 Yudai Nishide
Yudai Nishide Kohei Oguchi
Kohei Oguchi Maria Murakami
Maria Murakami Minoru Moriyama
Minoru Moriyama Ryuichi Koga
Ryuichi Koga Takema Fukatsu
Takema Fukatsu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 August 2022
Sec. Microbial Symbioses
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.962252
This article is part of the Research Topic Microbial Associates of Blood-Sucking Arthropods and Other Animals: Relevance to Their Physiology, Ecology and Evolution View all 12 articles
Insects exclusively feeding on vertebrate blood are usually dependent on symbiotic bacteria for provisioning of B vitamins. Among them, sucking lice are prominent in that their symbiotic bacteria as well as their symbiotic organs exhibit striking diversity. Here we investigated the bacterial diversity associated with the boar louse Haematopinus apri in comparison with the hog louse Haematopinus suis. Amplicon sequencing analysis identified the primary endosymbiont predominantly detected from all populations of H. apri with some minor secondary bacterial associates. Sequencing and phylogenetic analysis of bacterial 16S rRNA gene confirmed that the endosymbionts of the boar louse H. apri, the hog louse H. suis and the cattle louse Haematopinus eurysternus form a distinct clade in the Gammaproteobacteria. The endosymbiont clade of Haematopinus spp. was phylogenetically distinct from the primary endosymbionts of other louse lineages. Fluorescence in situ hybridization visualized the endosymbiont localization within midgut epithelium, ovarial ampulla and posterior oocyte of H. apri, which were substantially the same as the endosymbiont localization previously described in H. suis and H. eurysternus. Mitochondrial haplotype analysis revealed that, although the domestic pig was derived from the wild boar over the past 8,000 years of human history, the populations of H. apri constituted a distinct sister clade to the populations of H. suis. Based on these results, we discussed possible evolutionary trajectories of the boar louse, the hog louse and their endosymbionts in the context of swine domestication. We proposed ‘Candidatus Haematopinicola symbiotica’ for the distinct clade of the endosymbionts of Haematopinus spp.
Insects represent the majority of the biodiversity in the terrestrial ecosystem (Grimaldi and Engel, 2005), in which symbiotic microorganisms generally play important biological roles (Buchner, 1965; Bourtzis and Miller, 2003). In particular, insects utilizing nutritionally recalcitrant food resources are usually dependent on their specific microbial partners, wherein the representative microbial functions are digestion of plant cell wall polymers that are otherwise indigestible for many wood-feeding insects like termites, bark beetles, etc. (Brune, 2014; Biedermann and Vega, 2020), provisioning of essential amino acids that are lacking in plant vascular fluid for plant sap-sucking insects like aphids, cicadas, etc. (Moran et al., 2008; Douglas, 2009), and the synthesis of B vitamins that are deficient in vertebrate blood for blood-sucking insects like lice, bedbugs, etc. (Rio et al., 2016; Husnik, 2018).
Blood-sucking insects, such as mosquitoes, fleas, tsetse flies, lice, bedbugs, etc., are notorious not only for causing itch, wound, and inflammation to human skin but also for vectoring devastating human and veterinary pathogens (Lehane, 2005). Among them, those feeding on vertebrate blood throughout their life stages usually develop specialized symbiotic organs for hosting specific symbiotic bacteria: Wigglesworthia in tsetse flies (Aksoy, 1995; Akman et al., 2002), Aschnera in nycteribiid bat flies (Hosokawa et al., 2012), Wolbachia in bedbugs (Hosokawa et al., 2010; Nikoh et al., 2014), and others.
Sucking lice (Phthiraptera: Anoplura) live on vertebrate blood as the sole food source throughout their life cycle (Lehane, 2005), and most of them develop specialized symbiotic organs that harbor specific symbiotic bacteria (Ries, 1931; Buchner, 1965). Both histological inspection (Ries, 1931) and molecular phylogenetic survey (Hypša and Křížek, 2007) revealed that their symbiotic organs and associated bacterial symbionts are strikingly diverse among different lice lineages and likely of independent evolutionary origins: ‘Candidatus Riesia spp.’ are harbored in a distinct oval symbiotic organ located on the ventral side of the intestine called the “stomach disc” in human and primate lice Pediculus spp. and Pthirus spp. (Sasaki-Fukatsu et al., 2006; Allen et al., 2007; Perotti et al., 2007; Kirkness et al., 2010; Boyd et al., 2014); ‘Candidatus Puchtella pedicinophila’ is harbored in a specific region of the midgut epithelium in the monkey lice Pedicinus spp. (Fukatsu et al., 2009; Boyd et al., 2017); and ‘Candidatus Legionella polyplacis’ is in an intestine-associated symbiotic organ in a rodent louse Polyplax serrata (Říhová et al., 2017). In other lice lineages, the symbiotic organs are histologically not so distinct and the symbiotic associations might be either younger and/or casual: Sodalis and Rickettsia in a seal louse Proechinophthirus fluctus (Boyd et al., 2016); Neisseria/Snodgrassella-allied in rodent lice Hoplopleura spp. and Polyplax spp. (Říhová et al., 2021). Thus far, however, in vivo localization and developmental dynamics of the symbiotic organs and the symbiotic bacteria have been only briefly described in most cases, except for those in the human body louse Pediculus humanus and also those in the cattle louse Haematopinus eurysternus (Ries, 1931; Buchner, 1965).
The genus Haematopinus consists of some 20 species of ungulate lice, including the cattle louse H. eurysternus, the hog louse Haematopinus suis, the horse louse Haematopinus asini, and others (Durden and Musser, 1994). Historically, the endosymbiotic bacteria of Haematopinus spp. have been described as follows: the initial microscopic detection in H. suis (Sikora, 1919; Buchner, 1920; Florence, 1924); detailed histological inspection of in vivo localization and developmental dynamics in H. eurysternus (Ries, 1931); transmission electron microscopic observation in H. suis (Żelazowska and Biliński, 1999); and molecular phylogenetic analysis based on 16S rRNA gene sequences from H. suis, H. eurysternus and Haematopinus apri (Hypša and Křížek, 2007). These previous studies, mostly conducted on H. suis or H. eurysternus, have shown that the endosymbionts of Haematopinus spp. form a well-supported clade in the Gammaproteobacteria and exhibit peculiar localization patterns distinct from the other louse lineages: in both females and males, numerous bacteriocytes are scattered over the midgut epithelium, recognized as swellings into the midgut cavity, in which the symbiotic bacteria look like endocellular at a glance but actually reside in an extracellular space surrounded by the bacteriocyte cytoplasm; and specifically in females, the symbiotic bacteria also localize to the dorsal “depot bacteriomes,” from which the symbiotic bacteria migrate to the ovarial ampullae and infect to developing oocytes (Ries, 1931; Buchner, 1965). These early histological works, based on conventional light microscopy and illustrated by hand-drawn sketches, should be re-examined by modern histological techniques and sophisticated microscopy.
The boar louse H. apri parasitizes the wild boar and distributes across Eurasia (Durden and Musser, 1994). Reflecting the recent origin of the pig (Sus scrofa domestica) via domestication of the wild boar (Sus scrofa scrofa) over the past 8,000 years (Larson et al., 2005; Larson and Fuller, 2014; Frantz et al., 2016), the boar louse H. apri is morphologically and phylogenetically similar to the hog louse H. suis, although the former exhibits paler body color, relatively smaller legs, and smaller paratergites in comparison with the latter (Ferris, 1933). As for the endosymbiont of H. apri, only a bacterial 16S rRNA gene sequence (1,213 bp in size; DQ076665), has been reported and analyzed for inferring the molecular phylogenetic placement (Hypša and Křížek, 2007), while no other biological information is available. The evolutionary relationship between H. apri and H. suis is somewhat reminiscent of the relationship between the human head louse (P. humanus capitis) and the human body louse (P. humanus humanus) that has been argued in relation to human clothing and civilization (Kittler et al., 2003; Veracx and Raoult, 2012; Amanzougaghene et al., 2020). In this context, detailed characterization of the endosymbiont of H. apri in comparison with the endosymbiont of H. suis is of interest.
In an initial attempt toward this research goal, we investigated the bacterial diversity associated with multiple populations of the boar louse H. apri using amplicon sequencing, molecular phylogenetic and histological approaches, thereby identifying the primary endosymbiont and minor bacterial associates of H. apri. We discussed the relationship between the endosymbionts of H. apri and H. suis in the context of swine domestication, and proposed a candidate name for the endosymbiotic bacteria associated with the louse genus Haematopinus.
Table 1 and Supplementary Table S1 list the insect samples examined in this study. These insects were preserved either in ethanol or acetone (Fukatsu, 1999), or brought to the laboratory alive and processed immediately. For histological observations, the insects were dissected in phosphate-buffered saline (PBS; 0.8% NaCl, 0.02% KCl, 0.115% Na2HPO4, 0.02% KH2PO4) by using fine tweezers under a dissection microscope (M165FC; Leica).

Table 1. Samples of the boar louse H. apri and the hog louse H. suis examined in this study.
The samples were washed by soaking in 1% bleach and 0.1% Triton X-100 for 1 min and then rinsed twice with water. Then, the samples were individually crushed using BioMasher II (Nippi, Tokyo, Japan) and subjected to DNA extraction using DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). Insect mitochondrial cytochrome c oxidase subunit I (COI) gene was amplified by PCR using KOD FX Neo (TOYOBO, Osaka, Japan) with the primers modified-mtd6 (5′-GGA GGW TTY GGA AAT TGR TTA GTD CC-3′) and mtd11 (5′-ACT GTA AAY ATA YGR TGW GCT CA-3′) (Jiang et al., 2013) that were selected to match COI sequences of H. suis and H. apri (accession numbers HM241908 and KC814616). Bacterial 16S rRNA gene was amplified by PCR using Ex taq HS (TaKaRa, Shiga, Japan) with two sets of the primers 16SA1 (5′-AGA GTT TGA TCM TGG CTC AG-3′)—16SB2 (3′-CGA GCT GAC ARC CAT GCA-3′) and 16SA2 (5′-GTG CCA GCA GCC GCG GTA ATA C-3′)—16SB1 (5’-TAC GGY TAC CTT GTT ACG ACT T-3′) (Fukatsu and Nikoh, 1998). The PCR products were cloned using pGEM-T Easy Vector (Promega, Madison, United States), Mighty mix for DNA ligation (Takara) and Escherichia coli DH5 competent cells (Takara). The inserted plasmids were extracted from the transformed E. coli cells using QIAprep Spin miniprep kit (QIAGEN) and subjected to sequencing reactions using BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Massachusetts, United States) with the primers M13FW (5′-GTA AAA CGA CGG CCA GT-3′) and M13RV (5′-CAG GAA ACA GCT ATG AC-3′) targeting the flanking regions of the vector.
The molecular phylogenetic analyses were conducted by the maximum likelihood methods using MEGA 5.2 (Tamura et al., 2011). The optimum model was selected by model tests to describe each phylogenetic tree. Bootstrap probability values based on 1,000 replications are shown on the nodes. To analyze the genetic diversity of host COI sequences, we depicted the network diagrams of COI haplotypes using MEGA-X (Kumar et al., 2018), DnaSP 6 (Rozas et al., 2017) and Network 10 (fluxus-engineering.com).
The hypervariable V3/V4 region of bacterial 16S rRNA gene, around 0.4 kb in size, was targeted for amplicon sequencing analyses. Library construction was conducted by the 2-step tailed PCR procedure with the primers first_341_MIX (5′-ACA CTC TTT CCC TAC ACG CTC TTC CGA TCT NNN NNC CTA CGG GNG GCW GCA G-3′) and first_805r_MIX (5′-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TNN NNN GAC TAC HVG GGT ATC TAA TCC-3′), and then second_F (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC AC-Indexseq (8mer)-ACA CTC TTT CCC TAC ACG ACG C-3′) and second_R (5′-CAA GCA GAA GAC GGC ATA CGA GAT-Indexseq (8mer)-GTG ACT GGA GTT CAG ACG TGT G-3′). The libraries were measured for concentration using Synergy H1 (Bio Tec, Vermont, United States) and QuantiFluor dsDNA System (Promega), and quantified using Fragment Analyzer (Agilent, California, United States) and dsDNA 915 Reagent Kit (Agilent). Sequencing of the libraries was performed using Miseq (Illumina, California, United States). From the raw amplicon sequences obtained (accession number DRR360832-DRR360847), only the reads containing the tags in PCR primers were selected using FASTX-Toolkit (ver.0.0.14). The output reads were subjected to denoising and removal of chimeric sequences using the DADA2 plugin in the Quantitative Insights Into Microbial Ecology (QIIME2) v.2021.11 pipeline (Bolyen et al., 2019). Bacterial taxa were assigned to the representative amplicon sequences based on GreenGenes V.13_8 (McDonald et al., 2012) using the q2-feature-classifier QIIME2 plugin (Bokulich et al., 2018).
Whole-mount fluorescent in situ hybridization (FISH) targeting bacterial 16S rRNA was performed essentially as described previously (Koga et al., 2009). The dissected insect tissues were fixed in 4% paraformaldehyde in PBS for 3 h and then thoroughly washed in PBST (0.1% Tween 20 in PBS). The fixed insect tissues were washed twice in hybridization buffer (20 mM Tris–HCl [pH 8.0], 0.9 M NaCl, 0.01% SDS, 30% formamide). To specifically target 16S rRNA of the primary symbiont of Haematopinus spp., we designed the oligonucleotide probe Wbsym1181R (5′-ACC TTC GCA GGT TAG CTT-3′) labeled with fluorochrome Alexa Fluor 647 at the 5′ terminus. For universal detection of bacterial 16S rRNA, we used the probe EUB338 (5′-GCT GCC TCC CGT AGG AGT-3′) (Amann et al., 1990) labeled with Alexa Fluor 555 at the 5′ terminus. The samples were incubated in hybridization buffer containing 50 nM probe. After washed twice in PBST, host nuclear DNA and filamentous actin were stained with 4.5 μM 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) and 1.5 μM Alexa Fluor 488-labeled phalloidin (Thermo Fisher Scientific), respectively, for 1 h at room temperature. Then, the samples were washed with PBST again, mounted in 50% glycerol in PBS, and observed under a laser scanning confocal microscope (LSM 700; Carl Zeiss, Germany). As a control experiment for FISH detection, RNase digestion controls in which the tissue samples were treated with RNase A prior to hybridization were conducted (Supplementary Figure S1).
In order to grasp the bacterial diversity associated with H. apri, 13 insects representing six collection localities (see Supplementary Table S1) were subjected to amplicon sequencing analysis targeting the hypervariable V3/V4 region of bacterial 16S rRNA gene. In addition, three insects of H. suis collected from a locality were analyzed in the same way (see Supplementary Table S1). Figure 1 shows the composition of the amplicon sequences for each of the insect samples. The same bacterial taxon was predominantly detected from almost all the samples (except for Hyogo samples of H. apri), accounting for over 85% of the detected sequences for each of the samples. The remaining minor bacterial taxa were assigned as allied to Neisseria, Streptococcus, Serratia and Aeromonas for H. apri, and Turicibacter for H. suis (Figure 1). Exceptionally, two Hyogo samples of H. apri exhibited different patterns: in a sample, Enterobacter was highly predominant, accounting for over 80% of the detected sequences; in another sample, not only Enterobacter but also Citrobacter occupied substantial fractions of the microbiota (Figure 1). Notably, these Hyogo samples were shipped to our laboratory by refrigerated, not frozen, courier service, delayed in arrival, and then transferred to an ethanol vial for preservation. Hence, it seems likely that proliferation of the microbes in the dead insect bodies resulted in the exceptional microbial compositions in these samples. Taken together, these results strongly suggested that a single bacterial species constitutes the major microbial associate of H. apri and H. suis, which is likely the primary endosymbiont as described in previous histological and phylogenetic studies on H. suis (Florence, 1924; Żelazowska and Biliński, 1999; Hypša and Křížek, 2007).
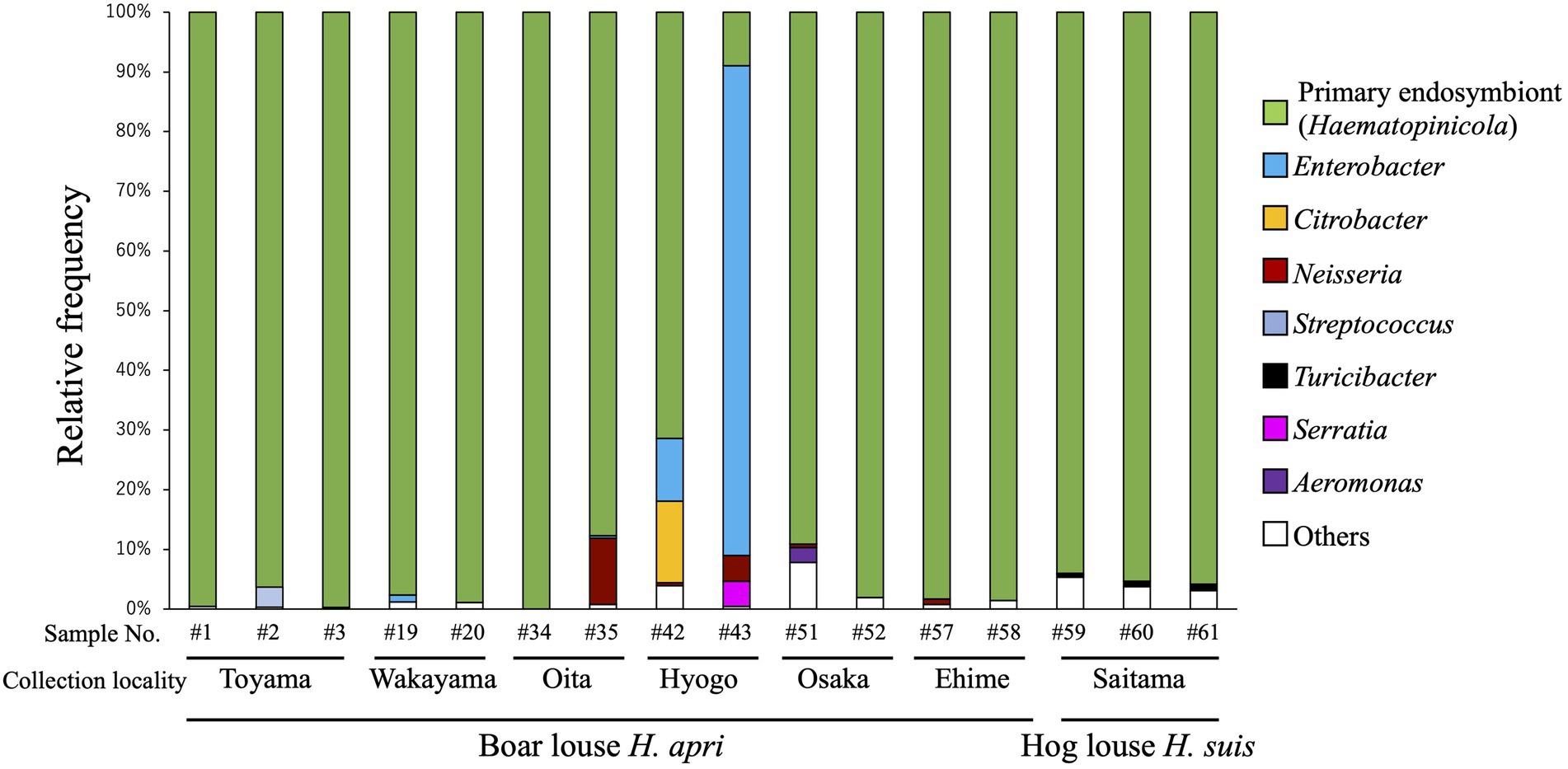
Figure 1. Bacterial diversity associated with the boar louse Haematopinus apri and the hog louse Haematopinus suis based on amplicon sequencing data of the hypervariable V3/V4 region of 16S rRNA gene. Assigned bacterial taxa are color-coded as shown on the right side. As for sample information and amplicon sequencing data, see Supplementary Table S1.
Ten insects of H. apri representing four collection localities in Japan (see Supplementary Table S1) were individually subjected to PCR, cloning and sequencing of bacterial 16S rRNA gene. All the sequences were completely identical to each other (accession number LC706254). DNA database searches using the sequence as query retrieved 16S rRNA gene sequences of endosymbionts of H. suis from United States (KX146200), H. suis from Czech (DQ076662), H. apri from Czech (DQ076665) and H. eurysternus from Czech (DQ076661) (Hypša and Křížek, 2007; Allen et al., 2016) as top hits. In addition, two insects of H. suis collected from a locality in Japan (see Supplementary Table S1) were subjected to the analysis, which yielded identical sequences (accession number LC706255) with 1 bp difference from the sequence obtained from H. apri. Supplementary Figure S2 A summarizes the sequence differences among the endosymbiont sequences of H. apri and H. suis from Japan, United States and Czech.
Figure 2 shows the phylogenetic relationship of the primary endosymbionts of H. apri and H. suis based on 16S rRNA gene sequences. The endosymbionts of H. apri from Japan, H. suis from Japan, H. suis from United States and H. suis from Czech formed a highly supported compact clade. Reflecting some nucleotide differences (see Supplementary Figure S2A), the endosymbiont of H. apri from Czech was placed outside the clade, and formed a highly supported clade together. Then, the endosymbiont of H. eurysternus was placed outside the clade of the endosymbionts of H. suis and H. apri, forming a highly supported clade together. The distinct clade of the endosymbionts of Haematopinus spp. was not allied to the endosymbionts of other insects including sucking lice, such as Riesia, Puchtella, Wigglesworthia, Blochmannia and Baumannia, in the Gammaproteobacteria.
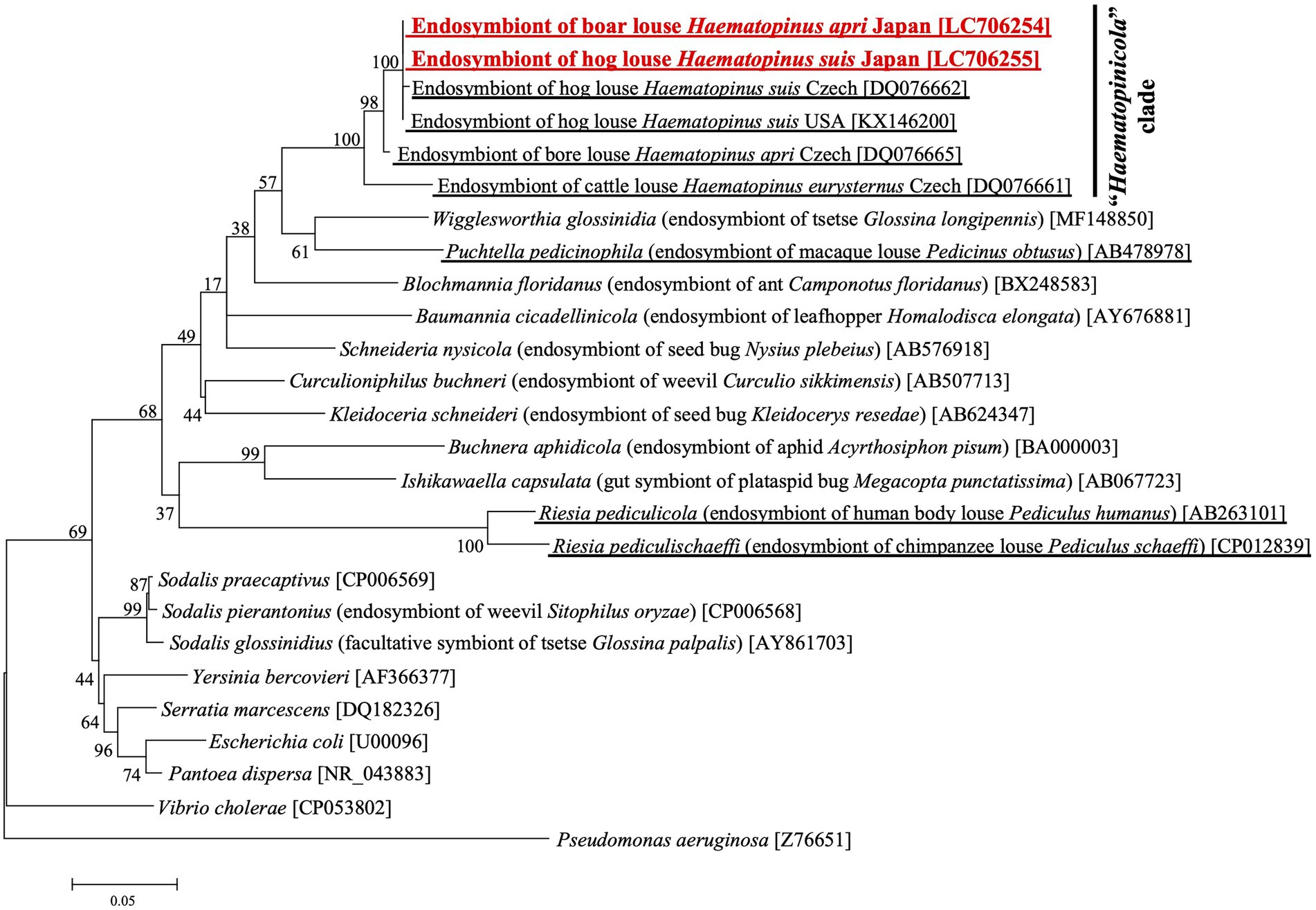
Figure 2. Phylogenetic placement of the primary endosymbionts of the boar louse H. apri and the hog louse H. suis based on 16S rRNA gene sequences. A maximum-likelihood phylogeny inferred from 1,013 aligned nucleotide sites is shown with bootstrap probability at each node (model: K2 + G + I). Host insect information is shown in parentheses, whereas accession number is indicated in brackets. The louse endosymbiont sequences determined in this study are highlighted in red, whereas the louse endosymbiont sequences reported in previous studies are underlined.
Figure 3 shows FISH visualization of the primary endosymbiont in H. apri. In adult females with mature ovaries, dense symbiont signals were detected in a specialized ovarial region lying between lateral oviduct and ovarioles, so-called ovarial ampulla (Buchner, 1965) (Figures 3B,C). In both females and males, dense symbiont signals were detected throughout the midgut epithelium in a scattered manner (Figures 3D,E). Each of the patchy bacteriocyte-like structures, which contained a dense population of the symbiont cells and protruded from the epithelial wall to the midgut cavity, were actually constituted by multiple epithelial cells that encased the symbiont cells within the extracellular cavity formed at the center (Figures 3F–H). Within the ovarial ampulla, by contrast, the symbiont cells were found within the host cytoplasm endocellularly (Figures 3I,J). In mature oocytes in the ovarioles, the symbiont cells localized to the posterior pole, where infecting bacterial cells through the follicle cell layer were often observed (Figure 3K). These in vivo localization and infection dynamics of the primary endosymbiont were generally in agreement with previous histological descriptions on H. eurysternus and H. suis (Ries, 1931; Buchner, 1965; Żelazowska and Biliński, 1999).
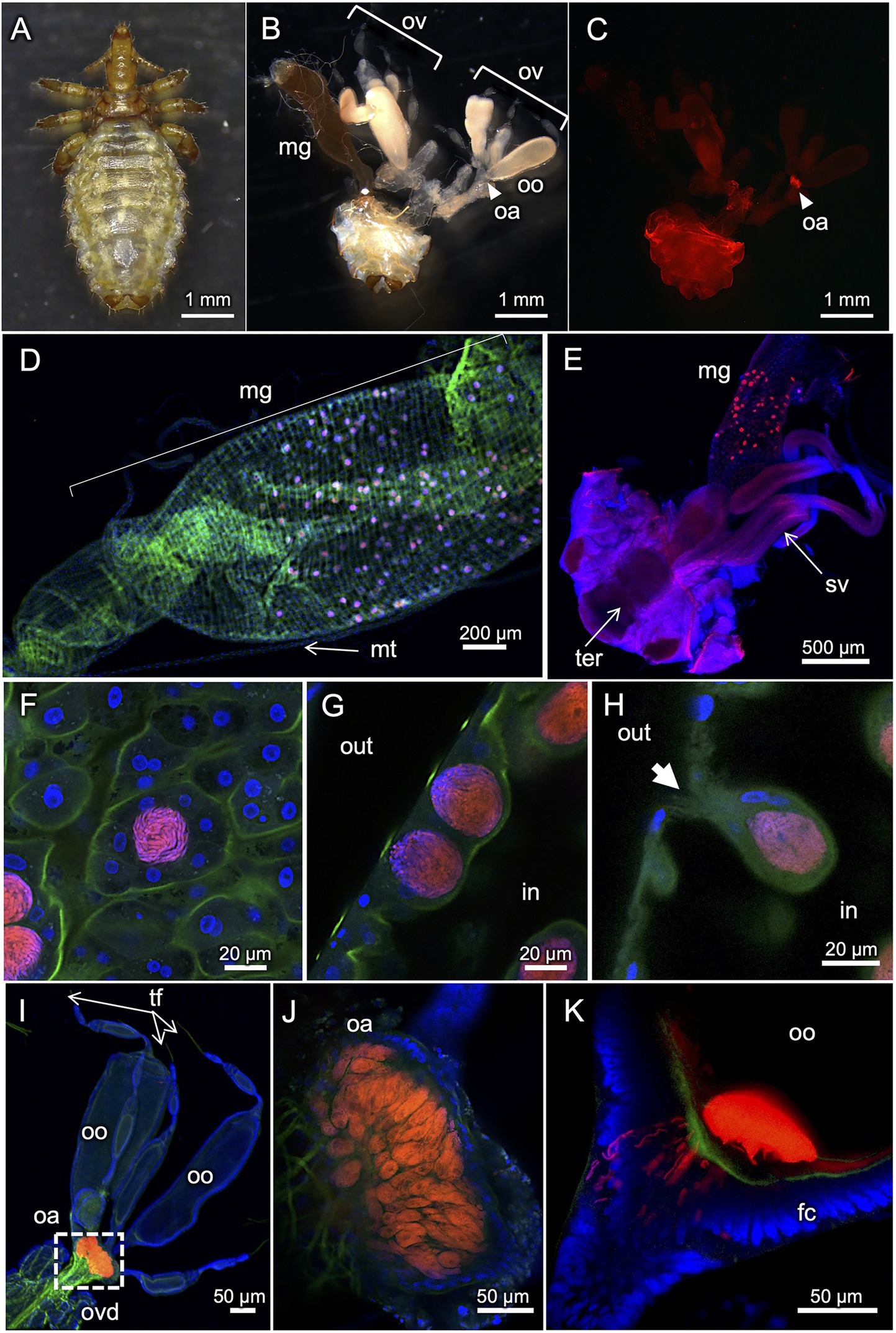
Figure 3. FISH visualization of in vivo localization of the primary endosymbiont in H. apri. (A) Dorsal view of an adult female. (B) Internal organs dissected from an adult female. (C–E) Whole-mount FISH visualization of the symbiont in dissected organs. (C) Localization of the symbiont in the ovarial ampulla of an adult female. The sample is the same as (B). (D) Scattered localization of the symbiont across the midgut epithelial region of an adult female. (E) Similar localization pattern of the symbiont in the midgut epithelium of an adult male. (F–H) Magnified confocal images of the symbiont in the midgut epithelium. (F) Optical section image parallel to the epithelial plane. (G) Optical section image perpendicular to the epithelial plane. (H) Optical section image crossing the epithelial plane, in which a bacteriocyte-like structure with an external pit is seen. The bacteriocyte-like structures are actually extracellular cavities round in shape, full of tubular bacterial cells, and surrounded by the midgut epithelial cells. (I–K) Localization of the symbiont in ovaries and oocytes. (I) Localization of the symbiont in the ovarial ampulla located at the interface of lateral oviduct and oocyte-containing ovarioles. (J) Optical section image of the ovarial ampulla, in which peculiar host cells are densely populated by the symbiont cells. (K) Optical section image of the posterior pole of an oocyte, to which the tubular symbiont cells are infecting through the follicle cell layer. Abbreviations: fc, follicle cell layer; hg, hindgut; mg, midgut; mt, Malpighian tubule; oa, ovarial ampulla; oo, oocyte; ov, ovary; ovd, oviduct; sv, seminal vesicle; ter, tergite; tf, terminal filament. In (B,C), arrowheads indicate the location of the ovarial ampulla. In (G,H), “out” and “in” indicate outside and inside of the midgut, respectively. In (H), an arrow highlights a pit associated with the bacteriocyte-like structure.
Recently, microbiome survey of Polyplax and Hoplopleura rodent lice identified Neisseria/Snodgrassella-allied bacteria as either obligatory or facultative microbial associates (Říhová et al., 2021). Since our amplicon sequencing analysis also detected Neisseria-allied bacterial associates from several H. apri samples at low frequencies (Figure 1), we cloned and sequenced 16S rRNA gene sequence of the bacteria from H. apri. DNA database searches using the sequence as query retrieved 16S rRNA gene sequences of Neisseriaceae bacteria from a rodent louse Hoplopleura acanthopus (CP046107) and a prairie dog flea Oropsylla hirsute (EU137419) as the top hits, and Snodgrassella alvi from bumble bees and honeybees (HM108703, HM113170 and others) as the next best hits. Figure 4 shows the phylogenetic relationship of Snodgrassella, Neisseria and allied 16S rRNA gene sequences, which indicated that the bacterium associated with H. apri is certainly placed in the Neisseriaceae, and the most closely related to the bacterial associates of the rodent louse Hoplopleura and the prairie dog flea Oropsylla.

Figure 4. Phylogenetic placement of the Neisseria/Snodgrassella-allied bacterium of the boar louse H. apri in the Neisseriaceae based on 16S rRNA gene sequences. A maximum-likelihood phylogeny inferred from 1,063 aligned nucleotide sites is shown with bootstrap probability at each node (model: HKY + G + I). Host insect information is shown in parentheses, whereas accession number is indicated in brackets. The louse derived sequence determined in this study is highlighted in red, whereas the louse derived sequence reported in a previous study is underlined.
In order to understand the population genetic structure of the host lice, mitochondrial COI gene was amplified by PCR and sequenced for 44 individuals of H. apri originating from five collection localities and also for two individuals of H. suis (see Supplementary Table S1). In total, 13 haplotypes were identified for the COI gene sequences: haplotypes 1–12 represented H. apri and closely related to each other, whereas haplotype 13 represented H. suis and was distant from the other haplotypes (Figure 5A). Supplementary Figure S2 B summarizes the sequence differences among mitochondrial COI gene sequences of H. apri and H. suis from Japan, China and Australia. In the DNA databases, a COI gene sequence of H. apri from Hyogo, Japan (KC814616), a COI gene sequence of H. apri from Hunan, China (ON000919), and two COI gene sequences of H. suis from Perth, Australia (HM241908, KC814607), were deposited. We performed molecular phylogenetic analysis of these COI gene sequences, and demonstrated that H. apri from Japanese and Chinese populations and H. suis from Japanese and Australian populations formed compact and well-supported sister clades, respectively (Figure 5B).
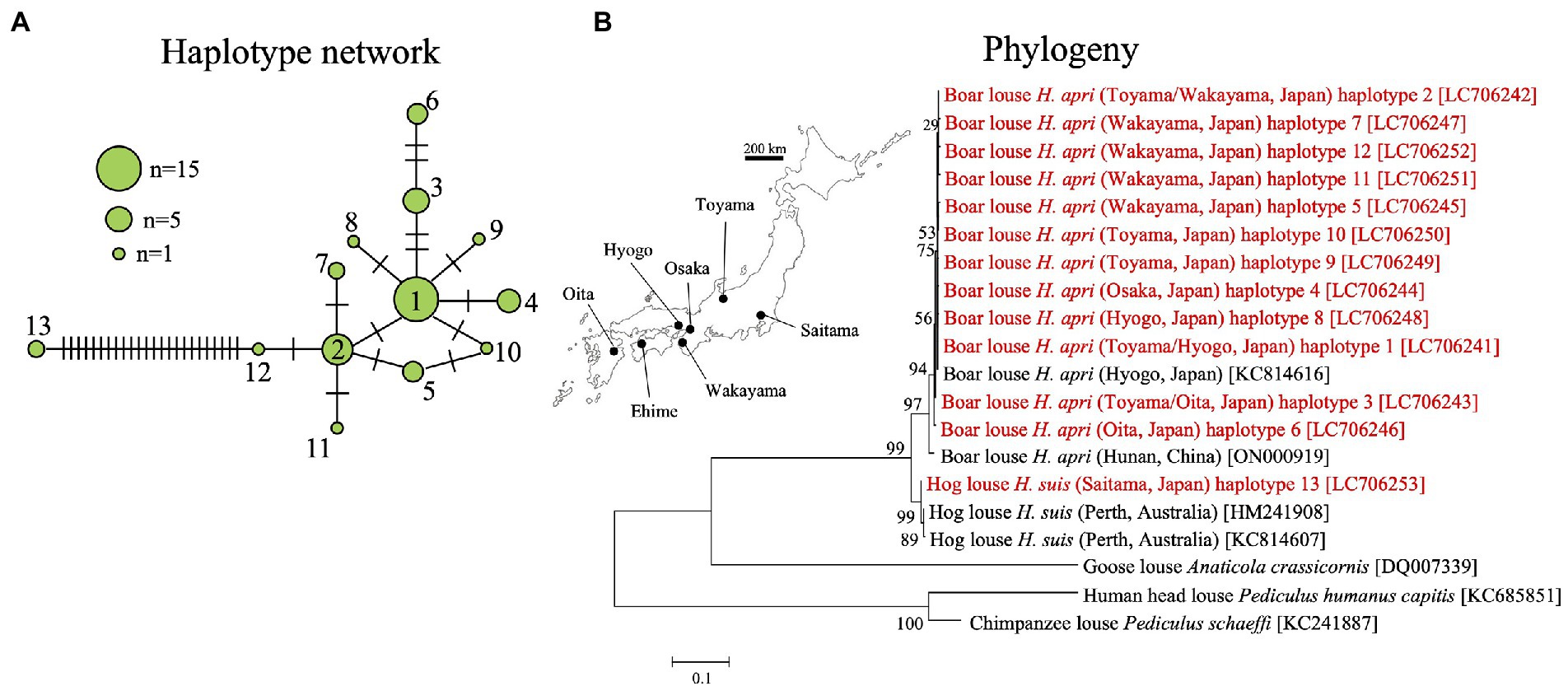
Figure 5. (A) Haplotype network of mitochondrial COI gene haplotypes representing 44 individuals from five collection localities of H. apri and two individuals from a collection locality of H. suis. (B) Phylogenetic relationship of the COI haplotypes of H. apri and H. suis. COI gene sequences of H. apri from Japan and China, and those of H. suis from Australia, were retrieved from the DNA databases and analyzed together. A maximum-likelihood phylogeny inferred from 516 aligned nucleotide sites is shown with bootstrap probability at each node (model: HKY + G + I). Collection locality information is shown in parentheses, whereas accession number is indicated in brackets. The collection localities are depicted on the map of Japan. The sequences determined in this study are highlighted in red.
In the recognized diversity of the symbiotic organs and the endosymbiotic bacteria among sucking lice (Ries, 1931; Buchner, 1965; Rio et al., 2016; Husnik, 2018), previous studies on the endosymbiotic system in Haematopinus ungulate lice were restricted to early histological descriptions on H. suis and H. eurysternus (Florence, 1924; Ries, 1931; Żelazowska and Biliński, 1999) and a brief molecular phylogenetic characterization for H. suis, H. eurysternus and H. apri (Hypša and Křížek, 2007). As for the boar louse H. apri, the only available information on its endosymbiont has been a single 16S rRNA gene sequence (Hypša and Křížek, 2007). In this study, we investigated the endosymbiotic microbiota of H. apri samples derived from multiple local populations using amplicon sequencing, molecular phylogenetic and FISH histological approaches, thereby unequivocally identifying microbiological nature, in vivo localization and infection dynamics of the boar louse endosymbiont.
Our amplicon sequencing analysis targeting the hypervariable V3/V4 region of bacterial 16S rRNA gene identified a specific bacterial associate predominantly found from all the local populations of H. apri (Figure 1). Molecular phylogenetic analysis based on bacterial 16S rRNA gene sequences showed that the bacterium is closely related to and forming a distinct clade with the endosymbionts of H. suis, H. apri and H. eurysternus whose 16S rRNA gene sequences have been reported previously (Hypša and Křížek, 2007) (Figure 2). Detailed histological observations by FISH (Figure 3) revealed that in vivo localization and infection dynamics of the bacterium are concordant with the detailed early histological descriptions on the endosymbiont localization in H. eurysternus (Ries, 1931), and, though less comprehensive, in H. suis (Florence, 1924; Żelazowska and Biliński, 1999). Therefore, we conclude that this bacterium is the primary endosymbiont of H. apri, and suggest that this bacterial clade may represent the primary endosymbiont clade of Haematopinus ungulate lice.
Here, however, we point out that the endosymbiont gene sequence of Japanese H. apri was considerably different, by 15 nucleotide sites, from that of Czech H. apri (Supplementary Figure S2 A). Notably, while the endosymbiont gene sequence of Japanese H. suis was identical to that of American H. suis, the endosymbiont gene sequence of Czech H. suis differed by 6 nucleotide sites in comparison with that of Japanese H. suis (Supplementary Figure S2 A). These patterns may be explained by the following alternative hypotheses: (i) the endosymbionts of Japanese and American H. apri and H. suis are genetically differentiated from the endosymbionts of European H. apri and H. suis, respectively; or (ii) the endosymbiont gene sequences of Czech H. apri and H. suis (Hypša and Křížek, 2007) contain sequencing errors. We note that the endosymbiont sequence of Czech H. apri (DQ076665) contains an ambiguous (N) nucleotide site. Collection and analysis of more samples of H. apri and H. suis from Europe and other regions in the world will clarify which of these hypotheses is more appropriate.
In this study, we found that, on the basis of mitochondrial COI gene sequences, Japanese H. apri samples from different local populations are, though with some genetic differences, genetically coherent, whereas they are genetically distinct from Japanese H. suis samples (Figure 5A). Molecular phylogenetic analysis of the COI gene sequences, together with those of Chinese H. apri and Australian H. suis retrieved from the DNA databases, revealed that H. apri and H. suis are in a sister clade relationship (Figure 5B). However, considering the limited number of samples derived from the limited number of original localities examined in this study, our result should be regarded as tentative and treated with caution, with the following conditions kept in mind.
Thus far, molecular phylogenetic, comparative genomic, and archeological studies have been extensively conducted to clarify how the pig originated from the wild boar via domestication. These studies uncovered that (i) domestication of boars into pigs occurred at least twice independently, in East Anatolia and China, about 8,000 years ago, (ii) since then, the domestic pigs spread across the world with human movements, with occasional hybridization with local wild boars, and (iii) therefore, the genomic architecture and the evolutionary trajectory of the domestic pigs are rather mosaic and reticulated (Larson et al., 2005; Frantz et al., 2015, 2016, 2019). In this context, it is of great interest how the boar louse H. apri, the hog louse H. suis, and their endosymbionts have evolved in the process of swine domestication. The phylogenetic relationship, the species status, and the evolutionary history of H. apri and H. suis should be established with more louse samples collected from all over the world.
Besides the primary endosymbiont, amplicon sequencing analysis detected relatively minor bacterial associates of H. apri and H. suis, which were assigned as Enterobacter, Citrobacter, Neisseria, Streptococcus, Serratia, Aeromonas, Turicibacter, etc. (Figure 1). On account of the low incidence in the amplicon sequencing data, these bacteria are plausibly facultative and/or casual associates for the host insects. Among them, the Neisseria-allied bacterium associated with H. apri is of particular interest, because it was recently reported that Neisseria/Snodgrassella-allied bacteria are associated with rodent lice of the genera Hoplopleura and Polyplax as their potential endosymbionts (Říhová et al., 2021). Molecular phylogenetic analysis verified that the Neisseria-like bacterium of H. apri is closely related to Neisseriaceae bacteria from the rodent louse H. acanthopus and the prairie dog flea Oropsylla hirsute, and also to the gut symbionts of bees Snodgrassella alvi (Figure 4). It is also notable that Neisseria species tend to be associated with mammals and birds, such as N. canis with dog, N. animaloris with racoon dog, N. zalophi with sea lion, N. arctica with goose, etc. (see Figure 4), as inhabitants of the mucous membranes of animals (Tønjum, 2015). These observations suggest the possibility that, although speculative, the blood-sucking lice may acquire the Neisseria-allied bacteria from their host animals either casually or as commensal associates, and some of them have established an obligatory association as observed in H. acanthopus (Říhová et al., 2021).
On the basis of the distinct microbiological, phylogenetic and histological features described in this and previous studies, we propose the designation ‘Candidatus Haematopinicola symbiotica’ for the hitherto unnamed endosymbiotic bacterial clade associated with ungulate lice of the genus Haematopinus. The generic name highlights the endosymbiotic association with Haematopinus spp., and the specific name indicates the endosymbiotic nature of the bacterial clade. Thus far, the cattle louse H. eurysternus (Ries, 1931; Hypša and Křížek, 2007), the hog louse H. suis (Sikora, 1919; Buchner, 1920; Florence, 1924; Ries, 1931; Żelazowska and Biliński, 1999; Hypša and Křížek, 2007; this study) and the boar louse H. apri (Hypša and Křížek, 2007; this study) have been shown to host ‘Ca. H. symbiotica’. Histologically, the horse louse H. asini seems likely to harbor ‘Ca. H. symbiotica’ (Buchner, 1965). Whether the other Haematopinus species are also associated with ‘Ca. H. symbiotica’ requires verification in future studies.
In conclusion, we characterized the primary endosymbiont of the boar louse H. apri in detail, uncovered its population genetic and phylogenetic aspects in relation to host’s local populations, and proposed the designation ‘Ca. H. symbiotica’ for the primary endosymbiont clade of the louse genus Haematopinus. The biological role of ‘Ca. H. symbiotica’ is expected as provisioning of B vitamins for the blood-sucking host lice, which should be verified by sequencing and analysis of the endosymbiont genomes. Extensive worldwide collection of H. apri and H. suis samples and genetic analysis of them and their endosymbionts would uncover how the boar louse, the hog louse, and their endosymbionts have evolved in the process of swine domestication in the human history.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
YN performed molecular phylogenetic and amplicon sequencing analyses with support by MaM and MiM. KO conducted histological analyses with support by RK. TF conceived the study and arranged collection of insect samples. YN and TF wrote the paper. All authors contributed to the article and approved the submitted version.
This study was supported by the Japan Science and Technology Agency (JST) ERATO Grant Numbers JPMJER1803 and JPMJER1902 to RK and TF, and by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP17H06388 to TF. KO was supported by the JSPS Research Fellowships for Young Scientists Number 21 J01321.
We thank Ai Takano, Shigehiko Uni, Takeo Yamauchi and Yuki Miyaoka for providing louse samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.962252/full#supplementary-material
SUPPLEMENTARY TABLE S1 | Samples of H. apri and H. suis used in this study with sequence accession numbers.
SUPPLEMENTARY FIGURE S1 | FISH detection and RNase digestion control of the primary endosymbiont in the midgut epithelium of H. apri.
SUPPLEMENTARY FIGURE S2 | (A) Nucleotide differences among 16S rRNA gene sequences of the primary endosymbionts of Haematopinus apri and Haematopinus suis from Japan, United States and Czech. Number of nucleotide differences out of 1100 aligned nucleotide sites is indicated for each of the sequence pairs. Note that the sequence of H. apri symbiont Czech [DQ076665] contains an ambiguous (N) site. (B) Nucleotide substitutions among mitochondrial COI gene sequences of H. apri and H. suis from Japan, Australia and China. Number of nucleotide differences out of aligned nucleotide sites is indicated for each of the sequence pairs. The sequences determined in this study are highlighted in red.
Akman, L., Yamashita, A., Watanabe, H., Oshima, K., Shiba, T., Hattori, M., et al. (2002). Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32, 402–407. doi: 10.1038/ng986
Aksoy, S. (1995). Wigglesworthia gen. nov. and Wigglesworthia glossinidia sp. nov., taxa consisting of the mycetocyte-associated, primary endosymbionts of tsetse flies. Int. J. Syst. Evol. Microbiol. 45, 848–851. doi: 10.1099/00207713-45-4-848
Allen, J. M., Reed, D. L., Perotti, M. A., and Braig, H. R. (2007). Evolutionary relationships of “Candidatus Riesia spp.,” endosymbiotic Enterobacteriaceae living within hematophagous primate lice. Appl. Environ. Microbiol. 73, 1659–1664. doi: 10.1128/AEM.01877-06
Allen, J. M., Burleigh, J. G., Light, J. E., and Reed, D. L. (2016). Effects of 16S rDNA sampling on estimates of the number of endosymbiont lineages in sucking lice. PeerJ 4:e2187. doi: 10.7717/peerj.2187
Amann, R. I., Krumholz, L., and Stahl, D. A. (1990). Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 172, 762–770. doi: 10.1128/jb.172.2.762-770.1990
Amanzougaghene, N., Fenollar, F., Raoult, D., and Mediannikov, O. (2020). Where are we with human lice? A review of the current state of knowledge. Front. Cell. Infect. Microbiol. 9:474. doi: 10.3389/fcimb.2019.00474
Biedermann, P. H. W., and Vega, F. E. (2020). Ecology and evolution of insect–fungus mutualisms. Annu. Rev. Entomol. 65, 431–455. doi: 10.1146/annurev-ento-011019-024910
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Boyd, B. M., Allen, J. M., de Crécy-Lagard, V., and Reed, D. L. (2014). Genome sequence of Candidatus Riesia pediculischaeffi, endosymbiont of chimpanzee lice, and genomic comparison of recently acquired endosymbionts from human and chimpanzee lice. G3 4, 2189–2195. doi: 10.1534/g3.114.012567
Boyd, B. M., Allen, J. M., Koga, R., Fukatsu, T., Sweet, A. D., Johnson, K. P., et al. (2016). Two bacterial genera, Sodalis and Rickettsia, associated with the seal louse Proechinophthirus fluctus (Phthiraptera: Anoplura). Appl. Environ. Microbiol. 82, 3185–3197. doi: 10.1128/AEM.00282-16
Boyd, B. M., Allen, J. M., Nguyen, N.-P., Vachaspati, P., Quicksall, Z. S., Warnow, T., et al. (2017). Primates, lice and bacteria: speciation and genome evolution in the symbionts of hominid lice. Mol. Biol. Evol. 34, 1743–1757. doi: 10.1093/molbev/msx117
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Buchner, P. (1920). Zur Kenntnis der Symbiose niederer pflanzlicher Organismen mit Pedikuliden. Biol. Zentralbl. 39, 535–540. doi: 10.5962/bhl.part.19203
Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. doi: 10.1111/j.1365-2435.2008.01442.x
Durden, L. A., and Musser, G. G. (1994). The mammalian hosts of the sucking lice (Anoplura) of the world: a host-parasite list. Bull. Soc. Vector Ecol. 19, 130–168.
Ferris, G. F. (1933). Contributions toward a monograph of the sucking lice. Part VII. Stanford Univ. Pub. Biol. Sci. 2, 415–634.
Florence, L. (1924). An intracellular symbiont of the hog louse. Am. J. Trop. Med. 4, 397–409. doi: 10.4269/ajtmh.1924.s1-4.397
Frantz, L. A. F., Haile, J., Lin, A. T., Scheu, A., Geörg, C., Benecke, N., et al. (2019). Ancient pigs reveal a near-complete genomic turnover following their introduction to Europe. Proc. Natl. Acad. Sci. U. S. A. 116, 17231–17238. doi: 10.1073/pnas.1901169116
Frantz, L., Meijaard, E., Gongora, J., Haile, J., Groenen, M. A. M., and Larson, G. (2016). The evolution of Suidae. Annu. Rev. Anim. Biosci. 4:61. doi: 10.1146/annurev-animal-021815-111155
Frantz, L. A. F., Schraiber, J. G., Madsen, O., Megens, H. J., Cagan, A., Bosse, M., et al. (2015). Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes. Nat. Genet. 47, 1141–1148. doi: 10.1038/ng.3394
Fukatsu, T. (1999). Acetone preservation: a practical technique for molecular analysis. Mol. Ecol. 8, 1935–1945. doi: 10.1046/j.1365-294x.1999.00795.x
Fukatsu, T., Hosokawa, T., Koga, R., Nikoh, N., Kato, T., Hayama, S., et al. (2009). Intestinal endocellular symbiotic bacterium of the macaque louse Pedicinus obtusus: distinct endosymbiont origins in anthropoid primate lice and the old world monkey louse. Appl. Environ. Microbiol. 75, 3796–3799. doi: 10.1128/AEM.00226-09
Fukatsu, T., and Nikoh, N. (1998). Two intracellular symbiotic bacteria from the mulberry psyllid Anomoneura mori (Insecta, Homoptera). Appl. Environ. Microbiol. 64, 3599–3606. doi: 10.1128/AEM.64.10.3599-3606.1998
Grimaldi, D., and Engel, M. S. (2005). Evolution of the Insects. Cambridge University Press, New York.
Hosokawa, T., Koga, R., Kikuchi, Y., Meng, X. Y., and Fukatsu, T. (2010). Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. U. S. A. 107, 769–774. doi: 10.1073/pnas.0911476107
Hosokawa, T., Nikoh, N., Koga, R., Satô, M., Tanahashi, M., Meng, X.-Y., et al. (2012). Reductive genome evolution, host–symbiont co-speciation and uterine transmission of endosymbiotic bacteria in bat flies. ISME J. 6, 577–587. doi: 10.1038/ismej.2011.125
Husnik, F. (2018). Host–symbiont–pathogen interactions in blood-feeding parasites: nutrition, immune cross-talk and gene exchange. Parasitology 145, 1294–1303. doi: 10.1017/S0031182018000574
Hypša, V., and Křížek, J. (2007). Molecular evidence for polyphyletic origin of the primary symbionts of sucking lice (Phthiraptera, Anoplura). Microb. Ecol. 54, 242–251. doi: 10.1007/s00248-006-9194-x
Jiang, H., Barker, S. C., and Shao, R. (2013). Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol. Evol. 5, 1298–1308. doi: 10.1093/gbe/evt094
Kirkness, E. F., Haas, B. J., Sun, W., Braig, H. R., Perotti, M. A., Clark, J. M., et al. (2010). Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. U. S. A. 107, 12168–12173. doi: 10.1073/pnas.1003379107
Kittler, R., Kayser, M., and Stoneking, M. (2003). Molecular evolution of Pediculus humanus and the origin of clothing. Curr. Biol. 13, 1414–1417. doi: 10.1016/S0960-9822(03)00507-4
Koga, R., Tsuchida, T., and Fukatsu, T. (2009). Quenching autofluorescence of insect tissues for in situ detection of endosymbionts. Appl. Entomol. Zool. 44, 281–291. doi: 10.1303/aez.2009.281
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Larson, G., Dobney, K., Albarella, U., Fang, M., Matisoo-Smith, E., Robins, J., et al. (2005). Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307, 1618–1621. doi: 10.1126/science.1106927
Larson, G., and Fuller, D. Q. (2014). The evolution of animal domestication. Annu. Rev. Ecol. Evol. Syst. 45, 115–136. doi: 10.1146/annurev-ecolsys-110512-135813
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Nikoh, N., Hosokawa, T., Moriyama, M., Oshima, K., Hattori, M., and Fukatsu, T. (2014). Evolutionary origin of insect–Wolbachia nutritional mutualism. Proc. Natl. Acad. Sci. U. S. A. 111, 10257–10262. doi: 10.1073/pnas.1409284111
Perotti, M. A., Allen, J. M., Reed, D. L., and Braig, H. R. (2007). Host-symbiont interactions of the primary endosymbiont of human head and body lice. FASEB J. 21, 1058–1066. doi: 10.1096/fj.06-6808com
Ries, E. (1931). Die Symbiose der Laüse und Federlinge. Z. Morphol. Ökol. Tiere 20, 233–367. doi: 10.1007/BF00444101
Říhová, J., Batani, G., Rodríguez-Ruano, S. M., Martinů, J., Vácha, F., Nováková, E., et al. (2021). A new symbiotic lineage related to Neisseria and Snodgrassella arises from the dynamic and diverse microbiomes in sucking lice. Mol. Ecol. 30, 2178–2196. doi: 10.1111/mec.15866
Říhová, J., Nováková, E., Husník, F., and Hypša, V. (2017). Legionella becoming a mutualist: adaptive processes shaping the genome of symbiont in the louse Polyplax serrata. Genome Biol. Evol. 9, 2946–2957. doi: 10.1093/gbe/evx217
Rio, R. V. M., Attardo, G. M., and Weiss, B. L. (2016). Grandeur alliances: Symbiont metabolic integration and obligate arthropod hematophagy. Trends Parasitol. 32, 739–749. doi: 10.1016/j.pt.2016.05.002
Rozas, J., Ferrer-Mata, A., Sánchez-Del Barrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sasaki-Fukatsu, K., Koga, R., Nikoh, N., Yoshizawa, K., Kasai, S., Mihara, M., et al. (2006). Symbiotic bacteria associated with stomach discs of human lice. Appl. Environ. Microbiol. 72, 7349–7352. doi: 10.1128/AEM.01429-06
Sikora, H. (1919). Vorläufige Mitteilungen über Mycetome bei Pediculiden. Biol. Zentralbl. 39, 287–288.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tønjum, T. (2015). Neisseria. In Bergey’s manual of systematics of Archaea and Bacteria (eds M. E. Trujillo, S. Dedysh, P. DeVos, B. Hedlund, and P. Kämpfer),, et al., 1–48, Hoboken, New Jersey: Wiley.
Veracx, A., and Raoult, D. (2012). Biology and genetics of human head and body lice. Trends Parasitol. 28, 563–571. doi: 10.1016/j.pt.2012.09.003
Keywords: Haematopinus apri, boar louse, Haematopinus suis, hog louse, symbiont, evolution, domestication, symbiotic organ
Citation: Nishide Y, Oguchi K, Murakami M, Moriyama M, Koga R and Fukatsu T (2022) Endosymbiotic bacteria of the boar louse Haematopinus apri (Insecta: Phthiraptera: Anoplura). Front. Microbiol. 13:962252. doi: 10.3389/fmicb.2022.962252
Edited by:
Jun-Bo Luan, Shenyang Agricultural University, ChinaReviewed by:
Xiaoli Bing, Nanjing Agricultural University, ChinaCopyright © 2022 Nishide, Oguchi, Murakami, Moriyama, Koga and Fukatsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yudai Nishide, bmlzaGl5dTBAYWZmcmMuZ28uanA=; Takema Fukatsu, dC1mdWthdHN1QGFpc3QuZ28uanA=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.