 Shaojun Tang
Shaojun Tang Lei Jin
Lei Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 September 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.967135
This article is part of the Research Topic Chromosome and Genome Biology of Fungi View all 7 articles
Inonotus hispidus (I. hispidus) is a medicinal macrofungus that plays a key role in anti-tumor and antioxidant functions. To further understand and enhance the value of I. hispidus, we performed whole-genome sequencing and an analysis of its strain for the first time. I. hispidus was sequenced using the Illumina NovaSeq high-throughput sequencing platform. The genome length was 35,688,031 bp and 30 contigs, with an average length of 1,189,601.03 bp. Moreover, database alignment annotated 402 CAZyme genes and 93 functional genes involved in regulating secondary metabolites in the I. hispidus genome to find the greatest number of genes involved in terpenes in that genome, thus providing a theoretical basis for its medicinal value. Finally, the phylogenetic analysis and comparative genomic analysis of single-copy orthologous protein genes from other fungi in the same family were conducted; it was found that I. hispidus and Sanghuangporus baumii have high homology. Our results can be used to screen candidate genes for the nutritional utilization of I. hispidus and the development of high-yielding and high-quality I. hispidus via genetic means.
Inonotus hispidus (I. hispidus) is known as Shaggy bracket or Shaggy polypore due to the hairy shape of its fruiting bodies (Song et al., 2021). I. hispidus is a valuable medicinal fungus that mainly grows on mulberry, ash, elm, poplar, and Japanese locust (Liu et al., 2019b); it is found in Hunan, Heilongjiang, Jilin, Liaoning, Inner Mongolia, and Xinjiang in China. Exploration of its chemical composition has shown that I. hispidus is rich in polysaccharides, polyphenols, steroids, terpenes, fatty acids, amino acids, and other active ingredients, and it is rich in pigment substances (Politi et al., 2007). Because of the richness of these active ingredients, I. hispidus plays an essential role in medicine (Benarous et al., 2021; Zhang et al., 2021). In Chinese history, it is recorded that I. hispidus, as a traditional Chinese medicine, has anti-inflammatory, hypoglycemic, hypolipidemic, and blood-activating and stasis-removing effects (Gründemann et al., 2016). This coincides with the research results of modern medicine, and many medical studies have explored the pharmacological mechanism of I. hispidus (Awadh Ali et al., 2003; Benarous et al., 2015). Gründemann et al. explored the effect of I. hispidus extract on different types of human immune cells using flow cytometry; the results showed that the extract exerted an immunomodulatory influence (Gründemann et al., 2016). Another study found that (4S,5S)-4-Hydroxy-3,5-dimethoxycyclohex-2-enone (HDE), isolated and purified from I. hispidus, can inhibit proliferation of HepG2 cells and has an anti-tumor effect (Yang et al., 2019). In addition to targeting liver cancer, I. hispidus extracts have inhibited the proliferation of various tumor cells and has significantly improved the immune function of the host organism (Wang et al., 2016; Li and Bao, 2022). The polysaccharides and polyphenols in I. hispidus exhibit excellent antioxidant and anti-inflammatory effects and can strongly scavenge DPPH and OH free radicals (Zan et al., 2011; Liu et al., 2019a).
At present, most of the related research on I. hispidus focuses on the extraction of its active ingredients or its functional roles, and there is very little content about its genome sequencing. In recent years, more studies have reported on the results of fungal genome sequencing. Genome-wide sequencing of fungi from Hymenochaetaceae has revealed their biological activity value (Huo et al., 2020; Jiang et al., 2021). The genome sequencing results also provide a strong research basis for our understanding of fungal function and ingenuity. Through the sequencing analysis of the genome library, we can learn about the gene regulatory network, biosynthetic pathway, and drug characteristics of these large fungi, which creates a good foundation for their subsequent commercial production and the enrichment of their medical value. In the study discussed in this paper, we developed a genome-wide map of I. hispidus (CGMCC 21046). The genes of I. hispidus were annotated through public and proprietary databases to identify the fungus’ potential medicinal functional roles. Then, I. hispidus was compared with the genome information of Pyrrhoderma noxium (P. noxium), Fomitiporia mediterranea (F. mediterranea), Sanghuangporus baumii (S. baumii), Phellopilus nigrolimitatus (P. nigrolimitatus), Phellinidium pouzarii (P. pouzarii), Schizopora paradoxa (S. paradoxa), Rickenella mellea (R. mellea), Wolfiporia cocos (W. cocos), Ganoderma sinense (G. sinense), Polyporus arcularius (P. arcularius), Fomes fomentarius (F. fomentarius), Trametes cingulate (T. cingulate), Trametes coccinea (T. coccinea) and Trametes cinnabarina (T. cinnabarina) belonging to the Hymenochaetales order of fungi to further reveal the evolution and species dynamics of I. hispidus.
The fruiting bodies of I. hispidus were collected from mulberry trees in Linqing, Shandong Province, China. Specimen was identified by morphologic and molecular analyses (Figure 1; Schwarzer et al., 2003). The specimen of I. hispidus was deposited in the China General Microorganism Culture Collection and Management Center (CGMCC 21046). The monokaryotic strain was germinated from one of the spores of the specimen and used for whole-genome sequencing. The mycelia of I. hispidus were cultured on an improved potato dextrose agar medium containing 5% wheat bran for ten days at 24°C in the dark and collected for genome sequencing (Tian et al., 2021).
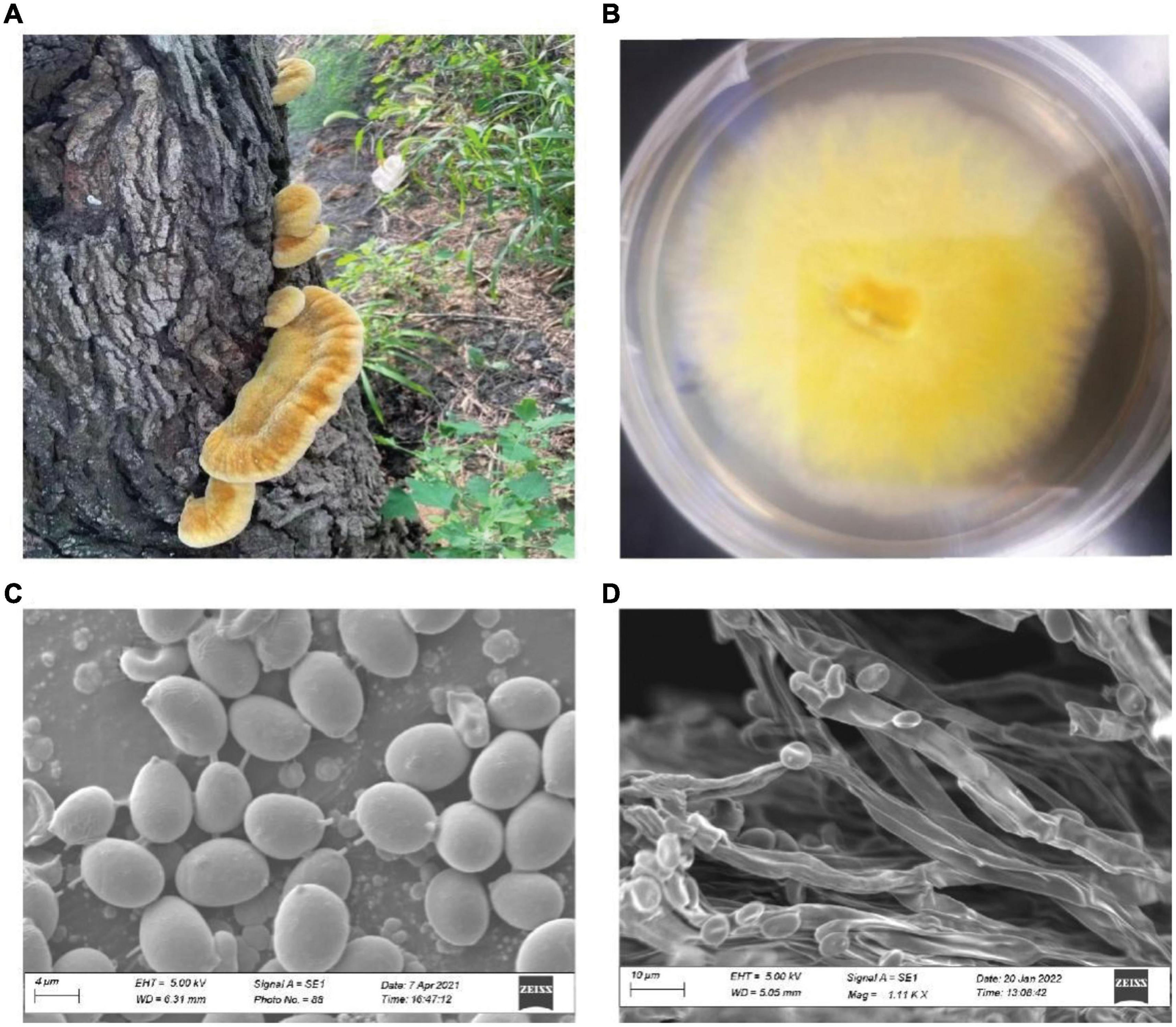
Figure 1. The fruiting bodies of I. hispidus and the monokaryotic strain used for genome sequencing. (A) The fruiting bodies of I. hispidus. (B) Mycelia of I. hispidus. (C) Scanning electron microscope image of I. hispidus spores. (D) Scanning electron microscope image of I. hispidus mycelia.
First, the sample was removed from liquid nitrogen, then 1 mL of B1 lysis buffer was added, and then vortexed and mixed. Next, 2 μL of RNaseA, 40 μL of lysozyme, and 45 μL of Proteinase K were added in sequence and mixed in a water bath at 37°C for 60 min. Then, 0.35 mL of B2 lysate was added and mixed in a water bath at 50°C for 60 min. After centrifugation at 12,000 rpm for 5 min, the supernatant was stored for later use.
Next, 2 mL of QBT (QBT: 43.83 g NaCl and 10.46 g MOPS (acid-free) were dissolved in 800 mL water, then the solution pH was adjusted to 7.0, 150 mL pure isopropanol and 15 mL 10 % Triton-100 were added to the final volume to 1 L.) was added to the equilibrated 20 G column to allow it to flow out by gravity; treated lysate was added to the column to allow it to flow out by gravity, then 1 mL of QC (QC: 58.44 g NaCl and 10.46 g MOPS were dissolved in 800 mL water, then pH was adjusted to 7.0, 150 mL pure isopropanol was added to the final volume to 1 L.) was added to the cleaning column to allow it to flow out by gravity for a total of three washes; 1 mL of QF (QF: 73.05 g NaCl and 6.06 g Tris-base are dissolved in 800 mL water, then the pH value of the solution is adjusted to 8.5, 150 mL pure isopropanol is added to the final volume to 1 L) was added and used to elute the DNA; 0.7-times the volume of isopropanol was added to precipitate the DNA; the sample was centrifuged at 12,000 rpm for 20 min at 4°C, then the supernatant was discarded, the DNA pellet was collected, and 1 mL of 70% ethanol was added to wash the pellet. The pellet was washed twice; an appropriate volume of TE (10 mmol/l Tris-HCl pH 8.0, 0.1 mmol/l EDTA) was added to dissolve the DNA, and it was incubated in a metal bath at 37°C for 60–120 min.
The following steps were used to extract high-quality DNA. After the samples passed the quality inspection, the genomic DNA was randomly interrupted; the large fragments of DNA were enriched and purified by magnetic beads, and the large fragments were recovered using cutting gel. End repair and 3′ end adding A was done the reaction product was purified; the fragment repair product was obtained and purified using sequencing-related adapters to obtain the final library. Qubit was used to accurately and quantitatively identify the constructed DNA library. A specific amount of the DNA from the library was mixed with the relevant reagents on the machine and added to the flow cell. Real-time single-molecule sequencing was performed on the PromethION sequencer to obtain the original sequencing data.
The raw data format of Nanopore PromethION sequencing data is a binary fast5 format containing all the raw sequencing signals. After base calling with Guppy v. 3.2.6 software, the fast5 format data were converted to the fastq format. After further filtering of the adapters, the low-quality data, and the short fragments (length < 2,000 bp), the total dataset was obtained.
The proportion of clean reads aligned to the reference genome accounts for the total number of clean reads. If the reference genome is properly selected and there is no contamination in the relevant experimental process, the alignment rate of the sequencing reads will be greater than 80%. Moreover, the alignment rate is related to the relative relationship of the reference genome, the quality of the reference genome assembly, and the quality of the reads sequencing. The more reads in the genome, the higher the alignment rate. The comparison uses Burrows-Wheeler Aligner (BWA) v. 0.7.10 software (Li and Durbin, 2009). We used BUSCO v. 2.0 software to assess the integrity of the fungal genome assembly (Simão et al., 2015).
We used four software (LTR_FINDER v. 1.05, MITE-Hunte v. 1.0.0r, RepeatScout v. 1.0.5, PILER-DF v. 2.4) to build a database of repeat sequences in I. hispidus genome based on the principle of structure prediction and ab-initio prediction (Edgar and Myers, 2005; Price et al., 2005; Xu and Wang, 2007; Han and Wessler, 2010). The repeat sequence database of the fungal genome was classified by PASTEClassifier v. 1.0, and then merged with the Repbase (19.06) database as the final repeat sequence database (Jurka et al., 2005; Wicker et al., 2007). Next, RepeatMasker v. 4.0.6 software was used and based on the constructed repeat sequence database, the repeat sequence of the fungus was predicted (Tarailo-Graovac and Chen, 2009).
The repetitive sequence is described in the general feature format (GFF), including the source of the repetitive sequence elements (mainly transposons), the locus in the genome, as well as the type, characteristics, and other information. For details, please refer to the description of GFF format at: http://www.sequenceontology.org/gff3.shtml.
Gene structure prediction mainly adopts de novo prediction, based on homologous protein and transcriptome evidence, and then integrates the three prediction results. Genscan v. 1.0 (Burge and Karlin, 1997), Augustus v. 2.4 (Stanke and Waack, 2003), GlimmerHMM v. 3.0.4 (Majoros et al., 2004), GeneID v. 1.4 (Blanco et al., 2007), SNAP (version 2006-07-28) (Korf, 2004) were used for de novo prediction; homologous protein-based prediction was done using GeMoMa v. 1.3.1. The reference transcript-based assembly was determined using Hisat2 v. 2.0.4 and Stringtie v. 1.2.3, and PASA v. 2. TransDecoder v. 2.0.0.2 was used to predict the UniGene sequence based on the transcriptome assembly; finally, EVM v. 1.1.1 was used to integrate the prediction results obtained by the three methods previously mentioned, and the results were modified with PASA v. 2.0.2. The number and average length of the protein-coding genes was determined, as were the number and average length of the introns and exons. Non-coding RNAs are RNAs that do not encode proteins, including RNAs with known functions, such as microRNA, rRNA, and tRNA. Depending on the structural characteristics of non-coding RNAs, different strategies are used to predict different non-coding RNAs. tRNA in the genome was predicted using tRNAscan-SE v. 1.3.1 software. The rRNA in the genome and ncRNAs other than tRNA and rRNA were predicted using Infernal v. 1.1 software based on the Rfam v. 12.0 database (Nawrocki and Eddy, 2013).
The predicted gene sequences were compared with the functional databases, such as Clusters of Orthologous Genes (COG) (Tatusov et al., 2000), Gene Ontology (GO) (Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2004), protein families (Pfam) (Finn et al., 2016), and Nr (Deng et al., 2006) for BLAST v. 2.2.29 to obtain the gene function annotation results. Gene function annotation mainly includes a sequence similarity search and a motif similarity search. For the sequence similarity search, diamond BlastP (version: 2.9.0) was carried out between the protein sequence encoded by the full-length gene and the existing protein database Uniprot, RefSeq, and NR. The metabolic pathway was determined using the KEGG database. The parameters (-evalue 1e-5) were compared to obtain functional information about the sequence and the metabolic pathway information that may be involved in the protein. KEGG annotation uses KOBAS (version: 3.0) to associate with KEGG orthology and pathway. The Uniprot database records the correspondence between each protein family and the functional nodes in GO, and it predicts the biological function of the protein sequence encoded by the gene. Based on the association between the databases (Uniprot/Swiss-Port), the annotation information of the eggNOG database is obtained, and the COG annotation results are selected for COG classification statistics and mapping. For the motif similarity search, proteins are generally composed of one or more functional regions, which are usually called domains. Different combinations of domains produce a variety of proteins. Therefore, the identification of protein domains is important for the analysis of protein functions. This was done using a hmmScan (version: 3.1; parameter: e-value).
Carbohydrates play an important role in many biological functions. A significant amount of meaningful biological information can be obtained by studying carbohydrate-related enzymes. CAZy (a database of Carbohydrate-Active enZYmes [CAZymes]) data focuses on analyzing the genome, structure, and biochemical information of carbohydrate enzymes. HMMER (version: 3.2.1, filter parameter e-value 0.35) was used to annotate the protein sequences based on the CAZy database (Cantarel et al., 2009).
Cytochrome P450 (CYP450) is a family of proteins supplemented by heme. They can catalyze the oxidation of many substrates. Due to the maximum absorption wavelength at 450 nm after the reduction state of the protein combined with CO, it is also known as P450. It participates in the metabolism of endogenous substances and exogenous substances, including drugs and environmental compounds. Diamond BlastP (version >: 2.9.0; parameter: –evalue 1e-5) was used to annotate the target protein sequence based on the Fungal Cytochrome P450 Database (FCPD) (Park et al., 2008).
Transporter Classification Database (TCDB) is used to classify membrane transport proteins. It develops a transporter classification (TC) system, which is similar to the Enzyme Commission (EC) system for enzyme classification; however, the TC system provides both functional and evolutionary information. TCDB provides a TC number for each transporter family. TC numbers consist of five digits or letters separated by decimals. SignalP (version: 5.0) software was used to analyze the protein sequences of all the predicted genes, and the proteins containing signal peptides were found. TMHMM (version: 5.0) software was used to analyze the protein sequences of all the predicted genes to identify the proteins that contain transmembrane helixes, namely transmembrane proteins. The proteins containing a transmembrane helix were removed from the predicted proteins containing a signal peptide. The remaining proteins are secreted proteins (Saier et al., 2006).
antiSMASH 3.01 was used to analyze the secondary metabolite biosynthetic gene clusters in the fungal genome. Thus, the query and prediction of natural product synthetic gene clusters between genomes can be realized. The parameter settings remain the default parameter values. To validate the predicted results, we manually checked the resulting gene clusters. The genome sequence of the secondary metabolite biosynthesis gene cluster of I. hispidus was predicted using the antiSMASH 3.0 database and it was compared with 12 other fungi under the phylum Hymenochaetales (Medema et al., 2011).
The protein sequences of all the genomes were predicted using Prodigal v. 2.6.3 software. Orthofinder v. 2.5.2 software was used to calculate the orthologous single-copy genes according to the above protein sequences. MAFFT v. 7.480 software was used for multiple sequence alignment of the direct homologous single-copy genes (Tian et al., 2021). Finally, Python v. 3.7. script is used to concatenate the aligned protein sequences belonging to each bacterium, and IQ-TREE v. 2.1.3 software was used to calculate the evolutionary tree with the fasta file concatenated mentioned above. The algorithm that was used is the maximum likelihood method. Collinearity analysis was performed using TBTools software, based on location information from the GFF3 files of I. hispidus, S. baumii, P. nigrolimitatus and P. pouzarii.
Based on all the amino acid sequences of the selected species, OrthoFinder software (version: 2.3.12; parameter: –Mesa) (Emms and Kelly, 2019) was used for gene family clustering, and BlastP software (version: 2.6.0; parameters: –evalue 1e-5 – outfmt 6) (Camacho et al., 2009) was used for the genome alignment. Finally, the gene family identification results were statistically analyzed. Perl script was used to count and map the Wayne diagram based on the clustering results of the protein sequences of the four selected species. The gene family common to all species is called a shared gene. Based on the shared gene and functional annotation (GO and KEGG), R package clusterProfiler was used for the GO and KEGG enrichment analysis. Among all the selected species, the specific gene family is called the unique gene family. Based on the unique gene and functional annotation (GO and KEGG), R package clusterProfiler was used for GO and KEGG enrichment analysis.
The length of the genome obtained from Mushroom is 35,688,031 bp, which contains 30 contigs; the average contig length is 1,189,601.03 bp, the longest contig is 4,604,913 bp, and the guanine-cytosine (GC) content is 48.53%. The specific data information of sequencing is shown in Table 1. The sequenced reads were analyzed using K-mer methods to estimate genome size and heterozygosity. The genome assembly results are shown in Table 2 and Figure 2. The map rate, average depth, and coverage in the sequencing are 99.33%, 152.52 and 99.75%, respectively. The K-mer results are shown in Figure 3. Finally, we used BUSCO software (version: 4.1.4) to assess the completeness of the genome assemblies based on the fungi database (fungi_odb10). The completeness of the genome assembly and the annotation of single-copy ortholog test results indicated that the annotation set was very complete, with 85.8% of the fungal BUSCO present in the RefSeq annotation set and 2% of the genes were fragmented (Figure 4).
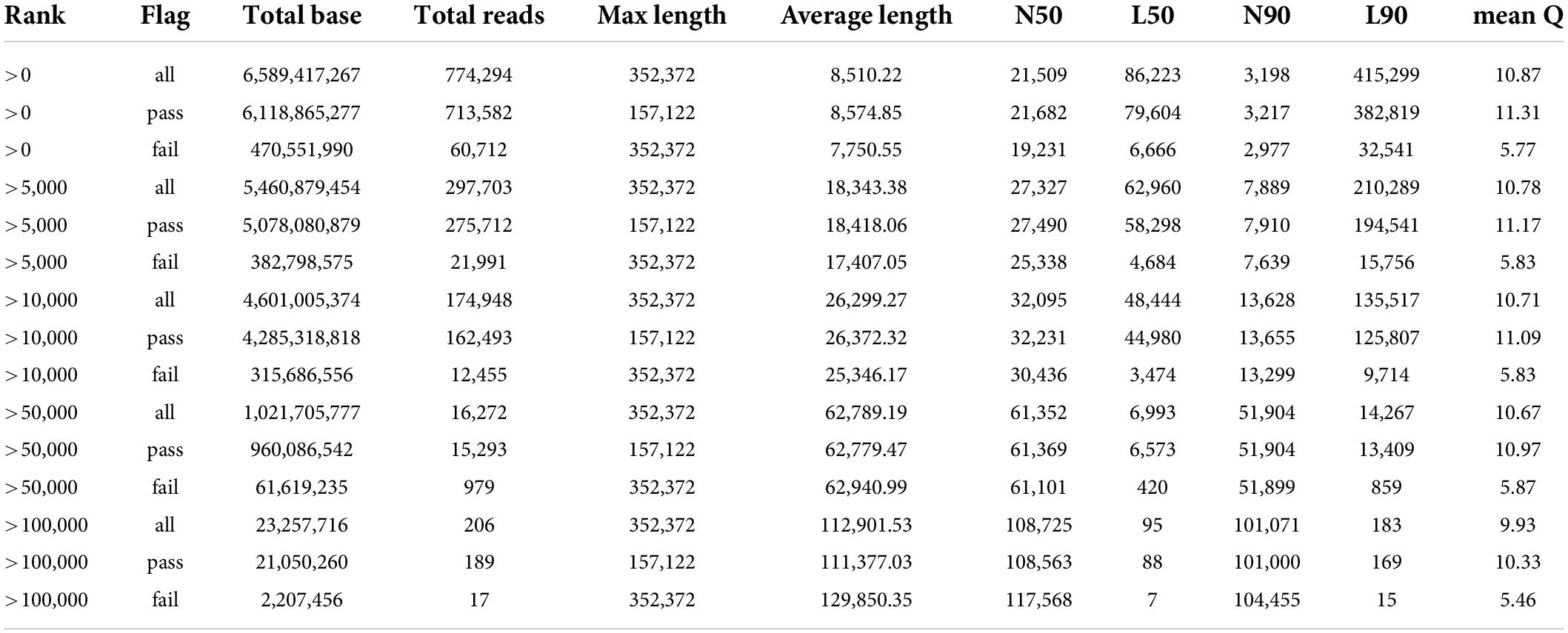
Table 1. Statistics of the data volume of the gene sequencing.

Table 2. Statistics of the assembly results.
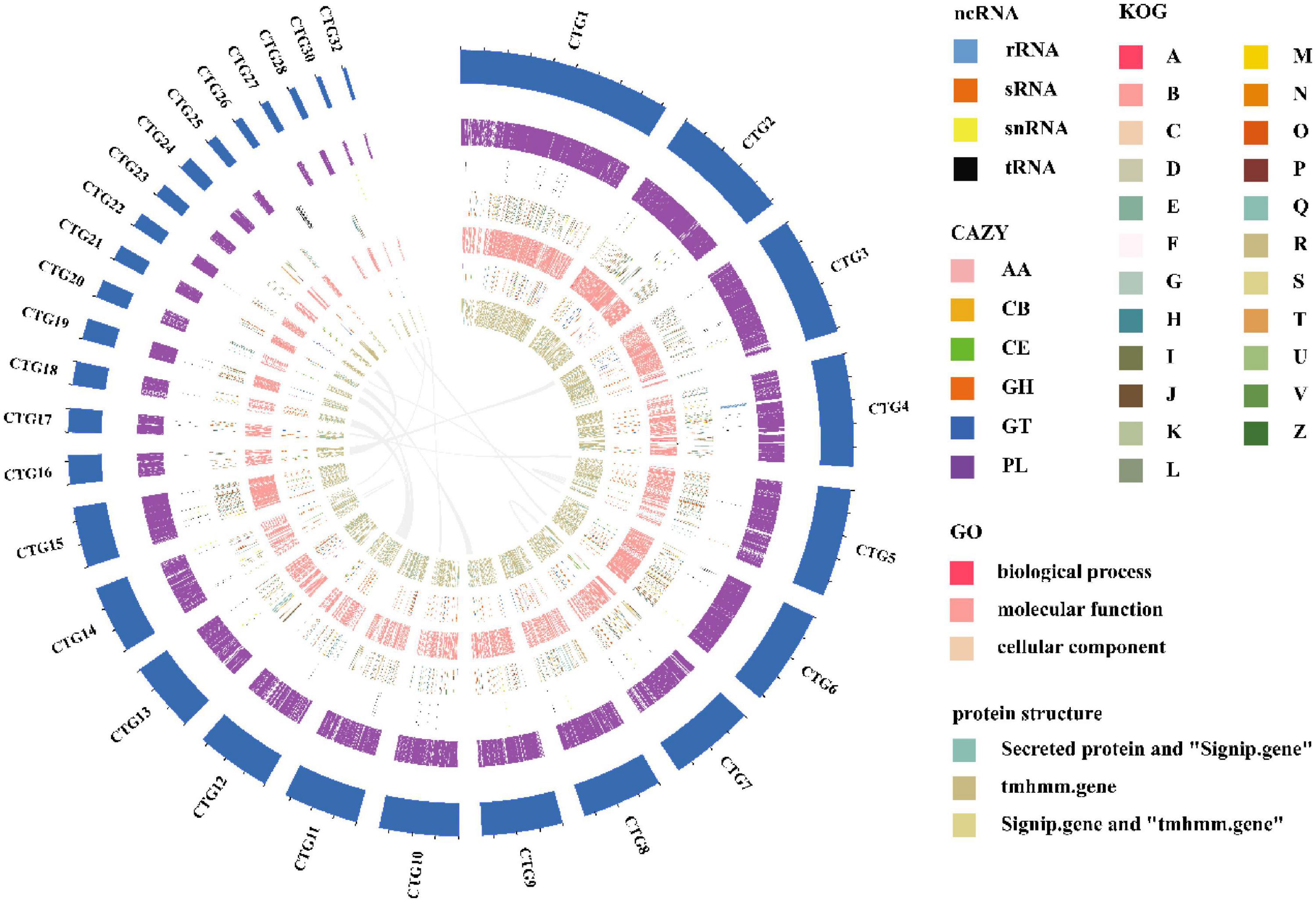
Figure 2. A circular genome map of I. hispidus. From outside to the center: circle 1: contig keyboard; circle 2: gene density; circle 3: ncRNA; circles 4–6: the predicted protein-coding genes using KOG, GO, and CAZy databases, respectively, where different colors represent different function classifications; 7th circle: large fragment duplication.
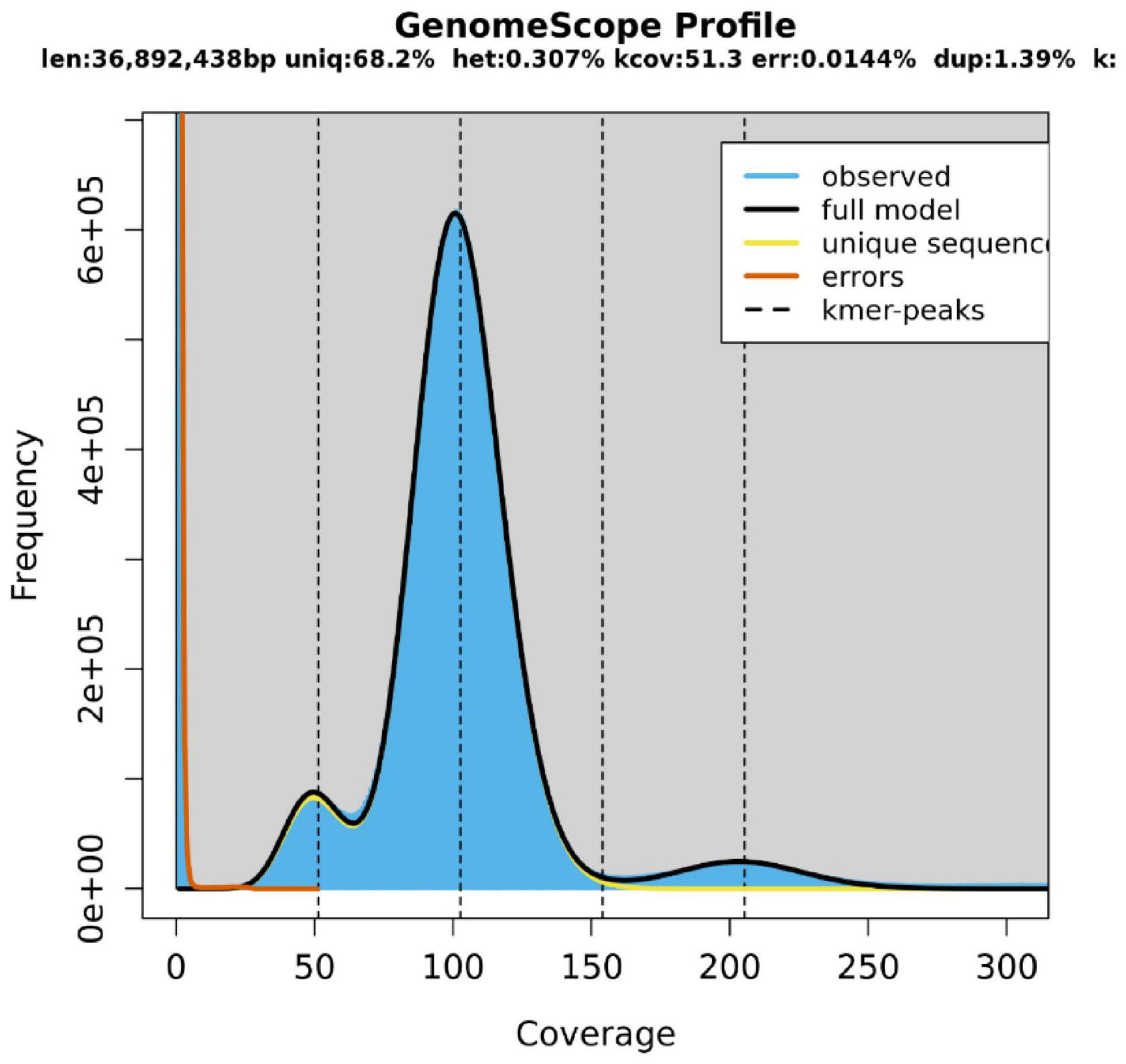
Figure 3. K-mer-Depth and K-mer Species—Frequency Distribution Plot.
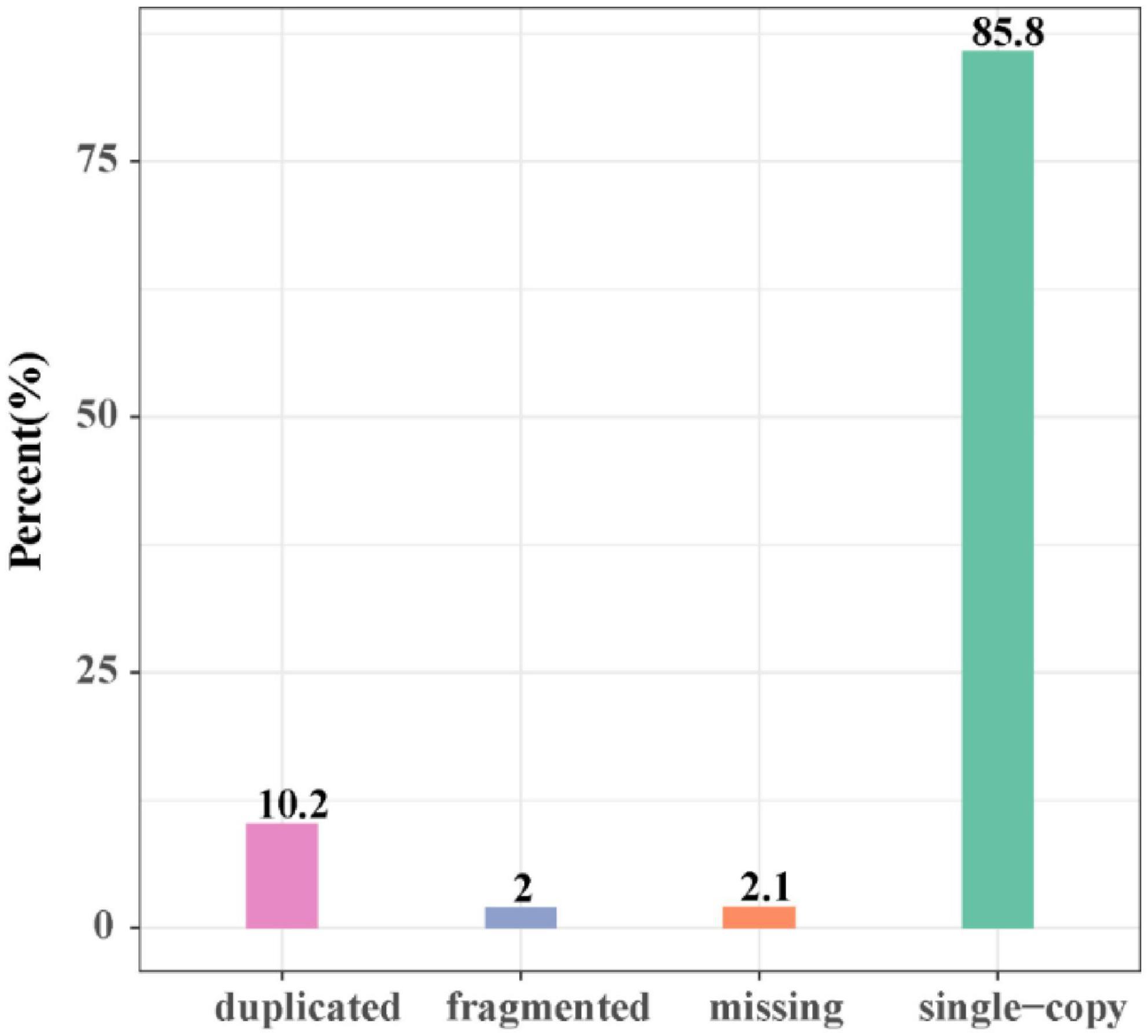
Figure 4. The map of the gene coding gene BUSCO evaluation results.
Gene prediction mainly uses BRAKER software (version: 2.1.4). First, Gene Mark-EX was used to train the model, and then AUGUSTUS was used for the prediction. The protein coding region contains a total of 12,671 genes, and the average length of the mRNA is 1,796.85 bp. The average number of exons per gene was 6.59, and the average lengths of exons and introns were 213.25 and 70.03 bp, respectively. For the non-coding RNAs, we predicted 17 rRNAs, 1 sRNA, 16 snRNAs, and 88 tRNAs, where the average length of r RNA is 1,836 bp.
The orthology of the I. hispidus genes was classified using the COG database. We found that the top five enriched categories were: “translation, ribosomal structure and biogenesis,” “posttranslational modification, protein turnover, chaperones,” “amino acid transport and metabolism,” “lipid transport and metabolism,” and “general function prediction only SIFunction unknown” (Figure 5A). Next, we simplified the GO annotation information to obtain the GOslim classification. After summarizing the functions of the genes from the three aspects of biological processes, cellular components, and molecular functions, we selected the top 20 annotations under each classification. The most abundant secondary classification of GOslim was drawn, and it was found that the “cell division” has the highest gene enrichment in the biological process, the “nucleus” has the highest gene enrichment in the cellular component, and the “ATP binding” has the highest gene enrichment in molecular (Figure 5B).
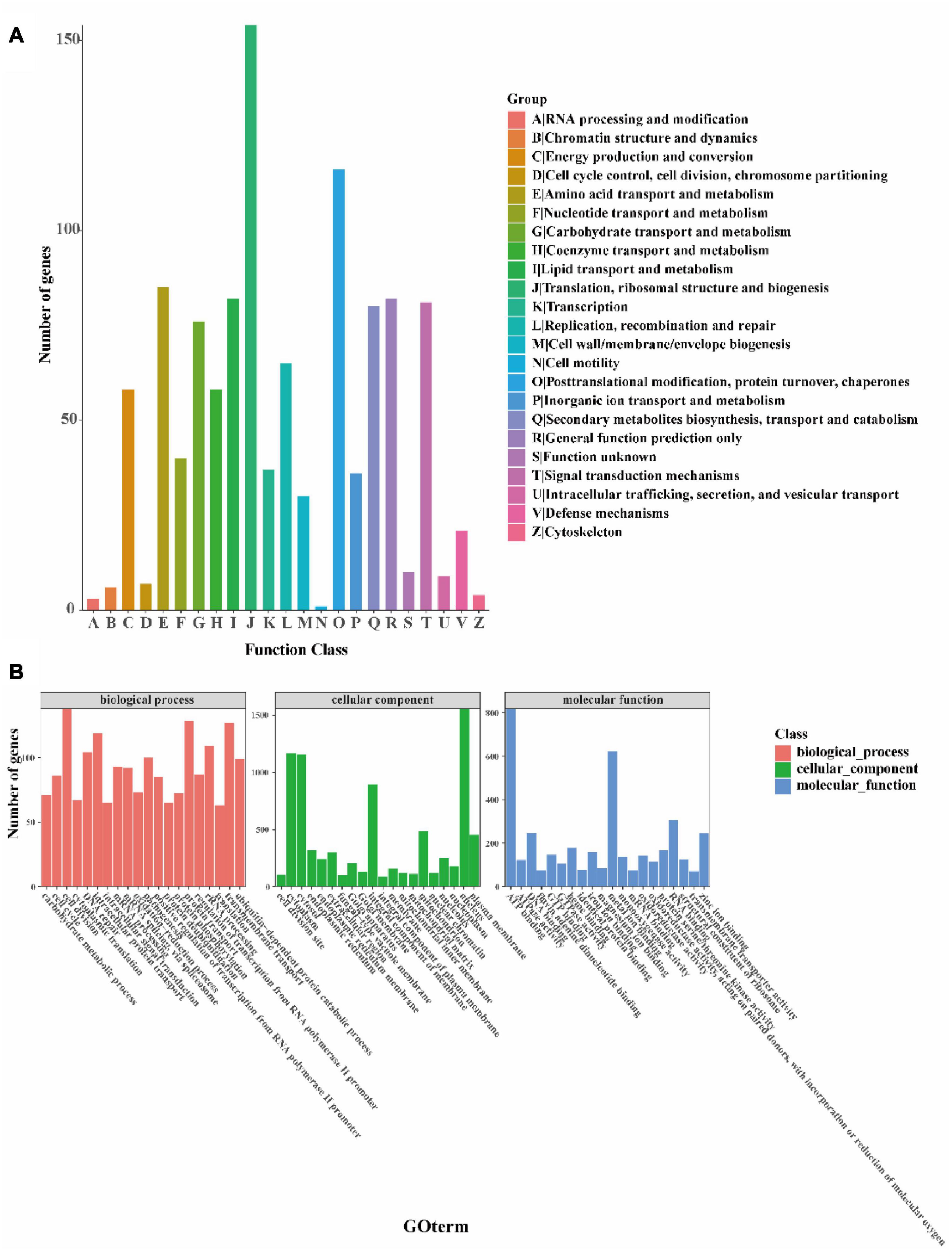
Figure 5. Functional annotation of the I. hispidus genome. (A) COG feature annotation classification chart. (B) GO functional annotation classification statistics plot.
Finally, to further understand the gene functions in I. hispidus, we annotated the sequences with KEGG and classified them according to the KEGG metabolic pathways in which they were involved. We found that the gene function with the highest enrichment in the organic systems classification is the “endocrine system,” the gene function with the highest enrichment in the metabolism classification is “carbohydrate metabolism,” and the gene function with the highest enrichment in the genetic information processing classification is “translation.” The most enriched gene function in the Environmental Information Processing classification is “signal transduction,” and the most enriched gene function in the Cellular Processes classification is “transport and catabolism” (Figure 6A). We also identified 266 Pkinases, 226 Pkinase Tyr, 186 F-box-like, etc., in the Pfam domain of the I. hispidus genome (Figure 6B).
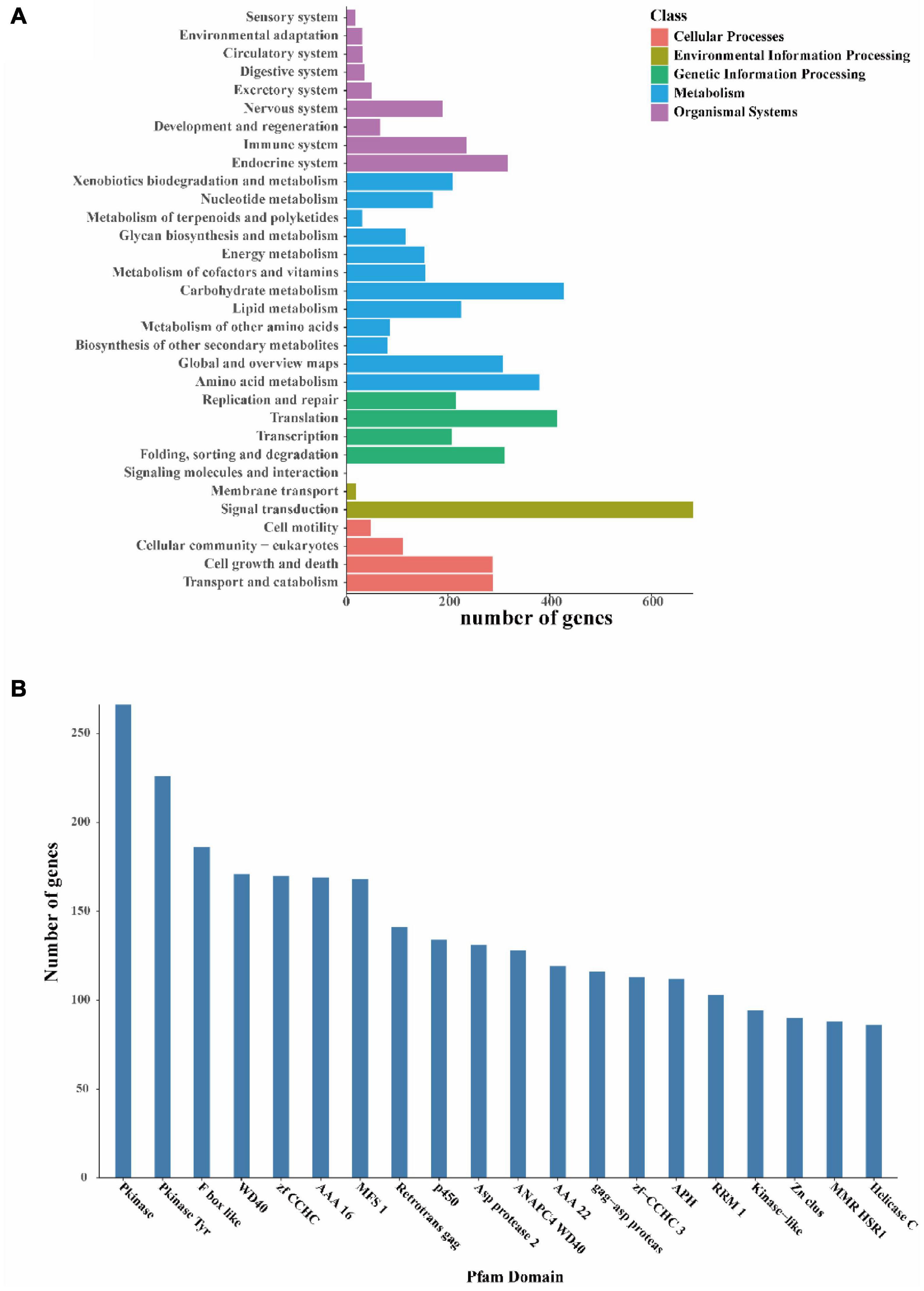
Figure 6. Functional annotation of the I. hispidus genome. (A) KEGG Pathway functional classification diagram. (B) Pfam functional annotation classification statistics plot.
We annotated the I. hispidus protein sequences based on the CAZyme database with HMMER (version: 3.2.1, filter parameters E-value < 1e-18; coverage > 0.35). The results showed that the genes were significantly enriched in the glycoside hydrolase family. A total of 402 CAZyme-encoding genes were annotated, including 121 superfamilies. Among the 402 CAZyme-encoding genes, 197 belong to glycoside hydrolases, 71 belong to glycosyl transferases, nine belong to polysaccharide lyases, 45 belong to carbohydrate esterase, 75 belong to auxiliary activities, and five belong to carbohydrate-binding modules (Figure 7).
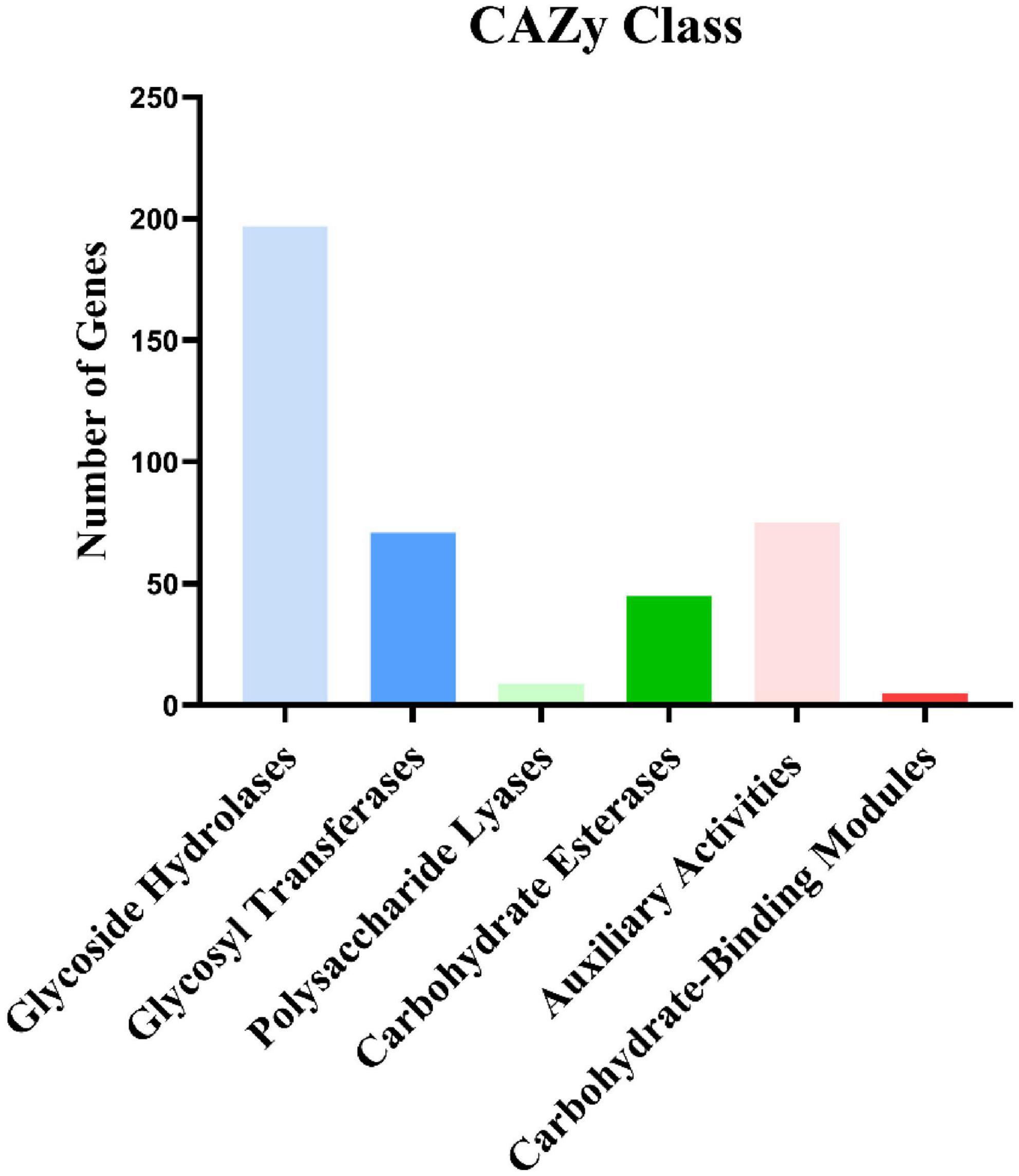
Figure 7. Carbohydrate activity enzyme annotation results.
We obtained 999 protein sequences by cytochrome P450 annotation. We obtained 1,408 membrane transport-related proteins through TCBD annotation and found 733 proteins containing signal peptides. We identified 2,305 transmembrane proteins and 495 secreted proteins (Table 3).

Table 3. Statistical table of protein structure prediction.
Prediction of the genome sequence of the secondary metabolite biosynthesis gene cluster using the AntiSMASH database found that 87 functional genes are involved in regulating the production of secondary metabolites. Among them, 33 functional genes regulated the production of NRPS-like, 35 functional genes regulated the production of terpenes, 12 functional genes regulated the production of T1PKS, and seven functional genes regulated NRPS. It was found that the other fungal genomes of Hymenochaetaceae mainly regulated the production of NRPS-like, terpene, T1PKS, and NRPS. However, some genes in other species could simultaneously regulate NRPS-like, T1PKS, or NRPS-like, terpene or NRPS, NRPS-like (Figure 8).
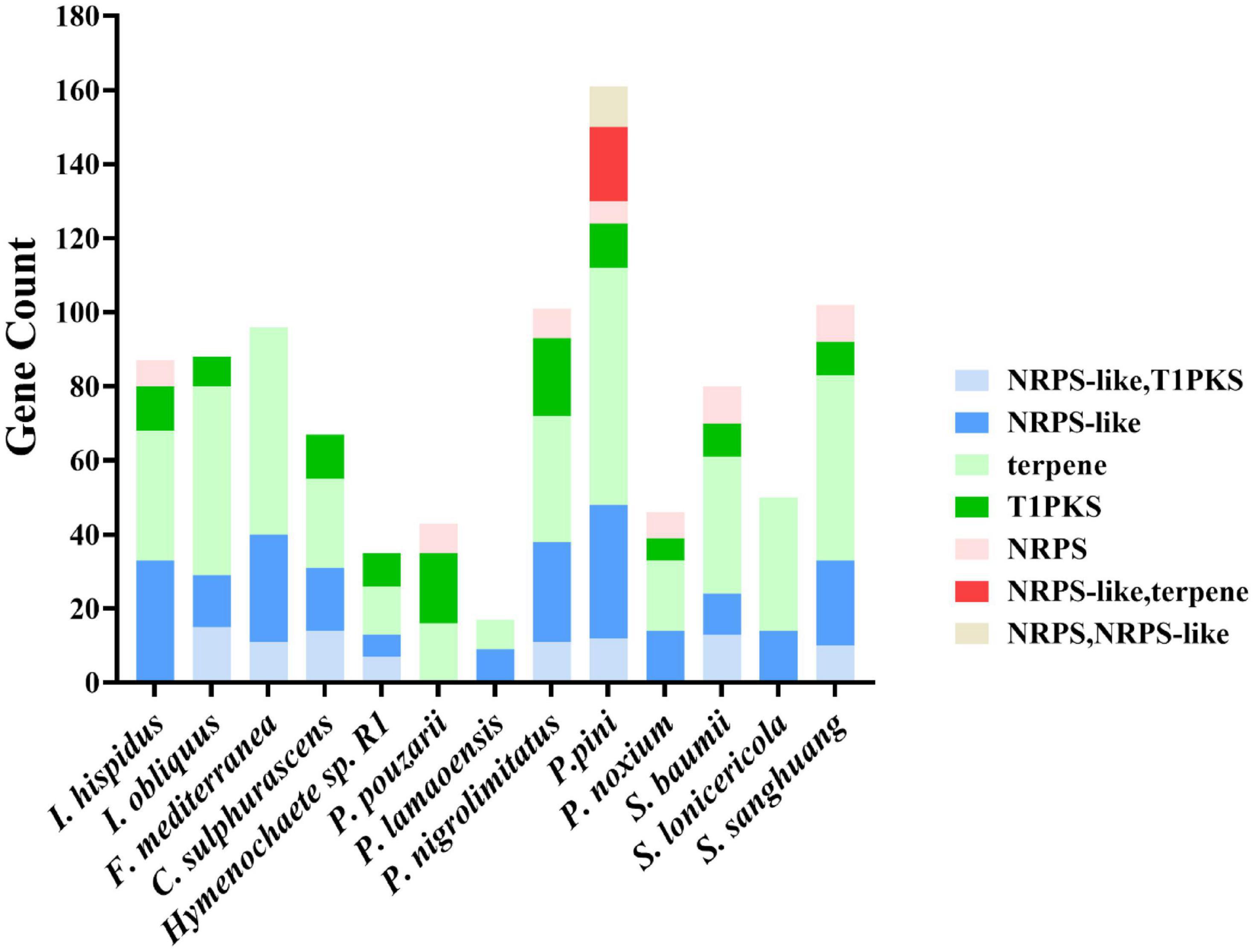
Figure 8. Genome sequence comparison of the secondary metabolite biosynthetic gene clusters.
We constructed a phylogenetic tree using single-copy orthologous protein genes from I. hispidus and 14 other fungal species. The 14 fungi all belonged to the Class Agaricomycetes, seven from Hymenochaetales, seven from Polyporaceae (Figure 9A). We also counted the homologous genes of I. hispidus and 14 other fungi species (Table 4). I. hispidus has 2,730 single-copy homologous genes and 1,323 multi-copy homologous genes in the species-shared gene family (Figure 9B). The gene family function enrichment analysis of I. hispidus and other 14 fungi showed that the top three enriched gene families of its unique GO annotated genes are “thiol-dependent deubiquitinase,” “cysteine-type endopeptidase activity,” and “ubiquitin-dependent protein catabolic process” (Figure 10A). A comparison of the KEGG database showed that the highest enriched gene families of I. hispidus are: “FoxO signaling pathway” (Figure 10B).
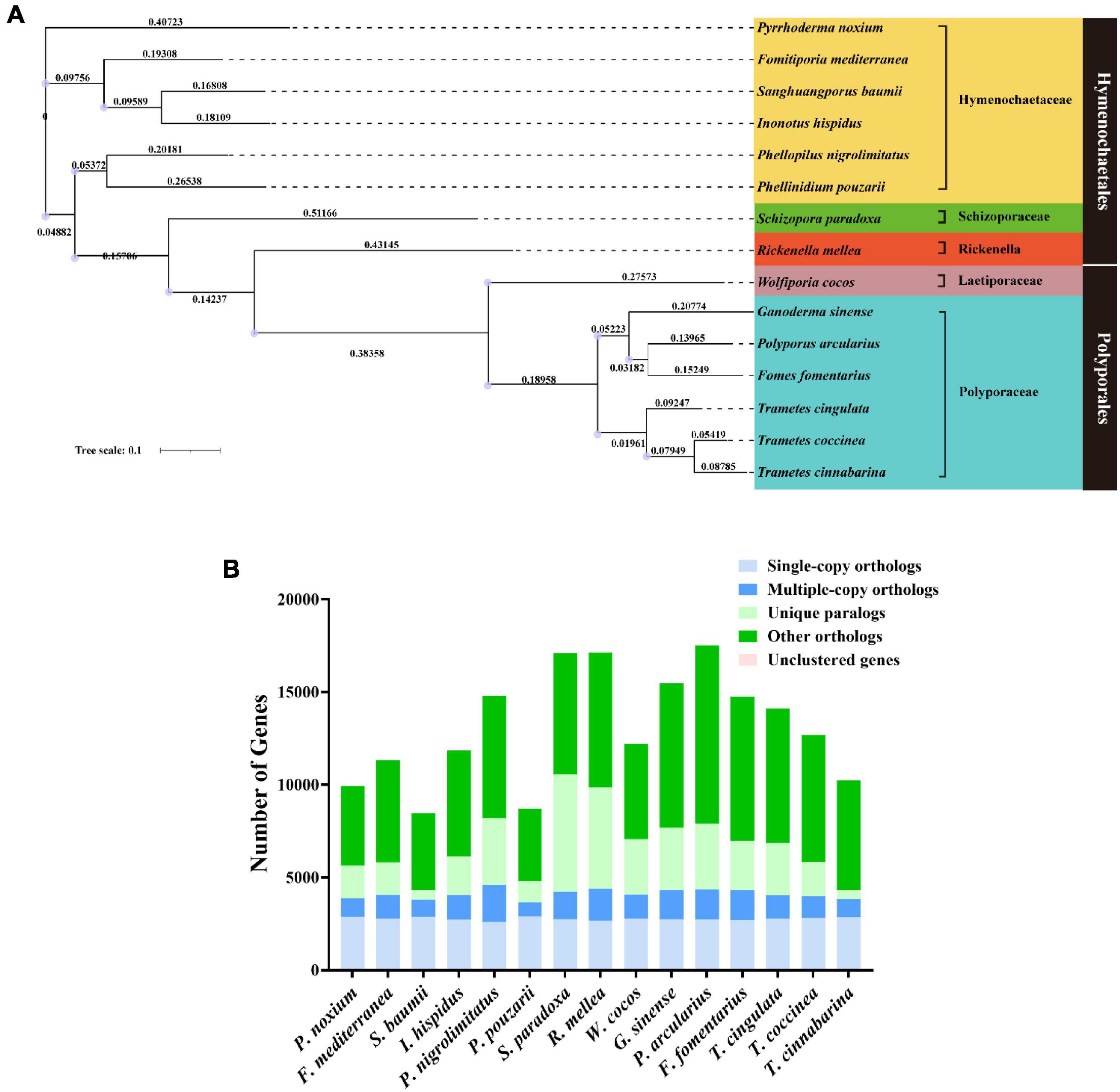
Figure 9. Comparative genomic analysis of I. hispidus and other 14 fungi. (A) Phylogenetic tree of I. hispidus and 14 other fungal species. (B) Homologous genes of I. hispidus and 14 other fungi species.
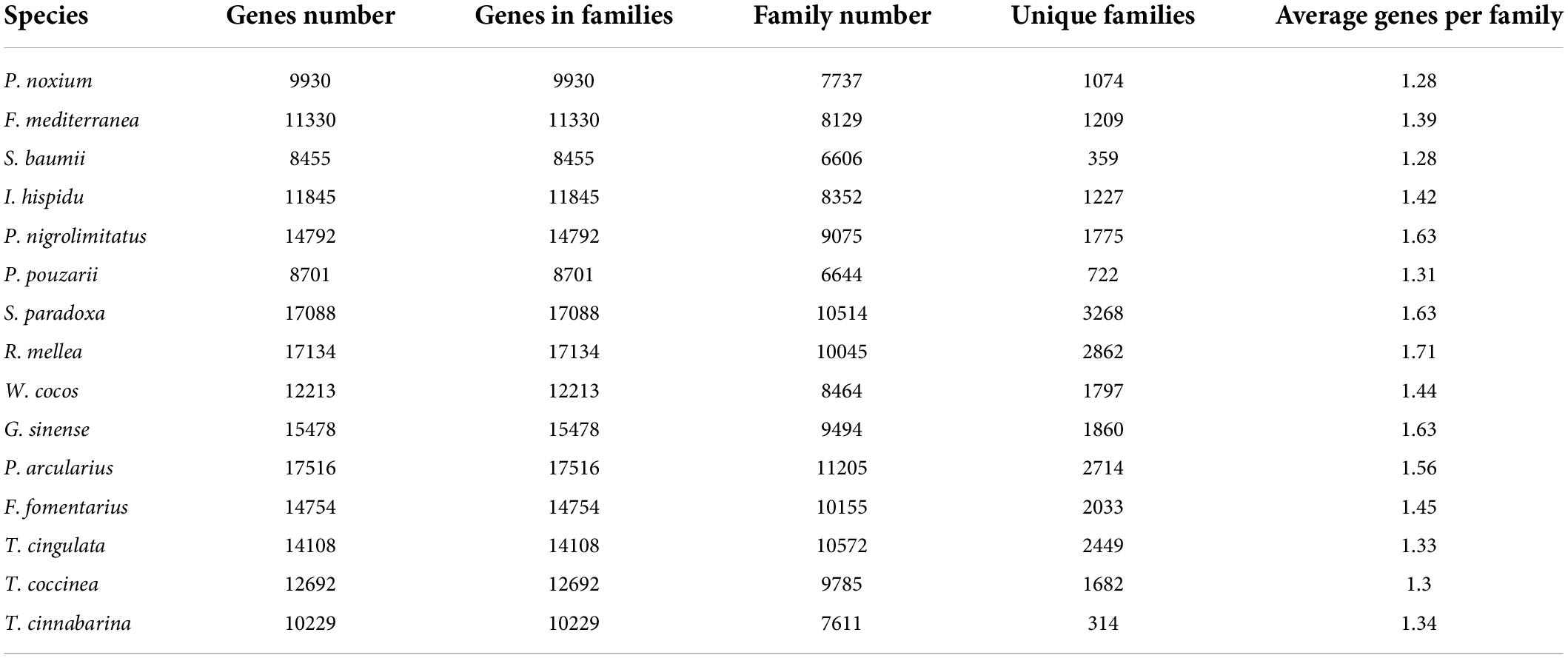
Table 4. Statistics table of gene family clustering results.
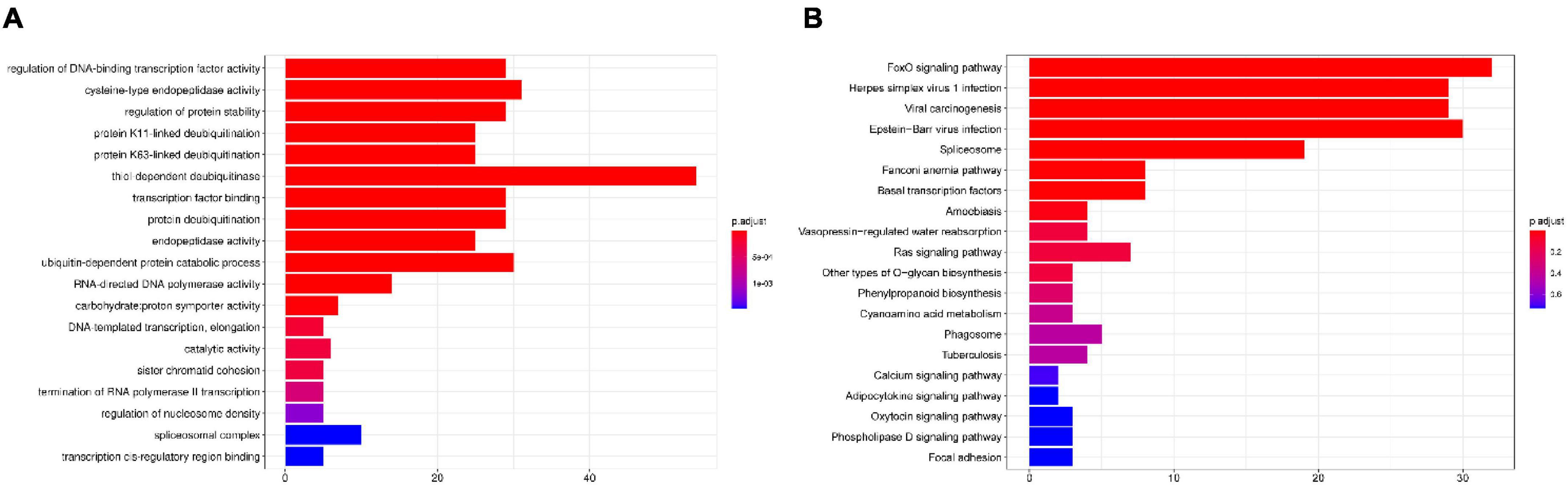
Figure 10. Gene family enrichment analysis of I. hispidus compared with other 14 fungi. (A) GO enrichment results of the share gene family. (B) KEGG enrichment results of the shared gene family.
The results showed the highest homology among I. hispidus, S. baumii, P. nigrolimitatus and P. pouzarii. Therefore, we further performed the genome synteny analysis of these four fungi (Figures 11A,B). The gene families shared by these four fungi were subjected to GO database functional annotation analysis; the results showed that the four categories with the highest enrichment of genes shared by the four fungi were: “transmembrane transporter activity,” “FAD binding,” “unfolded protein binding,” and “translation initiation factor activity” (Figure 12A). The gene families shared by these four fungi were functionally annotated by the KEGG Pathway database; the results showed that the two categories with the highest enrichment of genes shared by the four fungi were: “biosynthesis of amino acids” and “carbon metabolism” (Figure 12B). Compared with the other three fungal I. hispidus unique gene families, GO functional annotation showed that the highest enrichment categories were “thiol-dependent deubiquitinase” (Figure 12C). Compared with the other three fungal I. hispidus unique gene families, KEGG Pathway functional annotation showed that the highest enrichment categories were: “FoxO signaling pathway” (Figure 12D).
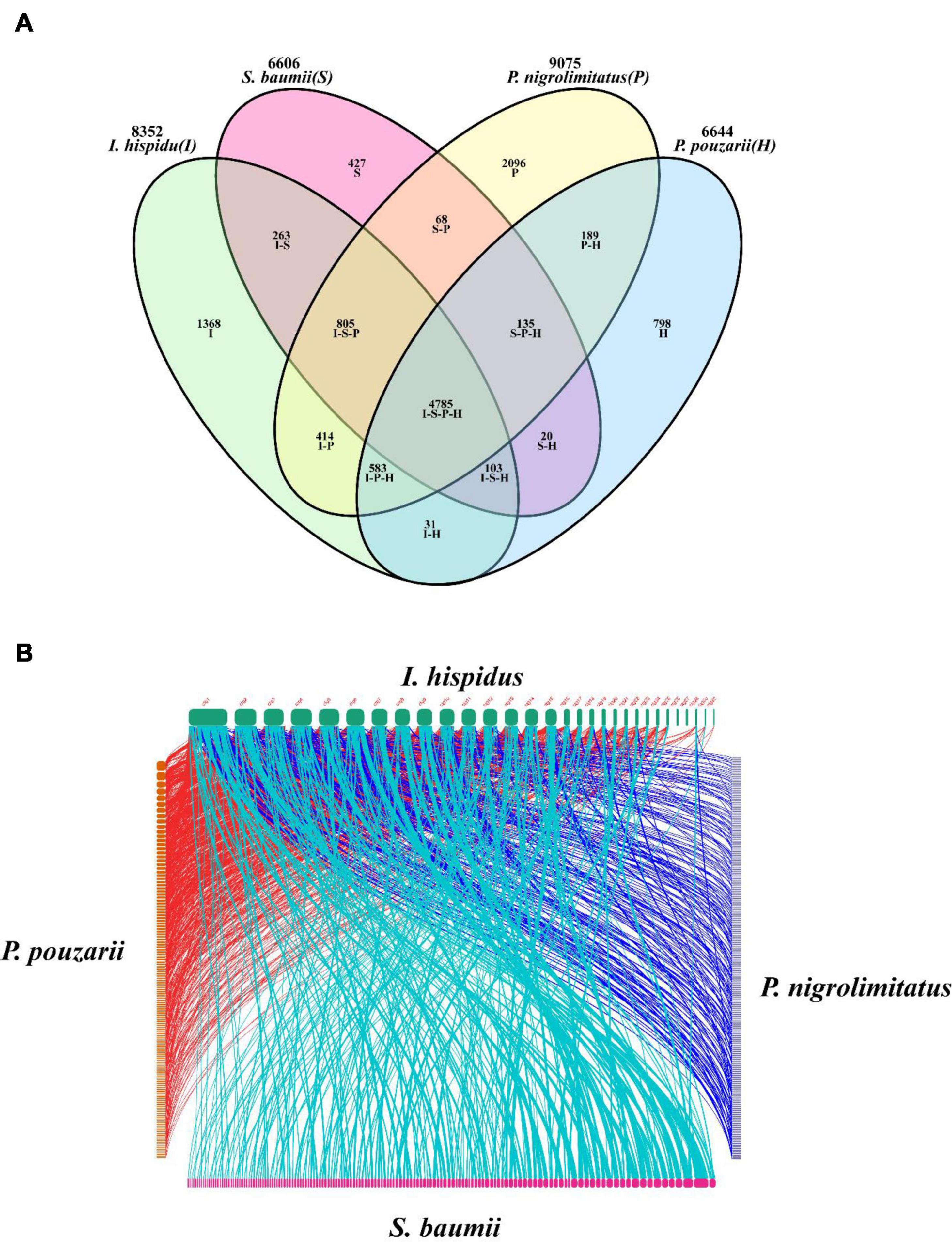
Figure 11. Comparative genomic analysis of I. hispidus and other three fungi. (A) Venn diagram of I. hispidus, S. baumii, P. nigrolimitatus, and P. pouzarii. (B) Genome synteny of I. hispidus, S. baumii, P. nigrolimitatus, and P. pouzarii.
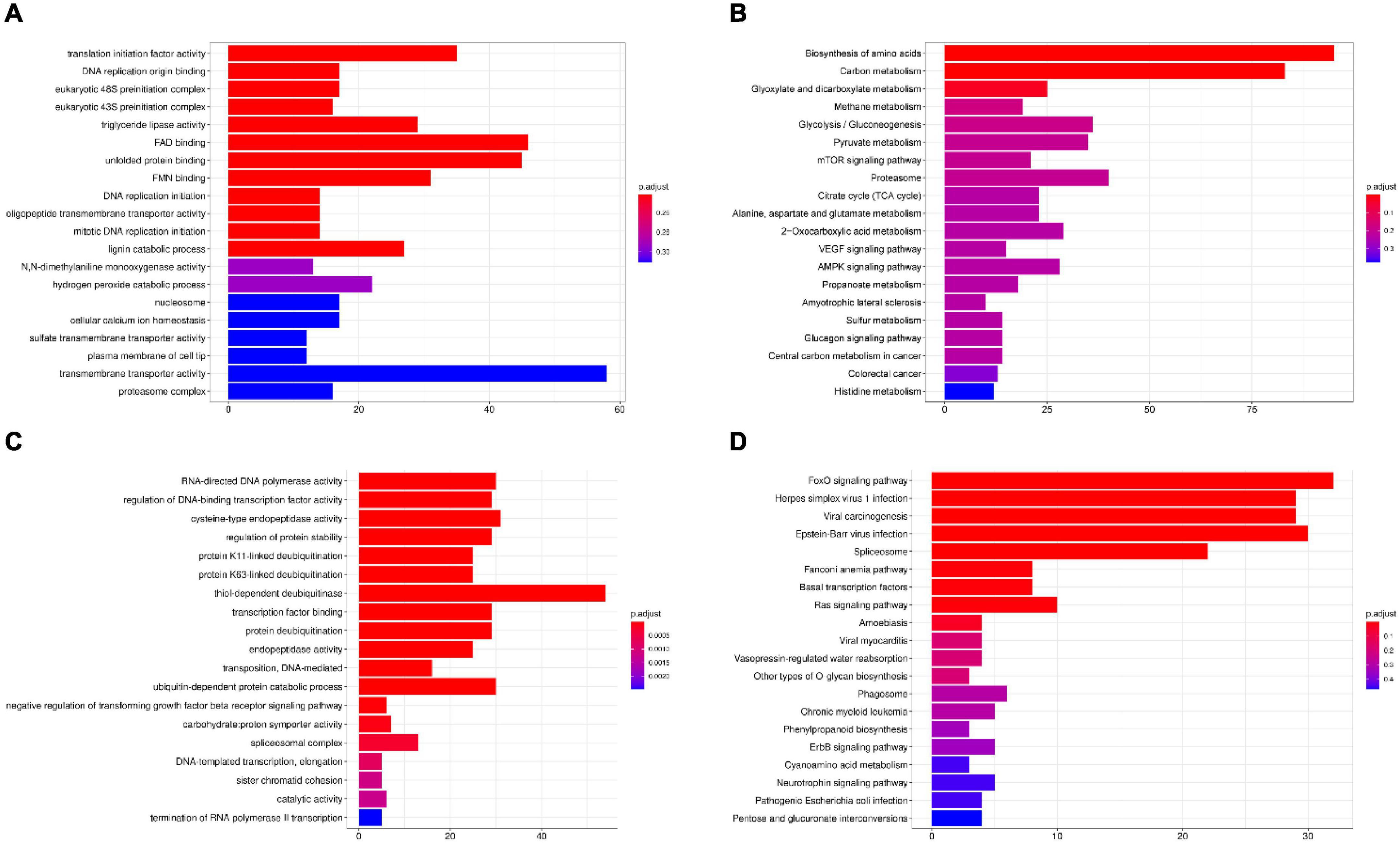
Figure 12. Gene family enrichment analysis of I. hispidus compared with S. baumii, P. nigrolimitatus and P. pouzarii. (A) GO enrichment results of the share gene family. (B) KEGG enrichment results of the shared gene family. (C) GO enrichment results of the unique gene family. (D) KEGG enrichment results of the unique gene family.
I. hispidus is one of the key commercial products in agricultural production. However, few studies have investigated the genome sequence and genetic structure of I. hispidus. The genetic information of fungi can provide agricultural industry with data on the molecular mechanisms of host function and interactions with plants (Prasad Singh et al., 2019). Therefore, in this study, the whole-genome map of the I. hispidus was developed to explore the expression of its genetic information and the annotation of related functional genes. We obtained 999 protein sequences by cytochrome P450 annotation. Cytochrome P450 enzymes are heme thiol proteins that have been widely demonstrated to be involved in primary and secondary metabolism in fungi (Crešnar and Petrič, 2011). P450 enzymes can also participate in the detoxification process of exogenous substances, such as CYP504A1 and CYP504B1, and can degrade the benzene ring and its hydrocarbon derivatives (Ferrer-Sevillano and Fernández-Cañón, 2007). Furthermore, the P450 enzyme system can use benzoate as a substrate and exhibit O-demethylation activity, which is particularly important for the detoxification process of antifungal substances (Podobnik et al., 2008). The annotation results of cytochrome P450 are helpful for the subsequent development of functional genes of I. hispidus. Through the analysis of CAZymes, we found that the glycoside hydrolase family genes were significantly enriched in I. hispidus. The study conducted by Tao et al. found that glycoside hydrolase-related genes play a key role in the growth and development of mushrooms, and they are involved in the expansion of the cap (Tao et al., 2013). The results of another study showed that glycoside hydrolase-related genes can be significantly involved in the growth and senescence of edible mushrooms (Ding et al., 2007). Most glycoside hydrolase family genes also have the function of starch degradation, which means that the enrichment of these genes indicates that the I. hispidus contributes to nutrient absorption and utilization, and can use diverse substrates as energy sources (Fang et al., 2020). This evidence indicates that, at the gene level, the enrichment of glycoside hydrolase family genes contributes to the growth and the diversification of nutrient substrate utilization of I. hispidus. The yield of mushrooms depends on the utilization of substrates, and the degradation of lignocellulose is the most important part of the solution to substrate utilization (Ling and Fungi, 2018). Thus, it is important to study the composition of CAZyme in I. hispidus and determine its mechanism of action to provide a theoretical basis for the yield improvement of I. hispidus at the genetic level. Therefore, we annotated the CAZyme gene information based on the I. hispidus genome sequence. Our results can be used for the subsequent screening of candidate genes for the nutritional utilization of I. hispidus and the development of high-yielding and high-quality I. hispidus by genetic means.
This study has identified many essential genes related to secondary metabolites, which endow I. hispidus with diverse biological activities. Genes involved in the regulation of terpenes were most often found in the genome of I. hispidus. Triterpene structures in fungi play a crucial role in their biological functions. For example, triterpenes in Ganoderma lucidum (G. lucidum) can inhibit the proliferation of some cancer cells (Artursson et al., 2001). It showed significant toxicity to lung cancer and liver cancer cells. In vitro studies have shown that these triterpenoids inhibit the invasion and metastasis of cancer cells in vitro and may be used as a potential drug for cancer treatment (Chen et al., 2010). G. lucidum also showed a strong anti-tumor effect. In the experiment conducted by Gao et al., triterpenoids extracted from G. lucidum fruiting bodies inhibited tumor growth in mice with Lewis lung cancer (Gao et al., 2006). Moreover, terpenes showed vigorous antioxidant activity, significantly increasing the activities of superoxide dismutase and catalase, ultimately eliminating destructive reactive oxygen species (Ajith et al., 2009; Smina et al., 2011). I. hispidus has 35 genes involved in regulating terpenes, which explains the anti-tumor mechanism of this fungus at the genetic level. Further homology analysis demonstrated that I. hispidus and S. baumii showed high genetic similarity. The S. baumii extract also showed excellent antioxidant, anti-inflammatory, and anti-tumor activities (Wang et al., 2020; Sun et al., 2021; Zheng et al., 2021). In particular, S. baumii extract significantly induced a potential mitochondrial membrane breakdown and mitochondrial-dependent apoptosis in A375 cells (Wang et al., 2020). This also suggests that these similar genomes have the same biological activity.
By comparison of gene family enrichment analysis, we found that I. hispidus, S. baumii, P. nigrolimitatus and P. pouzarii shared the main category of “transmembrane transporter activity,” which performs the function of transporting substances from one side to the other. The transmembrane transporter family is associated with fungal growth and development (Saier, 1999). In addition to the common genes that function in the daily growth of fungi, compared with S. baumii, P. nigrolimitatus and P. pouzarii, the unique genes of I. hispidus are enriched in “thiol-dependent deubiquitinase” and “FoxO signaling pathway.” The FoxO signaling pathway involves many cellular physiological events such as apoptosis, cell cycle control, glucose metabolism, anti-oxidative stress and longevity (Flachsbart et al., 2009). Accumulated studies have found that FOXO can remove excessive free radicals and prevent oxidative damage. FOXO activates the expression of antioxidant enzyme systems (including promoting the expression of SOD and CAT) so that free radicals can be more effectively removed (Anselmi et al., 2009; Dansen et al., 2009). These may also be why I. hispidus has a strong antioxidant capacity.
In summary, in this study, the whole-genome map of I. hispidus, a crucial medicinal fungus, was developed for the first time. The Illumina NovaSeq high-throughput sequencing platform was used for sequencing. After establishing the database, the flow cell was transferred to the Oxford Nanopore PromethION sequencing instrument for real-time single-molecule sequencing. Functional annotations of the I. hispidus genome were performed using public databases and proprietary databases. Finally, the genes related to secondary metabolites were predicted; it was found that most of the genes involved in terpenes in the I. hispidus genome provided a theoretical basis for its medicinal value.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA848015 and https://www.ncbi.nlm.nih.gov/, SAMN28983551.
ST performed the study and conducted data analysis. JX designed the research. LJ, PL, CS, SW, YY, YH, and RR provided assistance for the study. All authors read and revised the manuscript.
This research was funded by Hunan Province Key R&D Projects (2019NK2192), Provincial Natural Science Foundation of Hunan (2022JJ40233 and 2020JJ4049), and Changsha Key R&D Projects (kh2201216).
We are grateful to the National Natural Science Foundation of China and the Hunan Provincial Department of Science and Technology for their financial support for this experiment and the reviewers and editor for their constructive comments, which significantly improved our manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ajith, T. A., Sudheesh, N. P., Roshny, D., Abishek, G., and Janardhanan, K. K. (2009). Effect of Ganoderma lucidum on the activities of mitochondrial dehydrogenases and complex I and II of electron transport chain in the brain of aged rats. Exp. Gerontol. 44, 219–223. doi: 10.1016/j.exger.2008.11.002
Anselmi, C. V., Malovini, A., Roncarati, R., Novelli, V., Villa, F., Condorelli, G., et al. (2009). Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 12, 95–104. doi: 10.1089/rej.2008.0827
Artursson, P., Palm, K., and Luthman, K. (2001). Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 46, 27–43. doi: 10.1016/s0169-409x(00)00128-9
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Awadh Ali, N. A., Mothana, R. A., Lesnau, A., Pilgrim, H., and Lindequist, U. (2003). Antiviral activity of Inonotus hispidus. Fitoterapia 74, 483–485. doi: 10.1016/s0367-326x(03)00119-9
Benarous, K., Benali, F. Z., Bekhaoua, I. C., and Yousfi, M. (2021). Novel potent natural peroxidases inhibitors with in vitro assays, inhibition mechanism and molecular docking of phenolic compounds and alkaloids. J. Biomol. Struct. Dyn. 39, 7168–7180. doi: 10.1080/07391102.2020.1808073
Benarous, K., Bombarda, I., Iriepa, I., Moraleda, I., Gaetan, H., Linani, A., et al. (2015). Harmaline and hispidin from Peganum harmala and Inonotus hispidus with binding affinity to Candida rugosa lipase: In silico and in vitro studies. Bioorg. Chem. 62, 1–7. doi: 10.1016/j.bioorg.2015.06.005
Blanco, E., Parra, G., and Guigó, R. (2007). Using geneid to identify genes. Curr. Protoc. Bioinformatics Chapter 4:Unit4.3. doi: 10.1002/0471250953.bi0403s18
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 37:D233–D238. doi: 10.1093/nar/gkn663
Chen, N. H., Liu, J. W., and Zhong, J. J. (2010). Ganoderic acid T inhibits tumor invasion in vitro and in vivo through inhibition of MMP expression. Pharmacol. Rep. 62, 150–163. doi: 10.1016/s1734-1140(10)70252-8
Crešnar, B., and Petrič, S. (2011). Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta. 1814, 29–35. doi: 10.1016/j.bbapap.2010.06.020
Dansen, T. B., Smits, L. M., van Triest, M. H., de Keizer, P. L., van Leenen, D., Koerkamp, M. G., et al. (2009). Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 5, 664–672. doi: 10.1038/nchembio.194
Deng, Y., Jianqi, L. I., Songfeng, W. U., Zhu, Y., Chen, Y., and Fuchu, H. E. J. C. E. (2006). Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 32, 71–72.
Ding, S., Ge, W., and Buswell, J. A. (2007). Molecular cloning and transcriptional expression analysis of an intracellular beta-glucosidase, a family 3 glycosyl hydrolase, from the edible straw mushroom, Volvariella volvacea. FEMS Microbiol. Lett. 267, 221–229. doi: 10.1111/j.1574-6968.2006.00550.x
Edgar, R. C., and Myers, E. W. (2005). PILER: Identification and classification of genomic repeats. Bioinformatics 21:i152–i158. doi: 10.1093/bioinformatics/bti1003
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fang, M., Wang, X., Chen, Y., Wang, P., Lu, L., Lu, J., et al. (2020). Genome Sequence Analysis of Auricularia heimuer Combined with Genetic Linkage Map. J. Fungi 6:37. doi: 10.3390/jof6010037
Ferrer-Sevillano, F., and Fernández-Cañón, J. M. (2007). Novel phacB-encoded cytochrome P450 monooxygenase from Aspergillus nidulans with 3-hydroxyphenylacetate 6-hydroxylase and 3,4-dihydroxyphenylacetate 6-hydroxylase activities. Eukaryot Cell 6, 514–520. doi: 10.1128/ec.00226-06
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44:D279–D285. doi: 10.1093/nar/gkv1344
Flachsbart, F., Caliebe, A., Kleindorp, R., Blanché, H., von Eller-Eberstein, H., Nikolaus, S., et al. (2009). Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. U.S.A. 106, 2700–2705. doi: 10.1073/pnas.0809594106
Gao, J. J., Hirakawa, A., Min, B. S., Nakamura, N., and Hattori, M. J. J. O. N. M. (2006). In vivo antitumor effects of bitter principles from the antlered form of fruiting bodies of Ganoderma lucidum. J. Nat. Med. 60, 42–48.
Gründemann, C., Arnhold, M., Meier, S., Bäcker, C., Garcia-Käufer, M., Grunewald, F., et al. (2016). Effects of Inonotus hispidus Extracts and Compounds on Human Immunocompetent Cells. Planta Med. 82, 1359–1367. doi: 10.1055/s-0042-111693
Han, Y., and Wessler, S. R. (2010). MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199. doi: 10.1093/nar/gkq862
Huo, J., Zhong, S., Du, X., Cao, Y., Wang, W., Sun, Y., et al. (2020). Whole-genome sequence of Phellinus gilvus (mulberry Sanghuang) reveals its unique medicinal values. J. Adv. Res. 24, 325–335. doi: 10.1016/j.jare.2020.04.011
Jiang, J. H., Wu, S. H., and Zhou, L. W. (2021). The First Whole Genome Sequencing of Sanghuangporus sanghuang Provides Insights into Its Medicinal Application and Evolution. J. Fungi 7:787. doi: 10.3390/jof7100787
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32:D277–D280. doi: 10.1093/nar/gkh063
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Z., and Bao, H. (2022). Anti-tumor effect of Inonotus hispidus petroleum ether extract in H22 tumor-bearing mice and analysis its mechanism by untargeted metabonomic. J. Ethnopharmacol. 285:114898. doi: 10.1016/j.jep.2021.11489
Ling, Z., and Fungi, R. Z. J. A. E. (2018). Advances of Genomics-assisted Cultivation and Breedingof Edible and Medicinal Mushrooms. Acta Edulis Fungi 25, 93–106.
Liu, X., Hou, R., Xu, K., Chen, L., Wu, X., Lin, W., et al. (2019a). Extraction, characterization and antioxidant activity analysis of the polysaccharide from the solid-state fermentation substrate of Inonotus hispidus. Int. J. Biol. Macromol. 123, 468–476. doi: 10.1016/j.ijbiomac.2018.11.069
Liu, X., Hou, R., Yan, J., Xu, K., Wu, X., Lin, W., et al. (2019b). Purification and characterization of Inonotus hispidus exopolysaccharide and its protective effect on acute alcoholic liver injury in mice. Int. J. Biol. Macromol. 129, 41–49. doi: 10.1016/j.ijbiomac.2019.02.011
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39:W339–W346. doi: 10.1093/nar/gkr466
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Park, J., Lee, S., Choi, J., Ahn, K., Park, B., Park, J., et al. (2008). Fungal cytochrome P450 database. BMC Genomics 9:402. doi: 10.1186/1471-2164-9-402
Podobnik, B., Stojan, J., Lah, L., Krasevec, N., Seliskar, M., Rizner, T. L., et al. (2008). CYP53A15 of Cochliobolus lunatus, a target for natural antifungal compounds. J. Med. Chem. 51, 3480–3486. doi: 10.1021/jm800030e
Politi, M., Silipo, A., Siciliano, T., Tebano, M., Flamini, G., Braca, A., et al. (2007). Current analytical methods to study plant water extracts: The example of two mushrooms species, Inonotus hispidus and Sparassis crispa. Phytochem. Anal. 18, 33–41. doi: 10.1002/pca.949
Prasad Singh, P., Srivastava, D., Jaiswar, A., and Adholeya, A. (2019). Effector proteins of Rhizophagus proliferus: Conserved protein domains may play a role in host-specific interaction with different plant species. Braz. J. Microbiol. 50, 593–601. doi: 10.1007/s42770-019-00099-x
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21:i351–i358. doi: 10.1093/bioinformatics/bti1018
Saier, M. H. Jr. (1999). Eukaryotic transmembrane solute transport systems. Int. Rev. Cytol. 190, 61–136. doi: 10.1016/s0074-7696(08)62146-4
Saier, M. H. Jr., Tran, C. V., and Barabote, R. D. (2006). TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 34:D181–D186. doi: 10.1093/nar/gkj001
Schwarzer, D., Finking, R., and Marahiel, M. A. (2003). Nonribosomal peptides: From genes to products. Nat. Prod. Rep. 20, 275–287. doi: 10.1039/b111145k
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smina, T. P., De, S., Devasagayam, T. P., Adhikari, S., and Janardhanan, K. K. (2011). Ganoderma lucidum total triterpenes prevent radiation-induced DNA damage and apoptosis in splenic lymphocytes in vitro. Mutat. Res. 726, 188–194. doi: 10.1016/j.mrgentox.2011.09.005
Song, F., Su, D., Keyhani, N. O., Wang, C., Shen, L., and Qiu, J. (2021). Influence of selenium on the mycelia of the shaggy bracket fungus, Inonotus hispidus. J. Sci. Food Agric. 102, 3762–3770. doi: 10.1002/jsfa.11724
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19:ii215–ii225. doi: 10.1093/bioinformatics/btg1080
Sun, Y., Huo, J., Zhong, S., Zhu, J., Li, Y., and Li, X. (2021). Chemical structure and anti-inflammatory activity of a branched polysaccharide isolated from Phellinus baumii. Carbohydr. Polym. 268:118214. doi: 10.1016/j.carbpol.2021.118214
Tao, Y., Xie, B., Yang, Z., Chen, Z., Chen, B., Deng, Y., et al. (2013). Identification and expression analysis of a new glycoside hydrolase family 55 exo-β-1,3-glucanase-encoding gene in Volvariella volvacea suggests a role in fruiting body development. Gene 527, 154–160. doi: 10.1016/j.gene.2013.05.071
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4:Unit4.10. doi: 10.1002/0471250953.bi0410s25
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Tian, F., Li, C., and Li, Y. (2021). Genomic Analysis of Sarcomyxa edulis Reveals the Basis of Its Medicinal Properties and Evolutionary Relationships. Front. Microbiol. 12:652324. doi: 10.3389/fmicb.2021.652324
Wang, T., Bao, H. Y., Bau, T., and Li, Y. (2016). [Antitumor Effect of Solid State Fermentation Powder of Inonotus hispidus on H22 Bearing Mice]. Zhong Yao Cai 39, 389–394.
Wang, T., Sun, S., Liang, C., Li, H., Liu, A., and Zhu, H. (2020). Effective isolation of antioxidant Phelligridin LA from the fermentation broth of Inonotus baumii by macroporous resin. Bioproc. Biosyst. Eng. 43, 2095–2106. doi: 10.1007/s00449-020-02398-2
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Xu, Z., and Wang, H. (2007). LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35:W265–W268. doi: 10.1093/nar/gkm286
Yang, S., Bao, H., Wang, H., and Li, Q. (2019). Anti-tumour Effect and Pharmacokinetics of an Active Ingredient Isolated from Inonotus hispidus. Biol. Pharm. Bull. 42, 10–17. doi: 10.1248/bpb.b18-00343
Zan, L. F., Qin, J. C., Zhang, Y. M., Yao, Y. H., Bao, H. Y., and Li, X. (2011). Antioxidant hispidin derivatives from medicinal mushroom Inonotus hispidus. Chem. Pharm. Bull. 59, 770–772. doi: 10.1248/cpb.59.770
Zhang, F., Xue, F., Xu, H., Yuan, Y., Wu, X., Zhang, J., et al. (2021). Optimization of Solid-State Fermentation Extraction of Inonotus hispidus Fruiting Body Melanin. Foods 10:2893. doi: 10.3390/foods10122893
Keywords: Inonotus hispidus, functional annotation, CAZyme, secondary metabolites, phylogenetic analysis
Citation: Tang S, Jin L, Lei P, Shao C, Wu S, Yang Y, He Y, Ren R and Xu J (2022) Whole-genome assembly and analysis of a medicinal fungus: Inonotus hispidus. Front. Microbiol. 13:967135. doi: 10.3389/fmicb.2022.967135
Received: 12 June 2022; Accepted: 17 August 2022;
Published: 06 September 2022.
Edited by:
Lakxmi Subramanian, Queen Mary University of London, United KingdomReviewed by:
Chenyang Huang, Institute of Agricultural Resources and Regional Planning (CAAS), ChinaCopyright © 2022 Tang, Jin, Lei, Shao, Wu, Yang, He, Ren and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Xu, aG5zd3N3X3hqQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.