 Kaiyue Pang1,2,3†
Kaiyue Pang1,2,3† Dongwen Dai1,2,3†
Dongwen Dai1,2,3† Yingkui Yang1,2,3
Yingkui Yang1,2,3 Xun Wang1,2,3Shujie Liu1,2,3Weihua Huang1,2,3Bin Xue1,2,3
Xun Wang1,2,3Shujie Liu1,2,3Weihua Huang1,2,3Bin Xue1,2,3 Shatuo Chai1,2,3*ShuXiang Wang1,2,3*
Shatuo Chai1,2,3*ShuXiang Wang1,2,3*- 1Qinghai Academy of Animal Husbandry and Veterinary Sciences in Qinghai University, Xining, Qinghai, China
- 2Key Laboratory of Plateau Grazing Animal Nutrition and Feed Science of Qinghai Province, Xining, Qinghai, China
- 3Yak Engineering Technology Research Center of Qinghai Province, Xining, Qinghai, China
Ruminal microflora is closely correlated with the ruminant’s diet. However, information regarding the effect of high concentrate diets on rumen microflora in yaks is lacking. In the current study, 24 healthy male yaks were randomly assigned to two groups, each fed with different diets: less concentrate (LC; concentrate: coarse = 40: 60) and high concentrate (HC; concentrate: coarse = 80: 20) diets. Subsequently, a 21-day feeding trial was performed with the yaks, and rumen fluid samples were collected and compared using 16 s rRNA sequencing. The results showed that NH3-N, total VFA, acetate, butyrate, isobutyrate, and isovalerate were significantly higher in the HC group than that in the LC group (p < 0.05), while microbial diversity and richness were significantly lower in the HC group (p < 0.05). Principal coordinate analysis indicated that rumen microflora was significantly different in LC and HC groups (p < 0.05). In the rumen, phyla Firmicutes and Bacteroidota were the most abundant bacteria, with Firmicutes being more abundant, and Bacteroidota being less abundant in the HC group than those found in the LC group. Christensenellaceae_R-7_group and Prevotella are the highest abundant ones at the genus level. The relative abundance of Acetitomaculum, Ruminococcus, and Candidatus_Saccharimonas were significantly higher in the HC group than that in the LC group (p < 0.05), while the relative abundance of Olsenella was significantly lower in the HC group than in the LC group (p < 0.05). Compared to the LC group, the relative abundance of Prevotella, Ruminococcus, and Candidatus_Saccharimonas was significantly higher in the HC group. The relative abundances of Prevotella, Prevotellaceae_UCG-003, Olsenella, Ruminococcus, Acetitomaculum, Candidatus_Saccharimonas, and NK4A214_group were correlated with ruminal fermentation parameters (p < 0.05). Furthermore, PICRUSt 2 estimation indicated that microbial genes associated with valine, leucine, and isoleucine biosynthesis were overexpressed in the rumen microflora of yaks in the HC group (p < 0.05). Conclusively, our results suggest that high concentrate diets affect the microflora composition and fermentation function in yak rumen. The present findings would provide new insights into the health of yaks under high concentrate feeding conditions and serve as a potent reference for the short-term fattening processes of yaks.
Introduction
Yak (Bos grunniens), an endemic livestock species living on the Qinghai-Tibetan Plateau (QTP), is an important means of production and livelihood for local herders (Long et al., 2018). In recent years, yak breeding has gradually developed from traditional grazing to a large-scale short-term fattening process, where high concentrate feeding is a common practice. The proportion of concentrate in the diet is an important factor affecting ruminant nutrition, and an appropriate proportion of concentrate can improve rumen microbiota (Petrir et al., 2012; Nugroho et al., 2013). When high concentrate diets are fed for a long period, the concentrate will be rapidly fermented in the rumen, producing large amounts of volatile fatty acids and lactic acid causing rumen acidosis as well as nutritional metabolic diseases. Severe metabolic diseases can induce mortality in ruminants, resulting in reduced economic efficiency (Malekkhahi et al., 2016; Nagata et al., 2018).
The rumen is the main site of digestion and metabolism in ruminants, and it contains microorganisms such as protozoa, bacteria, and fungi (Qiu et al., 2020). These microflora degrade diet fibers, produce volatile fatty acids, and use nitrogenous substances to synthesize microbial proteins that provide protein and energy to animals and are essential for the growth and reproduction of ruminant animals (Shabat et al., 2016). It is known that diet structure is an important factor affecting the composition of the rumen microbiota (Petri et al., 2013; Yáñez-Ruiz et al., 2015). The response of the dietary structure to ruminal microbiota has been a research focus in recent years. For example, the study by Fernando et al. (2010) used multiple molecular approaches to provide a more comprehensive illustration of the structure of the ruminal microbial community in beef cattle during adaptation from a high-grain diet to a forage diet. Mao et al. (2013) found that the effect of subacute rumen acidosis (SARA) adaptation on the rumen microbiota of dairy cows was studied by feeding a high-concentrate ration. However, to date, there is limited understanding of the effects of high concentrate diets on ruminal fermentation and ruminal microbiota, and their interactions. The current comprehensive analysis could provide valuable information to the short-term yak fattening industry.
We hypothesized that high concentrate feeds would affect ruminal fermentation, rumen fiber-degrading bacteria species, and rumen metabolic function of yaks, which in turn would affect the rumen health of yaks. As comprehensive analyses of the effects of high concentrate diets on yak microbiota are absent, in this study, we have used 16 s rRNA sequencing technology to analyze the effects of high concentrate rations on ruminal fermentation and rumen microbiota of yaks extensively and discussed the possible correlation of these two factors.
Materials and methods
Animals, diets, and experimental design
The feeding trial was conducted in September 2019 at the Lao Zhaxi breeding base, Guinan County, Qinghai Province, China. Twenty-four healthy male yaks at 3 weeks of age, with uniform health conditions (weight: 164.46 ± 31.18 kg), were randomly divided into two groups and each group was fed a full mixed ration of different diets: low concentrate (LC; concentrate: coarse = 40: 60) and high concentrate (HC; concentrate: coarse = 80: 20) diets. The diets were formulated according to the Chinese Beef Cattle Feeding Standard (NY/T815-2004), and the composition and nutrient contents of the two diets are shown in Table 1. Yaks were selected from grazing pastures. All yaks were uniformly numbered and fed alone. The rations were fed daily from 8: 00–9: 00 and 17:00–18:00 with free access to water. The pretest period was 7 days, and the experimental period was 21 days.
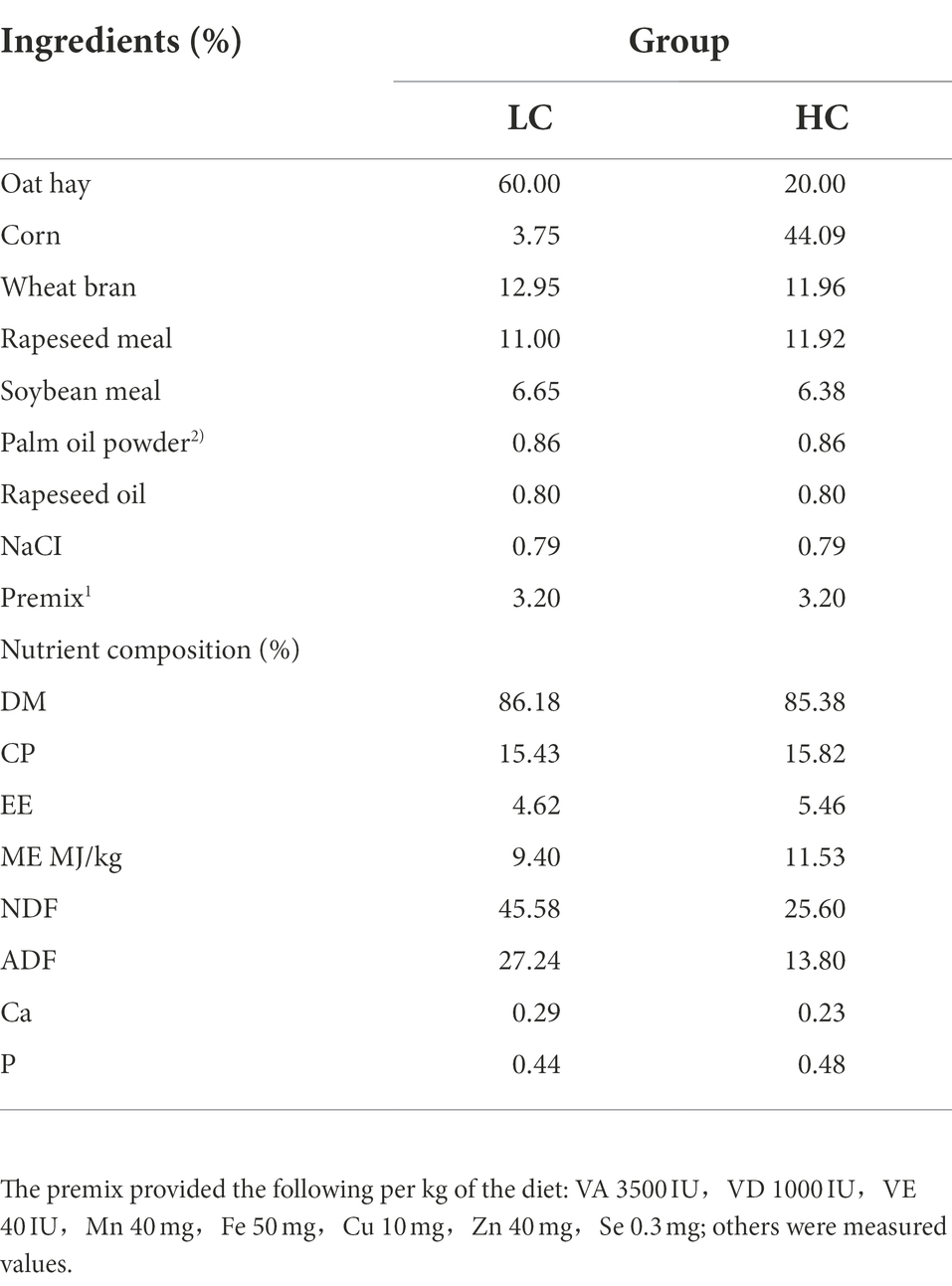
Table 1. Ingredients and nutritional composition of each diet.
Sample collection and measurements
At the end of the experiment, before feeding in the morning, rumen fluid was collected using a bendable oral gastric tube with a metal filter, which was pre-cleaned by rinsing with clean warm water. The first 100 ml of rumen fluid was discarded to eliminate saliva contamination. Finally, a 50 ml rumen fluid sample from each yak was collected and filtered through four layers of gauze before measuring rumen pH with a pH meter (Model HI221, HANNA, Italy). The samples were divided into 15 ml sterile centrifuge tubes and stored in liquid nitrogen for the determination of ruminal fermentation parameters and microbiota analysis.
The filtered rumen fluid was centrifuged (at 17,000 g for 30 min at 4°C) to obtain the supernatant, which was further analyzed for NH3-N using phenol hypochlorite analysis (Broderick and Kang, 1980). Freshly prepared metaphosphoric acid (25% w/v, 2 ml) was added to filtered rumen fluid (8 ml) and then centrifuged (at 17,000 g for 10 min at 4°C). Volatile Fatty Acids (VFAs) concentrations were determined using gas chromatography (GC-2014; Shimadzu Corporation, Japan) as described by Cao et al. (2008).
16 s rRNA gene amplification and MiSeq sequencing
Microbial DNA was extracted using the CTAB (Sigma-Aldrich, Milan, Italy) method according to the instructions provided by the manufacturer. The purity and concentration of DNA were checked using 1% agarose gel electrophoresis. An appropriate amount of DNA sample was taken in a centrifuge tube and the sample was diluted to 1 ng/μL with sterile water. Specific primers with barcodes were synthesized for the V3-V4 variable region of the bacterial 16 s rRNA gene. Common primer sequences were as follows: 515F (5′-GTGCCAGCMGCCGCGG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). PCR (polymerase chain reaction) was performed using a 25 μl amplification system; 1 μl each of 5 μmol/l upstream and downstream primers and 5 ng of template DNA. Equal amounts of purified amplicons were pooled together to construct paired-end sequencing libraries, which were sequenced by Beijing Ovison Gene Technology Co., Ltd. (Beijing, China) using a platform (Mixed PE 300) according to a standard protocol.
Sequence and rumen microflora processing
The raw sequencing data were processed and filtered for quality using Trimmomatic (Version 0.36; Bolger et al., 2014) software, and valid sequences were obtained by removing chimeras through VSearch software and species databases. In addition, sequences with ≥97% similarity were categorized as operational taxonomic units (OTUs) using UPARSE software (Uparse v7.0.1001; Edgar, 2013).1 To obtain species classification information of each out, representative sequences were compared and analyzed using the RDP classifier algorithm version 2.2 (Wang, 2007) and Silvadatabase1 (Quast et al., 2012.), allowing community annotation at the kingdom, phylum, class, order, family, and genus levels. Alpha Diversity (Chao1, Shannon, PD-whole-tree, and Observed-species) was calculated by QIIME 2 (version 1.9.0), and the richness of the community was analyzed using Chao1 richness The Chao1 richness index (Chao1), the Shannon index, the coverage index of PD-whole-tree were applied to analyze the diversity of the community. The number of OTUs was analyzed using the observed-species index. Beta diversity was calculated based on the unweighted UniFracdistance and visualized by principal coordinate analysis (PCoA). Linear discriminant analysis effect sizes (LEfSe, LDA > 3) were used to identify important bacteria in both groups (Miller et al., 2016). To predict microbiota function and explore differences between the two groups, PICRUSt 2 software was used (Douglas et al., 2019).
Statistical analysis
Independent samples t-test based on SAS (SAS, version 9.2) was applied to compare ruminal fermentation parameters between LC and HC groups, and the differences were considered statistically significant at p < 0.05. Microbial networks were generated using Gephi software (version 0.9.2)2 to calculate correlations between dominant taxa. Pearson correlation coefficients between the relative abundances of rumen bacteria (genera) and ruminal fermentation parameters were calculated using the heat map package in R software (version 4.0.2). Functional prediction of rumen microflora in yaks of LC and HC groups was studied with PICRUSt 2 and differences between the two groups in levels 1, 2, and 3 of the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway were determined.
Results
Ruminal fermentation parameters
The effect of high concentrate diets on ruminal fermentation parameters in yaks is shown in Table 2. NH3-N (p = 0.014), total VFA (p < 0.001), butyrate (p < 0.001), isobutyrate (p = 0.007), isovalerate (p < 0.001) were significantly higher in the HC group than those in the LC group (p < 0.05). The proportion of Acetate is higher in the LC group and the Propionate higher in the HC group.
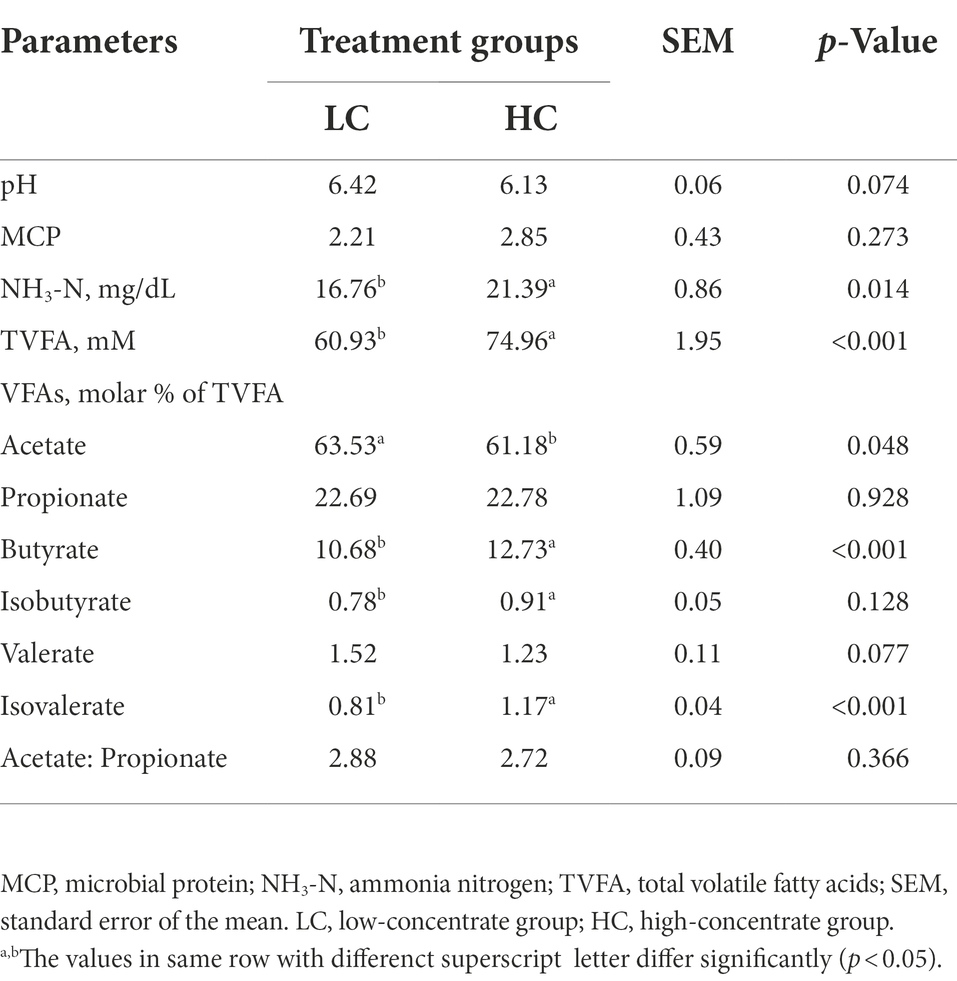
Table 2. Effect of high concentrate diets on ruminal fermentation parameters in yaks.
Richness, diversity estimates, and rumen bacteria composition
According to the Venn diagram, 1,352 OTUs were present in the rumen of yaks in LC and HC groups, with 448 and 138 unique OTUs, respectively (Figure 1A). According to PCoA (Figure 1B), significant differences were observed between the rumen microflora of the LC and HC groups. Alpha diversity calculations (Figure 2) showed significant differences in Chao1, Shannon, PD-whole-tree, and Observed-species between the two groups, indicating that Chao1, Shannon, PD-whole-tree, and Observed-species were significantly lower in the HC group than that in the LC group (p < 0.05).
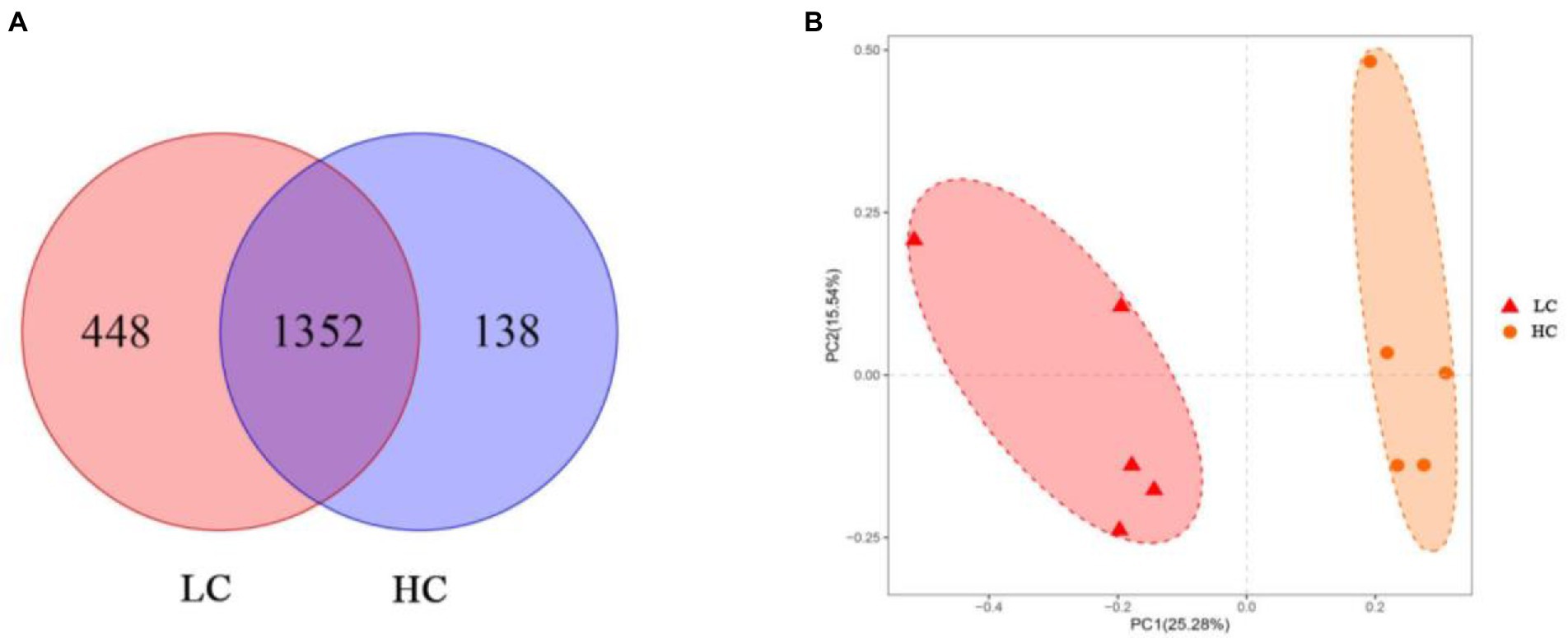
Figure 1. Differences in ruminal microbial communities and operational taxonomic units (OTUs) of yaks fed low concentrate (LC) and high concentrate diets (HC). Venn diagrams (A) show the specific and shared OTU between the two groups. Differences in rumen microflora between the two groups were calculated using weighted UniFrac distances (B) and coordinates were calculated using principal coordinate analysis (PCoA).
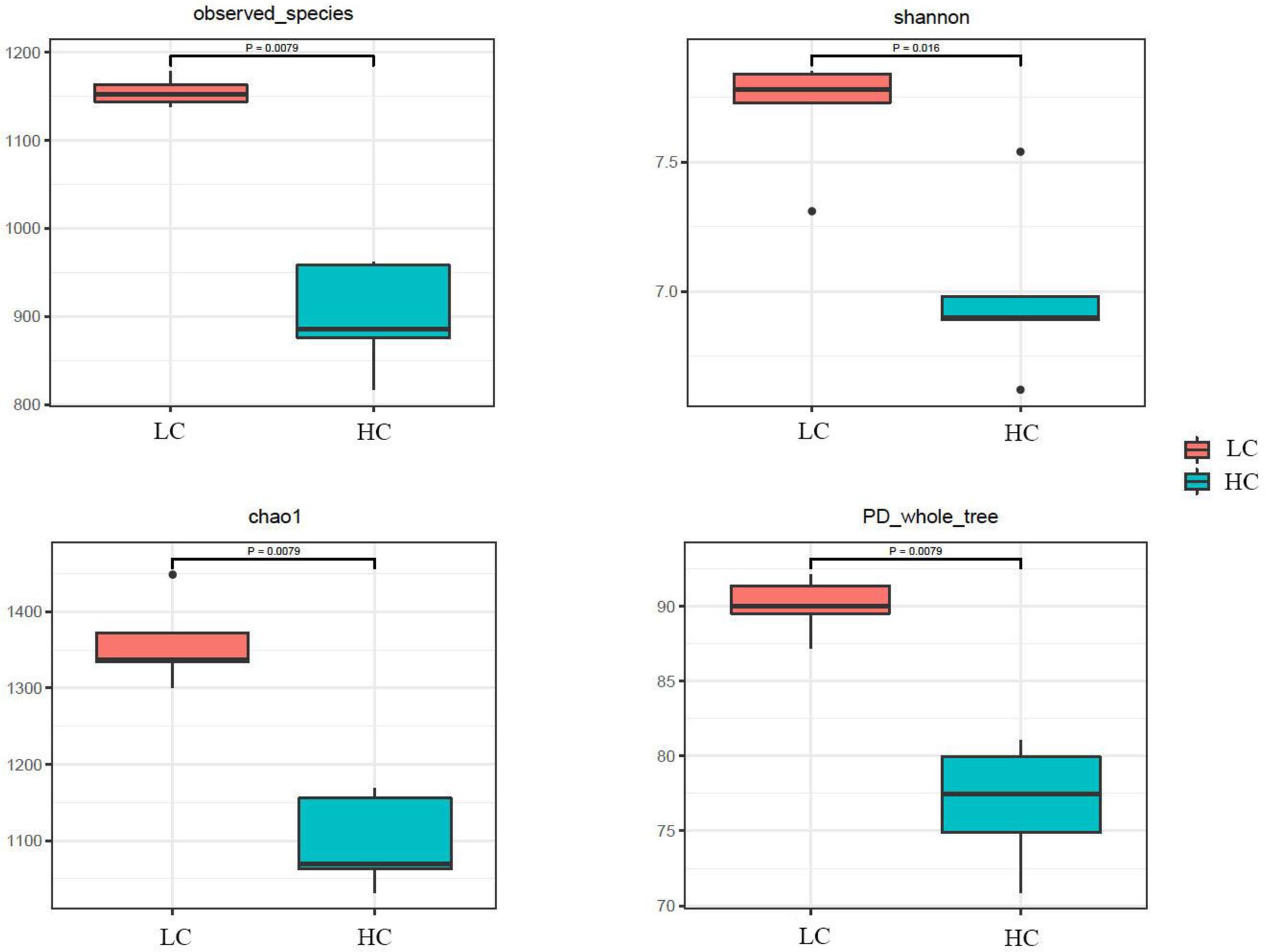
Figure 2. Diversity in the ruminal microbial community of yaks fed low concentrate (LC) and high concentrate diets (HC). p < 0.05 indicates a significant difference.
Bacterial compositions among treatment groups
We performed a taxonomic analysis of the 10 bacterial phyla identified. At the phylum level, among them, Firmicutes and Bacteroidota represented 61.55 and 30.69% of the total reads, respectively, followed by Actinobacteriota (3.65%), Patescibacteria (2.33%) and Verrucomicrobiota (0.46%; Figure 3A). The relative abundances of Patescibacteria and Firmicutes were higher in the HC group than that in the LC group, while a lower relative abundances of Bacteroidota, Actinobacteriota and Verrucomicrobiota were detected in the HC group (Figure 3B). A total of 167 genera was identified from rumen samples. Christensenellaceae_R-7_group (19.06%) and Prevotella (13.35%) were the most dominant genera, followed by NK4A214_group (10.17%), Rikenellaceae_RC9_ gut_group (4.97%), Ruminococcus (4.30%), Lachnospiraceae_NK3A20_group (4.11%), Acetitomaculum (3.60%), Candidatus_Saccharimonas (2.33%), Olsenella (2.18%), and Prevotellaceae_UCG-003 (1.11%; Figure 3C). Among them, the relative abundance of Acetitomaculum, Ruminococcus, and Candidatus_Saccharimonas were significantly higher in the HC group than that in the LC group (p < 0.05), whereas the relative abundance of Olsenella was significantly lower in the HC group than that in the LC group (p < 0.05; Figure 3D).
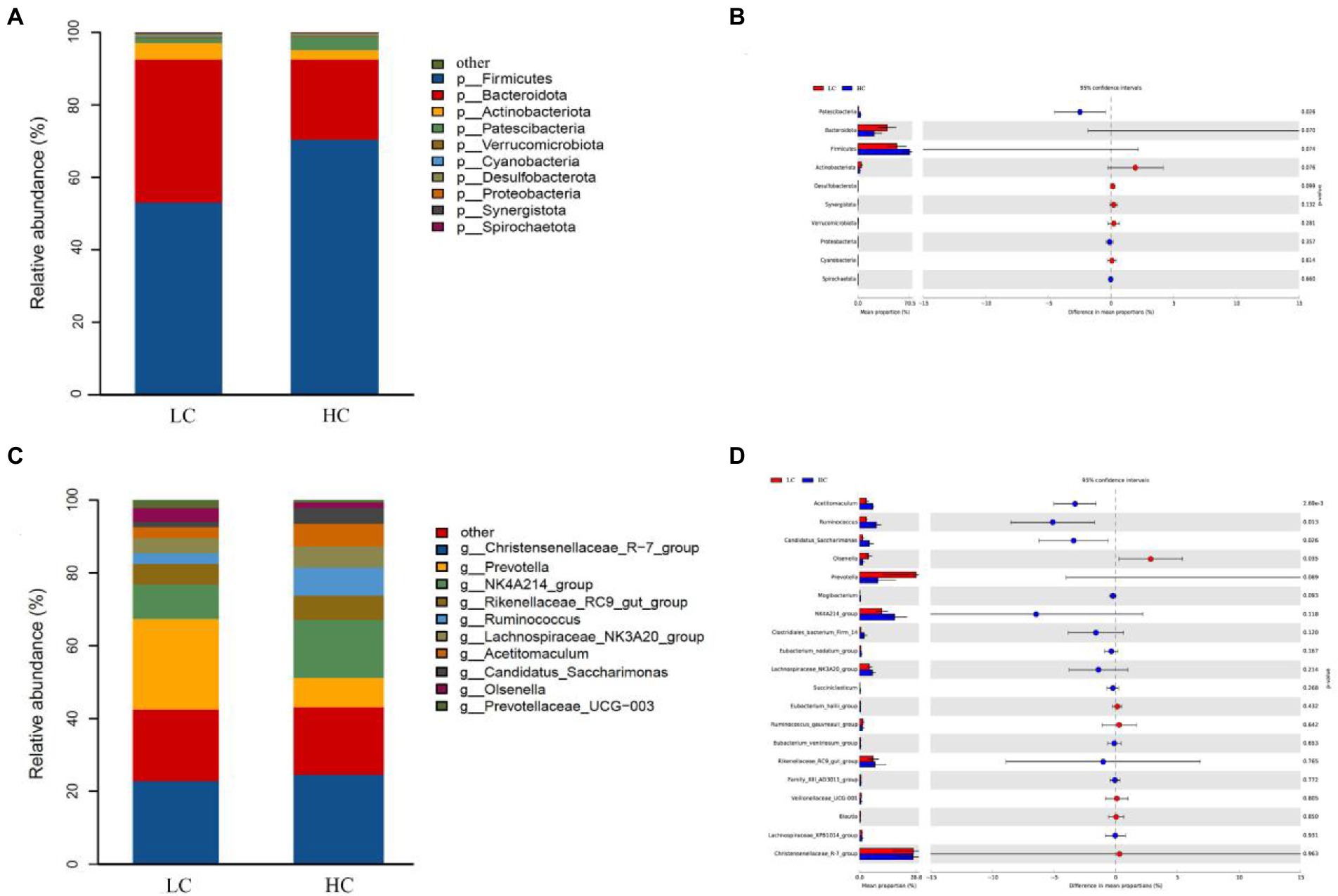
Figure 3. Classification of bacterial community composition in yaks fed low concentrate (LC) and high concentrate (HC) diets. (A) Phylum level. (C) Extended error bars showing the bacteria that differed between the two groups at the phylum level. (B) Genus level. (D) Extended error bars showing the two groups of bacteria that differed at the genus level.
To better understand the dominance of specific bacteria in HC and LC groups, we used the LEfSe method (Figure 4). Figure 4A depicts a representative cladogram of the predominant microbiome structure, showing the most remarkable differences in taxa in the two groups. The data comparing the two groups indicated that 11 clades were more abundant in the LC group and seven clades were more abundant in the HC group. Prevotellaceae, Prevotella, Atopobiaceae, Coriobacteriia, Coriobacteriales, Olsenella, Prevotellaceae _UCG-003, and Prevotella_ruminicola were abundant in the LC group. Clostridia_UCG_014, Patescibacteria, Candidatus_Saccharimonas, Saccharimonadia, Saccharimonadaceae, Saccharimonadales, Acetitomaculum, Ruminococcus, Ruminococcaceae, NK4A214_group, Oscillospirales, and Clostridia were in the HC group in an Overrepresentation (Figure 4B).
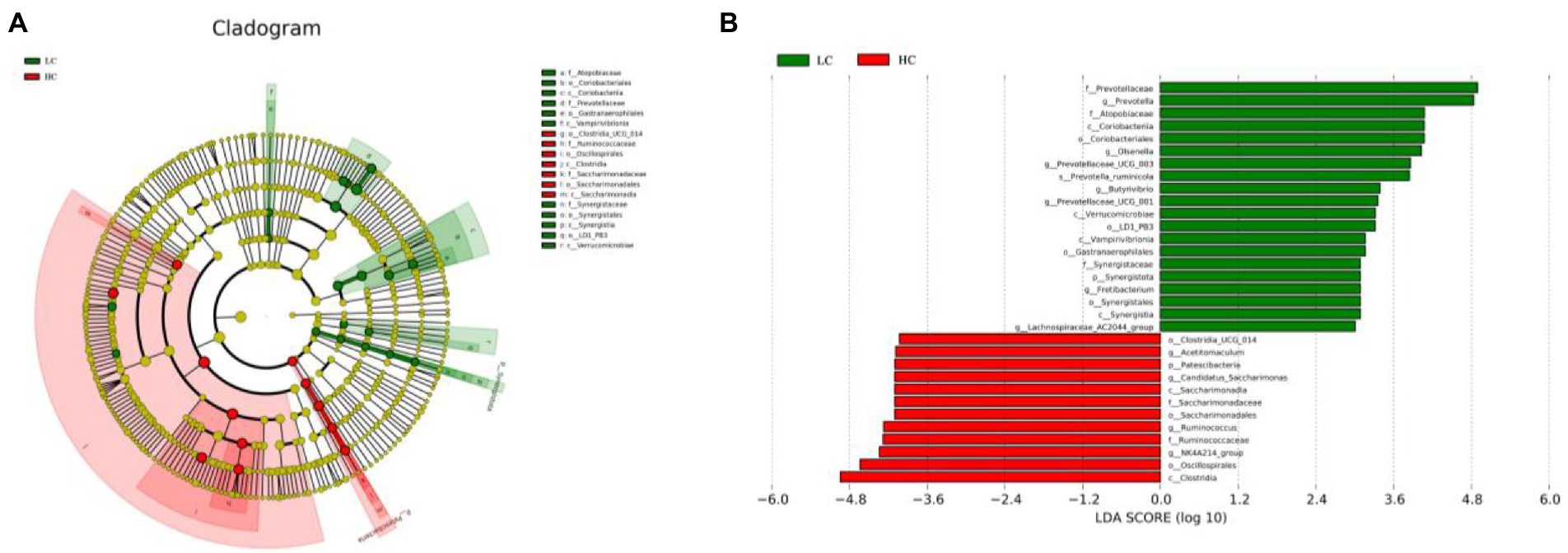
Figure 4. LEfSe analysis of rumen microflora of yaks fed low concentrate (LC) and high concentrate diets (HC). (A) Histogram of linear discriminant analysis scores based on categorical information. (B) Linear discriminant analysis effect size classification plot based on categorical information.
Network analysis of bacterial communities
Microbial interactions between rumen bacterial communities in yaks were analyzed using microbial networks. The results showed that the high concentrate diet altered the correlation within the microbiota (Figure 5), and we verified that the negative correlation was stronger in the HC group than that observed in the LC group.
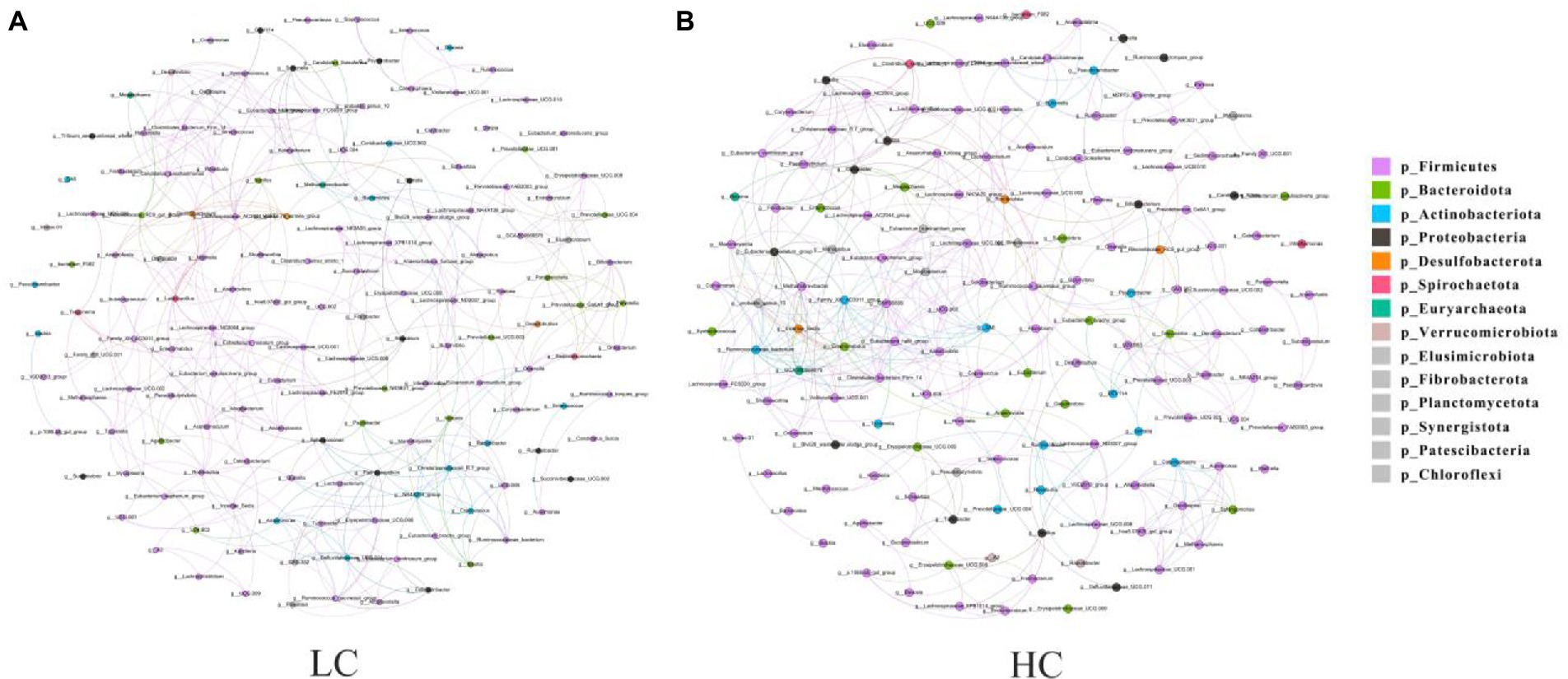
Figure 5. Interaction network of ruminal microflora. The ruminal microflora correlation network based on 16S rRNA genes showed statistically significant interactions with absolute values of correlation coefficients >0.6. The size of the nodes is scaled according to the abundance of each taxon in the microflora. The red line indicates a positive correlation and the green line indicates a negative correlation.
Correlations between rumen bacteria and ruminal fermentation parameters
Correlation analyses between rumen bacteria and ruminal fermentation parameters were based on Spearman’s correlation coefficients, and significantly influential rumen microflora (genus level) were significantly correlated with ruminal fermentation parameters (Figure 6). Prevotella was negatively correlated with NH3-N and isovalerate concentrations; Prevotellaceae_UCG-003 was negatively correlated with isovalerate; Olsenella was negatively correlated with Isobutyrate, isovalerate, and butyrate concentrations; Ruminococcus was positively correlated with Butyrate concentration; Acetitomaculum was positively correlated with acetate and butyrate concentrations. Positively correlation was observed for Candidatus_Saccharimonas with concentrations of sovalerate, MCP, propionate, acetate, and butyrate, and for NK4A214_group with isobutyrate, isovalerate, and acetate concentrations.
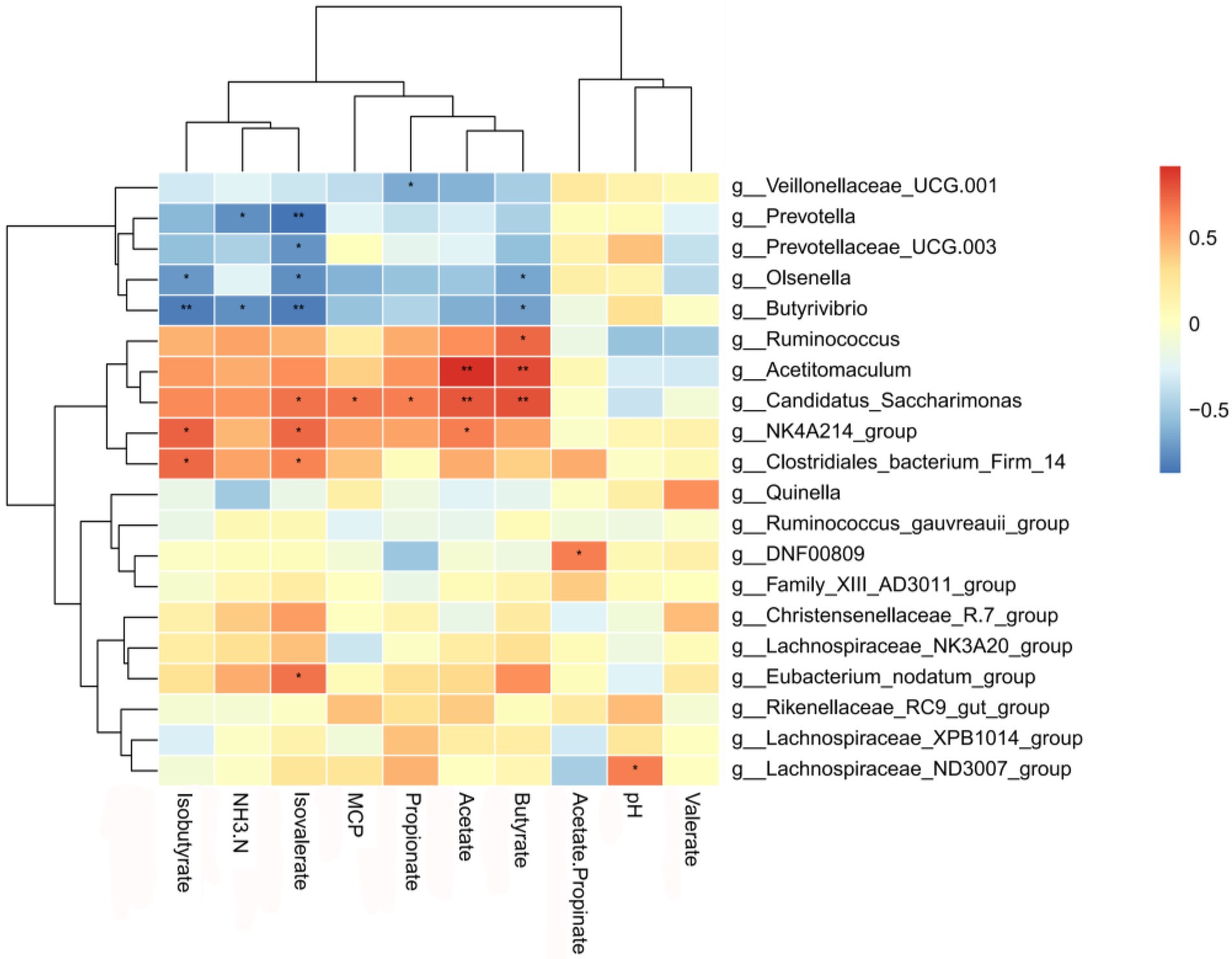
Figure 6. Correlation between bacteria and fermentation parameters in the rumen. Each row in the figure represents a genus, each column represents a metabolite, and each grid represents the Pearson correlation coefficient between a component and a metabolite. The red color represents a positive correlation, while the blue color represents a negative correlation. * and ** indicate significant levels of 0.05 and 0.01, respectively.
PICRUSt2 function prediction
PICRUSt 2 gene function assessment was used to predict the function of rumen microflora in LC and HC groups of yaks. The highest abundance of valine, leucine, and isoleucine biosyntheses (26.58%) was observed, followed by lysine biosynthesis (18.74%). PICRUSt 2 prediction software enriched 40 major pathways (relative abundance >1%) in the 3-level KEGG pathway, 19 of which showed significant differences between the LC and HC groups (p < 0.05; Figure 7). Notably, the relative abundance of metabolism of other amino acids and xenobiotics biodegradation were significantly increased in the HC group (p < 0.05).
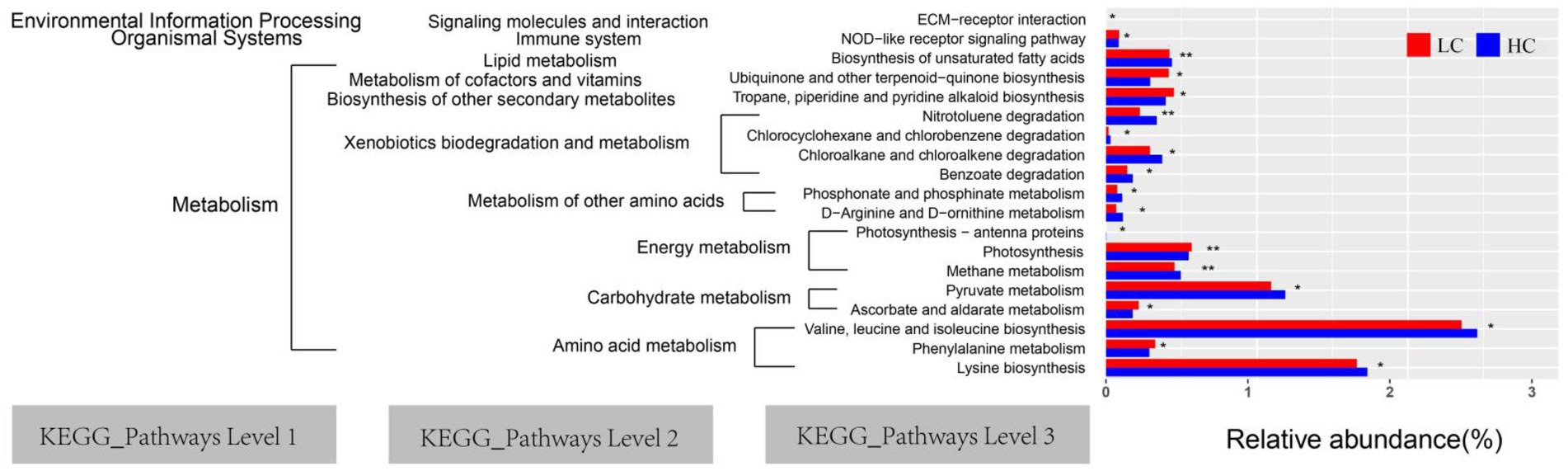
Figure 7. Yaks, which were fed low concentrate (LC) and high concentrate diets (HC), had significantly different functional predictions of the rumen microbiota of the KEGG pathway (p < 0.05). The graph shows the KEGG pathways for levels 1, 2, and 3. “*” and “**” indicate significance levels of 0.05 and 0.01, respectively.
Discussion
In this study, we investigated the effects of high concentrate diet on microflora and fermentation in rumen of yaks and interactions between these two factors. A stable intra-rumen environment is particularly important for ruminants; ruminal pH, NH3-N and VFA molar concentrations are important indicators of a stable intra-rumen environment and reflect the status of ruminal fermentation (Tomczak et al., 2019). A stable ruminal pH is a prerequisite for its normal function, and it is determined by volatile fatty acid and lactic acid accumulation (Tomkins et al., 2015). The present results showed that the pH decreasing tendency in the HC group. The study conducted by Ogata et al. (2019) found that ruminal pH was significantly lower in Japanese black wagyu cattle that were fed a high grain diet. Similar results were obtained in other studies (Kmicikewycz et al., 2015; Ramos et al., 2021). The reduction in pH may have resulted from the significant change in the ruminal bacterial community as a result of feeding high concentrates. The increased frequency of lactic acid producing bacteria and lactic acid utilizing bacteria in the rumen and an imbalance in the bacterial flora leads to the accumulation of lactic acid in the rumen, further reducing ruminal pH. Moreover, the number of fiber degrading bacteria also decreases drastically due to the low fiber content in the diet and the accumulation of lactic acid in the rumen (Mackie and Gilchrist, 1979; Tajima et al., 2001; Lee et al., 2019).
NH3-N is endogenous ruminal nitrogen, a product of fermentation and decomposition of diet proteins, and a major component for microbial synthesis of bacteriophage proteins in the rumen (Garcia-Gonzalez et al., 2010; Thao et al., 2014; Lv et al., 2020). Carlos et al., 2021 reported that the NH3-N concentration in the rumen of dairy goats that were fed high concentrate diets was significantly higher than that of the low concentrate diet group. NH3-N concentrations in the current HC group were comparable to the above results and with those of Jahan et al. (2018); a large amount of nitrogenous substances in the high concentrate diet may be the possible reason for this outcome. The more nutrients in the rumen are decomposed by ruminal microorganisms to produce a larger amount of NH3-N, increasing NH3-N concentration and even ammonia toxicity in severe cases (Vcdsa et al. 2020).
Volatile fatty acids are derived from the fermentation of carbohydrates and proteins in the diet by rumen microorganisms and are an important source of energy for ruminants (Li et al., 2016). Acetic acid, propionic acid, and butyric acid in the rumen are the main components of volatile fatty acids (Guilloteau et al., 2010). Acetic and propionic acids enter the portal circulation and are metabolized in the liver (Minamoto et al., 2019), acetic acid is the main synthetic precursor of fat in ruminants (Hou et al., 2020), and propionic acid is converted to glucose by gluconeogenesis or enters the tricarboxylic acid cycle for oxidative energy supply (Young, 1977; Jia et al., 2018), and butyric acid is an important source of energy for animals (Gorka et al., 2009); in this study, the concentrations of Total VFA, Acetate, Propionate and Butyrate were higher in the HC group than in the LC group, which is similar to Bevans et al. (2005), Sato (2016), and Nagata et al. (2018) studies were similar. Due to the high percentage of concentrate in the diet, the diet enters the rumen to produce a large amount of VFA (including acetic acid, propionic acid and butyric acid) by rapid fermentation, resulting in a lower absorption rate of VFA and excessive accumulation of VFA in the rumen (Goad et al., 1998), which affects ruminal fermentation in yaks, which in turn can lead to reduced feed intake as well as digestive problems affecting the health of yaks.
We further investigated the effect of high concentrate diets on rumen microflora of yaks using 16S rDNA high-throughput sequencing technology. In the present study, both alpha and beta diversity indices of microflora were significantly different between the two groups, indicating that the diversity of rumen microflora in yaks was closely related to the diet concentrate ratio. Our results showed that intra-ruminal bacterial diversity and richness were reduced in yaks fed a high-concentrate diet, which was comparable with the results obtained by Petri et al. (2013) and Zened et al. (2013). This suggests that feeding high concentrate diets to yaks causes a decrease in rumen pH and inhibition of cellulose-degrading bacteria, leading to a decrease in the diversity and abundance of ruminal bacteria. PCoA analysis showed that rumen microbial communities aggregated according to the concentrate ratio in diets, and significant differences between intra-ruminal microbial communities were observed, which were consistent with previous reports (Hu et al., 2019; Islam et al., 2021).
In the current study, as similar to previous studies, Bacteroidota and Firmicutes were the dominant phyla among ruminal microorganisms in yaks (Zhou et al., 2017; Bi et al., 2018; Liu C. et al., 2019a; Fan et al., 2020), indicating that these bacteria play an important role in the yak rumen. Previous studies have shown that Bacteroidota is mainly responsible for energy conversion and acquisition; while Firmicutes play an important role in the degradation of non-fibrous material (Evans et al., 2011; Reigstad and Kashyap, 2013; Ya-Bing et al., 2016). Among the two groups considered in this study, Bacteroidota was more abundant in the LC group, while Firmicutes were more abundant in the HC group, indicating that the number of bacteria involved in ruminal starch digestion and metabolism was significantly increased in yaks fed with high concentrate.
At the genus level, Christensenellaceae_R-7_group, Prevotella and NK4A214_group were the dominant bacteria in the rumen in this experiment. Christensenellaceae_R-7_group belongs to the phylum of thick-walled bacteria (Waters and Ley, 2019), which mainly catabolizes fibrous material (Evans et al., 2011). Prevotella is a protein-degrading bacterium of the rumen and gastrointestinal tract of ruminants; it mainly degrades the hemicellulose component of the food and promotes the degradation of non-fibrous polysaccharides and pectins (Purushe et al., 2010). In the present study, Christensenellaceae_R-7_group and Prevotella were more abundant in the LC group, which may be related to the low level of concentrate fed to the yaks. The NK4A214_group belongs to the family Rumenococcaceae, rumen bacteria are rich in endo-1, 4-beta-xylanase and Cellulase genes, these genes play an important role in the degradation of cellulose and hemicellulose, which are degraded to produce short-chain fatty acids that are available for use by the host. Thus, its relative abundance is related to the diet concentrate ratio (Koike and Kobayashi, 2009; Biddle et al., 2013). The relative abundance of NK4A214_group was found to be associated with isobutyl acid and isovaleric acid concentrations were positively correlated (Liu C. et al., 2019a). In the present study, NK4A214_group was more abundant in the HC group, similar to a previously report (Chen et al., 2021), this could explain the higher isobutyl acid and isovaleric acid concentration in the HC group. Some bacteria, including Ruminococcus, Acetitomaculum, Candidatus_Saccharimonas and NK4A214_group, were positively correlated with acetate, butyrate, isovalerate, propionate and isobutyrate concentrations, respectively, indicating that these bacteria may favor VFA production. For instance, Ruminococcus can ferment cellobiose or cellulose to produce butyric acid (Henderson et al., 2015). Acetitomaculum, which is mainly found in ruminants fed high concentrate diet, can produce acetic acid from monosaccharides (Hua et al., 2017). Due to the complex interactions between bacteria (Olotu et al., 2019), it is difficult for us to understand the bacterial activities that directly produce VFA (Mahowald et al., 2009).
Microbial flora has an important role in ecosystem function, and the relationships among microorganisms involved in ruminal fermentation ecosystems are very complex (Liu H. et al., 2019b). In this study, key microbial taxa with topological properties in the rumen of yaks were identified for the first time. The results showed that high concentrate diet altered the correlation between components of microbial flora. Network analysis can reveal interactions between species in both positive and negative ways (Liu et al., 2020). Negative interactions may weaken competitive relationships, while positive interactions may strengthen them back (Fan et al., 2018). In our study, compare to the low concentrate diet group, the high concentrate diet group revealed stronger negative correlations. We hypothesized that the cellulose-degrading bacteria in the rumen were inhibited due to the low cellulose content of the high concentrate feeds, which resulted in fewer bacteria. This further suggests that high concentrate rations can regulate the microbial dynamics of the rumen in yaks.
In the current study, we used PICRUSt 2 to predict the function of the ruminal microbial community in yaks. The data suggest that the assessment of gene functions of the ruminal microbiota is significantly influenced by the ration concentrate ratio. In most cases, genes involved in the metabolism of amino acids in the level 2 KEGG pathway were enhanced in yaks fed high concentrate diets. This suggests that the ruminal microflora of yaks fed high concentrates produce large amounts of protein, which provide the host with raw materials such as protein, which in turn sustains life and normal metabolism. It is worth noting that valine, leucine and isoleucine biosyntheses are the most expressed pathways, directly involved in amino acid metabolisms. In this study, valine, leucine and isoleucine biosyntheses were active at significantly higher levels in HC group, indicating that the increase in the percentage of concentrates increased the protein content of the rations, creating a more favorable environment for fermentation and the growth of cytolytic bacteria, which promote the participation of rumen microorganisms in the digestion and metabolism of nutrients. However, our results are based on predicted macrogenomics only and may not be representative of the actual function of ruminal bacteria. Further macrogenomic analyses are needed to explore the mechanism of these gene functions in feeding high concentrate diet to yaks.
Conclusion
In this study, ruminal fermentation parameters and microflora of yaks that were fed high concentrate diets were analyzed. In fact, high concentrate diets altered the ruminal fermentation pattern, and the structure and composition of the microflora of yaks, which in turn affected their functions. The promotion of Ruminococcus and Acetitomaculum growth led to increased acetic acid and butyric acid contents in the rumen of yaks fed high concentrate diets, and the fermentation produced large amounts of VFA, tending to decrease ruminal pH and affecting yak ruminal health.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI, PRJNA843719.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of the Qinghai University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
SW, SC, and KP contributed to the conception and design of the study. KP, SW, and XW collected the samples. SW, KP, YY, HW, XB, and SC conducted relevant experiments. KP and SW organized the database. XW and SL performed the statistical analysis. KP wrote the first draft of the manuscript. XW, YY, SL, HW, and SC wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Key Research and Development Program of China (grant no. 2018YFD0502300), Qinghai Provincial Science and Technology Department of China (grant no. 2020-ZJ-935Q) and Qinghai ProviProvince’susands of High-end Innovative Talents Plan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Bevans, D. W., Beauchemin, K. A., Schwartzkopf-Genswein, K. S., McKinnon, J. J., and McAllister, T. A. (2005). Effect of rapid or gradual grain adaptation on subacute acidosis and feed intake by feedlot cattle1,2. J. Anim. Sci. 83, 1116–1132. doi: 10.2527/2005.8351116x
Bi, Y. L., Zeng, S. Q., Zhang, R., Diao, Q. Y., and Tu, Y. (2018). Effects of dietary energy levels on rumen bacterial community composition in Holstein heifers under the same forage to concentrate ratio condition. BMC Microbiol. 18:69. doi: 10.1186/s12866-018-1213-9
Biddle, A., Stewart, L., Blanchard, J., and Leschine, S. (2013). Untangling the genetic basis of fibrinolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 5, 627–389640. doi: 10.3390/d5030627
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Broderick, G. A., and Kang, J. H. (1980). Automated simultaneous determination of ammonia and total amino acids in ruminal fluid and in vitro medial. Dairy Sci. 63, 64–75. doi: 10.3168/jds.S0022-0302(80)82888-8
Cao, Z. J., Li, S. L., Xing, J. J., Ma, M., and Wang, L. L. (2008). Effects of maize grain and lucerne particle size on ruminal fermentation, digestibility and performance of cows in midlactation. Anim. Physiol. Anim. Nutr. (Berl). 92, 157–167. doi: 10.1111/j.1439-0396.2007.00721.x
Carlos, F., Alberto, H., Julio, G. T., and Loor, J. J. (2021). Changes in nutrient balance, methane emissions, physiologic biomarkers and production performance in goats fed different forage-to-concentrate ratios during lactation. J. Anim. Sci. 99:7. doi: 10.1093/jas/skab114
Chen, H., Wang, C., Huasai, S., and Chen, A. (2021). Effects of dietary forage to concentrate ratio on nutrient digestibility, ruminal fermentation and rumen bacterial composition in angus cows. Sci. Rep. 11:17023. doi: 10.1038/s41598-021-96580-5
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., and Langille, M. (2019). PICRUSt2: an improved and extensible approach for metagenome inference. Cold Spring Harbor Laboratory. 38, 685. doi: 10.1101/672295
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Evans, N. J., Brown, J. M., Murray, R. D., Getty, B., Birtles, R. J., Hart, C. A., et al. (2011). Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis treponemes. Appl. Environ. Microbiol. 77, 138–147. doi: 10.1128/AEM.00993-10
Fan, Q., Wanapat, M., Yan, T., and Hou, F. (2020). Altitude influences microbial diversity and herbage fermentation in the rumen of yaks. BMC Microbiol. 20:370. doi: 10.1186/s12866-020-02054-5
Fan, K., Weisenhorn, P., Gilbert, J. A., Shi, Y., Bai, Y., and Chu, H. (2018). Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Elds. Soil Biol. Biochem. 121, 185–192. doi: 10.1016/j.soilbio.2018.03.017
Fernando, S. C., Purvis, H. T., Najar, F. Z., Sukharnikov, L. O., Krehbiel, C. R., Nagaraja, T. G., et al. (2010). Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 76, 7482–7490. doi: 10.1128/AEM.00388-10
Garcia-Gonzalez,, Gonzalez, J. S., and Lopez, (2010). Decrease of ruminal methane production in Rusitec fermenters through the addition of plant material from rhubarb (rheum spp.) and alder buckthorn (Frangula alnus). J. Dairy Sci. 93, 3755–3763. doi: 10.3168/jds.2010-3107
Goad, D. W., Goad, C. L., and Nagaraja, T. G. (1998). Ruminal microbial and fermentative changes associated with experimentally induced subacute acidosis in steers. J. Anim. Sci. 76, 234–241. doi: 10.2527/1998.761234x
Gorka, P., Kowalski, Z. M., Pietrzak, P., Kotunia, A., and Zabielski, R. (2009). Effect of sodium butyrate supplementation in milk replacer and starter diet on rumen development in calves. J. Physiol. Pharmacol. 102, 2563–2575. doi: 10.1152/jn.00345.2009
Guilloteau, P., Martin, L., Eeckhaut, V., Ducatelle, R., Zabielski, R., and Immerseel, F. V. (2010). From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr. Res. Rev. 23, 366–384. doi: 10.1017/S0954422410000247
Henderson, G., Cox, F., Ganesh, S., Jonker, A., Young, W., and Janssen, P. H. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Report. 5:14567. doi: 10.1038/srep14567
Hou, K., Tong, J., Zhang, H., Gao, S., Guo, Y., Niu, H., et al. (2020). Microbiome and metabolic changes in milk in response to artemisinin supplementation in dairy cows. AMB Express 10:154. doi: 10.1186/s13568-020-01080-w
Hu, R., Zou, H., Wang, Z., Cao, B., Peng, Q., Jing, X., et al. (2019). Nutritional interventions improved rumen functions and promoted compensatory growth of growth-retarded yaks as revealed by integrated transcripts and microbiome analyses. Front. Microbiol. 10:318. doi: 10.3389/fmicb.2019.00318
Hua, C., Tian, J., Tian, P., Cong, R., Luo, Y., Geng, Y., et al. (2017). Feeding a high concentration diet induces unhealthy alterations in the composition and metabolism of ruminal microbiota and host response in a goat model. Front. Microbiol. 8:138. doi: 10.3389/fmicb.2017.00138
Islam, M., Kim, S., Ramos, S. C., Mamuad, L. L., Son, A.-R., and Zhong, T. Y. (2021). Holstein and Jersey steers differ in rumen microbiota and enteric methane emissions even fed the same total mixed ration. Front. Microbiol. 12:601061. doi: 10.3389/fmicb.2021.601061
Jahan, R., Amin, M. R., Sarker, N. R., and Kamal, M. T. (2018). Feeding effects of total mixed ration on rumen metabolic profile in cattle. Bangladesh J. Animal Sci. 47, 35–39. doi: 10.3329/bjas.v47i1.39401
Jia, P., Cui, K., Ma, T., Wan, F., Wang, W., Yang, D., et al. (2018). Influence of dietary supplementation with bacillus licheniformis and Saccharomyces cerevisiae as alternatives to monensin on growth performance, antioxidant, immunity, ruminal fermentation and microbial diversity of fattening lambs. Sci. Rep. 8:16712. doi: 10.1038/s41598-018-35081-4
Kmicikewycz, A. D., Harvatine, K. J., and Heinrichs, A. J. (2015). Effects of corn silage particle size, supplemental hay, and forage-to-concentrate ratio on rumen pH, feed preference, and milk fat profile of dairy cattle. J. Dairy Sci. 98, 4850–4868. doi: 10.3168/jds.2014-9249
Koike, S., and Kobayashi, Y. (2009). Fibrolytic rumen bacteria: their ecology and functions. Asian Australasian J. Animal Sci. 22, 131–138. doi: 10.5713/ajas.2009.r.01
Lee, M., Jeong, S., Seo, J., and Seo, S. (2019). Changes in the ruminal fermentation and bacterial community structure by a sudden change to a high-concentrate diet in Korean domestic ruminants. Asian-Australas J Anim Sci. 32, 92–102. doi: 10.5713/ajas.18.0262
Li, D., Zhang, Y., Cui, Z., He, L., Chen, W., Meng, Q., et al. (2016). Effects of Phytoecdysteroids (PEDS) extracted from Cyanotis arachnoidea on rumen fermentation, enzyme activity and microbial efficiency in a continuous-culture system. PLoS One 11:e0153584. doi: 10.1371/journal.pone.0153584
Liu, H., Hu, L., Han, X., Zhao, N., and Xu, S. (2020). Tibetan sheep adapt to plant phenology in alpine meadows by changing rumen microbial community structure and function. Front. Microbiol. 11:587558. doi: 10.3389/fmicb.2020.5875583
Liu, C., Wu, H., Liu, S., Chai, S., and Zhou, Z. (2019a). Dynamic alterations in yak rumen bacteria community and metabolome characteristics in response to feed type. Front. Microbiol. 10:1116. doi: 10.3389/fmicb.2019.01116
Liu, H., Xu, T., Xu, S., Ma, L., and Zhao, X. (2019b). Effect of dietary concentrate to forage ratio on growth performance, rumen fermentation and bacterial diversity of Tibetan sheep under barn feeding on the Qinghai-Tibetan plateau. PeerJ. 7:e7462. doi: 10.7717/peerj.7462
Long, R., Ding, L., and Shang, Z. (2018). The yak grazing system on the Qinghai-Tibetan plateau and its status. Rangel. J. 30, 241–246. doi: 10.1071/RJ08012
Lv, F., Wang, X., Pang, X., and Liu, G. (2020). Effects of supplementary feeding on the rumen morphology and bacterial diversity in lambs. PeerJ. 8:e9353. doi: 10.7717/peerj.9353
Mackie, R. I., and Gilchrist, F. M. C. (1979). Changes in lactate-producing and lactate-utilizing bacteria in relation to pH in the rumen of sheep during stepwise adaptation to a high-concentrate diet. Appl. Environ. Microbiol. 38, 422–430. doi: 10.1007/BF00403678
Mahowald, M. A., Rey, F. E., Seedorf, H., Turnbaugh, P. J., Fulton, R. S., Wollam, A., et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. U. S. A. 106, 5859–5864. doi: 10.1073/pnas.0901529106
Malekkhahi, M., Tahmasbi, A. M., Naserian, A. A., Naserian, M., Danesh-Mesgaran, J. L., Kleen, O., et al. (2016). Effects of supplementation of active dried yeast and malate during sub-acute ruminal acidosis on rumen fermentation, microbial population, selected blood metabolites and milk production in dairy cows. Anim. Feed Sci. Technol. 213, 29–43. doi: 10.1016/j.anifeedsci.2015.12.018
Mao, S. Y., Zhang, R. Y., Wang, D. S., and Zhu, W. Y. (2013). Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 24, 12–19. doi: 10.1016/j.anaerobe.2013.08.003
Miller, G. E., Engen, P. A., Gillevet, P. M., Maliha, S., Masoumeh, S., Forsyth, C. B., et al. (2016). Lower neighborhood socioeconomic status associated with reduced diversity of the colonic microbiota in healthy adults. PLoS One 11:e148952. doi: 10.1371/journal.pone.0148952
Minamoto, Y., Minamoto, T., Isaiah, A., Sattasathuchana, P., Buono, A., Rangachari, V. R., et al. (2019). Fecal short-chain fatty acid concentrations and dysbiosis in dogs with chronic enteropathy. J. Vet. Intern. Med. 33, 1608–1618. doi: 10.1111/jvim.15520
Nagata, R., Kim, Y. H., Ohkubo, A., Kushibiki, S., Ichijo, T., and Sato, S. (2018). Effects of repeated subacute ruminal acidosis challenges on the adaptation of the rumen bacterial community in Holstein bulls. J. Dairy Sci. 101, 4424–4436. doi: 10.3168/jds.2017-13859
Nugroho, D., Sevilla, C. C., Angeles, A. A., and Sunarso, S. (2013). The effects of dietary neutral detergent fiber ratio from forage and concentrate on the dietary rumen degradability and growth performance of Philippine native goats (Capra hircus Linn.). Intern. J. Sci. Engineering. 6, 75–80. doi: 10.14334/jitv.v18i4.336
Ogata, T., Makino, H., Ishizuka, N., Iwamoto, E., Masaki, T., Ikuta, K., et al. (2019). Long-term high-grain diet altered the ruminal pH, fermentation, and composition and functions of the rumen bacterial community, leading to enhanced lactic acid production in Japanese black beef cattle during fattening. PLoS One 14:e0225448. doi: 10.1371/JOURNAL.PONE.0225448
Olotu, C., Lehmensiek, F., Koch, B., Kiefmann, M., Riegel, A. K., Hammerschmidt, S., et al. (2019). Streptococcus pneumoniae inhibits purinergic signaling and promotes purinergic receptor P2Y2internalization in alveolar epithelial cells. J. Biol. Chem. 294, 12795–12806. doi: 10.1074/jbc.RA118.007236
Petri, R. M., Tyler, S., Penner, G. B., Beauchemin, K. A., Forster, R. J., McKinnon, J. J., et al. (2013). Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS One 8:e83424.8. doi: 10.1371/journal.pone.0083424
Petrir, M., Forster, R. J., Yang, W., Mckinnon, J. J., and Mcallister, T. A. (2012). Characterization of rumen bacterial diversity and fermentation parameters in concentrate fed cattle with and without forage. J. Appl. Microbiol. 112:1152–1162. doi:doi: 10.1111/j.1365-2672.2012.05295.x
Purushe, J., Fouts, D. E., Morrison, M., White, B. A., and Mackie, R. I. (2010). Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb. Ecol. 60, 721–729. doi: 10.1007/s00248-010-9692-8
Qiu, Q., Gao, C., Rahman, M., Cao, B., and Su, H. (2020). Digestive ability,physiological characteristics,and rumen bacterial community of Holstein finishing steers in response to three nutrient density diets as fattening phases advanced. Microorganisms. 8:335. doi: 10.3390/microorganisms8030335
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., and Glckner, F. O. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ramos, S. C., Chang, D. J., Mamuad, L. L., Ho, S., and Sang, S. L. (2021). Diet transition from high-forage to high-concentrate alters rumen bacterial community composition, epithelial transcriptomes and ruminal fermentation parameters in dairy cows. Animals: an open access journal from MDPI. 11:838. doi: 10.3390/ani11030838
Reigstad, C. S., and Kashyap, P. C. (2013). Beyond phylotyping: understanding the impact of gut microbiota on host biology. Neurogastroenterol. Motil. 25, 358–372. doi: 10.1111/nmo.12134
Sato, S. (2016). Pathophysiological evaluation of subacute ruminal acidosis (SARA) by continuous ruminal pH monitoring. Anim. Sci. J. 87, 168–177. doi: 10.1111/asj.12415
Shabat, S. K., Sasson, G., Doron-Faigenboim, A., Durman, T., Yaacoby, S., Berg Miller, M. E., et al. (2016). Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958–2972. doi: 10.1038/ismej.2016.62
Tajima, K., Aminov, R. I., Nagamine, T., Matsui, H., Nakamura, M., and Benno, Y. (2001). Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl. Environ. Microbiol. 67, 2766–2774. doi: 10.1128/AEM.67.6.2766-2774.2001
Thao, N. T., Wanapat, M. A., et al. (2014). Effects of eucalyptus crude oils supplementation on rumen fermentation, microorganism and nutrient digestibility in swamp buffaloes. Asian Australasian J. Animal Sci. 27, 46–54. doi: 10.5713/ajas.2013.13301
Tomczak, D. J., Samuelson, K. L., Jennings, J. S., and Richeson, J. T. (2019). Oral hydration therapy with water and bovine respiratory disease incidence affects rumination behavior, rumen pH, and rumen temperature in high-risk, newly received beef calves. J. Anim. Sci. 97, 2015–2024. doi: 10.1093/jas/skz102
Tomkins, N. W., Denman, S. E., Pilajun, R., Wanapat, M., Mcsweeney, C. S., and Elliott, R. (2015). Manipulating rumen fermentation and methanogenesis using an essential oil and monensin in beef cattle fed a tropical grass hay. Anim. Feed Sci. Technol. 200, 25–34. doi: 10.1016/j.anifeedsci.2014.11.013
Vcdsa, C., Jdm, A., Edb, B., Klgca, A., Avld, A., Lmc, C., et al. (2020). Assessing amino acid utilization in young Nellore steers fed high-concentrate diets with different sources and levels of nitrogen. Anim. Feed Sci. Technol. 269:114642. doi: 10.1016/j.anifeedsci.2020.114642
Wang, Q. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Waters, J. L., and Ley, R. E. (2019). The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 17:83. doi: 10.1186/s12915-019-0699-4
Ya-Bing, C., Dao-Liang, L., Cheng, T., Xiao-Nong, Y., and Jian, L. (2016). Effect of DNA extraction methods on the apparent structure of yak rumen microbial communities as revealed by 16S rDNA sequencing. Pol. J. Microbiol. 64, 29–36. doi: 10.33073/pjm-2015-004
Yáñez-Ruiz, D. R., Leticia, A., and Newbold, C. J. (2015). Manipulating rumen microbiome and fermentation through interventions during early life:a review. Front. Microbiol. 6:1133. doi: 10.3389/fmicb.2015.01133
Young, J. W. (1977). Gluconeogenesis in cattle: significance and methodology. J. Dairy Sci. 60, 1–15. doi: 10.3168/jds.S0022-0302(77)83821-6
Zened, A., Combes, S., Cauquil, L., Mariette, J., Klopp, C., Bouchez, O., et al. (2013). Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 83, 504–514. doi: 10.1111/1574-6941.12011
Keywords: high concentrate, rations, yak, rumen fermentation, rumen microorganisms
Citation: Pang K, Dai D, Yang Y, Wang X, Liu S, Huang W, Xue B, Chai S and Wang S (2022) Effects of high concentrate rations on ruminal fermentation and microbiota of yaks. Front. Microbiol. 13:957152. doi: 10.3389/fmicb.2022.957152
Edited by:
Mostafa Sayed A. Khattab, National Research Centre (Egypt), EgyptReviewed by:
Susana P. Alves, University of Lisbon, PortugalHossam H. Azzaz, National Research Centre (Egypt), Egypt
Copyright © 2022 Pang, Dai, Yang, Wang, Liu, Huang, Xue, Chai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shatuo Chai, Y2hhaXNoYXR1b0AxNjMuY29t; ShuXiang Wang, MTU4NTA1OTQ4OTBAMTYzLmNvbQ==
†These authors have contributed equally to this work