 Aman Prakash
Aman Prakash- Department of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, Guwahati, India
In the genome of various Leptospira interrogans serovars, the subtype I-B locus of CRISPR-Cas possesses either one or multiple CRISPR arrays. In silico database (CRISPRCasdb) for predicting CRISPR-Cas reveals seven CRISPR arrays in L. interrogans serovar Lai positioned between the two independent cas-operons. Here, we present the redefined repeat-spacer boundaries of the CRISPR subtype I-B locus of serovar Lai. Such refinement of boundaries of arrays in serovar Lai was done after comparison with the characterized array of another serovar Copenhageni and the manual analysis of CRISPR flanking sequences. Using the reverse transcription-PCR (RT-PCR), we account that the seven CRISPR are transcriptionally active in serovar Lai. Our RT-PCR and quantitative real-time PCR analysis of transcripts in serovar Lai indicated that seven CRISPR of subtype I-B transcribe together as a single precursor unit. Moreover, the cleavage of the two miniature pre-crRNA of the subtype I-B by Cas6 demonstrates the biogenesis of the expected size of mature crRNA essential for the guided interference of foreign DNA. This study features insight into transcription direction and the crRNA biogenesis in serovar Lai essential for RNA-mediated interference of invading nucleic acids.
Introduction
CRISPR-Cas acquires the RNA-based adaptive immunity in prokaryotes against mobile foreign genetic material (Pourcel et al., 2005). A set of cas genes, a leader sequence, and the CRISPR array are the functional elements of the CRISPR-Cas immune system. A CRISPR comprises an array of direct repeats segregated by distinct spacer sequences, with a preceding leader sequence (Pourcel et al., 2005; Garneau et al., 2010; Yosef et al., 2012; Makarova et al., 2015). CRISPR immunity is led through three molecular stages: adaptation (or acquisition), expression (or CRISPR RNA biogenesis), and interference. An explicit region of foreign nucleic acid (protospacer) is embodied as a memory card at the leader’s proximal end during the adaptation stage (Yosef et al., 2012; Wang et al., 2015; Hille et al., 2018). In the expression stage, the CRISPR array is transcribed as a precursor CRISPR RNA (pre-crRNA) molecule and processed into mature crRNAs by the Cas endoribonuclease (Charpentier et al., 2015; Hochstrasser and Doudna, 2015). After that, the crRNA forms a complex with the Cas effector proteins in the final stage (interference) to interfere with the cognate alien nucleic acids (Barrangou et al., 2007; Hale et al., 2009; Garneau et al., 2010; Hille et al., 2018). The CRISPR-Cas require a protospacer adjacent motif (PAM) to recognize, differentiate, and eliminate specific foreign DNA (Westra et al., 2013). Although the length of PAMs can vary from two to six nucleotides, the most commonly reported PAM size is three to four nucleotides (Nishimasu et al., 2017). With the identification of a rising number of cas genes, the CRISPR-Cas systems have been divided into two classes (Class 1 and Class 2), six types (Type I-VI), and 33 subtype variants based on the different arrangements of cas genes and effector complex subunits (Makarova et al., 2020).
Several studies have shown the involvement of the CRISPR-cas system in regulating bacterial pathogenesis (Sampson et al., 2013; Sampson and Weiss, 2014). In a comparative genomics study among the saprophytic and infectious groups of Leptospira, CRISPR-Cas systems were identified mainly in the pathogenic Leptospira species (Fouts et al., 2016). The pathogenic species of the genus Leptospira causes leptospirosis, a zoonotic disease of global importance (Da and Levett, 2015). The presence of the CRISPR-Cas systems has been inferred as the virulence factor in pathogenic Leptospira (Fouts et al., 2016). Understanding Leptospira pathogenesis is still confined due to the lack of efficient genetic manipulation tools (Fernandes et al., 2021). With the advent of a shuttle vector (pMaOri), a new strategy to genetically manipulate Leptospira has been developed where episomal delivery of CRISPR-Cas9 (type II) was possible (Pappas et al., 2015). However, the type II application is limited to very few bacteria because the Cas9 induces double-strand breaks (DSBs) at the target DNA of the host, which must be repaired for cell viability (Lieber, 2010). In the Leptospira genome, owing to the absence of a non-homologous end-joining repair (NHEJ) system for DSBs, Cas9-induced DSBs were found to be lethal (Fernandes et al., 2019). To withstand the lethality, an inactive variant (dead) of CRISPR-dCas9 (CRISPRi) was employed for the targeted genetic manipulation in Leptospira spp. (Shapiro et al., 2018; Fernandes et al., 2021). However, the CRISPRi technology is limited to gene silencing (Zhao et al., 2017). Recently, CRISPR-Cas9 DSB lethality in Leptospira has been surpassed by the concomitant expression of the Mycobacterium NHEJ repair system (Fernandes and Nascimento, 2022). However, the tedious conjugation process and fastidious growth of Leptospira often lead to low efficiency of genetic manipulation (Fernandes et al., 2021).
Harnessing endogenous CRISPR-Cas types I and III systems of prokaryotes for genome editing is an attractive strategy to overcome the limitations of gene editing by type II Cas9 technology (Li et al., 2016). The pre-requisite of heterologous expression of Cas proteins (potentially toxic) can be surpassed inside the prokaryotes while exploiting the endogenous CRISPR-based method (Maikova et al., 2019). To date, an endogenous CRISPR-Cas system (subtypes I-A, I-B, or III-B) has been successfully applied for genome editing in several archaea and Clostridium spp. (Li et al., 2016; Pyne et al., 2016; Cheng et al., 2017; Maikova et al., 2019). Nevertheless, reprogramming the endogenous CRISPR-Cas system for genome editing relies on understanding the RNA-mediated immunity process. Thus, the molecular details of CRISPR arrays such as CRISPR transcription and orientation, repeat-spacer boundaries, and the PAM sequence are prerequisites to repurpose the endogenous CRISPR-Cas systems in Leptospira for genome editing. Such knowledge regarding the CRISPR-Cas system of pathogenic Leptospira will be advantageous in developing a genetic tool to understand gene function and pathogenesis at a higher efficiency. In the genus Leptospira and the genome of its infectious serovars (svs.), three variants of CRISPR/Cas type I systems (subtypes I-B, –C, and –E) are prevalent (Makarova et al., 2011; Xiao et al., 2019). In addition, recently CRISPR-Cas type V was recorded in a saprophytic Leptospira strain (L. biflexa) (Martínez Arbas et al., 2021). Serovars of L. interrogans harbor two subtypes (I-B and I-C) of the type I system; however, CRISPR arrays were identified only at the I-B locus (Xiao et al., 2019). Thus, it was speculated that the CRISPR-Cas I-B in L. interrogans might be sufficient for CRISPR immunity. In contrast, type I-C possibly carries out other unknown functions in L. interrogans (Xiao et al., 2019). In an in silico study of 41 Leptospira strains, 42% (48 out of 114) of the total identified CRISPR arrays were found more than 10 kb away from any cas gene (Xiao et al., 2019). Such isolated CRISPR arrays were referred to as orphan arrays (Zhang and Ye, 2017). In the genome of the pathogenic L. interrogans svs., such as Copenhageni and Lai, the two cas operons of the subtype I-B locus span a hypervariable region that contains either one or multiple CRISPR (Xiao et al., 2019). Previous studies from our group have defined the architecture CRISPR-Cas I-B locus in L. interrogans sv. Copenhageni strain Fiocruz L1-130 that comprises eight cas genes (cas1-cas8) and a single CRISPR (LIC_Cr2) (Dixit et al., 2016). The orientation of LIC_Cr2 pre-crRNA was along with the cas operons (Prakash and Kumar, 2021). Among the Cas proteins (LinCas1-LinCas8) associated with the I-B system of sv. Copenhageni, LinCas1 (Dixit et al., 2021b), LinCas2 (Dixit et al., 2016), LinCas4 (Dixit et al., 2021a), LinCas6 (Prakash and Kumar, 2021), and LinCas7 (Hussain and Kumar, 2022) have been characterized to date. In this study, in the genome of L. interrogans sv. Lai, the possible CRISPRs were predicted using the CRISPRCasdb database and was compared with the previously characterized I-B array of sv. Copenhageni. After that, the transcription of CRISPR I-B arrays in sv. Lai was analyzed using the reverse transcription-polymerase chain reaction (RT-PCR) technique. In addition, the processing of miniature pre-crRNA of sv. Lai has been studied using Cas6 to generate mature crRNAs.
Materials and methods
Bacterial strains and nucleic acid isolation
Leptospira strains (L. interrogans sv. Copenhageni strain Fiocruz L1-130 and L. interrogans sv. Lai strain 56601) were grown, maintained, and subcultured in the laboratory as described previously (Ghosh et al., 2018a,b). The cultures of leptospires strains were used in genomic DNA or total RNA isolation. The Escherichia coli strain (DH5α) was used for cloning and transformation.
Bioinformatics analysis
From the available genomic sequence of sv. Copenhageni and Lai, the CRISPRCasdb (Pourcel et al., 2020) defined CRISPR repeats and spacer sequences were extracted. Multiple sequence alignments (MSA) of repeats and spacers’ nucleotide sequences were performed using Clustal Omega (Madeira et al., 2019), and the graphic images of aligned sequences were obtained using the ESPript program (version 3.0) (Robert and Gouet, 2014). The WebLogo tool (Crooks et al., 2004) was used to construct a logo of eight nucleotides flanking (5′ and 3′) protospacers that were retrieved via the CRISPRTarget (Biswas et al., 2013) tool. The primers were designed manually or using the “OligoPerfect Primer Designer” tool of Thermo Fisher Scientific.
Reverse transcription-PCR (reverse transcription-polymerase chain reaction) and quantitative real-time PCR
The complementary DNA (cDNA) was synthesized from the total RNA (1 μg) of Leptospira (sv. Copenhageni or Lai) using the random hexamers or spacer-specific primer in a reverse transcription reaction as described previously (Ghosh et al., 2018a; Prakash and Kumar, 2021). Under similar conditions, an additional reaction was set up without the reverse transcriptase. Additional reaction served as a control (negative) in RT-PCR or qPCR to rule out the possibility of gDNA contamination in the purified RNA transcripts. The diluted (5-folds) and undiluted cDNA obtained were used as a template to perform qPCR and RT-PCR, respectively. Products of RT-PCR experiments were resolved onto 2% agarose gel. The qPCR analysis of CRISPR transcripts was performed according to established laboratory protocol (Ghosh et al., 2018a), where the transcripts of the target CRISPR were normalized with the 16S rRNA (rrs1) of Leptospira using the 2–ΔΔCT method (Livak and Schmittgen, 2001). The CRISPR transcripts were calculated per 106 copies of the 16S rRNA of respective Leptospira svs. For statistical analysis, two independent experiments were performed in quadruplets.
In vitro synthesis of precursor CRISPR RNAs
Two miniature CRISPR DNA corresponding to LA_Cr6 R2R4 (178 bp) and LA_Cr12 R2R3 (107 bp) were amplified through nested PCR using primer sets described in Table 1. The DNA fragments were cloned in a transcription vector pTZ57R/T between HindIII and KpnI restriction sites, and the generated plasmid constructs were outsourced for sequencing. Pre-crRNAs were synthesized in vitro after linearizing each plasmid construct with KpnI, as described previously (Prakash and Kumar, 2021). The two miniature pre-crRNA transcript [LA_Cr6 R2R4 (188 nt) and LA_Cr12 R2R3 (117 nt)] at its 5′ end also contains a vector-derived 10 nt sequence (5′GGGAAAGCUU3′).

Table 1. Oligos used in this study.
RNase assay with rLinCas6
The recombinant LinCas6 (rLinCas6) was purified using Ni-NTA (nitriloacetic acid) chromatography as described before (Prakash and Kumar, 2021). Cleavage assays with rLinCas6 ribonuclease were performed on the synthesized miniature pre-RNA substrates, as described previously (Prakash and Kumar, 2021). In brief, the synthesized miniature pre-crRNA (100 ng) was incubated with or without rLinCas6 (50-2000 nM) in a cleavage buffer (20 mM HEPES-KOH pH 8.0, 250 mM KCl, 1 mM DTT, and 2 mM MgCl2) for 1 h at 37°C. After that, cleavage reactions were terminated, resolved onto denaturing urea (8 M) 10% PAA gel, and were visualized with SYBR-Gold stain. Single-stranded RNA fragments of known sizes (24-500 nt) were used as a marker to estimate the size of the processed RNAs as described in a previous study (Prakash and Kumar, 2021).
Results
In silico analysis reveals seven CRISPR arrays at the subtype I-B locus of L. interrogans sv. Lai
In the genome of L. interrogans, the CRISPR subtype I-B locus is flanked by the two independent cas-operons (I and II) (Dixit et al., 2016) (Figure 1A). In this study, the intergenic region between cas2 and cas6 is called the hypervariable region that harbors the CRISPR I-B arrays of L. interrogans. Using the database CRISPRCasdb, a total of 11 CRISPR arrays (LIC_Cr1–11) were predicted in sv. Copenhageni. These CRISPR arrays provided by the CRISPRCasdb were numbered based on their serial order 1 to 11. Out of 11 CRISPR arrays (LIC_Cr1–11), a single CRISPR array (LIC_Cr2) (Prakash and Kumar, 2021) was predicted in the hypervariable region of L. interrogans sv. Copenhageni genome (Figure 1A and Supplementary Table 1). The remaining 10 arrays (LIC_Cr1 and LIC_Cr3–11) identified outside the hypervariable region were orphan CRISPR arrays. The array LIC_Cr2 comprises four repeats (36 nt each) interspaced by three unique spacers (Prakash and Kumar, 2021). On nucleotide BLAST analysis of LIC_Cr2 spacer sequences, no match was found in the other serovars of L. interrogans. This was consistent with a previous study (Xiao et al., 2019) where spacers were found variable across the serovars of L. interrogans. Moreover, a 100% nucleotides sequence identity was observed among the first three repeats, whereas the fourth repeat demonstrated variation at the 3′ end (Prakash and Kumar, 2021). Such variations persuaded us to analyze the repeats of CRISPR arrays in the hypervariable region of L. interrogans sv. Lai, another well-studied pathogenic serovar of L. interrogans. Unlike the genome of sv. Copenhageni, the sv. Lai harbored multiple arrays at the CRISPR-Cas I-B locus. Recently, in the CRISPR subtype I-B hypervariable region of sv. Lai, four CRISPR arrays with identical repeat consensus (28 nt) were predicted using the CRISPRFinder program (Xiao et al., 2019). In this study, an advanced version of the program CRISPRCasdb was used, which predicted 14 CRISPR arrays (LA_Cr1–14) in sv. Lai. Out of these 14 CRISPR arrays (LA_Cr1–14), 7 CRISPR arrays (LA_Cr6–12) were predicted in the hypervariable region of the sv. Lai genome (Figure 1A and Supplementary Table 1). The three extra arrays predicted in the hypervariable region of sv. Lai were LA_Cr8, Cr11, and Cr12. In sv. Lai, 7 (LA_Cr1–5 and LA_Cr13–14) out of the 14 arrays identified outside the hypervariable region were orphan CRISPR arrays. The CRISPRCasdb revealed that each repeat of the predicted CRISPR arrays (LA_Cr6–12) measured the size of 28 nucleotides. For any predicted CRISPR array, the CRISPRCasdb, by default, computes repeat consensus. Repeat consensus represents a sequence based on the occurrence of each nucleotide of repeats. The repeat consensus sequences provided by CRISPRCasdb of these arrays (LA_Cr6–12) were 100% identical (Supplementary Table 1). Interestingly, on alignment of the repeat consensus of the seven arrays, LA_Cr6–12 (28 nt) and LIC_Cr2 (36 nt), we noticed that at the 3′ end, each repeat of seven arrays (LA_Cr6–12) is deficit of eight nucleotides (Figure 1B). Compared to the 36 nt long repeats in array LIC_Cr2, shorter repeats (28 nt) in seven arrays LA_Cr6–12 incited to explore the spacer sequences of these seven arrays LA_Cr6–12. From the seven arrays LA_Cr6–12, a total of 17 spacer sequences were retrieved using the CRISPRCasdb. The retrieved 17 spacer sequences were used to perform MSA. In MSA, the 5′ ends of each of the 17 spacers were conserved by eight nucleotides (TTGAGCAC) (Figure 1C). This was in contrast to the observation where conserved repeats of the CRISPR array are separated by unique spacers (Mohamadi et al., 2020). Hence, to uphold the individuality in spacers and sequence conservation among repeats of seven arrays LA_Cr6–12, the conserved spacers sequence in this study has been redefined as part of adjacent repeats (Figure 1C). Such manual redefining of the spacers and repeats composition in the seven arrays (LA_Cr6–12) directed to increase repeats size (28 nt; program-defined) to 36 nucleotides. In addition, in order to maintain the consistency of repeat size (36 nt), the terminal repeats of each CRISPR array in the sv. Lai genome were extended at the 3′ end by eight nucleotides.
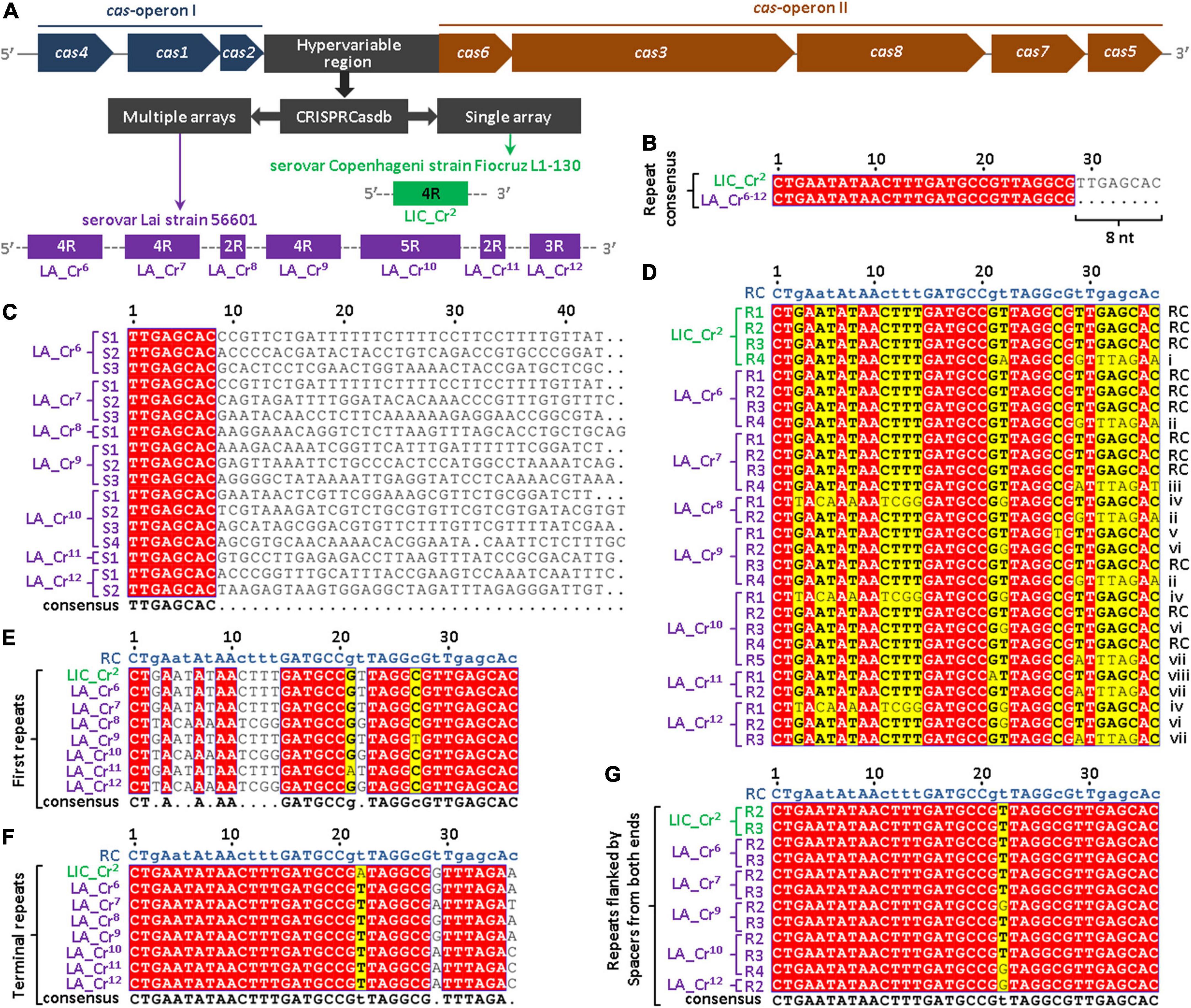
Figure 1. Multiple sequence alignments (MSA) of spacers and repeats of CRISPRs predicted in the I-B locus of L. interrogans serovar Lai strain 56601. (A) CRISPRCasdb database analysis of the hypervariable region in I-B locus of serovar Lai. The generalized architecture of the CRISPR-Cas I-B locus harbored by different strains of L. interrogans shows the hypervariable region that contains single or multiple CRISPRs, as observed in serovar Copenhageni (LIC_Cr2; Prakash and Kumar, 2021) or Lai (LA_Cr6– 12; in this study), respectively. Each CRISPR is represented by the number of associated repeats predicted by the CRISPRCasdb. (B) Nucleotide alignment of repeat consensus sequences of LIC_Cr2 and LA_Cr6– 12. Nucleotide alignment between database-defined repeat consensus of LIC_Cr2 and LA_Cr6– 12 showed a deficit of 8 nt at the 3′ end of LA_Cr6– 12 repeats. (C) MSA of spacer sequences. Spacer sequences (n = 17) of CRISPRs I-B (LA_Cr6– 12) in serovar Lai were retrieved from CRISPRCasdb and aligned using Clustal Omega. The alignment shows conserved 8 nt sequences at the 5′ end of each spacer (red filled box). (D) MSA of repeat sequences. Previously characterized and manually curated repeat sequences of CRISPRs I-B in serovar Copenhageni (LIC_Cr2) and Lai (LA_Cr6– 12), respectively, were aligned. The alignment shows conservation or polymorphism in the 1st to 36th nucleotide position of each repeat. Repeat consensus (RC) and repeat variants (i-viii) are indicated at the right to the respective sequences in the alignment. MSA of first repeats (E), terminal repeats (F), and repeats that are flanked by spacers at both ends (G). Red and yellow colored nucleotides in the alignments represent 100% and more than 70% conservation, respectively, at that position among total sequences. Consensus sequences of each MSA were shown below the alignment where upper and lower cases denote conserved and semi-conserved nucleotides, respectively. Bold letter code in the alignments indicates consensus nucleotide. Dot in consensus sequences indicates no nucleotide conservation at that particular position.
The direction of pre-crRNA transcription of LIC_Cr2 is not defined as per the CRISPRCasdb (Supplementary Table 1) (Prakash and Kumar, 2021). In a recent study, the direction of pre-crRNA transcription of LIC_Cr2 was demonstrated through RT-PCR and found to align with the direction of associated cas operon (Prakash and Kumar, 2021). We thus hypothesized that CRISPR arrays and cas genes of the subtype I-B system might also be co-directional among other Leptospira serovars or strains. However, CRISPRCasdb predicted the direction of pre-crRNA transcription for the seven arrays LA_Cr6–12 opposite to the cas genes (Supplementary Table 1). Thus, the CRISPRCasdb erred in projecting a defined and correct orientation of pre-crRNA in serovar Copenhageni (Prakash and Kumar, 2021) and Lai (in this study), respectively.
In this study, the previously characterized repeats (n = 4) of array LIC_Cr2 (Prakash and Kumar, 2021) and the repeats (n = 24) of seven arrays LA_Cr6–12 were manually curated and aligned to address the variation in the repeats. MSA of repeats (n = 4 + 24) from eight arrays (LIC_Cr2 and LA_Cr6–12) generated a repeat consensus (RC) of 36 nucleotides (Figure 1D). Analysis of the RC’s nucleotides demonstrated that each nucleotide is conserved by more than 70%. The nucleotides forming the stem-loop of the RC (Prakash and Kumar, 2021) were conserved in most of the repeat sequences (Figure 1D). Although 12 out of 28 repeats were identical to RC, variants of the repeat (RVs; n = 8) were also identified in sv. Copenhageni (i) and sv. Lai (ii-viii) at the hypervariable region (Figure 1D). Out of eight RVs (i-viii) identified, three RVs (v, vi, and viii) had a single nucleotide polymorphism. In contrast, the five RVs (i-iv and vii) showed variations of five to nine nucleotides, all coincidentally located either at first or terminal repeats of the arrays. Hence, a separate MSA of first and terminal repeats was conducted as shown in Figures 1E,F, respectively. The MSA with the RC demonstrated variation in nucleotides sequence primarily at 5′ of the first repeats (Figure 1E) and 3′ ends of terminal repeats (Figure 1F). In this study, thus the most conserved repeats of sv. Copenhageni and Lai genomes were the ones that possessed spacers at either end (Figure 1G).
Comparative analysis of the spirochete genome at the hypervariable region
Identification of multiple CRISPR arrays at the hypervariable region of sv. Lai’s genome prompted us to perform a comparative analysis of the nucleotides sequences with the genome of sv. Copenhageni. The hypervariable sequences (3,792 bp) of the sv. Lai genome were obtained from the NCBI database, which encompasses the region between cas2 (3′ end) and cas6 (5′ end) of CRISPR-Cas subtype I-B (Supplementary Figure 1A). In the hypervariable region, the inter-array sequences range from 112 to 229 nucleotides, while each array is 108 to 323 nucleotides in size. The alignment of the five inter-array sequences (LA_Cr6-Cr7, LA_Cr7-Cr8, LA_Cr8-Cr9, LA_Cr9-Cr10, and LA_Cr10-Cr11) show similarity in the first 155 to 158 nucleotides from the 5′ end (Supplementary Figure 1B). However, beyond 155 to 158 nucleotides, three inter-arrays (LA_Cr6-Cr7, LA_Cr8-Cr9, and LA_Cr10-Cr11) demonstrated to possess additional repeat (∼94% identity) and the spacer-like sequences (36 nt). The repeat-like sequences were identical to one proposed RV (iv). Therefore, these newly identified repeat- and the spacer-like sequences (36 nt) were redefined as first-repeat and –spacers for the three (LA_Cr7, Cr9, and Cr11) arrays (Supplementary Figures 1B, 2A, and Supplementary Table 2). Such redefining of repeat and spacers resulted in the addition of two more RVs (ix and x) at the hypervariable region of sv. Copenhageni and Lai (Table 2). We also generated an identity matrix of RC and various RVs, where the RC sequence shared ∼69 to 97% identity with the list of RVs denoted as i-x (Figure 2B). However, the identity perceived among RVs ranges from ∼56 to 100%.
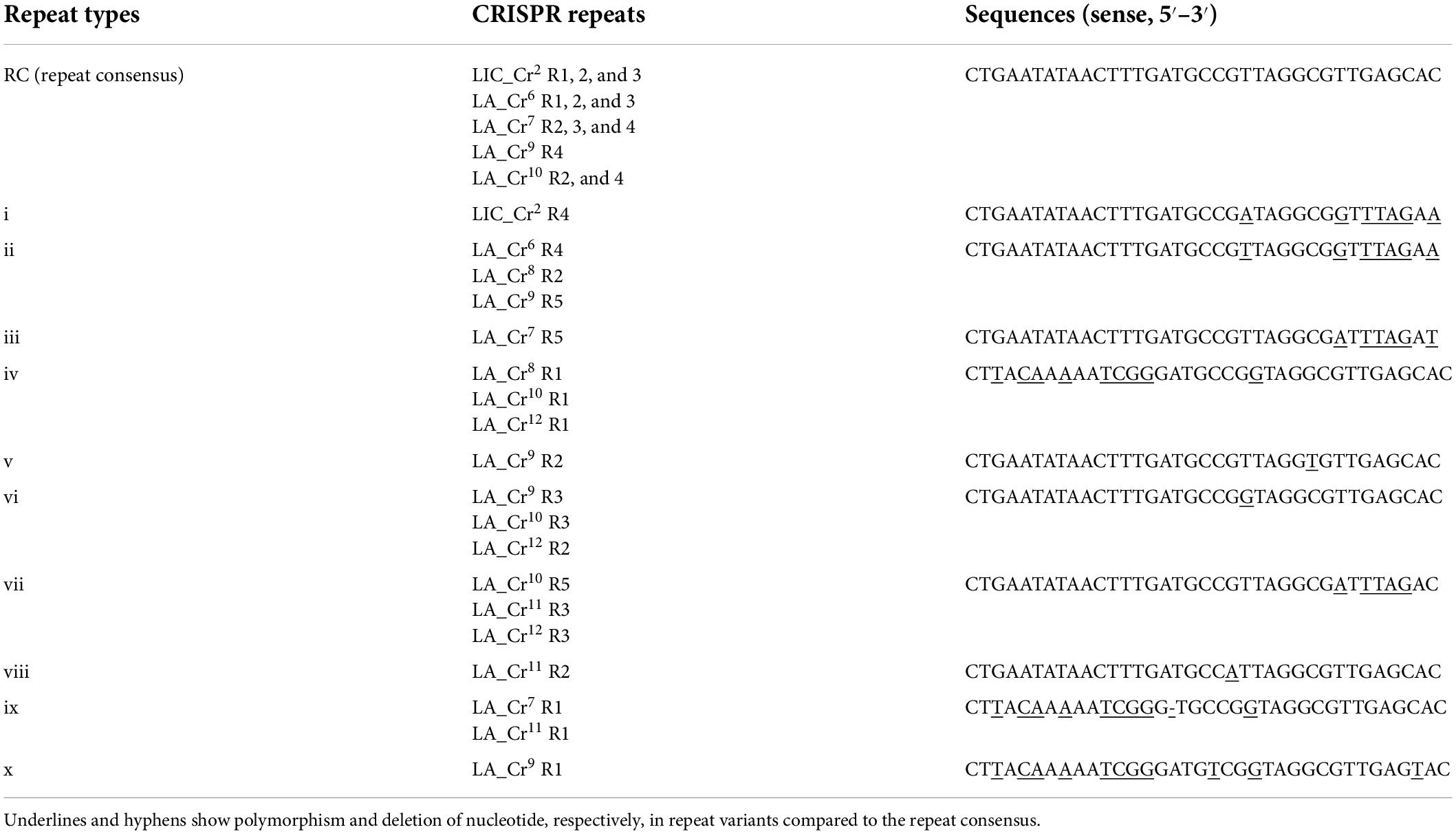
Table 2. CRISPR I-B repeats variants in L. interrogans svs. Copenhageni and Lai (in redefined CRISPR locus).
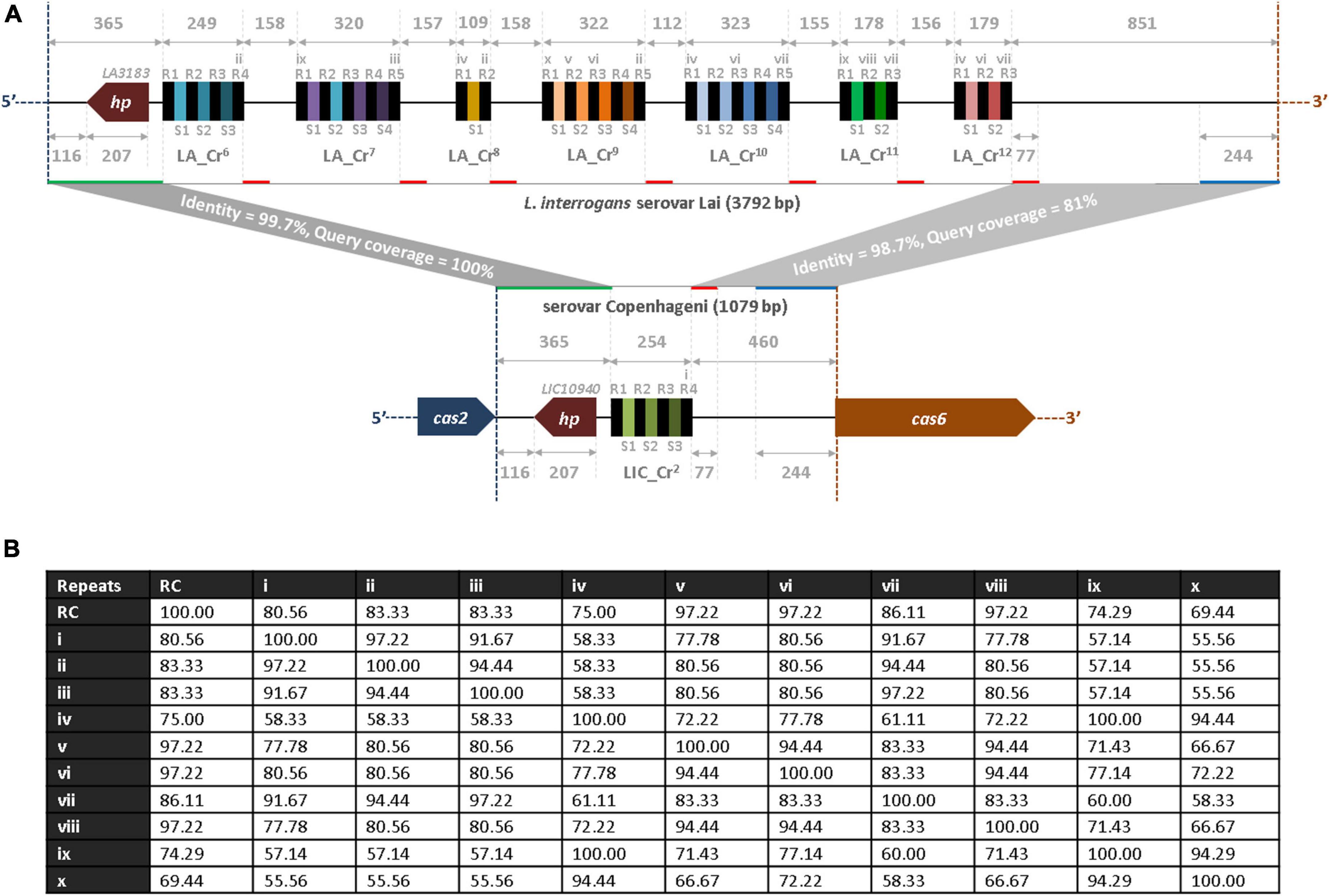
Figure 2. Bioinformatics analysis of hypervariable region at subtype I-B loci between L. interrogans sv. Lai and Copenhageni. (A) Comparison of the hypervariable region between L. interrogans serovars, Lai and Copenhageni. The region between cas2 (LA3182 and LIC10941) and cas6 (LA3189 and LIC10939) orthologs in serovar Lai (3,792 bp; top panel) and Copenhageni (1,079 bp; bottom panel) are drawn (manually) to scale in the 5′-3′ direction of the CRISPR-Cas I-B. Highly similar regions between serovar Lai and Copenhageni are shown by the same color-coded horizontal lines (green, red, and blue) along the lines (solid gray) that correspond to the hypervariable region in Leptospira serovars. Identical genes (hp; LIC10940 and LA3183) in both strains are denoted by the dark red color-filled pentagon. Black and unique color-filled rectangles represent similar repeats and spacer regions in all CRISPRs. Different variants of repeats (i-x), except the repeats identical to the consensus sequence, are indicated over the black color-filled rectangles in the architecture. (B) Identity matrix of repeat consensus (RC) and variants. A total of 11 repeat sequences (RC and variants i-x) were aligned and an identity matrix was generated using the MAFFT program. The numbers in the matrix correspond to the percent identity between the two respective repeat sequences.
At the hypervariable locus of sv. Lai and Copenhageni, the region between cas2 and its proximal repeat (R1 of LA_Cr6 and LIC_Cr2) is 365 bp long and is highly conserved (99.7% sequence identity and 100% query coverage), as illustrated in Figure 2A. Whereas, the region between cas6 and its proximal repeat (R3 of LA_Cr12 and R4 of LIC_Cr2) are 851 and 460 bp long with 98.7% sequence identity (81% query coverage). Within this region, around 244 bp upstream to cas6 and 77 bp downstream to a proximal repeat of cas6 are highly similar between serovar Lai and Copenhageni (Figure 2A). Interestingly, the DNA segment of 77 bp downstream to LIC_Cr2 aligned with high similarity to the downstream DNA segment of each seven arrays LA_Cr6–12 (Figure 2A). Such an identical feature in the hypervariable region suggests that the downstream DNA segment (77 bp, denoted by the red line) of each CRISPR array in serovar Lai and Copenhageni is conserved (Figure 2A).
CRISPR arrays at the hypervariable region of serovar Lai are transcriptionally active
The transcripts of seven CRISPR arrays (LA_Cr6–12) at the hypervariable region were determined by RT-PCR. The primer pairs used in RT-PCR were synthesized such that they anneal to the first and terminal spacers of each array (Table 1 and Figure 3A). The specificity of these primer sets was first tested by PCR using the genomic DNA of sv. Lai as a template. Expected DNA amplicons of 177 bp (LA_Cr6 S1S3), 249 bp (LA_Cr7 S1S4), 250 bp (LA_Cr9 S1S4), 251 bp (LA_Cr10 S1S4), and 107 bp (LA_Cr11 S1S2 and LA_Cr12 S1S2) were obtained using genomic DNA (Figures 3A,B, top panel). In agreement, similar sizes of amplicons were obtained from the two-step RT-PCR reaction, where cDNA was made using a random hexamer (Figure 3B, middle panel). No template amplification in another control RT-PCR reaction devoid of reverse transcriptase suggested RNA was free of DNA contamination (Figure 3B, bottom panel). Thus, we confirmed the active transcription of seven CRISPR arrays (LA_Cr6–12) at the hypervariable region of sv. Lai. However, due to long inter-array regions (112 to 158 nucleotides), it was unclear whether these seven CRISPR arrays (LA_Cr6–12) are transcribed as a single long pre-crRNA or as multiple independent pre-crRNA. Therefore, another set of PCR was performed to amplify consecutive arrays with a partial overlapping CRISPR region, as presented graphically in Supplementary Figure 2A. The partial overlapping region was amplified using the primer pairs enlisted in Supplementary Table 3 and the cDNA (random hexamer) as a template. The amplicons of size (655, 955, and 685 bp) could be detected for three overlapping CRISPR regions of LA_Cr6 S1-Cr7 S4, LA_Cr7–9, and LA_Cr9–10, respectively (Supplementary Figure 2B). We additionally substantiated our finding by generating another set of cDNA, where instead of random hexamer (RH), we used a single primer (12S2r) to the spacer region of the terminal array (LA_Cr12). After that, PCR was performed with the specific primer pair (6S1f and 6S3r) of the first array (LA_Cr6) and the new set of cDNA (12S2r) as a template. A DNA amplicon of 177 bp was detected (Figure 3C), and thus, transcription of seven CRISPR arrays (LA_Cr6–12) in sv. Lai as a single long pre-RNA of CRISPR subtype I-B was ascertained from two independent approaches.
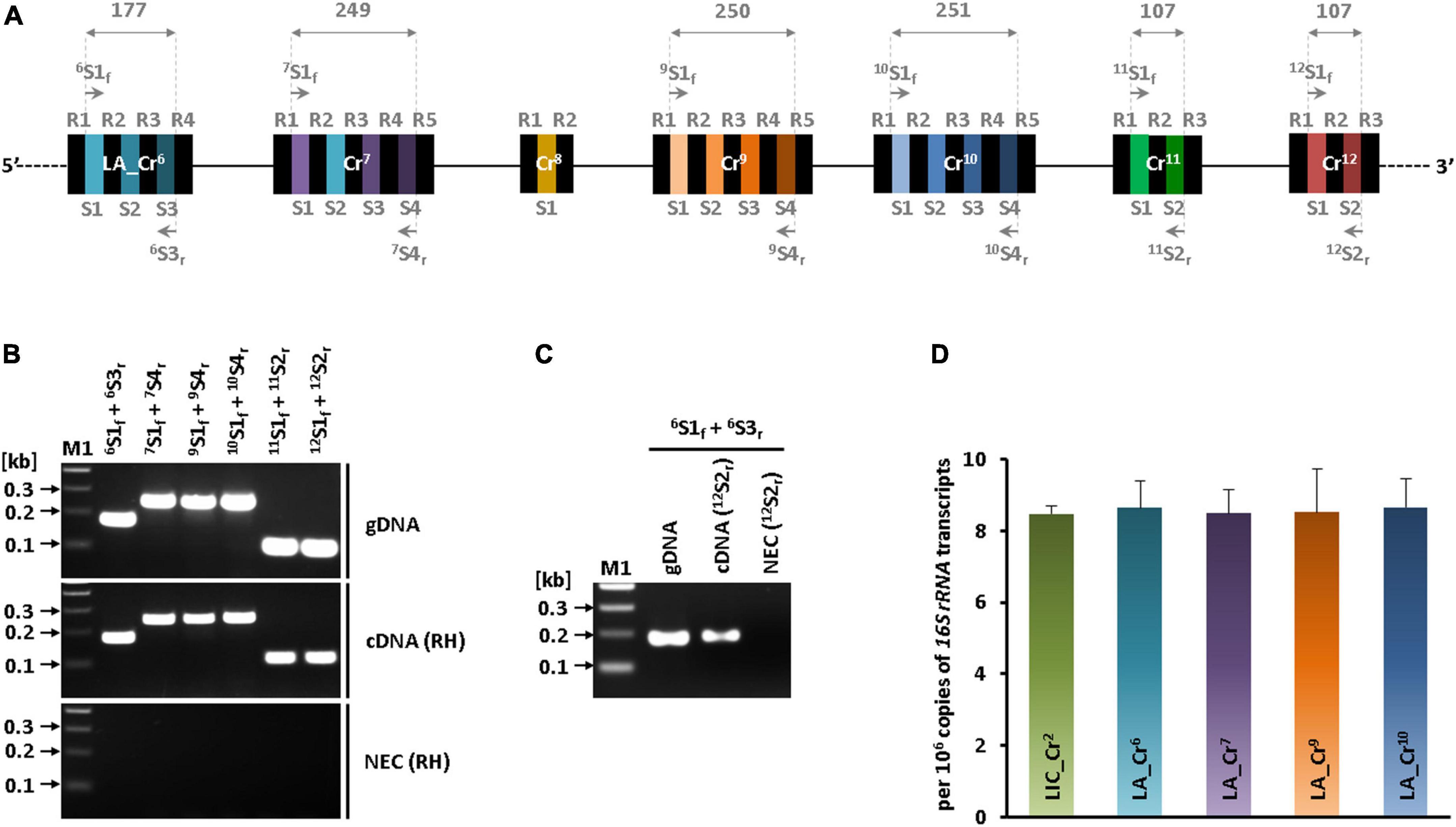
Figure 3. RT-PCR and qPCR of subtype I-B CRISPRs. (A) Schematic representation of primer pairs position used in the study. Spacer-specific primer pairs used in the RT-PCR experiment are denoted on the hypervariable region (I-B) of serovar Lai. CRISPR regions and length of fragments that were expected to get amplified with primers used in the study are indicated by vertical dashed lines (gray) and numbers (in bp) given at the apex of the double arrowhead over the architecture. (B) Identification of subtype I-B CRISPRs transcription in serovar Lai. PCR using genomic DNA (gDNA; positive control) of serovar Lai with the spacer (first and terminal) specific primer pair of each CRISPR (except LA_Cr8) (top panel). PCR with cDNA template synthesized from total RNA of serovar Lai using random hexamers (middle panel). PCR with RNA (cDNA synthesis reaction without reverse transcriptase) is a no enzyme control (NEC) of the experiment (bottom panel). PCR products were resolved on 2% agarose gel. “M1” denotes the DNA marker used for size estimation of PCR-amplified DNA fragments. (C) RT-PCR of first CRISPR using cDNA prepared using spacer-specific primer of terminal CRISPR of LA_Cr6– 12 series. A reverse transcriptase reaction was performed using a terminal spacer-specific reverse primer of LA_Cr12. This cDNA template was used in PCR for amplification of LA_Cr6. (D) Quantification of transcripts of CRISPR I-B arrays of sv. Copenhageni and Lai. Transcripts of CRISPR arrays (LIC_Cr2, LA_Cr6, Cr7, Cr9, and Cr10) are quantified using qRT-PCR per 106 copies of 16 S rRNA transcripts of respective Leptospira serovars. Results are indicative of two independent experiments, each performed in quadruplets.
Next, the abundance of pre-crRNA of CRISPR subtype I-B in sv. Copenhageni and Lai were assessed using quantitative real-time PCR. The cDNA used was synthesized using random hexamers, and the primer pairs designed gave amplicon in the range of ∼180 to 250 bp size of subtype I-B arrays (LIC_Cr2 of sv. Copenhageni, LA_Cr6, Cr7, Cr9, and Cr10 of sv. Lai). The quantified number of pre-crRNA in both serovars was more than 8 per 106 copies of 16S rRNA of L. interrogans (Figure 3D). The relative number of precursor RNA transcripts (CRISPR subtype I-B) in serovar Lai substantiates the RT-PCR analysis results in this study that demonstrated transcription CRISPR cluster jointly as a single precursor unit.
Recombinant LinCas6 of serovar Copenhageni processes the pre-crRNA transcripts of serovar Lai
Using the endoribonuclease activity of rLinCas6, mature crRNAs can be generated from pre-crRNA (LIC_Cr2) of sv. Copenhageni, as described previously (Prakash and Kumar, 2021). In LinCas6 (LIC10939), potential catalytic residues and glycine rich-loop (G-loop) are crucial for self-folding and RNA substrate recognition (Prakash and Kumar, 2021). LinCas6 in sv. Copenhageni (LIC10939) and Lai (LA3189) share 96.2% amino acid sequence identity with 100% query coverage (210 residues). Pairwise alignment of LIC10939 and LA3189 revealed a mismatch at eight residues (T36, Q65, T124, I143, Q146, K149, V184, and S202) in LA3189. These mismatches in LA3189 were not observed at the potential catalytic triad and G-loop. Therefore, to address whether the CRISPR arrays in sv. Lai can be processed to yield mature crRNAs; we opted to use rLinCas6 (LIC10939) endonuclease. Previously, a miniature form of pre-crRNA has been successfully used in the RNase assay of Cas6 protein (Reimann et al., 2017). Similarly, an in vitro cleavage assay was set up in which miniature pre-crRNA of the LA_Cr6 array (R2R4) was incubated with increasing concentrations of rLinCas6. For clarity, the total feasible RNA fragments (n = 9) after processing the miniature pre-crRNA (188 nt) of LA_Cr6 (R2R4) with rLinCas6 were mapped for cleavage reaction analysis (Figure 4A, top panel). These transcript fragments have been categorized as incompletely processed (IP; n = 5, 79-180 nt) and completely processed (CP; n = 4, 8-71 nt) fragments, as described previously (Prakash and Kumar, 2021). The miniature pre-crRNA cleavage by rLinCas6 on denaturing urea-PAGE revealed six distinct bands (Figure. 4A, bottom panel). Larger IP fragments of pre-crRNA were identified when rLinCas6 was employed at a lower range of concentrations (50–250 nM), whereas at a higher concentration of rLinCas6 (500-2000 nM), an increase in the intensity of CP fragments of partial pre-crRNA was observed. RNA fragments of 71 nucleotides in CP products illustrate the generation of mature crRNAs from the miniature LA_Cr6 transcript. Similarly, rLinCas6-mediated mature crRNA biogenesis was also investigated on the miniature LA_Cr12 (R2R3) array (Figure 4B). Alike the miniature LA_Cr6 processing, RNA fragments of 71 nucleotides in CP products could be detected for the miniature LA_Cr12 (R2R3) array. Thus, with the processing results of the miniature versions of LA_Cr6 and LA_Cr12, we infer that rLinCas6 may process the remaining five array transcripts of serovar Lai to yield mature crRNAs.
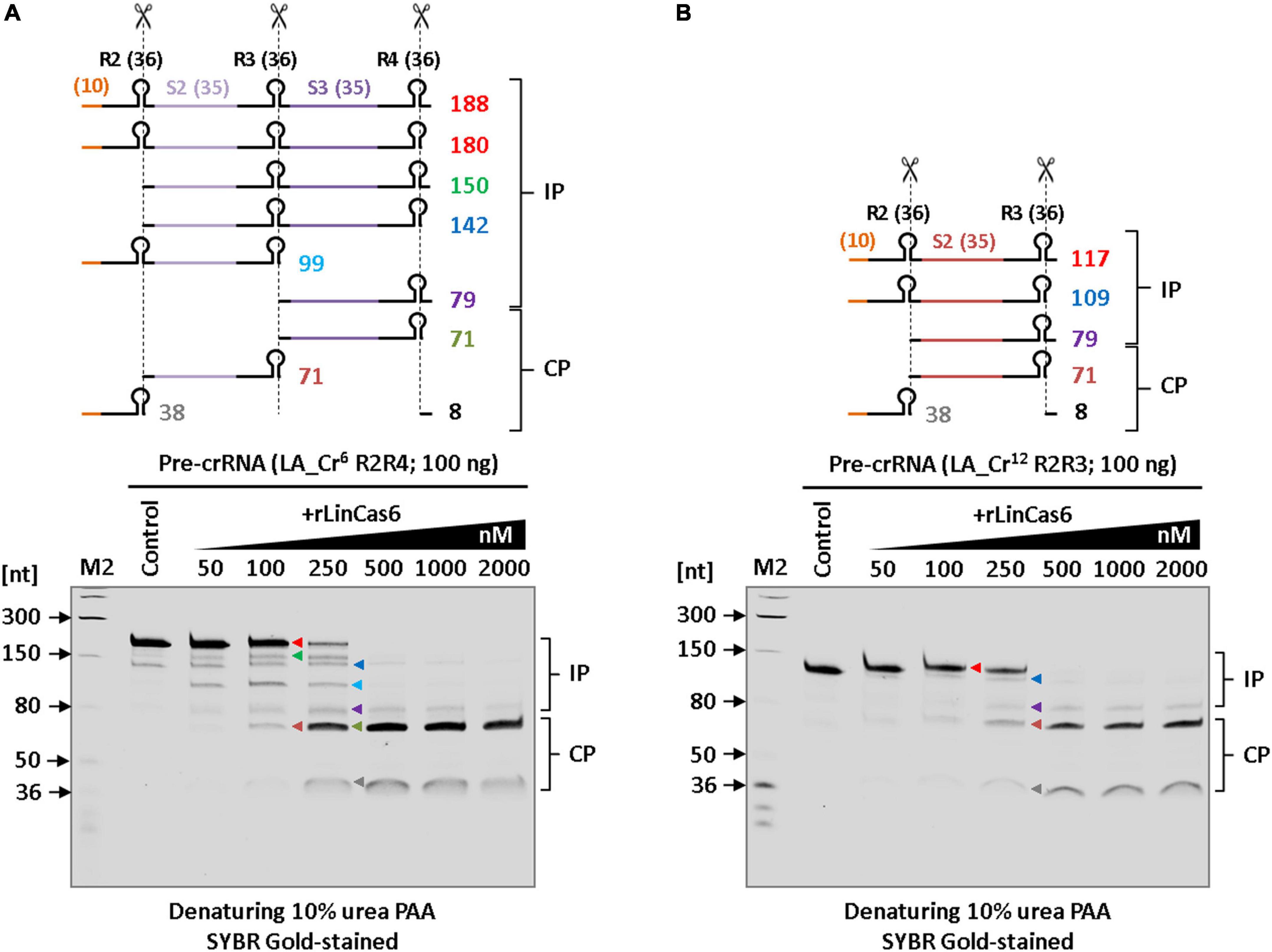
Figure 4. In vitro processing of the miniature CRISPR I-B transcript of serovar Lai with rLinCas6 endoribonuclease. (A) Nuclease activity of rLinCas6 on pre-crRNA of LA_Cr6. In vitro synthesized LA_Cr6 miniature transcript (R2R4; 188 nt) was incubated with increasing concentrations of rLinCas6 (50–2,000 nM). The possible pre-crRNA derived cleaved fragments (n = 9) by rLinCas6 are illustrated as incompletely processed (IP; n = 5) and completely processed (CP; n = 4) fragments (top panel). The reaction products were analyzed on denaturing urea gel after staining with SYBR Gold. At 100 to 250 nM of rLinCas6, six bands of different molecular lengths, each marked with unique color, were detected (bottom panel). (B) Nuclease activity of rLinCas6 on miniature pre-crRNA of LA_Cr12 (R3R4). IP and CP fragments on the gel were mapped and indicated right to the gel images. Repeats and spacers are shown by black and unique colors, respectively, in the pre-crRNA outline. The orange line denotes a vector-derived additional 10 nt at its 5′ end of each pre-crRNA. All reactions, including controls (no protein), were incubated for 1 h at 37°C. “M2” denotes RNA markers used for the size estimation of RNA fragments observed on the gels.
In silico analysis of spacers of I-B array identified a consensus PAM
CRISPR-Cas systems rely on protospacer adjacent motif (PAM) to differentiate between self (spacer) and non-self (protospacer) DNA sequences (Westra et al., 2013). In CRISPR-Cas type I systems, PAMs are often reported to be present at the 5′-end of the protospacers sequence (Deveau et al., 2008; Mojica et al., 2009). In this study, the spacer sequences and CRISPR boundaries of sv. Lai’s genome has been redefined manually. Therefore, the rectified spacer sequences (Supplementary Table 2) were utilized to identify PAMs for the subtype I-B system of Leptospira by in silico approach. Using the CRISPRTarget program, analysis of rectified spacers of the I-B array obtained 53 hits as possible protospacers (with a cut-off score of 25). These hits aligned with viral genome fragments derived from metagenomic samples and Leptospira phages (LinZ_10, Lin_34, LbrZ_5399, LnoZ_CZ214). Further, by the upstream of a majority of predicted protospacers (37 out of 53; 70% of hits), a trinucleotide ATG was conserved, as evident in the sequence logo of nucleotides flanking the protospacers (Figure 5). Therefore, we speculate that one of the PAM (5′-ATG-3′) may be employed for the interference study against mobile genetic elements in Leptospira.
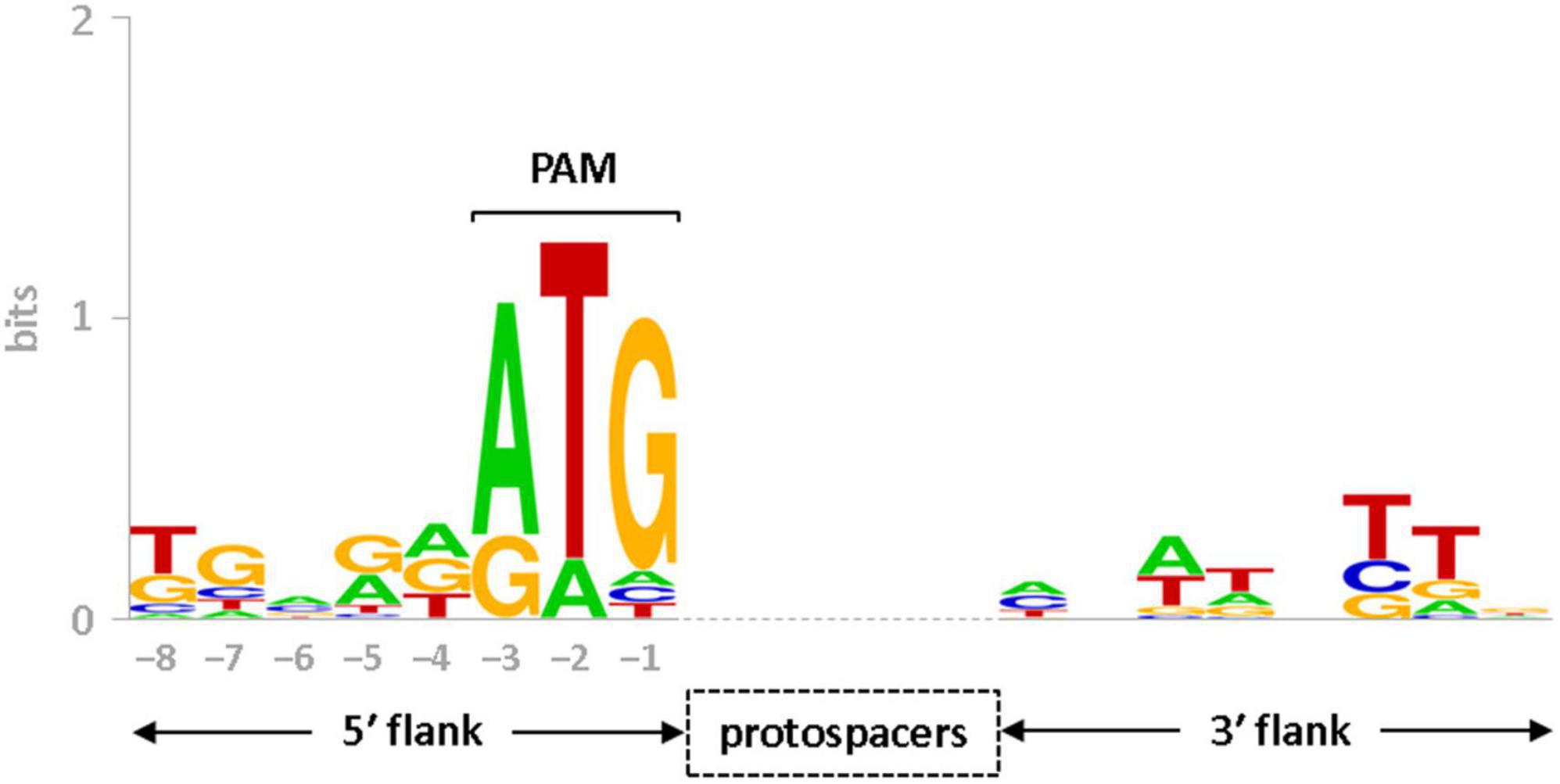
Figure 5. Sequence logo of protospacer flanks. Conservation of nucleotides at the 5′ and 3′ flanks (8 nt each) of protospacers, corresponding to the spacers of seven CRISPR arrays (LA_Cr6– 12) in serovar Lai, are presented in the form of sequence logo. Conserved nucleotides at –3 (A), –2 (T), and –1 (G) positions in the upstream of protospacers represent consensus PAM sequence (5′-ATG-3′). The vertical axis indicates the information content of the sequence position in bits. The height of each letter in the logo represents the conservation of that nucleotide at each position.
Discussion
Understanding the CRISPR transcript orientation and the repeat-spacer junctions is valuable for the evolving genome editing techniques, selective killing, and gene expression modulation (Zheng et al., 2020). Two of the most accustomed programs for searching CRISPR arrays or Cas loci in prokaryotic genomes are the CRISPRFinder (Grissa et al., 2007) and CRISPRCasFinder (Pourcel et al., 2020). Using the CRISPRFinder program, recently, four CRISPR arrays (LA_Cr6, Cr7, Cr9, and Cr10) have been reported in the hypervariable region of L. interrogans sv. Lai genome (Xiao et al., 2019). On the other hand, in this study, using the more robust CRISPRCasdb program, three more CRISPR arrays (LA_Cr8, Cr11, and Cr12) were predicted. Although the CRISPRCasdb program accurately predicted the repeat-spacer junctions of the CRISPR subtype I-B array in the sv. Copenhageni, it failed to provide the correct repeat-spacer junctions or the orientation of arrays in sv. Lai genome.
Integrating protospacer DNA into the CRISPR array is an essential and primary stage of the CRISPR-Cas-mediated adaptive immunity for developing memory against mobile genetic elements (Mosterd et al., 2021). Such integration depends on the length of leader elements driving the array or the consensus within repeat nucleotides. The length of the leader sequences is reported from 100 to 500 nucleotides (Yosef et al., 2012; Carte et al., 2014); however, 10 to 43 nucleotides of the leader at the leader-repeat junction are critical for adaptation (Yosef et al., 2012; Wei et al., 2015). Also, mutation of the eight nucleotides of the repeat at the leader junction disrupts the adaptation process (Grainy et al., 2019). In sv. Lai, identifying transcripts from seven CRISPR arrays (LA_Cr6 to Cr12) by RT-PCR suggested that a single leader controls these arrays. In agreement, the nucleotide sequences flanking the 5′ end of the first array (LA_Cr6), where a leader is expected, differ from the remaining six CRISPR arrays (LA_Cr7–12) present in the hypervariable region.
In a study done elsewhere (Barrangou et al., 2007), it is observed that repeats are often highly conserved within a given CRISPR array except for the terminal repeat. In contrast, multiple (n = 9) repeat variants (RV) were found in the CRISPR arrays of sv. Lai. The first repeat of array LA_Cr6 resembles the RC; however, in the remaining six CRISPR arrays (LA_Cr7–12), the first repeats differed from the RC at the 5′ end. Therefore, such variations at the first repeats of six CRISPR arrays (LA_Cr7–12) in sv. Lai may disrupt the adaptation process, as reported elsewhere (Grainy et al., 2019). Based on earlier reported work (Grainy et al., 2019), a hypothesis thus can be drawn that during a new spacer integration in CRISPR-Cas subtype I-B of serovar Lai, adaptation may exclusively occur at the first CRISPR array (LA_Cr6). The biogenesis of crRNA from pre-crRNA (miniature LA_Cr6 and Cr12) and the conservation of the stem-loop of repeat RNA indicate that the interference process from all seven arrays (LA_Cr6–12) may still be possible in Leptospira. Such adaptation and interference study, however, needs to be experimentally proven and can be an exciting subject for future study.
The biogenesis of crRNAs in a CRISPR-Cas type I and its subtypes from pre-crRNA requires Cas6 endoribonucleases except for the subtype I-C where processing is done by Cas5 (Charpentier et al., 2015; Hochstrasser and Doudna, 2015). After processing, crRNA is loaded into a multi-protein effector complex, forming a “Cascade” that guides crRNA to the target (He et al., 2020). Cascade recruits Cas3 (signature protein of Type I) to form the “Interference” complex at the target and degrades the specific DNA (He et al., 2020). In general, Cas3 protein possess an N-terminal histidine-aspartate (HD) nuclease domain and a C-terminal superfamily 2 (SF2) helicase domain (Jackson et al., 2014; He et al., 2020). The SF2 domain contains a DEAD/DEAH box region. Multiple proteins having the DEAD-box family (SF2) of RNA helicases have been shown to resolve RNA secondary structures and unfold RNA hairpins in vitro (Rogers et al., 1999; Marsden et al., 2006; König et al., 2013). Interestingly, in L. interrogans sv. Linhai, Cas3 has been reported naturally fused with Cas6 (Prakash and Kumar, 2021). Therefore, Cas3 may also have some role during the expression stage, if not for all, at least in Leptospira. The existence of single long pre-crRNA arrays in sv. of L. interrogans may have some evolutionary role for the fusion stage of LinCas3 and LinCas6. It is likely that the long pre-crRNA may form a stable and compact structure as reported elsewhere (König et al., 2013) and thus may not be uniformly accessible to LinCas6. Thus, it is plausible that the processing of lengthy pre-crRNA may require another helper protein(s) to maintain a susceptible structure of pre-crRNA for LinCas6. We speculate that the helicase activity of LinCas3 may unwind the long pre-crRNA so that repeat RNA segments are accessible to LinCas6. However, further work is warranted to validate this hypothesis.
In addition to the eight CRISPR arrays (LIC_Cr6 and LA_Cr6–12) identified in the I-B loci of svs. Copenhageni and Lai, 17 CRISPR arrays were qualified as orphan arrays. It has been suggested that orphan CRISPR arrays can be non-functional or associated with a remotely located cas locus in the same genome (Zhang and Ye, 2017). However, the biological relevance of these orphan CRISPR arrays in L. interrogans remains to be elucidated via transcriptional analysis and processing by host Cas endoribonucleases.
In sv. Lai, using the redefined spacer sequences, we identified a conserved PAM sequence (5′-ATG-3′) for CRISPR-Cas subtype I-B. Another weakly conserved PAM sequence (5′-TAC-3′) was identified through the same approach in the L. interrogans subtype I-B elsewhere (Xiao et al., 2019). Such inconsistency in the prediction of consensus PAM could be due to feeding different spacer lengths or its orientation during in silico analysis. Recently, a computational pipeline (Vink et al., 2021) has predicted the functional orientation of spacer and its associated PAM (5′-ATG-3′) for the subtype I-B system of Leptospira, which agrees well with our results presented in this study and elsewhere (Prakash and Kumar, 2021).
Understanding the endogenous CRISPR-Cas system may be advantageous in developing a genetic tool to understand gene function in pathogenic Leptospira like other infectious organisms described before (Li et al., 2016; Pyne et al., 2016; Cheng et al., 2017; Maikova et al., 2019). In this study, we deciphered the transcription of CRISPR RNA in one of the reference serovars of L. interrogans (sv. Lai strain 56601) harboring CRISPR-Cas subtype I-B in its genome. Although various robust in silico CRISPR prediction tools have been developed to project the CRISPR direction and boundaries, we failed to apply the same in L. interrogans sv. Lai and other serovars. Using RT-PCR, we present the transcriptional analysis of a cluster of CRISPR arrays at the I-B locus in L. interrogans sv. Lai. Our study suggests that these CRISPR arrays are transcriptionally active and controlled by a single leader. We also demonstrated the generation of mature crRNAs from selective pre-crRNA suggesting crRNA biogenesis associated with the CRISPR-Cas I-B in sv. Lai. In addition, a conserved PAM sequence was predicted using the spacer sequences of sv. Lai. This study comprehends the knowledge of CRISPR locus transcription and its processing of spirochetes and will be valuable in the future study of the endogenous CRISPR-Cas system in Leptospira.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
AP performed the research work. MK and AP wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This present work was financially supported by the Department of Biotechnology, Government of India, bearing project numbers BT/PR25083/NER/95/1002/2017 and BT/PR36586/GET/119/361/2020.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.960559/full#supplementary-material
References
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Biswas, A., Gagnon, J. N., Brouns, S. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827. doi: 10.4161/rna.24046
Carte, J., Christopher, R. T., Smith, J. T., Olson, S., Barrangou, R., Moineau, S., et al. (2014). The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in S treptococcus thermophilus. Mole. Microbiol. 93, 98–112. doi: 10.1111/mmi.12644
Charpentier, E., Richter, H., van der Oost, J., and White, M. F. (2015). Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 2015:fuv023. doi: 10.1093/femsre/fuv023
Cheng, F., Gong, L., Zhao, D., Yang, H., Zhou, J., Li, M., et al. (2017). Harnessing the native type IB CRISPR-Cas for genome editing in a polyploid archaeon. J. Genet. Gen. 44, 541–548. doi: 10.1016/j.jgg.2017.09.010
Crooks, G. E., Hon, G., Chandonia, J.-M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Gen. Res. 14, 1188–1190. doi: 10.1101/gr.849004
Da, H., and Levett, P. N. (2015). Leptospirosis in humans. Curr. Top. Microbiol. Immunol. 387, 65–97. doi: 10.1007/978-3-662-45059-8_5
Deveau, H., Barrangou, R., Garneau, J. E., Labonté, J., Fremaux, C., Boyaval, P., et al. (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390–1400. doi: 10.1128/JB.01412-07
Dixit, B., Anand, V., Hussain, M. S., and Kumar, M. (2021a). The CRISPR-associated Cas4 protein from Leptospira interrogans demonstrate versatile nuclease activity. Curr. Res. Microb. Sci. 2:100040. doi: 10.1016/j.crmicr.2021.100040
Dixit, B., Prakash, A., Kumar, P., Gogoi, P., and Kumar, M. (2021b). The core Cas1 protein of CRISPR-Cas IB in Leptospira shows metal-tunable nuclease activity. Curr. Res. Microb. Sci. 2:100059. doi: 10.1016/j.crmicr.2021.100059
Dixit, B., Ghosh, K. K., Fernandes, G., Kumar, P., Gogoi, P., and Kumar, M. (2016). Dual nuclease activity of a Cas2 protein in CRISPR–Cas subtype I-B of Leptospira interrogans. FEBS Lett. 590, 1002–1016. doi: 10.1002/1873-3468.12124
Fernandes, L., and Nascimento, A. (2022). A Novel Breakthrough in Leptospira spp. Mutagenesis: knockout by Combination of CRISPR/Cas9 and Non-homologous End-Joining Systems. Front. Microb. 2022:13. doi: 10.3389/fmicb.2022.915382
Fernandes, L. G. V., Guaman, L., Vasconcellos, S. A., Heinemann, M. B., Picardeau, M., and Nascimento, A. L. T. O. D. (2019). Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): a new tool for genetic engineering in Leptospira. Sci. Rep. 9, 1–14. doi: 10.1038/s41598-018-37949-x
Fernandes, L. G. V., Hornsby, R., Nascimento, A. L. T. O. D., and Nally, J. (2021). Genetic manipulation of pathogenic Leptospira: CRISPR interference (CRISPRi)-mediated gene silencing and rapid mutant recovery at 37 C. Sci. Rep. 11, 1–12. doi: 10.1038/s41598-021-81400-7
Fouts, D. E., Matthias, M. A., Adhikarla, H., Adler, B., Amorim-Santos, L., Berg, D. E., et al. (2016). What makes a bacterial species pathogenic?: comparative genomic analysis of the genus Leptospira. PLoS Negl. Trop. Dis. 10:e0004403. doi: 10.1371/journal.pntd.0004403
Garneau, J. E., Dupuis, M.-E., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67.
Ghosh, K. K., Prakash, A., Balamurugan, V., and Kumar, M. (2018a). Catecholamine-modulated novel surface-exposed adhesin LIC20035 of Leptospira spp. binds host extracellular matrix components and is recognized by the host during infection. Appl. Environ. Microbiol. 84, e2360–e2317. doi: 10.1128/AEM.02360-17
Ghosh, K. K., Prakash, A., Shrivastav, P., Balamurugan, V., and Kumar, M. (2018b). Evaluation of a novel outer membrane surface-exposed protein, LIC13341 of Leptospira, as an adhesin and serodiagnostic candidate marker for leptospirosis. Microbiology 164, 1023–1037. doi: 10.1099/mic.0.000685
Grainy, J., Garrett, S., Graveley, B. R., and Terns, M. P. (2019). CRISPR repeat sequences and relative spacing specify DNA integration by Pyrococcus furiosus Cas1 and Cas2. Nucleic Acids Res. 47, 7518–7531. doi: 10.1093/nar/gkz548
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35(Suppl._2), W52–W57.
Hale, C. R., Zhao, P., Olson, S., Duff, M. O., Graveley, B. R., Wells, L., et al. (2009). RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956.
He, L., St John James, M., Radovcic, M., Ivancic-Bace, I., and Bolt, E. L. (2020). Cas3 protein—a review of a multi-tasking machine. Genes 11:208. doi: 10.3390/genes11020208
Hille, F., Richter, H., Wong, S. P., Bratoviè, M., Ressel, S., and Charpentier, E. (2018). The biology of CRISPR-Cas: backward and forward. Cell 172, 1239–1259.
Hochstrasser, M. L., and Doudna, J. A. (2015). Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 40, 58–66. doi: 10.1016/j.tibs.2014.10.007
Hussain, M. S., and Kumar, M. (2022). Assembly of Cas7 subunits of Leptospira on the mature crRNA of CRISPR-Cas IB is modulated by divalent ions. Gene 818:146244. doi: 10.1016/j.gene.2022.146244
Jackson, R. N., Lavin, M., Carter, J., and Wiedenheft, B. (2014). Fitting CRISPR-associated Cas3 into the helicase family tree. Curr. Opin. Struct. Biol. 24, 106–114. doi: 10.1016/j.sbi.2014.01.001
König, S. L., Liyanage, P. S., Sigel, R. K., and Rueda, D. (2013). Helicase-mediated changes in RNA structure at the single-molecule level. RNA Biol. 10, 133–148. doi: 10.4161/rna.23507
Li, Y., Pan, S., Zhang, Y., Ren, M., Feng, M., Peng, N., et al. (2016). Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res. 44, e34–e34.
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu. Rev. Biochem. 79:181.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 25, 402–408.
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Maikova, A., Kreis, V., Boutserin, A., Severinov, K., and Soutourina, O. (2019). Using an endogenous CRISPR-Cas system for genome editing in the human pathogen Clostridium difficile. Appl. Env. Microbiol. 85, e1416–e1419. doi: 10.1128/AEM.01416-19
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 9, 467–477.
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 13:722.
Makarova, K. S., Wolf, Y. I., Shmakov, S. A., Liu, Y., Li, M., and Koonin, E. V. (2020). Unprecedented diversity of unique CRISPR-cas-related systems and cas1 homologs in asgard archaea. CRISPR J. 3, 156–163. doi: 10.1089/crispr.2020.0012
Marsden, S., Nardelli, M., Linder, P., and McCarthy, J. E. (2006). Unwinding single RNA molecules using helicases involved in eukaryotic translation initiation. J. Mole. Biol. 361, 327–335. doi: 10.1016/j.jmb.2006.06.016
Martínez Arbas, S., Narayanasamy, S., Herold, M., Lebrun, L. A., Hoopmann, M. R., Li, S., et al. (2021). Roles of bacteriophages, plasmids and CRISPR immunity in microbial community dynamics revealed using time-series integrated meta-omics. Nat. Microb. 6, 123–135. doi: 10.1038/s41564-020-00794-8
Mohamadi, S., Bostanabad, S. Z., and Mirnejad, R. (2020). CRISPR arrays: a review on its mechanism. Journal of Applied Biotechnology Reports 7, 81–86.
Mojica, F. J., íez-Villaseñor, C. D., García-Martínez, J., and Almendros, C. (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740.
Mosterd, C., Rousseau, G. M., and Moineau, S. (2021). A short overview of the CRISPR-Cas adaptation stage. Can. J. Microb. 67, 1–12. doi: 10.1139/cjm-2020-0212
Nishimasu, H., Yamano, T., Gao, L., Zhang, F., Ishitani, R., and Nureki, O. (2017). Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mole. Cell 67, 139.–147.
Pappas, C. J., Benaroudj, N., and Picardeau, M. (2015). A replicative plasmid vector allows efficient complementation of pathogenic Leptospira strains. Appl. Env. Microbiol. 81, 3176–3181. doi: 10.1128/AEM.00173-15
Pourcel, C., Salvignol, G., and Vergnaud, G. (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. doi: 10.1099/mic.0.27437-0
Pourcel, C., Touchon, M., Villeriot, N., Vernadet, J.-P., Couvin, D., Toffano-Nioche, C., et al. (2020). CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res. 48, D535–D544. doi: 10.1093/nar/gkz915
Prakash, A., and Kumar, M. (2021). Characterizing the transcripts of Leptospira CRISPR IB array and its processing with endoribonuclease LinCas6. Internat. J. Biol. Macromole. 182, 785–795. doi: 10.1016/j.ijbiomac.2021.04.066
Pyne, M. E., Bruder, M. R., Moo-Young, M., Chung, D. A., and Chou, C. P. (2016). Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium. Sci. Rep. 6, 1–15. doi: 10.1038/srep25666
Reimann, V., Alkhnbashi, O. S., Saunders, S. J., Scholz, I., Hein, S., Backofen, R., et al. (2017). Structural constraints and enzymatic promiscuity in the Cas6-dependent generation of crRNAs. Nucleic Acids Res. 45, 915–925. doi: 10.1093/nar/gkw786
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Rogers, G. W., Richter, N. J., and Merrick, W. C. (1999). Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem. 274, 12236–12244.
Sampson, T. R., Saroj, S. D., Llewellyn, A. C., Tzeng, Y.-L., and Weiss, D. S. (2013). A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497, 254–257.
Sampson, T. R., and Weiss, D. S. (2014). CRISPR-Cas systems: new players in gene regulation and bacterial physiology. Front. Cell. Infect. Microb. 4:37.
Shapiro, R. S., Chavez, A., and Collins, J. J. (2018). CRISPR-based genomic tools for the manipulation of genetically intractable microorganisms. Nat. Rev. Microbiol. 16, 333–339.
Vink, J. N., Baijens, J. H., and Brouns, S. J. (2021). PAM-repeat associations and spacer selection preferences in single and co-occurring CRISPR-Cas systems. Gen. Biol. 22, 1–25. doi: 10.1186/s13059-021-02495-9
Wang, J., Li, J., Zhao, H., Sheng, G., Wang, M., Yin, M., et al. (2015). Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR-Cas systems. Cell 163, 840–853. doi: 10.1016/j.cell.2015.10.008
Wei, Y., Chesne, M. T., Terns, R. M., and Terns, M. P. (2015). Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 43, 1749–1758. doi: 10.1093/nar/gku1407
Westra, E. R., Semenova, E., Datsenko, K. A., Jackson, R. N., Wiedenheft, B., Severinov, K., et al. (2013). Type IE CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 9:e1003742. doi: 10.1371/journal.pgen.1003742
Xiao, G., Yi, Y., Che, R., Zhang, Q., Imran, M., Khan, A., et al. (2019). Characterization of CRISPR-Cas systems in Leptospira reveals potential application of CRISPR in genotyping of Leptospira interrogans. Apmis 127, 202–216. doi: 10.1111/apm.12935
Yosef, I., Goren, M. G., and Qimron, U. (2012). Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40, 5569–5576. doi: 10.1093/nar/gks216
Zhang, Q., and Ye, Y. (2017). Not all predicted CRISPR–Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinform. 18:1–12. doi: 10.1186/s12859-017-1512-4
Zhao, C., Shu, X., and Sun, B. (2017). Construction of a gene knockdown system based on catalytically inactive (“dead”) Cas9 (dCas9) in Staphylococcus aureus. Appl. Env. Microb. 83, e291–e217. doi: 10.1128/AEM.00291-17
Keywords: Leptospira, CRISPR I-B, repeats, transcription, pre-crRNA, Cas6 endoribonuclease, crRNA biogenesis, mature crRNA
Citation: Prakash A and Kumar M (2022) Transcriptional analysis of CRISPR I-B arrays of Leptospira interrogans serovar Lai and its processing by Cas6. Front. Microbiol. 13:960559. doi: 10.3389/fmicb.2022.960559
Received: 03 June 2022; Accepted: 05 July 2022;
Published: 29 July 2022.
Edited by:
Ernesto Perez-Rueda, Universidad Nacional Autónoma de México, MexicoReviewed by:
Luis G. V. Fernandes, Butantan Institute, BrazilWenlong Zhang, Jilin University, China
Copyright © 2022 Prakash and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manish Kumar, mkumar1@iitg.ac.in