
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 29 July 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.948633
 Xiaoyu Tang1†Zhen Yang1†Ke Dai1†Geyan Liu1
Xiaoyu Tang1†Zhen Yang1†Ke Dai1†Geyan Liu1 Yung-Fu Chang2Xinwei Tang1Kang Wang1Yiwen Zhang1Bangdi Hu1
Yung-Fu Chang2Xinwei Tang1Kang Wang1Yiwen Zhang1Bangdi Hu1 Sanjie Cao1Xiaobo Huang1
Sanjie Cao1Xiaobo Huang1 Qigui Yan1Rui Wu1
Qigui Yan1Rui Wu1 Qin Zhao1
Qin Zhao1 Senyan Du1
Senyan Du1 Yifei Lang1Xinfeng Han1Yong Huang1Xintian Wen1
Yifei Lang1Xinfeng Han1Yong Huang1Xintian Wen1 Yiping Wen1*
Yiping Wen1*Natural transformation is a mechanism by which a particular bacterial species takes up foreign DNA and integrates it into its genome. The swine pathogen Glaesserella parasuis (G. parasuis) is a naturally transformable bacterium. The regulation of competence, however, is not fully understood. In this study, the natural transformability of 99 strains was investigated. Only 44% of the strains were transformable under laboratory conditions. Through a high-resolution melting curve and phylogenetic analysis, we found that genetic differences in the core regulator of natural transformation, the tfoX gene, leads to two distinct natural transformation phenotypes. In the absence of the tfoX gene, the highly transformable strain SC1401 lost its natural transformability. In addition, when the SC1401 tfoX gene was replaced by the tfoX of SH0165, which has no natural transformability, competence was also lost. These results suggest that TfoX is a core regulator of natural transformation in G. parasuis, and that differences in tfoX can be used as a molecular indicator of natural transformability. Transcriptomic and proteomic analyses of the SC1401 wildtype strain, and a tfoX gene deletion strain showed that differential gene expression and protein synthesis is mainly centered on pathways related to glucose metabolism. The results suggest that tfoX may mediate natural transformation by regulating the metabolism of carbon sources. Our study provides evidence that tfoX plays an important role in the natural transformation of G. parasuis.
Natural transformation, a horizontal gene transfer (HGT) mode of bacteria, allows bacteria to actively uptake foreign DNA under natural conditions and integrate it into the genome through homologous recombination (Lorenz and Wackernagel, 1994). Natural transformation plays a vital role in the rapid spread of pathogenic virulence factors and resistance genes, resulting in the emergence of multidrug-resistant or highly pathogenic strains (Domingues, Harms et al., 2012; Domingues, Nielsen et al., 2012; Domingues et al., 2019). Unlike electroporation or chemical transformation (Mell et al., 2012), natural transformation is an active process that relies on proteins encoded by the recipient cell. Competence is the physiological state in which the natural transformation of bacteria can occur (Fontaine et al., 2015). The primary process of natural transformation can be divided into the uptake of foreign genes and their integration into the bacterial DNA (Chen and Dubnau, 2004). The transformation process requires the precise coordination of multiple competence genes, which are highly conserved in transformable bacteria. The encoded proteins perform the functions of DNA uptake, transport, and recombination processing that allow foreign DNA to enter and integrate into the genome of the recipient bacteria (Chen and Dubnau, 2004; Dubnau and Blokesch, 2019). For gram-negative bacteria, the type IV pilus system plays a vital role in the uptake of exogenous DNA (Costa et al., 2021). Double-stranded DNA (dsDNA) is captured by the extracellular fimbriae protein PilA, and through the contraction of the pilus, the dsDNA passes through the outer membrane through the activity of the secretory porin PilQ. Then the dsDNA binds to the DNA-binding protein ComEA in the periplasm of the cell and delivers the dsDNA to the inner membrane channel protein ComEC. The dsDNA is then unwound into single-stranded DNA (ssDNA); one strand enters the cytoplasm through ComEC, and the other is degraded. The ssDNA entering the cytoplasm can be bound to an ssDNA-binding protein encoded by ssb, protecting it from endogenous nuclease degradation. SsB presents the ssDNA to DprA, a DNA-processing protein. Subsequently, the ssDNA and genome integration are promoted through the synergistic action of RecA and a DNA helicase encoded by comM (Antonova and Hammer, 2015; Hepp and Maier, 2016).
Due to different growth environments, bacteria have evolved in various ways to induce competence, and nutritional conditions are the critical factors of many pathogenic bacteria (Seitz and Blokesch, 2013). As an essential element of microbial growth, the carbon source is involved in a variety of metabolic processes and regulatory pathways, and its effects on natural transformation have also been reported in many bacteria. For example, the natural transformation frequency of E. coli could be increased by adding a common carbon source in the intestinal culture medium (Guo et al., 2015); the formation of competence of Haemophilus influenzae was influenced by the PTS carbon source (Macfadyen and Redfield, 1996).
Although competence genes are well-conserved in bacteria, it has been commonly reported that the transformability of different isolates of competent species is variable. For instance, nearly 60% of H. influenzae and Pseudomonas stutzeri isolates are non-transformable, and the transformation frequency was also significantly different (Maughan and Redfield, 2009; Durieux et al., 2019). Comparable variation has also been found in Glaesserella parasuis (G. parasuis), a naturally transformable, gram-negative, pleomorphic bacterium of the Pasteurellaceae group (Bigas et al., 2005; Dai et al., 2021). Although a comparison of the entire genome sequence of G. parasuis isolates published in NCBI showed that all strains contained the identified homologous genes required for natural transformation, such as pilA, comEA, comM, etc., many G. parasuis isolates are non-transformable (Dai et al., 2016, 2018b). The molecular mechanism of such diversity is unclear, however, it may due to significant differences in the regulatory systems of the different strains. TfoX (encoded by tfoX, the orthologous gene is sxy) is a core regulatory factor of natural transformation in a variety of Gram-negative bacteria (Metzger et al., 2019; Sondberg et al., 2019). TfoX-deficient mutant strains of H. influenzae, Vibrio cholerae and Vibrio fischeri have lost their intrinsic natural transformability (Sinha et al., 2013; Lo Scrudato et al., 2014). The regulatory pathways of TfoX are diverse in different bacteria, but they all ultimately act on downstream competence genes. TfoX interacts synergically with the cAMP-CRP complex to activate transcription of the competence genes containing noncanonical CRP binding sites (CRP-S), thus regulating the natural transformation of bacteria (Sinha et al., 2012). In our previous studies, we analyzed the biologic functions of a homolog tfoX in SC1401, a G. parasuis strain with a high natural transformation efficiency (Dai et al., 2016). We found that other genes (e.g., comABCDEF, pilABCD, cyaA, crp, etc.) associated with natural transformation were highly conserved with 90% nucleotide sequence identity, while the DNA sequence of tfoX from G. parasuis SC1401 to that of strain SH0165 had only 74% nucleotide sequence identity. A proline residue was found inserted into G. parasuis SC1401 TfoX at codon 64th. Moreover, a secondary structure comparison showed that SC1401TfoX harbors two more α-helices (Dai et al., 2018a). Interestingly, SH0165 is a non-transformable strain (Zhang et al., 2015). The heterogenicity of tfoX among G. parasuis strains suggests that G. parasuis is unique in its regulation of natural transformation, but the molecular mechanism is still to be determined.
The mechanism of the regulation of natural transformation in G. parasuis occurs largely via the product of tfoX, a gene shown to have significant heterogenicity among G. parasuis strains. In this study, we analyzed the tfoX genes from 99 strains of G. parasuis using a High-Resolution Melting (HRM) analysis and gene sequencing to measure the nucleotide sequence identity of this locus. We then measured the natural transformation frequency of these strains and found that sequence variations led to two distinct natural transformation phenotypes, suggesting that differences in tfoX could play an important role in determining the natural transformability of various G. parasuis strains.
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1, and primers are listed in Supplementary Table S2. Escherichia coli (E. coli) DH5α, used to propagate the plasmids, was cultured in Luria-Bertani (LB, Difco, United States) broth with shaking or LB agar (Invitrogen, China) plates supplemented with 50 μg ml−1 kanamycin (Sigma-Aldrich, United States) when required. G. parasuis was cultured in Tryptic Soy Broth (TSB, Difco, United States) with shaking or Tryptic Soy agar (TSA, Difco, United States) supplemented with 5% inactivated bovine serum (Solarbio, China) and 0.1% (w/v) nicotinamide adenine dinucleotide (NAD, Sigma-Aldrich, United States; TSB++ and TSA++). When necessary, these media were supplemented with 50 μg ml−1 of kanamycin or 100 μg ml−1 erythromycin. All strains were grown at 37°C. Serotypes of G. parasuis were identified using PCR-based molecular serotyping (Howell et al., 2015).
The specific protocol for natural transformation was performed as previously described (Dai et al., 2021). Briefly, the recipient bacteria were isolated into 3 ml TSB++ and shaken at 37°C for 14 h to an optical density 600 (OD600) = 1.8; late-stationary phase. The bacteria were then spotted on TSA++, cultured for 13 h at 37°C, and resuspended in 30 μl TSB++ adjusted to a density of 5 × 1010 cfu/ml. A 20 μl aliquot was extracted and supplemented with 1 μl of cAMP (final concentration of 8 mM) and 2 μg of donor DNA [genomic DNA of SC1401ΔhtrA::Kan (Zhang et al., 2016), SC1401Δcdt::Erm or a recombinant plasmid constructed in this study]. The mixture was incubated for 10 min at 37°C and then spotted on TSA++ again, and incubated for 5 h at 37°C to induce the expression of antibiotic resistance. Afterwards, bacteria were harvested and resuspended in 100 μl of TSB++ and plated on a selective medium after serial dilution. The diluted bacteria (about 10−7) were plated on TSA++. Bacteria were incubated for 1–2 days. The formula of natural transformation frequencies (NTF) is kanamycin/erythromycin-resistant colony-forming units (CFU) ml/total CFU as previously described (Humbert et al., 2011).
DNA samples were isolated from G. parasuis clinical isolates using the TIANamp Bacteria DNA Kit (Tiangen, China) according to the manufacturer’s instructions. The HRM primers (P1, P2) were designed to amplify the sequences (approximately 100 bp) surrounding the target site. The annealing temperatures of the HRM primers were optimized to approximately 60°C and purified by HPLC.
A sample of 20 ng of the genomic DNA of a G. parasuis strain was used as a template in a 20 μl PCR reaction using a LightCycler® 480 High Resolution Melting Master kit (Roche), with 1× LightCycler® 480 High Resolution Melting Master Mix, and final concentrations of 200 nM of forward and reverse primers and 3 mM MgCl2. A total of 108 bp PCR products surrounding the target site were amplified in the LightCycler® 96 instrument (Roche), using the following cycling conditions: pre-incubation 10 min 95°C, 45 cycles 3-step amplification (95°C for 10 s, 57°C for 10 s, 72°C for 20 s) followed by one cycle of high resolution melting [95°C for 60 s, 40°C for 60 s, 65°C for 1 s, and finally using a ramp rate 0.07°C/s and an acquisition mode setting of 15 readings/s to reach the target temperature 95°C (1 s)]. Raw data were analyzed using the LightCycler® 96 software 1.1 (Roche), which sorted the samples into groups based on their normalized melting curves. Each sample included 2–3 technical replicates.
PCR fragments containing the tfoX gene were amplified from the genomic DNA of G. parasuis using the primers P3 and P4. The PCR products were submitted to Sangon Biotech for sequencing. Phylogenetic trees were made by MEGA v6 and visualized using the online server iTOL v41 (Letunic and Bork, 2019). Neighbor-joining (NJ) method was utilized to analyze sequence distances among different G.parasuis strains. In all analyses, the Phylogeny reconstruction used maximum likelihood, and the phylogeny was tested with the bootstrap method, using 500 bootstrap replications.
To construct an in-frame nonpolar tfoX mutant, the target vector pK18-tfoX was built as follows: first, the 653-bp upstream homologous region and the 656-bp downstream homologous region of the in-situ tfoX gene were amplified using the primers P5/P6 and P7/P8 from the genomic DNA of G. parasuis SC1401, then the 935-bp kanamycin-resistant cassette (KanR, aminoglycoside phosphotransferase Aph (3′)-IIa gene) was amplified by PCR from pKD4 using the primers P9/P10. Subsequently, these three fragments were purified using a Qiaquick spin column kit (Qiagen, Hilden, Germany) and were ligated into the linearized vector pK18 mobSacB. Similarly, the tfoX gene of SC1401 and SH0165 were amplified, and the four fragments were ligated into pK18 mobSacB to construct the target vector pK18-SC1401tfoX-Kan and pK18-SH0165tfoX-Kan. Finally, the resulting plasmids were transformed into the G. parasuis strain SC1401 using a natural transformation technique. Transformant bacteria were incubated for 36 h. The resultant kanamycin-resistant transformants were cultured on a large scale in TSB++/Kan for further identification by PCR.
RNA samples were isolated from the G. parasuis strains or SC1401 derivatives (cultured in TSB++ to obtain a bacterial culture volume equivalent to 1 ml of OD600 = 1.8 or on TSA++ for 13 h, respectively) using the UNIQ-10 Column Total RNA Purification Kit (Sangon, China) according to the manufacturer’s instructions. Two-step RT-PCR was performed using the PrimeScriptTM RT reagent Kit with the gDNA Eraser (Takara, Japan). The competence gene transcripts were then analyzed using quantitative reverse transcription PCR (qRT-PCR) with the reagent of SYBR Premix EX TaqTM II (Tli RNaseH Plus; Takara, Japan). The 2 [–ΔΔC(T)] method was used to relatively quantitate the expression of multiple genes compared to the stably expressed 16S RNA reference gene. Runs of qPCR were performed with a Lightcycler96 (Roche, Switzerland) system. The experiments were performed in independent triplicates.
Strains SC1401 and SC1401ΔtfoX::Kan were cultivated with three replicates in TSB++ at 220 rpm and 37°C. According to the manufacturer’s instructions, when the cultures had grown to the late-stationary phase, the total RNA of each sample was extracted using the Trizol reagent (Invitrogen, Carlsbad, CA, United States). After dissolving the RNA samples in RNase-free water, the total RNA degradation and contamination levels were monitored on 1% agarose gels. RNA quantification, library preparation, clustering, and sequencing were performed at Novogene Bioinformatics Technology Co., Ltd. (Beijing, PR China). The clean data (clean reads) were acquired by removing reads containing adapter sequence, reads containing poly-N, and low-quality reads from raw data. The G. parasuis SC1401 genome (CP015099.1) and the gene model annotation files were downloaded2 and used to build the index and to align clean reads of the G. parasuis SC1401 genome (CP015099.1) with Bowtie2-2.2.3 (Langmead et al., 2009).
To quantify the gene expression, the read numbers mapped to each gene were counted using the HTSeq v0.6.1 (Anders et al., 2015). The FPKM (expected number of Fragments Per Kilobase of transcript sequence per Million base pairs sequenced) of each gene was used to analyze the gene expression variations. In our study, differential expression analysis of G. parasuis SC1401 and G. parasuis SC1401ΔtfoX was performed using the DEGSeq R package (version 1.20.0; Anders and Huber, 2010). The resulting p-values were adjusted using the Benjamini and Hochberg approach to control the false discovery rate. Genes with an adjusted p-value <0.05 were considered to be significantly differentially expressed. The GOseq R package implemented a Gene Ontology (GO) enrichment analysis of differentially expressed genes to correct for gene length bias (Young et al., 2010). GO terms with a corrected p-value less than 0.05 were considered significantly enriched by differentially expressed genes. We used the KOBAS software (Center for Bioinformatics, Peking University & Institute of Computing Technology, Chinese Academy of Sciences) to test the statistical enrichment of differential expression genes in KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (Kanehisa et al., 2008; Young et al., 2010).
Three replicates of the strains SC1401 and SC1401ΔtfoX::Kan were cultivated with in TSB++ at 220 rpm and 37°C. When the cultures had grown to the late-stationary phase, the total protein of each sample was extracted as previously described (Wisniewski et al., 2009). Protein concentrations were determined using the Bradford protein assay (Bio-Rad, United States). In addition, 10 μg of the total extracted proteins were analyzed by SDS-PAGE. Peptide preparation, TMT labeling, HPLC fractionation, and LC–MS/MS analysis of protein samples were performed at Novogene Bioinformatics Technology Co., Ltd. (Beijing, PR China).
The resulting spectra from each fraction were searched separately against the database3 using Proteome Discoverer 2.2 (PD 2.2, Thermo, United States). The confidence levels for the identified proteins were determined by the false discovery rate (FDR). Proteins containing similar peptides that could not be distinguished based on MS/MS analysis were grouped separately. Reporter Quantification (TMT) was used for TMT quantification. The protein quantitation results were statistically analyzed by the Mann–Whitney test, and the significance ratios, defined as p < 0.05 and |log2FC| > 1.2 were used to screen the differentially expressed proteins (DEP) as previously described.
To deepen our understanding of the potential biological functions of our tfoX genes, GO and KEGG enrichment analyses on the differentially expressed proteins were performed using hypergeometric tests to discover the biological processes and pathways involved. More specifically, biological process terms from the GO and KEGG pathways were chosen from the annotation resources provided to functionally characterize the driver genes identified.
Transformation frequencies were determined from the number of antibiotic-resistant cfu mL−1 divided by the total cfu mL−1 scored on non-selective agar. Statistical analyses were performed using the GraphPad Prism 6 (GraphPad software, CA, United States). Statistical significance was evaluated using Student’s t-test, a one-way ANOVA, or a two-way ANOVA. The value of p < 0.05 represented statistically significant differences. Error bars in all figures represent the standard deviations of three independent experiments.
Despite the genes required for natural transformation in all sequenced G. parasuis strains and the nucleotide sequence homology of these genes exceeding 95%, many strains still appeared non-transformable under laboratory conditions. To gain insight into these extensive variations, we evaluated the natural competence and transformation ability of 15 standard strains and 84 clinical isolates. As shown in Figure 1A, 44 competent strains, and 55 strains with no natural transformability were identified under laboratory conditions. Furthermore, these transformable strains showed varying levels of natural transformation; some strains had high transformation frequencies (about 1.0 × 10−4), while others had low transformation frequencies (about 1.0 × 10−7). In brief, like many other competent bacteria, the natural transformability of G. parasuis varied greatly under the same experimental conditions.
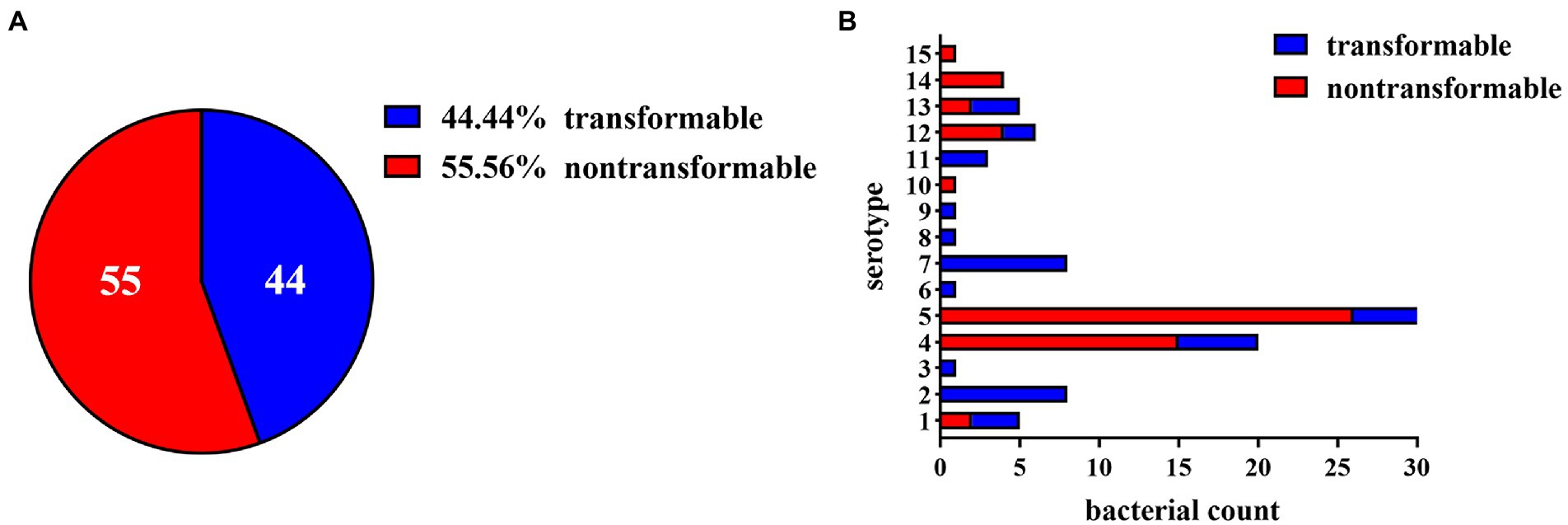
Figure 1. Distribution of transformable strains in Glaesserella parasuis. (A) Pie chart of the proportion of transformable and non-transformable isolates in 99 G. parasuis strains. (B) The distribution of transformable and non-transformable strains of different serotypes. The horizontal axis represents the number of bacteria, and the vertical axis represents the serotype. Regions in blue or red depict transformable or non-transformable strains, respectively. When the natural transformation frequency is greater than 1 × 10−8, we considered the strain to be transformable.
To explore whether the natural transformability of G. parasuis was related to bacterial virulence, we analyzed the serotypes of the 84 clinical isolates mentioned above. As shown in Figure 1B, serotypes 4 and 5 comprise the largest proportion of all of the strains investigated because these two serotypes are the most frequent in China. Most strains of these two serotypes have no transformability. In addition, serotypes 2 and 7 were abundant, and all isolates were found to have transformability. The remaining serotypes are relatively infrequent because they are rarely present in China. The results indicated that all of the low virulence strains (serotypes 3, 6, 7, 8, 9, 11) were naturally transformable. In contrast, only 45% of the moderate virulence strains (serotypes 2, 4, 15) and 29% of the high virulence strains (serotypes 1, 5, 10, 12, 13, 14) had natural transformability. This result demonstrates that the virulence of G. parasuis does not completely determine its natural transformability status. However, there were significantly more naturally transformable strains in the medium and low virulence strain groups, suggesting that the virulence factors of G. parasuis may be related to the natural transformability of these bacteria.
In our previous study, we sequenced the highly transformable strain SC1401 and compared it to the G. parasuis reference strain SH0165, which is non-transformable under all of the experimental conditions we tried. Intriguingly, we found that the nucleotide sequence identity of tfoX gene between the two strains was 74%, and the amino acid sequence identity was 73%. Further comparison with tfoX genes of the sequenced strains showed that sequence differences are widely found in G. parasuis strains. In contrast, tfoX is conserved in E. coli and in H. influenzae (Dai et al., 2018a). To explore the distribution of the tfoX gene variants in G. parasuis, the 99 G. parasuis isolates referred to in Section “The Natural Transformation Frequency Is Variable in G. parasuis Wild Strains” were genotyped using a high-resolution melting (HRM) analysis. As shown in Figure 2A, the tfoX gene of all tested strains could be divided into two categories, one with a melting peak Tm value between 72°C and 74°C, similar to SC1401, and another with a melting peak Tm value between 75°C and 77°C, similar to SH0165.
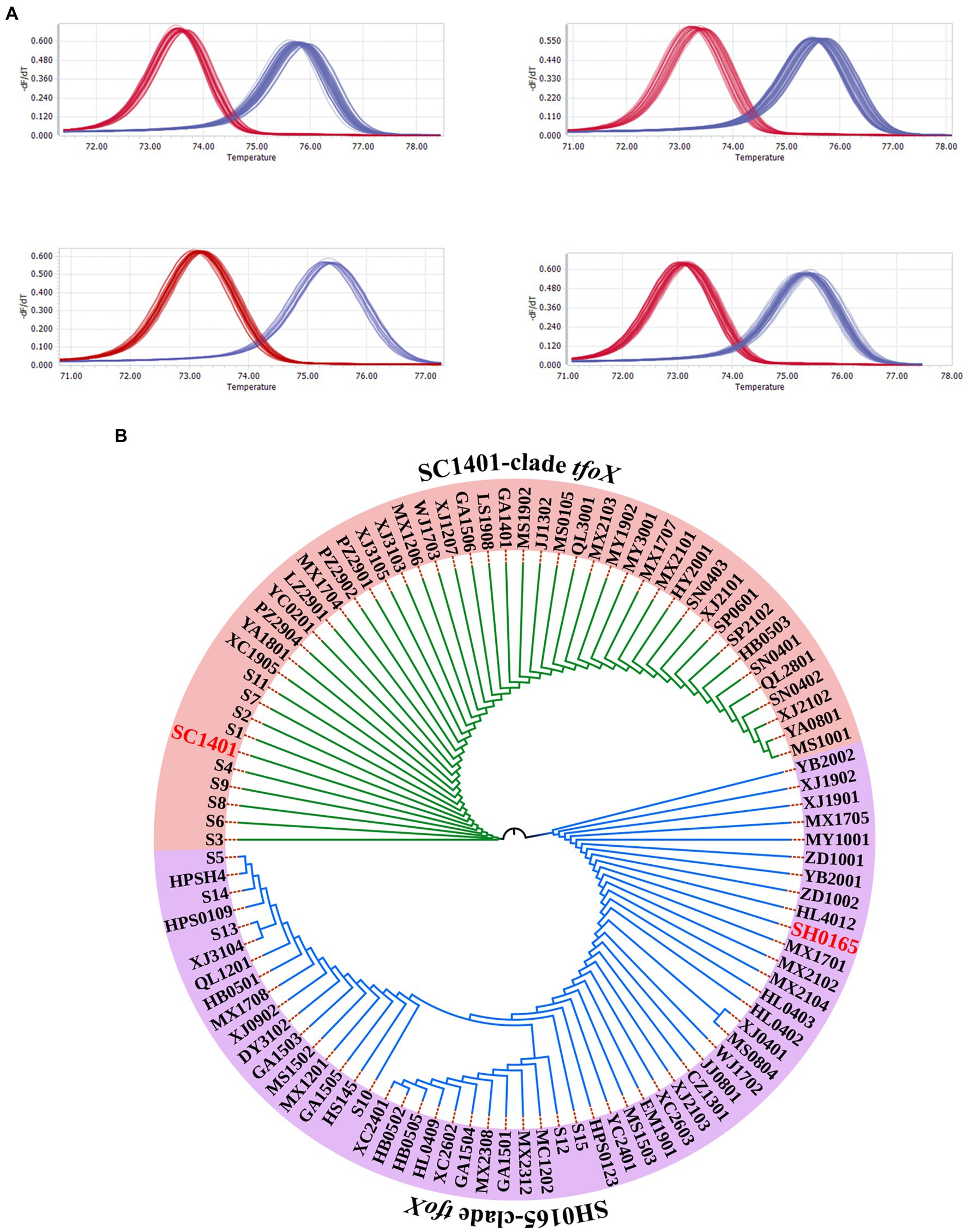
Figure 2. tfoX molecular differences in Glaesserella parasuis. (A) High resolution melting curves of the tfoX genes of 99 G. parasuis strains. A HRM analysis showed that two types of melting curves occurred: the red and blue curves are consistent with SC1401tfoX, or SH0165tfoX, respectively. (B) Phylogenetic tree of tfoX in G. parasuis. A neighbor-joining (NJ) tree demonstrated the overall diversity of tfoX from G. parasuis. The results are consistent with the HRM analysis, and all tfoX genes are evolutionarily divided into two clades.
We next sequenced the tfoX gene of each G. parasuis strain, and compared and analyzed the sequence homology via a phylogenetic analysis. The results showed that tfoX evolved into two clades, SC1401 and SH0165, consistent with the HRM results (Figure 2B). According to tfoX gene variants, we divided the 99 G. parasuis strains into SC1401 tfoX strains and SH0165 tfoX strains, and these strains represented two different genotypes of G. parasuis. Since the tfoX gene has been identified as a core regulator in other bacterial species, we speculated that this molecular level difference could be a key determinant of natural competence.
The tfoX typing and natural transformability of all strains were assessed to determine the relationship between the tfoX gene sequence and the natural transformation phenotype. It is worth mentioning that the natural transformation capacity was consistent with the tfoX genotype. We found that the natural transformation frequencies of G. parasuis with different tfoX variants were significantly different. The SH0165 tfoX groups were not naturally transformable, while SC1401 tfoX group exhibited varying degrees of natural transformability (Supplementary Table S3). Therefore, we concluded that tfoX is a core regulatory gene for the natural transformation of G. parasuis. The molecular level difference of tfoX gene can be used as a molecular indicator of whether G. parasuis has natural transformability.
To further explore the role of tfoX vis-á-vis the natural transformation of G. parasuis, a tfoX gene deletion strain was constructed by naturally transforming with the homologous fragments harboring the suicide plasmid pK18-tfoX into SC1401 (Figure 3A). PCR analyses were performed to confirm the proper construction of the ΔtfoX::Kan (Figure 3B). As shown in Figure 4A, deletion of tfoX leads to a loss of natural competence. However, the ΔtfoX::Kan strain did not show significant growth defects in vitro when compared with the wildtype SC1401 strains (Figure 4B). These results indicate that tfoX is an indispensable factor in the regulation of natural transformation in G. parasuis. To further confirm that the SH0165tfoX gene leads to the failure of the natural transformation in G. parasuis, we constructed the tfoX gene differential replacement strains ΔtfoX::SH0165tfoX-Kan and ΔtfoX::SC1401tfoX-Kan using SC1401 by the same method as described in Figure 3A above. The results showed that when the tfoX gene of SC1401 was replaced with the tfoX gene of SH0165, the strain was unable to undergo natural transformation. Similarly, neither of these replacement strains had different growth characteristics in vitro (Figures 4A,B). These results confirmed that SC1401tfoX is a vital molecular regulatory factor of the natural transformability of G. parasuis.
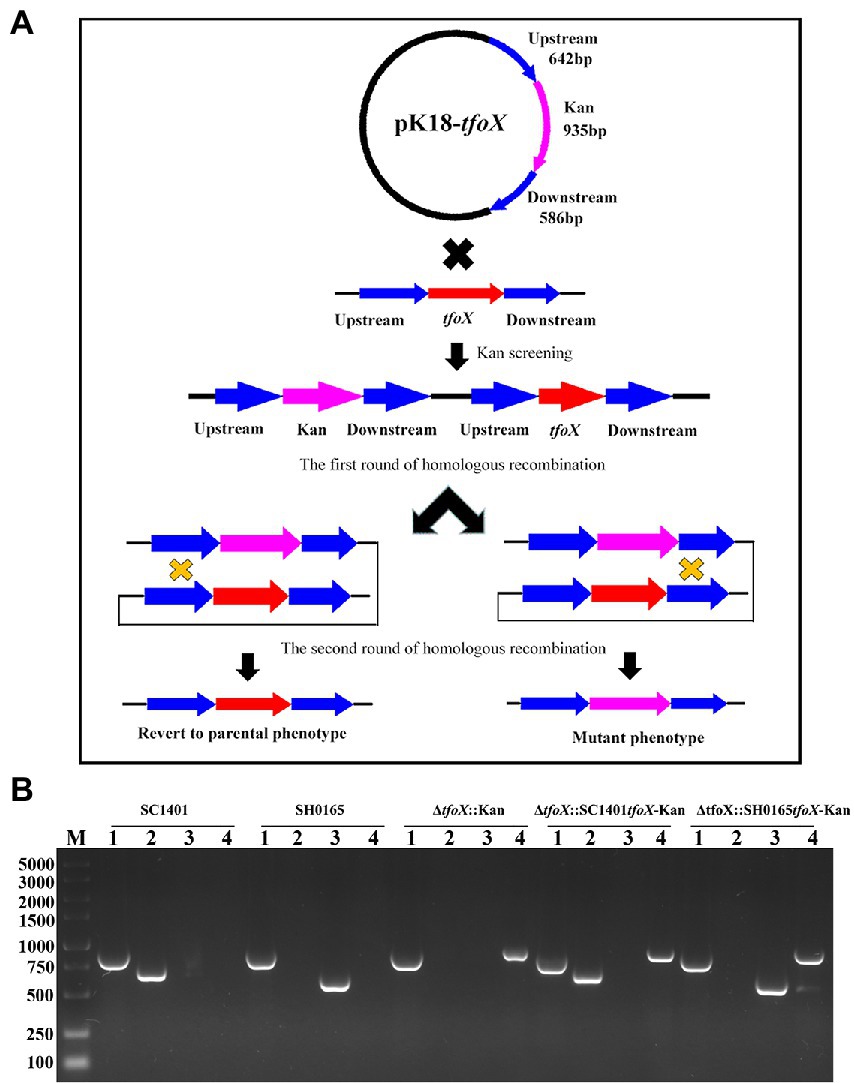
Figure 3. The derivative strains of SC1401, ΔtfoX::Kan, ΔtfoX::SC1401tfoX-Kan, and ΔtfoX::SH0165tfoX-Kan were constructed by a natural transformation method. (A) The schematic diagram of pK18-tfoX plasmid and the host genome were recombined in two rounds of homologous recombination to construct tfoX gene deletion strains. In the first round of single exchange, the whole plasmid was recombined into the genome. There were both target and resistance genes in the genome of the recipient bacteria; in the second round of recombination, the tfoX gene was replaced by Kan. (B) PCR verification of SC1401, ΔtfoX::Kan, ΔtfoX::SC1401tfoX-Kan and ΔtfoX::SH0165tfoX-Kan constructs. Four primer pairs (P17/P18, P9/P10, P19/P20, P21/P22) were used to amplify the Glaesserella parasuis 16S rRNA fragment, the kan fragment, the SC1401tfoX and SH0165tfoX, respectively. Lane 1: G. parasuis 16S rRNA was amplified from these four strains, respectively; Lane 2: the SC1401tfoX gene fragment was amplified from these four strains, respectively; Lane 3: the SH0165tfoX gene fragment was amplified from these four strains, respectively; Lane 4: the kanamycin resistance cassette was amplified from these four strains, respectively.
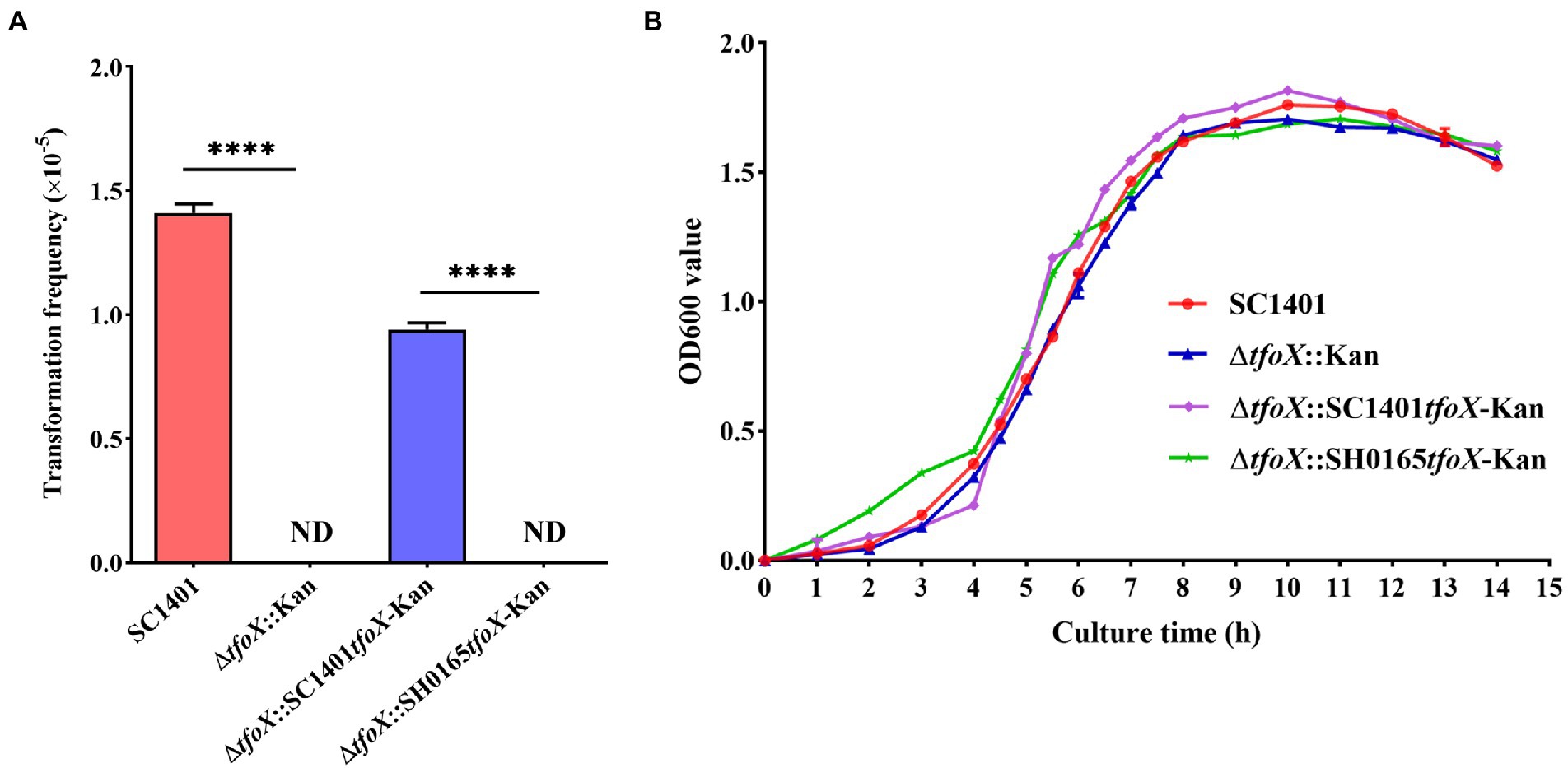
Figure 4. Natural transformation frequency and phenotype of tfoX mutant and growth curves of SC1401, ΔtfoX::Kan, ΔtfoX::SC1401tfoX-Kan, and ΔtfoX::SH0165tfoX-Kan. (A) The natural transformation frequency of the wild type and mutant strains. **** value of p <0.0001. (B) The growth curves of the Glaesserella parasuis SC1401 and its derivatives. Bacterial growth was monitored by measuring the optical density at 600 nm at each time point. The results are representative of three independent experiments. Error bars denote the standard deviation.
We can confirm that the molecular structure differences of tfoX lead to diversity in the natural transformation phenotype, however, the molecular mechanism of natural transformation regulation of G. parasuis is unknown. To explore whether tfoX variants would result in expression levels of TfoX in SH0165tfoX strains significantly lower than those of the SC140tfoX strains, in addition to SH0165 and SC1401, three of each SH0165tfoX and SC140 tfoX strains were selected for the analysis of tfoX gene transcription levels by fluorescence quantitative PCR. The results showed that not all SC1401 tfoX strains have higher tfoX gene transcription levels than that of SH0165 tfoX strains. Some have lower tfoX gene transcription levels (Figure 5A), indicating that such tfoX variants are independent of the expression level. Moreover, our results suggested that in different SC1401 tfoX strains, the expression level of tfoX was not proportional to the natural transformation frequency. Some strains had a high expression level of tfoX, but the natural transformation level was low (Figure 5B). This result indicates that the transcription level of tfoX alone does not determine the level of natural transformation in different strains. The difference in natural transformation phenotype caused by tfoX molecular level difference is not due to the tfoX expression level. This result also indicated that tfoX is not the only regulatory factor, and many elements also regulate the level of natural transformability.
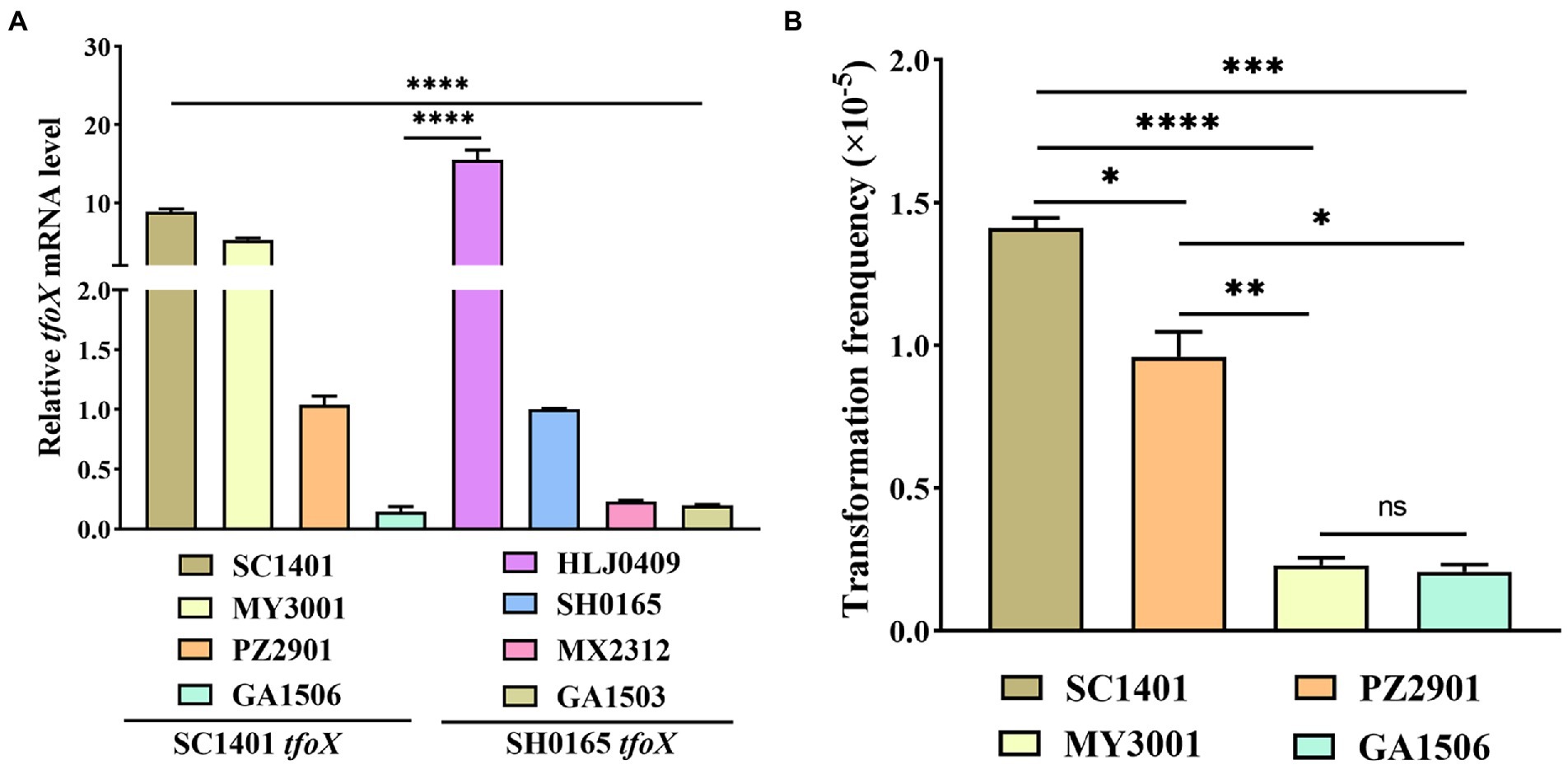
Figure 5. Transcriptional levels of tfoX and natural transformation frequency of Glaesserella parasuis wild strains. (A) Quantitative real-time PCR analysis of the relative transcription of tfoX in four each SC1401tfoX and SH0165tfoX isolates. (B) The natural transformation frequency of four SC1401tfoX isolates. **** value of p <0.0001; *** value of p <0.001; ** value of p <0.01; * value of p <0.05.
To further analyze the role of the tfoX gene in G. parasuis, transcriptomic sequencing was performed on the wildtype strain G. parasuis SC1401 and the tfoX mutant strain G. parasuis SC1401∆tfoX::Kan. Three biological replicates were performed. The Total Mapped Reads were all greater than 98%, and Multiple Mapped Reads were all lower than 5% (Supplementary Table S4), indicating that the samples were not contaminated and that the RNA-Seq quality was acceptable. The squared Pearson correlation coefficient (R2) between the replicate samples was always greater than 0.95 (Supplementary Figure S1), which assured the reliability of the test experiment and the rationality of sample selection. A total of 12 genes were selected for qRT-PCR to test the expression of these genes to validate the reproducibility and repeatability of DEGs identified from transcriptome sequencings (Supplementary Figure S2). The fold changes calculated by qRT-PCR were generally consistent with the trend of the RNA-Seq results, which confirmed the reliability of the RNA-Seq results.
A differential gene expression analysis was performed on the readcount data obtained from the gene expression level analysis to determine the changes in the gene expression profile after deletion of the tfoX gene. As shown in the volcano plots (Figure 6), 963 genes were differentially expressed, of which 516 were upregulated and 447 were down-regulated. Supplementary Table S5 shows the readcount of candidate competence genes homologous to that of H. influenzae of SC1401 and SC1401∆tfoX::Kan. As listed in Supplementary Table S6, an analysis of the differentially expressed genes (DEGs) revealed 8 competence genes. It is worth noting that almost all were significantly downregulated among the 8 genes except the upregulated gene ssb. The 6 downregulated genes, which play crucial roles in DNA uptake, pilA, pilB, A4U84_02730, pilC, comEA, and pilF in SC1401ΔtfoX were nearly 7.4-, 4.3-, 3,9-, 2.1-, 2.8-, and 1.4-fold lower than that of in SC1401, respectively. The comM gene, which is involved in the transformation process, was downregulated 5.3-fold. Only one gene, ssb, which encodes a single-stranded DNA-binding protein that protects DNA from degradation, was slightly upregulated. These competence genes play an essential role in the bacterial uptake of foreign DNA and integration into its genome, suggesting that TfoX may regulate the natural transformability of G. parasuis by directly or indirectly activating the expression of these competence genes.
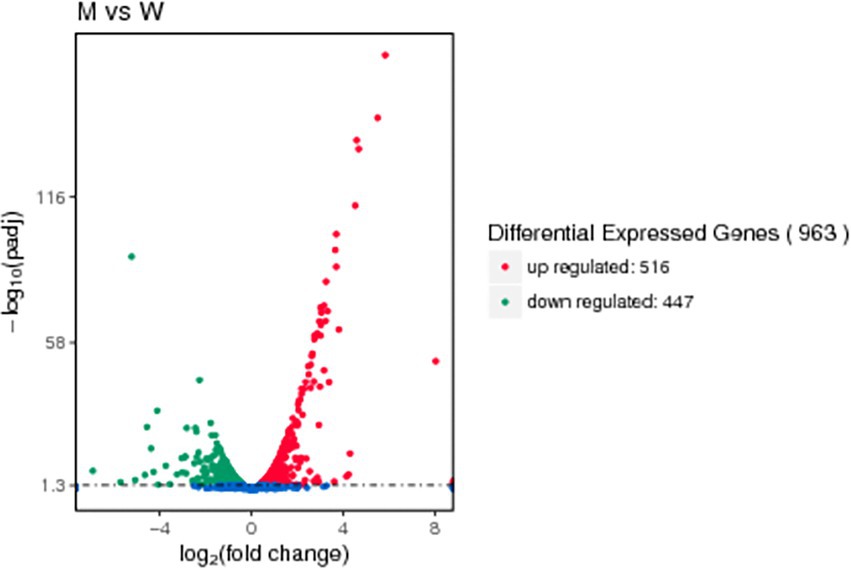
Figure 6. Volcano map of DEGs. Genes with significant differential expression are represented by red dots (upregulated) and green dots (down-regulated), while blue dots represent genes with no significant differences in expression. The abscissa represents the quantitative change in gene expression in the different samples. The ordinate represents the statistical significance of the change in the gene expression levels.
Gene ontology (GO) is an international standard classification system of gene functions, which aims to establish linguistic standards for defining and describing the functions of genes and proteins applicable to all species. GO is divided into molecular function, biological process, and cellular components. We analyzed the distribution of DEGs in GO terms to clarify the expression of sample differences in gene function. The 30 GO terms with the most significant enrichment of DEGs are shown in Figure 7A. The most dominant subcategories were oxidation–reduction process, electron transfer activity, protein-containing complexes, energy derivation by oxidation of organic compounds, and anion transmembrane transporter activity. In organisms, different genes coordinate to perform their biological functions, and significant enrichment of pathways can determine the main biochemical metabolic and signal transduction pathways involved by DEGs. The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database of systematic analysis of gene functions and their pathway networks. A KEGG analysis showed that DEGs were significantly enriched for pathways involved in carbon metabolism, methane metabolism, pyruvate metabolism, and microbial metabolism in diverse environments (Figure 7B).
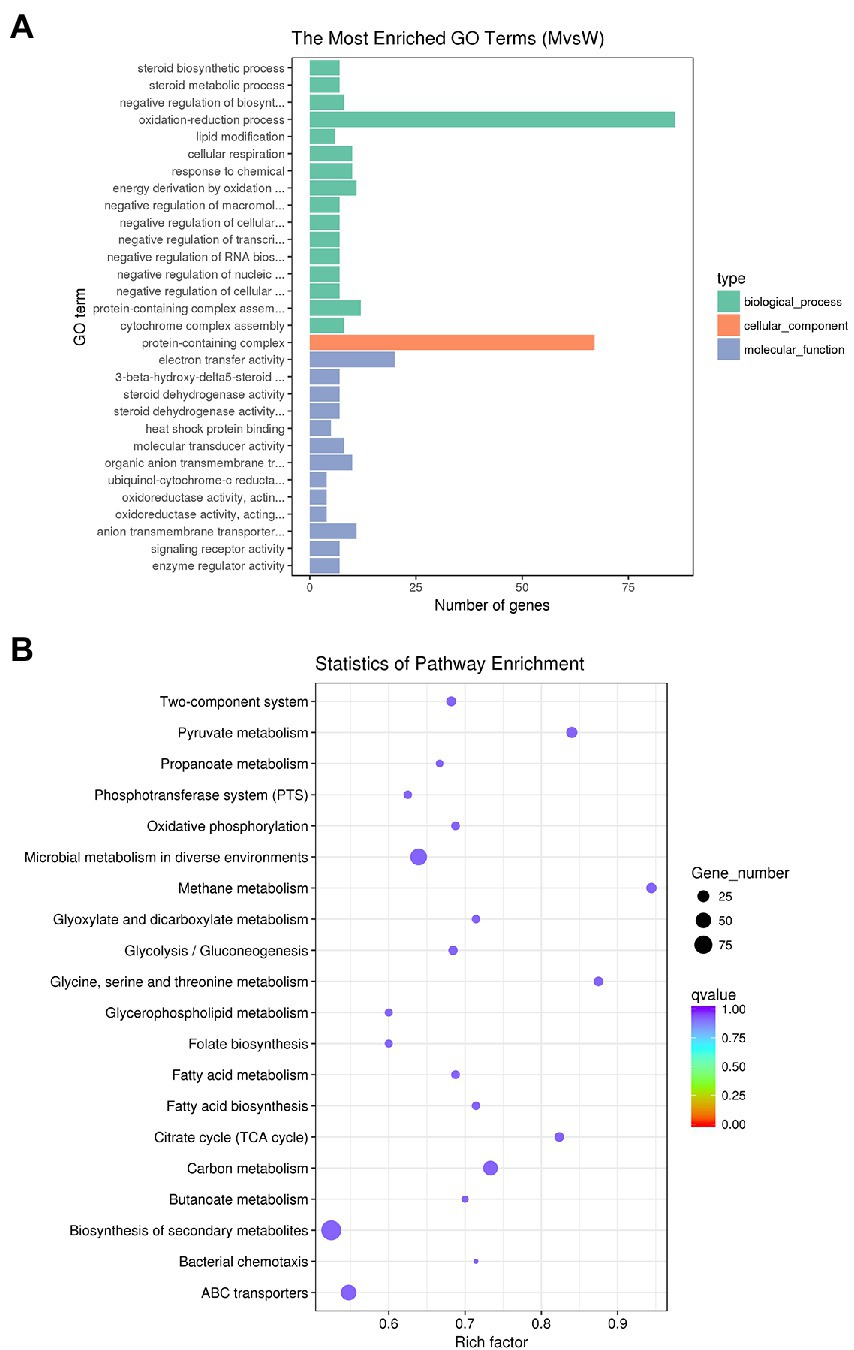
Figure 7. GO and KEGG analysis of DEGs. (A) GO enrichment histogram of DEGs. The ordinate is the enriched GO term, and the abscissa is the number of differential genes in this term. Different colors are used to distinguish biological processes, cellular components, and molecular functions. (B) Rich distribution points of DEGs in KEGG pathway. The vertical axis represents the pathway name, the horizontal axis represents the enrichment coefficient, the size of the dots represents the number of differentially expressed genes in the pathway, and the dot color corresponds to different qvalue ranges.
We further performed a comparative proteomic analysis on the SC1401 and ∆tfoX::Kan. As shown in Figure 8, a total of 180 differentially expressed proteins (DEPs) were identified, of which 70 were upregulated (FC ≥ 1.2, p-value ≤ 0.05) and 110 were downregulated (FC ≤ 0.83, p-value ≤ 0.05).
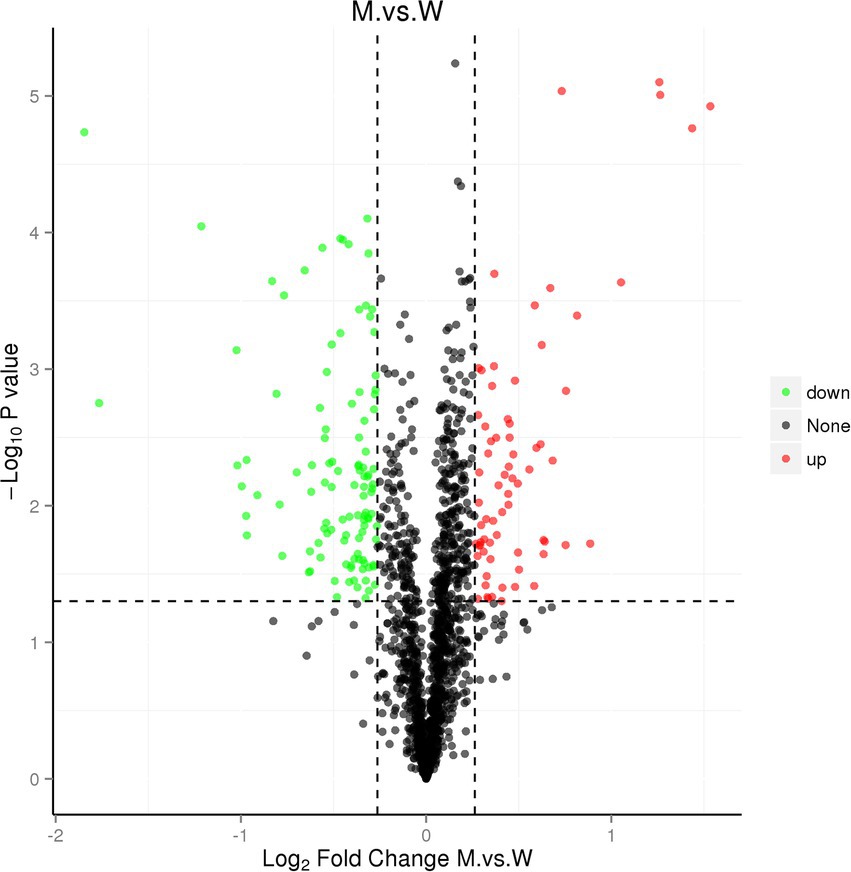
Figure 8. Volcano map of DEPs. The horizontal axis represents the fold change (log2 value) of the DEPs, the vertical axis represents the value of p (−log10 value), black represents proteins with no significant differences, red represents upregulated proteins, and the green represents downregulated proteins.
The DEPs were analyzed by GO enrichment and KEGG enrichment. As shown in Figure 9A, all DEPs were grouped into three GO categories (biological process, cellular component, and molecular function). The GO items enriched by DEPs mainly include single-organism, small molecule metabolic and oxidation–reduction processes in the biological pathways. The GO entries are mainly oxido-reductase activity, ion binding, and cation binding in the molecular function. The GO entries are mainly integral components of the plasma membrane in the cellular component. As shown in Figure 9B, KEGG analysis showed that the main enrichment pathways of DEPs were carbon metabolism, microbial metabolism in diverse environments, fructose and mannose metabolism, citrate cycle, pyruvate, and phosphotransferase system.
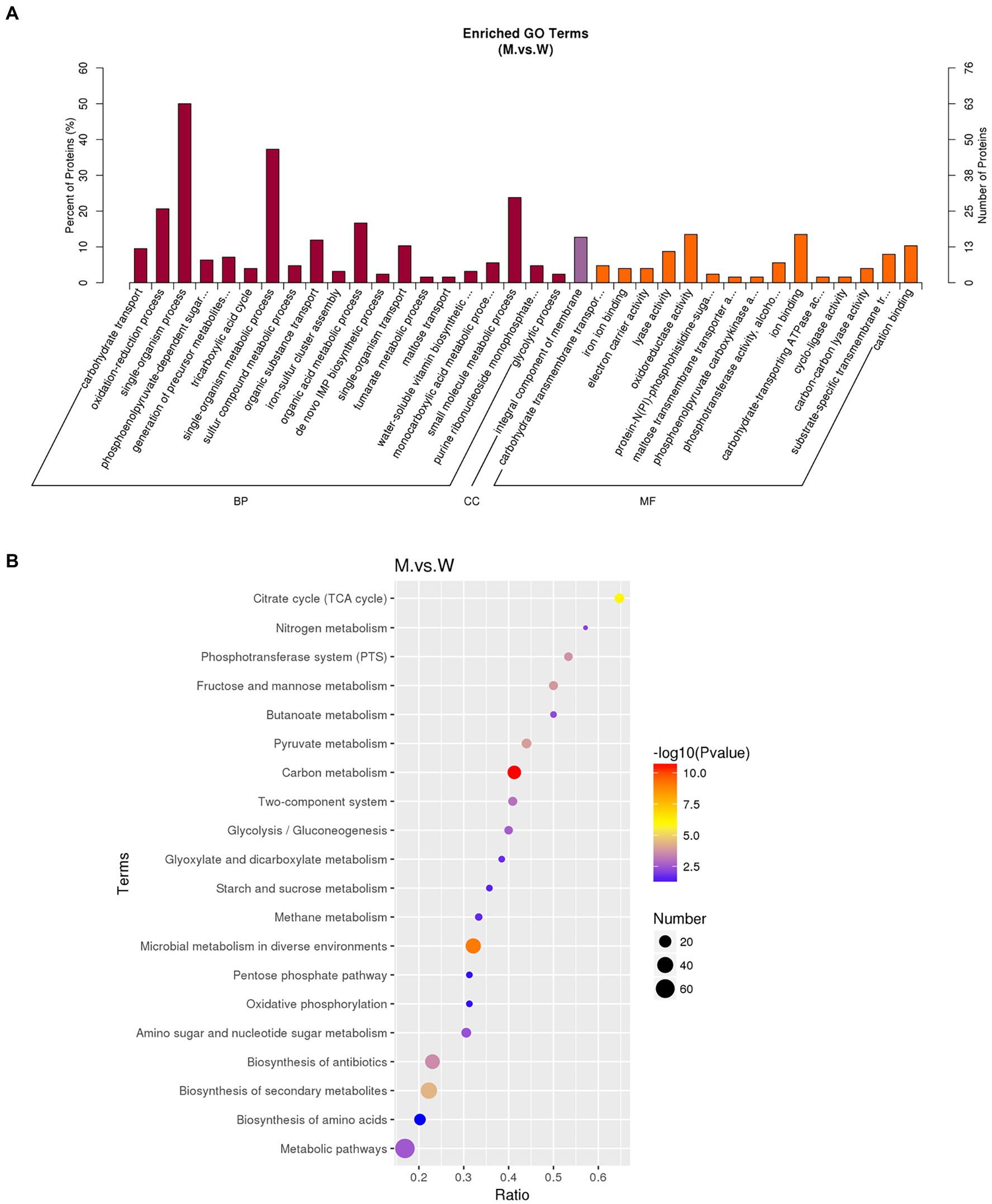
Figure 9. GO and KEGG analysis of DEGs. (A) GO enrichment histogram of DEPs. The abscissa is the enriched GO term, and the ordinate is the number of DEPs in this term. The percentage on the ordinate represents the ratio of the number of differential proteins associated with this GO to the number of differential proteins annotated by GO. (B) KEGG enrichment bubble diagram of DEPs. The horizontal axis is the ratio of the number of DEPs in the corresponding pathway to the number of total proteins identified. The vertical axis represents the name of the pathway. The color of the point represents the value of p of the hypergeometric test; The size of the dots represents the number of differential proteins in the corresponding pathway.
Since the first discovery of natural transformation in Streptococcus pneumoniae in 1928, many other bacteria have been found to be naturally transformable (Griffith, 1928; Seitz and Blokesch, 2013; Huang et al., 2021). So far, at least 83 species of bacteria have been reported to be capable of natural transformation. However, not all isolates of the transformable species can transform (Evans and Rozen, 2013; Zhang et al., 2021). We found that more than half of G. parasuis in laboratory conditions were non-transformable. This result suggests that G. parasuis, like H. influenzae and S. pneumoniae, has a wide range of phenotypic differences related to transformation (Maughan and Redfield, 2009; Evans and Rozen, 2013). It is worth mentioning that the induction conditions are a critical factor for the development of natural transformability of bacteria; for example, high transformability in H. influenzae requires induction in starvation M-IV medium. Although the induction conditions for the natural transformation of G. parasuis are unknown, it should be emphasized that the environmental conditions in which we showed that some G. parasuis isolates were non-transformable were all under laboratory conditions. These strains may develop natural transformability under other specific growth conditions in the field. It was previously suggested that the homologous recombination efficiency of high virulence strains was very low when constructing gene deletion strains by the natural transformation method. It has been speculated that the highly virulent strains have a more restrictive modification system. The foreign DNA was mostly sheared rather than recombined with the target gene to integrate it into the bacterial genome (Zhang et al., 2019). Serotypes have been used as virulence markers in G. parasuis (Yu et al., 2014). We found that the percentage of transformable isolates in a group of serotypes that represent high virulence was very low. This result suggests that the natural transformability of bacteria may be related to their virulence. Considering the limited number of experimental strains, further studies are needed to explore the deeper relationship between bacterial virulence and natural transformation.
The natural transformation process occurs due to the precise regulation of many genes, including the core regulatory factors tfoX (sxy), crp, and cyaA (Antonova and Hammer, 2015; Simpson et al., 2019). The systematic regulation of the natural transformability of different bacteria, especially gram-negative bacteria, is inconsistent, making it more challenging to study the mechanism of natural transformation (Stutzmann and Blokesch, 2020). With the recent emergence of advanced genomics and transcriptomic sequencing, researchers have gradually discovered the internal mechanism of differential natural transformation phenotypes in several strains (Cohen et al., 2021). For instance, Durieux et al. used genome-wide association studies and found that the loss of the transformation of Legionella pneumophila was associated with the loss of a conjugative plasmid. (Attaiech et al., 2016). SC1401tfoX and SH0165tfoX have significant molecular level differences, only 74 and 73% homology of the DNA and amino acid sequences, respectively. Such molecular diversity of tfoX has not been reported in bacteria such as H. influenzae or E. coli. We speculated that the differential phenotypes of natural transformation of G. parasuis were caused by molecular level differences in tfoX, as SH0165 failed to transform under laboratory culture conditions.
High resolution melting (HRM) technology plays a vital role in genotyping (Li et al., 2019; Soejima and Koda, 2021). Through HRM and sequencing analyses, we determined that all tfoX genes can be divided into two types. Further study showed that all of SH0165tfoX laboratory strains were non-transformable, while SC1401tfoX strains had different natural transformability levels. These results indicate that G. parasuis may have a unique natural transformation regulatory system, and tfoX can be used as a molecular marker to distinguish whether a particular G. parasuis isolate will be naturally transformable. In addition, the previously reported G. parasuis molecular classification method may be somewhat complex and not applicable to specific strains. Meanwhile, all G. parasuis strains could be divided into two types using tfoX molecular level differences, namely, SC1401tfoX and SH0165tfoX, respectively representing strains with or without natural transformability. Such results provide a new perspective for G. parasuis molecular typing.
The complete loss of the natural transformability of ΔtfoX::Kan and ΔtfoX::SH0165tfoX-Kan indicated that SC1401TfoX is an indispensable factor in the regulation of natural transformation. This result showed that SH0165 failed to undergo natural transformation which was mediated independently by tfoX. Previous studies on the natural transformation of H. influenzae showed that the transcription and translation efficiency of a sxy gene mutation strain was greatly improved. However, our experiments have shown that the tfoX variants did not affect expression levels. The diversity in the natural transformability of G. parasuis is due to the structure of tfoX per se, which leads tfoX genes to perform different functions of activating or inhibiting downstream genes, or the activation function of SH0165tfoX is affected by its structure. The natural competence of S. pneumoniae is regulated by the ComD protein, whose histidine kinase activity is regulated by the competence stimulating peptide (CSP; Yang et al., 2017). The majority of S. pneumoniae strains produce one of two variants of CSP, namely CSP1 or CSP2, and the cognate receptors ComD1 and ComD2, respectively (Yang and Tal-Gan, 2019). In E. coli, TfoX could directly interact with cAMP Receptor Protein (CRP; Sondberg et al., 2019). However, CRP is conserved in G. parasuis. Identifying the proteins that interact with TfoX will be a key step in understanding the molecular mechanism of how the two TfoX proteins regulate natural transformation. In addition, the expression level of tfoX was not positively correlated with the natural transformation frequency in SC1401tfoX strains. The reason is unknown. It could be that the regulation of natural transformation is a complex network, and different strains could be affected by many other factors.
Multiple competence genes were significantly down-regulated in tfoX mutant strains compared to the wildtype, including pilA, comM, pilB, A4U84_02730, comEA, pilC, and pilF. Proteins encoded by those genes play an essential role in the uptake and transport of foreign DNA (Nero et al., 2018; Dalia and Dalia, 2019). Our results indicated that SC1401tfoX can activate transcription of downstream genes associated with natural transformation in G. parasuis. In various bacteria, TfoX synergistically binds the cAMP-CRP complex to the CRP binding site in the promoter region of the competence gene, thereby initiating transcription (Jaskolska and Gerdes, 2015). CRP is a cyclic adenosine monophosphate (cAMP) receptor protein widely existing in the γ-proteobacteria (Redfield et al., 2005; Cameron and Redfield, 2008). In G. parasuis, the interaction between TfoX and CRP is unknown. However, we found that CRP proved to be an indispensable factor in regulating natural transformation in G. parasuis SC1401 in an independent experiment. When the crp gene was deleted, natural transformability was lost. Further, if the adenylate cyclase coding gene cyaA was deleted, the natural transformation level of G. parasuis decreased to a very low level (data not shown). These results suggest that the cAMP-CRP complex also plays a vital role in G. parasuis natural transformation. Further studies to dissect the binding affinity of the two TfoX proteins to the cAMP-CRP complex is needed.
A transcriptomic analysis showed that 936 genes were differentially expressed and a proteomics analysis showed that 180 proteins were differentially expressed. These results indicated that TfoX had an overall regulatory effect in the bacterium. Differentially expressed genes and protein accumulation pathways mostly revolve around sugar metabolism. As a fundamental element of microbial growth, the carbon source participates in various metabolic processes and regulatory pathways (Macfadyen and Redfield, 1996). The effect of carbon sources on natural transformation has been reported in many other bacteria. For example, transformable states of H. influenzae are affected by the PTS carbon source coupled cAMP (Macfadyen et al., 1996; Macfadyen and Redfield, 1996; Sinha et al., 2013). Carbon source metabolism raises the intracellular cAMP content via PTS, and then influences the expression of downstream receptor genes initiated by the transcriptional regulator cAMP-CRP (Guo et al., 2015). Regulatory effects of TfoX on carbon metabolism has not been reported in other bacteria. Still in the triple regulatory network of natural transformation in V. cholerae, tfoX transcription is induced by the chitin pathway, and TfoX expression in E. coli is directly regulated by the cAMP-CRP complex (Sondberg et al., 2019; Stutzmann and Blokesch, 2020). Whether there is a bidirectional regulation mechanism between TfoX and carbon source metabolic pathways or cAMP must be determined in future studies.
An in-depth analysis of the biological significance of natural transformation in bacteria can provide a new perspectives for studies of bacterial virulence changes, the horizontal spread of antibiotic resistance cassettes and genetic evolution. G. parasuis is a naturally transformable bacterium, and has been mainly used currently to create a set of gene manipulation techniques and to construct gene deletion strains for functional studies (Zhang et al., 2015, 2019; Jiang et al., 2020). However, the natural transformation mechanism in G. parasuis is unknown. In this study, we focused on the genetic heterogenicity of tfoX as represented by SC1401tfoX and SH0165tfoX, and discovered that it is an essential determinant for the natural transformation of G. parasuis. We have also preliminarily explored tfoX regulatory pathways. Future studies to understand the structural differences of the two TfoX variants and their regulatory functions via á vis the natural transformation phenotypes of G. parasuis are warranted.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA813391; https://www.iprox.cn/, IPX0004196000.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
XT, ZY, and KD: designed the experiments. Y-FC, SC, XHu, RW, QZ, YH, SD, YL, XW, and XHa: performed the experiments with assistance from GL, XT, YZ, and BH. XT, ZY, YW, and KD: analyzed the data and wrote the paper. All the authors reviewed and approved the final manuscript.
This work was funded by the Chengdu Science and Technology Bureau key R&D Support Plan (2022-YF05-00817-SN).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.948633/full#supplementary-material
1. ^https://itol.embl.de/ (Accessed July 16, 2022)
2. ^https://www.ncbi.nlm.nih.gov/nuccore/CP015099.1 (Accessed July 16, 2022)
3. ^https://www.ncbi.nlm.nih.gov/bioproject/PRJNA317522 (Accessed July 7, 2022)
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Antonova, E. S., and Hammer, B. K. (2015). Genetics of natural competence in Vibrio cholerae and other Vibrios. Microbiol. Spectr. 3, VE-0010-2014.
Attaiech, L., Boughammoura, A., Brochier-Armanet, C., Allatif, O., Peillard-Fiorente, F., Edwards, R. A., et al. (2016). Silencing of natural transformation by an RNA chaperone and a multitarget small RNA. Proc. Natl. Acad. Sci. U. S. A. 113, 8813–8818. doi: 10.1073/pnas.1601626113
Bigas, A., Garrido, M. E., de Rozas, A. M., Badiola, I., Barbe, J., and Llagostera, M. (2005). Development of a genetic manipulation system for Haemophilus parasuis. Vet. Microbiol. 105, 223–228. doi: 10.1016/j.vetmic.2004.10.015
Cameron, A. D., and Redfield, R. J. (2008). CRP binding and transcription activation at CRP-S sites. J. Mol. Biol. 383, 313–323. doi: 10.1016/j.jmb.2008.08.027
Chen, I., and Dubnau, D. (2004). DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2, 241–249. doi: 10.1038/nrmicro844
Cohen, J. J., Eichinger, S. J., Witte, D. A., Cook, C. J., Fidopiastis, P. M., Tepavcevic, J., et al. (2021). Control of competence in Vibrio fischeri. Appl. Environ. Microbiol. 87:e01962-20. doi: 10.1128/AEM.01962-20
Costa, T. R. D., Harb, L., Khara, P., Zeng, L., Hu, B., and Christie, P. J. (2021). Type IV secretion systems: advances in structure, function, and activation. Mol. Microbiol. 115, 436–452. doi: 10.1111/mmi.14670
Dai, K., He, L., Chang, Y. F., Cao, S., Zhao, Q., Huang, X., et al. (2018a). Basic characterization of natural transformation in a highly transformable Haemophilus parasuis strain SC1401. Front. Cell. Infect. Microbiol. 8:32. doi: 10.3389/fcimb.2018.00032
Dai, K., Jin, J., Wen, Y., Wen, X., He, L., Cao, S., et al. (2016). Complete genome sequence of highly virulent Haemophilus parasuis serotype 11 strain SC1401. Genome Announc. 4:e00628-16. doi: 10.1128/genomeA.00628-16
Dai, K., Wen, X., Chang, Y. F., Cao, S., Zhao, Q., Huang, X., et al. (2018b). A streptomycin resistance marker in H. parasuis based on site-directed mutations in rpsL gene to perform unmarked in-frame mutations and to verify natural transformation. PeerJ 6:e4253. doi: 10.7717/peerj.4253
Dai, K., Yang, Z., Ma, X., Chang, Y. F., Cao, S., Zhao, Q., et al. (2021). Deletion of polyamine transport protein PotD exacerbates virulence in Glaesserella (Haemophilus) parasuis in the form of non-biofilm-generated bacteria in a murine acute infection model. Virulence 12, 520–546. doi: 10.1080/21505594.2021.1878673
Dalia, A. B., and Dalia, T. N. (2019). Spatiotemporal analysis of DNA integration during natural transformation reveals a mode of nongenetic inheritance in bacteria. Cell 179, 1499–1511. doi: 10.1016/j.cell.2019.11.021
Domingues, S., Harms, K., Fricke, W. F., Johnsen, P. J., da Silva, G. J., and Nielsen, K. M. (2012). Natural transformation facilitates transfer of transposons, integrons and gene cassettes between bacterial species. PLoS Pathog. 8:e1002837. doi: 10.1371/journal.ppat.1002837
Domingues, S., Nielsen, K. M., and da Silva, G. J. (2012). Various pathways leading to the acquisition of antibiotic resistance by natural transformation. Mob. Genet. Elem. 2, 257–260. doi: 10.4161/mge.23089
Domingues, S., Rosario, N., Candido, A., Neto, D., Nielsen, K. M., and Da Silva, G. J. (2019). Competence for natural transformation is common among clinical strains of resistant Acinetobacter spp. Microorganisms 7:30. doi: 10.3390/microorganisms7020030
Dubnau, D., and Blokesch, M. (2019). Mechanisms of DNA uptake by naturally competent bacteria. Annu. Rev. Genet. 53, 217–237. doi: 10.1146/annurev-genet-112618-043641
Durieux, I., Ginevra, C., Attaiech, L., Picq, K., Juan, P. A., Jarraud, S., et al. (2019). Diverse conjugative elements silence natural transformation in Legionella species. Proc. Natl. Acad. Sci. U. S. A. 116, 18613–18618. doi: 10.1073/pnas.1909374116
Evans, B. A., and Rozen, D. E. (2013). Significant variation in transformation frequency in Streptococcus pneumoniae. ISME J. 7, 791–799. doi: 10.1038/ismej.2012.170
Fontaine, L., Wahl, A., Flechard, M., Mignolet, J., and Hols, P. (2015). Regulation of competence for natural transformation in streptococci. Infect. Genet. Evol. 33, 343–360. doi: 10.1016/j.meegid.2014.09.010
Griffith, F. (1928). The significance of pneumococcal types. J. Hyg. (London) 27, 113–159. doi: 10.1017/s0022172400031879
Guo, M., Wang, H., Xie, N., and Xie, Z. (2015). Positive effect of carbon sources on natural transformation in Escherichia coli: role of low-level cyclic AMP (cAMP)-cAMP receptor protein in the derepression of rpoS. J. Bacteriol. 197, 3317–3328. doi: 10.1128/JB.00291-15
Hepp, C., and Maier, B. (2016). Kinetics of DNA uptake during transformation provide evidence for a translocation ratchet mechanism. Proc. Natl. Acad. Sci. U. S. A. 113, 12467–12472. doi: 10.1073/pnas.1608110113
Howell, K. J., Peters, S. E., Wang, J., Hernandez-Garcia, J., Weinert, L. A., Luan, S. L., et al. (2015). Development of a multiplex PCR assay for rapid molecular serotyping of Haemophilus parasuis. J. Clin. Microbiol. 53, 3812–3821. doi: 10.1128/JCM.01991-15
Huang, L., Liu, M., Zhu, D., Xie, L., Huang, M., Xiang, C., et al. (2021). Natural transformation of Riemerella columbina and its determinants. Front. Microbiol. 12:634895. doi: 10.3389/fmicb.2021.634895
Humbert, O., Dorer, M. S., and Salama, N. R. (2011). Characterization of Helicobacter pylori factors that control transformation frequency and integration length during inter-strain DNA recombination. Mol. Microbiol. 79, 387–401. doi: 10.1111/j.1365-2958.2010.07456.x
Jaskolska, M., and Gerdes, K. (2015). CRP-dependent positive autoregulation and proteolytic degradation regulate competence activator Sxy of Escherichia coli. Mol. Microbiol. 95, 833–845. doi: 10.1111/mmi.12901
Jiang, C., Cheng, Y., Cao, H., Zhang, B., Li, J., Zhu, L., et al. (2020). Effect of cAMP receptor protein gene on growth characteristics and stress resistance of Haemophilus parasuis serovar 5. Front. Cell. Infect. Microbiol. 10, 19. doi: 10.3389/fcimb.2020.00019
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, S., Yu, Y. P., Liu, S. M., Fu, H. W., Huang, J. Z., Shu, Q. Y., et al. (2019). Identifying mutations by high resolution melting in a TILLING population of rice. J. Vis. Exp. e59960. doi: 10.3791/59960
Lo Scrudato, M., Borgeaud, S., and Blokesch, M. (2014). Regulatory elements involved in the expression of competence genes in naturally transformable Vibrio cholerae. BMC Microbiol. 14, 327. doi: 10.1186/s12866-014-0327-y
Lorenz, M. G., and Wackernagel, W. (1994). Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58, 563–602. doi: 10.1128/mr.58.3.563-602.1994
Macfadyen, L. P., and Redfield, R. J. (1996). Life in mucus: sugar metabolism in Haemophilus influenzae. Res. Microbiol. 147, 541–551. doi: 10.1016/0923-2508(96)84010-1
Macfadyen, L. P. D. I., Reizer, J., Saier, M. H. Jr., and Redfield, R. J. (1996). Regulation of competence development and sugar utilization in Haemophilus influenzae Rd by a phosphoenolpyruvate:fructose phosphotransferase system. Mol. Microbiol. 21, 941–952. doi: 10.1046/j.1365-2958.1996.441420.x
Maughan, H., and Redfield, R. J. (2009). Extensive variation in natural competence in Haemophilus influenzae. Evolution 63, 1852–1866. doi: 10.1111/j.1558-5646.2009.00658.x
Mell, J. C., Hall, I. M., and Redfield, R. J. (2012). Defining the DNA uptake specificity of naturally competent Haemophilus influenzae cells. Nucleic Acids Res. 40, 8536–8549. doi: 10.1093/nar/gks640
Metzger, L. C., Matthey, N., Stoudmann, C., Collas, E. J., and Blokesch, M. (2019). Ecological implications of gene regulation by TfoX and TfoY among diverse Vibrio species. Environ. Microbiol. 21, 2231–2247. doi: 10.1111/1462-2920.14562
Nero, T. M., Dalia, T. N., Wang, J. C., Kysela, D. T., Bochman, M. L., and Dalia, A. B. (2018). ComM is a hexameric helicase that promotes branch migration during natural transformation in diverse gram-negative species. Nucleic Acids Res. 46, 6099–6111. doi: 10.1093/nar/gky343
Redfield, R. J., Cameron, A. D., Qian, Q., Hinds, J., Ali, T. R., Kroll, J. S., et al. (2005). A novel CRP-dependent regulon controls expression of competence genes in Haemophilus influenzae. J. Mol. Biol. 347, 735–747. doi: 10.1016/j.jmb.2005.01.012
Seitz, P., and Blokesch, M. (2013). Cues and regulatory pathways involved in natural competence and transformation in pathogenic and environmental gram-negative bacteria. FEMS Microbiol. Rev. 37, 336–363. doi: 10.1111/j.1574-6976.2012.00353.x
Simpson, C. A., Podicheti, R., Rusch, D. B., Dalia, A. B., and van Kessel, J. C. (2019). Diversity in natural transformation frequencies and regulation across Vibrio species. mBio 10:e02788-19.
Sinha, S., Mell, J. C., and Redfield, R. J. (2012). Seventeen Sxy-dependent cyclic AMP receptor protein site-regulated genes are needed for natural transformation in Haemophilus influenzae. J. Bacteriol. 194, 5245–5254. doi: 10.1128/JB.00671-12
Sinha, S., Mell, J., and Redfield, R. (2013). The availability of purine nucleotides regulates natural competence by controlling translation of the competence activator Sxy. Mol. Microbiol. 88, 1106–1119. doi: 10.1111/mmi.12245
Soejima, M., and Koda, Y. (2021). High-resolution melting analysis for detection of fusion allele of FUT2. Electrophoresis 42, 315–318. doi: 10.1002/elps.202000241
Sondberg, E., Sinha, A. K., Gerdes, K., and Semsey, S. (2019). CRP interacts specifically With Sxy to activate transcription in Escherichia coli. Front. Microbiol. 10, 2053. doi: 10.3389/fmicb.2019.02053
Stutzmann, S., and Blokesch, M. (2020). Comparison of chitin-induced natural transformation in pandemic Vibrio cholerae O1 El Tor strains. Environ. Microbiol. 22, 4149–4166. doi: 10.1111/1462-2920.15214
Wisniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Yang, Y., Koirala, B., Sanchez, L. A., Phillips, N. R., Hamry, S. R., and Tal-Gan, Y. (2017). Structure-activity relationships of the competence stimulating peptides (CSPs) in Streptococcus pneumoniae reveal motifs critical for intra-group and cross-group ComD receptor activation. ACS Chem. Biol. 12, 1141–1151. doi: 10.1021/acschembio.7b00007
Yang, Y., and Tal-Gan, Y. (2019). Exploring the competence stimulating peptide (CSP) N-terminal requirements for effective ComD receptor activation in group1 Streptococcus pneumoniae. Bioorg. Chem. 89:102987. doi: 10.1016/j.bioorg.2019.102987
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Yu, J., Wu, J., Zhang, Y., Du, Y., Peng, J., Chen, L., et al. (2014). Identification of putative virulence-associated genes among Haemophilus parasuis strains and the virulence difference of different serovars. Microb. Pathog. 77, 17–23. doi: 10.1016/j.micpath.2014.10.001
Zhang, L., Huang, L., Huang, M., Wang, M., Zhu, D., Wang, M., et al. (2021). Effect of nutritional determinants and TonB on the natural transformation of Riemerella anatipestifer. Front. Microbiol. 12:644868. doi: 10.3389/fmicb.2021.644868
Zhang, B., Ku, X., Zhang, X., Zhang, Y., Chen, G., Chen, F., et al. (2019). The AI-2/luxS quorum sensing system affects the growth characteristics, biofilm formation, and virulence of Haemophilus parasuis. Front. Cell. Infect. Microbiol. 9:62. doi: 10.3389/fcimb.2019.00062
Zhang, L., Li, Y., Dai, K., Wen, X., Wu, R., Huang, X., et al. (2015). Establishment of a successive Markerless mutation system in Haemophilus parasuis through natural transformation. PLoS One 10:e0127393. doi: 10.1371/journal.pone.0127393
Keywords: Glaesserella parasuis, natural transformation, tfoX, molecular diversity, competence gene
Citation: Tang X, Yang Z, Dai K, Liu G, Chang Y-F, Tang X, Wang K, Zhang Y, Hu B, Cao S, Huang X, Yan Q, Wu R, Zhao Q, Du S, Lang Y, Han X, Huang Y, Wen X and Wen Y (2022) The molecular diversity of transcriptional factor TfoX is a determinant in natural transformation in Glaesserella parasuis. Front. Microbiol. 13:948633. doi: 10.3389/fmicb.2022.948633
Edited by:
Baolei Jia, Chung-Ang University, South KoreaReviewed by:
Douglas B Rusch, Saurashtra University, IndiaCopyright © 2022 Tang, Yang, Dai, Liu, Chang, Tang, Wang, Zhang, Hu, Cao, Huang, Yan, Wu, Zhao, Du, Lang, Han, Huang, Wen and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiping Wen, d3lwQHNpY2F1LmVkdS5jbg==.
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.