
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 02 September 2022
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.949272
 Betsy Anaid Peña-Ocaña1
Betsy Anaid Peña-Ocaña1 Yuki Hoshiko2
Yuki Hoshiko2 Mayel Silva-Flores1
Mayel Silva-Flores1 Toshinari Maeda2Israel Pérez-Torres3
Toshinari Maeda2Israel Pérez-Torres3 Rodolfo García-Contreras4
Rodolfo García-Contreras4 Wilbert Gutiérrez-Sarmiento5Luz Hernández-Esquivel1Álvaro Marín-Hernández1Rosina Sánchez-Thomas1
Wilbert Gutiérrez-Sarmiento5Luz Hernández-Esquivel1Álvaro Marín-Hernández1Rosina Sánchez-Thomas1 Emma Saavedra1José Salud Rodríguez-Zavala1
Emma Saavedra1José Salud Rodríguez-Zavala1 Ricardo Jasso-Chávez1*
Ricardo Jasso-Chávez1*A challenge in the study of gastrointestinal microbiota (GITm) is the validation of the genomic data with metabolic studies of the microbial communities to understand how the microbial networks work during health and sickness. To gain insights into the metabolism of the GITm, feces from healthy and sick rats with cancer were inoculated in a defined synthetic medium directed for anaerobic prokaryote growth (INC-07 medium). Significant differences between cultures of healthy and sick individuals were found: 1) the consumption of the carbon source and the enzyme activity involved in their catabolism (e.g., sucrase, lactase, lipases, aminotransferases, and dehydrogenases); 2) higher excretion of acetic, propionic, isobutyric, butyric, valeric, and isovaleric acids; 3) methane production; 4) ability to form biofilms; and 5) up to 500 amplicon sequencing variants (ASVs) identified showed different diversity and abundance. Moreover, the bowel inflammation induced by cancer triggered oxidative stress, which correlated with deficient antioxidant machinery (e.g., NADPH-producing enzymes) determined in the GITm cultures from sick individuals in comparison with those from control individuals. Altogether, the data suggested that to preserve the microbial network between bacteria and methanogenic archaea, a complete oxidation of the carbon source may be essential for healthy microbiota. The correlation of 16S rRNA gene metabarcoding between cultures and feces, as well as metabolomic data found in cultures, suggest that INC-07 medium may be a useful tool to understand the metabolism of microbiota under gut conditions.
Gastrointestinal microbiota (GITm) studies have gained relevance due to the findings that it can be associated with the integral health; indeed, changes in composition and microbial abundance of the microbiota have been to the development of some diseases (Malard et al., 2021). The microbial communities found in microbiota show symbiotic and mutualistic behaviors not only among them but also with host cells. Dysbiosis is a term that has been applied to an unhealthy condition that involves the imbalance, dysfunction, or alteration of the GITm, and it has been correlated with the development of different types of cancer, metabolic and heart diseases, infections, etc., (Wei et al., 2021). In this regard, there is evidence that some pathologies, such as inflammatory bowel disease or metabolic syndrome, provoke important alterations in the gastrointestinal microbiota composition since they occur with variations in the O2 concentration in the large intestine, which in turn induce changes in the expression levels of HIF-1α, which has a role in the regulation of iron absorption by epithelial cells (Zheng et al., 2015; Henson and Phalak, 2017). Therefore, under pathological conditions, the increase in Fe2+ and other essential ions, such as Cu2+ or Zn2+, in the colon may induce the production of reactive oxygen species (ROS) affecting the anaerobic microbiota, thus leading to dysbiosis (Kohzadi et al., 2017; Pinho et al., 2021; Sohrabi et al., 2021).
Different approaches have been used to study the composition of the GITm (Namsolleck et al., 2004); colonic fermentation is the most frequently used technique to grow microorganisms from fecal samples, followed by 16S RNA gene metabarcoding analysis (this technique manages to cultivate 20-25 microbial species), metagenomic (shotgun) analysis, and fluorescence in situ hybridization (Blatchford et al., 2015; Vogtmann and Goedert, 2016; Gutiérrez-Sarmiento et al., 2020; Barros de Medeiros et al., 2021; Zhou et al., 2021). Quantitative imaging has also been used to study the spatial organization of gut microbiota (Earle et al., 2015). Despite these important advances, integral biochemical studies (culturomics, proteomics, ionomics, and kinetics) to explore the physiology of the gastrointestinal microbiota are still scarce. The co-culture of bacteria and archaea extracted from extremophile environments has been successfully implemented in our laboratory. Crenarchaeota and Euryarchaeota, as well as Thermotogales and Aquificae, from the Archaea and Bacteria domains, respectively, were mainly identified. Furthermore, with the development in the culture approach, metabolic interactions were identified between prokaryotes (Peña-Ocaña et al., 2022).
In this work, cultivation (culturomics) of anaerobic bacteria and methanogenic archaea present in fecal samples from healthy control (CR) and hepatoma-bearing rats (HR) was achieved to 1) understand the changes associated in microbial abundance and composition of GITm in an animal model of disease and 2) deepen in the physiology of the microbiota community. The biomass obtained was used for 16S rRNA gene metabarcoding and metabolic and enzymatic analyses. Culturomics indicated impairment of the antioxidant response and loss of microbial diversity in GITm as a consequence of bowel inflammation.
For the experiment, 3-month-old female Wistar rats of 200–250 g weight were injected intraperitoneally with 4 mL ascites hepatoma AS30D cells, while control rats were not injected with any innocuous solution as previously reported (Marín-Hernández et al., 2020). The animals were kept in cages with food and fresh water ad libitum, and 8 days after inoculation, the hepatoma was fully developed. Feces were collected just before animals were euthanized. All animals were manipulated according to the guidelines NOM-062-ZOO-1999 (Norma Oficial Mexicana) for the care and use of laboratory animals, and the project received approval from the Instituto Nacional de Cardiología (CICUAL) with number INC/CICUAL/006/2021.
The culture medium was prepared based on the high-salt medium designed for methanogens, modified for the cultivation of anaerobic extremophiles, and redesigned for anaerobic gastrointestinal microbiota cells (Sowers et al., 1993; Peña-Ocaña et al., 2022). Because one of the aims of this study was to evaluate the activity of enzymes involved in organic matter degradation of the cultured microbiota, insoluble fiber was not added; instead, specific concentrations of a wide range of substrates, such as carbohydrates (mono- and disaccharides), soluble triacetylglycerol (TAG), organic acids, and amino acids, were added. The concentrations of carbon sources used were 5 mM glycerol, 10 mM TAG, 10 mM glucose, 5 mM lactose, 1 mM maltose, 0.5 mM DL-lactate, and 0.1 % (w/v) amino acids from enzymatic digested casein. To promote the growth of methanogens, 50 mM acetate and 50 mM methanol were added; in addition, 100 mM NaCl and mineral and vitamin solutions were added (Peña-Ocaña et al., 2022). The culture medium was buffered with 10 mM NaHCO3 and 25 mM HEPES (final pH 6.8–7.0). Except for acetate, all stock solutions of carbon sources were sterilized by filtration under anaerobic conditions and were added before cell inoculation. The culture medium was bubbled in an anaerobic chamber (COY Instruments, OH, USA) filled with 80:15:5 ratio of N2, CO2, and H2, respectively. After 2 h, 0.5 mM cysteine was added to reach complete anaerobiosis, using resazurin 0.01% (w/V) as redox indicator. To promote the growth of sulfate-reducing bacteria, 0.05 mM FeSO4 was added, instead of sulfide. Afterward, 50 mL of the culture medium were poured in 100-mL bottles and sealed with Teflon stoppers and aluminum collars and sterilized by autoclaving. This medium was further named INC-07 (patent registration in process; file MX/u/2022/000169).
To minimize the contact of the feces with air, feces were collected immediately after animal defecation, transferred to a sterile test tube containing 1 mL of INC-07 fresh medium, vortexed for a few seconds, and 0.2 mL of the vortexed sample was injected into culture bottles containing the INC-07 medium. The complete process lasted less than 20 s (Figure 1). It was determined that up to four subsequent transfer/passages using 2–3 mL inoculum, there was no loss in their ability to produce methane; therefore, the results showed here were from cultures with three passages. Microbiota cultures of control rats (CRC) and rats with hepatoma (HRC) were incubated in darkness at 37°C without shaking. Growth was monitored by measuring OD600; methane production was determined and used as a parameter of the complete oxidation of the carbon source consumed. As control, cultures of feces from rats before hepatoma inoculation were carried out, and no differences in methane production were attained between hepatoma and control rats; similarly, culture of feces from rats with hepatoma until 6 days after inoculation did not show differences in methane production with respect to control rats (not shown).
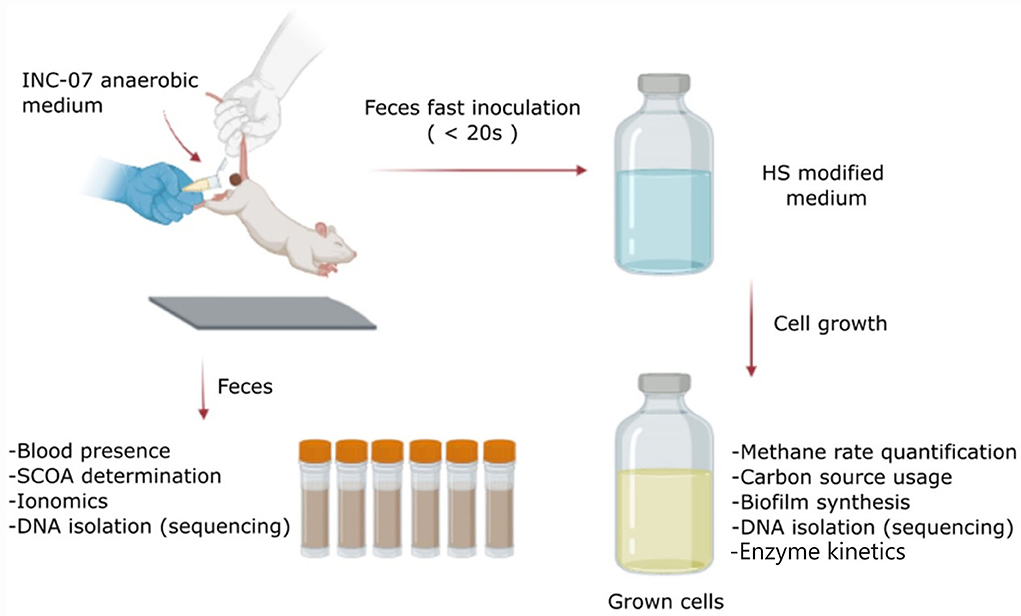
Figure 1. Scheme of the protocol followed in this study. Feces were collected from control and hepatoma rats just before euthanasia. The samples were used to determine changes in short-chain organic acids (SCOAs), ionomics, blood presence, and metagenomic analysis using 16S rRNA gene. On the other hand, a fraction of the feces was used for a quick inoculation in a medium designed for the growth of anaerobic microorganisms (INC-07 medium), to determine changes in abundance and variability of cultured species by analysis of 16S rRNA gene, biofilm synthesis, metabolites (SCOA production and carbon source consumption), enzyme kinetics, and ROS production. Created with BioRender.com.
Microbiological, biochemical, and 16S rRNA metabarcoding methodologies such as histopathological analysis carried out in this work are described in detail in the Supplementary material S1 text file.
Alpha diversity based on ASVs was calculated using the vegan R studio package (version 1.2.1335, Inc., Boston, MA, USA) (Oksanen et al., 2020). To determine the microbial rarefaction curves, Chao1, ACE, Simpson, and Shannon indexes, the analysis was performed based on the mean values of the ASV data in triplicate. Graphic visualization of the 16S rRNA metabarcoding results was carried out with a relative abundance of the domain, phyla, and genus levels > 0.05% from the ASV count. For other culturomic results, Student's t-test was carried out. P < 0.05 was interpreted as significant.
To analyze the intestinal environment from the control and hepatoma rats in which microbiota developed before cultivation, histological analysis of the large intestine was carried out. The control rats (CR) showed normal intestine histology, flat mucosal surface with abundant vertically oriented Lieberkühn crypts, and well-defined muscular zone (Supplementary Figures S1A,C). By contrast, the rats with hepatoma (HR) showed superficial erosion of mucosa with severe atrophy and irregular microvilli and thickened irregular muscular zone (Supplementary Figures S1B,D). The highly stained regions observed after colloidal iron staining suggested an increment of total iron in the intestine of HR (Supplementary Figures S1C,D). The fecal occult blood test was positive in feces from HR, which was 4.2-fold higher than that of feces from the CR, as judged by the intense blue color from the complex formed by chromogen and hemoglobin (Figure 2A). Hence, oxygen (bound to hemoglobin) and iron may be present in the intestinal lumen of the HR due to tissue disruption.
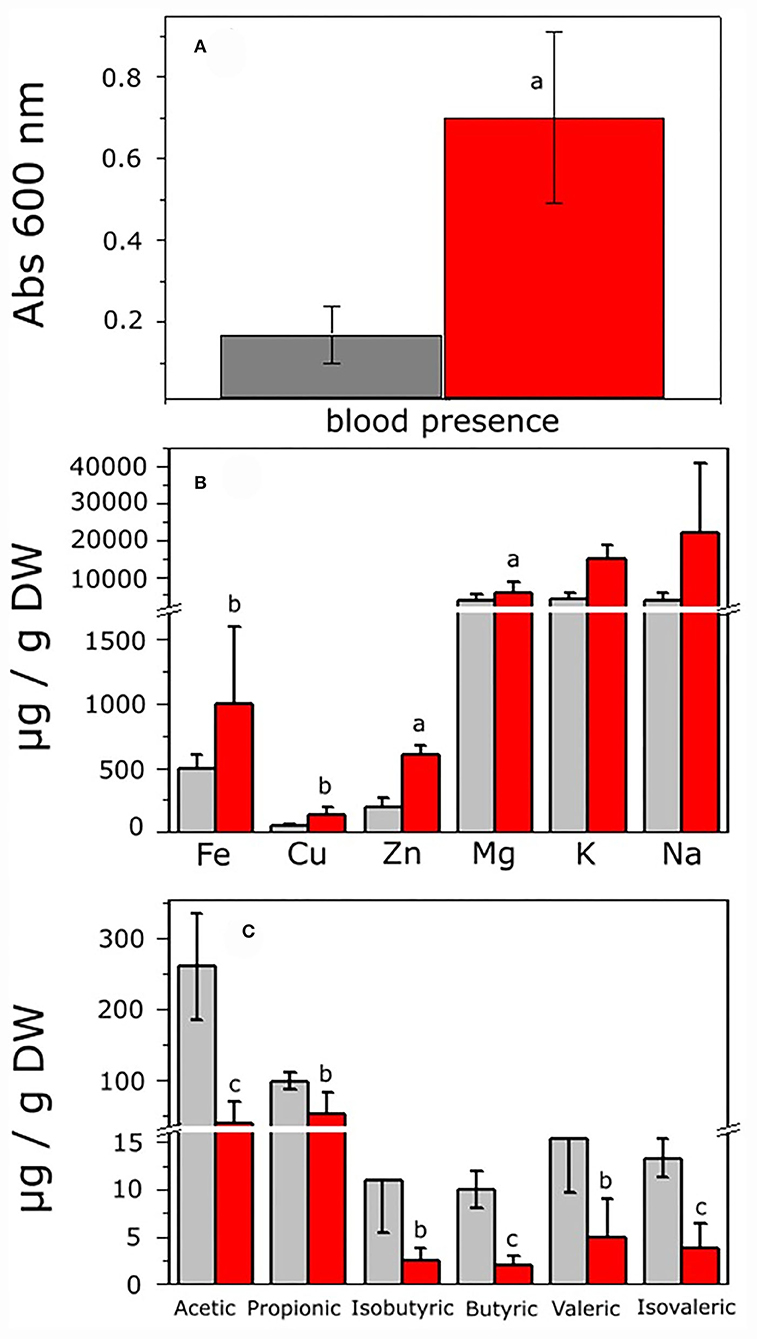
Figure 2. Analysis of feces from control and hepatoma-bearing rats. Feces were collected from control and rats with hepatoma and processed as indicated in the Methods section. (A) Detection of blood in feces as determined by the toluidine blue method. (B) Fe, Cu, Zn, Mg, K, and Na were determined by ICP-OES analysis. (C) SCOAs were extracted and acetic, propionic, isobutyric, butyric, valeric, and isovaleric acids quantified by gas chromatography, as described in the Supplementary materials. Values shown are the mean ± SD of six (for blood detection), five (for SCOA determination), and 12 (for ionomics) different rats, respectively. aP < 0.01; bP < 0.05; cP < 0.001 vs feces from control rats.
Disruption of the integrity of the epithelial barrier resulted in entrance of metals from blood to the lumen; the content of relevant pro-oxidant metals showed a significant increase of 2.3, 2.8, and 3 times higher for Fe, Cu, and Zn, respectively, in the same way, K increased 3.6 times, whereas Mg and Na remained roughly constant in HR vs. CR (Figure 2B). Hence, a potential oxidative stress condition might alter the GITm composition, leading to dysbiosis. To evaluate the latter, changes in the content of short-chain organic acids (SCOAs) produced by microbial metabolism as a parameter of microbiota diversity were determined in feces from the CR and HR. Acetic and propionic acids in the samples of CR were far more abundant than isobutyric, butyric, valeric, and isovaleric acids, but most of SCOAs showed a drastic decrement in samples from the HR (Figure 2C).
These data indicated that cancer provoked bowel inflammation and changes in Fe2+ levels in feces, possibly promoting a prooxidant environment on mostly the anaerobic microbiota cells present in the lumen of the intestine and a change in its composition in the diseased animals. However, further metabolic and biochemical analyses were impaired by the scarcity of the biological material found in the fecal samples, which made establishing a culturomic approach necessary to achieve such studies.
As described in Materials and Methods, the modified INC-07 medium was developed for culturing microbiota from fecal samples to obtain enough biological material for biochemical analyses in vitro (section Growth conditions of anaerobic microbiota). Figure 3 summarizes the results obtained from GITm grown in the INC-07 medium. No significant difference in cell growth was determined in microbiota of control rat fecal cultures (CRC) and hepatoma rat fecal cultures (HRC); however, methane production from GITm of HRC was completely diminished in comparison to CRC (Figure 3A). The CRC also produced 2.5–3 times more biofilm than HRC (Figure 3B). Lower biofilm synthesis in HRC suggested that the GITm of the rats suffered dysbiosis status due to cancer invasion.
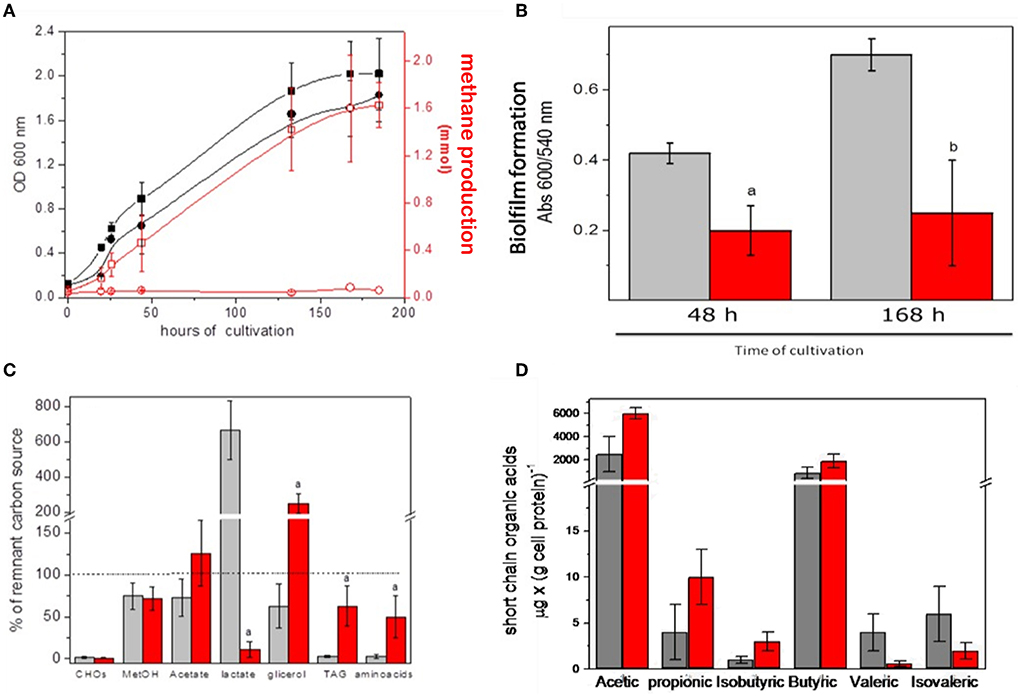
Figure 3. Gastrointestinal microbiota cultured in the INC-07 medium. (A) Growth curves and methane formation, (B) biofilm synthesis, (C) carbon source consumption, and (D) SCOAs excreted to the culture medium. CRC growth (black squares), methane production (open squares), HRC growth (red circles), and methane production (open circles) were determined throughout time. CRC are given in gray columns and HRC in red columns. Values are the mean ± SD of five different cultures. aP < 0.001 vs. control samples. bP < 0.05.
Changes in CO2 production/consumption were also found (not shown), suggesting active cell metabolism. The preference of CRC for carbon source consumption was carbohydrates equal to TAG and amino acids (90%) > methanol, acetate, and glycerol (25–40%) and overproduction of lactate; in turn, HRC consumed carbohydrates > lactate (90 %) > methanol, TAG, and amino acids (30–50%) and overproduced acetate and glycerol (Figure 3C). Metabolism of the GITm cultured in the INC-07 medium also produced SCOAs as previously determined in the fecal samples, although at different levels. Acetic acid was by far the most abundant acid determined in the CRC and HRC samples, but mostly due to external addition (as carbon source); the butyric acid content (which is probably the most important short-chain organic acid involved for human wellbeing) was abundant in both types of cultures. In turn, propionic and isobutyric acids were higher in the HRC than in the CRC, but valeric and isovaleric acids were higher in CRC (Figure 3D). These data suggested different metabolic performance due to diversity and abundance of cultured GITm between the healthy control and rats with cancer.
High yield of biomass allowed determination of enzyme activities involved in carbon catabolism in cellular fractions from both types of cultures (Table 1). Significantly higher enzyme activities of lipases and glycerol dehydrogenase were found in the CRC, which correlated with higher consumption of TAG and glycerol in these cultures. Activities of hexokinase (glucokinase), maltase (alpha-glucosidase), and lactase (beta-galactosidase) were similar between the CRC and HRC, which correlated with the efficient utilization of carbohydrates in both cultures. Acetate kinase, which catalyzes the reversible reaction (production of acetate and ATP in bacteria and acetyl-P and ADP in acetoclastic methanogenesis in archaea), was very active in both cultures. In turn, the CRC and HRC showed high activity of soluble NAD+-lactate dehydrogenase but lower activity of membrane-bound NAD+-independent lactate dehydrogenase. ALAT and ASAT transaminases, key enzymes in amino acid metabolism, were also present, with remarkably 15 times higher ALAT activity in the soluble fraction from the HRC.
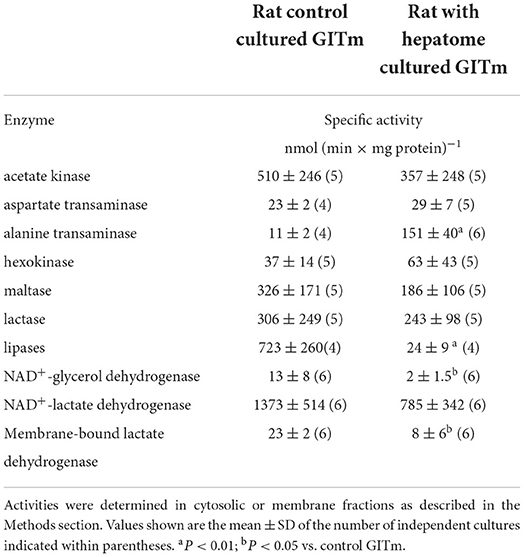
Table 1. Enzyme activities related to carbon sources consumption in GITm.
The occurrence of blood in the feces of HR suggested the presence of O2 and extra Fe in the lumen of the gastrointestinal tract, which may disturb the otherwise strict anaerobic environment. Indeed, when CRC microbiota were incubated for 3 and 7 days in a combination of air (1%) plus a mixture of ions (Fe2+, Cu2+, Zn2+, and K+), simulating the conditions found in the bowel of the HR, methane production was strongly affected (Supplementary Figure S2).
Furthermore, the rate of ROS production was determined in cell suspensions, showing that cells of the HRC produced two times more ROS than cells from the CRC; GR activity was also significantly higher in HRC cells, whereas GPx activity was similar respect to CRC cells (Table 2). However, activities of the NADPH-producing enzymes (IDH, ME, and Glc6PDH) were strongly affected in HRC cells, which may impede the efficient provision of reducing equivalents for ROS detoxification (Table 2).
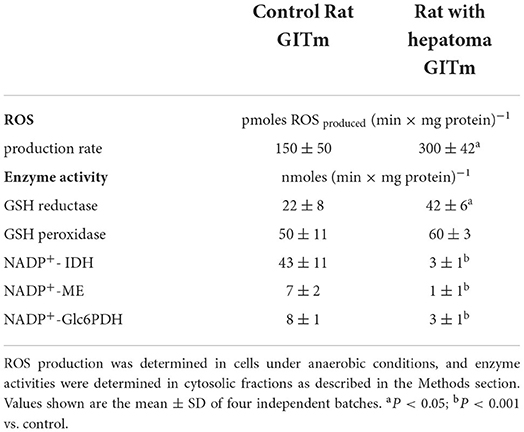
Table 2. ROS production and antioxidant enzymes in rat microbiota cultures.
Yields of DNA extracted showed non-significant differences between the CRC and HRC, whereas the amount of DNA recovered was 2–3-times higher in feces from CR than in feces from HR (n = 3, P < 0.01). In CR feces and CRC (after three cultivation transfers), 1,167 and 236 unique amplicon sequence variants (ASVs) were identified at 0.05% relative abundance, respectively, whereas in HR feces and HRC, 510 and 500 ASVs were identified, respectively. Relative abundances at different values are shown in Table A1. The ASV CR/CRC ratio was 4.92, and the HR/HRC ratio was 1.02. The number of ASVs of high-quality sequences and adequate length are shown in the rarefaction curves (Supplementary Figure S3). Asymptotes of CRC, HRC, and HR curves indicated a satisfactory identification of GITm, whereas non-saturating curves from CR samples indicate an incomplete GITm representation, which may improve with higher sequencing steps. Data supported the occurrence of taxonomic changes induced by the growth of the hepatoma cells. Although ASVs can detect small changes in some regions of 16S rRNA, they may also produce technical errors, especially when data quality is insufficient or the set of data is too small (Joos et al., 2020). To avoid over-interpretation of identified ASVs, the alpha diversity estimators, which describe the number of different species that should be found in each sample, were emphasized (Calle, 2019) (Figure 4). The observed index showed higher diversity of the microbial species in feces (CR and HR) than in samples from cultures (CRC and HRC) (P < 0.01). Absolute values for the CR and CRC were 696 ± 165 and 98 ± 2, respectively, and for HR and HRC, 278 ± 27and 61 ± 6, respectively (Figure 4A). CR showed a Chao1 index value of 1,217 ± 225, whereas CRC and HRC, as well as HR, showed a lower Chao1 index values, ranging from 300 to 500; a higher value suggested the presence of a higher number of rare microorganisms or with small representation. In agreement with the ACE, Simpson, and InvSimpson, the Shannon–Weaver index (H) indicated that the GITm analyzed from feces from the CR and HR is highly diverse (H = 4.5 ± 0.2 and H =3.63 ± 0.11, respectively); in turn, CRC microbiota was more diverse (H = 1.46 ± 0.15) than HRC microbiota (H = 0.56 ± 0.3). Therefore, after carefully revision, the final depuration of ASVs from CRC and HRC resulted in the identification of 98 and 61 different species (bacteria and archaea), respectively, as many ASVs were identified as the same species. In the same way, to estimate the overall difference of the microbial communities, we evaluated the beta diversity using the Jaccard index (Hu et al., 2018). The diversity identified in HR was 40% lower with respect to CR, and 79% lower for HRC with respect to CRC. This decrement in diversity in HRC could be a sign of the proliferation of the hepatoma cells affecting the intestinal microbiota. In turn, CRC diversity was 67% lower with respect to CR, as HRC was 58% lower as for HR. The latter is indicative of the efficiency of the INC-07 growth medium.
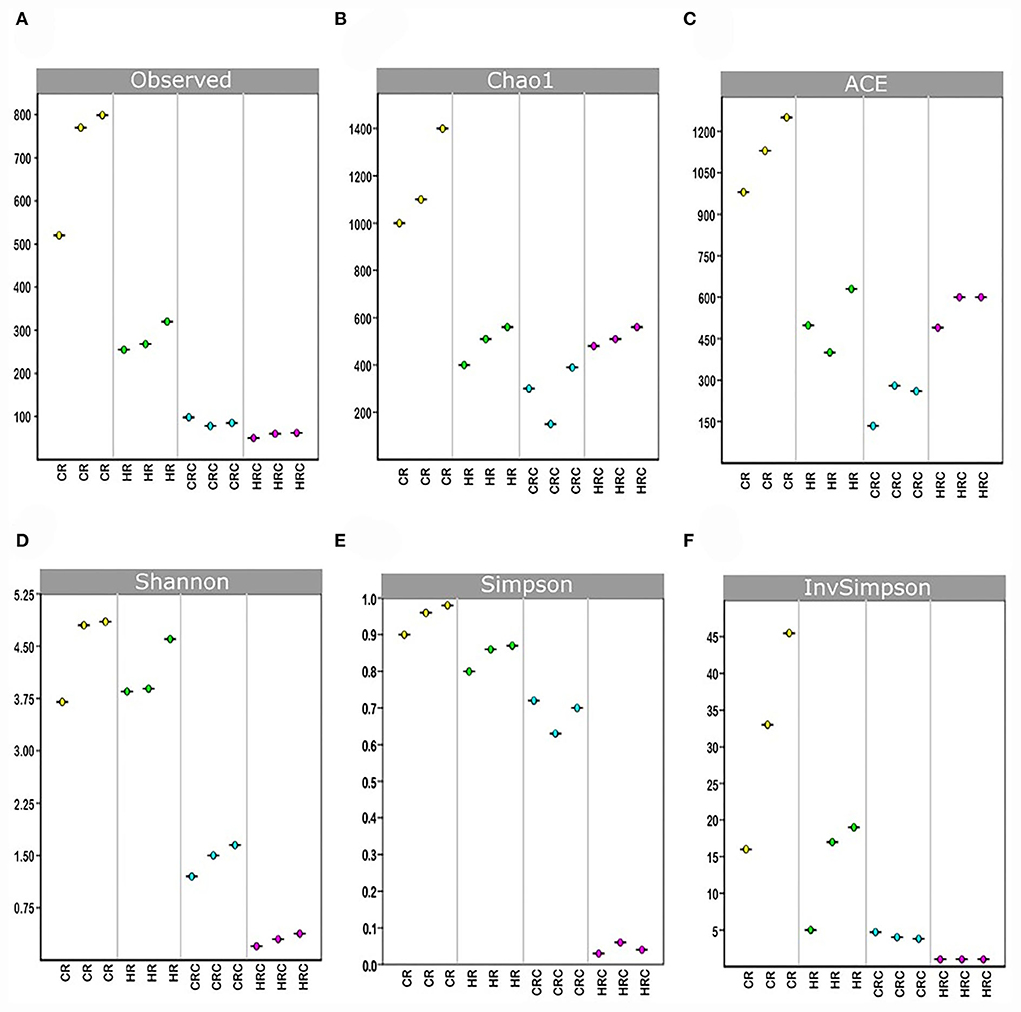
Figure 4. Indexes of richness. Observed (A), Chao1 (B), and ACE (C) and indexes of uniformity Shannon (D), Simpson (E), and 1/Simpson (F), were plotted based on the complete ASV data set determined in fecal samples from control (CR) and hepatoma (HR) rats, and their respective microbiota cultures CRC and HRC. P < 0.01, ANOVA unidirectional and the LSD multiple range test.
The use of the V3-V4 regions of the 16S rRNA gene to perform the taxonomic assignment of prokaryotes allowed for the simultaneous identification of bacteria and archaea in feces and its cultures. In control GITm (CR and CRC), archaea was represented by the phylum Euryarchaeota, with 2–4% abundance; by contrast, in HR and HRC, the absence of archaea and methane was evident (Figure 5A). Bacteria and Archaea were identified, with 39 genera distributed in 13 phyla (Figure 5B); the more abundant microbes in CR feces were Bacteroidetes 53% and Firmicutes 42%, and there was marginal presence of Actinobacteria, Fibrobacteres, and Spyrochetes (5%). The phyla mainly represented by the next genera (Figure 5C) were Prevotella (38.14%), Alloprevotella (5.65%), Lactobacillus (15%), Lachnospiraceae (7.7%), Roseburia (1.35%), Butyricicoccus (1%), and Ruminococcus (7%); two methanogenic archaea were identified: Methanobrevibacter and Methanomassiliicoccus (4%). In HR feces, phyla identified were 61% Bacteroidetes, 19% Firmicutes, 12% Proteobacteria, and 5% Patescibacteria, Actinobacteria, and Epsilonbacteraeota. The predominant genera identified were Bacteroides (28.7 %), Lachnospiraceae (10.7%), Alloprevotella (8.7%), Prevotella (3.35%), Tannerellaceae_unclassified (13.22%), Helicobacter (1.26%), Ruminococcus (3.8%), and Salmonella-Shigella (5.8 %). Data obtained from the cultures were as follows: in the CRC samples, phyla identified were Firmicutes (65%), Lachnospiraceae (41%), Streptococcus (15%), Coprococcus (7.57%), and Actinobacteria (35%), which was mainly represented by Bifidobacterium (34.6%). In HRC samples, Firmicutes represented mainly by Streptococcus (98%) prevailed, and Bacteroidetes, Actinobacteria, Bifidobacterium (0.78%), and Prevotella (0.23%) were the most abundant.

Figure 5. Microbial diversity in feces and culture samples. Microbiota derived from feces of control rats (CR), rats with hepatoma (HR), and direct from rats (R) and cultures (C). Microorganisms were analyzed by Domain (A) Euryarchaeota (dark blue) and Bacteria (cyan), by phylum (B), and by genus (C). Only microorganisms with a relative abundance >0.05 % were considered and evaluated by 16S rRNA analysis.
Comparison of the control and hepatoma conditions revealed that control microbiota showed beneficial prokaryotes such as Bifidobacterium, Faecalibacterium, Prevotella, Lactobacillus, Methanobrevibacter, and Methanomassiliicoccus of clinical relevance; therefore, they may be proposed as marker organisms of a healthy microbiota. By contrast, in the dysbiotic HR microbiota, their abundance was much lower, allowing bacteria considered potentially pathogenic such as Salmonella, Shigella, and Streptococcus to proliferate (Figure 6).
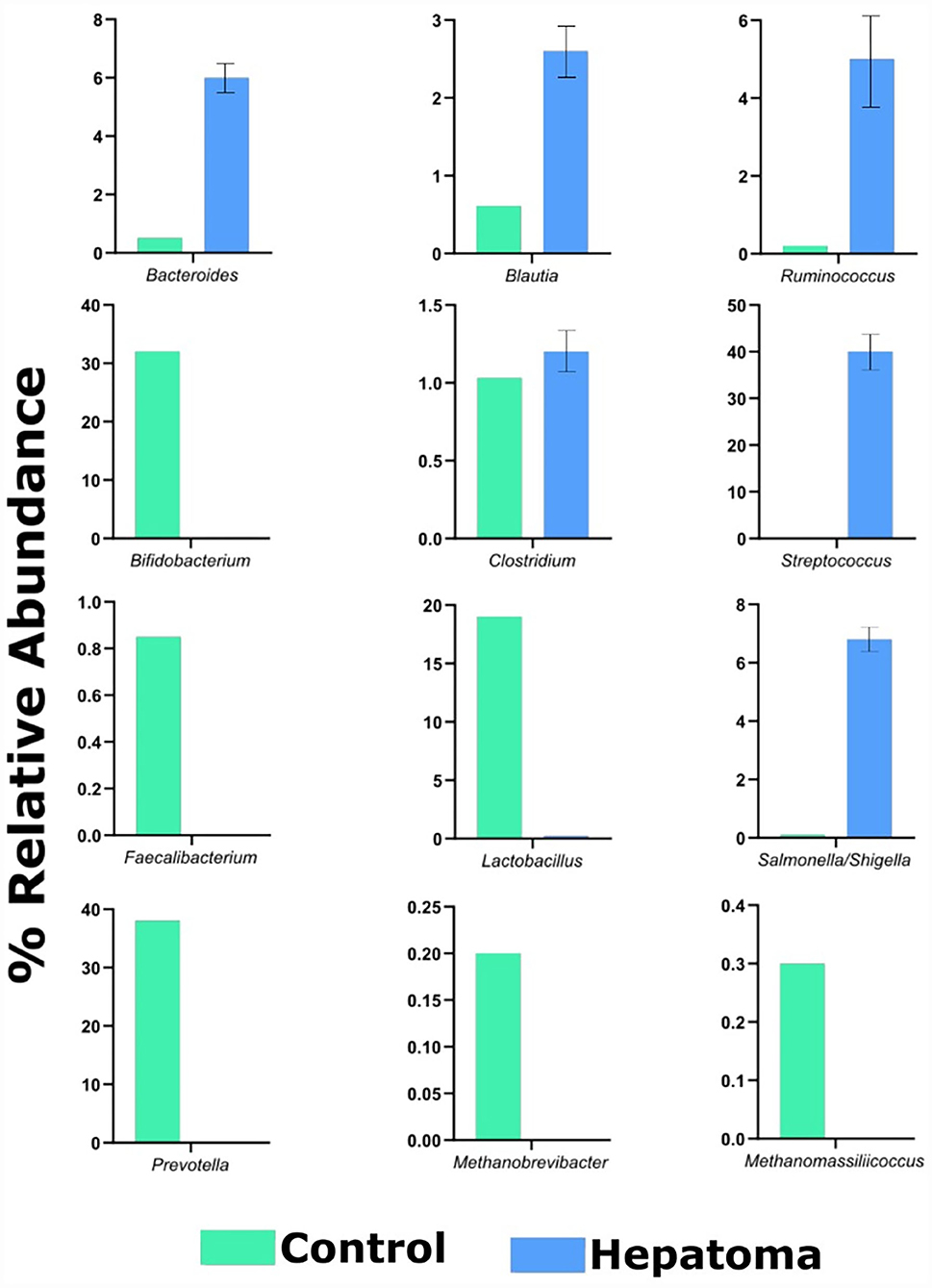
Figure 6. Bar plot of relative abundance of cultured microorganisms with clinical relevance. Analysis of relative abundances of beneficial and pathogenic ASVs was carried out in CRC (green columns) and HRC (blue columns). All species showed significance of P < 0.001 vs. control samples.
The microbial community abundance was used to predict the metabolic changes of the GITm due to the presence of the hepatoma in rats. For this purpose, a phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) was conducted. The predicted pathways in control conditions (CR and CRC), as well as in the samples from the rats with hepatoma (HR and HRC), were grouped in archaea and bacteria (Supplementary Figure S4).
A total of 131 pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) from the 16S rRNA functional ortholog sequences (KO) were analyzed. Fifty nine pathways of KO level 3 with a value higher to 1% of abundance were found, of these 38 pathways were gathered in pathways of KO level 2. These abundant KOs represented 60% of the total processes predicted, and they corresponded to the KOs for thiamine metabolism, Krebs cycle, and metabolism of amino acids in archaea, whereas for bacteria, the most abundant processes were for fatty acids, carbohydrates, and amino acid metabolism.
Finally, the canonical correspondence analysis (CCA) was carried out to evaluate the degree of correlation between metabolic capabilities established (consumption of carbon sources and production of short-chain organic acids, methane and CO2) and microbial communities found (most abundant taxonomy). A 69% relationship between microbial species and metabolic variables with a permutability analysis of 999 and a Monte Carlo permutation linear relationship test of 0.089 was determined. The two-dimensional map of axes 1 and 3 showed a strong relationship between the biological models of healthy and diseased states.
Hence, Faecalibacterium, Bifidobacterium, Methanomassiliicoccus, Methanobrevibacter, among others were identified in the healthy GITm, as well as high amounts of SCOAs such as butyric, valeric, and propionic acids and their isoforms. The INC-07 culture system favored methanogenic metabolism, observing a strong correlation with the production of methane and higher production of acetate and CO2 (Supplementary Figure S5). On the other hand, the HRC correlated with low consumption of carbohydrates and amino acids and a high consumption of the TAG available in the culture medium. Remarkably, undesirable bacteria such as Enterobacteria (Salmonella/Shigella), Helicobacter, Enterococcus, and Streptococcus in the disease model were abundant.
The AS30D ascites tumor cells are of epithelial origin, and the ascitic fluid becomes increasingly hemorrhagic after 5 days of tumor growth (Smith et al., 1970). In this work, rats after 8 days of hepatoma developed severe bowel inflammation, presence of blood in feces (with concomitant increase of O2), and an increase in pro-oxidants Fe, Cu, and Zn, as well as K, in feces. These events induced dysbiosis in the gastrointestinal tract of the rats due to the sensitivity of anaerobic microorganisms to oxidative conditions. The abundance of Bifidobacterium, Faecalibacterium, Prevotella, Lactobacillus, Methanobrevibacter, and Methanomassiliicoccus was highly present the feces from HR presumably due to oxidative stress. These prokaryotes are important in the production of acetate and propionate, which can be used by mitochondria from the intestinal epithelium cells (Dugas et al., 2018; Jackson and Theiss, 2020). In the lumen of the intestine, hyperbaric O2 alters the gut microbiota composition, mainly affecting the strict anaerobes, which have been cataloged as catalase-positive bacteria, although enzymatic activity was not determined (Albenberg et al., 2014).
Difficulties in co-cultivation of bacteria and archaea are the main limitation for further experimental approaches such as proteomic, metabolomic, kinetic, and fluxomic analyses, as well as metagenomics. These studies are required to understand the subtle balance between nutrient-consuming microorganisms and benefit producers; the disruption of this balance promotes dysbiosis (Albenberg et al., 2014; Zheng et al., 2015; Henson and Phalak, 2017; Kapoore et al., 2021; Xu et al., 2021; Zhang et al., 2021). The design of cultures may help evaluate the complex interaction between different microorganisms present in microbiota and the effect of external stressors such as oxygen (Smith et al., 2019). It has been reported that 1 μM O2 can induce inflammatory bowel disease in rats, resulting in a decrease in Faecalibacterium prausnitzii in the gut and a large increase in E. coli. The authors concluded that dysbiosis was consistent with disease progression and could be qualitatively predicted solely based on metabolic differences between the species (Henson and Phalak, 2017). However, this conclusion was based by analyzing a community of only three species. Studies on more complex microbiota communities are restricted due to complications in cultivation in vitro. A strategy to circumvent this limitation was one of the main objectives of the present study, which was successfully achieved by the design of the INC-07 medium, which allows growth of bacteria and archaea. Our results demonstrated the high susceptibility of the microbial network to an O2 pulse plus a mixture of Fe, Cu, and Zn, which dramatically decreased methane synthesis in control cultures.
It is reported that the presence of blood in feces increases the abundance of Bacteroides uniformis, Collinsella aerofaciens, Eggerthella lenta, and Clostridium symbiosum, while decreases the abundance of Prevotella copri, Coprococcuseu tactus and catus, Faecalibacterium prausnitzii, Roseburia faecis, Blautia obeum, Gemmiger formicilis, and Clostridium celatum (Chénard et al., 2020).
Methane production detected from GITm cultured in the INC-07 medium and the identification by metabarcoding of hydrogenotrophic Methanobrevibacter and methylotrophic Methanomassiliicoccus led to two main interpretations: i) H2 production by bacteria-metabolizing carbohydrates and lactate, and ii) sulfide production by sulfate-reducing bacteria since they thrived and produced enough sulfide for the successful growth of methanogens (Smith et al., 2019). The presence of methylotrophic methanogens may be relevant for the consumption of cardiotoxic methylamines from the gut or methanol present in the INC-07 medium, but H2 is required for their metabolism (Kurth et al., 2020). The successful co-culture of bacteria with archaea demonstrated the importance of methanogens in a healthy GITm and that their absence may generate dysbiosis (Djemai et al., 2021; Miller et al., 2021). Hence, secondary metabolites produced by the complete oxidation of the carbon source consumed were H2, CO2, sulfide, methane, and SCOAs.
The presence, induction, and activities of enzymes related to the catabolism of nutrients and xenobiotics and their changes due to illness have been determined not only in the serum, liver, and intestine from human but also in gastrointestinal microbiota. Acetate kinase, sucrase, lactase, and lipase have been extensively studied (Kushkevych, 2014; Lin et al., 2022; Zeinali et al., 2022). On the other hand, enzymes related to protection against oxidative stress have also been determined in microbiota studies (Yu et al., 2021; Panigrahi et al., 2022; Zhu et al., 2022). From GITm cultured in the INC-07 medium, it was feasible to prepare cytosolic and membrane fractions to identify key components of the enzymatic machinery involved in the degradation of carbon sources added, as well as those that contend against oxidative stress. Differences in catabolic enzymes and in the capacity of NADPH production, indispensable to maintain reduced GSH (or other thiol-containing molecules) to contend with ROS, were lower in dysbiotic GITm.
Butyrate-producing organisms proliferate in the INC-07 medium. This metabolite has been involved in i) the synthesis of some interleukins (IL6 and IL10) by healthy intestinal tissues; ii) the regulation of the levels of the brain-derived neurotrophic factor (BDNF), which plays a vital role in neuronal survival and growth; and iii) the synthesis of gamma-aminobutyric acid, which is transported to the brain to anxiety-like behavior (Romo-Araiza et al., 2018; Hao et al., 2019; Silva et al., 2020). Production of SCOAs (preferentially butyrate) was low in fecal samples of rats with hepatoma (HR), probably due to an imbalance in the abundance between butyrate-producing/consuming microorganisms. The family of Lachnospiraceae belonging to the Clostridiales order shows metabolic capacities for polysaccharides and short-chain alcohol and acid fermentation, such as acetate and butyrate. It has been reported that high levels of butyrate are related to a protective effect against colon cancer incidence (Meehan and Beiko, 2014; Ai et al., 2019). A low content of SCOAs in feces of HR might be related to growth of AS30D cells since it has been demonstrated that acetate and propionate are consumed by cancer cells (Rodríguez-Enríquez et al., 2021). Probably, many other enzymes and metabolic pathways can be studied in detail with these cultures without the necessity of purification from feces, which facilitates further experimental approaches.
Hepatoma induced a decrement of beneficial bacteria such as Prevotella, Faecalibacterium, Lachnospiraceae, Bifidobacterium, and Lactobacillus; these organisms have been proposed as essential in the maintenance of a healthy microbiota because they can improve glucose metabolism and digestion of complex fibers, and some of these represent biomarkers of a healthy intestine and are related to the modulation of the immune system (Meance et al., 2003; Kovatcheva-Datchary et al., 2015; Vuillermin et al., 2020). These organisms were substituted by genera Bacteroides, Parabacteroides (Tannerellaceae) Helicobacter, Streptococcus, and Salmonella-Shigella in microbiota of HR. Most of these bacteria are related to dysbiosis, infectious diseases, and cancer (Gao et al., 2018; Xie et al., 2019; Guo et al., 2020; Sha et al., 2020; Devi et al., 2021; Cao et al., 2022).
According to different authors, the core of the gastrointestinal microbiota is constituted preferentially by Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria, as well as other taxonomic divisions of minor abundance (Duda-Chodak et al., 2015). Interestingly, Kim et al. (2020) analyzed fecal samples from ~900 persons from Korea, where archaea represented 10% of all microorganisms with most members of the methane-producing archaea. In this work, in the GITm of healthy rats, Archaea of the Euryarchaeota phylum represented by the genera Methanomassiliicoccus and Methanobrevibacter constituted 2–4%. The hydrogenotrophic Methanobrevibacter is the main methane producer found in the digestive tract of humans with its colonization starting after birth, and it is also abundant in the gut microbiota in athletes, suggesting that in rat and human, methanogens are present in a presumably healthy individuals (Grine et al., 2017; Scheiman et al., 2019; Wosinska et al., 2019; Mohr et al., 2020).
Information about the relevance of archaea in the gastrointestinal microbial network is scarce. Most microbiota studies are focused on bacteria, and although some have begun the search and identity of eukaryotic microorganisms (i.e., fungi), the group of archaea is still largely ignored mainly due to technical difficulties in their experimental handling, due to their lower percentage than other groups, and because no archaeal pathogens have been identified (Pausan et al., 2019). Nevertheless, methane production and archaeal presence only in healthy rats suggested an important and unknown role for a healthy microbiota. Therefore, in the microbiota cultured in INC-07 medium developed here, methane is proposed as a biomarker of intestinal health. Parkar et al. (2021) in an in vitro hindgut model reported 11 microbial genera at the same percentage that it was used in this work (0.05%), whereas in the INC-07 system, 16 genera corresponding to 98–150 species were identified. When the relative abundance at 0.1% was carried out, 54 and 80 species were identified in the INC-07 medium for HRC and CRC, respectively, whereas Blatchford et al. (2015) reported 23 species in the colonic fermentation in vitro. Therefore, INC-07 medium was highly effective when comparing the recovered diversity indexes with other previously reported results obtained by in vitro colonic fermentation.
The INC-07 medium was a successful alternative for growing a huge diversity of microbial components from the gastrointestinal tract (H2 producers/consumers, sulfate reducers, methanogens), including those of clinical relevance. This cultivation system allowed for the validation (or contrasting) of predictive modeling and inferences in the metabolic dynamics derived from the use of the 16S rRNA. Thanks to the wide diversity of microorganisms cultured, it was possible to carry out metabolic and kinetic analyses in isolated cytosolic fractions. This study provides a new approach to evaluate the relationship between diseases and the gastrointestinal microbiota and the development of strategies, which allow for the restoration of a healthy microbiota. In the same way, this cultivation system may be highly effective for studying the effects of factors that disturb the delicate equilibrium of the gastrointestinal microbiota.
The datasets presented in this study can be found online at https://www.ncbi.nlm.nih.gov/ with the accession number KFUR00000000.
The animal study was reviewed and approved by the guidelines NOM-062-ZOO-1999 (Norma Oficial Mexicana) for the care and use of laboratory animals and the project received approval from Instituto Nacional de Cardiología CICUAL with number INC/CICUAL/006/2021.
RJ-C: conceptualization and methodology. RJ-C, ÁM-H, TM, and RG-C: resources. RJ-C, BA-PO, YH, MS-F, IP-T, WG-S, LH-E, ÁM-H, and RS-T: investigation. R-JC, BA-PO, JS-RZ, ÁM-H, ES, IP-T, RS-T, RG-C, LH-E, and WG-S: formal analysis and data interpretation. RJ-C, BA-PO, YH, MS-F, JS-RZ, ÁM-H, RG-C, RS-T, and ES: original draft. RJ-C, BA-PO, JS-RZ, TM, and ES: writing—review and editing. All authors confirm that the work reported in this article and have approved the final submitted version.
This work was supported by grants [CPC2022 #320299 (BA-PO) and CONACYT CB 2017–2018 # A1-S-8530 and PAPITT UNAM # IN214218 (RG-C) and # A1-S-40481 (ÁM-H)] from CONACyT, Mexico, and by the Sakura Science Exchange Program, Japan Science and Technology Agency [# P2020A0228092 (TM)]. MS-F was supported by CONACYT-Mexico fellowship (No. 1084917) from the program 000237-Maestría en Ciencias Bioquímicas, Facultad de Química, UNAM.
We thank Mr. Fernando Limones-Martínez for his technical assistance and Mr. Benito Chávez Rentería for his technical support in the histological studies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.949272/full#supplementary-material
Supplementary Figure S1. Histological section of control (A,C) and with hepatoma (B,D), rat colon stained with hematoxylin-eosin (A,B) or with colloidal iron (C,D). Image analysis was made at 20X. Figure is representative of a total of 6 rat samples.
Supplementary Figure S2. Effect of stressors in methane production in cultured microbiota. Control microbiota was cultured in the absence (control, white column) or in the presence of a metal mixture (0.5 mM FeSO4, 0.1 mM CuSO4, 1 mM ZnCl2 and 50 mM KCl) plus 1% (v/v) of O2. Methane production was determined at day 3 (light gray column) and day 7 (gray column) of growth. Values shown are the mean ± SD of 3 independent cultures. a P < 0.001 vs. control cultures.
Supplementary Figure S3. Rarefaction plot of amplicon sequences variants (ASVs) from CRC, CR, HRC, and HR. The x axis indicates the number of sequences and y axis indicates the observed microorganisms in ASVs. Each curve represents a different sample replicate.
Supplementary Figure S4. Heat map of metabolic function predicted by archaea (A) and bacteria (B) based on community phylogenetic research by reconstructing Unobserved State Analysis (PICRUSt) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). The rows indicate the KEGG level 2 pathway (> 1% abundance) and the columns show the hierarchical groups of the in vivo control model (CR), the AS30D hepatoma in vivo model (HR), the control culture (CRC) and the culture of the microbiota of rats with hepatoma (HRC).
Supplementary Figure S5. Canonical correspondence analysis (CCA) of gut microbial communities and metabolic parameters ASV with relative abundance higher than 0.01% and which is also a marker for microorganisms of intestinal metabolism were applied for CCA, as environmental conditions, the metabolic variables of organic acids, CO2 and methane production, and carbon sources consumption were considered.
SCOAs, short-chain organic acids; CR, control rats; HR, rats with hepatoma; CRC, fecal cultures of control rats; HRC, fecal cultures of rats with hepatoma; GITm, gastrointestinal microbiota; TAG, triacetylglycerol; GPX, glutathione peroxidase; GR, glutathione reductase; NADP+-IDH, NADP+-isocitrate dehydrogenase; NADP+-ME, NADP+-malic enzyme; NADP+-Glc6PDH, NADP+-glucose-6-phosphate dehydrogenase; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Ai, D., Pan, H., Li, X., Gao, Y., Liu, G., and Xia, L. C. (2019). Identifying gut microbiota associated with colorectal cancer using a zero-inflated lognormal model. Front. Microbiol. 10, 826. doi: 10.3389/fmicb.2019.00826
Albenberg, L., Esipova, T. V., Judge, C. P., Bittinger, K., Chen, J., Laughlin, A., et al. (2014). Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–1063.E8. doi: 10.1053/j.gastro.2014.07.020
Barros de Medeiros, V. P., Salgaço, M. K., Pimentel, T. C., Rodrigues da Silva, T. C., Sartoratto, A., Lima, M. D. S., et al. (2021). Spirulina platensis biomass enhances the proliferation rate of Lactobacillus acidophilus 5 (La-5) and combined with La-5 impact the gut microbiota of medium-age healthy individuals through an in vitro gut microbiome model. Food Res. Int. 154, 110880. doi: 10.1016/j.foodres.2021.110880
Blatchford, P., Stoklosinski, H., Walton, G., Swann, J., Gibson, G., Gearry, R., et al. (2015). Kiwifruit fermentation drives positive gut microbial and metabolic changes irrespective of initial microbiota composition. Bioact. Carbohydr. Diet. Fibre. 6, 37–45. doi: 10.1016/j.bcdf.2015.07.001
Calle, M. L. (2019). Statistical analysis of metagenomics data. Genomics Inform. 17, e6. doi: 10.5808/GI.2019.17.1.e6
Cao, D., Pang, M., Wu, D., Chen, G., Peng, X., Xu, K., et al. (2022). Alterations in the gut microbiota of tibetan patients with Echinococcosis. Front. Microbiol. 13, 860909. doi: 10.3389/fmicb.2022.860909
Chénard, T., Malick, M., Dub,é, J., and Mass,é, E. (2020). The influence of blood on the human gut microbiome. BMC Microbiol. 20, 44. doi: 10.1186/s12866-020-01724-8
Devi, T. B., Devadas, K., George, M., Gandhimathi, A., Chouhan, D., Retnakumar, R. J., et al. (2021). Low Bifidobacterium abundance in the lower gut microbiota is associated with Helicobacter pylori-related gastric ulcer and gastric cancer. Front. Microbiol. 12, 631140. doi: 10.3389/fmicb.2021.631140
Djemai, K., Drancourt, M., and Tidjani-Alou, M. (2021). Bacteria and methanogens in the human microbiome: a review of syntrophic interactions. Microb. Ecol. 83, 536–554. doi: 10.1007/s00248-021-01796-7
Duda-Chodak, A., Tarko, T., Satora, P., and Sroka, P. (2015). Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: a review. Eur. J. Nutr. 54, 325–341. doi: 10.1007/s00394-015-0852-y
Dugas, L. R., Lie, L., Plange-Rhule, J., Bedu-Addo, K., Bovet, P., Lambert, E. V., et al. (2018). Gut microbiota, short chain fatty acids, and obesity across the epidemiologic transition: the METS-microbiome study protocol. BMC Public Health. 18, 978. doi: 10.1186/s12889-018-5879-6
Earle, K. A., Billings, G., Sigal, M., Lichtman, J. S., Hansson, G. C., Elias, J. E., et al. (2015). Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe. 18, 478–488. doi: 10.1016/j.chom.2015.09.002
Gao, J. J., Zhang, Y., Gerhard, M., Mejias-Luque, R., Zhang, L., Vieth, M., et al. (2018). Association between gut microbiota and Helicobacter pylori-related gastric lesions in a high-risk population of gastric cancer. Front. Cell. Infect. Microbiol. 8, 202. doi: 10.3389/fcimb.2018.00202
Grine, G., Boualam, M. A., and Drancourt, M. (2017). Methanobrevibacter smithii, a methanogen consistently colonising the newborn stomach. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2449–2455. doi: 10.1007/s10096-017-3084-7
Guo, Y., Zhang, Y., Gerhard, M., Gao, J. J., Mejias-Luque, R., Zhang, L., et al. (2020). Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut 69, 1598–1607. doi: 10.1136/gutjnl-2019-319696
Gutiérrez-Sarmiento, W., Sáyago-Ayerdi, S. G., Goñi, I., Gutiérrez-Miceli, F. A., Abud-Archila, M., Rejón-Orantes, J. C., et al. (2020). Changes in intestinal microbiota and predicted metabolic pathways during colonic fermentation of mango (Mangifera indica L.)-based bar indigestible fraction. Nutrients 12, 683. doi: 10.3390/nu12030683
Hao, Z., Wang, W., Guo, R., and Liu, H. (2019). Faecalibacterium prausnitzii (ATCC 27766) has preventive and therapeutic effects on chronic unpredictable mild stress-induced depression-like and anxiety-like behavior in rats. Psychoneuroendocrinology 104, 132–142. doi: 10.1016/j.psyneuen.2019.02.025
Henson, M. A., and Phalak, P. (2017). Microbiota dysbiosis in inflammatory bowel diseases: in silico investigation of the oxygen hypothesis. BMC Syst. Biol. 11, 145. doi: 10.1186/s12918-017-0522-1
Hu, Y. L., Pang, W., Huang, Y., Zhang, Y., and Zhang, C. J. (2018). The gastric microbiome is perturbed in advanced gastric adenocarcinoma identified through shotgun metagenomics. Front. Cell. Infect. Microbiol. 8, 433. doi: 10.3389/fcimb.2018.00433
Jackson, D. N., and Theiss, A. L. (2020). Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes. 11, 285–304. doi: 10.1080/19490976.2019.1592421
Joos, L., Beirinckx, S., Haegeman, A., Debode, J., Vandecasteele, B., Baeyen, S., et al. (2020). Daring to be differential: metabarcoding analysis of soil and plant-related microbial communities using amplicon sequence variants and operational taxonomical units. BMC Genomics 21, 733. doi: 10.1186/s12864-020-07126-4
Kapoore, R. V., Padmaperuma, G., Maneein, S., and Vaidyanathan, S. (2021). Co-culturing microbial consortia: approaches for applications in biomanufacturing and bioprocessing. Crit. Rev. Biotechnol. 42, 46–72. doi: 10.1080/07388551.2021.1921691
Kim, J. Y., Whon, T. W., Lim, M. Y., Kim, Y. B., Kim, N., Kwon, M. S., et al. (2020). The human gut archaeome: identification of diverse haloarchaea in Korean subjects. Microbiome 8, 114. doi: 10.1186/s40168-020-00894-x
Kohzadi, S., Sheikhesmaili, F., Rahehagh, R., Parhizkar, B., Ghaderi, E., Loqmani, H., et al. (2017). Evaluation of trace element concentration in cancerous and non-cancerous tissues of human stomach. Chemosphere 184, 747–752. doi: 10.1016/j.chemosphere.2017.06.071
Kovatcheva-Datchary, P., Nilsson, A., Akrami, R., Lee, Y. S., De Vadder, F., Arora, T., et al. (2015). Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 22, 971–982. doi: 10.1016/j.cmet.2015.10.001
Kurth, J. M., Op den Camp, H. J. M., and Welte, C. U. (2020). Several ways one goal-methanogenesis from unconventional substrates. Appl. Microbiol. Biotechnol. 104, 6839–6854. doi: 10.1007/s00253-020-10724-7
Kushkevych, I. V. (2014). Acetate kinase activity and kinetic properties of the enzyme in Desulfovibrio piger Vib-7 and Desulfomicrobium sp. Rod-9 intestinal bacterial strains. Open Microbiol. J. 8, 138-43. doi: 10.2174/1874285801408010138
Lin, G., Liu, Q., Wang, L., Li, H., Zhao, J., Zhang, H., et al. (2022). The comparative analysis of genomic diversity and genes involved in carbohydrate metabolism of eighty-eight Bifidobacterium pseudocatenulatum isolates from different niches of China. Nutrients. 14, 2347. doi: 10.3390/nu14112347
Malard, F., Dore, J., Gaugler, B., and Mohty, M. (2021). Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 14, 547–554. doi: 10.1038/s41385-020-00365-4
Marín-Hernández, Á., Gallardo-Pérez, J. C., Reyes-García, M. A., Sosa-Garrocho, M., Macías-Silva, M., Rodríguez-Enríquez, S., et al. (2020). Kinetic modeling of glucose central metabolism in hepatocytes and hepatoma cells. Biochim. Biophys. Acta Gen. Subj. 1864, 129687. doi: 10.1016/j.bbagen.2020.129687
Meance, S., Cayuela, C., Raimondi, A., Turchet, P., Lucas, C., and Antoine, J. M. (2003). Recent advances in the use of functional foods: effects of the commercial fermented milk with Bifidobacterium animalis Strain DN-173 010 and yoghurt strains on gut transit time in the Elderly. Microb. Ecol. Health Dis. 15, 15–22. doi: 10.1080/08910600310015565
Meehan, C. J., and Beiko, R. G. (2014). A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 6, 703–713. doi: 10.1093/gbe/evu050
Miller, B. M., Liou, M. J., Lee, J. Y., and Bäumler, A. J. (2021). The longitudinal and cross-sectional heterogeneity of the intestinal microbiota. Curr. Opin. Microbiol. 63, 221–230. doi: 10.1016/j.mib.2021.08.004
Mohr, A. E., Jäger, R., Carpenter, K. C., Kerksick, C. M., Purpura, M., Townsend, J. R., et al. (2020). The athletic gut microbiota. J. Int. Soc. Sports Nutr. 17, 24. doi: 10.1186/s12970-020-00353-w
Namsolleck, P., Thiel, R., Lawson, P., Holmstrøm, K., Rajilic, M., Vaughan, E. E., et al. (2004). Molecular methods for the analysis of gut microbiota. Microb. Ecol. Health Dis. 16, 71–85. doi: 10.1080/08910600410032367
Oksanen, J., Simpson, G. L., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., et al. (2020). Vegan: community ecology package. R package version 2.5–6. Available online at: https://cran.r-project.org/package=vegan (accessed November 27, 2021).
Panigrahi, M. K., Kaliaperumal, V., Akella, A., Venugopal, G., and Ramadass, B. (2022). Mapping microbiome-redox spectrum and evaluating Microbial-Redox Index in chronic gastritis. Sci. Rep. 12, 8450. doi: 10.1038/s41598-022-12431-x
Parkar, S. G., Frost, J., Rosendale, D., Stoklosinski, H. M., Jobsis, C. M. H., Hedderley, D. I., et al. (2021). The sugar composition of the fibre in selected plant foods modulates weaning infants' gut microbiome composition and fermentation metabolites in vitro. Sci. Rep. 11, 9292. doi: 10.1038/s41598-021-88445-8
Pausan, M. R., Csorba, C., Singer, G., Till, H., Schöpf, V., Santigli, E., et al. (2019). Exploring the archaeome: detection of archaeal signatures in the human body. Front. Microbiol. 10, 2796. doi: 10.3389/fmicb.2019.02796
Peña-Ocaña, B. A., Ovando-Ovando, C. I., Puente-Sánchez, F., Tamames, J., Servín-Garcidueñas, L. E., González-Toril, E., et al. (2022). Metagenomic and metabolic analyses of poly-extreme microbiome from an active crater volcano lake. Environ. Res. 203, 111862. doi: 10.1016/j.envres.2021.111862
Pinho, S. C. M., Faria, M. A., Melo, A., Pinto, E., Almeida, A., Alves, R., et al. (2021). Effect of skimmed milk on intestinal tract: Prevention of increased reactive oxygen species and nitric oxide formation. Int Dairy J. 118:105046. doi: 10.1016/j.idairyj.2021.105046
Rodríguez-Enríquez, S., Robledo-Cadena, D. X., Gallardo-Pérez, J. C., Pacheco-Velázquez, S. C., Vázquez, C., Saavedra, E., et al. (2021). Acetate promotes a differential energy metabolic response in human HCT 116 and COLO 205 colon cancer cells impacting cancer cell growth and invasiveness. Front. Oncol. 11, 697408. doi: 10.3389/fonc.2021.697408
Romo-Araiza, A., Gutiérrez-Salmeán, G., Galván, E. J., Hernández-Fausto, M., Herrera-López, G., Romo-Parra, H., et al. (2018). Probiotics and prebiotics as a therapeutic strategy to improve memory in a model of middle-aged rats. Front. Aging Neurosci. 10, 416. doi: 10.3389/fnagi.2018.00416
Scheiman, J., Luber, J. M., Chavkin, T. A., MacDonald, T., Tung, A., Loc-Duyen, P., et al. (2019). Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat. Med. 25, 1104–1109. doi: 10.1038/s41591-019-0485-4
Sha, S., Ni, L., Stefil, M., Dixon, M., and Mouraviev, V. (2020). The human gastrointestinal microbiota and prostate cancer development and treatment. Investig. Clin. Urol. 61, S43–S50. doi: 10.4111/icu.2020.61.S1.S43
Silva, Y. P., Bernardi, A., and Frozza, R. L. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 11, 25. doi: 10.3389/fendo.2020.00025
Smith, D. F., Walborg, E. F. Jr., and Chang, J. P. (1970). Establishment of a transplantable ascites variant of a rat hepatoma induced by 3'-Methyl-1-4-dimethylamino azobenzene. Cancer Res. 30, 2306–2309.
Smith, N. W., Shorten, P. R., Altermann, E. H., Roy, N. C., and McNabb, W. C. (2019). Hydrogen cross-feeders of the human gastrointestinal tract. Gut Microbes. 10, 270–288. doi: 10.1080/19490976.2018.1546522
Sohrabi, M., Nikkhah, M., Sohrabi, M., Farimani, A. R., Shahi, M. M., Ziaie, H., et al. (2021). Evaluating tissue levels of the eight trace elements and heavy metals among esophagus and gastric cancer patients: A comparison between cancerous and non-cancerous tissues. J. Trace Elem. Med. Biol. 68, 126761. doi: 10.1016/j.jtemb.2021.126761
Sowers, K. R., Boone, J. E., and Gunsalus, R. P. (1993). Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl. Environ. Microbiol. 59, 3832–3839. doi: 10.1128/aem.59.11.3832-3839.1993
Vogtmann, E., and Goedert, J. J. (2016). Epidemiologic studies of the human microbiome and cancer. Br. J. Cancer 114, 237–242. doi: 10.1038/bjc.2015.465
Vuillermin, P. J., O'Hely, M., Collier, F., Allen, K. J., Tang, M. L. K., Harrison, L. C., et al. (2020). Maternal carriage of Prevotella during pregnancy associates with protection against food allergy in the offspring. Nat. Commun. 11, 1452. doi: 10.1038/s41467-020-14552-1
Wei, S., Bahl, M. I., Baunwall, S. M. D., Hvas, C. L., and Licht, T. R. (2021). Determining gut microbial dysbiosis: a review of applied indexes for assessment of intestinal microbiota imbalances. Appl. Environ. Microbiol. 87, e00395–e00321. doi: 10.1128/AEM.00395-21
Wosinska, L., Cotter, P. D., O'Sullivan, O., and Guinane, C. (2019). The potential impact of probiotics on the gut microbiome of athletes. Nutrients 11, 2270. doi: 10.3390/nu11102270
Xie, X., He, Y., Li, H., Yu, D., Li, N., Sun, T., et al. (2019). Effects of prebiotics on immunologic indicators and intestinal microbiota structure in perioperative colorectal cancer patients. Nutrition 61, 132–142. doi: 10.1016/j.nut.2018.10.038
Xu, K., Gao, X., Xia, G., Chen, M., Zeng, N., Wang, S., et al. (2021). Rapid gut dysbiosis induced by stroke exacerbates brain infarction in turn. Gut 70, 1486–1494. doi: 10.1136/gutjnl-2020-323263
Yu, Y., Zhu, H., Shen, M., Yu, Q., Chen, Y., and Xie, J. (2021). Sulfation modification enhances the intestinal regulation of Cyclocarya paliurus polysaccharides in cyclophosphamide-treated mice via restoring intestinal mucosal barrier function and modulating gut microbiota. Food Funct. 12, 12278–12290. doi: 10.1039/D1FO03042F
Zeinali, L. I., Giuliano, S., Lakshminrusimha, S., and Underwood, M. A. (2022). Intestinal dysbiosis in the infant and the future of lacto-engineering to shape the developing intestinal microbiome. Clin. Ther. 44, 193–214.e1. doi: 10.1016/j.clinthera.2021.11.005
Zhang, J., Huang, Y. J., Yoon, J. Y., Kemmit, J., Wright, C., Schneider, K., et al. (2021). Primary human colonic mucosal barrier crosstalk with super oxygen-sensitive Faecalibacterium prausnitzii in continuous culture. Med. 2, 74–98.e9. doi: 10.1016/j.medj.2020.07.001
Zheng, L., Kelly, C. J., and Colgan, S. P. (2015). Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 309, C350–360. doi: 10.1152/ajpcell.00191.2015
Zhou, Q., Fu, X., Dhital, S., Zhai, H., Huang, Q., and Zhang, B. (2021). In vitro fecal fermentation outcomes of starch-lipid complexes depend on starch assembles more than lipid type. Food Hydrocolloids 120, 106941. doi: 10.1016/j.foodhyd.2021.106941
Zhu, Y. Y., Thakur, K., Feng, J. Y., Zhang, J. G., Hu, F., Cespedes-Acuña, C. L., et al. (2022). Riboflavin bioenriched soymilk alleviates oxidative stress-mediated liver injury, intestinal inflammation, and gut microbiota modification in b2 depletion-repletion mice. J. Agric. Food Chem. 70, 3818–3831. doi: 10.1021/acs.jafc.2c00117
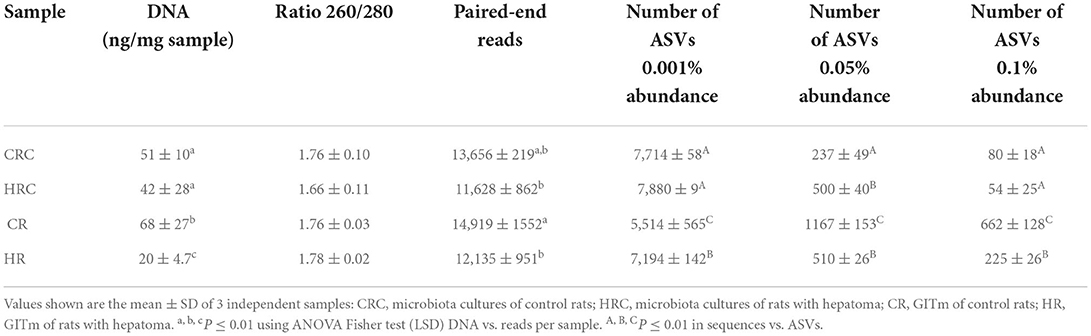
Table A1. Illumina MiSeq sequencing summary and amplicon sequencing variants (ASVs) identified per treatment.
Keywords: anaerobic culture, ionomics, methane production, oxidative stress, biofilm, metabarcoding, kinetic analysis
Citation: Peña-Ocaña BA, Hoshiko Y, Silva-Flores M, Maeda T, Pérez-Torres I, García-Contreras R, Gutiérrez-Sarmiento W, Hernández-Esquivel L, Marín-Hernández Á, Sánchez-Thomas R, Saavedra E, Rodríguez-Zavala JS and Jasso-Chávez R (2022) Cultivation of gastrointestinal microbiota in a new growth system revealed dysbiosis and metabolic disruptions in carcinoma-bearing rats. Front. Microbiol. 13:949272. doi: 10.3389/fmicb.2022.949272
Received: 20 May 2022; Accepted: 01 August 2022;
Published: 02 September 2022.
Edited by:
Emanuel Vamanu, University of Agricultural Sciences and Veterinary Medicine, RomaniaReviewed by:
Ankur Naqib, Rush University, United StatesCopyright © 2022 Peña-Ocaña, Hoshiko, Silva-Flores, Maeda, Pérez-Torres, García-Contreras, Gutiérrez-Sarmiento, Hernández-Esquivel, Marín-Hernández, Sánchez-Thomas, Saavedra, Rodríguez-Zavala and Jasso-Chávez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ricardo Jasso-Chávez, cmljYXJkby5qYXNzb0BjYXJkaW9sb2dpYS5vcmcubXg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.