 Sven Holtrup
Sven Holtrup Maximilian Greger
Maximilian Greger Benjamin Mayer
Benjamin Mayer Mara Specht1
Mara Specht1 Barbara Waidner
Barbara Waidner
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 August 2022
Sec. Microbial Physiology and Metabolism
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.929194
One important factor that promotes the colonization of the upper digestive system of the human pathogen Helicobacter pylori is its helical cell shape. The bacteria cell shape is predominantly defined by its peptidoglycan cell wall. In rod-shaped species, PG synthesis is mediated by two dynamic molecular machines that facilitate growth along the perpendicular axis and the septum, called the elongasome and the divisome, respectively. Furthermore, many bacteria evolved additional mechanisms to locally change PG synthesis patterns to generate diverse cell shapes. Recent work characterizing cell shape mutants of Helicobacter pylori revealed a novel mechanism for the generation of a twisted helix from a rod, including PG-modifying enzymes as well as additional proteins such as the bactofilin homolog CcmA or the membrane proteins Csd5 and Csd7. In this study, we investigate the localization and dynamics of CcmA and Csd7 using live-cell imaging. We also address the question of how these change in the presence or absence of the putative interaction partners.
Bacterial cells are usually associated with characteristic morphology and size, and many plausible models regarding the functional significance of cell morphology have emerged (Young, 2006; Yang et al., 2016). The ε-proteobacterium Helicobacter pylori is known for its colonization of the human stomach where it potentially causes gastric inflammations leading to ulcers and gastric cancer (Marshall and Warren, 1984). Among other pathogenicity factors, its typical cell shape has been described to enhance efficient stomach colonization (Bonis et al., 2010; Sycuro et al., 2010). Studies on the molecular basis of H. pylori have shed light on a novel mechanism of cell shape control as reviewed in Salama (2020). As such, a set of peptidoglycan endopeptidases, as well as cytoskeletal proteins, has been identified to act in concert and promote helical cell shape by locally relaxing and cross-linking the PG sacculus (Bonis et al., 2010; Sycuro et al., 2010, 2012). Factors predicted to work within this complex are the cell shape determining proteins Csd1/2, Csd5, and Csd7 as well as the bactofilin homolog CcmA and the PG precursor synthase MurF (Blair et al., 2018; Yang et al., 2019). The M23-type metallopeptidase Csd1 forms a heterodimer with its enzymatically inactive counterpart Csd2 which cleaves PG tetra-penta cross-links (An et al., 2016). Further factors found to be involved in helical shape generation are Csd3/HpdA, Csd4, and Csd6 (Sycuro et al., 2013). Csd5 is a single-pass inner membrane protein that has a short cytoplasmatic N-terminal domain which was found to interact with CcmA and MurF and a C-terminal SH3 domain which interacts with the PG (Blair et al., 2018). Another direct connection between cytoplasmatic factors and the periplasm is the multi-transmembrane domain protein Csd7, which was found to interact with Csd2 in the periplasm and was found to co-purify with CcmA and MurF in co-immunoprecipitation, respectively (Yang et al., 2019). The combination of these results in H. pylori cells suggests the existence of one or more the so-called Shapeosome complexes, for which a schematic model was postulated and presented in Salama (2020).
Bactofilins resemble a widespread family of bacterial polymer-forming proteins. Interestingly, some examples have recently been described in the eukaryotic stramenopiles clade (Deng et al., 2019). Bactofilins are defined by a central beta-helical domain (DUF583), which enables polymerization into filaments or 2D crystalline sheets based on hydrophobic interactions. This central domain is usually flanked by mainly unstructured N- and C-terminal regions that are thought to mediate protein–protein interactions. Filaments are largely insensitive to pH, salt, or chelating agents (Koch et al., 2011; Deng et al., 2019; Holtrup et al., 2019). Diverse functions have been described for bactofilins, ranging from directing PG modifications (Kühn et al., 2010; Sycuro et al., 2010) to chromosome segregation (Lin et al., 2017) to flagella synthesis and motility (El Andari et al., 2015). In the stalked budding bacterium C. crescentus, the two homologs BacA and BacB cooperate to form a sheet-like structure that mediates the localization of a PG synthesis to the stalk (Kühn et al., 2010). Analogously, in its close relative Asticaccaulis biprosthecum, the bactofilin homolog BacA is recruited by the morphogen SpmX to organize zonal PG synthesis at the stalks (Caccamo et al., 2020). Interestingly, bactofilin genes in both organisms are within an operon with M23-type endopeptidases, as csd1/2 and ccmA. This is also true for the Thermus thermophilus homolog TtBacA, of which cryo-EM data gave insights into the structure of bactofilin filaments, demonstrating that monomers polymerize in a head–head-like manner (Deng et al., 2019).
Despite an increasing knowledge on bactofilins, the role of CcmA in generating H. pylori helical shape is still under the investigation. According to the latest model, a core of the putative shape complex is likely to be formed by the membrane-spanning non-enzymatic protein Csd7, which interacts with Csd2 in the periplasm, and may be bound to the membrane-associated CcmA, which in turn is thought to interact directly with the inner membrane proteins MurF and Csd5. However, it is still unclear how these proteins work together to promote cell shape. Subcellular localization is one of the most important aspects in determining protein function. For this reason, we have previously shown by immunofluorescence that the H. pylori bactofilin CcmA localizes as subcellular accumulations in varying numbers adjacent to the cell membrane (Holtrup et al., 2019). A recent study, also using immunofluorescence, confirmed this finding and showed that the H. pylori bactofilin of strain LSH100 is present in cells as numerous spots with the preference of the major axis. Here, the authors propose a model where CcmA and MreB promote PG synthesis at areas of increased positive and negative Gaussian curvatures, respectively (Taylor et al., 2020).
In this study, we focus on the subcellular in vivo localization and dynamics of the bactofilin CcmA in two different H. pylori strains using fluorescent fusions of CcmA at the native gene locus. This allowed us to perform epifluorescence, super-resolution microscopy, and single-particle (molecule) tracking (SPT/SMT) (Rösch et al., 2018). We also addressed the question of whether the absence of the putative interaction partners Csd5 and Csd7 changes the dynamics of CcmA and shows Csd7 in vivo localization and dynamics both in the presence and in the absence of CcmA.
Subcellular localization of the H. pylori bactofilin has previously been described by us and others using immunofluorescence. By that, it was shown that CcmA preferentially localizes to the areas of increased positive Gaussian curvature and that altered polymerization properties lead to the loss of helical shape (Taylor et al., 2020). To analyze in vivo dynamics of CcmA, we generated strains in which a C-terminal Gfp-tagged version of ccmA was located under the native promotor in the chromosome. H. pylori isolates display diverse morphologies (Martinez et al., 2016). Since a major question is how CcmA affects cell shape, we used the laboratory strains G27 and 26695, as both strains are well characterized and exhibit a different morphological plasticity, with G27 cells being a twisted helix, whereas 26695 cells have predominantly curved rod morphology. Since the loss of CcmA leads to a drastic defect in cell shape, we were able to verify the influence of the Gfp-tag on our ccmA_gfp strain by qualitative and quantitative shape analyses. For qualitative analysis, we acquired phase-contrast images of both wt strains and compared them with their respective ccmA_gfp derivatives (Figure 1A). Qualitative comparisons of these images gave the impression that both ccmA_gfp strains showed similar morphology to the wild type in both strains, with tendency toward a slightly less curved shape (Figure 1A, upper panels). To more accurately assess cell shape and highlight potential shape defects, we induced filamentous growth by treatment with the drug aztreonam (Figure 1A, lower panels). Thereby, the effects on the spiral cell shape were highlighted, thus facilitating the interpretation of the images. We confirmed the correct size of our fusion product, as well as semiquantitatively monitored the protein amount by western blot analysis over the growth phases (Figure 1B), thus demonstrating that our Gfp fusion did not show degradation of CcmA and Gfp in either strain (Figure 1B and Supplementary Figure 1). However, the signal appears to be somewhat weaker in the Gfp fusion cells. This could partially result from the increase in molecular weight and associated transfer effects during blotting; however, we cannot rule out that protein levels are affected. Further, no change in signal intensity during the cell cycle could be detected in wt and ccmA_gfp fusion strains, as normalization to the loading control (UreB) demonstrated (Supplementary Figure 1).
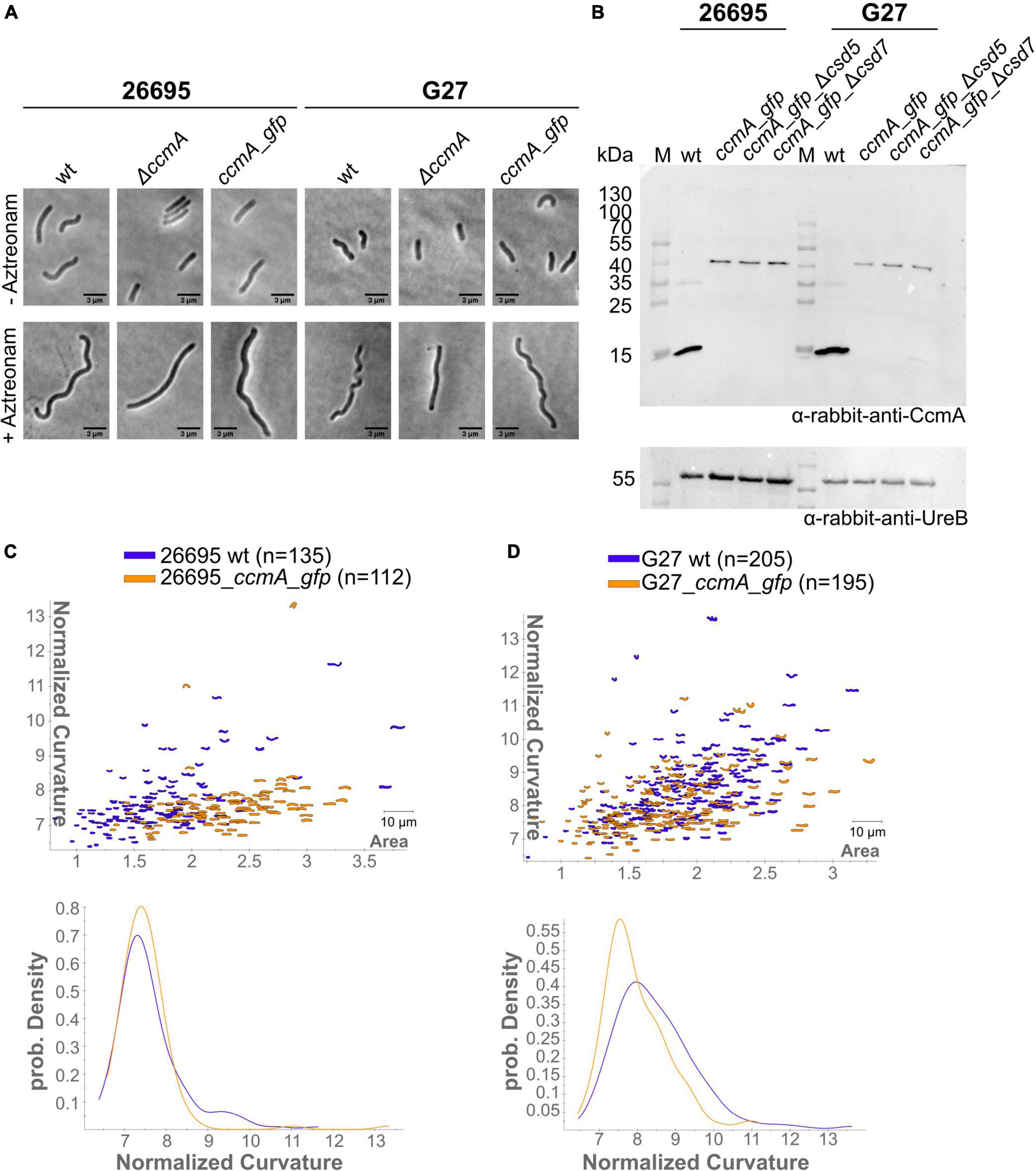
Figure 1. (A) Exemplary phase-contrast images illustrating the effect of the insertion of ccmA_gfp at the native locus on the cell shape of H. pylori 26695 and G27. For a better comparison, filamentous growth was induced by treating cells with Aztreonam. CcmA_Gfp cells are slightly less curved as compared to the wt but clearly not as straight as the ccmA knockout. (B) Western blot of H. pylori strains was used in this study. The membrane was first probed with CcmA-specific antibodies. Afterward, the membrane was stripped and again probed with UreB-specific antibodies, serving as a loading control. (C) Comparison of 26695 wt and 26695_ccmA_gfp by principal component analysis and probability density plot of normalized curvature. (D) Comparison of G27 wt and G27_ccmA_gfp by principal component analysis and probability density plot of normalized curvature.
For quantitative analysis of the phase-contrast images, the CellTool software described in the study of Pincus and Theriot (2007) was used. In this procedure, the contours of the cells were extracted from the phase-contrast images and plotted by area against curvature normalized by cell length. By comparing 26695 wt cells to 26695 ccmA_gfp cells, both cell populations share a large portion of similarly shaped cells. However, 26695 wt cells additionally exhibit a small population of cells with higher curvature which is not present in ccmA_gfp cells (Figure 1C). This observation is more pronounced in strain G27 (Figure 1D). In summary, our results show that the distribution of CcmA_Gfp cells in both strains strongly overlaps with that of the corresponding wt cells. In both cases, however, the wt cells show a slightly broader distribution toward higher curvature, reflecting the rather small influence of the protein fusion. Taken together, we concluded that the ccmA_gfp fusions in both strains are capable of replacing most of the wt bactofilin function. However, some limitations due to spherical effects could not be excluded.
To get detailed insights into the subcellular localization of CcmA, we performed structured illumination microscopy (SIM) with exponentially growing cells of our generated ccmA_gfp fusion strains using multi-color SIM mode with three different excitation wavelengths and three rotations (Figure 2A). CcmA_Gfp was found to localize to the inner membrane in both strains as small foci as shown by projections of reconstructed Z-stacks (Figure 2A).
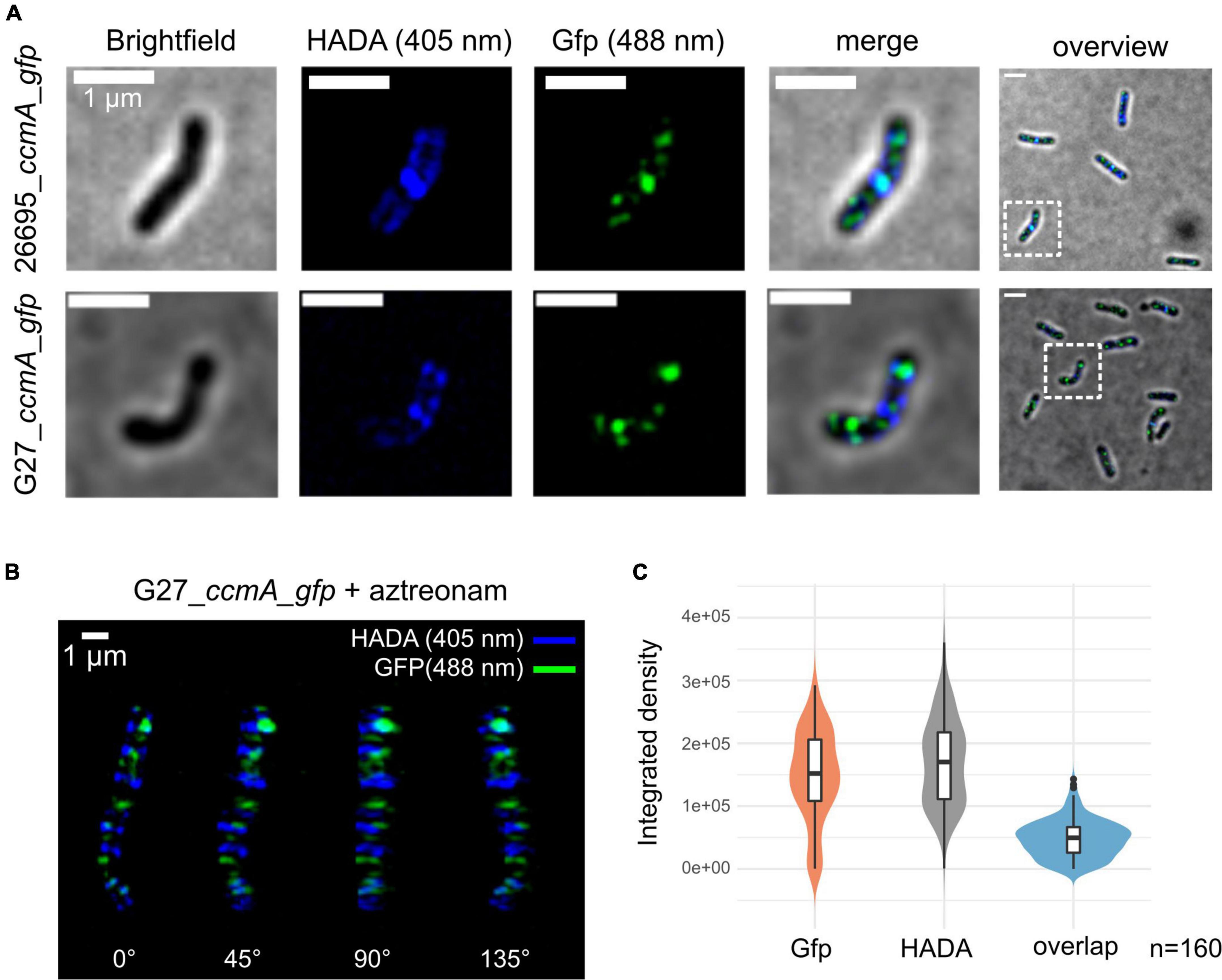
Figure 2. Structured illumination microscopy of 26695_ccmA_gfp and G27_ccmA_gfp. Exponentially growing cells were pulse-labeled with HADA and images were reconstructed from three rotation angles (A) Brightfield, HADA (blue) and GFP (green) channels of exemplary 26695_ccmA_gfp (upper lane) and G27_ccmA_gfp (lower lane) cells. (B) 3D-view of aztreonam-treated G27_ccmA_gfp cell containing multiple growth rings. (C) Violin plot of integrated density distributions of the HADA- and Gfp signals compared to overlapping regions of 26695_ccmA_gfp cells.
Because the current model describes CcmA as the part of a dynamic PG-modifying complex preferentially localized at positive super curvature (Taylor et al., 2020), we were interested in whether these patches would localize to the areas of increased peptidoglycan insertion. Therefore, we visualized the zones of increased PG synthesis by pulsed incubation with the fluorescently labeled D-alanine derivative HADA (3-[[(7-hydroxy-2-oxo-2H-1-benzopyran-3-yl)carbonyl]amino]-D-alanine). Initial inspection indicated that the CcmA_Gfp signal, although broadly distributed across the cell, was often present close to the HADA signal but did not appear to overlap with it (Figure 2A). To facilitate visualization and analysis of this pattern, we induced filamentous growth by treatment with aztreonam (Figure 2B). Because aztreonam inhibits Pbp2A at the septum and thus blocks septation, treatment results in multiple growth rings in elongated cells. This points out the observed pattern illustrated by the projection of the 3D reconstructed filamentous cells G27_ccmA_gfp (Figure 2B). Also, the signals from CcmA_Gfp and HADA appear to be only close to each other, without being colocalized (Figure 2B). We complemented this observation with a quantification of the signal overlap by subtracting Z-projections of individual cells (n = 160) from three independent replicates and measuring the respective integrated densities. As visualized by violin plots, we found only very few signals to overlap (Figure 2C). Furthermore, to examine the correlation of the two signals, we used the Pearson correlation coefficient (PCC), which is a measure of the linear relationship between two sets of data. The signal correlation was performed using the Fiji (Schindelin et al., 2012) plugin JACoP (Bolte and Cordelières, 2006). This yielded a Pearson correlation coefficient of r = 0.103 and Manders coefficients of M1 = 0.007 (proportion of Gfp signal overlapping with HADA proportion) and M2 = 0.004 (HADA proportion overlapping with Gfp proportion), indicating that the two signals are neither correlated nor overlapping.
To study the localization of CcmA in terms of dynamics, we used the established single-molecule tracking approach (SMT) of exponentially growing cells. In this approach, cells are first bleached with a slim-field laser until individual fluorophore movements can be observed, which has been shown to be a powerful tool for analyzing dynamic processes in living cells [reviewed in Rösch et al. (2018)]. It was shown that freely diffusing Gfp molecules can only be tracked accurately with very fast acquisition rates (e.g., 5 ms; Stracy et al., 2014; Schibany et al., 2018). Since we wanted to focus on slower diffusing molecules and exclude these background activities from our analysis, we set our acquisition rate at 50 ms. Using this approach, we confirmed that the individual CcmA_Gfp molecules were localized in clusters near the membrane along the cell. Qualitative motion analysis using the Fiji (Schindelin et al., 2012) plugin TrackMate (Tinevez et al., 2017) revealed a picture of signal clusters (Figure 3A), in which individual molecules occasionally jumped from one cluster to another. To characterize these arbitrarily shaped clusters, we analyzed the data using the density-based spatial clustering of applications with noise (DBSCAN) algorithm, which is commonly used to quantitatively assess subcellular assemblies. Clusters are defined based on the search radius (R) and the minimum number of single-molecule localizations (SMLs) within that radius (N). As such, the DBSCAN algorithm was applied to the tracking data obtained from the same movies but with the Fiji package TrackMate (Tinevez et al., 2017), and we used the R packages DBSCAN (Hahsler et al., 2019) and fpc (Hennig, 2020) for the analysis. Using the identical parameters, an average value of 6.84 ± 2.65 and 6.91 ± 2.64 clusters per cell was obtained for strains 26695_ccmA_gfp and G27_ccmA_gfp, respectively (Supplementary Figure 3). In addition, we also wanted to get a picture of the dynamics within the cell-shaping complex. Therefore, we focused on the two proteins potentially interacting with CcmA, Csd5, and Csd7 (Blair et al., 2018). Csd7 is a multi-transmembrane domain protein that is thought to act as a link between cytoplasmic factors and the periplasmic LytM domain of the endopeptidase Csd1. Csd5 is a single membrane-bound protein with a periplasmic SH3b domain and a short cytoplasmic appendage that has been shown to interact directly with CcmA (Blair et al., 2018). Therefore, we generated deletion strains of csd5 and csd7 in the ccmA_gfp background of both wt strains. First, we also examined the CcmA clusters in these strains using the DBSCAN algorithm. The result showed that the average number of clusters was almost the same for all strains (Supplementary Figure 3).
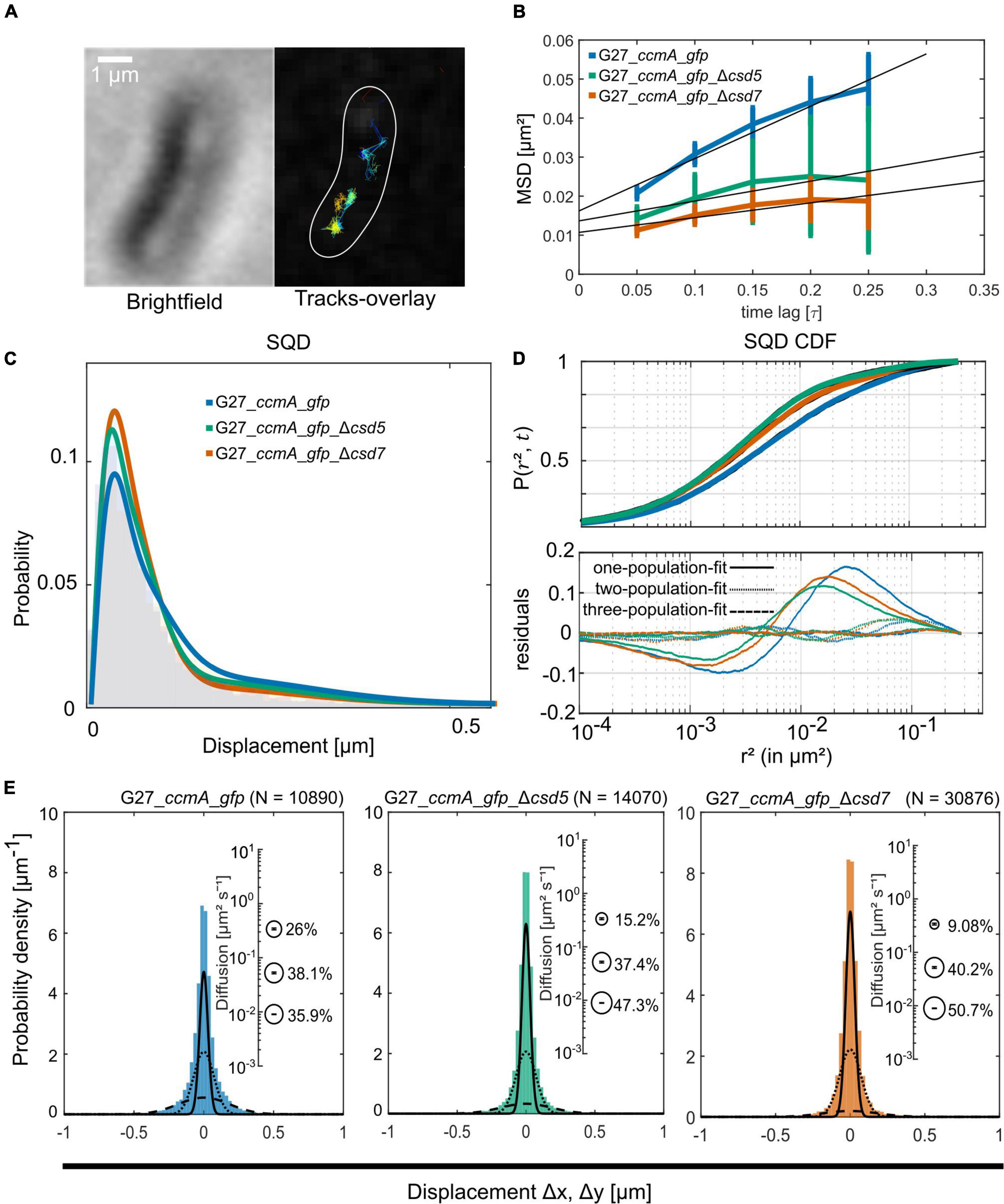
Figure 3. Single-molecule dynamics of CcmA_Gfp in wt, Δcsd5, and Δcsd7 background. (A) Brightfield image of an example cell and respective CcmA_Gfp tracks. Scale bar = 1 μm. (B) Mean-squared displacement of single-molecule tracks of CcmA_Gfp in different strain backgrounds, plotted against the time lag. (C) Density plot illustrating the probability for different squared displacement lengths for CcmA_Gfp. (D) The cumulative distribution function of CcmA_Gfp squared displacements. The probability for a molecule to stay within a certain radius r2 at a certain time t is plotted against the radius. The residue plot in the lower panel illustrates the differences of the applied models (single, double, or triple fit) to the data. (E) Probability densities of single-molecule displacements fitted by Gaussian-mixture models. Population sizes estimated from a triple-fit model are illustrated as bubble plots. We observed a slight decrease of CcmA_Gfp single-step distances in the deletion strains as compared to wt background.
For further SMT analysis, bleaching curves were generated in the G27 strains and the movies were cropped to single-molecule level. Detection of single-molecule tracks was performed with u-track (Jaqaman et al., 2008), and cell edges were labeled with Oufti (Paintdakhi et al., 2016). These data were combined in the SMTracker software as described in the study of Rösch et al. (2018). All single-molecule data analyses were done using SMTracker (Rösch et al., 2018), which uses R2 as a measure for goodness of fit and several statistical tests (Kolmogorov–Smirnov goodness-of-fit, null hypothesis significance) to determine whether acquired distributions of molecule movements can be explained by single or multiple fractions.
A common approach to describe the molecular motion of individual particles is the mean square displacement (MSD). Here, molecular 2D displacements are averaged over several steps (τ). For our analysis, only continuous tracks of at least five steps were considered. As a result, we obtained an overall diffusion constant of D = 0.044 μm2/s for CcmA_Gfp in the presence and 0.02 μm2/s and 0.019 μm2/s in the absence of Csd5 and Csd7, respectively (Figure 3B and Table 1). Thus, the results showed a decrease in the overall diffusion of CcmA_Gfp in the absence of the putative interaction partners Csd5 and Csd7.
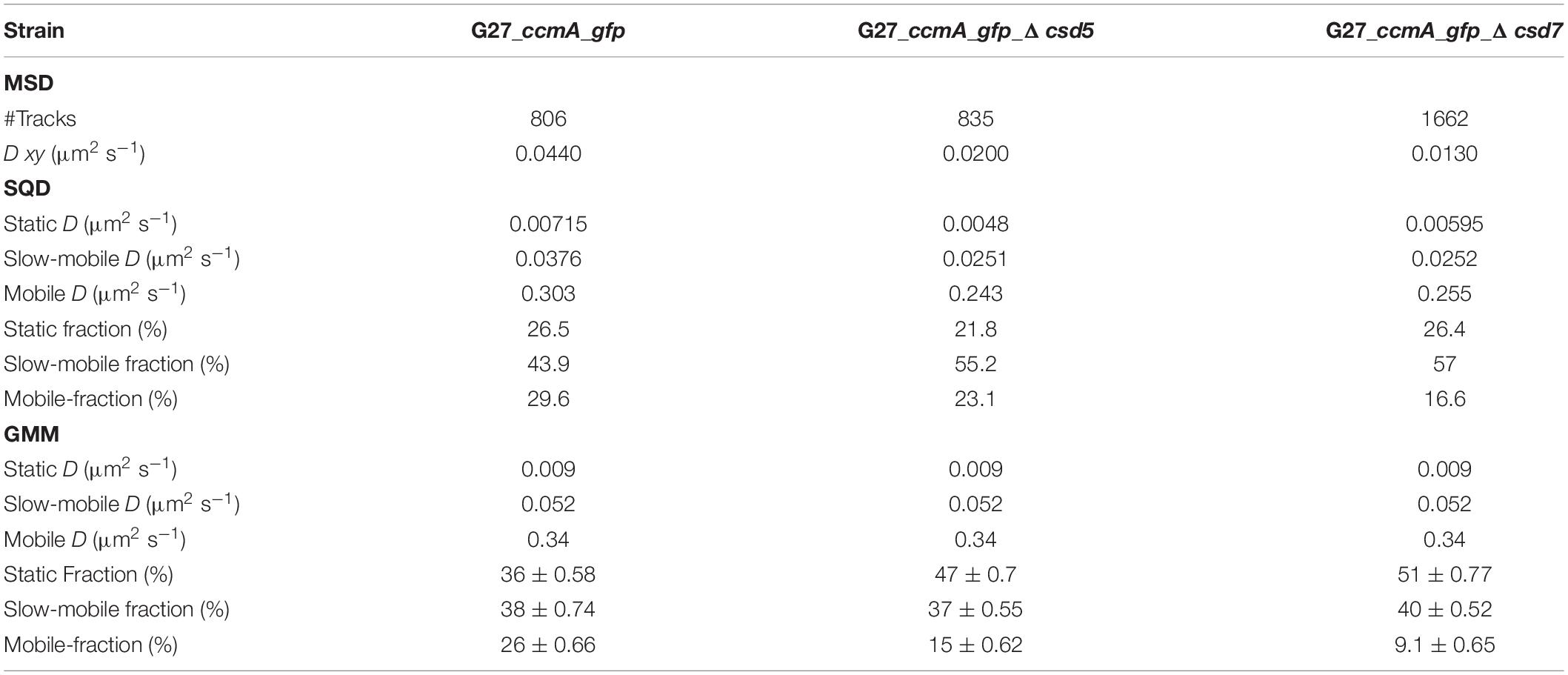
Table 1. Summary of CcmA_Gfp single-molecule dynamics.
Next, we analyzed the diffuse behavior at subpopulation levels. Therefore, we applied a squared displacement (SQD) analysis that scores the probability density of the squared x/y-displacements of certain molecules (Figure 3C). The cumulative distribution function (CDF) of these squared displacements (r2) represents the probability P (r2, t) of a molecule remaining in a circle of radius r in time t. To determine whether the recorded distributions of molecular motions could be represented by single or multiple fractions, the SMTracker software used R2 as the goodness of several statistical tests (Kolmogorov–Smirnov goodness-of-fit, null hypothesis significance) (Figure 3D). To avoid overfitting, the approximate number of diffuse groups is determined using the Bayes information criterion. The bottom panel of Figure 3D shows the differences (“residuals”) between the modeled data and the measured values with respect to random diffusion, represented by the baseline (“0”). The smallest deviation from our model occurred when using a triple fit, indicating that this best describes the situation of our fusion protein. However, it should be noted that the SMTracker software does not consider higher-order adjustments. In brief, the static population of G27_ccmA_gfp represents 26.5% of all captured movements (D = 0.018 μm2/s), the slow-mobile 43.9% (D = 0.038 μm2/s), and the mobile fraction 29.6% (D = 0.303 μm2/s). In the absence of Csd5 or Csd7, we observe that the mobile population shifts to the smaller ones, which is consistent with the MSD analysis (Table 1). In addition, this analysis also suggests an increase in shorter tracks for CcmA_Gfp in csd5- and csd7-deficient cells. Individual analysis of three replicates showed only minor day-specific differences and further validated the analysis of the datasets (Supplementary Figure 3). Another commonly used strategy for subpopulation analysis is the Gaussian mixture model (GMM). Unlike the SQD method, the GMM is based on the probability that molecules take a number of steps. The Kolmogorov–Smirnov goodness-of-fit (KS GoF) implemented in SMTracker 2.0 rejected two and one population fits, whereas three populations were accepted for the three datasets (Figure 3E and Table 1). Both the fraction sizes and the corresponding diffusion coefficients derived from this GMM analysis show a decrease in the fast fraction whereas the static fraction size increases in csd5- and csd7-deficient cells, supporting the previous results of the SQD and MSD analysis (refer to bubble bots Figure 3D and Table 1). However, the observed effect on the CcmA diffusion of the two missing proteins is rather small.
As another control for our CcmA fusion, we generated a CcmA_mNG fusion as mNeonGreen (mNG) was reported as the brightest monomeric green or yellow fluorescent protein (Shaner et al., 2013). Furthermore, mNG exhibits increased photostability and a shorter maturation time (Shaner et al., 2013). In addition, we placed a linker between the bactofilin and the fluorophore to address concerns about potentially impaired interaction of the individual CcmA monomers (please refer to Supplementary Figure 4 for a comparison of the two structures). Also, we created a mNG fusion of the point mutant CcmA_I55A, for which polymerization defects and a diffuse in vivo localization as foci were reported (Taylor et al., 2020). The correct size and stability of both fusion products were confirmed by western blot analysis (Supplementary Figure 5). Structural illumination microscopy (SIM) images of exponentially growing cells confirmed the helical shape of the CcmA_mNG (Figure 4A) fusion and exhibited a similar in vivo localization as compared to CcmA_Gfp (Figure 1A). No preference for areas of positive or negative curvature was observed (Figures 3D, 4A visualization in Supplementary Movie 1), which we also not observed for CcmA_Gfp. As expected, the point mutation showed a straight phenotype, and in vivo localization analysis revealed a signal distributed throughout the cell without any preferences (Figure 4B). Furthermore, the polymeric state of CcmA is thought to be the active functional form (Taylor et al., 2020). In addition, also, the observed dynamics of the CcmA_mNG fusion using the SQD analysis represented the results similar to the CcmA_Gfp fusion (Figure 4D). As SMT analysis of the non-polymerizing mutant allows us to distinguish which of the observed subpopulations represents the active functional fraction, we subsequently performed SMT analyses with both variants. Based on the observed signal distributed in the cell and the known lack of polymerization ability of the CcmAI55A_mNG fusion, we expected the changes to occur mainly in the region of fast-moving molecules. Thus, to better account for this fraction of proteins in the molecules, we moved to an exposure time of 20 ms. Only a small shift in population sizes toward faster molecules or CcmA_mNG in the SQD analysis was shown by studying the difference between the two exposure times (Figures 4C,D and Table 2). Subsequently, we compared the CcmA_mNG fusion with the CcmAI55A_mNG fusion using the mean-squared displacement (MSD) analysis. In fact, MSD analysis of at least three independent SMT experiments with 20-ms exposure time demonstrated that the CcmAI55A_mNG derivative moved faster than CcmA_mNG (Figure 4C). To answer the question of which of the subpopulations might represent the polymeric and non-polymeric fractions, we compared the dynamics of CcmA_mNG and CcmAI55A_mNG using the provided apparent diffusion (AppD) analyses of the SMT software. AppD determines the diffusion constants based on the time-averaged MSD. Figure 4F shows the estimated population and diffusion coefficients (Table 3) illustrated by bubble plots. While 42.3% of the CcmA_mNG molecules showed static behavior, only 19% of the molecules in the CcmAI55A_mNG variant belonged to this fraction. Accordingly, the proportion of fast-moving molecules of CcmAI55A_mNG increased to 49.3%. With 34.5% for CcmA_mNG and 31.7% for CcmAI55A_mNG, almost no difference was observed in the slow-moving fraction. Based on these results, we can assume that the polymeric, and thus presumably functional, state of CcmA is represented by the static population.
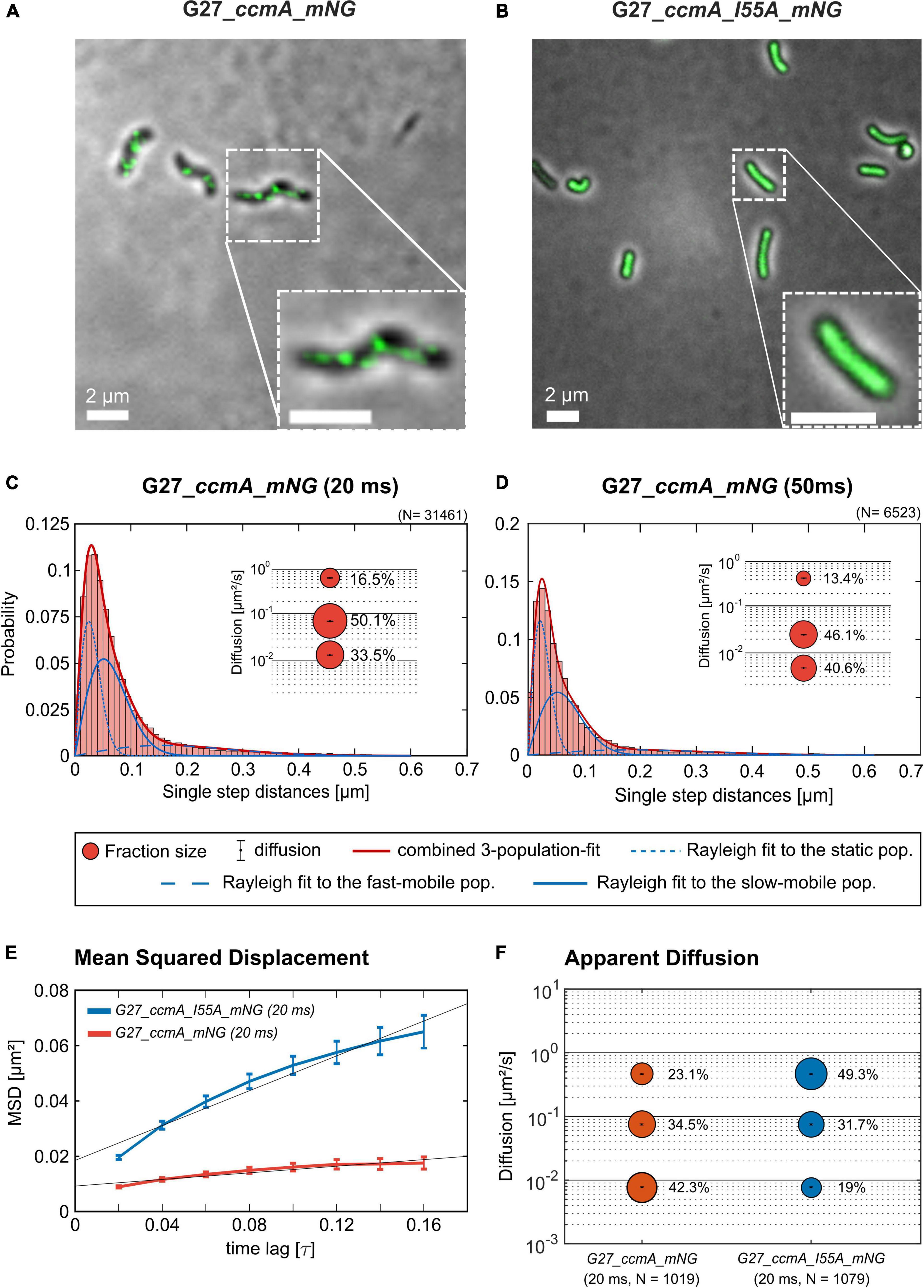
Figure 4. Structured illumination microscopy imaging of CcmA_mNG (A) and CcmA_I55A_mNG (B) in G27 at exponential phase, scale = 2 μm. Polymeric CcmA forms distinct foci at the membrane as shown by Z-projection while its mutated derivate CcmA_I55A is distributed equally over the cell (microscope settings mNG: 488 nm laser, 10.0%, exp. time: 20 ms, 3 rotations, EMCCD gain: 50.0). (C,D) Squared displacement analysis (SQD) of CcmA_mNG with an exposure time of 20 (C) and 50 ms (D) per frame of single-molecule tracks as obtained from slim-field bleaching microscopy. Diagrams display the probability for each detected jump distance with non-simultaneous SQD curve fitting. Data acquired with 50-ms exposure time represent one replicate. (E) Mean-squared displacement curves with an aligned linear fitting to the first 9 time lags showing a general diffusion regardless of subdiffusion. The MSD analysis demonstrates that the inhibited polymerization of CcmA_I55A results in an overall faster movement. (F) Apparent diffusion analysis of CcmA_mNG and CcmA_I55A_NG in G27 of single-molecule tracks obtained from slim-field bleaching microscopy. Bubble plots show the estimated population and diffusion coefficients as identified by simultaneous APPD curve fitting. Data derived from at least three biological triplicates with an exposure time of 20 ms per frame. CcmA_I55A_mNG (126 cells, N = 1,079 tracks) exhibited a strong shift to the mobile population as compared to CcmA_mNG (113 cells, N = 1,019 tracks) while the slow-mobile population remain unaffected, suggesting a functional state represented by the static population.
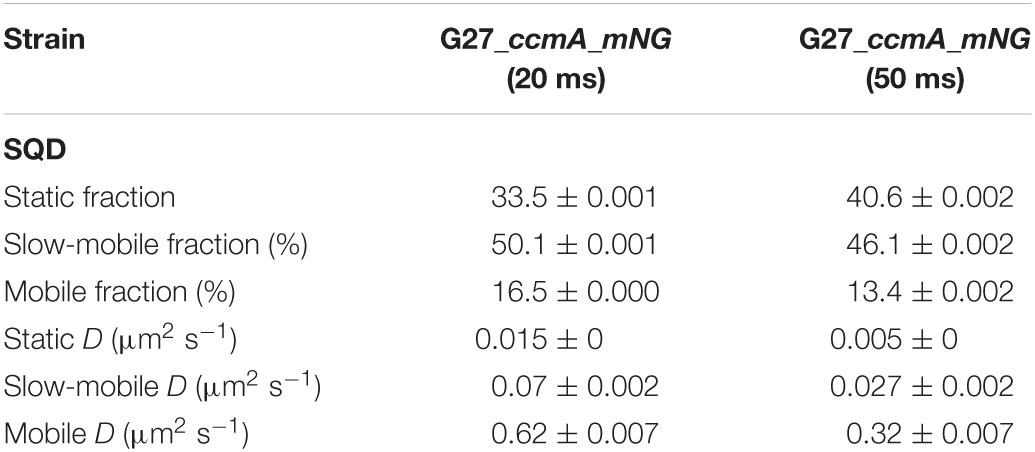
Table 2. Summary of CcmA_mNG single-molecule dynamics acquired with 20- and 50-ms exposure time according to squared displacement analysis (SQD).
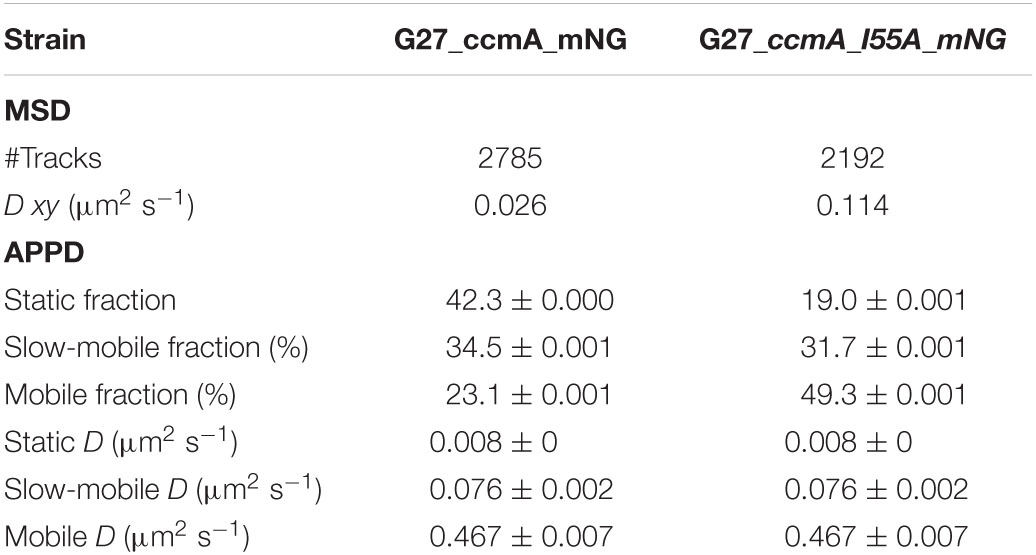
Table 3. Summary of CcmA_mNG single-molecule dynamics according to mean-squared displacement (MSD) and apparent diffusion (APPD) analysis.
Next, we also wanted to characterize the potential interacting partner Csd7 of CcmA and additionally investigate this suggested interaction of Csd7 and CcmA from the other perspective. The exact function of the membrane protein Csd7 is still unknown. Therefore, we constructed a Csd7_mNG fusion both in wt strains G27 and 26695 and the corresponding CcmA deletions. Considering that the loss of Csd7 also leads to altered cell morphology (Yang et al., 2019), we verified the functionality using the quantitative cell shape analysis according to the analysis of the CcmA_Gfp fusion strains performed. For this purpose, the CellTool software was used (Pincus and Theriot, 2007) and the results were visualized as cell contours on a coordinate plane (Figures 5A,B). Because the spiral morphology is more pronounced in strain G27, the differences among wild-type cells (gray), the Csd7_mNG fusions (blue), and the bactofilin-deficient derivatives (red) are more apparent in this strain than in the less curved strain 26695. However, in both cases, the Csd7_mNG fusion strains (blue) had similar curvatures to the wild type (gray) and were only slightly shorter, reflecting a rather minor influence of the protein fusion. Bactofilin-deficient cells (red) exhibited the expected straighter morphology, demonstrating the successful introduction of the bactofilin deletion in the Csd7_mNG fusion strains. In addition, western blot analysis demonstrated that the Csd7 protein fusion was neither degraded nor resulted in altered CcmA expression in either strain (Figures 5C,D). Detailed in vivo localization analyses using structured illumination microscopy (SIM) images of the Csd7_mNG fusion in both strains revealed extensive filamentous structures with clear membrane association (Figures 5E, 6A). Subjective observation of these structures gave the impression of a perpendicular arrangement. Relatively abundantly, in exponentially growing cells appearing to be close to division, a filamentous structure is seen spiraling along the membrane from the pole to the septum. To illustrate this result, an exemplary cell of strain G27 is shown as a projected 3D image and as a movie (Figure 5E and Supplementary Movie 2).
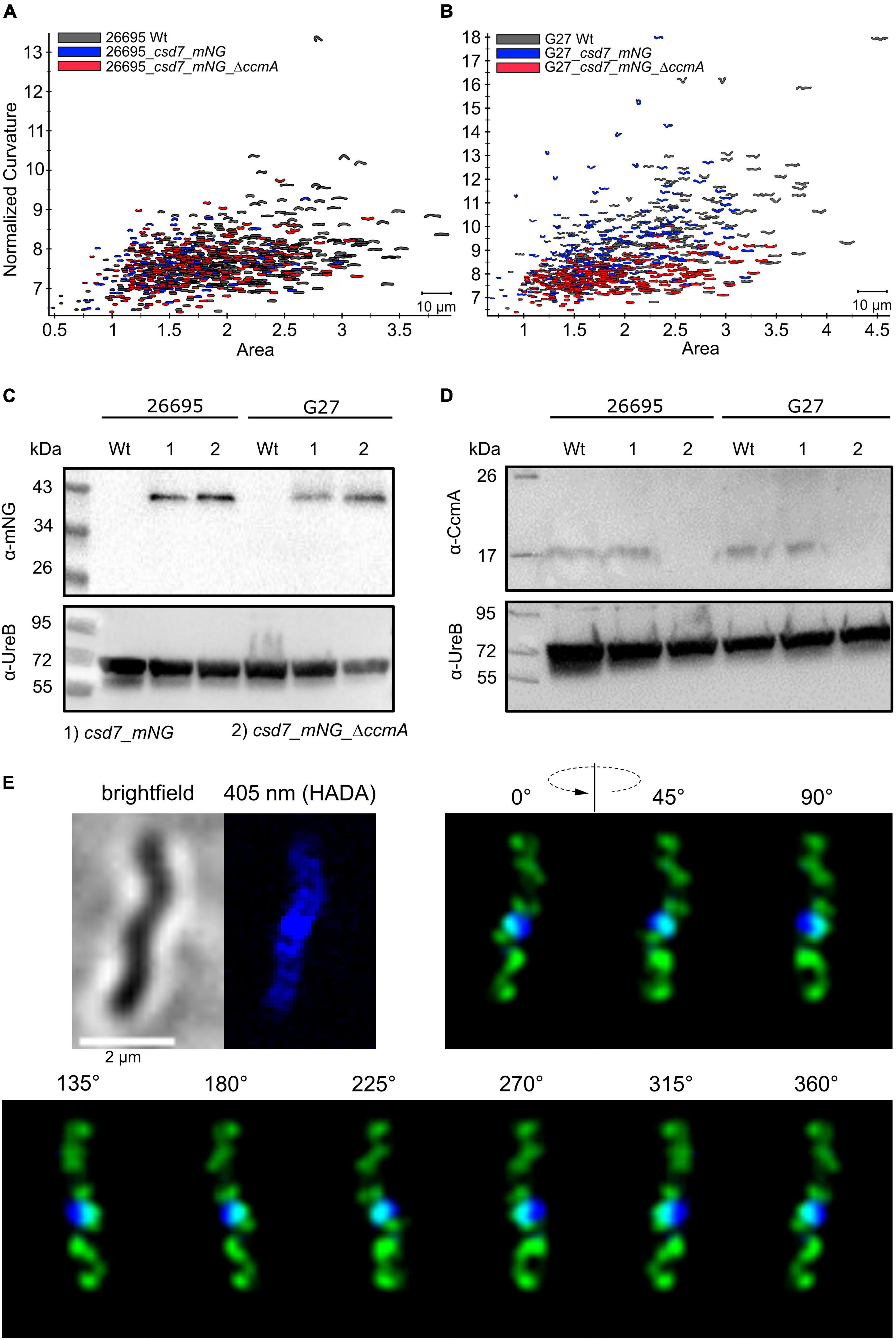
Figure 5. (A,B) Morphological comparison of the wild-type strains (26695, G27-gray) to the respective csd7_mNG (blue) strains and their bactofilin knockout variants (red) by principal component analysis. Cells are illustrated in a scatterplot according to their length (x-axis) and side curvature (y-axis). G27 wild-type, n = 253; G27_csd7_mNG, n = 251; G27_csd7_mNG_ΔccmA, n = 258; 26695 wild-type, n = 278; 26695_csd7_mNG, n = 272; 26695_csd7_mNG_ΔccmA, n = 270. Western blot depicting stable fluorescent fusions of mNG with Csd7 in 26695 and G27 (C) and verifies the correct bactofilin deletion in Csd7_mNG strains (D). The usage of urease B-antibody functions as a loading control for each western blot. (E) Structured illumination microscopy of the Csd7_mNG fusion protein in H. pylori G27. Exemplary cell with Csd7_mNG (green) and newly incorporated peptidoglycan visualized by pulse labeling (20 min) with HADA (blue). Cells were acquired via Z-stack and projected to a 3D-image. Montage shows cells from different angles. Csd7_mNG fusion forms membrane-associated filamentous structures along with the cell. Scale bar = 2 μm.
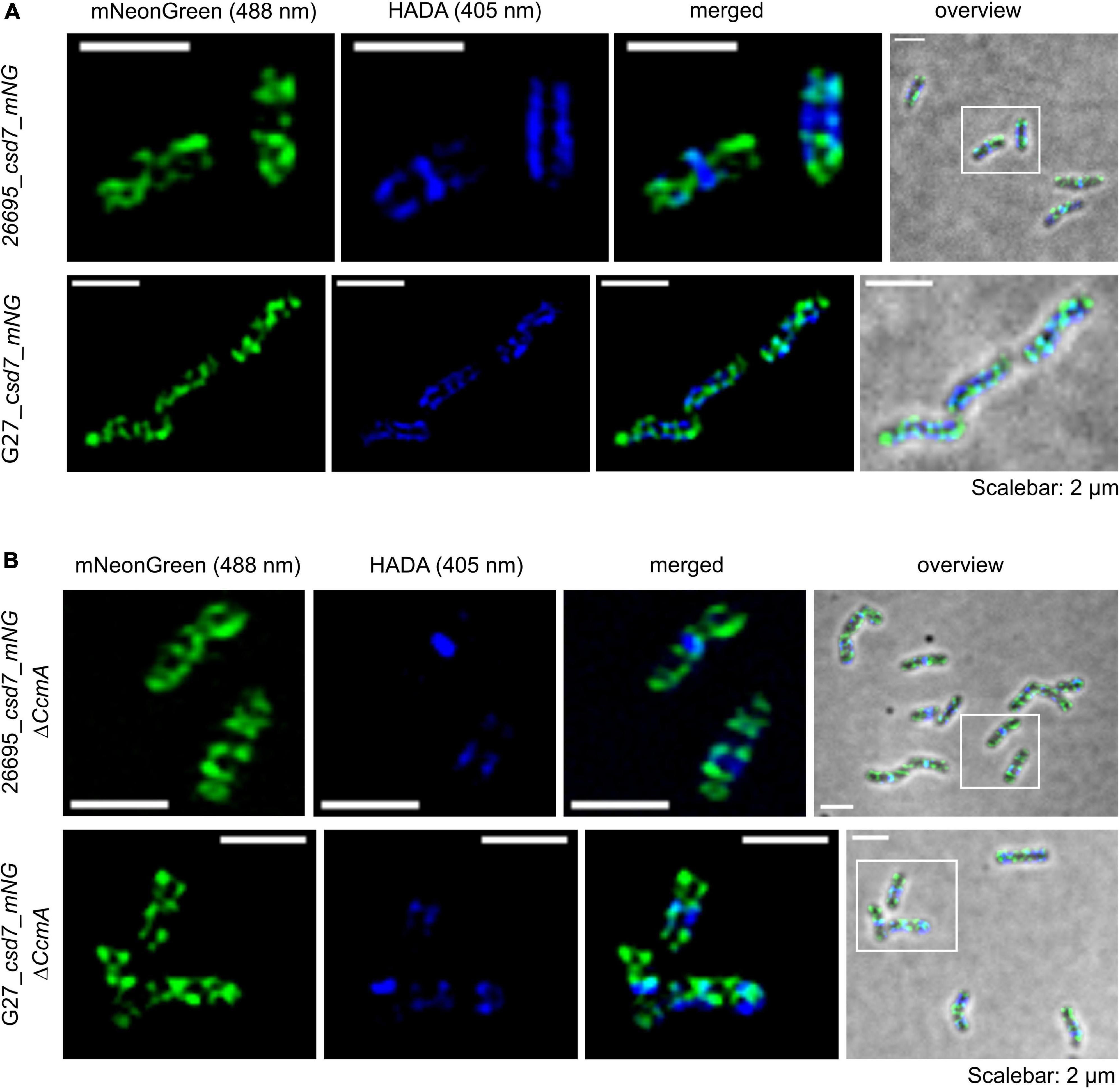
Figure 6. Structured illumination microscopy of the Csd7_mNG fusion protein in H. pylori G27 and 26695 under wild-type conditions (A) and bactofilin deletion background (B). Exemplary 26695 and G27 cells with Csd7_mNG (green) and newly incorporated peptidoglycan visualized by pulse labeling (20 min) with HADA (blue). Cells were acquired via Z-stack and projected by the average intensity. Scale = 2 μm. The signal was located at the membrane with a predominant localization at the cell termini. The comparison revealed no altered signal localization pattern (microscope settings mNG: 488 nm laser, 10.0%, exp. time: 20 ms; HADA: 405 nm laser, 10.0%, exp. time: 50 ms; 3 rotations, EMCCD gain: 50.0).
We next examined the potential colocalization of Csd7_mNG with the areas of increased peptidoglycan incorporation by pulsed incubation with the fluorescently labeled D-alanine derivative HADA (3-[[(7-hydroxy-2-oxo-2H-1-benzopyran-3-yl)carbonyl]amino]-D-alanine). Initial qualitative visual inspection of the Csd7_mNG fusion in both wt strains gave the impression that both signals were often present in close proximity to each other but did not clearly overlap (Figures 5E, 6A), complicating interpretation of colocalization status. As with CcmA_Gfp, we therefore quantified the signals and used the Pearson correlation coefficient to measure the statistical relationship. Pearson correlation analysis (PCC) yielded a Pearson correlation coefficient of r = 0.499 and r = 0.544 for strains G27_csd7_mNG and 26695_csd7_mNG, respectively. These results suggest that about 50% of the two signals overlap, so that at least some colocalization has occurred (Figure 2C). Interestingly, the localization of Csd7_mNG does not appear to be altered in the CcmA-deficient strains from either wt (Figure 6B). Moreover, the Pearson correlation coefficients of the CcmA-deficient strains were r = 0.609 (G27_cds7_mNG_ΔccmA) and r = 0.575 (26695_cds7_mNG_ΔccmA), giving approximately the same or slightly higher probability of colocalization with newly synthesized peptidoglycan (Figure 6B).
Next, we addressed the dynamics of Csd7 both in the presence and in the absence of CcmA using the single-molecule tracking (SMT) approach with an exposure time of 50 ms. As such, the SQD analysis of Csd7_mNG resulted in (at least) three populations of diffusive molecules (Figure 7A) as identified by residual analysis (Figure 7A). According to SQD analysis, 36.2% of Csd7_mNG molecules were assigned to the mobile fraction, with a diffusion coefficient of 0.36 μm2/s, 38.6% of molecules showed an intermediate average diffusion coefficient of 0.03 μm2/s, and 25.2% comprised the static fraction with D = 0.005 μm2/s (Figure 7B left panel and Table 4). Only minor changes were observed with respect to ccmA deletion (Figure 7 and Table 4). In strain G27, the main change was observed in the slow mobility population. However, these data show that the size of the slow-moving population of Csd7 decreases very slightly in the absence of the potential interaction partner CcmA. In each of the other two populations, there was a minimal increase. Even smaller variation was noted in the SQD analysis in the same constellation of strain 26695 (Table 5 and Supplementary Figure 6). Likewise, further mean-squared displacement (MSD) analysis revealed that the deletion of the bactofilin had no effect on the overall diffusion behavior of the Csd7_mNG in both strains (Figure 7C and Supplementary Figure 6B). Thus, including all single-molecule tracking data, it can be noted that the absence of bactofilin CcmA has only a marginal effect on the dynamics and diffusion behavior of membrane protein Csd7 in strains G27 and 26695.
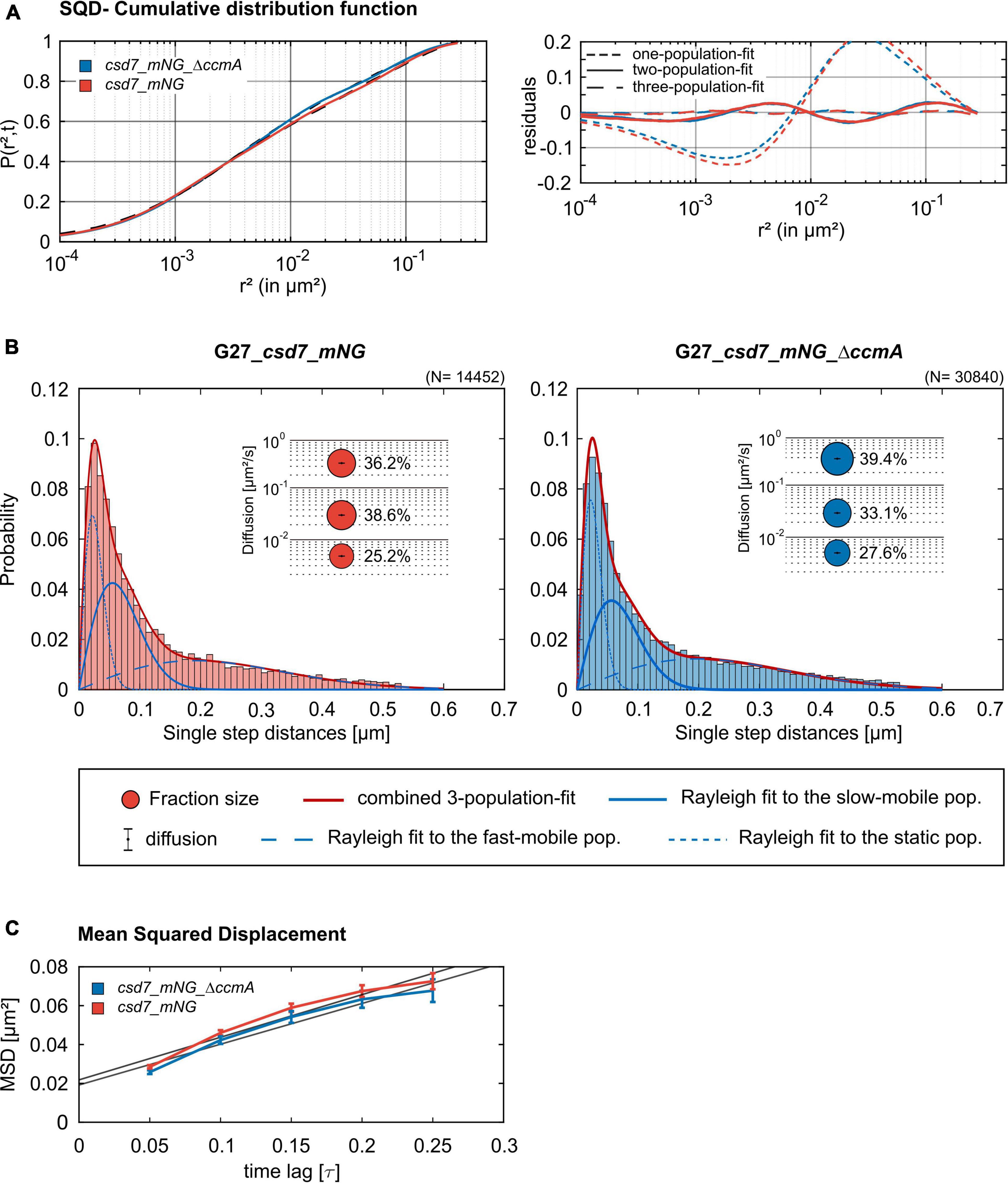
Figure 7. Single-molecule tracking analysis of the G27_csd7_mNG strain and the bactofilin-knockout strain. Data represent a biological triplicate with 95 cells, 2,056 tracks for G27_csd7_mNG and 110 cells, 4,326 Tracks for G27_csd7_mNG_ΔccmA. The movies were acquired with an exposure time of 50 ms per frame. (A) The cumulative distribution function (left panel) indicates the best fit for three populations. Residual analysis (right panel) is a quality indicator of the SQD analysis: the difference between the measured data (colored line) and modeled data (“zero” line) confirms three population assumptions. According to the data, we detected marginal effects of the bactofilin deletion in G27. With diffusion coefficients barely changing, a small fraction of the slow-mobile population shifts to the mobile and static populations. (B) Squared displacement analysis (SQD) of Csd7_mNG in G27 wt and CcmA deletion background. Diagrams display the probability for each detected jump distance. Bubble plots show the estimated population and diffusion coefficients as identified by non-simultaneous SQD curve fitting. N, number of steps. (C) Mean-squared displacement curves show a general diffusion regardless of subdiffusion. According to the MSD, the deletion of CcmA did not affect the overall diffusion of Csd7.
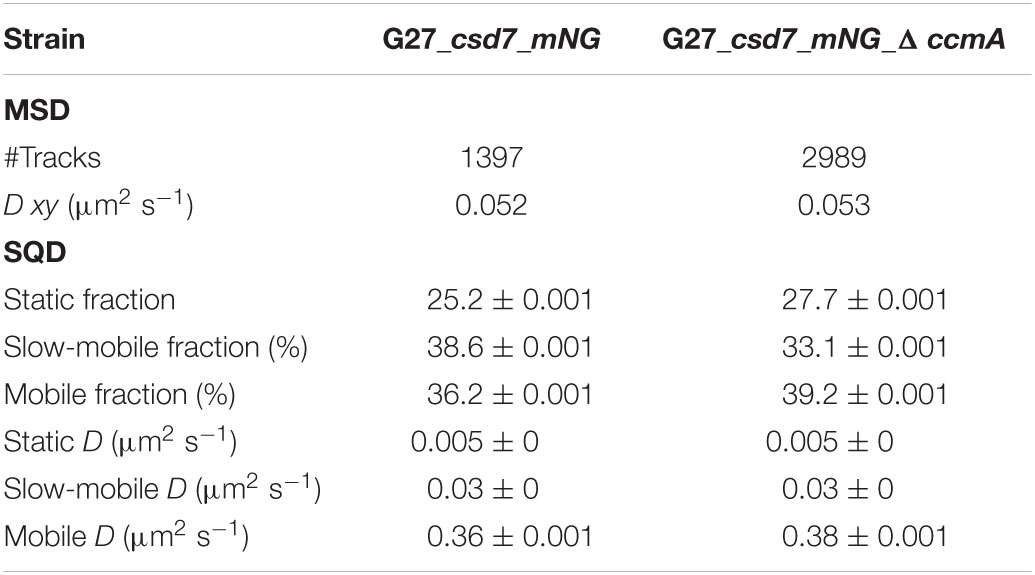
Table 4. Summary of Csd7_mNG single-molecule dynamics in H. pylori G27.
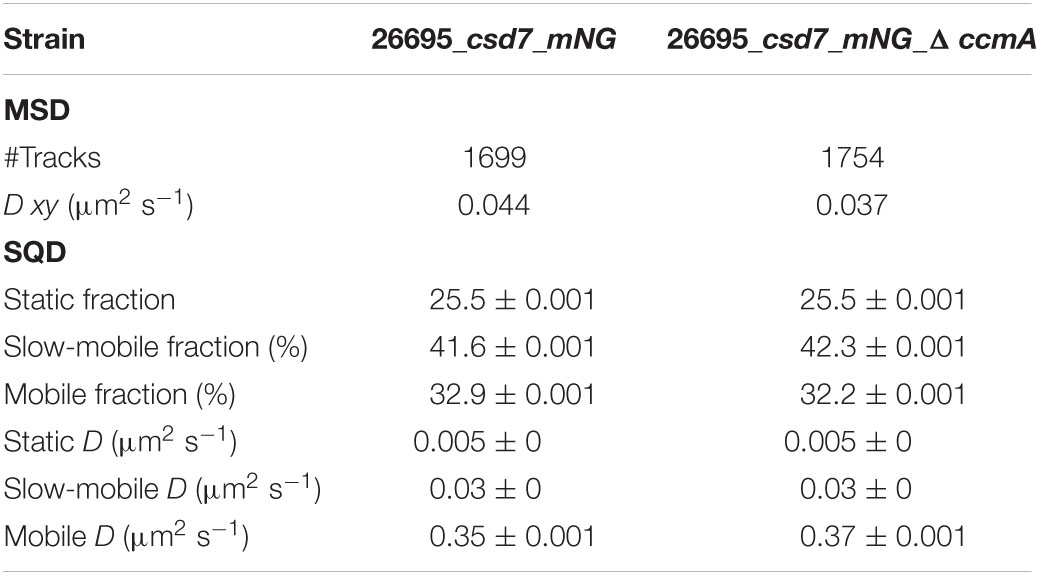
Table 5. Summary of Csd7_mNG single-molecule dynamics in H. pylori 26695.
Recent studies shed light on a novel mechanism of bacterial shape generation in the helically twisted epsilon-proteobacterium H. pylori. This novel mechanism has been shown to rely on the activity of several PG-modifying endopeptidases and non-enzymatic factors such as the membrane-spanning proteins Csd5 and Csd7 (Blair et al., 2018; Yang et al., 2019), the membrane-associated bactofilin CcmA (Blair et al., 2018). Additionally, a set of coiled-coil-rich proteins (Ccrps) has been shown to influence the cell shape (Specht et al., 2011). The current working model assumes that at least some of these factors act in a dynamic complex to facilitate helical cell shape by local PG modifications (Taylor et al., 2020). Even though a relatively detailed image of this complex has been drawn in the literature, concrete information on dynamics and the interplay of these factors remain elusive. We focused on the in vivo localization and dynamics of the bactofilin CcmA of H. pylori and its putative interacting partners Csd7. In this context, we also addressed the question of whether both the number of CcmA clusters and CcmA dynamics is affected in the absence of a further putative interaction partner, Csd5. The ability of bactofilins to polymerize appears to be important for function, and polymerization is thought to be mediated via hydrophobic interactions between conserved hydrophobic residues in the core DUF583 domain (Vasa et al., 2015). Work on T. thermophilus bactofilin showed that the N-terminal region is involved in membrane binding (Deng et al., 2019). Our approach was based on strains expressing a ccmA_gfp fusion from the native ccmA locus under native expression conditions, resulting in a C-terminally tagged fusion protein. Although Gfp is almost two times the size of CcmA, we managed to obtain a fusion with an almost wild-type cell shape in both H. pylori strains 26695 and G27, as shown by 2D shape analysis. In previous studies, C-terminal bactofilin fusions had already been described as functional. For instance, the C-terminal eYfp fusion of BacA in C. crescentus still retained the ability to polymerize (Kühn et al., 2010), and in A. biprosthecum, the functional C-terminal BacA-mVenus fusion exhibited normal stalk formation (Caccamo et al., 2020). In addition to our Gfp fusions, we also generated a CcmA_mNG fusion carrying a linker sequence between the C-terminus and the fluorophore. Interestingly, this fusion appeared to even better restore the wild-type phenotype, indicating a potential role of the C-terminus in protein–protein interactions (S4). Both constructs localize similarly, as shown by the 3D SIM analysis, and also exhibit an analogous dynamic behavior. Moreover, the fact that in both strains, the CcmA fluorophore fusions localize to the membrane contributes to the fusion protein functionality as Taylor et al. (2020) demonstrated that CcmA mutants lacking polymerization do not associate with the membrane. This is also demonstrated by a comparison to our polymerization-deficient CcmAI55A_mNG fusion. For this point mutation, a limited function has already been described in a previous study (Taylor et al., 2020). As expected, the localization observed in vivo was detached from the membrane, thereby validating our obtained results. Analysis of the subcellular localization of CcmA with both fluorophores, which was performed using structural illumination microscopy (SIM) with exponentially growing cells, showed that CcmA was localized as small foci at the inner membrane in both strains, similar to published immunofluorescence data (Taylor et al., 2020). This confirmed the localization of CcmA previously observed by immunofluorescence (Taylor et al., 2020) in vivo. However, no clear localization preference for a specific curvature or defined pattern was observed, and CcmA was localized to both positive and negative curvatures. The same study found CcmA to be enriched in the areas of increased peptidoglycan (PG) synthesis, especially in the areas of negative Gaussian curvature and the major axis of the twisted helix (Taylor et al., 2020). Here, we used the fluorescently labeled D-Ala derivative HADA for probing PG synthesis (Kuru et al., 2019). We mainly observed signals at the septa and in sub-polar regions, which has also been described in the literature. Modified-D-Ala is incorporated into the pentapeptide through the action of PG transpeptidases where they are mainly found at the fifth position in H. pylori (Kuru et al., 2019). The HADA-labeling efficiency may be sensitive to local PG modifications and provides more knowledge about the septal growth zone. In the same context, only weak labeling of laterally dispersed PG insertion by fluorescently labeled D-Ala, as compared to other labeling approaches such as labeling the glycan backbone, has been reported for H. pylori. We found that CcmA frequently neighbored and partially overlapping with PG growth rings. Nevertheless, as indicated by Pearson correlation coefficient, this overlapping is neither positively nor negatively related. These growth rings can serve as landmarks to approximate the relative localization of CcmA in the further studies.
Interestingly, the recently described bactofilin homolog BacA from A. biprosthecum has been described as a recruitment platform and diffusion barrier (Giacomelli et al., 2022). In this organism, the bactofilin homolog BacA was demonstrated to interact with the PG-modifying enzyme SpmX to form pseudostalks. There, BacA presumably subsequently recruits other cell wall-modifying enzymes. In addition, BacA appears to retain SpmX and other factors at the base of the growing pseudostalk, preventing diffusion into the stalk. If we apply these findings to the shaping complex of H. pylori, a similar role of CcmA as a recruitment platform seems very likely. In this context, the interaction of CcmA with other cell shape-determining factors postulated in previous studies, such as Csd1/Csd2, Csd7, and Csd5, is also consistent. However, how these factors interact with each other and how this is spatially and temporally controlled is rather unclear, as the interactions were inferred from in vitro data such as immunoprecipitation and bacterial two-hybrid data. In addition, recent studies have changed the assumption that nucleotide-independent scaffold proteins of the cytoskeleton to which bactofilins belong constitute rigid scaffolds (Giacomelli et al., 2022). In B. subtilis, for example, the molecular function of DivIVA as a scaffolding platform undergoes modulation by other protein factors and is targeted to a specific molecular task that makes either a more stable complex dynamic or a more dynamic complex stable in response to protein–protein interactions (Feddersen et al., 2021). To answer the question of the dynamics of CcmA in living cells using a cell biology approach, we investigated this with single-molecule tracking using the SMT software (Rösch et al., 2018). In this context, we assume that if other shaping factors are controlled by CcmA, their diffusion behavior would change accordingly in the presence or absence of CcmA and vice versa.
By analyzing single-molecule steps using the squared displacement model (SQD) and the Gaussian mixture model, we identified at least three diffusive subpopulations for CcmA due to the limitation of the software. In both models, diffusion constants and population sizes were in the same range. We termed the slowest population static and the more mobile populations slow-mobile and fast-mobile fractions. Both models illustrate that the static and the fast-mobile fractions represent the largest and smallest populations, respectively.
Based on our previous knowledge of CcmA, we assume that the static fraction represents the sheet-like polymer structure and the fast-mobile fraction represents the freely diffusive units, whereas the intermediate fraction most likely represents an intermediate form of these two states. A comparison of CcmA with CcmAI55A under the same imaging conditions confirmed that the static fraction corresponds to the polymeric state, leading to the further conclusion that this fraction is the one required for cell shaping. Previous studies have proposed a speculative model in which the two non-enzymatic putative scaffold proteins, Csd5 and Csd7, and Csd1-3 and MurF, work together to promote helical twisting of the cell shape.
Previous studies postulated that the N-terminal and transmembrane domains of Csd5 and the integral membrane protein Csd7 interact with CcmA to be the part of a multiprotein transmembrane complex to connect the cytoplasm to the periplasm. However, according to our observations, CcmA molecules tend to diffuse only slightly more slowly in the absence of both putative interaction partners. Moreover, neither the absence of Csd5 nor Csd7 has any effect on the formation of CcmA clusters. Interestingly, the same tendency of Csd7 dynamics was observed in the absence of CcmA. However, a closer examination of the localization pattern of Csd7 using structured illumination microscopy (SIM) images of the generated Csd7_mNG fusion strain in both strain 26695 and G27 revealed a very different pattern from that observed in the CcmA fusion strains. As such, extensive filamentous structures were observed with clear membrane association and the impression of a perpendicular arrangement. Similar to CcmA, Csd7_mNG was found near PG growth rings. Interestingly, as indicated by Pearson correlation coefficient, the Csd7 fusion was lightly co-localized with the PG machinery, unlike CcmA. Since no direct interaction between Csd7 and CcmA has been found so far, we hypothesize that they might interact indirectly in the same dynamic complex. However, we have not observed major changes in the dynamics of CcmA in the absence of Csd7 or vice versa, so it seems more likely that the two act in different subcomplexes. As described previously, CcmA and Csd7 share the PG precursor synthase MurF as an interaction partner (Yang et al., 2019). MurF normally acts on ATP-driven synthesis of PG precursors in the cytoplasm and interacts directly with the elongasome via MurG (Favini-Stabile et al., 2013), MraY (White et al., 2010), and MreB (Mohammadi et al., 2007). Interestingly, four N-terminal transmembrane domains were identified for H. pylori MurF (Blair et al., 2018). Also, in agreement with our 3D-SIM data, it would be conceivable that Csd7 also prefers elongasome-like localization, whereas CcmA has more localized foci. While we did not find large changes in the dynamics of CcmA or Csd7, we did detect a slight tendency of both molecules to diffuse more slowly when putative interaction partners are absent. A possible explanation for this could also be that the proteins are uncoupled from the driving force of the main complex. Another possibility with respect to CcmA is that fewer molecules are released from the complex when interaction partners are absent. However, both ideas are highly speculative and require further investigation.
In summary, our work provides the first in vivo localization and dynamics of the bactofilin CcmA and its putative interaction partner Csd7 and thus makes an important contribution to the model of how CcmA might act within the postulated shapeosome complex. Based on our results, we can now also question whether CcmA could be a linker and thus possibly a regulatory factor between different complexes, as the interaction with both putative interaction partners seems to be temporally limited, if it occurs at all. Thus, this study provides further insight into the strategy of H. pylori to regulate its helical cell shape, which is critical for its virulence.
Helicobacter pylori strains were routinely cultivated on Dent blood agar in a microaerobic atmosphere as described earlier (Schätzle et al., 2015). Growth experiments were performed in Brucella broth with 5% fetal calf serum (BBF). All growth experiments were performed in triplicate and were repeated at least three times. E. coli strains were grown aerobically at 37°C in Luria-Bertani media. When needed, growth media were supplemented with 50 μg/l ampicillin, 20 μg/l kanamycin, or 20 μg/l chloramphenicol, respectively. Ellipsoid testing was performed according to the manufacturer’s instructions. For visualization of cell wall synthesis, we utilized the fluorescently labeled D-amino acid derivative HADA according to Kuru et al. (2019). HADA was added to a final concentration of 0.5 mM, and the samples were incubated under a micro-aerophilic atmosphere for 30 min at 37°C, 150 rpm. Subsequently, two parts of ice-cold ethanol were added and incubated for 10 min on ice. Cells were washed three times in PBS before mounting and imaging.
Cloning was performed in E. coli according to standard protocols. Restriction and modifying enzymes (New England Biolabs, United States) were used according to the manufacturer’s instructions. Plasmids were isolated from E. coli with the QIAprep Spin MiniPrep Kit (QIAGEN, Germany), E.Z.N.A. Plasmid DNA Mini Kit (Omega Bio-Tek, United States), and Monarch Plasmid Miniprep Kit (NEB). The isogenic H. pylori csd5 and csd7 deletion mutants were constructed as described earlier (Waidner et al., 2009; Schätzle et al., 2015). Briefly, the resistance marker gene (kan) was fused to upstream and downstream DNA regions of mutagenized genes using a modified version of the megaprimer polymerase chain reaction (PCR) protocol (Sarkar and Sommer, 1990) and primers listed in Table 6. Subsequently, marker exchange mutagenesis of H. pylori was performed according to the standard procedures (Leying et al., 1992). H. pylori mutants carrying the resistance genes inserted into the chromosome were selected by growth on Dent blood agar containing kanamycin at concentrations of 20 mg/l. The correct insertions were verified by PCR and sequencing. Notably, downstream of csd5 is aroE and encodes shikimate dehydrogenase, which plays an essential role in basic metabolism. Since deletion of Csd5 did not result in growth restriction, a polar effect on the aroE gene is not likely. Csd7 is localized at the end of an operon. The isogenic H. pylori ccmA_gfp strain was generated using the double crossover integration of the pRDX-C (Smeets et al., 2000) in which the rdxA coding regions were replaced by the ccmA_gfp fusion and the coding region of the downstream gene. Construction of ccmA_mNG fusion was performed by replacing Gfp from already existing pRDX-C_ccmA_gfp_1541 plasmid with mNG via vector-PCR and Gibson assembly (New England Biolabs). CcmA mutant I55A was separately inserted in pRDX-C_ccmA_mNG according to the published method (Edelheit et al., 2009).
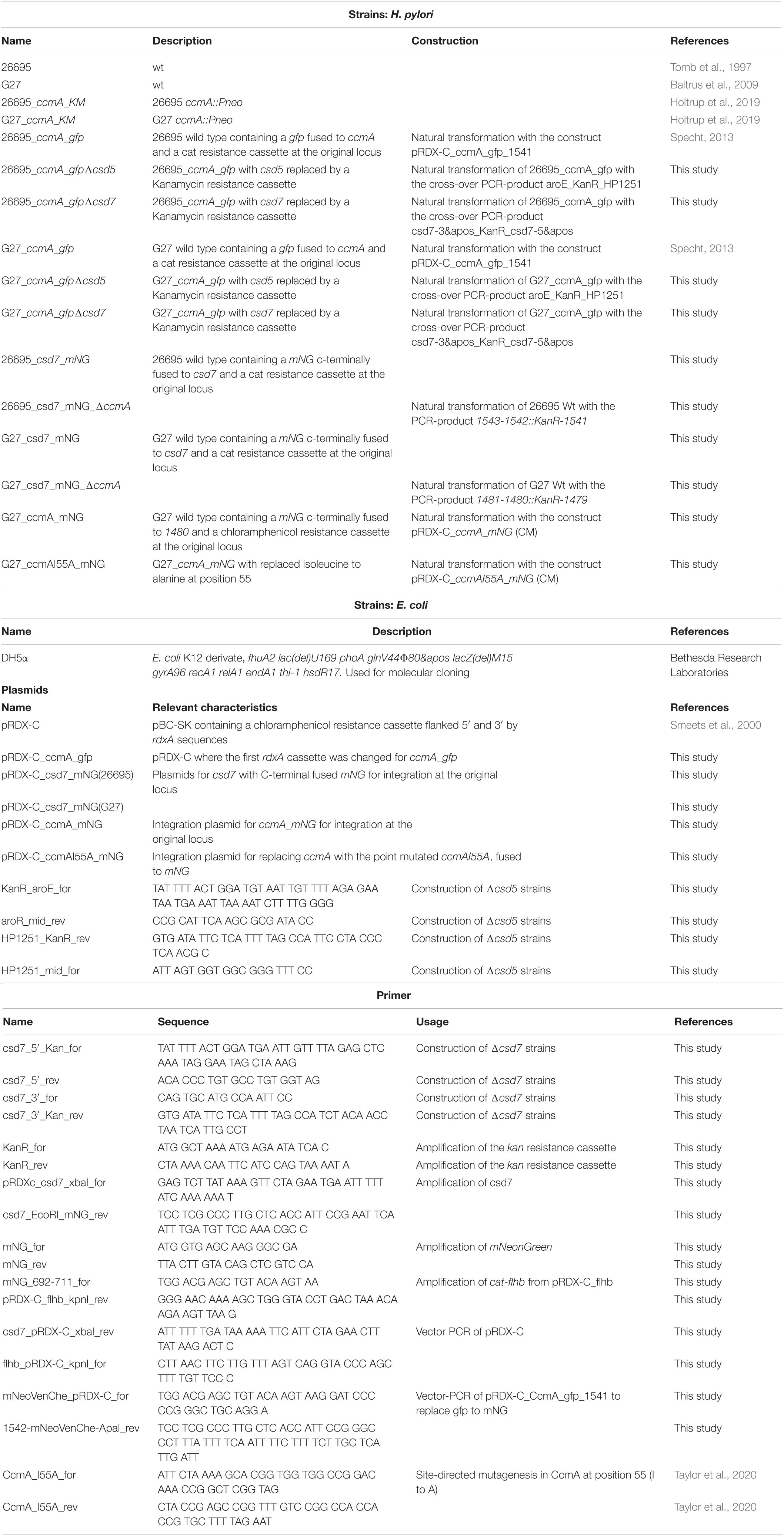
Table 6. List of strains, primers, and plasmids used in this study.
Csd7_mNG fusion constructions were conducted by amplifying the csd7 gene from the respective chromosomal wild-type DNA and cat-flhb from pRDX-C_flhb (rdxAII was replaced with flhb) and were integrated with mNG into pRDX-C by Gibson assembly (New England Biolabs) according to the manufacturer’s manual. H. pylori strains 26695 and G27 were naturally transformed with the resulting plasmids, creating a C-terminal fusion of the csd7 gene with mNG at the original locus.
About 1 ml of exponentially growing cells was pelleted and subsequently stored at –80°C until usage. Equal amounts of total cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes by western blotting (Hybond C Extra, GE Healthcare Life Sciences). For immunodetection, membranes were incubated with appropriately diluted primary antibodies and then probed with secondary antibodies conjugated with horseradish peroxidase. Bound antibodies were detected with enhanced chemiluminescent (ECL) detection reagents as substrates followed by incubation for 2 min and chemiluminescence detection with ChemiDoc MP System (Bio-Rad).
Phase-contrast images were obtained using an Olympus AX-20 microscope (objective: 100x/NA 1.30, oil, Ph) and a CoolSNAP HQ2 (Photometrics) camera. For quantitative shape analysis, binary images were generated using Fiji (Schindelin et al., 2012) and subsequently analyzed using the CellTool software (Pincus and Theriot, 2007).
High-resolution images of H. pylori were acquired by a ZEISS ELYRA PS.1 system using multi-color SIM (SR-SIM Mode) with three different excitation wavelengths: (I) 488 nm line, 10% intensity, gain 50, 100 ms (Gfp), 20 ms, and 50 ms (mNG); (II):405 nm line, 10% intensity, gain 50, 50 ms (HADA), and an EMCCD camera (ANDOR Solis EMCCD). SIM reconstructions were processed using ZEN-Black by ZEISS. Processing was done in Fiji (Schindelin et al., 2012) version 1.52p. Quantitative analysis was done using workflows based on R-statistics and R-studio version 3.6.0 using the packages tidyverse and ggplot2 (Wickham et al., 2019). Signal correlation in SIM images was quantified by using Pearson and Manders colocalization coefficients in the Fiji Plugin JACoP (Bolte and Cordelières, 2006).
Clusters of tracks as identified by TrackMate (Tinevez et al., 2017) were identified using the R packages DBSCAN (Hahsler et al., 2019) and fpc (Hennig, 2020).
For obtaining single-molecule dynamic information, we used a slim-field set up on a customized Nikon Ti Eclipse microscope (objective: 100x/NA 1.49, oil). Samples were illuminated using a 488 nm diode at max 160 W/cm2 on the image plane to initially bleach most fluorophores and subsequently observe single-molecule tracks using a EMCCD camera (ImagEM X2, Hamamatsu, Japan) with an exposure time of 50 (Gfp) and 20 ms (mNG). The movies were processed using ImageJ2/FIJI to remove the initial bleaching phase. Single-molecule tracks obtained using u-track (Jaqaman et al., 2008) and cell meshes generated by using Oufti (Paintdakhi et al., 2016) were formatted and loaded in our custom software SMTracker (Rösch et al., 2018) for analysis of single-molecule behavior using SMMTrack (Schenk et al., 2017). Localization errors are determined by the software according to Michalet (2010).
The data supporting the findings of this study are included in the publication/Supplementary Material, further inquiries may be directed to the corresponding author.
SH performed the experiments, analyzed the data shown in Figures 1–3 and Supplementary Figures 1–4, and helped in writing of the manuscript. BM and SH developed the principal component analysis tool to visualize different cell shape populations. BM helped to set up the SIM. MG performed the experiments, analyzed the data shown in Figures 4–7, Supplementary Figures 5, 6, and Supplementary Movies 1, 2, and helped in writing of the manuscript. MS constructed the Helicobacter pylori CcmA_Gfp strains. BW devised the study, supervised all experiments, helped to analyze the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche Forschungsgemeinschaft (https://www.dfg.de) WA2574/2 to BW. Open Access funding provided by the Open Acess Publication Fund of Philipps-Universität Marburg with support of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Rebecca Hinrichs for her support in Western Blotting.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.929194/full#supplementary-material
An, D. R., Im, H. N., Jang, J. Y., Kim, H. S., Kim, J., Yoon, H. J., et al. (2016). Structural basis of the heterodimer formation between cell shape-determining proteins Csd1 and Csd2 from Helicobacter pylori. PLoS One 11:e0164243. doi: 10.1371/journal.pone.0164243
Baltrus, D. A., Amieva, MR, Covacci, A., Lowe, T. M., Merrell, D. S., Ottemann, K. M., et al. (2009). The Complete Genome Sequence of Helicobacter pylori Strain G27. J. Bacteriol. 191, 447–448.
Blair, K. M., Mears, K. S., Taylor, J. A., Fero, J., Jones, L. A., Gafken, P. R., et al. (2018). The Helicobacter pylori cell shape promoting protein Csd5 interacts with the cell wall, MurF, and the bacterial cytoskeleton. Mol. Microbiol. 110, 114–127. doi: 10.1111/mmi.14087
Bolte, S., and Cordelières, F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. doi: 10.1111/j.1365-2818.2006.01706.x
Bonis, M., Ecobichon, C., Guadagnini, S., Prévost, M.-C., and Boneca, I. G. (2010). A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol. Microbiol. 78, 809–819. doi: 10.1111/j.1365-2958.2010.07383.x
Caccamo, P. D., Jacq, M., VanNieuwenhze, M. S., and Brun, Y. V. (2020). A division of labor in the recruitment and topological organization of a bacterial morphogenic complex. Curr. Biol. 30, 3908–3922. doi: 10.1016/j.cub.2020.07.063
Deng, X., Llamazares, A. G., Wagstaff, J. M., Hale, V. L., Cannone, G., McLaughlin, S. H., et al. (2019). The structure of bactofilin filaments reveals their mode of membrane binding and lack of polarity. Nat. Microbiol. 4, 2357–2368. doi: 10.1038/s41564-019-0544-0
Edelheit, O., Hanukoglu, A., and Hanukoglu, I. (2009). Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9:61. doi: 10.1186/1472-6750-9-61
El Andari, J., Altegoer, F., Bange, G., and Graumann, P. L. (2015). Bacillus subtilis bactofilins are essential for flagellar hook-and filament assembly and dynamically localize into structures of less than 100 nm diameter underneath the cell membrane. PLoS One 10:e0141546. doi: 10.1371/journal.pone.0141546
Favini-Stabile, S., Contreras-Martel, C., Thielens, N., and Dessen, A. (2013). M re B and M ur G as scaffolds for the cytoplasmic steps of peptidoglycan biosynthesis. Environ. Microbiol. 15, 3218–3228.
Feddersen, H., Würthner, L., Frey, E., and Bramkamp, M. (2021). Dynamics of the Bacillus subtilis Min system. mBio 12:e00296-21.
Giacomelli, G., Feddersen, H., Peng, F., Martins, G. B., Grafemeyer, M., Meyer, F., et al. (2022). Subcellular Dynamics of a Conserved Bacterial Polar Scaffold Protein. Genes 13:278.
Hahsler, M., Piekenbrock, M., and Doran, D. (2019). dbscan: Fast density-based clustering with R. J. Stat. Softw. 91, 1–30.
Holtrup, S., Heimerl, T., Linne, U., Altegoer, F., Noll, F., and Waidner, B. (2019). Biochemical characterization of the Helicobacter pylori bactofilin-homolog HP1542. PLoS One 14:e0218474. doi: 10.1371/journal.pone.0218474
Jaqaman, K., Loerke, D., Mettlen, M., Kuwata, H., Grinstein, S., Schmid, S. L., et al. (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. doi: 10.1038/nmeth.1237
Koch, M. K., McHugh, C. A., and Hoiczyk, E. (2011). BacM, an N-terminally processed bactofilin of Myxococcus xanthus, is crucial for proper cell shape. Mol. Microbiol. 80, 1031–1051. doi: 10.1111/j.1365-2958.2011.07629.x
Kühn, J., Briegel, A., Mörschel, E., Kahnt, J., Leser, K., Wick, S., et al. (2010). Bactofilins, a ubiquitous class of cytoskeletal proteins mediating polar localization of a cell wall synthase in Caulobacter crescentus. EMBO J. 29, 327–339. doi: 10.1038/emboj.2009.358
Kuru, E., Radkov, A., Meng, X., Egan, A., Alvarez, L., Dowson, A., et al. (2019). Mechanisms of incorporation for D-amino acid probes that target peptidoglycan biosynthesis. ACS Chem. Biol. 14, 2745–2756. doi: 10.1021/acschembio.9b00664
Leying, H., Suerbaum, S., Geis, G., and Haas, R. (1992). Cloning and genetic characterization of a Hellcobacter pylori flagellin gene. Mol. Microbiol. 6, 2863–2874. doi: 10.1111/j.1365-2958.1992.tb01466.x
Lin, L., Valeriano, M. O., Harms, A., Søgaard-Andersen, L., and Thanbichler, M. (2017). Bactofilin-mediated organization of the ParAB S chromosome segregation system in Myxococcus xanthus. Nat. Commun. 8:1817. doi: 10.1038/s41467-017-02015-z
Marshall, B., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 323, 1311–1315. doi: 10.1016/s0140-6736(84)91816-6
Martinez, L. E., Hardcastle, J. M., Wang, J., Pincus, Z., Tsang, J., Hoover, T. R., et al. (2016). H elicobacter pylori strains vary cell shape and flagellum number to maintain robust motility in viscous environments. Mol. Microbiol. 99, 88–110. doi: 10.1111/mmi.13218
Michalet, X. (2010). Mean square displacement analysis of single-particle trajectories with localization error: brownian motion in an isotropic medium. Phys. Rev. E Stat. Nonlin. Soft. Matter Phys. 82:041914. doi: 10.1103/PhysRevE.82.041914
Mohammadi, T., Karczmarek, A., Crouvoisier, M., Bouhss, A., Mengin-Lecreulx, D., and Den Blaauwen, T. (2007). The essential peptidoglycan glycosyltransferase MurG forms a complex with proteins involved in lateral envelope growth as well as with proteins involved in cell division in Escherichia coli. Mol. Microbiol. 65, 1106–1121. doi: 10.1111/j.1365-2958.2007.05851.x
Paintdakhi, A., Parry, B., Campos, M., Irnov, I., Elf, J., Surovtsev, I., et al. (2016). Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol. Microbiol. 99, 767–777. doi: 10.1111/mmi.13264
Pincus, Z., and Theriot, J. (2007). Comparison of quantitative methods for cell-shape analysis. J. Microsc. 227, 140–156.
Rösch, T. C., Oviedo-Bocanegra, L. M., Fritz, G., and Graumann, P. L. (2018). SMTracker: a tool for quantitative analysis, exploration and visualization of single-molecule tracking data reveals highly dynamic binding of B. subtilis global repressor AbrB throughout the genome. Sci. Rep. 8:15747. doi: 10.1038/s41598-018-33842-9
Salama, N. R. (2020). Cell morphology as a virulence determinant: lessons from Helicobacter pylori. Curr. Opin. Microbiol. 54, 11–17. doi: 10.1016/j.mib.2019.12.002
Sarkar, G., and Sommer, S. S. (1990). The” megaprimer” method of site-directed mutagenesis. Biotechniques 8, 404–407.
Schätzle, S., Specht, M., and Waidner, B. (2015). Coiled coil rich proteins (Ccrp) influence molecular pathogenicity of Helicobacter pylori. PLoS One 10:e0121463. doi: 10.1371/journal.pone.0121463
Schenk, K., Hervás, A. B., Rösch, T. C., Eisemann, M., Schmitt, B. A., Dahlke, S., et al. (2017). Rapid turnover of DnaA at replication origin regions contributes to initiation control of DNA replication. PLoS Genet. 13:e1006561. doi: 10.1371/journal.pgen.1006561
Schibany, S., Kleine Borgmann, L. A., Rösch, T. C., Knust, T., Ulbrich, M. H., and Graumann, P. L. (2018). Single molecule tracking reveals that the bacterial SMC complex moves slowly relative to the diffusion of the chromosome. Nucleic Acids Res. 46, 7805–7819. doi: 10.1093/nar/gky581
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Shaner, N. C., Lambert, G. G., Chammas, A., Ni, Y., Cranfill, P. J., Baird, M. A., et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409. doi: 10.1038/nmeth.2413
Smeets, L. C., Bijlsma, J. J., Boomkens, S. Y., Vandenbroucke-Grauls, C. M., and Kusters, J. G. (2000). comH, a novel gene essential for natural transformation of Helicobacter pylori. J. Bacteriol. 182, 3948–3954. doi: 10.1128/JB.182.14.3948-3954.2000
Specht, M. (2013). The Helical Life of a Bacterium: Cell Shape and Cell Division of H. pylori. Ph. D. Dissertation. Freiburg i. Br: Albert-Ludwigs-Universität.
Specht, M., Schätzle, S., Graumann, P. L., and Waidner, B. (2011). Helicobacter pylori possesses four coiled-coil-rich proteins that form extended filamentous structures and control cell shape and motility. J. Bacteriol. 193, 4523–4530. doi: 10.1128/JB.00231-11
Stracy, M., Uphoff, S., de Leon, F. G., and Kapanidis, A. N. (2014). In vivo single-molecule imaging of bacterial DNA replication, transcription, and repair. FEBS Lett. 588, 3585–3594.
Sycuro, L., Rule, C., Petersen, T., Wyckoff, T., Sessler, T., Nagarkar, D., et al. (2013). Flow cytometry-based enrichment for cell shape mutants identifies multiple genes that influence Helicobacter pylori morphology. Mol. Microbiol. 90, 869–883. doi: 10.1111/mmi.12405
Sycuro, L. K., Pincus, Z., Gutierrez, K. D., Biboy, J., Stern, C. A., Vollmer, W., et al. (2010). Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell 141, 822–833. doi: 10.1016/j.cell.2010.03.046
Sycuro, L. K., Wyckoff, T. J., Biboy, J., Born, P., Pincus, Z., Vollmer, W., et al. (2012). Multiple peptidoglycan modification networks modulate Helicobacter pylori’s cell shape, motility, and colonization potential. PLoS Pathog. 8:e1002603. doi: 10.1371/journal.ppat.1002603
Taylor, J. A., Bratton, B. P., Sichel, S. R., Blair, K. M., Jacobs, H. M., DeMeester, K. E., et al. (2020). Distinct cytoskeletal proteins define zones of enhanced cell wall synthesis in Helicobacter pylori. eLife 9:e52482. doi: 10.7554/eLife.52482
Tinevez, J.-Y., Perry, N., Schindelin, J., Hoopes, G. M., Reynolds, G. D., Laplantine, E., et al. (2017). TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90. doi: 10.1016/j.ymeth.2016.09.016
Tomb, J.-F., White, O., Kerlavage, A. R., Clayton, R. A., Sutton, G. G., Fleischmann, R. D., et al. (1997). The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547.
Vasa, S., Lin, L., Shi, C., Habenstein, B., Riedel, D., Kühn, J., et al. (2015). β-Helical architecture of cytoskeletal bactofilin filaments revealed by solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 112, E127–E136. doi: 10.1073/pnas.1418450112
Waidner, B., Specht, M., Dempwolff, F., Haeberer, K., Schaetzle, S., Speth, V., et al. (2009). A novel system of cytoskeletal elements in the human pathogen Helicobacter pylori. PLoS Pathog. 5:e1000669. doi: 10.1371/journal.ppat.1000669
White, C. L., Kitich, A., and Gober, J. W. (2010). Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol. Microbiol. 76, 616–633. doi: 10.1111/j.1365-2958.2010.07108.x
Wickham, H., Averick, M., Bryan, J., Chang, W., McGowan, L. D., François, R., et al. (2019). Welcome to the Tidyverse. J. Open Source softw. 4:1686.
Yang, D. C., Blair, K. M., and Salama, N. R. (2016). Staying in shape: the impact of cell shape on bacterial survival in diverse environments. Microbiol. Mol. Biol. Rev. 80, 187–203. doi: 10.1128/MMBR.00031-15
Yang, D. C., Blair, K. M., Taylor, J. A., Petersen, T. W., Sessler, T., Tull, C. M., et al. (2019). A genome-wide Helicobacter pylori morphology screen uncovers a membrane-spanning helical cell shape complex. J. Bacteriol 201:e724-18. doi: 10.1128/JB.00724-18
Keywords: bactofilin, single-molecule tracking, Helicobacter pylori, cell shape, structured illumination microscopy
Citation: Holtrup S, Greger M, Mayer B, Specht M and Waidner B (2022) Insights Into the Helical Shape Complex of Helicobacter pylori. Front. Microbiol. 13:929194. doi: 10.3389/fmicb.2022.929194
Received: 26 April 2022; Accepted: 13 June 2022;
Published: 24 August 2022.
Edited by:
Patrick Eichenberger, New York University, United StatesReviewed by:
Ramanujam Srinivasan, National Institute of Science Education and Research (NISER), IndiaCopyright © 2022 Holtrup, Greger, Mayer, Specht and Waidner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Waidner, YmFyYmFyYS53YWlkbmVyQHN5bm1pa3JvLnVuaS1tYXJidXJnLmRl
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.