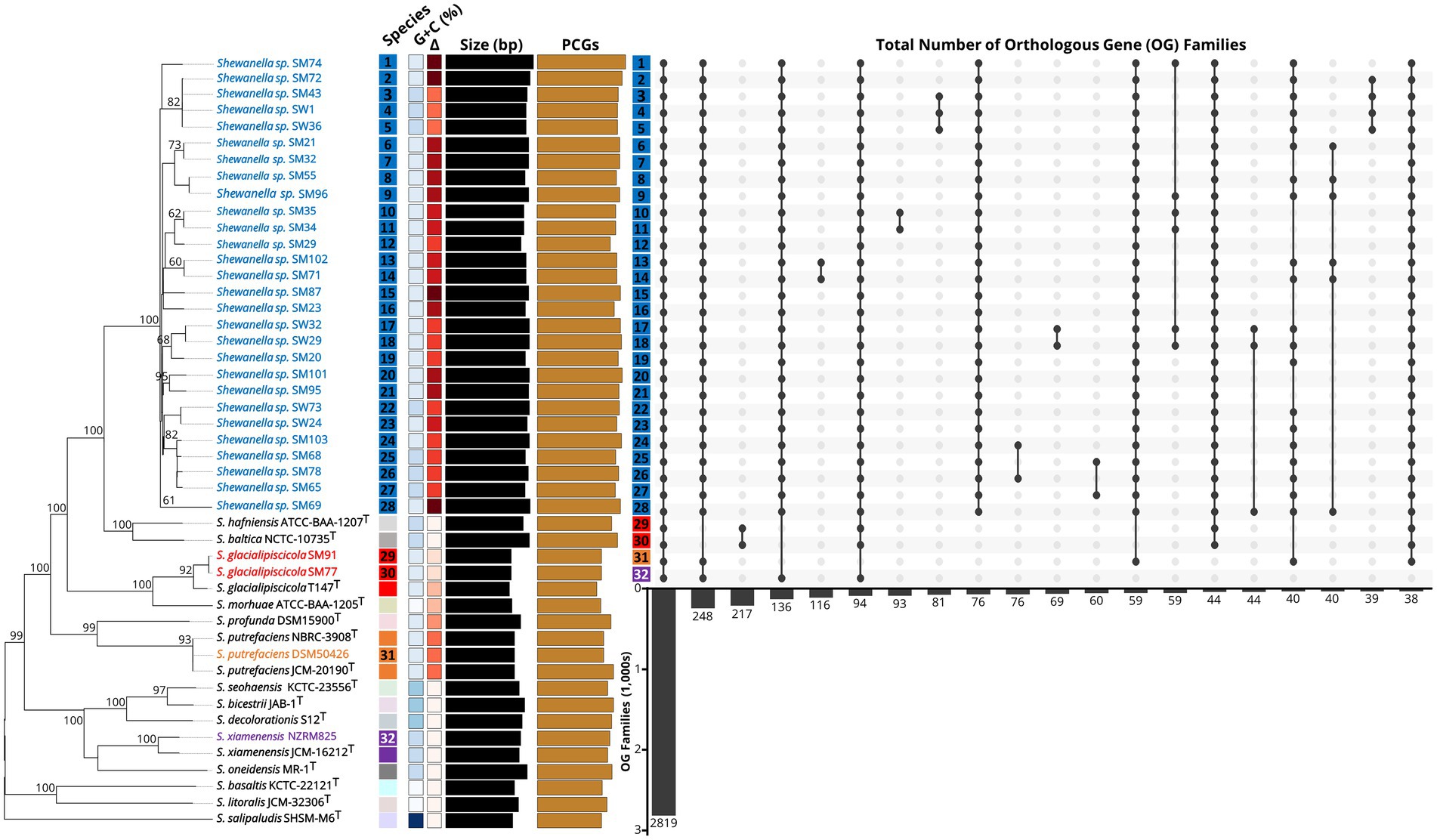
Figure 1. Phylogenomic species-tree reconstruction and homologous gene families of the Shewanella genomes. The phylogenomic tree (left) inferred with GBDP displays the pseudo-bootstrap support values from 100 replications above the branches. Type species are indicated with a superscripted T. The colored boxes to the right of each name refer to the species determined by dDDH cutoffs of 70 and 79%, respectively. The gradients further right indicate G + C content (blue) and Δ statistics (red) indicate overall low (light) to high (dark) accuracy of phylogenetic likeness. Black bars represent the approximate genome size and brown bars the protein-coding genes (PCGs). Genome names are colored to represent putative species grouping. The top 20 orthologous gene (OG) families across all 32 Shewanella proteomes is shown on the far right. Dark circles represent presence of OGs across genomes and total number of gene families shown in numbers below the bars.
Genome-wide replication events in Shewanella have been considered as a mechanism for diversification and adaptation to the environment. To analyze the diversity and evolutionary conservation of the 32 Shewanella genomes, we compared the protein sets for each genome in a pairwise manner to identify sequence similarities and orthologous genes between the different species. Our comparative analysis of the expansions and contractions of gene families revealed markedly different orthology profiles and models of gene gain or loss, but particularly for the major Shewanella cluster (Figure 1). In total, we identified 2,819 orthogroups with all species present, 2,683 single-copy conserved orthologs, 836 one-to-many, and 434 many-to-many co-orthologous genes, with 50 species-specific orthogroups consisting of 106 species-specific genes among the 32 Shewanella genomes, indicating that these genes have expanded to varying degrees. While the comparative analysis of the Shewanella gene families will be helpful to identify the retention and loss of key gene family members after the differentiation of various species, further phenotypic characterization will be required to routinely discriminate between closely related species that might exist within any given meat sample.
The spoilage potential of Shewanella spp. has been associated with sulfur, putrescine, and trimethylamine metabolism (Remenant et al., 2015). To better understand the spoilage potential of Shewanella spp. strains in the spoiled vacuum-packaged refrigerated lamb, genes were annotated against the KEGG database using the EGGNOG mapper of the Blast2GO suite (Figures 2, 3).
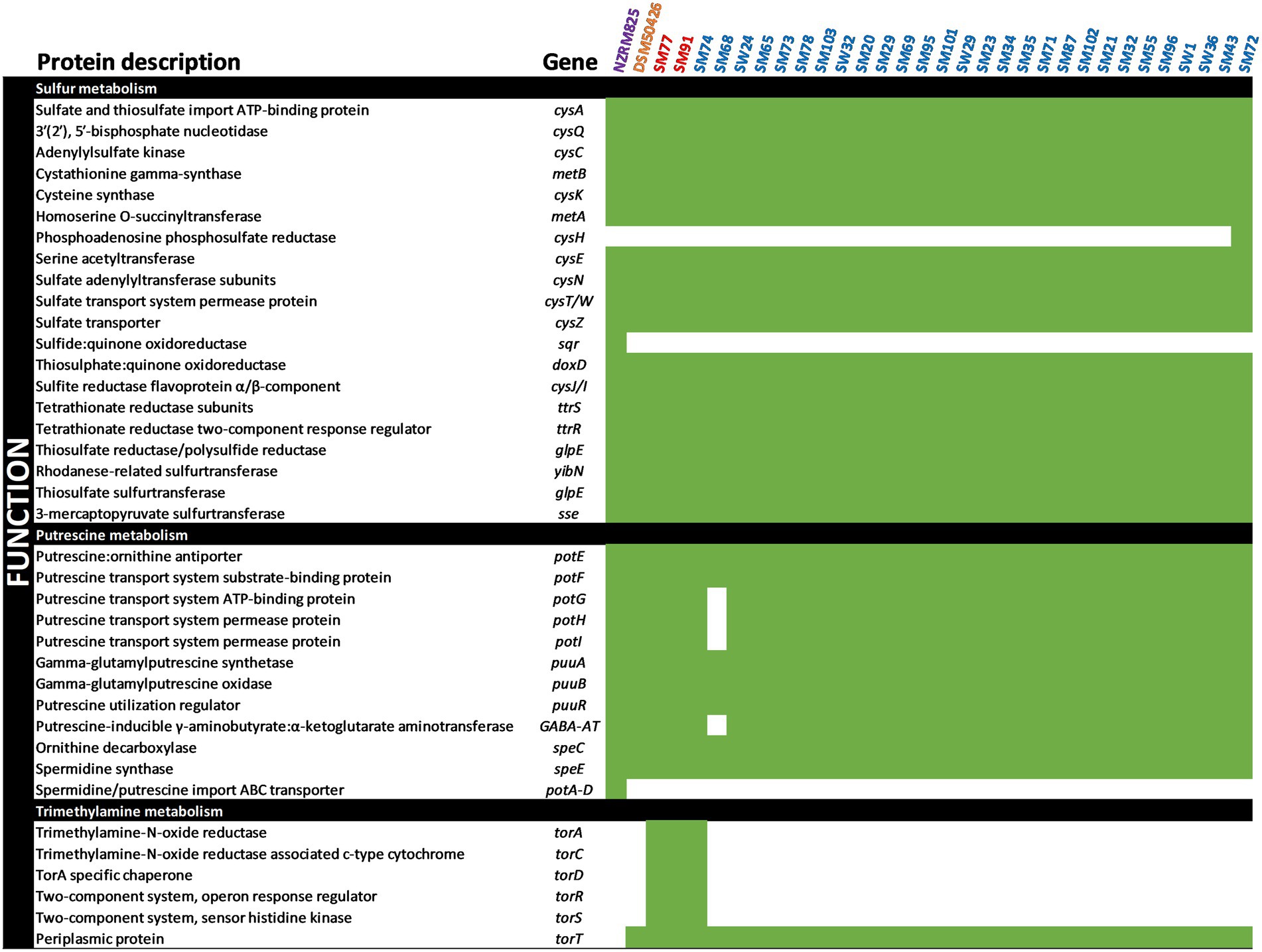
Figure 2. Occurrence of spoilage-related genes in Shewanella genomes. Presence (green) or absence (white) for each gene is depicted.
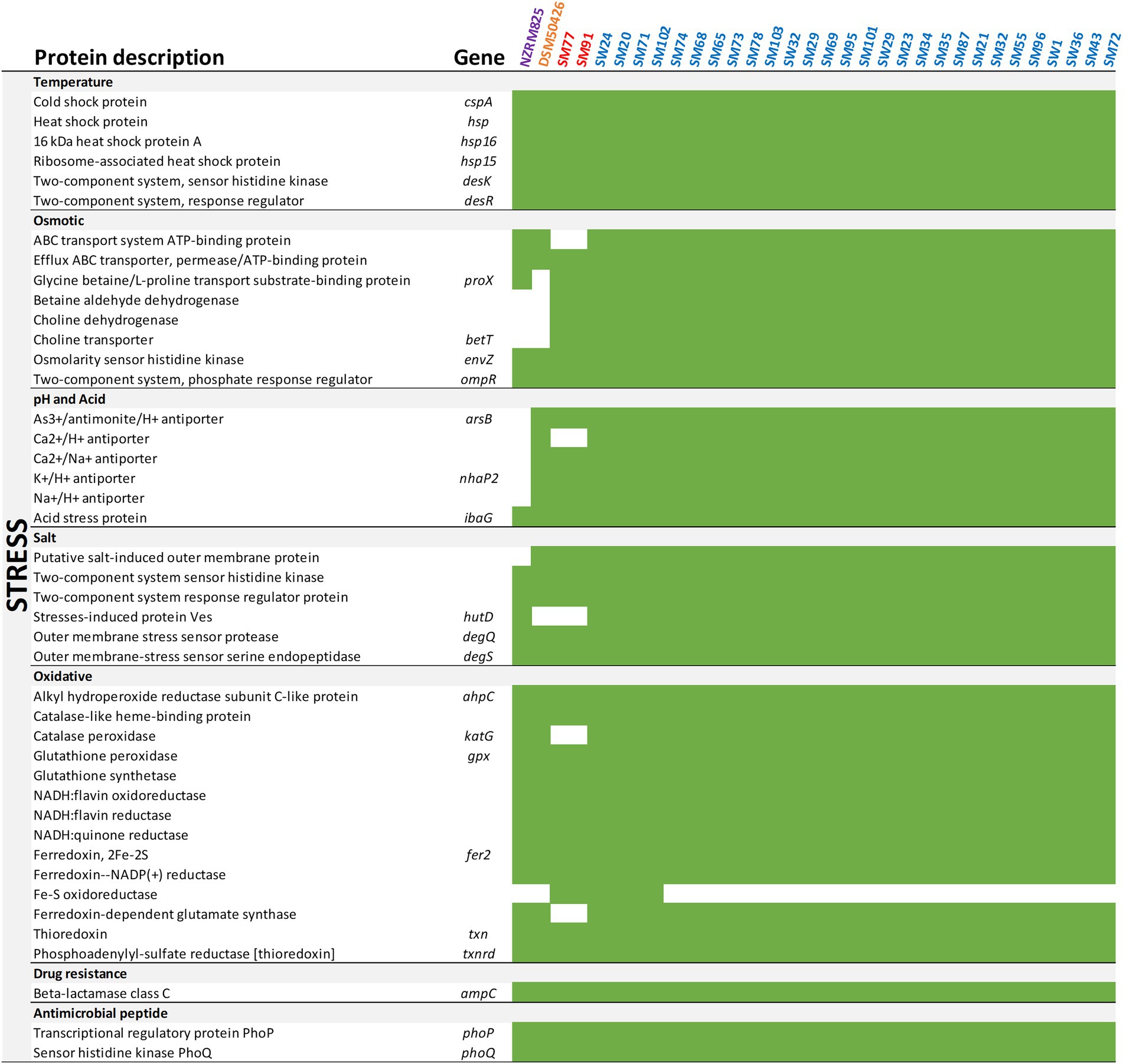
Figure 3. Presence of stress-related genes of Shewanella. Presence (green) or absence (white) for each gene is depicted.
The specific bacterial spoilage of vacuum-packaged meat products has been associated with sulfur metabolism, and the production of hydrogen sulfide gas (H2S) responsible for putrid off-odors in such products has been associated with the presence of Shewanella putrefaciens (Borch et al., 1996). As shown in Figure 2, a series of the cys genes involved in sulfur metabolism and responsible for H2S production were identified in the Shewanella genomes. Genes encoding sulfate adenylyltransferase (cysN, EC 2.7.7.4), adenylylsulfate kinase (cysC, EC 2.7.1.25), and sulfite reductase (cysJ/I EC 1.8.1.2) were found, suggesting that the Shewanella spp. strains have good potential for sulfur metabolism. However, only Shewanella spp. strain SM72 was found to encode a phosphoadenosine phosphosulfate reductase (cysH, EC 1.8.4.8) that reduces 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to sulfite. Notably, our genome analysis identified genes encoding tetrathionate reductase (ttrS, EC 1.8.5.5) and cysteine synthase B (cysK, EC 2.5.1.47), suggesting that tetrathionate may also be preferentially reduced to sulfide by all Shewanella spp. strains.
Biogenic amines such as putrescine and spermidine are produced via decarboxylation of various amino acids and their transport systems have also been described (Remenant et al., 2015). A series of pot genes encoding putrescine transport systems and permease proteins (potE-I), as well as the putrescine:ornithine antiporter (potE) were found in all Shewanella genomes, except Shewanella spp. strain SM68 that lacked potG-I. In addition, genes encoding L-ornithine decarboxylase (speC, EC4.1.1.17) and spermidine synthase (speE, 2.5.1.16) were also found in all Shewanella genomes. The occurrence of these key enzymes indicates that the reported Shewanella spp. strains could produce putrescine from L-ornithine and spermidine.
Recently, the genomic potential of Shewanella species for their ability to decompose trimethyl-amine-N-oxide (TMAO) into trimethylamine (TMA) resulting in the production of a strong odor has been elucidated (Li et al., 2020). The full complement of genes required for de-oxygenation of TMAO into TMA and subsequent metabolism, including trimethylamine-N-oxide reductase (torA), TorA specific chaperone (torD), sensor histidine kinase (torS), periplasmic protein TorT, were found in only Shewanella spp. strains SM74, SM77, and SM91. Our findings provide evidence that the regulatory genes of trimethylamine metabolism may be highly conserved across Shewanella species associated with spoilage of a variety of meat products and warrant further investigation.
Ultimately, the success of spoilage microorganisms is dependent on their ability to survive under some critical environmental stresses associated with the storage and processing of food. Figure 3 summarizes the occurrence of key stress-related genes that regulate the adaptability of Shewanella spp. to temperature, osmotic, salt, oxidative, antimicrobial peptide, and drug resistance stresses. The presence of genes encoding cold shock protein (cspA), sensor histidine kinase (desK), and response regulator (desR) may allow Shewanella spp. to perform transcription during prolonged cold exposure and improve their tolerance to low temperatures. Also, Shewanella spp. all contain genes encoding ABC-type transport system permease and transport proteins, osmolarity sensor histidine kinase (envZ), and phosphate regulon response regulator (ompR) related to osmotic pressure. Furthermore, genes encoding sodium-proton antiporters (Na+/H+) responsible for the maintenance of homeostasis and salt tolerance were abundant across all strains. For example, degS functions as the principal regulator of degradative enzymes and encodes the outer membrane-stress sensor serine endopeptidase. Lastly, ampC that encodes β-lactamase class C involved in drug resistance and phoP/Q associated with antimicrobial peptide resistance have been identified in all Shewanella spp. strains. Overall, the Shewanella spp. genome sequences are a valuable resource for future investigations aiming to develop new detection and prevention strategies targeting food spoilage bacteria.
The genome sequence and associated data for Shewanella strains have been deposited under the GenBank BioProject accession number PRJNA574489. Detailed information can be found in Table 1 regarding BioSample and Sequence Read Archive (SRA) accession numbers: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA574489.
GB and JM conceived the study and acquired the funding. FP and AG collected the samples and conducted the experiments. NP performed the bioinformatics analysis, collated the data, and wrote the manuscript with input from all other authors. All authors contributed to the article and approved the submitted version.
This research was supported by the AgResearch Ltd. Strategic Science Investment Fund (grant PRJ0331443).
We thank Paul Maclean and Vince Carbone for reviewing an earlier version of the manuscript and for their helpful feedback.
NP, FP, AG, GB, and JM were employed by AgResearch Ltd.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. ^https://www.R-project.org/ (Accessed September 2, 2022).
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge, UK: Babraham Bioinformatics, Babraham Institute.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borch, E., Kant-Muermans, M.-L., and Blixt, Y. (1996). Bacterial spoilage of meat and cured meat products. Int. J. Food Microbiol. 33, 103–120. doi: 10.1016/0168-1605(96)01135-X
Coil, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Edwards, U., Rogall, T., Blöcker, H., Emde, M., and Böttger, E. C. (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 17, 7843–7853. doi: 10.1093/nar/17.19.7843
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Fang, Y., Wang, Y., Liu, Z., Lu, B., Dai, H., Kan, B., et al. (2017). Shewanella carassii sp. nov., isolated from surface swabs of crucian carp and faeces of a diarrhoea patient. Int. J. Syst. Evol. Microbiol. 67, 5284–5289. doi: 10.1099/ijsem.0.002511
Gill, C. (1986). “The control of microbial spoilage in fresh meats,” in Advances in Meat Research. Vol. 2. eds. A. Pearson and T. Dutson (Westport: AVI Publishing), 49–88.
Gill, C., and Newton, K. (1979). Spoilage of vacuum-packaged dark, firm, dry meat at chill temperatures. Appl. Environ. Microbiol. 37, 362–364. doi: 10.1128/aem.37.3.362-364.1979
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Gram, L., Trolle, G., and Huss, H. H. (1987). Detection of specific spoilage bacteria from fish stored at low (0°C) and high (20°C) temperatures. Int. J. Food Microbiol. 4, 65–72. doi: 10.1016/0168-1605(87)90060-2
Holt, H., Gahrn-Hansen, B., and Bruun, B. (2005). Shewanella algae and Shewanella putrefaciens: clinical and microbiological characteristics. Clin. Microbiol. Infect. 11, 347–352. doi: 10.1111/j.1469-0691.2005.01108.x
Ivanova, E. P., Flavier, S., and Christen, R. (2004). Phylogenetic relationships among marine Alteromonas-like proteobacteria: emended description of the family Alteromonadaceae and proposal of Pseudoalteromonadaceae fam. Nov., Colwelliaceae fam. Nov., Shewanellaceae fam. Nov., Moritellaceae fam. Nov., Ferrimonadaceae fam. Nov., Idiomarinaceae fam. Nov. and Psychromonadaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 54, 1773–1788. doi: 10.1099/ijs.0.02997-0
Janda, J. M., and Abbott, S. L. (2014). The genus Shewanella: from the briny depths below to human pathogen. Crit. Rev. Microbiol. 40, 293–312. doi: 10.3109/1040841X.2012.726209
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Lemaire, O. N., Méjean, V., and Iobbi-Nivol, C. (2020). The Shewanella genus: ubiquitous organisms sustaining and preserving aquatic ecosystems. FEMS Microbiol. Rev. 44, 155–170. doi: 10.1093/femsre/fuz031
Li, J., Yu, H., Yang, X., Dong, R., Liu, Z., and Zeng, M. (2020). Complete genome sequence provides insights into the quorum sensing-related spoilage potential of Shewanella baltica 128 isolated from spoiled shrimp. Genomics 112, 736–748. doi: 10.1016/j.ygeno.2019.05.010
MacDonell, M., and Colwell, R. (1985). Phylogeny of the Vibrionaceae, and recommendation for two new genera, Listonella and Shewanella. Syst. Appl. Microbiol. 6, 171–182. doi: 10.1016/S0723-2020(85)80051-5
Martín-Rodríguez, A. J., and Meier-Kolthoff, J. P. (2022). Whole genome-based taxonomy of Shewanella and Parashewanella. Int. J. Syst. Evol. Microbiol. 72:005438. doi: 10.1099/ijsem.0.005438
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14, 1–14. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-10210-3
Nealson, K. H., and Scott, J. (2006). Ecophysiology of the genus Shewanella. Prokaryotes 6, 1133–1151. doi: 10.1007/0-387-30746-X_45
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parte, A. C., Carbasse, J. S., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Reis, M. M., and Rosenvold, K. (2014). Early on-line classification of beef carcasses based on ultimate pH by near infrared spectroscopy. Meat Sci. 96, 862–869. doi: 10.1016/j.meatsci.2013.10.016
Remenant, B., Jaffrès, E., Dousset, X., Pilet, M.-F., and Zagorec, M. (2015). Bacterial spoilers of food: behavior, fitness and functional properties. Food Microbiol. 45, 45–53. doi: 10.1016/j.fm.2014.03.009
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Keywords: Shewanella, genome sequence, meat spoilage, bacteria, stress adaptation
Citation: Palevich N, Palevich FP, Gardner A, Brightwell G and Mills J (2022) Genome collection of Shewanella spp. isolated from spoiled lamb. Front. Microbiol. 13:976152. doi: 10.3389/fmicb.2022.976152
Edited by:
Yiannis Kourkoutas, Democritus University of Thrace, GreeceReviewed by:
Foteini F. Parlapani, University of Thessaly, GreeceCopyright © 2022 Palevich, Palevich, Gardner, Brightwell and Mills. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikola Palevich, bmlrLnBhbGV2aWNoQGFncmVzZWFyY2guY28ubno=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.