
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 November 2022
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.969961
 Charlotte Huber1Silver A. Wolf2
Charlotte Huber1Silver A. Wolf2 Wilma Ziebuhr3Mark A. Holmes4Julia Assmann1
Wilma Ziebuhr3Mark A. Holmes4Julia Assmann1 Antina Lübke-Becker5Andrea Thürmer2
Antina Lübke-Becker5Andrea Thürmer2 Torsten Semmler2Julian Brombach5
Torsten Semmler2Julian Brombach5 Astrid Bethe5
Astrid Bethe5 Markus Bischoff6
Markus Bischoff6 Lothar H. Wieler7
Lothar H. Wieler7 Lennard Epping2
Lennard Epping2 Birgit Walther1*
Birgit Walther1*Previous research on methicillin susceptible Staphylococcus aureus (MSSA) belonging to livestock-associated (LA-) sequence type (ST) 398, isolated from pigs and their local surroundings, indicated that differences between these MSSA and their methicillin resistant predecessors (MRSA) are often limited to the absence of the staphylococcal cassette chromosome mec (SCCmec) and few single nucleotide polymorphisms. So far, our understanding on how LA-MRSA endure the environmental conditions associated with pig-farming as well as the putative impact of this particular environment on the mobilisation of SCCmec elements is limited. Thus, we performed in-depth genomic and transcriptomic analyses using the LA-MRSA ST398 strain IMT38951 and its methicillin susceptible descendant. We identified a mosaic-structured SCCmec region including a putative replicative SCCmecVc which is absent from the MSSA chromosome through homologous recombination. Based on our data, such events occur between short repetitive sequences identified within and adjacent to two distinct alleles of the large cassette recombinase genes C (ccrC). We further evaluated the global transcriptomic response of MRSA ST398 to particular pig-farm associated conditions, i.e., contact with host proteins (porcine serum) and a high ammonia concentration. Differential expression of global regulators involved in stress response control were identified, i.e., ammonia-induced alternative sigma factor B-depending activation of genes for the alkaline shock protein 23, the heat shock response and the accessory gene regulator (agr)-controlled transcription of virulence factors. Exposure to serum transiently induced the transcription of distinct virulence factor encoding genes. Transcription of genes reported for mediating the loss of methicillin resistance, especially ccrC, was not significantly different compared to the unchallenged controls. We concluded that, from an evolutionary perspective, bacteria may save energy by incidentally dismissing a fully replicative SCCmec element in contrast to the induction of ccr genes on a population scale. Since the genomic SCCmec integration site is a hot-spot of recombination, occasional losses of elements of 16 kb size may restore capacities for the uptake of foreign genetic material. Subsequent spread of resistance, on the other hand, might depend on the autonomous replication machinery of the deleted SCCmec elements that probably enhance chances for reintegration of SCCmec into susceptible genomes by mere multiplication.
Since the mid-2000s, sequence type (ST)398 and its closely related descendants represent the predominant clonal complex (CC) of livestock-associated methicillin resistant Staphylococcus aureus (LA-MRSA) throughout Europe (Butaye et al., 2016). ST398 LA-MRSA most likely originate from a lineage of human methicillin susceptible S. aureus (MSSA) and have since adapted to livestock (particularly to pig farming) via acquisition of resistance genes against tetracycline and beta-lactam antibiotics (Price et al., 2012). ST398 LA-MRSA are also capable of causing infections in humans (Williamson et al., 2014; Sieber et al., 2019; Lu et al., 2021), a problem that is of particular concern across regions with high pig farming density (Köck et al., 2009; Vanderhaeghen et al., 2010; Gómez et al., 2020). Indeed, some MRSA are prone to cross species barriers (Sheppard et al., 2018) and are capable of rapid adaptation to different habitats and environmental conditions (Larsen et al., 2022). Adaptation processes were mainly studied on the genomic level and include either changes of the core genome (Huber et al., 2020) or an uptake of mobile genetic elements (MGEs; Mccarthy and Lindsay, 2013; Walther et al., 2018; Sieber et al., 2019; Huber et al., 2020; Leinweber et al., 2021).
Genome alterations also affect the staphylococcal cassette chromosome mec (SCCmec) including its mec complex which confers broad-spectrum β-lactam resistance in S. aureus and whose spontaneous loss was reported for ST398 isolates (Chlebowicz et al., 2010; Gómez et al., 2020). SCCmec elements represent large genomic islands of various sizes and compositions, which are inserted into the highly conserved region between the rlmH (previously orfX) and dus (previously orfY) genes of staphylococcal chromosomes (Semmler et al., 2016). Loss of SCCmec elements can either result from homologous recombination (HR) events (involving the recombination apparatus of the cell) or through precise excision of the element from its integration site. The latter process is driven by the SCCmec-encoded site-specific serine recombinases (i.e., ccrA/B or ccrC) which also mediate integration of the element into the rlmH gene (Hanssen and Ericson Sollid, 2006; Chlebowicz et al., 2010; Wang and Archer, 2010; Liu et al., 2017; Gómez et al., 2020).
Interestingly, loss of SCCmec is thought to provide an advantage for S. aureus in challenging environments, for example when exposed to antibiotics (Noto et al., 2008) and/or the host immune system (Read et al., 2018; Tickler et al., 2020); and in LA-MRSA, the specific environmental conditions surrounding pig farming were proposed as factors that might facilitate deletion of the element (Nübel et al., 2010; Vandendriessche et al., 2014).
In industrial pig farming, the toxic gas ammonium hydroxide (NH3) is released from manure during (bacterial) deamination of proteins fed to the animals. The gas subsequently accumulates in the air of the barn and, due to its high solubility in fluids, it is harmful to airway mucosal surfaces of pig farm workers and exposed pigs (Urbain et al., 1996; Wang et al., 2020) Thus, it is unavoidable that MRSA colonizing moist mucosal surfaces of the anterior nostrils of pigs are exposed to increased concentrations of ammonia. Therefore, exposure to high concentrations of ammonia is a condition commonly encountered by LA-MRSA ST398, and a recent study demonstrated that these bacteria indeed survive under such conditions (Astrup et al., 2021).
Regarding cytosolic pH, ammonia is the preferred nitrogen source of most microorganisms, and once in the cell, is directed to glutamine synthesis (Gruswitz et al., 2007). However, information on ammonia-induced (metabolic) gene expression patterns in LA-MRSA are currently scarce. Aside from exposure to the harsh barn environment, LA-MRSA remain associated with pigs as their primary host, which illustrates another condition the bacteria are required to adapt to.
Taking these different environmental conditions into account, the aims of this study were: (i) to reconstruct the region downstream of rlmH in an ST398 MRSA and its methicillin susceptible descendant (MSSA) in order to reveal the mechanisms involved in the loss of SCCmec; (ii) to evaluate the global transcriptomic response of MRSA ST398 exposed to particular LA environmental conditions, i.e., contact with host proteins (porcine serum) and enhanced ammonia concentrations, with a special focus on (iii) transcription levels of genes previously reported as being associated with the acquisition or loss of SCCmec elements.
Strain MRSA IMT38951, isolated from a nose swab of a pig in 2016, and its isogenic MSSA descendant IMT38951_42 (Busche et al., 2018) were used throughout this study. The MSSA isolate was discovered during serial-dilutions of overnight grown broth cultures [Mueller-Hinton (MH) bouillon (Becton Dickinson, Heidelberg, Germany)] of IMT38951. Presence and absence of methicillin resistance was determined according to CLSI standards (Clinical and Laboratory Standards Institute, 2013, 2014) and verified by PCR (Merlino et al., 2002). MSSA IMT38951_42 was enclosed to comparatively analyze the chromosomal integration site downstream of rlmH (Boundy et al., 2013), as described previously (Chlebowicz et al., 2010; Wang et al., 2017; Wu et al., 2018; Gómez et al., 2020).
Genomic bacterial DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s instructions. Isolates were pre-incubated with 0.1 mg/ml Lysostaphin and 0.1 mg/ml Proteinase K (both from Sigma-Aldrich, St. Louis, Missouri, United States). NanoDrop Spectrophotometer (Thermo Fisher Scientific, United States) as well as the Qubit 2.0 Fluorometer (Invitrogen, United States) were used to evaluate the DNA quality and quantity, respectively.
Whole-genome sequencing (WGS) was performed using an Illumina MiSeq which resulted in 300 bp paired-end reads with an obtained coverage >90X. Furthermore, size selection was conducted on the DNA using a 10 K Blue Pippin kit for additional sequencing with the Single Molecule Real-Time (SMRT) Sequencing Technology on a PacBio RS II by GATC Biotech AG (Konstanz, Germany). PacBio raw data was assembled by Flye v2.8.2-b1689 (Kolmogorov et al., 2019). Adapter-trimmed Illumina short reads were used for assembly correction utilizing the polishing procedure of unicycler v0.4.8 (Wick et al., 2017). De novo assembled genomes were annotated using Prokka v1.14.6 (Seemann, 2014). Both genomes were comparatively investigated with respect to single nucleotide polymorphisms (SNPs), insertions and deletions by read mapping utilizing botwie2 v2.3.0 (Langmead and Salzberg, 2012). Putative sequence variants were defined as divergent nucleotides with a minimum coverage of 10X and an allele frequency of >80%. ResFinder-4.1 (Zankari et al., 2012; Bortolaia et al., 2020) was used to identify resistance genes, and further genomic sites of interest were investigated using Geneious 11.1.5 (Biomatters Ltd., Australia; Kearse et al., 2012). PHASTER (Arndt et al., 2016) was utilized to identify putative (pro-) phages across both genomes.
Cells of IMT38951 were grown in 60 ml MH II and incubated overnight at 37°C and 250 rpm and, on the following day, diluted to an OD600 of 0.1 in 100 ml of fresh MH II and further incubated at 37°C and 250 rpm.
Based on previous research (Anderson et al., 2010; Malachowa et al., 2011; Cardile et al., 2014; Pinon et al., 2015) we used an ammonia concentration of 0.3% (Th. Geyer, Renningen, Germany), 10% porcine serum (Innovative Research Inc., Peary Court Novi, Michigan, United States) as well as a combination of both in order to mimic environmental conditions associated with pig-farming. Sampling time points were selected based on results of Stojanov et al. (2015) on SCCmec excision and the major regulative activity of the accessory gene regulator (agr)-system (Wang and Muir, 2016; Huber et al., 2020). At an OD600 of 0.4, the broths were supplemented with either porcine serum (10%), ammonia (0.3%), or both. To ensure the viability of the bacteria in the supplemented media, the viable plate count was determined every 15 min (0–90 min after media supplementation). Media from controls were not supplemented.
A previously published protocol (Lavoie and Summers, 2018) was used to select the samples for RNA sequencing to guarantee overall comparability. Briefly, for each of the experiments, seven biological replicates were initially grown alongside seven corresponding controls. The OD600 was determined for each set of samples and controls (Supplementary Table 1). Then, three out of the seven samples that exhibited the most similar OD600 values per growth condition and time point (10 and 60 min after media supplementation) were selected for subsequent RNA isolation (n = 18 samples). In addition, the three most similar control samples per growth condition and time point were selected as well (n = 18; written in bold in Supplementary Table 1). Each aliquot was centrifuged at 20,000 g and 4°C for 3 min. The supernatant was aspirated, and the pellet was immediately frozen at −80°C following previously published protocols (Mäder et al., 2016; Lavoie and Summers, 2018).
In total, 36 cell pellets (18 samples and 18 associated controls) were shipped to LGC Genomics GmbH (Berlin, Germany), where the RNA was isolated using the RNASnap method (Stead et al., 2012). RNA sequencing was performed on an Illumina NextSeq 500/550 V2, resulting in one channel paired-end reads. Details of the company’s standard protocols for quality control, RNA extraction and rRNA depletion using Ribo-Zero (Epicentre Biozym, Hessisch Oldendorf, Germany) are available online.1 cDNA synthesis, library generation, indexing and cluster generation were performed using Illumina technology (TruSeq RNA Sample Preparation Kit v2).
SCORE v1.0.2 (Wolf et al., 2020) was used to perform differential RNA sequencing analysis by comparing the transcriptomic profiles of selected challenges (ammonia, porcine serum or the combination of them) with control samples, resulting in the identification of differentially expressed genes (DEGs) across the sample set. Initial analysis steps including read preprocessing (trimming of low-quality ends with Phred score < 20), mapping to the genomic reference, transcript quantification (genes below 10 counts were discarded), statistical identification of differentially expressed genes between sample groups (corrected p ≤ 0.05), overrepresentation analysis of associated Gene Ontologies (GO) and visualization of affected Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were performed within SCORE. An additional fold change filter (log2FC > 1) was applied to ensure biological relevancy of the DEGs (Slany et al., 2017). Genes not fulfilling these criteria were counted as not being differentially expressed between conditions. The resulting expression tables were subsequently merged and additionally characterized using CD-Search v3.18 (Marchler-Bauer and Bryant, 2004) and eggNOG-mapper v2.0.5 (Huerta-Cepas et al., 2019). Principal component analysis (PCA) was performed based on normalized transcripts per million (TPM) expression values to assess inter-sample distances (Scholes and Lewis, 2020).
To allow determination of the precise genomic sites flanking the loss of a mecA-containing sequence in MSSA IMT38951_42, a finished whole-genome reference sequence was required. PacBio long reads were utilized to assemble the draft genomes using Flye, resulting in a coverage of >335-times and > 331-times. Short-read polishing of the genomes was performed, providing an additional 161X and 187X sequence coverage to the reconstructed genomes of IMT38951 (MRSA) and IMT38951_42, respectively (Figure 1).
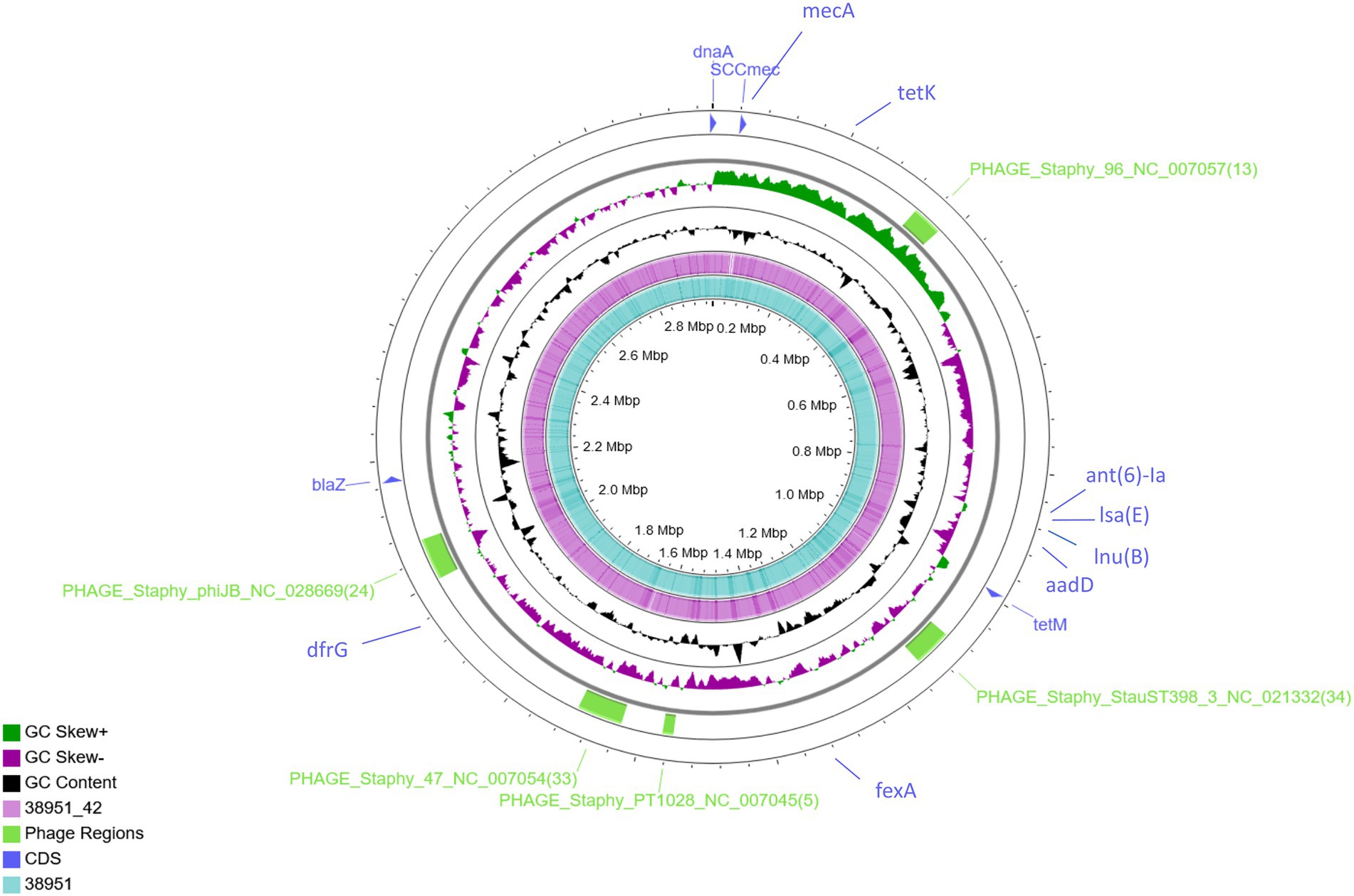
Figure 1. Genome-wide comparison of MRSA ST398 (IMT38951) and its descendant MSSA (IMT38951_42). A combinatorial approach including long reads (PacBio) and short reads (Illumina sequencing) allowed precise reconstruction of both genomes. Five regions harbouring phages or phage-associated genes identified using PHASTER are indicated in green. Location of resistance genes identified using ResFinder 2.1 and the SCCmec intergration site are indicated in blue. Comparative genomics determined a difference of 10 single nucleotide polymorphisms in total (excluding phage regions).
Further comparative WGS analysis revealed an overall genome size of 3,022,864 bases for IMT38951 and 3,011,968 bases for IMT38951_42. The former contains 2,896 coding sequences (CDS), 19 rRNA and 61 tRNAs, while the latter harbors 30 CDS less according to the annotation. Initial strain typing of IMT38951 and IMT38951_42 assigned both isolates to LA-ST398 and spa type t12359. In addition to the mecA gene present in IMT38951, both genomes harbour a similar set of further resistance genes [aadD, fexA, blaZ, dfrG, tet(K), tet(M), lnu(B) and lsa(E); Figure 1]. Prediction of prophages by PHASTER based on sequence similarity-based searches revealed three hits for complete phages, PHAGE_Staphy_47_NC_007054 (72.2 kb in size), PHAGE_Staphy_phiJB_NC_028669 (60 kb) and PHAGE_Staphy_StauST398_3_NC_021332 (66.9 kb) and two further incomplete phages of the lengths 16.6 kb (PHAGE_Staphy_PT1028_NC_007045) and 41.2 kb (PHAGE_Staphy_47_NC_007054), respectively (Figure 1). Both strains harbour a small plasmid of 5,359 bp size that resembles the Staphylococcus hyicus plasmid pKKS966 (99.82% identity, 73% coverage GenBank: FN677368.1) carrying the trimethoprim resistance gene dfrK (Kadlec et al., 2012).
Pairwise distance and relatedness (Figure 1) was calculated, revealing a total difference of 10 SNPs (excluding phage regions) and the lack of a 16 kb sequence downstream of the SCCmec integration site (rlmH) in IMT38951_42 (Figure 2). A closer inspection of the region revealed the loss of a complete SCCmecVc element (Figure 2) comprising the ccrC8 type recombinase, the C2 mec complex and a recently described putative operon containing an A-family DNA polymerase (CCPol) together with a small protein lacking conserved domains (“middle protein,” MP) as well as a putative helicase (Cch2; referred to as “CCPol-MP-Cch2” I & II in Figures 2, 3), as recently described by Bebel et al. (2020). The region downstream of rlmH in IMT38951 includes a notable repertoire of homologues sequences, i.e., two distinct but closely related ccrC variants and two nearly homologues intergenic spacer regions of 224 bp length, four transposases (tnp) of insertion sequence (IS) element 431 (IS431) and two variants of the replication machinery CCPol-MP-Cch2 (Figure 2; Supplementary Table 2) (region rlmH – dus, highlighted in green). The data suggest that the deletion of SCCmecVc occurred during HR between either ccrC8 and ccrC1 (first 48 bp are 100% identical) or the terminal 38 bp (identity: 100%) of the two 224 bp-comprising intergenic spacer regions upstream of the ccrC variants, respectively (Figure 3; Supplementary Figure 1). Both loci harbour short repetitive sequences (“TAAAA,” A & B and C & D in Figure 3) that provide at least two distinct possibilities for HR without any further sequence alterations apart from the loss of the SCCmec element within this region. Both intergenic spacer sequences present in the MRSA variant harbor the original (identical) promoter region of ccrC variants, including the 19 bp SOS box described by Liu et al. (Liu et al., 2017; Supplementary Figure 1). Interestingly, the deleted SCCmecVc element still harbours its putative autonomous replication apparatus, since all loci involved (direct repeats, inverted repeats, origin of replication, CCPol-MP-Cch2 II) remain completely unaffected by the proposed recombination event (Supplementary Figure 1).
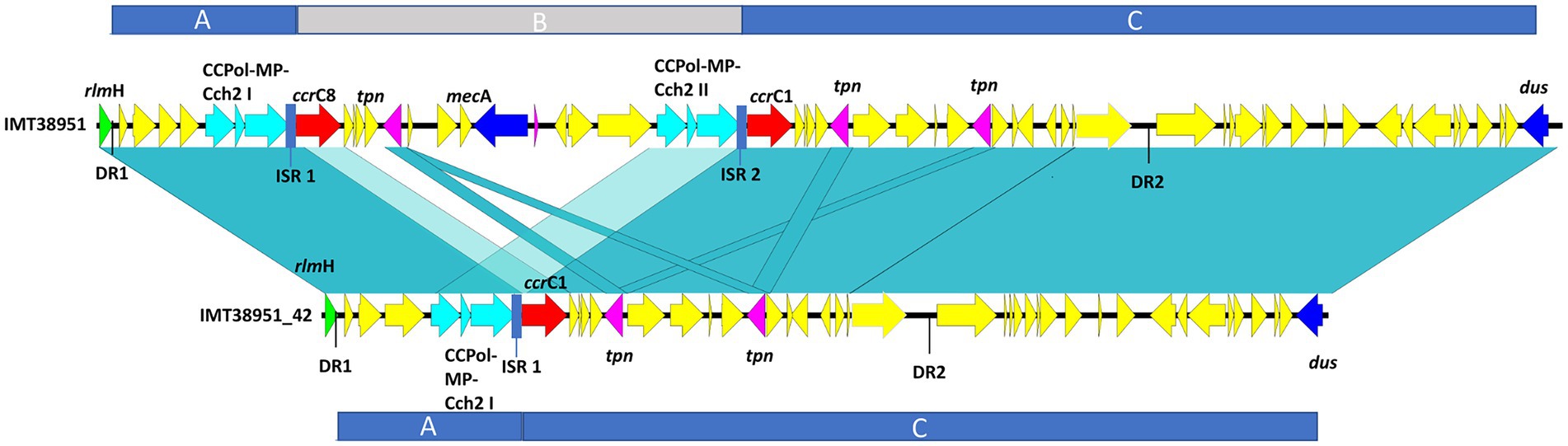
Figure 2. Sequence comparison of IMT38951 and IMT38951_42 from rlmH (previously: orfX) to dus (previously orfY). Repetitive elements and homologues regions challenged the precise reconstruction of MRSA IMT38951 and its isogenic descendant lacking SCCmecVc (region B). Since sequence identities of genes and intergenic spacer regions present in this region range from 98/99% (light blue-green) to 100% (blue-green), including sequences and genes that flank (regions A and C) the missing SCCmec element in IMT38951_42. DR, direct repeat of 15 bp length; CCPol-MP-Cch2, operon containing an A-family DNA polymerase (CCPol) together with a small protein lacking conserved domains (“middle protein,” MP) as well as a putative helicase (Cch2) (Bebel et al., 2020); ccrC, chromosomal cassette recombinase C; tpn, transposase associated with insertion element (IS) 431; ISR, intergenic spacer region of 224 bp length.
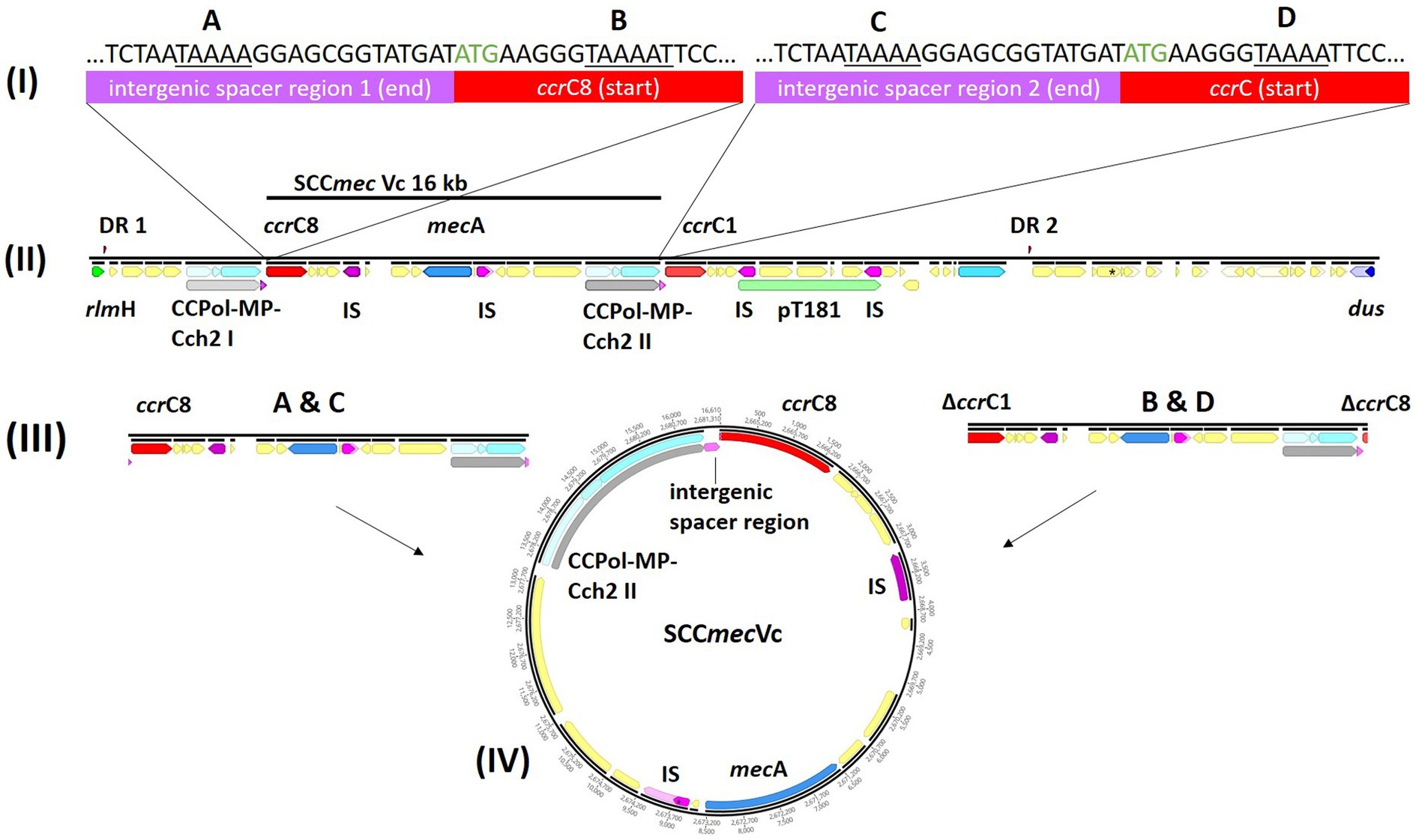
Figure 3. Tracking the loss of SCCmecVc. MRSA ST398 IMT38951: Illustration of the SCCmec integration site downstream of rlmH. (A) Comparison of the terminal sequences of the intergenic sequences spacer region 1 (I) and 2 (III) and the initial basepairs of ccrC8 (II) and ccrC1 (IV) (start codon: green) revealed two possibilities of homologues recombination after deletion of SCCmec: A & C, intergenic regions 1 and 2; and B & D, ccrC8 and ccrC1. (B) The region between rlmH (previously “orfX“) and dus (previously orfY) carries various repetitive DNA sequences. (C) Both recombination events would allow deletion of an identical and complete SCCmecVc element. (D) Circular SCCmec element with putative functional recombinase ccrC8 and a CCPol-MP-Cch2 II complex (Bebel et al., 2020) conferring its autonomous replication capabilities. ccr, chromosomal cassette recombinase; IS, insertion sequence; DR, directs repeats associated with ccr-mediated integration and excision of SCCmec elements (Semmler et al., 2016).
A ccr-mediated integration and excision, on the other hand, is known to change the number of 15 bp DR in the region downstream of rlmH (Semmler et al., 2016). Here, the total number and location of the two distinct 15 bp repeats present in the MSSA descendant is unchanged compared to its MRSA predecessor (Figure 2), arguing against recombinase driven loss of SCCmecVc.
Further sequence comparison with the type V SCCmec (GenBank: AB512767) genes and intergenic regions of a strain (TSGH17) belonging to another livestock-associated genetic background (ST59), used by Bebel et al. (2020) to describe the CCPol-MP complex and their Cch2 helicases, revealed a sequence identity of 94.9% and 80 SNPs, two larger insertions/deletions of 40 bp and 1,275 bases with 4 single insertions/deletions proving continuous evolvement of the region downstream of rlmH.
The transcriptomic profiles of IMT38951 triplicate samples challenged by environment-mimicking conditions such as an increased ammonia concentration (pH 9.0), porcine serum (pH 7.3) and a combination of them (pH 8.9) were compared to corresponding unchallenged control samples (pH 7.2) after 10 and 60 min of exposure. Cell viability and growth kinetics were assessed from immediate exposure to 90 min afterwards (Supplementary Figure 2). In addition, exposure to each of the environment mimicking conditions seemed to require a characteristic cellular response, as demonstrated by the principal component analysis carried out to ensure limited variation between individual samples (Supplementary Figure 3). A global and summarized overview on gene expression in transcripts per million (TPM) and DEGs (corrected p ≤ 0.05), is provided in Supplementary Table 2. DEGs were divided into upregulated (log2FC > 1) and downregulated genes (log2FC < −1), revealing characteristic transcription patterns for each sampling time point and growth condition (global overview provided in Figure 4). Based on the heatmap revealing the highest differentially expressed genes across all sample conditions investigated (Figure 5), the most important mechanisms and pathways involved in transcriptomic responses have been further evaluated.
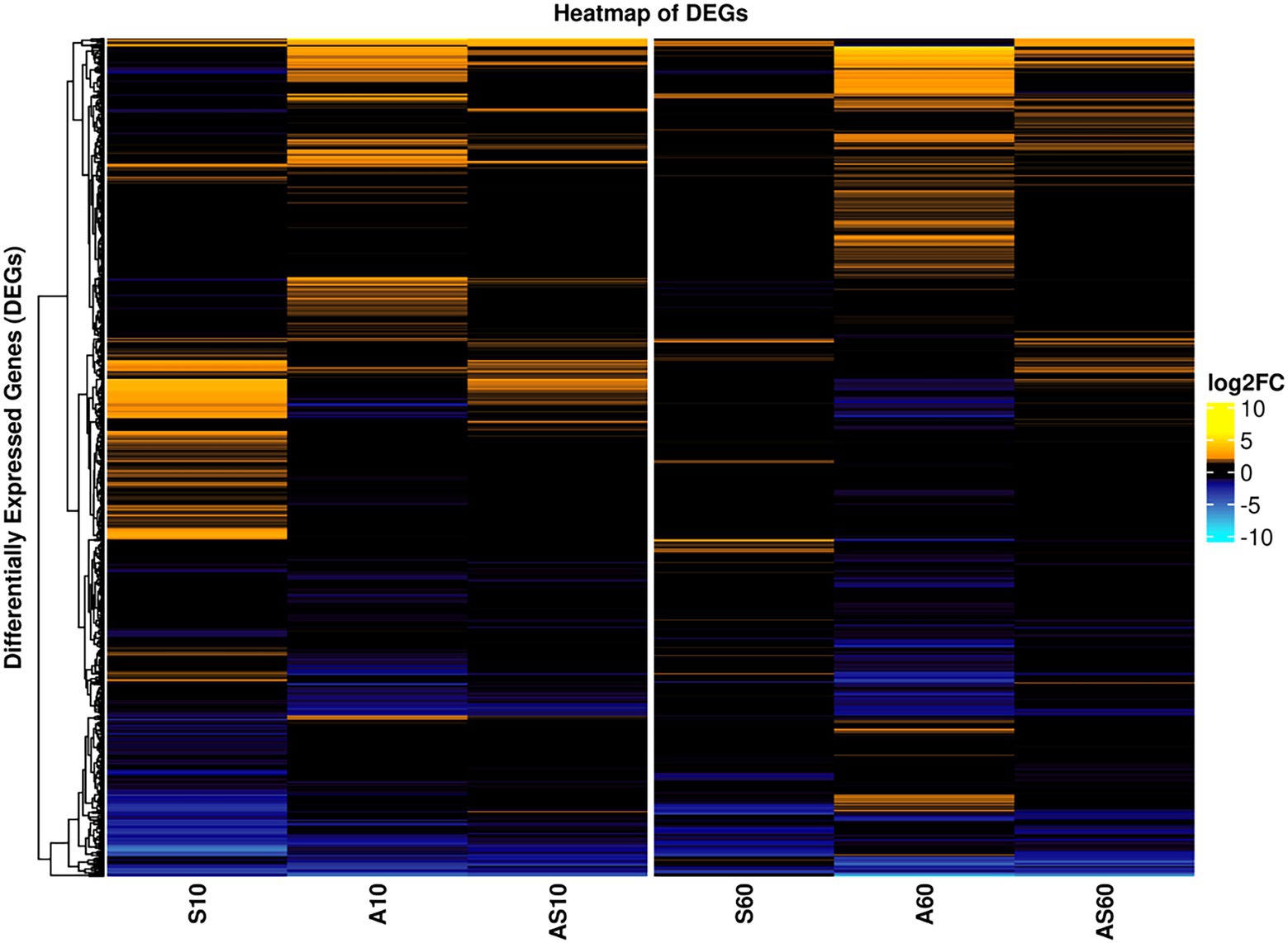
Figure 4. Global overview on differentially expressed genes for samples and conditions after 10 and 60 min of exposure. Pattern analysis of differentially expressed genes (DEGs) are visualized based on their log2 fold-change (log2FC) compared to the corresponding control samples, ranging from up- (yellow) to downregulated (blue). Each column corresponds to a distinct growth condition (S = serum, A = ammonia, AS = ammonia & serum) and time point (10 min and 60 min of exposure). Genes are clustered based on their respective expression across the tested conditions (left). Expression ranges from-10 log2FC to +10 log2FC.
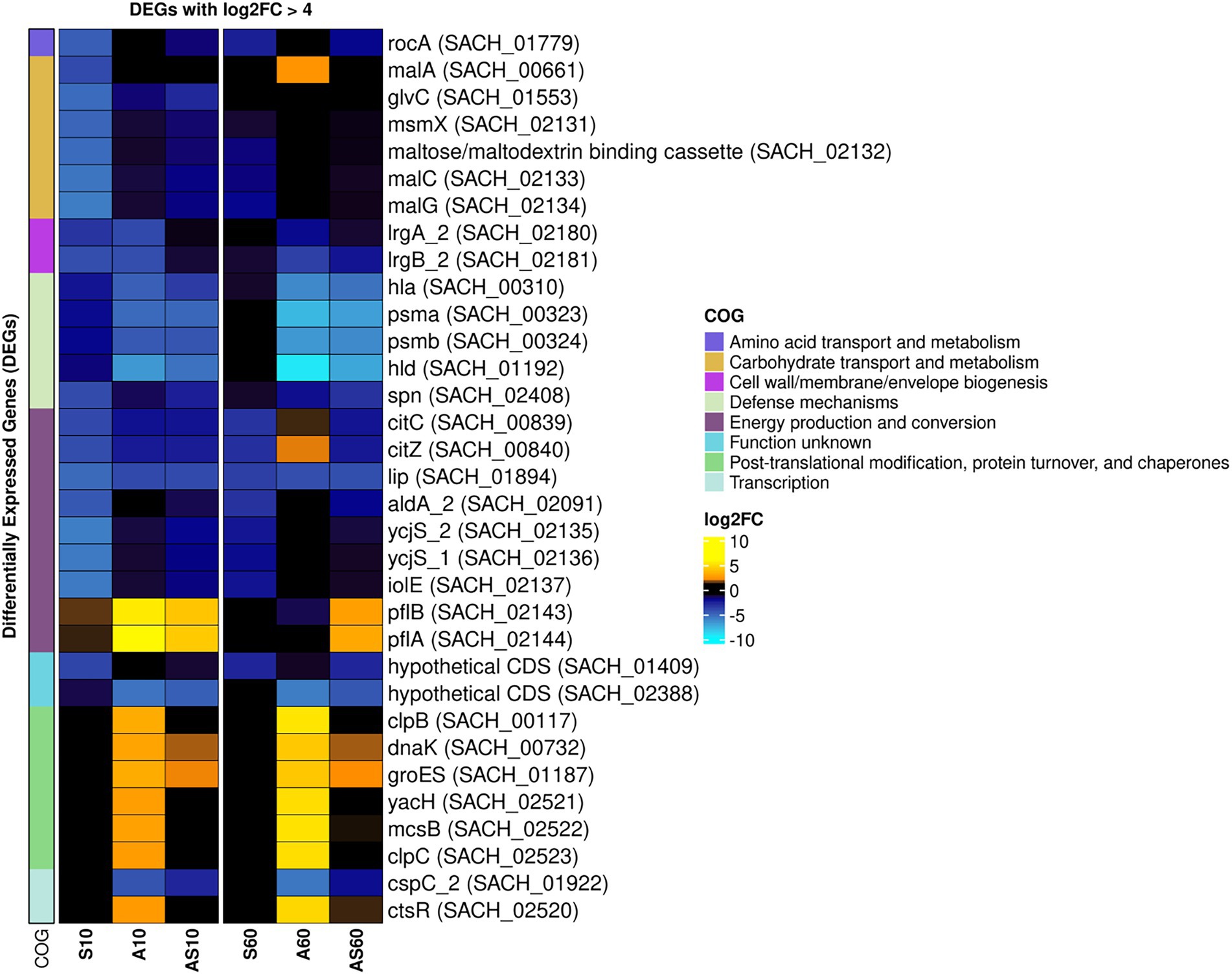
Figure 5. Heatmap of the top differentially expressed genes across all sample conditions. Top differentially expressed genes (DEGs) (n = 41) are visualized based on their respective log2 fold-change (log2FC) compared to the corresponding control samples. Each column corresponds to a stress condition (S = serum, A = ammonia, AS = ammonia & serum) and time point (10 min and 60 min). Genes are either strongly upregulated with log2FC > 5 (yellow) or downregulated with log2FC < −4 (blue). Genes are furthermore annotated with corresponding COG ontologies (left side).
Global regulators that might be affected by the growth conditions were the class I heat shock responses comprising the HrcA regulon (hrcA-grpE-dnaK-dnaJ and groESL) that is embedded within the CtsR regulon (Michel et al., 2006) which was mostly induced, especially in the presence of ammonia (Table 1).
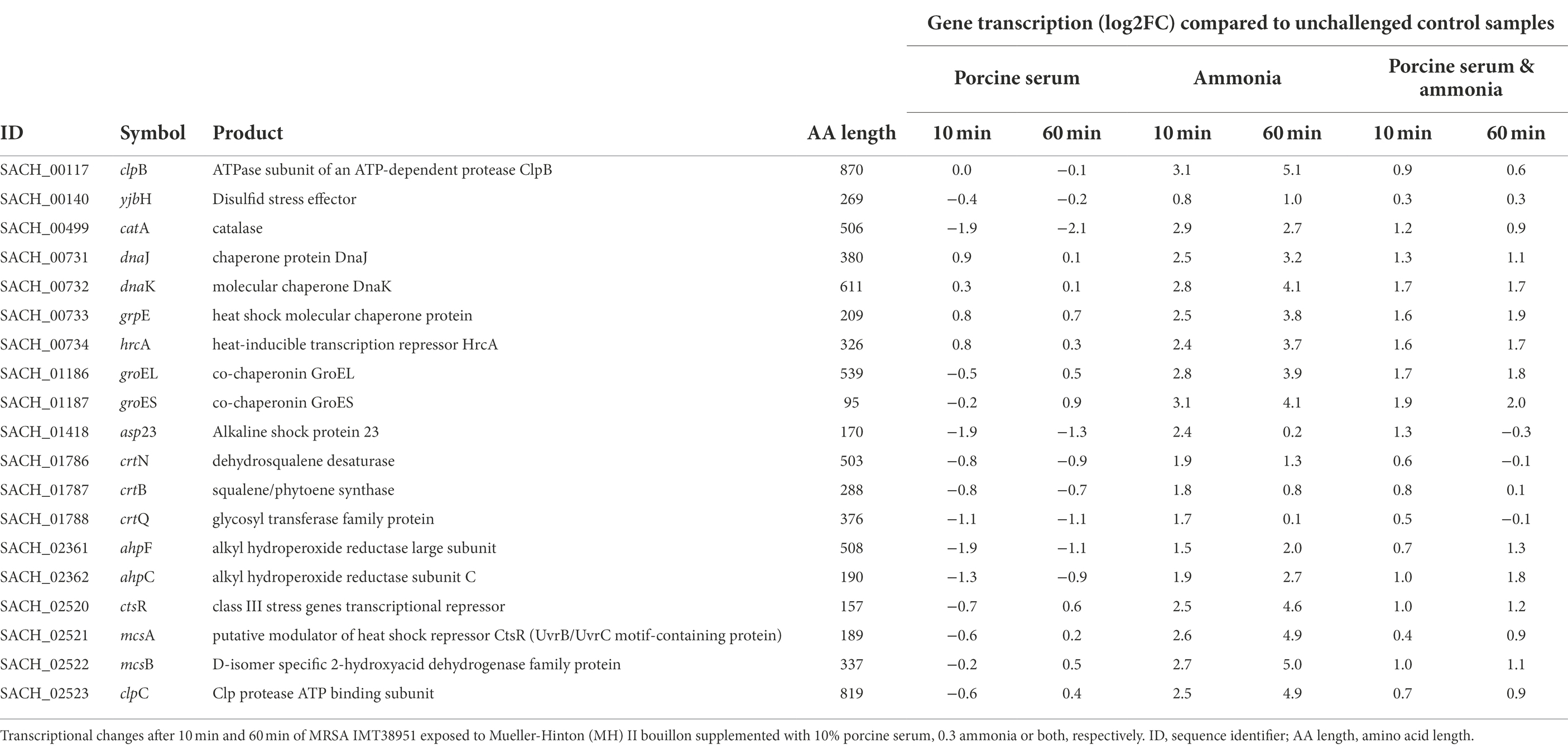
Table 1. Transcription of genes associated with coping mechanisms in livestock-associated MRSAST398.
The center of the regulatory network in S. aureus consists of quorum sensing through the accessory gene regulator (agr) system, which concerts virulence factor transcription and synthesis, beyond others (Novick et al., 1993; Gaupp et al., 2016). We noticed a decrease in transcription of the agr locus compared to the respective unchallenged control samples for all experiments supplemented with ammonia (up to −4.7 log2FC; Table 2), a result likely related to the reduced growth kinetics associated with these conditions (Supplementary Figure 2). The addition of ammonia reduced the activity of the response regulator of the agr locus, AgrA. There was also down-regulation of AgrA-dependent genes/operons such as the phenol soluble modulins (PSMs) encoding operon (psma and psmb), as well as of the agr-inherent effector molecule RNAIII (a larger sRNA which also includes the delta-hemolysin open reading frame (hld)) noticed in ammonia-challenged cells (Supplementary Table 2). Reduced levels of RNAIII, are likely to decrease the transcription of toxin encoding genes. This is exemplified by decreased transcript levels of the α-toxin encoding gene hla (Supplementary Table 2), presumably via reduced degradation of rot transcripts, encoding the repressor of toxins (Boisset et al., 2007), which were found to be enhanced by 1.4 to 1.9 log2FC in presence of ammonia, as compared to the controls (Table 2). Reduced RNAIII levels may also explain the decreased transcript levels seen for mgrA (encoding the global transcriptional regulator MgrA) in presence of ammonia (Table 2), as RNAIII is known to stabilize the mgrA mRNA (Gupta et al., 2015).
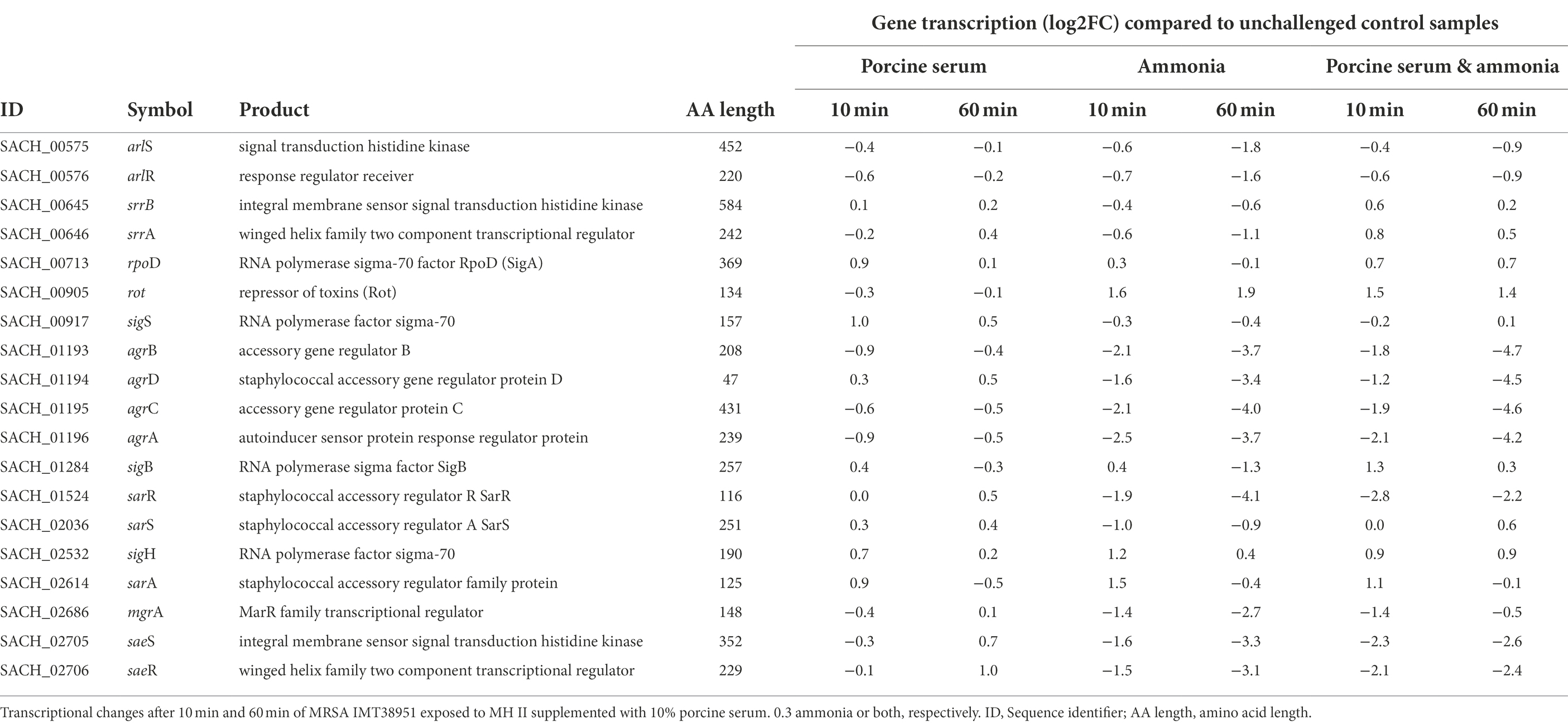
Table 2. Transcriptional changes of global regulators in livestock-associated MRSAST398 exposed to ammonia porcine serum or both.
When IMT38951 cultures were challenged with ammonia (in presence or absence of serum), clear reductions in sae (S. aureus exoprotein expression) transcript rates were noticed for both time points monitored (Table 2). Another potential factor contributing to the decrease in sae transcripts seen in presence of ammonia is SigB. This stress-induced alternative sigma factor is known to decrease sae transcription (Geiger et al., 2008), and is activated among others by a rapid increase in pH (Pané-Farré et al., 2009). Although the sigB operon is partially autoregulated, SigB activity is best followed by monitoring alkaline shock protein 23 (asp23) transcription (Giachino et al., 2001). Here, the sigma-B regulated asp23 gene (Kuroda et al., 1995) was induced after 10 min of exposure to ammonia alone (2.4 log2FC) or the combination of ammonia and porcine serum (1.2 log2FC; Table 1). In addition, transcription of only a part of the SarA regulon was found to be altered by ammonia supplementation in a relevant manner (i.e., hla, hlgA, lrgAB, nrdG, psma, but not aur, esxA, icaADBC, nuc, sdrD, and sodM; Supplementary Table 2).
Although the equilibrium of the chemical reaction NH3 + H2O ⇌ NH4+ + OH-shifts towards the left site (especially at pH 9), the rather limited number of free protons in bacterial cells (Saito and Kobayashi, 2003) may be captured by the hydroxyl radicals (OH−). Therefore, the formation of a small fraction of hydroxyl radicals (OH−) in all samples exposed to ammonia is expected (Saito and Kobayashi, 2003). Here, we noted a transient induction of genes (maximum at 10 min of exposure) known for their response to the presence of reactive oxygen species (ROS) such as hydroxyl radicals (Pandey et al., 2019) including parts of the machinery involved in staphyloxanthin biosynthesis such as the crtOPQMN operon (Pelz et al., 2005), a carotenoid thought to protect S. aureus from oxidative stress (Clauditz et al., 2006). Similarly, genes encoding catalase (katA) and especially alkyl hydroperoxide reductase (ahpC) were most effectively induced after 10 & 60 min of exposure to ammonia alone (Table 1).
Exposure to serum or even the combination of serum and ammonia did not affect transcription of the gene encoding the ribosome-associated translation inhibitor (raiA) after either 10 or 60 min, although 60 min of exposure to ammonia alone lead to a 2.0 log2FC, indicating the necessity to decrease overall protein production to cope with the presence of enhanced ammonia concentrations for more than an hour (Supplementary Table 2).
Of note, transcription of several virulence-and defence associated genes was transiently induced by exposure of MRSA ST398 to porcine serum only, i.e., genes encoding the von Willebrand-factor binding protein (10/60 min 2.2/1.4 log2FC), the chromosomal staphylococcal complements inhibitor (scn; 10/60 min 1.0/0.2 log2FC), an additional pathogenicity island-associated scn variant (10/60 min 1.5/1.6 log2FC), staphylocoagulase (coa; 10/60 min 2.2/0.6 log2FC) and immunoglobulin G-binding protein (sbi; 10/60 min 1.1/0.7 log2FC; Supplementary Table 2).
In S. aureus, the SOS response following the occurrence of ssDNA is tightly regulated by the interplay of the DNA damage-inducible repressor LexA and the recombination protein RecA (Butala et al., 2009; Maslowska et al., 2019; Podlesek and Žgur Bertok, 2020). Activation of RecA by binding to single-stranded DNA promotes transcriptional upregulation of genes belonging to the SOS-regulon by activation of LexA self-cleavage from the respective promoter sites. Although actual activation of the synthesized proteins cannot be followed by transcriptional analysis, some of the genes associated with SOS response in S. aureus are transcriptionally induced, i.e., the topoisomerase IV genes parE and parC at 10 and 60 min (1.3 /0.6 and 1.7 /0.8 log2FC) after exposure to porcine serum (Supplementary Table 2). The LexA-regulated low-fidelity and error-prone DNA polymerase V (Ha and Edwards, 2021) umuC (1.7 log2FC) and genes involved in nucleotide excision repair (uvrA/B; 1.5/1.2 log2FC) are significantly induced after 60 min of exposure to ammonia. Since a similar SOS-mediated induction of ccr genes by cleavage of LexA from its promoter sequence was reported previously (Liu et al., 2017), the transcriptional response of both ccr variants was evaluated. Both ccr allelic variants in IMT38951 lacked significant induction over the time points and conditions evaluated here, although a minimal induction was noted after 60 min of exposure to ammonia (between 0.1 and 0.2 log2FC; Figure 6). However, this observation seems in overall congruence with the limited but notable induction of genes belonging to the SOS-response after 60 min of exposure to ammonia described above. The transcription of lexA, on the other hand, is 0.7 log2FC after 10 and 1.5 log2FC after 60 min of exposure to ammonia, a rate that may indicate that LexA has been consumed by SOS-induction. Of note, recA transcription levels lack significant changes compared to the unchallenged controls at all conditions investigated (Supplementary Table 2).
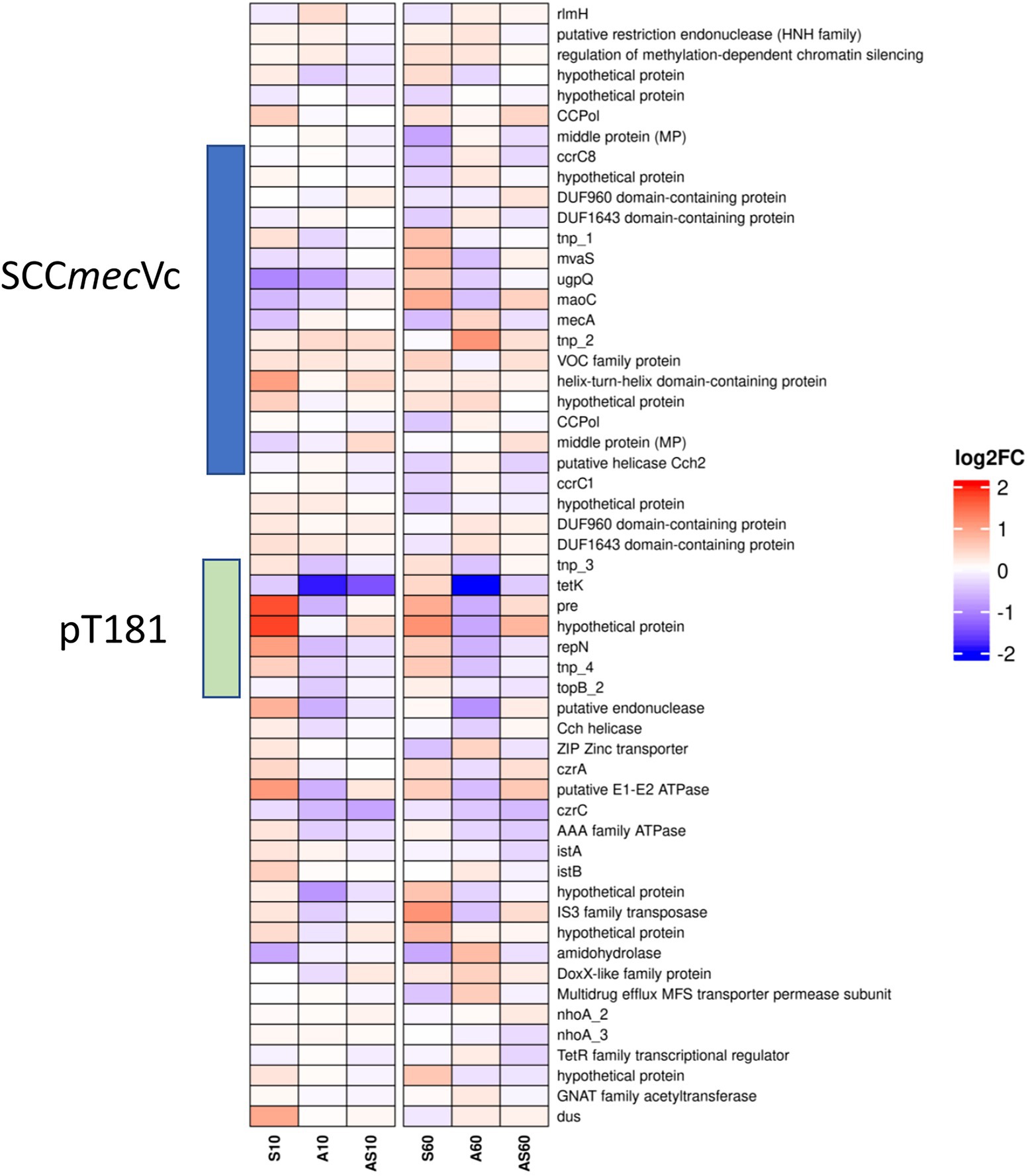
Figure 6. Heatmap illustrating transcription of genes at the chromosomal SCCmec integration site from rlmH (“orfX”) to dus (“orfY”). Gene transcripts are visualized based on their log2 fold-change (log2FC) with either red (upregulated), blue (downregulated) or white (no changes) compared to the corresponding control samples. Each column corresponds to a growth condition (S = serum, A = ammonia, AS = ammonia & serum) and time point (10 min and 60 min). Overall expression ranges from-2 log2FC to +2 log2FC.
Data on up-or downregulation of predicted CDSs within the genomic region harboring SCCmec, starting with the rRNA large subunit methyltransferase (rlmH) and ending with tRNA dihydrouridine synthase (dus), is provided in Figure 6 and Supplementary Table 2 (highlighted in green). Considering genes putatively involved in mobilization and recombination of transferrable MGEs present in this particular region, we noticed an increased transcription of transposase tnp_2 associated with IS431, located adjacent to mecA, after 10 min (0.3 log2FC) and 60 min (1.0 log2FC) of exposure to ammonia (Figure 6). Moreover, the replication initiation gene repN (10/60 min 1.0/0.5 log2FC) and the gene encoding for a plasmid recombination protein (pre; 10/60 min 1.7/0.8 log2FC), both belonging to the tetracycline resistance gene (tetK)-carrying plasmid pT181 that integrated into the SCCmec region downstream of ccrC1 (Figure 3A), showed a transcriptional increase after exposure to porcine serum (Figure 6; Supplementary Table 2).
Genes associated with both variants (I and II Figure 3) of the replication machinery CCPol-MP-Cch2 were not significantly induced by the growth conditions investigated here (Supplementary Table 2; Figure 6).
Following the loss of methicillin resistance in IMT38951 genomic investigations were hindered by multiple repetitive sequences in the region downstream of rlmH. This was overcome by a combination of long read sequences polished by short reads to allow for an accurate genomic inspection of this particular region, including a detailed reconstruction of the process most likely leading to the loss of the complete SCCmec element from its chromosomal integration site, outlined in Figure 3.
Previous research indicated that deletion followed by HR, or excision driven by large serine recombinases, allow complete SCCmec elements to detach from their chromosomal integration site downstream of rlmH (Hanssen and Ericson Sollid, 2006; Chlebowicz et al., 2010; Wang and Archer, 2010; Liu et al., 2017; Gómez et al., 2020). This likely requires the involvement of the serine recombinases (ccrA/B or ccrC) that facilitate chromosomal integration and excision of SCCmec, or other factors belonging to the bacterial recombination machinery. Since SCCmec elements are typically flanked by “direct repeats” (DR) of 15 bp length that are recognized by the respective large serine recombinases, ccr-driven integration or loss of these elements always leads to changes in the number of DRs at the chromosomal integration site (Ito et al., 1999; Semmler et al., 2016). However, since the number and position of the two direct repeats present in the MRSA predecessor remained unchanged in the descendent, MSSA IMT38951_42, ccr-mediated excision of the SCCmec element, as described previously (Noto et al., 2008; Wang and Archer, 2010; Zhang et al., 2015, 2016), is unlikely.
A further study identified recombination between the putative primase-encoding (now: helicase Cch2, according to Bebel et al., 2020) genes belonging to the CCPol-MP-Cch2 complex in the ST398 lineage (Gómez et al., 2020). Other authors have linked the loss of SCCmec with recombination between ccrC allelic variants (Chlebowicz et al., 2010; Vandendriessche et al., 2014), a possibility that cannot be ruled out for the isogenic pair belonging to ST398 in our study (Figure 3, recombination between B and C). However, the actual region prone to recombination described above comprises the end of intergenic spacer region 1 and the initial 48 bp of both ccr genes (Figure 3A), a region that harbors four identical short repetitive sequences. Interestingly, both recombination events (A & C and B & D) events would produce an identical SCCmec circular element (Figure 3D). Consequently, occasional deletion followed by HR might be of greater biological importance for the spread of methicillin resistance than previously considered, as Bebel et al. reported autonomous replication capabilities for exactly these elements (Bebel et al., 2020) that contribute not only to the maintenance of the circular SCCmec intermediates, but also to their proliferation within an extra-chromosomal space. Subsequently, novel SCCmec elements might be available for horizontal transfer to other suitable recipients - a hypothesis that should be addressed in future research.
Recently, the ability of the MRSA ST398 lineage to survive in pig manure has been demonstrated (Astrup et al., 2021). This particular LA environment is known for high natural ammonia levels - a major selective factor for bacterial community structures (Nordgård et al., 2017). In S. aureus, early responses to environmental challenges are often associated with changes in the activity of global regulators (Crosby et al., 2016; Ranganathan et al., 2020; Párraga Solórzano et al., 2021). Activation of components belonging to the heat shock response seems to be among the most important coping mechanisms involved with respect to ammonia-rich environments. The latter is a protective mechanism considered crucial for bacterial survival and adaptation to hostile environmental conditions by degradation of misfolded and denatured proteins and prevention of protein misfolding events (Roncarati and Scarlato, 2017).
The number of free protons present in a bacterial cell depends on the pH: At an alkaline pH above 9, the number of free protons is reduced to less than one per bacterium (calculated for Escherichia coli), a fact that hinders the protonation of specific sites in enzymes required for their activity (Saito and Kobayashi, 2003). Consequently, cellular stress including disturbances of pH-sensitive enzymatic reactions, energy metabolism and secondary protein structures were expected to feature in the transcriptomic responses of S. aureus cells exposed to ammonia at an alkaline pH (Gutierrez et al., 2022). Moreover, the data suggests that the electron pair of the NH3 molecule might interfere with hybrid bonds of peptides and/or proteins under these conditions, since addition of more protein (i.e., presence of porcine serum) reduces the effects associated with exposure to ammonia alone, arguing against a pH dependent effect alone. In line with this, the initial pHs of the ammonia-supplemented samples differed only slightly (ammonia: pH 9.0/ammonia & porcine serum: pH 8.9) and did not change significantly over time.
A reduction of the bacterial transcription machinery prevents excessive energy loss from protein synthesis (Roncarati and Scarlato, 2017). As shown in Supplementary Figure 2, viable cell numbers had the smallest increase over time in samples exposed to ammonia only, suggesting that down-regulation of the translation machinery (Supplementary Table 2) contributes to survival and adaptation in ammonia rich environments.
In addition, the asp23 expression that is controlled by the alternate sigma factor of RNA polymerase (SigB) was upregulated after 10 min of exposure to 0.3% ammonia and the combination of ammonia and porcine serum, an increase that has been reported for sodium hydroxide-induced pH increases previously (Anderson et al., 2010). However, after 60 min, the asp23 transcription rates were comparable to the control, suggesting that ammonia supplementation induces a short-lived activation of SigB. This is in line with earlier observations reporting that SigB activity is only transiently induced by stresses such as heat shock (Gertz et al., 1999; Giachino et al., 2001). Further support for a stimulating effect of ammonia on SigB activity is given by our findings of increased transcript rates for several SigB-regulated genes known to be directly controlled by this factor such as the genes crtMN, whose products are involved in staphyloxanthin biosynthesis, a carotenoid thought to protect S. aureus from oxidative stress (Clauditz et al., 2006), spoVG, a transcription factor acting downstream of SigB (Meier et al., 2007), and sarA (Bischoff et al., 2001). Notably, SarA is also reported to act as regulatory protein responsive to redox and pH (Fujimoto et al., 2009), and to repress the expression of rot (Manna and Ray, 2007).
The observed reduction in sae transcription in all samples except those exposed to porcine serum for at least 60 min might be, at least in part, due to increases in SigB and Rot-activities, which were shown to decrease sae transcription from the sae P1 and P3 promoters, respectively (Geiger et al., 2008; Li and Cheung, 2008), and by a reduction in RNAIII transcripts, which are known to enhance sae transcription (Novick and Jiang, 2003; Geiger et al., 2008).
Although not directly regulated by agr, gene transcription of a few virulence factors located on mobile genetic elements that can be considered as a specific response to host (porcine) proteins such as scn and the von Willebrand Factor binding protein were decreased in all samples exposed to ammonia with or without porcine serum, indicating that the pH of 8.9–9.0 and/or the NH3 molecule interferes with (host-)protein recognition, requires energy saving or both.
Transcription of genes involved in capsular polysaccharide biosynthesis in ammonia supplemented media was not recorded, although their upregulation due to increased alkaline conditions has been demonstrated before (Anderson et al., 2010).
Taken together, LA-MRSA ST398 seems fully equipped to endure pig-farm associated environmental factors such as ammonia and porcine serum, helping to support their long-term survival within that environment.
In S. aureus, the genomic region between origin-of-replication (ori) and the SCC generally seems prone to HR, with the integration site for SCC representing a particular “hotspot” in this regard (Everitt et al., 2014; Semmler et al., 2016). A recent study investigated the stability of four different SCCmec elements (I, II, III, IV) during 3 months of serial subculturing at room temperature, revealing that SCCmec stability was influenced both by internal mobile elements (IS431) as well as the cell environment (Scharn et al., 2022). Exposure to pig-farming associated environmental challenges did not significantly alter the expression ratios of genes likely to be involved in the mobilization of complete or partial SCCmec elements, including transposases associated with IS431 and the ccr recombinases (Wang and Archer, 2010; Wang et al., 2017; Scharn et al., 2022) in this study when compared to unchallenged controls. These results suggest that the aforementioned conditions do not strongly contribute to SCCmec mobility to a large extent - at least not within the timeframe investigated here.
Moreover, exposure to ammonia, porcine serum or both failed to significantly induce recA transcription that would lead to de-repression of LexA-controlled genes including allelic variants of ccrC, as reported before (Liu et al., 2017). Although some genes tightly regulated by LexA such as uvrAB and umuC showed a clear log2FC increase after 60 min of exposure, the corresponding transcription rate noted for both ccr genes (0.1/0.2 log2FC) lacked significant difference compared to the controls. Since autocatalysis of LexA is required for derepression of some genes within the SOS regulon (Ha and Edwards, 2021) including lexA, others are only derepressed when the N-terminal domain is further digested by Clp proteases including ClpCP. Here we noted an induction between 2.5 log2FC after 10 min and 4.9 log2FC after 60 min for clpC in cells exposed to ammonia only (Table 1), once more suggesting a fine-tuned activation of the SOS machinery in MRSA ST398 strain IMT38951. Therefore, a massive cleavage of LexA from the ccrC promoter region may depend on more invasive DNA damaging conditions, i.e., the presence of distinct antibiotics, as reported before (Cirz et al., 2007; Liu et al., 2017). It seems worth considering that DNA-damaging effects attributed to ammonia rich environments and further challenging circumstances, for instance, exposure to antibiotics, may accumulate in vivo. In light of the recent results of Scharn et al. (2022), we speculate that prolonged exposure to livestock-associated conditions might induce a more prominent transcription of the ccr genes.
Here we followed the deletion of a complete and fully functional SCCmec Vc element from its chromosomal integration site downstream of rlmH in an MRSA ST398 isolate (IMT38951) and highlighted the most important transcriptional responses of the bacteria required to endure pig-farm associated environmental challenges such as increased ammonia concentrations and porcine serum. A detailed reconstruction of SCCmec loss from its genomic integration site raises concerns with respect to the autonomous replication capabilities of the predicted circular SCCmec element and its potential to spread to other susceptible bacterial hosts. It seems likely that this particular method of SCCmec conservation and transmission might have been underestimated, especially considering that, at least so far, only severe DNA-damaging events seem to trigger an increased activity of the ccrC variants at the population level. However, further studies are required to evaluate which factors and co-factors may trigger and support the autonomous replication machinery present in the excised elements.
The datasets presented in this study can be found in online repositories. Raw sequencing reads, including RNA-Seq and long-read genomic data, were uploaded to NCBI and deposited within BioProjects PRJNA891722 and PRJNA449454.
CH, AL-B, LW, LE, SW, and BW contributed to conception and design of the study. CH, JB, JA, and BW performed laboratory experiments and analysis. Genomics and transcriptomics were carried out by LE, SW, AT, and TS. CH, MB, MH, AB, WZ, and BW analysed the results. CH and BW wrote the first draft of the manuscript. LE, SW, WZ, and MB wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the German Federal Ministry of Education and Research (BMBF) for #1Health-PREVENT (grant nos. 01KI2009D and 01KI2009F) and PAC-CAMPY (grant no. 01KI2007F) within the German Research Network of Zoonotic Diseases. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. CH received a research doctoral fellowship from Akademie für Tiergesundheit e.V. (AfT; Germany).
We thank our colleagues from the Advanced Light and Electron Microscopy (ZBS 4) department of the Robert Koch Institute for their individual contribution and support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.969961/full#supplementary-material
Anderson, K. L., Roux, C. M., Olson, M. W., Luong, T. T., Lee, C. Y., Olson, R., et al. (2010). Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol. Med. Microbiol. 60, 208–250. doi: 10.1111/j.1574-695X.2010.00736.x
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Astrup, L. B., Hansen, J. E., and Pedersen, K. (2021). Occurrence and survival of livestock-associated MRSA in pig manure and on agriculture fields. Antibiotics (Basel) 10:10. doi: 10.3390/antibiotics10040448
Bebel, A., Walsh, M. A., Mir-Sanchis, I., and Rice, P. A. (2020). A novel DNA primase-helicase pair encoded by SCCmec elements. elife 9:e55478. doi: 10.7554/eLife.55478
Bischoff, M., Entenza, J. M., and Giachino, P. (2001). Influence of a functional sigB operon on the global regulators Sar and agr in Staphylococcus aureus. J. Bacteriol. 183, 5171–5179. doi: 10.1128/JB.183.17.5171-5179.2001
Boisset, S., Geissmann, T., Huntzinger, E., Fechter, P., Bendridi, N., Possedko, M., et al. (2007). Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator rot by an antisense mechanism. Genes Dev. 21, 1353–1366. doi: 10.1101/gad.423507
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Boundy, S., Safo, M. K., Wang, L., Musayev, F. N., O'Farrell, H. C., Rife, J. P., et al. (2013). Characterization of the Staphylococcus aureus rRNA methyltransferase encoded by orfX, the gene containing the staphylococcal chromosome cassette mec (SCCmec) insertion site. J. Biol. Chem. 288, 132–140. doi: 10.1074/jbc.M112.385138
Busche, T., Hillion, M., Van Loi, V., Berg, D., Walther, B., Semmler, T., et al. (2018). Comparative Secretome analyses of human and zoonotic Staphylococcus aureus isolates CC8, CC22, and CC398. Mol. Cell. Proteomics 17, 2412–2433. doi: 10.1074/mcp.RA118.001036
Butala, M., Zgur-Bertok, D., and Busby, S. J. (2009). The bacterial LexA transcriptional repressor. Cell. Mol. Life Sci. 66, 82–93. doi: 10.1007/s00018-008-8378-6
Butaye, P., Argudín, M. A., and Smith, T. C. (2016). Livestock-associated MRSA and its current evolution. Current Clinical Microbiology Reports 3, 19–31. doi: 10.1007/s40588-016-0031-9
Cardile, A. P., Sanchez, C. J. Jr., Samberg, M. E., Romano, D. R., Hardy, S. K., Wenke, J. C., et al. (2014). Human plasma enhances the expression of staphylococcal microbial surface components recognizing adhesive matrix molecules promoting biofilm formation and increases antimicrobial tolerance in vitro. BMC. Res. Notes 7:457. doi: 10.1186/1756-0500-7-457
Chlebowicz, M. A., Nganou, K., Kozytska, S., Arends, J. P., Engelmann, S., Grundmann, H., et al. (2010). Recombination between ccrC genes in a type V (5C2&5) staphylococcal cassette chromosome mec (SCCmec) of Staphylococcus aureus ST398 leads to conversion from methicillin resistance to methicillin susceptibility in vivo. Antimicrob. Agents Chemother. 54, 783–791. doi: 10.1128/AAC.00696-09
Cirz, R. T., Jones, M. B., Gingles, N. A., Minogue, T. D., Jarrahi, B., Peterson, S. N., et al. (2007). Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J. Bacteriol. 189, 531–539. doi: 10.1128/JB.01464-06
Clauditz, A., Resch, A., Wieland, K. P., Peschel, A., and Götz, F. (2006). Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 74, 4950–4953. doi: 10.1128/IAI.00204-06
Clinical and Laboratory Standards Institute. (2013). “Vet01-S2 performance standards for antimicrobial disk and dilution susceptibility for bacteria isolated from animals. Second international supplement ”. (Wayne, PA).
Clinical and Laboratory Standards Institute. (2014). Performance standards for antimicrobial susceptibility testing: Twenty-fourth informational supplement M100-S24. Wayne, PA: CLSI (Clinical and Laboratory Standards Institute).
Crosby, H. A., Schlievert, P. M., Merriman, J. A., King, J. M., Salgado-Pabón, W., and Horswill, A. R. (2016). The Staphylococcus aureus global regulator MgrA modulates clumping and virulence by controlling surface protein expression. PLoS Pathog. 12:e1005604. doi: 10.1371/journal.ppat.1005604
Everitt, R. G., Didelot, X., Batty, E. M., Miller, R. R., Knox, K., Young, B. C., et al. (2014). Mobile elements drive recombination hotspots in the core genome of staphylococcus aureus. Nat. Commun. 5:3956. doi: 10.1038/ncomms4956
Fujimoto, D. F., Higginbotham, R. H., Sterba, K. M., Maleki, S. J., Segall, A. M., Smeltzer, M. S., et al. (2009). Staphylococcus aureus SarA is a regulatory protein responsive to redox and pH that can support bacteriophage lambda integrase-mediated excision/recombination. Mol. Microbiol. 74, 1445–1458. doi: 10.1111/j.1365-2958.2009.06942.x
Gaupp, R., Wirf, J., Wonnenberg, B., Biegel, T., Eisenbeis, J., Graham, J., et al. (2016). RpiRc is a pleiotropic effector of virulence determinant synthesis and attenuates pathogenicity in Staphylococcus aureus. Infect. Immun. 84, 2031–2041. doi: 10.1128/IAI.00285-16
Geiger, T., Goerke, C., Mainiero, M., Kraus, D., and Wolz, C. (2008). The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 190, 3419–3428. doi: 10.1128/JB.01927-07
Gertz, S., Engelmann, S., Schmid, R., Ohlsen, K., Hacker, J., and Hecker, M. (1999). Regulation of sigmaB-dependent transcription of sigB and asp23 in two different Staphylococcus aureus strains. Mol. Gen. Genet. 261, 558–566. doi: 10.1007/s004380051001
Giachino, P., Engelmann, S., and Bischoff, M. (2001). Sigma(B) activity depends on RsbU in staphylococcus aureus. J. Bacteriol. 183, 1843–1852. doi: 10.1128/JB.183.6.1843-1852.2001
Gómez, P., Aspiroz, C., Hadjirin, N. F., Benito, D., Zarazaga, M., Torres, C., et al. (2020). Simultaneous nasal carriage by methicillin-resistant and methicillin susceptible Staphylococcus aureus of lineage ST398 in a live pig transporter. Pathogens 9:4019. doi: 10.3390/pathogens9050401
Gruswitz, F., O’connell, J. 3rd, and Stroud, R. M. (2007). Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 a. Proc. Natl. Acad. Sci. U. S. A. 104, 42–47. doi: 10.1073/pnas.0609796104
Gupta, R. K., Luong, T. T., and Lee, C. Y. (2015). RNAIII of the Staphylococcus aureus agr system activates global regulator MgrA by stabilizing mRNA. Proc. Natl. Acad. Sci. U. S. A. 112, 14036–14041. doi: 10.1073/pnas.1509251112
Gutierrez, A., Havelaar, A. H., and Schneider, K. R. (2022). Antimicrobial efficacy of un-ionized ammonia (NH(3)) against salmonella typhimurium in buffered solutions with variable pH, NH(3) concentrations, and urease-producing bacteria. Microbiol Spectr 10:e0185021. doi: 10.1128/spectrum.01850-21
Ha, K. P., and Edwards, A. M. (2021). DNA repair in Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 85:e0009121. doi: 10.1128/MMBR.00091-21
Hanssen, A. M., and Ericson Sollid, J. U. (2006). SCCmec in staphylococci: genes on the move. FEMS Immunol. Med. Microbiol. 46, 8–20. doi: 10.1111/j.1574-695X.2005.00009.x
Huber, C., Stamm, I., Ziebuhr, W., Marincola, G., Bischoff, M., Strommenger, B., et al. (2020). Silence as a way of niche adaptation: mecC-MRSA with variations in the accessory gene regulator (agr) functionality express kaleidoscopic phenotypes. Sci. Rep. 10:14787. doi: 10.1038/s41598-020-71640-4
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–d314. doi: 10.1093/nar/gky1085
Ito, T., Katayama, Y., and Hiramatsu, K. (1999). Cloning and nucleotide sequence determination of the entire mec DNA of pre-methicillin-resistant Staphylococcus aureus N315. Antimicrob. Agents Chemother. 43, 1449–1458. doi: 10.1128/AAC.43.6.1449
Kadlec, K., Feßler, A. T., Couto, N., Pomba, C. F., and Schwarz, S. (2012). Unusual small plasmids carrying the novel resistance genes dfrK or apmA isolated from methicillin-resistant or-susceptible staphylococci. J. Antimicrob. Chemother. 67, 2342–2345. doi: 10.1093/jac/dks235
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Köck, R., Harlizius, J., Bressan, N., Laerberg, R., Wieler, L. H., Witte, W., et al. (2009). Prevalence and molecular characteristics of methicillin-resistant Staphylococcus aureus (MRSA) among pigs on German farms and import of livestock-related MRSA into hospitals. Eur. J. Clin. Microbiol. Infect. Dis. 28, 1375–1382. doi: 10.1007/s10096-009-0795-4
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kuroda, M., Ohta, T., and Hayashi, H. (1995). Isolation and the gene cloning of an alkaline shock protein in methicillin resistant Staphylococcus aureus. Biochem. Biophys. Res. Commun. 207, 978–984. doi: 10.1006/bbrc.1995.1281
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Larsen, J., Raisen, C. L., Ba, X., Sadgrove, N. J., Padilla-González, G. F., Simmonds, M. S. J., et al. (2022). Emergence of methicillin resistance predates the clinical use of antibiotics. Nature 602, 135–141. doi: 10.1038/s41586-021-04265-w
Lavoie, S. P., and Summers, A. O. (2018). Transcriptional responses of Escherichia coli during recovery from inorganic or organic mercury exposure. BMC Genomics 19:52. doi: 10.1186/s12864-017-4413-z
Leinweber, H., Sieber, R. N., Larsen, J., Stegger, M., and Ingmer, H. (2021). Staphylococcal phages adapt to new hosts by extensive attachment site variability. MBio 12:e0225921. doi: 10.1128/mBio.02259-21
Li, D., and Cheung, A. (2008). Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect. Immun. 76, 1068–1075. doi: 10.1128/IAI.01069-07
Liu, P., Wu, Z., Xue, H., and Zhao, X. (2017). Antibiotics trigger initiation of SCCmec transfer by inducing SOS responses. Nucleic Acids Res. 45, 3944–3952. doi: 10.1093/nar/gkx153
Lu, H., Zhao, L., Si, Y., Jian, Y., Wang, Y., Li, T., et al. (2021). The surge of Hypervirulent ST398 MRSA lineage with higher biofilm-forming ability is a critical threat to clinics. Front. Microbiol. 12:636788. doi: 10.3389/fmicb.2021.636788
Mäder, U., Nicolas, P., Depke, M., Pané-Farré, J., Debarbouille, M., Van Der Kooi-Pol, M. M., et al. (2016). Staphylococcus aureus transcriptome architecture: from laboratory to infection-mimicking conditions. PLoS Genet. 12:e1005962. doi: 10.1371/journal.pgen.1005962
Malachowa, N., Whitney, A. R., Kobayashi, S. D., Sturdevant, D. E., Kennedy, A. D., Braughton, K. R., et al. (2011). Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6:e18617. doi: 10.1371/journal.pone.0018617
Manna, A. C., and Ray, B. (2007). Regulation and characterization of rot transcription in Staphylococcus aureus. Microbiology Reading 153, 1538–1545. doi: 10.1099/mic.0.2006/004309-0
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331. doi: 10.1093/nar/gkh454
Maslowska, K. H., Makiela-Dzbenska, K., and Fijalkowska, I. J. (2019). The SOS system: a complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 60, 368–384. doi: 10.1002/em.22267
Mccarthy, A. J., and Lindsay, J. A. (2013). Staphylococcus aureus innate immune evasion is lineage-specific: a bioinfomatics study. Infect. Genet. Evol. 19, 7–14. doi: 10.1016/j.meegid.2013.06.012
Meier, S., Goerke, C., Wolz, C., Seidl, K., Homerova, D., Schulthess, B., et al. (2007). sigmaB and the sigmaB-dependent arlRS and yabJ-spoVG loci affect capsule formation in Staphylococcus aureus. Infect. Immun. 75, 4562–4571. doi: 10.1128/IAI.00392-07
Merlino, J., Watson, J., Rose, B., Beard-Pegler, M., Gottlieb, T., Bradbury, R., et al. (2002). Detection and expression of methicillin/oxacillin resistance in multidrug-resistant and non-multidrug-resistant Staphylococcus aureus in Central Sydney, Australia. J. Antimicrob. Chemother. 49, 793–801. doi: 10.1093/jac/dkf021
Michel, A., Agerer, F., Hauck, C. R., Herrmann, M., Ullrich, J., Hacker, J., et al. (2006). Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J. Bacteriol. 188, 5783–5796. doi: 10.1128/JB.00074-06
Nordgård, A. S. R., Bergland, W. H., Vadstein, O., Mironov, V., Bakke, R., Østgaard, K., et al. (2017). Anaerobic digestion of pig manure supernatant at high ammonia concentrations characterized by high abundances of Methanosaeta and non-euryarchaeotal archaea. Sci. Rep. 7:15077. doi: 10.1038/s41598-017-14527-1
Noto, M. J., Fox, P. M., and Archer, G. L. (2008). Spontaneous deletion of the methicillin resistance determinant, mecA, partially compensates for the fitness cost associated with high-level vancomycin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 52, 1221–1229. doi: 10.1128/AAC.01164-07
Novick, R. P., and Jiang, D. (2003). The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiology (Reading) 149, 2709–2717. doi: 10.1099/mic.0.26575-0
Novick, R. P., Ross, H. F., Projan, S. J., Kornblum, J., Kreiswirth, B., and Moghazeh, S. (1993). Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 12, 3967–3975. doi: 10.1002/j.1460-2075.1993.tb06074.x
Nübel, U., Dordel, J., Kurt, K., Strommenger, B., Westh, H., Shukla, S. K., et al. (2010). A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 6:e1000855. doi: 10.1371/journal.ppat.1000855
Pandey, S., Sahukhal, G. S., and Elasri, M. O. (2019). The msaABCR operon regulates the response to oxidative stress in Staphylococcus aureus. J. Bacteriol. 201:19. doi: 10.1128/JB.00417-19
Pané-Farré, J., Jonas, B., Hardwick, S. W., Gronau, K., Lewis, R. J., Hecker, M., et al. (2009). Role of RsbU in controlling SigB activity in Staphylococcus aureus following alkaline stress. J. Bacteriol. 191, 2561–2573. doi: 10.1128/JB.01514-08
Párraga Solórzano, P. K., Shupe, A. C., and Kehl-Fie, T. E. (2021). The sensor histidine kinase ArlS is necessary for Staphylococcus aureus to activate ArlR in response to nutrient availability. J. Bacteriol. 203:e0042221. doi: 10.1128/JB.00422-21
Pelz, A., Wieland, K. P., Putzbach, K., Hentschel, P., Albert, K., and Götz, F. (2005). Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem. 280, 32493–32498. doi: 10.1074/jbc.M505070200
Pinon, A., Decherf, S., Malet, G., Cupferman, S., and Vialette, M. (2015). Bactericidal activity of ammonia and monoethanolamine on Pseudomonas aeruginosa and Staphylococcus aureus strains of various origins. Int. J. Cosmet. Sci. 37, 207–211. doi: 10.1111/ics.12183
Podlesek, Z., and Žgur Bertok, D. (2020). The DNA damage inducible SOS response is a key player in the generation of bacterial Persister cells and population wide tolerance. Front. Microbiol. 11:1785. doi: 10.3389/fmicb.2020.01785
Price, L. B., Stegger, M., Hasman, H., Aziz, M., Larsen, J., Andersen, P. S., et al. (2012). Staphylococcus aureus CC398: host adaptation and emergence of methicillin resistance in livestock. MBio 3:11. doi: 10.1128/mBio.00305-11
Ranganathan, N., Johnson, R., and Edwards, A. M. (2020). The general stress response of Staphylococcus aureus promotes tolerance of antibiotics and survival in whole human blood. Microbiology (Reading) 166, 1088–1094. doi: 10.1099/mic.0.000983
Read, T. D., Petit, R. A. 3rd, Yin, Z., Montgomery, T., Mcnulty, M. C., and David, M. Z. (2018). USA300 Staphylococcus aureus persists on multiple body sites following an infection. BMC Microbiol. 18:206. doi: 10.1186/s12866-018-1336-z
Roncarati, D., and Scarlato, V. (2017). Regulation of heat-shock genes in bacteria: from signal sensing to gene expression output. FEMS Microbiol. Rev. 41, 549–574. doi: 10.1093/femsre/fux015
Saito, H., and Kobayashi, H. (2003). Bacterial responses to alkaline stress. Sci. Prog. 86, 271–282. doi: 10.3184/003685003783238635
Scharn, C. R., Tickler, I. A., Tenover, F. C., and Goering, R. V. (2022). Characterization of SCCmec instability in methicillin-resistant Staphylococcus aureus affecting adjacent chromosomal regions, including the gene for staphylococcal protein a (spa). Antimicrob. Agents Chemother. 66:21. doi: 10.1128/aac.02374-21
Scholes, A. N., and Lewis, J. A. (2020). Comparison of RNA isolation methods on RNA-Seq: implications for differential expression and meta-analyses. BMC Genomics 21:249. doi: 10.1186/s12864-020-6673-2
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Semmler, T., Harrison, E. M., Lübke-Becker, A., Ulrich, R. G., Wieler, L. H., Guenther, S., et al. (2016). A look into the melting pot: the mecC-harboring region is a recombination hot spot in staphylococcus stepanovicii. PLoS One 11:e0147150. doi: 10.1371/journal.pone.0147150
Sheppard, S. K., Guttman, D. S., and Fitzgerald, J. R. (2018). Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. doi: 10.1038/s41576-018-0032-z
Sieber, R. N., Larsen, A. R., Urth, T. R., Iversen, S., Møller, C. H., Skov, R. L., et al. (2019). Genome investigations show host adaptation and transmission of LA-MRSA CC398 from pigs into Danish healthcare institutions. Sci. Rep. 9:18655. doi: 10.1038/s41598-019-55086-x
Slany, M., Oppelt, J., and Cincarova, L. (2017). Formation of Staphylococcus aureus biofilm in the presence of sublethal concentrations of disinfectants studied via a transcriptomic analysis using transcriptome sequencing (RNA-seq). Appl. Environ. Microbiol. 83:17. doi: 10.1128/AEM.01643-17
Stead, M. B., Agrawal, A., Bowden, K. E., Nasir, R., Mohanty, B. K., Meagher, R. B., et al. (2012). RNAsnap™: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 40:e156. doi: 10.1093/nar/gks680
Stojanov, M., Moreillon, P., and Sakwinska, O. (2015). Excision of staphylococcal cassette chromosome mec in methicillin-resistant Staphylococcus aureus assessed by quantitative PCR. BMC. Res. Notes 8:828. doi: 10.1186/s13104-015-1815-3
Tickler, I. A., Goering, R. V., Dewell, S., Le, V. M., Johar, L., Obradovich, A. E., et al. (2020). Mobile genetic elements responsible for discordant Staphylococcus aureus phenotypes and genotypes in the same blood culture bottle. Diagn. Microbiol. Infect. Dis. 98:115175. doi: 10.1016/j.diagmicrobio.2020.115175
Urbain, B., Gustin, P., Charlier, G., Coignoul, F., Lambotte, J. L., Grignon, G., et al. (1996). A morphometric and functional study of the toxicity of atmospheric ammonia in the extrathoracic airways in pigs. Vet. Res. Commun. 20, 381–399. doi: 10.1007/BF00366545
Vandendriessche, S., Vanderhaeghen, W., Larsen, J., De Mendonca, R., Hallin, M., Butaye, P., et al. (2014). High genetic diversity of methicillin-susceptible Staphylococcus aureus (MSSA) from humans and animals on livestock farms and presence of SCCmec remnant DNA in MSSA CC398. J. Antimicrob. Chemother. 69, 355–362. doi: 10.1093/jac/dkt366
Vanderhaeghen, W., Hermans, K., Haesebrouck, F., and Butaye, P. (2010). Methicillin-resistant Staphylococcus aureus (MRSA) in food production animals. Epidemiol. Infect. 138, 606–625. doi: 10.1017/S0950268809991567
Walther, B., Klein, K. S., Barton, A. K., Semmler, T., Huber, C., Merle, R., et al. (2018). Equine methicillin-resistant sequence type 398 Staphylococcus aureus (MRSA) harbor Mobile genetic elements promoting host adaptation. Front. Microbiol. 9:2516. doi: 10.3389/fmicb.2018.02516
Wang, L., and Archer, G. L. (2010). Roles of CcrA and CcrB in excision and integration of staphylococcal cassette chromosome mec, a Staphylococcus aureus genomic island. J. Bacteriol. 192, 3204–3212. doi: 10.1128/JB.01520-09
Wang, B., and Muir, T. W. (2016). Regulation of virulence in Staphylococcus aureus: molecular mechanisms and remaining puzzles. Cell chemical biology 23, 214–224. doi: 10.1016/j.chembiol.2016.01.004
Wang, X., Wang, M., Chen, S., Wei, B., Gao, Y., Huang, L., et al. (2020). Ammonia exposure causes lung injuries and disturbs pulmonary circadian clock gene network in a pig study. Ecotoxicol. Environ. Saf. 205:111050. doi: 10.1016/j.ecoenv.2020.111050
Wang, A., Zhou, K., Liu, Y., Yang, L., Zhang, Q., Guan, J., et al. (2017). A potential role of transposon IS431 in the loss of mecA gene. Sci. Rep. 7:41237. doi: 10.1038/srep41237
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Williamson, D. A., Bakker, S., Coombs, G. W., Tan, H., Monecke, S., and Heffernan, H. (2014). Emergence and molecular characterization of clonal complex 398 (CC398) methicillin-resistant Staphylococcus aureus (MRSA) in New Zealand. J. Antimicrob. Chemother. 69, 1428–1430. doi: 10.1093/jac/dkt499
Wolf, S. A., Epping, L., Andreotti, S., Reinert, K., and Semmler, T. (2020). SCORE: smart consensus of RNA expression - a consensus tool for detecting differentially expressed genes in bacteria. Bioinformatics 37, 426–428. doi: 10.1093/bioinformatics/btaa681
Wu, Z., Zhang, L., Qiao, D., Xue, H., and Zhao, X. (2018). Functional analyses of cassette chromosome recombinase C2 (CcrC2) and its use in eliminating methicillin resistance by combining CRISPR-Cas9. ACS Synth. Biol. 7, 2590–2599. doi: 10.1021/acssynbio.8b00261
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, S., Ma, R., Liu, X., Zhang, X., and Sun, B. (2015). Modulation of ccrAB expression and SCCmec excision by an inverted repeat element and SarS in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 6223–6232. doi: 10.1128/AAC.01041-15
Keywords: methicillin resistant Staphylococcus aureus, livestock associated, SCCmec, transcriptome analysis, recombination, deletion, ammonium, manure (litter)
Citation: Huber C, Wolf SA, Ziebuhr W, Holmes MA, Assmann J, Lübke-Becker A, Thürmer A, Semmler T, Brombach J, Bethe A, Bischoff M, Wieler LH, Epping L and Walther B (2022) How to survive pig farming: Mechanism of SCCmec element deletion and metabolic stress adaptation in livestock-associated MRSA. Front. Microbiol. 13:969961. doi: 10.3389/fmicb.2022.969961
Edited by:
Karsten Becker, University Medicine Greifswald, GermanyReviewed by:
Richard V. Goering, Creighton University, United StatesCopyright © 2022 Huber, Wolf, Ziebuhr, Holmes, Assmann, Lübke-Becker, Thürmer, Semmler, Brombach, Bethe, Bischoff, Wieler, Epping and Walther. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Birgit Walther, d2FsdGhlcmJAcmtpLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.