 Balaram Mohapatra
Balaram Mohapatra Harshit Malhotra
Harshit Malhotra Prashant S. Phale
Prashant S. Phale
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 06 July 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.928848
This article is part of the Research TopicMulti-omics profiling of unique niches to reveal the microbial and metabolite compositionView all 12 articles
Comparative genomic and functional analyses revealed the presence of three genomic islands (GIs, >50 Kb size): ICEnahCSV86, Pseudomonas bharatica genomic island-1 (PBGI-1), and PBGI-2 in the preferentially aromatic-degrading soil bacterium, Pseudomonas bharatica CSV86T. Site-specific genomic integration at or near specific transfer RNAs (tRNAs), near-syntenic structural modules, and phylogenetic relatedness indicated their evolutionary lineage to the type-4 secretion system (T4SS) ICEclc family, thus predicting these elements to be integrative conjugative elements (ICEs). These GIs were found to be present as a single copy in the genome and the encoded phenotypic traits were found to be stable, even in the absence of selection pressure. ICEnahCSV86 harbors naphthalene catabolic (nah-sal) cluster, while PBGI-1 harbors Co-Zn-Cd (czc) efflux genes as cargo modules, whereas PBGI-2 was attributed to as a mixed-function element. The ICEnahCSV86 has been reported to be conjugatively transferred (frequency of 7 × 10–8/donor cell) to Stenotrophomonas maltophilia CSV89. Genome-wide comparative analyses of aromatic-degrading bacteria revealed nah-sal clusters from several Pseudomonas spp. as part of probable ICEs, syntenic to conjugatively transferable ICEnahCSV86 of strain CSV86T, suggesting it to be a prototypical element for naphthalene degradation. It was observed that the plasmids harboring nah-sal clusters were phylogenetically incongruent with predicted ICEs, suggesting genetic divergence of naphthalene metabolic clusters in the Pseudomonas population. Gene synteny, divergence estimates, and codon-based Z-test indicated that ICEnahCSV86 is probably derived from PBGI-2, while multiple recombination events masked the ancestral lineage of PBGI-1. Diversifying selection pressure (dN-dS = 2.27–4.31) imposed by aromatics and heavy metals implied the modular exchange-fusion of various cargo clusters through events like recombination, rearrangement, domain reshuffling, and active site optimization, thus allowing the strain to evolve, adapt, and maximize the metabolic efficiency in a contaminated niche. The promoters (Pnah and Psal) of naphthalene cargo modules (nah, sal) on ICEnahCSV86 were proved to be efficient for heterologous protein expression in Escherichia coli. GI-based genomic plasticity expands the metabolic spectrum and versatility of CSV86T, rendering efficient adaptation to the contaminated niche. Such isolate(s) are of utmost importance for their application in bioremediation and are the probable ideal host(s) for metabolic engineering.
Horizontal transfer of gene(s) “en masse” through mobile genetic elements [MGEs: genomic islands (GIs)/integrative conjugative elements (ICEs), prophages, plasmids, etc.] encoding adaptive or niche colonization traits is the decisive factor for species diversification, evolution, and pan-genomic stability in prokaryotes (Burrus et al., 2002; Rodríguez-Beltrán et al., 2020). Amongst MGEs, GIs are strain-specific segments of the bacterial genome, which are often found to be present at limited chromosomal locations. Depending on the functions they encode and the specific lifestyle of the bacterium, GIs can be classified into metabolic, pathogenicity, symbiosis, fitness, or resistance islands (Dobrindt et al., 2004; Juhas et al., 2009). GIs located on chromosomes are typically flanked by direct repeats and are inserted at the 3′ end of a transfer RNA (tRNA) gene. Further, these elements harbor transposase(s) or integrase(s) that are required for chromosomal integration and excision along with other mobile genes, in a strain or clone-specific manner. GIs are usually differentiated from the rest of the core genome by their atypical G + C contents and oligonucleotide compositions, with steep gradients at their boundaries (Reva and Tümmler, 2005). Comparative genetic mapping and evolutionary analyses have uncovered diverse GIs in bacterial taxa, particularly amongst proteobacterial members, with pathogenicity islands of Enterobacteriaceae being the most studied GIs (Hacker and Carniel, 2001).
Genomic islands and ICEs have structural similarities, such as the presence of integrase/excisionase or conserved core gene regions that belong to a common group of MGEs, often termed as ICElands (van der Meer and Sentchilo, 2003). While GIs might be self-transmissible and/or unstable, ICEs are distinguished by their conjugative, integrative, and stable/consistent behavior (Wozniak and Waldor, 2010; Delavat et al., 2017). Classically, the core module(s) of ICEs comprise (i) an integrase (xerC, responsible for ICE excision to form circular intermediate), often assisted by an excisionase or recombination directionality factors (RDFs), (ii) type-4 secretion system (T4SS) (for conjugative transfer), and (iii) partition (par)- maintenance cluster, ensuring successful horizontal transmission amongst near-relatives (Burrus, 2017; Liu et al., 2019). Most importantly, the “cargo gene array(s)” which differ from ICE to ICE, encode diverse metabolic traits which provide survival fitness to the host in a particular niche (Wozniak and Waldor, 2010). Although both GIs and ICEs are synonymous, there are no clear demarcations on their evolution and/or mode of mobilization.
Genomic islands or ICEs harboring genetic modules for the degradation of aromatics and tolerance/resistance to heavy metals (as functional cargo) are of immense scientific interest due to their potential application in bioremediation and metal detoxification/recovery. With large-scale discharge, resonance-stabilized structures, hydrophobicity, and persistent nature of aromatics and toxicity of metals, selection pressure is exerted on the microbial communities, leading to the evolution of efficient metabolic pathways or tolerance mechanisms (Nojiri et al., 2009; Fuentes et al., 2014; Phale et al., 2019). This requires a high degree of genetic exchange and recombination events within the community mediated by mobile genetic elements through horizontal gene transfer (HGT) process. Till date, few GIs and ICEs, such as phn island (phenanthrene metabolism), clc GI (later classified as ICEclcB13; chlorocatechol metabolism), ICEclcLB400 (chlorinated biphenyl, chlorocatechol), ICEclcJB2 (o-halobenzoates and o-hydroxybenzoate), ICEXTD (xylene-toluene), ICEbph-sal (biphenyl-salicylate), and ICETn4371 family (Co, Zn, Cd, Cr, As, and Hg resistance) have been characterized (ICEBerg 2.0 database1; Liu et al., 2019). ICEclcB13 (105 Kb) of Pseudomonas knackmussii B13 encoding ortho-cleavage of chlorocatechol (clc) and aminophenol (amp) is a functionally characterized prototypical element (Gaillard et al., 2006; Miyazaki et al., 2015). However, the coexistence of multiple GIs (either of similar or hybrid nature) encoding degradation/tolerance traits in a single bacterium has been rarely observed (Badhai and Das, 2016; Liu et al., 2019).
An Indian isolate, strain CSV86T (formerly Pseudomonas putida, recently effectively described as Pseudomonas bharatica), isolated from petrol-station soil contaminated with petroleum products preferentially metabolizes various aromatic compounds over glucose and displays the following carbon source utilization hierarchy: aromatics ≥ organic acid > glucose (Basu et al., 2006; Phale et al., 2021; Mohapatra et al., 2022). This unique preferential naphthalene degradation trait is stable, chromosomally encoded, and was found to be transferred to Stenotrophomonas maltophilia CSV89 through conjugation at a very low frequency (7 × 10–8/donor cell) (Basu and Phale, 2008). Besides, strain CSV86T is able to tolerate up to 2 mM concentration of heavy metals (Cd, Zn, Co, Cu, and As) during growth on aromatics (Paliwal et al., 2014). In addition to these unique traits, the absence of plasmid and other pathogenicity and virulence factors renders strain CSV86T an ideal candidate for pollutant bioremediation and host/chassis for metabolic engineering applications (Phale et al., 2021).
Initial genomic analyses of strain CSV86T revealed the presence of a large extent of MGEs (GIs, prophages, probable ICEs, and insertion sequences), amounting to ∼19% of its total genomic content (6.79 Mb), responsible for the genomic plasticity of this isolate (Mohapatra et al., 2022). To delineate the extent, functionality, and evolutionary significance of MGEs in strain CSV86T, combined comparative- and functional-genome minings and molecular analyses were performed. Analyses revealed the role and significance of MGEs in adaptation, niche colonization, and species competitive behavior of strain CSV86T at the contaminated site.
Pseudomonas bharatica CSV86T (= MTCC 2445T = ICMP 13424T = JCM 34937T) which metabolizes benzoate, naphthalene, and phenylpropanoids, amongst others was used in this study (Mahajan et al., 1994; Mohapatra et al., 2022). The strain was routinely grown on a minimal salt medium (MSM, 150 mL, pH 7.5; Basu et al., 2003) supplemented aseptically with naphthalene or benzoate (0.1%, w/v) or glucose (0.25%, v/v) as the sole source of carbon and energy at 30°C for 12 h at 200 rpm.
The whole-genome shotgun sequencing project of strain CSV86T was previously deposited at DDBJ/EMBL/GenBank under the accession number AMWJ00000000 with the first/standard version as AMWJ01000000 (Paliwal et al., 2014). The hybrid PacBio-GS FLX hybrid assembly based genome sequencing project has been deposited as the updated advanced version: AMWJ02000000 with BioProject: PRJNA65243 and BioSample: SAMN02472262 (Mohapatra et al., 2022). The GI replicons (corresponding nucleotide positions) are represented in the genome AMWJ02000000.
To identify the regions of genomic plasticity (RGPs) in strain CSV86T, advanced version of the genomic draft (AMWJ02000000) of CSV86T was analyzed by Microscope-Genoscope (MaGe2) and IslandViewer 4.03 using IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander prediction methods. The RGPs were manually curated to filter biases (false-positive and negative) using the following criteria: (i) presence of mobility genes (integrases/recombinases, and transposases); (ii) structural RNAs (tRNAs); (iii) atypical genomic content (GC mol% content, GC skewness, and codon usage); and (iv) presence of phenotypic trait cassettes (antibiotic/toxic compounds resistance, virulence, or substrate metabolism). To categorize RGPs, different methods were employed: (i) BLASTn searches against the ICEBerg database 2.04 and PHASTER5 with the E-value ≤ 10–5 and sequence coverage ≥50%; (ii) homology search against nr- and RefSeq-database; (iii) evaluation of t-RNAs (tRNA Scan-SE) followed by site-specific integrase/relaxase; and (iv) presence of mobile element and hypothetical proteins. Besides, repeats (direct/inverted) were identified to define the boundaries of these elements. RGPs were identified as GIs and suspected to be ICEs, thus annotated using the RAST-tk pipeline and compared with the reported reference ICEs in ICEBerg 2.0 database (Table 1). Genes located on these suspected ICEs were categorized into orthologous groups using OrthoVenn26 with an E-value cut-off of 10–5. Localized reciprocal best-hit of ICE genes was performed using the orthoANI tool (OAT) of the Ez-BioCloud server.7 Other genomic features (gene synteny, GC skewness, GC content mol%, and pairwise comparisons) were analyzed using Artemis Comparison Tool (ACT, release 18.1.0) and Mauve progressive alignment in MAUVE [version 20150226 build 10 (c)]. To confirm the integrative nature of the predicted ICEs, the origin of replication/transfer (oriC/T) was identified using both the DoriC database8 and manual curation using Pseudomonas spp. and Escherichia coli consensus sequences. To validate the genomic prediction and appropriate organization of the suspected ICEs, genome drafts of strain CSV86T (AMWJ01 and AMWJ02) were aligned, gaps were amplified using custom-designed primers (Supplementary Table 1 and Supplementary Figure 1), and sequenced. For details of the methodology, refer to “Supplementary Material.”
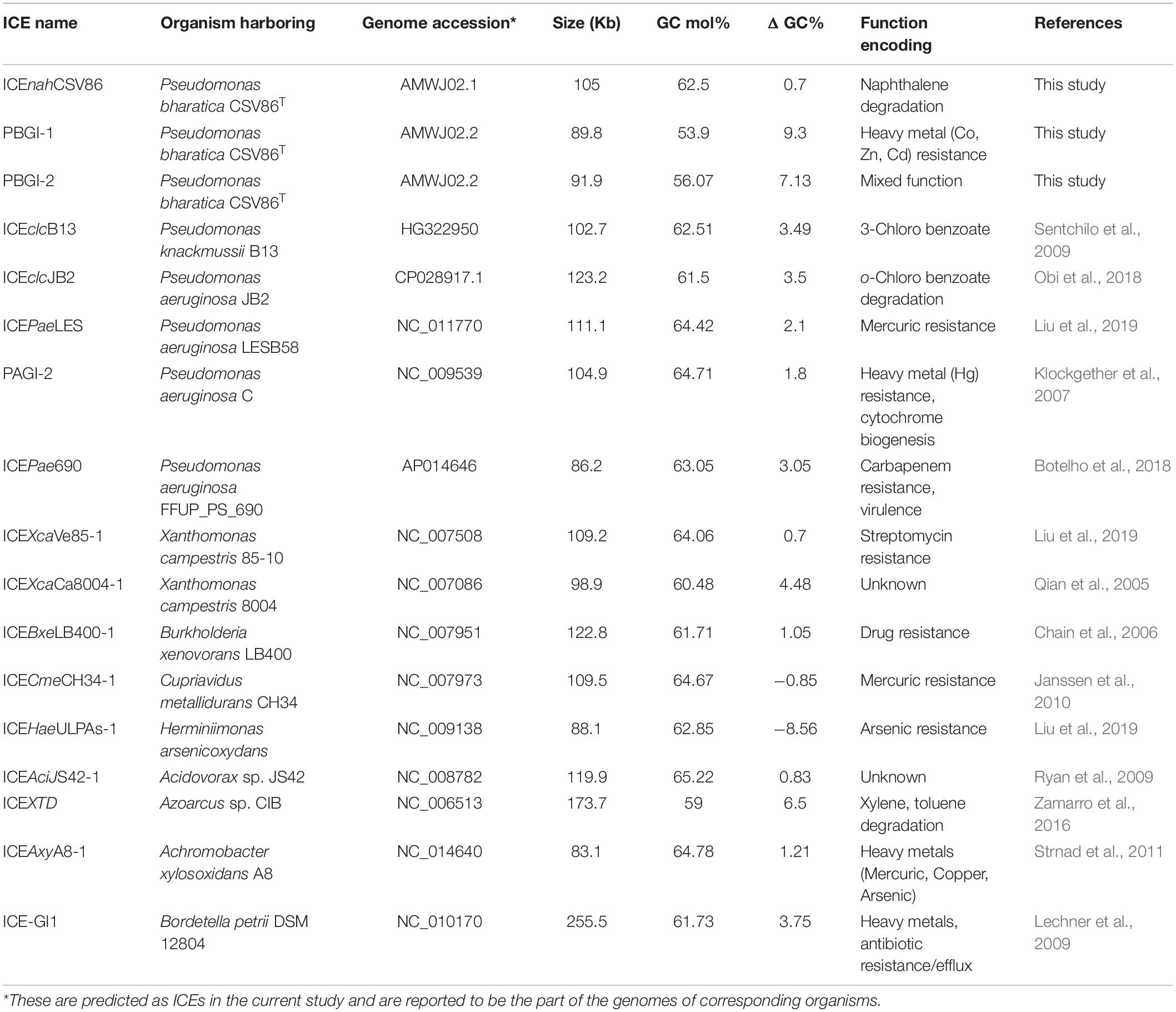
Table 1. Comparative analysis of three GIs, suspected to be ICEs, from Pseudomonas bharatica CSV86T and syntenic members of ICEclc family used in this study.
The three suspected ICEs and their similar/syntenic homologs were subjected to multiple alignment using CLUSTALW (without gaps and including all positions) in MEGA 7.0 (Kumar et al., 2016). Phylogenetic reconstruction was performed using the neighbor-joining (NJ) method based on 1,000 bootstrap iterations using Jukes–Cantor distance model, where the robustness of the trees was assessed using maximum-likelihood (ML) and minimum-evolution (ME) algorithms. Owing to high synteny and conservedness of core genes within ICE classes, amino acid sequences were subjected to phylogenetic reconstruction. Multiple alignment of concatenated amino acid sequences of T4SS-T4CP was performed using MUSCLE, and phylogeny was deciphered through the ML method involving Jones–Thornton–Taylor (JTT) substitution matrix with 1,000 bootstrap revaluation in MEGA 7.0. Additionally, individual ICE core proteins like integrase, transposase as well as cargo proteins (nah-sal cluster, NahR regulator, and czc heavy metal tolerance cluster) were used for phylogenetic interpretations using Poisson’s correction and Kimura-2-parameter models involving ML algorithm in MEGA 7.0. The robustness and reliability of phylogenetic trees were evaluated by 1,000 bootstrap replications. Further, overall ICE-related indices (average nucleotide-/amino acid identity, pattern disparity index, and substitution estimation) were calculated using the OrthoANI (OAT) tool of the EzBiocloud server,9 GGDC tool of Type Strain Genome Server (TYGS10) with recommended settings, and MEGA 7.0, respectively. Codon-based Z-Test of selection (diversifying/purifying-/neutral- selection) was performed by codon-by-codon alignment in MEGA 7.0 followed by dN-dS and dN/dS vs. dS computation to obtain the mean divergence rate. To validate the genomic divergence, the total GC mol% of the genomes and the mean deviation (ΔG + C mol%) were calculated using the G + C calculator. Nucleotide sequences of OriC and associated elements (parB, parA, gidB, yidC, rnpA, dnaA, dnaN, recF, and gyrB) were concatenated, aligned using CLUSTALW, and phylogeny was deciphered in MEGA 7.0.
The genetic and phenotypic stability of these elements in strain CSV86T was evaluated by performing growth experiments in the absence of selection pressure for 60 generations [corresponding to ∼6 generations in a single growth cycle in batch culture (td = 1.11 h), thus a total of 10 batch transfers]. Strain CSV86T was grown on MSM (150 mL, pH 7.5) amended with benzoate (0.1%, w/v) as the sole source of carbon and energy. Culture grown on benzoate (growth OD540 nm = 1.5–1.7) for 0th (control condition), 30th (5 batch transfers), and 60th (10 batch transfers) generations was transferred to MSM (150 mL, pH 7.5) containing: (i) naphthalene (0.1%, w/v) as the sole source of carbon, and (ii) benzoate plus heavy metals (CZC: Co2+, Zn2+, and Cd2+, each at 1 mM). Culture growth was assessed by measuring the optical density (OD540 nm) at periodic intervals for naphthalene-amended conditions. While for heavy metals, growth was monitored at 12 and 24 h intervals. Minimum inhibitory concentration (MIC) was determined by tube dilution method, where cells of strain CSV86T were inoculated into Luria broth (g/L: peptone, 10.0; yeast extract, 5.0, NaCl, 10.0, pH 7.2) containing increasing concentration of Co2+, Zn2+, or Cd2+ (0, 0.5, 1, 1.5, 2, 2.5, and 3 mM) and incubated for 48 h at 30°C (Wiegand et al., 2008). The lowest concentration of metals that inhibited growth (OD540 nm) completely was considered as MIC. The activity (at 10 h) of catechol 2,3-dioxygenase (C23DO, an enzyme catalyzing meta-ring cleavage of catechol in naphthalene metabolism) was monitored in the cell-free supernatant spectrophotometrically by measuring an increase in the absorbance at 375 nm due to the formation of 2-hydroxymuconic semialdehyde (375 = 33,000 M–1 cm–1) as described by Basu et al. (2006). Protein was estimated by the Bradford method using BSA as the standard (Bradford, 1976). All experiments were performed thrice in duplicates and the mean with the standard deviations are tabulated.
In order to assess the functionality of the cargo operons of ICEnahCSV86, the promoter regions present upstream of the nah and sal operons were predicted using BPROM and SAPPHIRE, whereas the NahR binding site was predicted using multiple sequence alignment (for details, refer to “Supplementary Material”). The polycistronic nature of the nah and sal operons was validated using co-transcription analysis (refer to “Supplementary Material” and Supplementary Figure 2). In order to validate the functionality and strength of the predicted promoters and the NahR regulator; Pnah, Psal, and Psal/nahR promoters were fused to gene (mcbC) encoding 1-naphthol 2-hydroxylase (1NH) as a reporter gene in pSEVA and the expression of 1NH was tested in E. coli BL21 (DE3) (Supplementary Figure 3). The details of the construct strategy and methodology are presented in the “Supplementary Material.”
An advanced version of the draft genome of P. bharatica CSV86T (AMWJ02000000: 6.79 Mb, 99.9% completeness, 0.03% heterogeneity) revealed the presence of three GIs of greater than 50 kb size on two of the largest contigs (Contig_001 and Contig_008, Figure 1). BLAST-based bidirectional hits of GIs against experimentally validated and genomically predicted ICE families in the ICEBerg 2.0 database predicted these GIs to be discrete, complete, and intact ICEs (Figure 1). Presence of genes encoding: (i) tRNA at either of the extreme ends of ICEs, (ii) integrase, and (iii) VirB4 homologs (hallmark gene of T4SS) having the highest similarity (72–95%) to ICEclc (ICEclcB13, ICEXTD, ICEBxeLB400-1) confirmed affiliation of these elements to mating-pair type T4SS-ICEs (Wozniak et al., 2009; Poulin-Laprade et al., 2015). Genome analyses indicated that all three predicted ICEs in strain CSV86T were present as a single copy. These elements were predicted to have no origin of replication (oriC). A single oriC site (831 bp with eight 9-mer DnaA boxes, constituting 48.01% AT content = 6,355,999 AT disparity, on contig_001) (Supplementary Figure 4) was found elsewhere in the genome of the strain CSV86T. Phylogenetic analysis of oriC and its associated elements indicated its lineage to non-ICE bearing members (Pseudomonas japonica WL, Pseudomonas parafulva CRS01-1) (Supplementary Figure 4). Further, the absence of any plasmid in strain CSV86T confirms the chromosomal nature of these GIs (Basu and Phale, 2008). Similar to T4SS-ICEs, these elements were found to encode the main core gene modules harboring integrases, T4SS-T4CP, and DNA partitioning cluster (par, PFG-1) (Figure 2A). These three elements: ICEnahCSV86 (∼105 Kb), Pseudomonas bharatica genomic island-1 (PBGI-1) (∼89.8 Kb), and PBGI-2 (∼91.9 Kb) were found to encode 127, 112, and 136 RAST subsystem features, respectively. Amongst these, ICEnahCSV86 has been reported to be successfully transferred through conjugation to a recipient, S. maltophilia CSV89 with a frequency of 7 × 10–8 (Basu and Phale, 2008). Thus, this element has been classified as a probable ICE (ICEnahCSV86), based on the nomenclature system proposed (Burrus et al., 2002; Liu et al., 2019). A minor average G + C skew was observed in ICEnahCSV86 (62.5 mol%), i.e., close to chromosomal G + C content (63.2 mol%) of strain CSV86T. While significant deviations (ΔG + C = 9.3 and 7.13 mol%) were observed for PBGI-1 (G + C content = 53.9 mol%) and PBGI-2 (G + C content = 56.07 mol%), respectively (Table 1 and Figure 2A). A low synteny at the core gene modules and no synteny for cargo modules were noted for ICEnahCSV86 vs. PBGI-1 and PBGI-1 vs. PBGI-2 (Supplementary Figure 5A). Whereas ICEnahCSV86 and PBGI-2 shared moderate synteny at the core gene level (Supplementary Figure 5B). It has been established that genes (encoded on any MGEs, like GIs, including ICEs) originate from a common ancestor during specification events and are usually syntenic between closely related species, hence orthologous in nature. Sequences that show greater divergence from other species are more likely to perform distinct functions (Fang et al., 2010; Xu et al., 2019). Therefore, genes encoded on ICEs were analyzed for orthologous clusters using the OrthoVenn2 platform and annotated through the COG database with the WebMGA server (data not shown). A total of 83 orthologous clusters for ICEnahCSV86 were observed, while ICEclcB13 formed the maximum, i.e., 126 clusters (Supplementary Figure 6). Both PBGI-1 and PBGI-2 showed the highest number of singletons/unique clusters (n = 94), which were categorized into COG classes of unknown function/hypothetical proteins (I), general function (S), transcription (T), and defense mechanisms (V). Overall, a higher number of integrases (n = 3), hypothetical proteins (77), and conjugative proteins (9) were observed in PBGI-2, while a higher number of repeats (5) and transposases (8) were observed in PBGI-1.
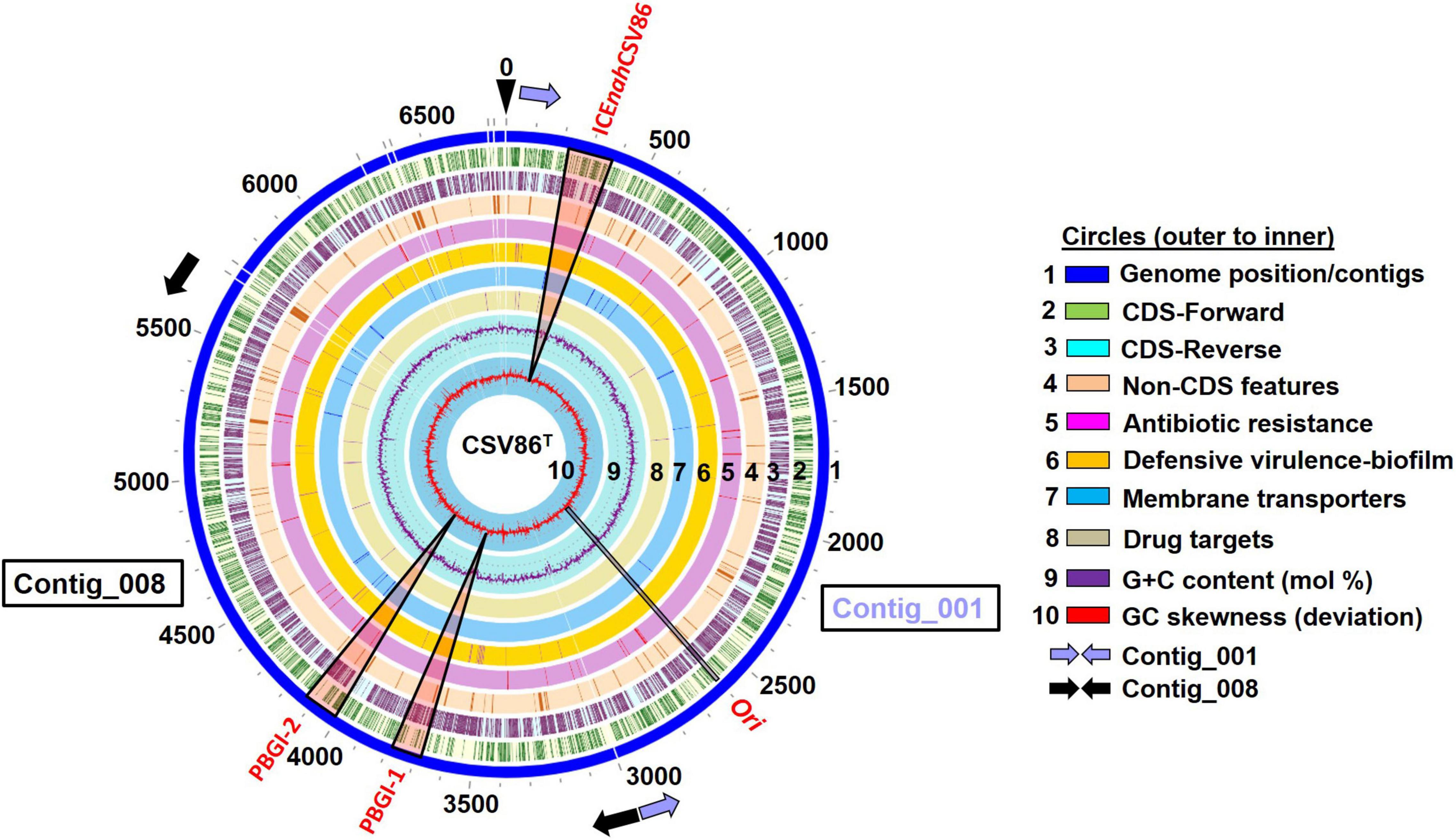
Figure 1. Circular genomic map (0–6,975 Kb) depicting the location of three genomic islands (GIs), predicted to be integrative conjugative elements (ICEs: ICEnahCSV86, PBGI-1, and PBGI-2 in the genome of Pseudomonas bharatica CSV86T along with various genomic attributes (GC mol%, skewness, CDS features). The genome map was reconstructed using PATRIC 3.7.2.2. Elongated triangular boxes (light red color) indicate the positions of three GIs on the genome. Ori, the origin of replication and replicative gene catamers are depicted by black rectangle. The start and the end positions of Contig_001 and Contig_008 are depicted by light blue and black filled arrows, respectively.
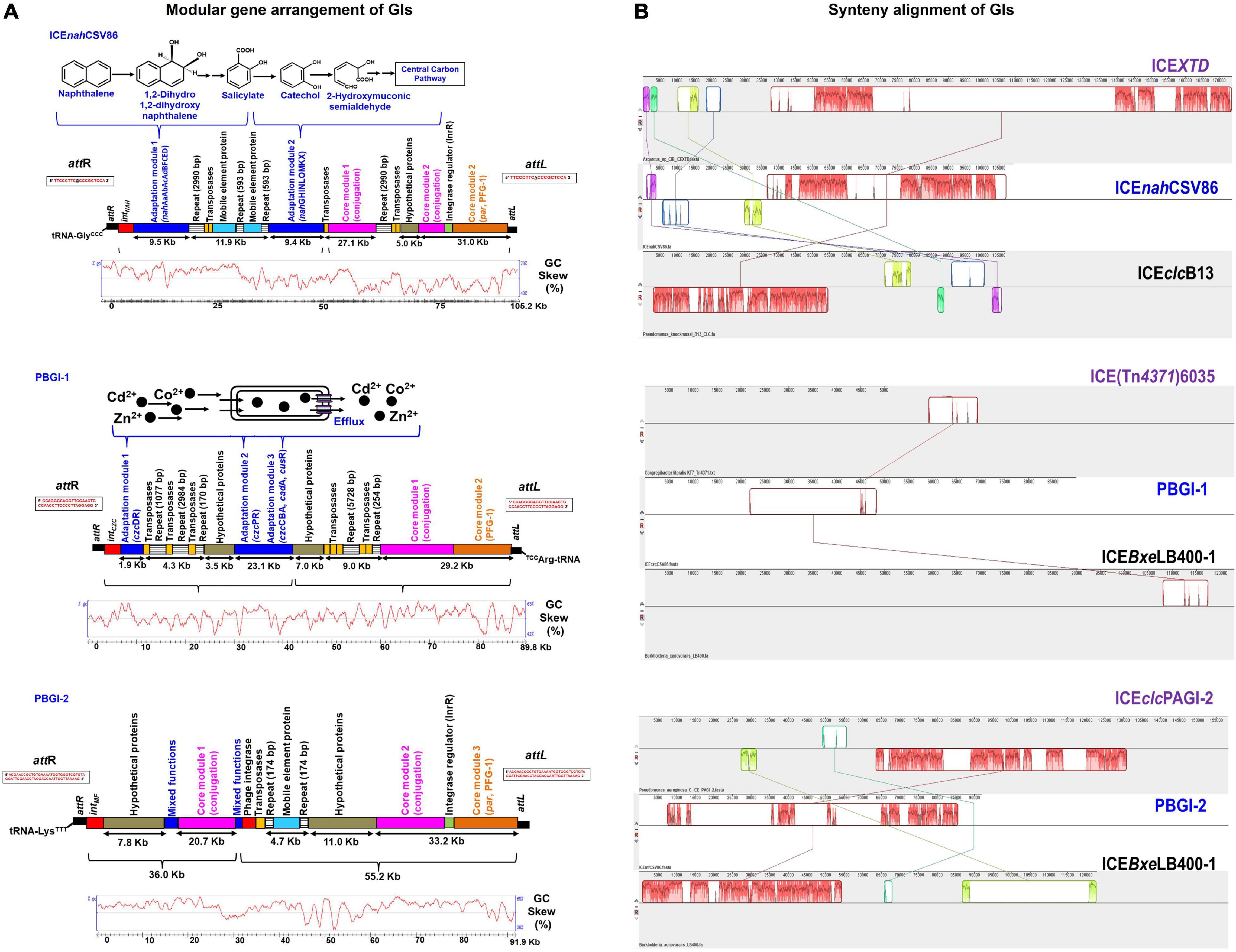
Figure 2. Comparative analyses of the three full-length GIs: ICEnahCSV86, PBGI-1 and PBGI-2 from Pseudomonas bharatica CSV86T. (A) Structural/modular organization of the three GIs, suspected to be ICEs. The color codes and depiction are as follows: cargo gene modules (blue), integration/excision (red), integrase regulator (green), conjugation (purple), partition modules (orange), regions containing repeats (lined boxes), genes encoding transposases (yellow), hypothetical proteins (gray), and mobile element proteins (light blue). Flanking attL and attR sites are indicated at the extreme ends (black solid rectangles) of the GIs and its sequences are depicted within the rectangular boxes. The functions encoded by respective cargo modules are depicted above the gene arrangement. The GC disparity plots (mol% skewness/deviations) for respective GIs are indicated below the gene arrangement. (B) Synteny alignment plots of predicted ICEs with the nearest similar ICEs from other bacteria obtained using progressive MAUVE alignment method. The blocks in different colors indicate linear conserved blocks (LCBs) and criss-cross connecting lines indicate the homologous LCBs amongst ICEs. For gene annotation and ORF details, refer to Supplementary Tables 3–5.
ICEnahCSV86, a probable ICE, is located on the contig_001 (length 105,221 bp; nucleotide positions: 274,076 to 379,297) and has tRNA-GlyCCC gene (74 bp) at 5′-end. The 18 bp sequence at the 3′-end of tRNA-Gly (TTCCCTTCGCCCGCTCCA) forms the attL site (direct repeat), while the attR site (18 bp long, TTCCCTTCGCCCACTCCA) is located at 3′-end of ICEnahCSV86 with a single base substitution (A with G), defining the structural borders (Figure 2). The gene annotation of ICEnahCSV86 is summarized in Supplementary Table 3 and Supplementary Figure 7A. This element harbors typical T4SS core modules and cargo genes responsible for naphthalene metabolism. Strain CSV86T utilizes naphthalene as the sole source of carbon and energy (Mahajan et al., 1994). Genes encoding enzymes for naphthalene metabolism were found to be a part of this element only and not elsewhere on the genome, hence designated as ICEnahCSV86. It showed the highest synteny with ICEclc elements [ICEclcB13 (102.7 Kb) and ICEXTD (173.7 Kb)]. ICEnahCSV86 exhibits a dual structure, i.e., characteristic of both ICEclcB13 and ICEXTD. However, unlike ICEclcB13, which is present in dual copies (Ravatn et al., 1998b), ICEnahCSV86 is present as a single copy, based on the genome analyses. ICEnahCSV86 displayed 76–83% nucleotide identity with core modules of various ICEclc members and displayed nearly similar gene synteny as observed in ICEXTD (Figure 2B). ICEnahCSV86 possesses a single oriT at position 41,464 to 41,838 Kb (375 bp), adjacent to an open reading frame (ORF) encoding SNF2 superfamily II DNA/RNA helicase (Supplementary Table 3). The oriT showed the highest homology with oriT of ICEXTD (92.8% identity) and ICEclcB13 (67.5%). Signature repeat motifs of 6 bp long (CCCTTT or GCCTTT, each 2 motifs) in oriT of ICEnahCSV86 were observed and found to be identical to ICEclcB13 (Supplementary Figure 8). The frequencies of repeats at nucleotide positions from 41,464 to 41,838 bp were 20.8- and 45.4-fold higher, respectively, than their random distribution on whole ICEnahCSV86, thus implying their recognition by helicase and oriT-processing proteins for DNA unwinding and conjugative transfer (Lawley et al., 2004). The presence of oriT and associated elements as a part of ICEnahCSV86 supports the successful conjugative transfer of naphthalene degradation property to S. maltophilia CSV89 (Basu and Phale, 2008).
Synteny alignment showed the occurrence of substantial rearrangement events (deletions and insertions) in ICEnahCSV86 as compared to ICEclc relatives (Figure 2B). Adjacent to tRNA-GlyCCC, gene encoding integrase (intNAH) responsible for catalyzing the site-specific integration and excision of ICEnahCSV86 was present (Supplementary Table 3). Similar to ICEclc (ICEclcB13), where integrase and its regulatory module are located at the two extreme ends of the ICE (Sentchilo et al., 2009), in ICEnahCSV86, InrR, an integrase regulator along with a DNA binding protein was found to be present ∼100.8 Kb downstream of intNAH and is likely to be involved in the regulation of intNAH expression (Figure 2A, Supplementary Table 3, and Supplementary Figure 7A). In strain CSV86T, naphthalene is metabolized to salicylate (upper pathway; nah operon: nahABFCED), which is further channeled to TCA cycle intermediates via catechol (lower pathway; sal cluster: nahGHINLOMKX; Figure 2A). These cargo genes [nah (9.5 Kb) and sal (10.7 Kb) clusters] were found to be located on the coding strand of ICEnahCSV86, while nahR (encoding LysR type transcription regulator, which regulates both clusters) is located on the complementary strand. Most of the core genes (∼68 Kb) of ICEnahCSV86 are present on the complementary strand, except for some hypothetical proteins (Figure 2A and Supplementary Figure 7A). ICEnahCSV86 harbors core genes: VirB4, ∼19 Kb downstream to nah-cluster, murein hydrolase, and VirD4 at ∼42 Kb downstream of nah cluster (Supplementary Table 3). These three key proteins: murein hydrolase (degradation of the peptidoglycan for forming mating channel), VirB4-ATPase (DNA transfer by T4SS), and VirD4 coupling protein (DNA transfer by mating channel) are reported to be the essential components for ICE transfer (Christie et al., 2014). The ParAB locus in the PFG-1 core gene cluster was found to be present ∼66 Kb downstream of the attL site in ICEnahCSV86. This locus is reported to be involved in stabilization and the maintenance of ICEs (Bignell and Thomas, 2001). The entire core module (T4SS, T4CP, and PFG-1) of ICEnahCSV86 formed a large synteny with ICEXTD, while no synteny was observed for cargo modules. In ICEnahCSV86, insertion of the repeat (2,990 bp), transposases (1.12 Kb), and the stretch of hypothetical proteins (5 Kb) segregated the core module (67.2 Kb) into two sub-modules: Core-I (27.1 Kb) and Core-II (31.0 Kb). Similar insertion (of 2,990 bp repeat and 1.12 Kb transposase) along with an additional region comprising mobile element proteins and 593 bp repeat (in alternate) segregated cargo modules into “nah” and “sal” clusters (Figure 2A). BLASTn search of 2,990 bp repeats (of both cargo and core module) showed 99–100% identity with repeats of Cycloclasticus zancles 78-ME (obligate marine PAH degrading isolate), Acinetobacter baumannii (antibiotic resistant strains 7835 and 9201), and Pseudomonas aeruginosa strain AK6U. Interestingly, repeats found in both ICEnahCSV86 and C. zancles 78-ME genome encodes an ISL3-TnpA family transposase, guarded by Type-4 pilus assembly proteins. The 593 bp repeat harboring IS5 family transposase showed the highest similarity (99–100%) to probable ICE-bearing naphthalene degrading Pseudomonas citronellolis strain SJTE-3, isolated from waste sludge. Other transposases (IS66 and IS5) and mobile element proteins located on ICEnahCSV86 are affiliated to Pseudomonas spp. (95–99% similarity). An abundance of repeats and transposases within core and cargo modules serves as hotspots for possible recombination events and implied their acquisition from other organisms (Burrus, 2017). A significant deviation in GC skewness (Figure 2A, 12–13 GC mol%) of this stretch further supported the observation of the possibility of horizontal gene transfer events.
Pseudomonas bharatica genomic island-1 is located on the contig_008 (length 89,869 bp; nucleotide positions: 6,34,598 to 7,25,367), and has tRNA-ArgCCT gene (75 bp) at 3′-end followed by a repeat of 38 bp sequence that forms the attL site. An identical repeat is located at 5′-end (attR), thus defining the borders of this suspected ICE (Figure 2A). PBGI-1 is present as a single copy in the genome and its annotation is depicted in Supplementary Table 4 and Supplementary Figure 7B. Strain CSV86T tolerates 2 mM concentration of various heavy metals (Co2+, Zn2+, Cd2+: CZC) supplemented during the growth of aromatics (Paliwal et al., 2014). Genes encoding efflux pump proteins/transporters for heavy metal cations (CZC) are found to be part of this GI as intact clusters, hence designated as PBGI-1. Compared to ICEnahCSV86, PBGI-1 exhibited a mixed structure, i.e., characteristic of more than one ICEs [ICEclcLB400 (122.2 Kb responsible for polychlorinated biphenyl metabolism), ICEclcCH34 (109.5 Kb for mercuric resistance), and ICE(Tn4371)6035 (50 Kb for acriflavine resistance)]. Synteny analysis of PBGI-1 showed linear conserved block with respect to a short stretch of core module (∼10 Kb) against ICEclcLB400 and ICE(Tn4371)6035 (Figure 2B). This element showed significantly poor similarity (<30%) with previously reported ICEs [ICEPaeLESB58-1 (P. aeruginosa), ICECmeCH34-1 (Cupriavidus metallidurans), ICEHaeULPAs1-1 (Herminiimonas arsenicoxydans), ICEAxyA8-1 (Achromobacter arsenitoxydans)] encoding heavy metal resistance (Cd, Zn, Co, Hg, As, and Cu). Overall, a different integration site (proximity to tRNA-Arg), the presence of hypothetical proteins within core modules, and Co-Zn-Cd resistance (czc operon) as a cargo function demarcated the uniqueness of this suspected ICE amongst reported ICEs for the heavy metal tolerance (Figure 2).
Downstream of tRNA-Arg, two core modules (conjugation and partition: T4SS, T4CP: a total of 29.2 Kb) were distributed conjointly on both the strands. Insertion of alternating repeats (sizes: 254 and 5,728 bp) and transposases (two Il-IS-2, size: 1,163 bp) separated the core modules from czc cargo modules (Figure 2A). Two membrane proteins (YraQ family) and ArsR transcription regulator were present upstream of these repeats and transposases (Supplementary Figure 7B). BLASTn searches (against RefSeq) of these observed repeats indicated 99–100% identity with repeats of Cycloclasticus zancles 78-ME (obligate marine PAH degrading isolate) and A. baumannii 7835 (antibiotic resistant), similar to ICEnahCSV86. These repeats were found to encode transposases (ISL3, TnpA family). Genes encoding Tn7 transposition proteins (TsnBC) are located next to the repeats. These proteins catalyze transposition by promoting DNA bending to form a highly organized protein-DNA complex (Arciszewska and Craig, 1991). In the vicinity of this region, a stretch of hypothetical proteins (homologous to conserved hypothetical proteins of Pseudomonas spp.) was observed (Figure 2A). Like ICEnahCSV86, the cargo module (total ∼25 Kb) harboring genes for the tolerance of Co-Zn-Cd is located at the 5′-end (Figure 2A). Strikingly, PBGI-1 harbors all the reported systems (and the associated mechanisms) for Co2+, Zn2+, and Cd2+ resistance-efflux, which are localized into three sub-modules: (I) cation diffusion facilitator (CDF) CzcD based efflux cluster, (II) PIB4-type ATPase CzcP, and (III) HME-RND (Heavy MEtal-Resistance-Nodulation-Division)-driven CzcCBA cluster. Three alternating Il-IS-2 transposases (size: 560, 716, and 1,163 bp) followed by repeats (sizes: 1.077, 2.984, 170 bp), and stretch of hypothetical proteins segregated czc cargo clusters into two sub-modules (I and II). Module I comprising of czc regulator (czcR) and efflux resistance (czcD) showed the maximum homology (99%) with marine bacterial taxa, Module II comprising a single component transporting ATPase (czcP), and multi-component RND-resistance efflux pumps (czcABC: transmembrane, membrane fusion, and outer membrane) showed affiliation with Pseudomonas spp. (Supplementary Table 4). This contrasting observation indicated probable differential gene acquisition events mediated through different sets of mobile elements. It is interesting to note that all of the mobile genes on PBGI-1, including repeats and transposases showed lineage to both aromatic degrading and metal-transforming halophilic marine bacteria (Cycloclasticus zancles 78-ME, Halomonas axialensis, Salinicola peritrichatus, and Marinomonas spp.) and Pseudomonas spp. (Supplementary Table 4). This signifies possible horizontal acquisition of mobile genes from these taxa. Codon-based Z-test for the selection also displayed diversifying selection (dN-dS vs. dS = 1.397, at 0.05% level) for czcDR. This observation indicates that in the presence of heavy metals (Co, Zn, and Cd), the tolerance pathway is likely to get stabilized with more non-synonymous substitutions than synonymous substitutions and tends to be retained in the population (Verma et al., 2014). A sigma factor RpoS and an integrase (intCZC) were found to be present upstream (5′-end) of the czc cluster (Supplementary Figure 7B). The RpoS is reported to be involved in making bistability decisions by the stationary phase cells to proceed for ICE transfer in ICEclcB13 (Miyazaki et al., 2012). It is also involved in activating promoters for regulated expression of integrase (int) and its regulator (InrR), thus mediating the high rate of ICE transfer to the recipient (Miyazaki et al., 2012). The presence of RpoS as a part of PBGI-1 could be attributed to regulating the expression of integrase in the absence of an integrase regulator (InrR).
The GI, P. bharatica genomic island-2 (PBGI-2) is also located on the contig_008 (length 91,907 bp; nucleotide positions: 947,073 to 1,038979) with tRNA-LysTTT gene (75 bp) at 3′-end followed by a repeat of 61 bp sequence forming attL border and identical sequence repeat at 5′-end forms attR borders (Figure 2A). The PBGI-2 is present as a single copy in the genome and its gene annotation is summarized in Supplementary Table 5 and Supplementary Figure 7C. It showed structural similarity with ICEclc (ICEclcLB400), but maximum homology with GIs of P. aeruginosa (PAGI-5, PAGI-2 of P. aeruginosa C) encoding virulence, metal-resistance, and cytochrome biogenesis traits (Klockgether et al., 2007; Battle et al., 2009). Based on the maximum overall similarity observed with PAGI of P. aeruginosa, this GI is designated as PBGI-2 (Figure 2B). The core module (53.9 Kb) of PBGI-2 was found to be intact and segregated into three sub-modules (conjugation, par, and PFG-1) by two successive repeats of 174 bp flanked by mobile element proteins. Additionally, two long stretches of hypothetical proteins (7.8 and 11 Kb) are present upstream of the core modules (Figure 2A). Genes for DNA replication repair, membrane protein-TonB, short-chain dehydrogenase, methyl-accepting chemotaxis protein, disulfide isomerase, DNA-binding regulators, phage proteins, etc., were found to be present on PBGI-2, indicating mixed function nature of its cargo module. It was also found to harbor an additional P4 phage-type integrase and the associated regulator (InrR) near the 5′-end of the core module for regulating intPBGI-2 expression. BLASTN/P similarity searches showed a maximum homology of all cargo and core genes with P. aeruginosa strains (Supplementary Table 5).
The National Center for Biotechnology Information (NCBI) RefSeq-based homology search of PBGI-2 showed the presence of multiple copies of syntenic GIs in Beta- and Gamma-proteobacteria genomes. Whereas PAGI-2 type islands were reported in the genome of various (total of 31) P. aeruginosa and P. putida strains, of which 12 strains harbor one, 11 strains two, 7 strains three, and 1 strain with four PAGI elements. These are further grouped into 10 sub-types based on differential DNA-hybridization patterns (Klockgether et al., 2007). The presence of these ICE-like GIs in numerous taxa suggests that they probably form a family with a deep evolutionary origin. Based on the structural synteny with ICEclc members and higher homology with PAGI-2 of P. aeruginosa strain C, it can be hypothesized that PBGI-2 is an intermediate between an ICE and GI. Many PAGI elements are reported to excise spontaneously at a frequency of 10–5 to 10–4 (Rajanna et al., 2003; Lesic et al., 2004), although mutations, deletions, and genome rearrangements are likely to be responsible for the inability of such elements to achieve precise excision and mobilization. On the other hand, PAGI-2 does not spontaneously excise from its host chromosome during its growth in vitro and has been shown to transfer to other proteobacterial members at a low frequency (10–5 to 10–4) (Ravatn et al., 1998a; Klockgether et al., 2007). For other similar elements, like pKLC102 and PAPI-1, the frequencies of excision are of 1–3 orders of magnitude higher. Similarly, PBGI-2 harboring the phage-type integrase, plasmid-related genes, a type IV pilus biogenesis gene cassette, and a syntenic set of conserved ORFs similar to those detected in PAGI-2 and PAGI-3 indicate its ability to excise from the chromosome and form an extra-chromosomal circular intermediate during transfer.
Bidirectional alignment and phylogenetic analyses of ICEnahCSV86, PBGI-1, and PBGI-2 of strain CSV86T, as well as its modular components, were performed to delineate their ancestral root/origin and phylogenetic relatedness. ICEnahCSV86 formed a coherent cluster with ICEXTD as its nearest neighbor and corroborated the pairwise comparison data (Figure 3A). Interestingly, PBGI-2 was found to be a near-distant relative of ICEclc members, including ICEnahCSV86, whereas PBGI-1 deviated from all and formed a separate clade, as an out group [Figure 3A (i)]. Bidirectional hit-based DNA-homology (% similarity) data also demarcated ICEXTD to be the overall nearest relative of ICEnahCSV86, whereas, PBGI-1 and PBGI-2 were congruent with ICEclcLB400 (51% similarity) and PAGI-2 (48%), respectively [Figure 3A (ii)]. Phylogenetic analysis of integrases revealed their non-coherent nature, forming three distinct clusters (Supplementary Figure 9A). The intNAH formed a coherent association with integrases of Pseudomonas, Betaproteobacteria, and intbph-sal (cluster-I), whereas intPAGI-2 of P. aeruginosa C served to be a distant member. The IntPBGI-2 formed a monophyletic grouping with integrases of non-ICE Pseudomonads, whereas intXTD and intB13 of ICEclc were found to be the nearest ICE relatives. Unlike the other two, intCZC was a distant relative of all ICE-encoded integrases reported so far (Supplementary Figure 9A).
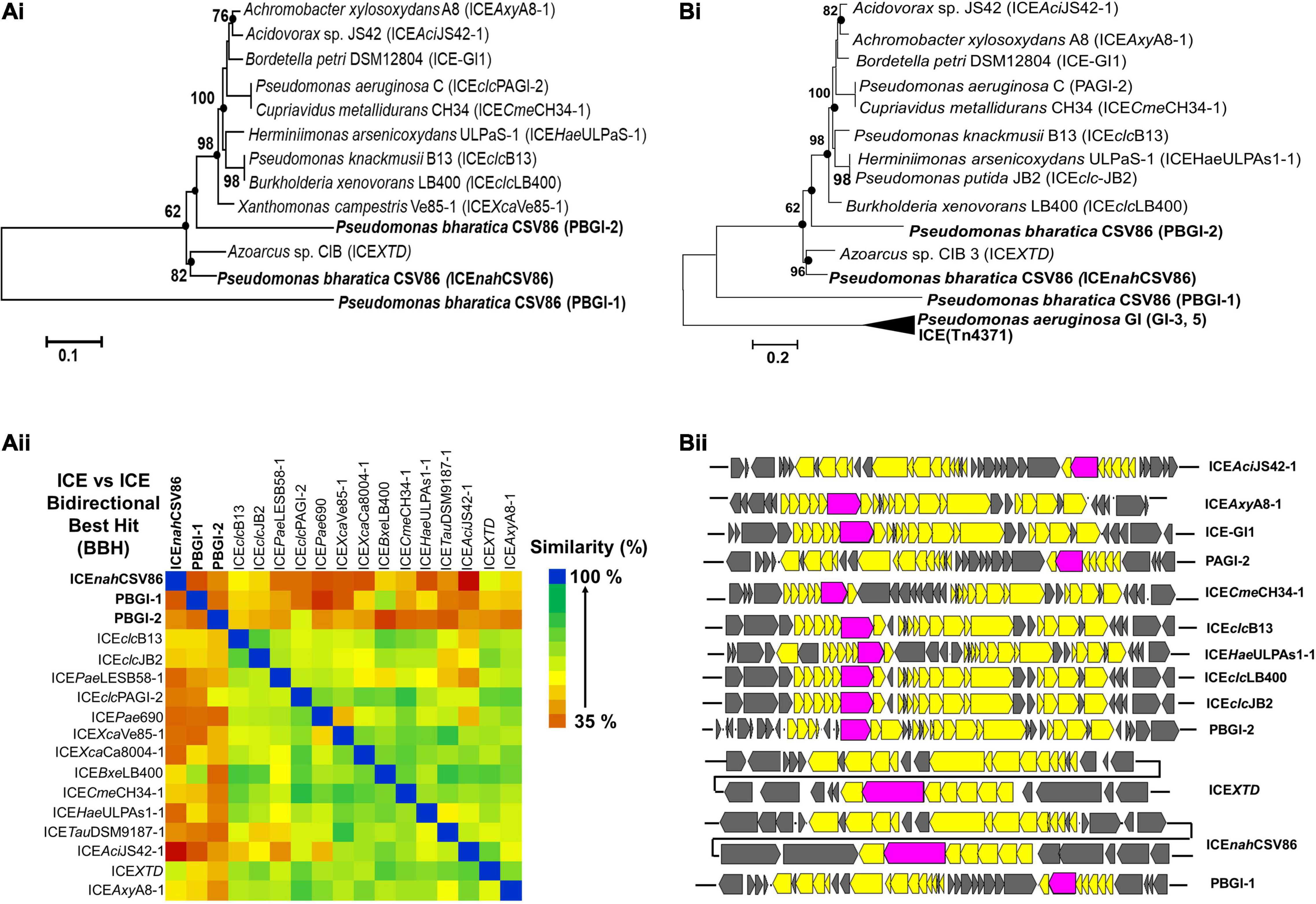
Figure 3. Evolutionary inferences and overall ICE related indices for three GIs, predicted to be ICEs from Pseudomonas bharatica CSV86T. [A (i)] Neighbor-joining (NJ) phylogenetic tree reconstructed using full-length nucleotide sequences of predicted ICEs and its nearest similar ICEs, and [A (ii)] Heat-map depicting bidirectional best hit-based average nucleotide identity (DNA–DNA homology level) of compared ICEs. [B (i)] Maximum-likelihood (ML)-based phylogeny of concatenated amino acid sequences of core gene modules (T4SS, T4CP), and [B (ii)] structural arrangement of core gene modules of various compared ICEs shown in [B (i)]. Trees were generated with 1,000 bootstrap iterations and the values at each node are represented as percentage replicates. The black solid circles represent generation of trees from all tree-making algorithms (NJ, ML, and ME).
Phylogeny of concatenated core proteins (T4SS: VirB4, VirD4, TraG, TraI, and ParAB) showed two distinct branches. ICEnahCSV86 and PBGI-2 were very closely related and clustered with ICEXTD, while PBGI-1 was distantly related to ICEclc and others (Tn4371), thus forming a discrete branch [Figure 3B (i)]. The structural arrangement of T4SS core modules from various ICEs also corroborated the observation indicating a genetic coherence between ICEnahCSV86 and ICEXTD, while a mixed pattern was observed for PBGI-2 and PBGI-1 [Figure 3B (ii)]. Similar results were obtained for relaxases, where PBGI-1 remained out-cladded as a distant relative of all compared ICEs (data not shown). All three predicted ICEs from strain CSV86T harbor a variable number of transposases, which are reported to be a key component for exchanging DNA under specific environmental conditions (Jurka et al., 2007). All these transposases were found to be distinct and showed a poor sequence homology (<30% similarity at 50% coverage) amongst each other. The transposases formed a mixed clustering (three distinct groups) and showed lineages with marine bacterial taxa (Halomonas spp., Marinomonas, and Shewanella fodinae) reported for encoding complex organic carbon metabolism and heavy metal resistance traits (Supplementary Figure 9B). Overall, phylogenomics and synteny mapping revealed that both ICEnahCSV86 and PBGI-2 might be speciated from a common ancestral lineage, i.e., ICEXTD/LB4001-1 like ICEclc members. Codon-based Z-test and the rate of diversifying selection (dN-dS, dN/dS vs. dS) between syntenic core gene modules of ICEnahCSV86 and PBGI-2 showed a very high positive dN-dS values (4.07–8.85) (Supplementary Figure 5B), indicating a diversifying selection (non-synonymous substitution vs. synonymous substitution) of ICEnahCSV86 with respect to PBGI-2. With an intact and complete set of core modules and a conserved synteny of PBGI-2 with PAGI, we hypothesize that this element might be the precursor from which ICEnahCSV86 has been diverged after acquiring and stabilizing cargo genes (nah-sal) under selection pressure. The PBGI-1 might have undergone high degrees of recombination events, that have masked its evolution from a common ancestor. The presence of similar intermediary ICEs with higher recombination events has also been postulated (Wozniak et al., 2009; Baharoglu et al., 2013).
The presence of functionally active naphthalene degradation and Co2+, Zn2+, and Cd2+ transport-efflux genes on two different GIs in a single bacterium is a novel observation. Many naphthalene-degrading bacterial spp. are reported to harbor naphthalene metabolic clusters either on the plasmid (conjugative/non-conjugative) or transposon (Mohapatra and Phale, 2021). To date, no ICEs have been functionally characterized and reported for encoding naphthalene degradation traits. It is well established that aromatics including naphthalene and its derivatives have become ubiquitous in the environment (Duttagupta et al., 2020; Mohapatra and Phale, 2021). It has also been observed that many aromatic ring-hydroxylating dioxygenases like naphthalene dioxygenase (NDO, the first enzyme of the naphthalene degradation pathway) display “catalytic promiscuity” toward structurally related yet distinct compounds (Verma et al., 2019; Phale et al., 2020). Noticeably, such catalytic promiscuity of metabolic enzymes has been reported for a number of enzymes like biphenyl dioxygenase, benzoate dioxygenase, and toluene monooxygenase involved in the degradation of aromatics. Based on this information, it can be hypothesized that the ubiquitous presence of aromatics like naphthalene has imposed a substantial positive selection pressure on strain CSV86T or similar spp. (from diverse biogeography) to acquire and evolve genes and enzymes for naphthalene metabolism through horizontal gene transfer events (by MGEs). Further, the genomic integration of these elements, re-organization, domain shuffling, and active-site optimization (for structurally related xenobiotic compounds) might have occurred to fine-tune the pathway, thus providing survival benefits to the recipient.
To validate this hypothesis, detailed analyses were performed for nah and sal clusters from genomes of naphthalene or aromatic degrading Pseudomonas spp. reported from diverse biogeographic origins (Table 2). Mega-BLAST analysis of nah-sal clusters of ICEnahCSV86 revealed similar and syntenic regions (of size 100–150 Kb, >70% coverage, 95% similar) in P. citronellolis SJTE-3, Pseudomonas balearica DSM 6083, Pseudomonas benzenivorans DSM 8628, Pseudomonas bauzanensis W13Z2, Pseudomonas stutzeri KOS6, etc. and some plasmids (pNAH7, pND6, pAK5, AN10, and pAS1) of Pseudomonas (Table 2).
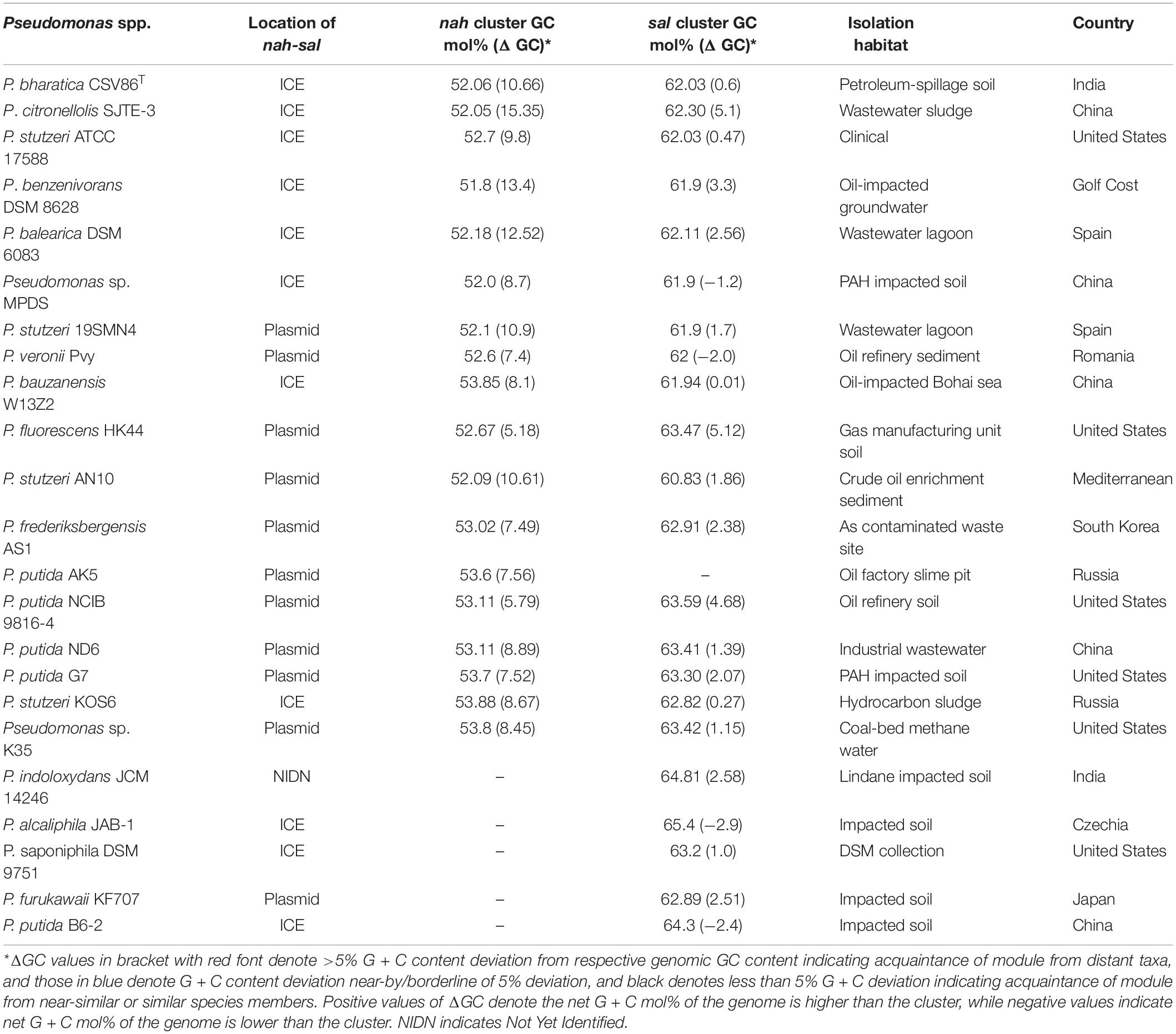
Table 2. GC content and deviation (mol%, from genomic GC) of nah and sal cluster from different predicted ICEs and plasmids of various naphthalene degrading Pseudomonas spp. isolated from diverse biogeographic regions.
Interestingly, RAST annotation identified the presence of 5’ tRNA-GlyCCC, border repeats (attL/R), integrase as well as cargo genes (nah-sal clusters), and core modules, suggesting these genomic regions to be probable ICEs, similar to that observed in strain CSV86T (Figure 4). The synteny alignment for all these hypothesized ICEs and ICEnahCSV86 indicated highly conserved cargo and core gene modules (as linearly conserved blocks, LCBs) (Figure 4A), thus denoting identical structural features, including oriT (homologous to ICEnahCSV86 and ICEXTD). Barring strain CSV86T (Basu and Phale, 2008), none of them have been experimentally validated for conjugative transfer. Based on this analysis, we propose ICEnahCSV86 to be a prototypical element for naphthalene degradation with consensus features. A monophyletic and coherent clustering was observed for ICEnahCSV86 and all probable ICEs (Figure 4B). Further, a highly conserved consensus attL (except substitution of A with G in the last 13 bp in two members), coherent tRNA-Gly, and integrase (syntenic to ICEnahCSV86) supported ICEnahCSV86 to represent a prototypical element (Supplementary Figure 10). A modular synteny plot confirmed ICEXTD and ICEclcB13 as the nearest relatives, thus classifying these probable ICEs as members of ICEclc (Figure 4C). It is observed that the typical core and structural elements were highly syntenic (60–76% identical with LCBs) in all three ICEs. Out of two cargo modules (representing upper pathway and lower pathway), a moderate synteny (45–52% identical) was observed in the lower pathway of ICEnahCSV86, ICEclcB13, and ICEXTD. Interestingly, the upper pathway in all three ICEs: nah cluster in ICEnahCSV86, clc cluster in ICEclcB13, and tod cluster in ICEXTD were non-syntenic (<20% similar). Guarding these upper pathways, the presence of various mobile elements (multiple repeats and transposases) suggested a separate modular fusion of the upper cluster/pathway within the ICE frame.
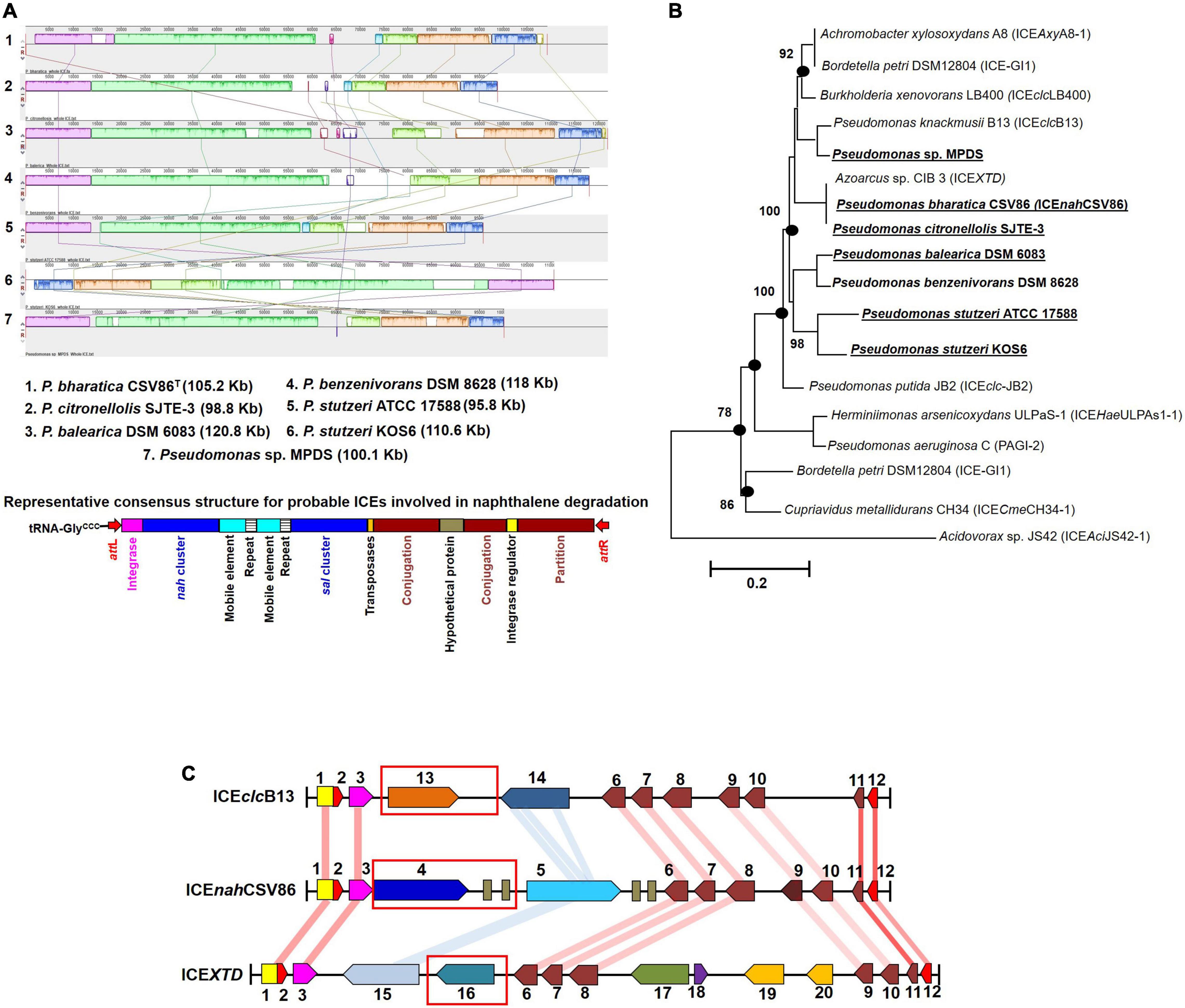
Figure 4. Comparative-functional genomic analyses of nah-sal clusters including adjacent up/down-stream sequences from various naphthalene/aromatic degrading Pseudomonas spp. (A) Synteny mapping of probable ICEs harboring nah-sal clusters and its comparison with ICEnahCSV86. The representative consensus structure for all naphthalene degradation encoding probable ICEs has been depicted at the bottom of the synteny plot. (B) ML phylogenetic analysis of ICEnahCSV86 with probable ICEs, belonging to the ICEclc family, from naphthalene and aromatic degrading bacteria. Taxa in bold indicate the probable naphthalene degrading bacteria while bold with underline indicate reported naphthalene utilization/degradation phenotype. Black dots at each node represent robustness of clade formed in all tree-making algorithms. Values near each branch of the phylogenetic tree indicate percent bootstrap re-iteration and bar below the trees indicate substitution (0.1–0.2%). (C) Artemis comparison-based synteny mapping of prototypical ICEnahCSV86 and its phylogenetically related neighbors (ICEclcB13 and ICEXTD). The connecting lines between LCBs indicate percent similarity between various modules: blue (<60%), light red (60–70%), and deep red (>70%). Rectangular red boxes represent the upper pathway cargo modules of the respective ICEs.
The p-distance based pairwise divergence estimate showed that nah cluster of ICEnahCSV86 has the highest homology with P. citronellolis SJTE-3, P. stutzeri ATCC 17588 (0.000 ± 0.001 distance), P. benzenivorans DSM 8628 (0.002 ± 0.001 distance), and P. balearica DSM 6083 (0.008 ± 0.002 distance) (Figure 5A). ML-based phylogenetic reconstruction corroborated this observation, where nah cluster of ICEnahCSV86 cladded with other probable ICE-bearing members and deviated from plasmid-encoded nah clusters as distant relatives (Figure 5A). Similar results were observed for sal cluster (Figure 5B). It is interesting to note that, a congruency/commonality was observed in terms of their habitat/ecotype, where all the probable naphthalene-ICE bearing members are isolated from oil, petroleum, or waste-impacted habitat, implying the role of the selection pressure; thus, helping strains to acquire and evolve these elements (Table 2). It has been reported that a broad GC content (>5%) change (distinct GC content of a cluster from the host genome) in stretches of prokaryotic genomes is indicative of the exchange of DNA/acquisition of gene(s) through HGT (Bohlin et al., 2010; Hildebrand et al., 2010). GC skewness analysis suggests that nah cluster displays higher order of mol% deviation as compared to sal cluster (Table 2). For nah cluster, the lowest deviation (5.18 mol%) was observed for Pseudomonas fluorescens HK44 and the highest (15.35 mol%) for P. citronellolis SJTE-3, whereas ICEnahCSV86 (10.66%) lied close to probable ICE of P. stutzeri AN10 (10.61%). On the contrary, the G + C mol% of sal cluster was significantly close to genomic G + C content, except for P. fluorescens HK44 (5.12% deviation). We have observed that the core genetic module of ICEnahCSV86 shows a GC content (62.95%) close to the genome (63.2% GC), whereas adaptation modules deviated sharply, i.e., module-1 (nah, 59.2% GC), module-2 (sal, 58.9% GC). Insertion of mobile elements (repeats and transposases) flanking nah-sal clusters further suggested the acquisition of cargo modules by different horizontal transfer events (Bosch et al., 1999; Hirose et al., 2021). To further validate these observations, dN-dS vs. dS (rate of non-synonymous over synonymous substitution) was performed. Positive dN-dS value (5.439–5.731) for nah cluster indicated higher non-synonymous substitutions than synonymous substitutions. This observation indicated its diversifying selection within the population and its still-evolving nature, which can be attributed to differential substrate selection pressure at impacted niches, i.e., naphthalene/structurally related compounds (oil-derived). Whereas lower dN-dS (2.139–2.247) of sal cluster indicated its intermediary/near-stable nature within the population. Additionally, the regulatory gene (nahR: LysR type regulators) of strain CSV86T showed 100% similarity with P. stutzeri AN10 and phylogenetic closeness to other ICE-encoded NahR, out-cladding it from plasmid encoded members (Supplementary Figure 11). Interestingly, the presence of transposases upstream of nahR in both CSV86T and AN10 and the absence of such gene in other plasmid-bearing members further confirms similar horizontal acquisition behavior.
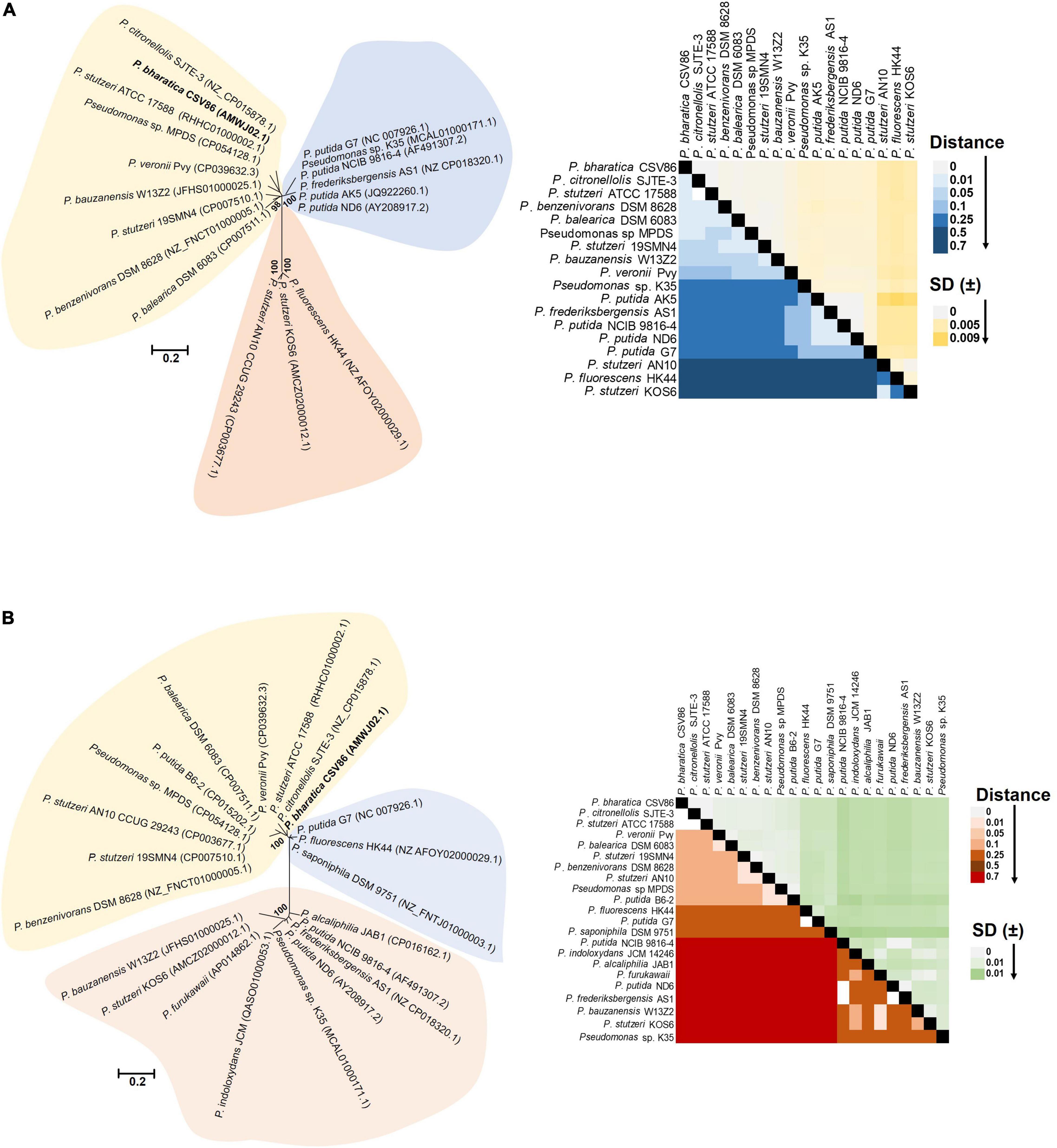
Figure 5. Phylogenomic analyses of cargo gene modules (nah-sal cluster) of ICEnahCSV86 and other mobile elements (probable ICEs, plasmids, transposons) from naphthalene degrading Pseudomonas spp. Phylogenetic trees of (A) nah cluster and (B) sal cluster are reconstructed using ML-based algorithm and validated using NJ, ME methods with 1,000 bootstrap iterations. The tree is optimized using interactive Tree of Life (iTOL), and the best optimum tree is depicted here. The color-shaded areas around the clades depict demarcation of differential cladding of clusters present on probable ICEs (light yellow), plasmids (light blue), and mixed elements (light pink, which include conjugative/non-conjugative transposons and intermediate forms). The heat-maps depict the p-distance based pairwise similarity of (A) nah clusters and (B) sal clusters located on various mobile genetic elements.
It has been hypothesized that catabolic pathways for aromatic metabolism have evolved through the modular fusion of at least two to three components (upper, middle, and lower pathways routes). For example, catechol meta-cleavage route is predominant and homologous in many bacterial taxa and is predicted to fuse with lower operon gene module (generating TCA intermediates). Whereas upper (peripheral) pathway operon might have been acquired as a separate module at a later stage, thus increasing the catabolic versatility of the recipient strain. Further to fine-tune metabolism, regulatory elements were recruited from the evolved bacterial genome(s) and were modified subsequently to optimize the expression of the catabolic enzymes (Harayama, 1994; Gerischer, 2002). The G + C content and codon usage analyses of the nah operon of P. stutzeri AN10 support this modular fusion and recombination-based evolutionary theory. It has been shown that G + C content and the codon usage patterns of P. stutzeri AN10 are different for both the upper and lower naphthalene pathway genes. A similar pattern is also observed in strain CSV86T. The occurrence of non-coding (repeat-transposases) DNA region, flanking the conserved nah upper pathway has been suggested to be involved in the mobilization and further rearrangements of this entire catabolic module (Eaton, 1994). Overall, these observations strengthen our hypothesis of modular fusion of nah cluster in ICEnahCSV86 upstream to sal operon. The modular regulation of enzymes involved in aromatic degradation (benzyl alcohol, hydroxybenzyl alcohol, and other aromatics) has been demonstrated at the biochemical level in CSV86T (Basu et al., 2003). Carbon source dependent metabolic studies also indicate that the degradation of aromatic alcohol and naphthalene involves two regulons (Basu et al., 2003; Mohan and Phale, 2017). Cellular respiration (nmol O2 consumed min–1 mg–1) studies of strain CSV86T grown on naphthalene, salicylate, and glucose on substrate naphthalene, salicylate, and catechol showed differential induction of modules/metabolic regulons. Similarly, we observed the metabolite-dependent induction of three regulons for the metabolism of naphthalene-based pesticide Carbaryl in Pseudomonas spp. (Singh et al., 2013). These results indicate that the end metabolite generated from the upper operon induces the enzymes involved in the middle or lower operons in a cascading manner, achieving metabolic efficiency.
Integrative conjugative elements-encoded degradation or resistance traits in bacteria provide advantages over plasmid-encoded traits. ICEs are able to integrate into the host chromosome in a site-specific manner, resulting in stable phenotypic property. Further, this eliminates the need for constant replication and maintenance as well as inherent variability in the copy number, which is observed in most plasmids. In addition, ICEs are reported to excise and transfer to new host(s) in response to a variety of signals like RecA-dependent SOS response to DNA damage, secretion of signaling molecules from recipients, the growth phase of the host, and additional mechanisms related to the expression of cargo genes (Johnson and Grossman, 2015). Interestingly, ICEs are also reported to facilitate the transfer of other mobile elements, thus providing genetic bioaugmentation/plasticity in the community for better adaptability and evolution (Delavat et al., 2017; Liu et al., 2019).
A strong promoter (designated Pnah, LDF = 6.03, P-value = 0.0023) in the upstream and several weaker promoters throughout the nah cluster were predicted (Figure 6A). A single putative strong promoter (LDF = 3.73) could also be detected upstream of the sal cluster (Figure 6A). NahR, a LysR family transcription regulator (LTTR), is found to be located in between nah and sal cluster on ICEnahCSV86 with its own promoter (LDF = 5.87) and transcribed in the opposite direction of nah cluster. NahR binding site is found to be located 60 bp upstream of the transcriptional start site of nah and sal clusters (Supplementary Table 6). The promoters (-10 box and -35 box) and NahR binding site were found to be similar to other naphthalene degradation operons (Supplementary Figure 12). Co-transcription analysis confirmed the polycistronic nature of the nah and sal operons, thus validating the presence of strong promoters (Pnah and Psal).
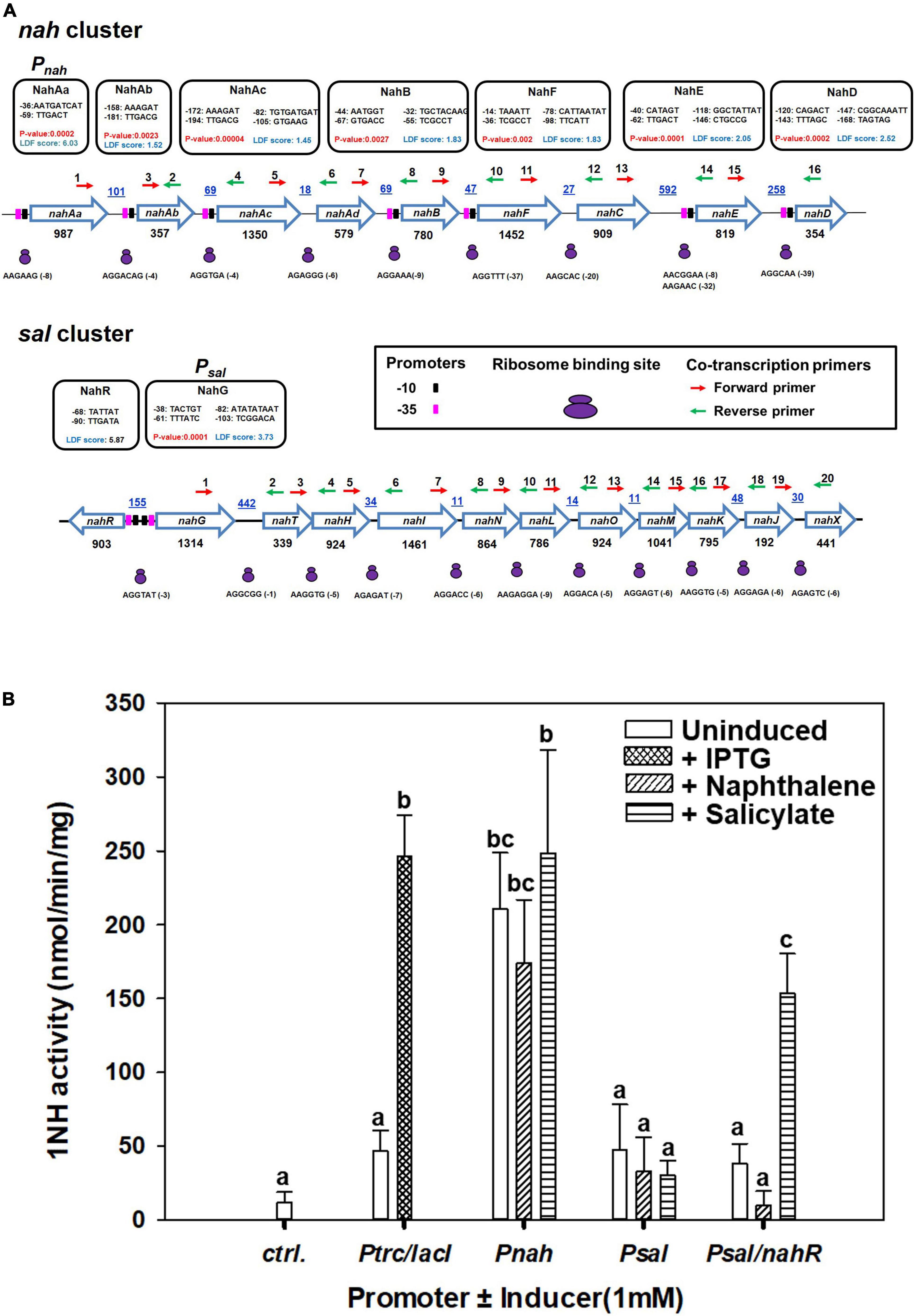
Figure 6. Analysis of the upstream regions of nah and sal operons present on ICEnahCSV86 of Pseudomonas bharatica CSV86T. (A) Operonic arrangement of upper (nah) and lower pathway (sal) genes and identification of putative promoter sequences [-10 (black filled rectangles) and -35 (pink filled rectangles) boxes], ribosome binding sites (RBS, as small and large purple ovals), and the location of primers [forward (red) and reverse (green) arrows] used for co-transcription analysis. The putative promoter sequences are depicted in the black box. The promoters with LDF scores have been predicted using BPROM and P-values were predicted using SAPPHIRE. The intergenic distances (bp) are indicated by blue color with underline, while gene lengths (in bp) are denoted below each gene. (B) Specific activity of 1-naphthol 2-hydroxylase (1NH) from various constructs expressed in E. coli BL21 (DE3) using different promoters and inducers. The bar represents the mean value with standard deviation (n = 3). Statistically significant differences (P = 0.05) among different promoter systems are denoted by a, b, c (Tukey’s post hoc test).
Amongst the promoters (Pnah, Psal, and Psal/NahR) fused with 1NH, as a reporter gene, the activity from Pnah was significantly higher than that of Psal, which showed expression only in the presence of NahR regulator and the inducer, salicylate (Figure 6B), while the expression from IPTG-induced Ptrc was similar to Pnah. The Psal/nahR system has also been studied in both Pseudomonas and E. coli systems and has been found to be inducible (de Lorenzo et al., 1993; Calero et al., 2016),11 while the Pnah has been reported to show leaky/constitutive expression (Burlage et al., 1990; Neilson et al., 1999). These observations provide an essential insight into the evolutionary logic driving the regulation of both operons. The presence of a strong but leaky promoter upstream to the upper nah operon is essential to generate the inducer (salicylate; the end product of the nah enzymes) at a sufficient concentration from the substrate (naphthalene). The lower operon harbors a relatively weak promoter which is tightly regulated and gets subsequently induced by a specific inducer metabolite generated by the action of upper pathway (nah) enzymes.
Strain CSV86T grown in the absence of GI-encoded selection pressure for 60 generations did not depict a significant change in the naphthalene utilization property and heavy metal tolerance phenotype as compared to a culture grown under selection pressure. Specifically, the growth rate, cell biomass yield, or enzyme activity (catechol 2,3-dioxygenase) in naphthalene amended conditions were similar (Supplementary Table 7). The growth in the presence of heavy metals (Co2+, Zn2+, Cd2+) and MIC values also remained unaltered (Supplementary Table 7). Therefore, these GIs are stably integrated/maintained in the genome of strain CSV86T and are not lost even in the absence of selection pressure. As a consequence, such elements have established themselves in diverse populations, playing an important role in bacterial evolution (Miyazaki et al., 2015; Delavat et al., 2017). Moreover, aromatic impacted habitats often contain a variety of toxic compounds like heavy metals, thus hindering the bioremediation process. Therefore, acquiring heavy metal tolerance trait is highly beneficial for a bacterium to survive in such contaminated niches (Jiménez et al., 2004; Phale et al., 2021). ICEnahCSV86 and PBGI-1 provide a survival advantage to strain CSV86T in the presence of naphthalene and/or Co2+-Zn2+-Cd2+.
Pseudomonas bharatica CSV86T harbors three GIs (>50 Kb), suspected to be ICEs as single copies on the genome, belonging to the T4SS-ICE family: ICEnahCSV86 (105.2 Kb), PBGI-1 (89.8 Kb), and PBGI-2 (91.9 Kb), and are syntenic to ICEclc (ICEXTD), ICETn4371, and PAGI-2, respectively. Based on the cargo genes present on these GIs and phenotypic traits observed in strain CSV86T, ICEnahCSV86 was attributed for naphthalene degradation, PBGI-1 for heavy metal (Co2+, Zn2+, Cd2+) resistance-efflux, and PBGI-2 for mixed/unknown functions. ICEnahCSV86 is the only functionally and genomically characterized prototypical element (predicted to be an ICE) for naphthalene degradation with a low conjugative transfer frequency. PBGI-2 is hypothesized to be an intermediate element between ICE and GI and proposed to be an ancestor for ICEnahCSV86. The nah cluster is predicted to be integrated through modular fusion upstream to the sal cluster followed by rearrangement and domain reshuffling to achieve metabolic efficiency. These elements are stably integrated into the genome and are not lost even in the absence of selection pressure, rendering advantageous survival benefits and adaptive lifestyle to the strain CSV86T under varied environmental conditions. The promoters of nah-sal on ICEnahCSV86 were transcriptionally active and found to be suitable for heterologous protein expression in E. coli. Functional genomic and genome-wide comparative analyses of multiple GIs in a single organism provide crucial insights into the evolution and distribution of metabolic traits, such as naphthalene degradation and metal tolerance through horizontal gene transfer, contributing to genomic plasticity, metabolic diversity, and niche colonization abilities to bacterial species. Further, the role of core modules in the acquisition of diverse cargo functions (as suited to the habitat) mediated through modular exchange and fusion has been hypothesized.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
PP, BM, and HM designed the study, performed data curation, validation, and interpretation, and agreed for the submission of the final version of the manuscript. BM performed the genome finishing, genomic data mining, comparative genomics, ICE-related experiments, and promoter prediction. HM conducted the promoter analyses, gene construct designing, co-transcription, and expression analyses. BM and HM wrote the manuscript drafts. PP managed the research funds and finalized the manuscript draft. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BM acknowledges IIT Bombay for providing a Post-Doctoral Fellowship. HM thank CSIR, Government of India for the Senior Research Fellowship. PP thank DST, Government of India for the research funding.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.928848/full#supplementary-material
Arciszewska, L. K., and Craig, N. L. (1991). Interaction of the Tn7-encoded transposition protein TnsB with the ends of the transposon. Nucleic Acids Res. 19, 5021–5029.
Badhai, J., and Das, S. K. (2016). Characterization of three novel SXT/R391 integrating conjugative elements ICEMfuInd1a and ICEMfuInd1b, and ICEMprChn1 identified in the genomes of Marinomonas fungiae JCM 18476T and Marinomonas profundimaris strain D104. Front. Microbiol. 7:1896. doi: 10.3389/fmicb.2016.01896
Baharoglu, Z., Garriss, G., and Mazel, D. (2013). Multiple pathways of genome plasticity leading to development of antibiotic resistance. Antibiotics 2, 288–315.
Basu, A., Apte, S. K., and Phale, P. S. (2006). Preferential utilization of aromatic compounds over glucose by Pseudomonas putida CSV86. Appl. Environ. Microbiol. 72, 2226–2230. doi: 10.1128/AEM.72.3.2226-2230.2006
Basu, A., Dixit, S. S., and Phale, P. S. (2003). Metabolism of benzyl alcohol via catechol ortho-pathway in methylnaphthalene-degrading Pseudomonas putida CSV86. Appl. Microbiol. Biotechnol. 62, 579–585. doi: 10.1007/s00253-003-1305-8
Basu, A., and Phale, P. S. (2008). Conjugative transfer of preferential utilization of aromatic compounds from Pseudomonas putida CSV86. Biodegradation 19, 83–92. doi: 10.1007/s10532-007-9117-7
Battle, S. E., Rello, J., and Hauser, A. R. (2009). Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 290, 70–78.
Bignell, C., and Thomas, C. M. (2001). The bacterial ParA-ParB partitioning proteins. J. Biotechnol. 91, 1–34.
Bohlin, J., Snipen, L., Hardy, S. P., Kristoffersen, A. B., Lagesen, K., Dønsvik, T., et al. (2010). Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genomics 11:464. doi: 10.1186/1471-2164-11-464
Bosch, R., Garcıìa-Valdés, E., and Moore, E. R. (1999). Genetic characterization and evolutionary implications of a chromosomally encoded naphthalene-degradation upper pathway from Pseudomonas stutzeri AN10. Gene 236, 149–157. doi: 10.1016/s0378-1119(99)00241-3
Botelho, J., Grosso, F., and Peixe, L. (2018). Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES–6 carbapenemase. J. Antimicrob. Chemother. 73, 77–83. doi: 10.1093/jac/dkx337
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Burlage, R. S., Sayler, G. S., and Larimer, F. (1990). Monitoring of naphthalene catabolism by bioluminescence with nah-lux transcriptional fusions. J. Bacteriol. 172, 4749–4757. doi: 10.1128/jb.172.9.4749-4757.1990
Burrus, V. (2017). Mechanisms of stabilization of integrative and conjugative elements. Curr. Opin. Microbiol. 38, 44–50.
Burrus, V., Pavlovic, G., Decaris, B., and Guédon, G. (2002). Conjugative transposons: the tip of the iceberg. Mol. Microbiol. 46, 601–610. doi: 10.1046/j.1365-2958.2002.03191.x
Calero, P., Jensen, S. I., and Nielsen, A. T. (2016). Broad-host-range ProUSER vectors enable fast characterization of inducible promoters and optimization of p-coumaric acid production in Pseudomonas putida KT2440. ACS Synth. Biol. 5, 741–753. doi: 10.1021/acssynbio.6b00081
Chain, P. S., Denef, V. J., Konstantinidis, K. T., Vergez, L. M., Agulló, L., Reyes, V. L., et al. (2006). Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc. Natl. Acad. Sci. U.S.A. 103, 15280–15287. doi: 10.1073/pnas.0606924103
Christie, P. J., Whitaker, N., and González-Rivera, C. (2014). Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta. Mol. Cell Res. 1843, 1578–1591.
de Lorenzo, V., Eltis, L., Kessler, B., and Timmis, K. N. (1993). Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123, 17–24. doi: 10.1016/0378-1119(93)90533-9
Delavat, F., Miyazaki, R., Carraro, N., Pradervand, N., and van der Meer, J. R. (2017). The hidden life of integrative and conjugative elements. FEMS Microbiol. Rev. 41, 512–537.
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424.
Duttagupta, S., Mukherjee, A., Bhattacharya, A., and Bhattacharya, J. (2020). Wide exposure of persistent organic pollutants (PoPs) in natural waters and sediments of the densely populated Western Bengal basin, India. Sci. Total Environ. 717:137187. doi: 10.1016/j.scitotenv.2020.137187
Eaton, R. W. (1994). Organization and evolution of naphthalene catabolic pathways: sequence of the DNA encoding 2-hydroxychromene-2-carboxylate isomerase and trans-o-hydroxybenzylidenepyruvate hydratase-aldolase from the NAH7 plasmid. J. Bacteriol. 176, 7757–7762. doi: 10.1128/jb.176.24.7757-7762.1994
Fang, G., Bhardwaj, N., Robilotto, R., and Gerstein, M. B. (2010). Getting started in gene orthology and functional analysis. PLoS Comput. Biol. 6:e1000703. doi: 10.1371/journal.pcbi.1000703
Fuentes, S., Méndez, V., Aguila, P., and Seeger, M. (2014). Bioremediation of petroleum hydrocarbons: catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 98, 4781–4794. doi: 10.1007/s00253-014-5684-9
Gaillard, M., Vallaeys, T., Vorhölter, F. J., Minoia, M., Werlen, C., Sentchilo, V., et al. (2006). The clc element of Pseudomonas sp. strain B13, a genomic island with various catabolic properties. J. Bacteriol. 188, 1999–2013. doi: 10.1128/JB.188.5.1999-2013.2006
Gerischer, U. (2002). Specific and global regulation of genes associated with the degradation of aromatic compounds in bacteria. J. Mol. Microbiol. Biotechnol. 4, 111–121.
Hacker, J., and Carniel, E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity. EMBO Rep. 2, 376–381.
Harayama, S. (1994). Codon usage patterns suggest independent evolution of two catabolic operons on toluene-degradative plasmid TOL pWW0 of Pseudomonas putida. J. Mol. Evol. 38, 328–335. doi: 10.1007/BF00163150
Hildebrand, F., Meyer, A., and Eyre-Walker, A. (2010). Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 6:e1001107. doi: 10.1371/journal.pgen.1001107
Hirose, J., Watanabe, T., Futagami, T., Fujihara, H., Kimura, N., Suenaga, H., et al. (2021). A new ICEclc subfamily integrative and conjugative element responsible for horizontal transfer of biphenyl and salicylic acid catabolic pathway in the PCB-degrading strain Pseudomonas stutzeri KF716. Microorganisms 9:2462. doi: 10.3390/microorganisms9122462
Janssen, P. J., Van Houdt, R., Moors, H., Monsieurs, P., Morin, N., Michaux, A., et al. (2010). The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS One 5:e10433. doi: 10.1371/journal.pone.0010433
Jiménez, J. I., Minambres, B., García, J. L., and Díaz, E. (2004). “Genomic insights in the metabolism of aromatic compounds in Pseudomonas,” in Pseudomonas, ed. J. L. Ramos (Boston, MA: Springer), 425–462.
Johnson, C. M., and Grossman, A. D. (2015). Integrative and conjugative elements (ICEs): what they do and how they work. Ann. Rev. Genet. 49, 577–601. doi: 10.1146/annurev-genet-112414-055018
Juhas, M., Van Der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393.
Jurka, J., Kapitonov, V. V., Kohany, O., and Jurka, M. V. (2007). Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 8, 241–259.
Klockgether, J., Würdemann, D., Reva, O., Wiehlmann, L., and Tummler, B. (2007). Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J. Bacteriol. 189, 2443–2459. doi: 10.1128/JB.0168806
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lawley, T., Wilkins, B. M., and Frost, L. S. (2004). Bacterial conjugation in gram-negative bacteria. Plasmid Biol. 13, 203–226.
Lechner, M., Schmitt, K., Bauer, S., Hot, D., Hubans, C., Levillain, E., et al. (2009). Genomic island excisions in Bordetella petrii. BMC Microbiol. 9:141. doi: 10.1186/1471-2180-9-141
Lesic, B., Bach, S., Ghigo, J. M., Dobrindt, U., Hacker, J., and Carniel, E. (2004). Excision of the high-pathogenicity island of Yersinia pseudotuberculosis requires the combined actions of its cognate integrase and Hef, a new recombination directionality factor. Mol. Microbiol. 52, 1337–1348. doi: 10.1111/j.1365-2958.2004.04073.x
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–D665. doi: 10.1093/nar/gky1123
Mahajan, M. C., Phale, P. S., and Vaidyanathan, C. S. (1994). Evidence for the involvement of multiple pathways in the biodegradation of 1-and 2-methylnaphthalene by Pseudomonas putida CSV86. Arch. Microbiol. 161, 425–433. doi: 10.1007/BF00288954
Miyazaki, R., Bertelli, C., Benaglio, P., Canton, J., De Coi, N., Gharib, W. H., et al. (2015). Comparative genome analysis of Pseudomonas knackmussii B13, the first bacterium known to degrade chloroaromatic compounds. Environ. Microbiol. 17, 91–104. doi: 10.1111/1462-2920.12498
Miyazaki, R., Minoia, M., Pradervand, N., Sulser, S., Reinhard, F., and Van Der Meer, J. R. (2012). Cellular variability of RpoS expression underlies subpopulation activation of an integrative and conjugative element. PLoS Genet. 8:e1002818. doi: 10.1371/journal.pgen.1002818
Mohan, K., and Phale, P. S. (2017). Carbon source-dependent inducible metabolism of veratryl alcohol and ferulic acid in Pseudomonas putida CSV86. Appl. Environ. Microbiol. 83:e03326-16. doi: 10.1128/AEM.03326-16
Mohapatra, B., Nain, S., Sharma, R., and Phale, P. S. (2022). Functional genome mining and taxono-genomics reveal eco-physiological traits and species distinctiveness of aromatic-degrading Pseudomonas bharatica sp. nov. Environ. Microbiol. Rep. 14, 464–474. doi: 10.1111/1758-2229.13066
Mohapatra, B., and Phale, P. S. (2021). Microbial degradation of naphthalene and substituted naphthalenes: metabolic diversity and genomic insight for bioremediation. Front. Bioeng. Biotechnol. 9:602445. doi: 10.3389/fbioe.2021.602445
Neilson, J. W., Pierce, S. A., and Maier, R. M. (1999). Factors influencing expression of luxCDABE and nah genes in Pseudomonas putida RB1353 (NAH7, pUTK9) in dynamic systems. Appl. Environ. Microbiol. 65, 3473–3482. doi: 10.1128/AEM.65.8.3473-3482.1999
Nojiri, H., Sota, M., and Shintani, M. (2009). “Catabolic plasmids involved in the degradation of polycyclic aromatic hydrocarbons and heteroaromatic compounds,” in Microbial Megaplasmids, ed. E. Schwartz (Berlin: Springer), 55–87.
Obi, C. C., Vayla, S., De Gannes, V., Berres, M. E., Walker, J., Pavelec, D., et al. (2018). The integrative conjugative element clc (ICEclc) of Pseudomonas aeruginosa JB2. Front. Microbiol. 9:1532. doi: 10.3389/fmicb.2018.01532
Paliwal, V., Raju, S. C., Modak, A., Phale, P. S., and Purohit, H. J. (2014). Pseudomonas putida CSV86: a candidate genome for genetic bioaugmentation. PLoS One 9:e84000. doi: 10.1371/journal.pone.0084000
Phale, P. S., Malhotra, H., and Shah, B. A. (2020). Degradation strategies and associated regulatory mechanisms/features for aromatic compound metabolism in bacteria. Adv. Appl. Microbiol. 112, 1–65. doi: 10.1016/bs.aambs.2020.02.002
Phale, P. S., Mohapatra, B., Malhotra, H., and Shah, B. A. (2021). Eco-physiological portrait of a novel Pseudomonas sp. CSV86: an ideal host/candidate for metabolic engineering and bioremediation. Environ. Microbiol. 1:15694. doi: 10.1111/1462-2920.15694
Phale, P. S., Shah, B. A., and Malhotra, H. (2019). Variability in assembly of degradation operons for naphthalene and its derivative, carbaryl, suggests mobilization through horizontal gene transfer. Genes 10:569. doi: 10.3390/genes10080569
Poulin-Laprade, D., Carraro, N., and Burrus, V. (2015). The extended regulatory networks of SXT/R391 integrative and conjugative elements and IncA/C conjugative plasmids. Front. Microbiol. 6:837. doi: 10.3389/fmicb.2015.00837
Qian, W., Jia, Y., Ren, S. X., He, Y. Q., Feng, J. X., Lu, L. F., et al. (2005). Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15, 757–767. doi: 10.1101/gr.3378705
Rajanna, C., Wang, J., Zhang, D., Xu, Z., Ali, A., Hou, Y. M., et al. (2003). The Vibrio pathogenicity island of epidemic Vibrio cholerae forms precise extrachromosomal circular excision products. J. Bacteriol. 185, 6893–6901. doi: 10.1128/JB.185.23.6893-6901.2003
Ravatn, R., Zehnder, A. J., and van der Meer, J. R. (1998b). Low-frequency horizontal transfer of an element containing the chlorocatechol degradation genes from Pseudomonas sp. strain B13 to Pseudomonas putida F1 and to indigenous bacteria in laboratory-scale activated-sludge microcosms. Appl. Environ. Microbiol. 64, 2126–2132. doi: 10.1128/AEM.64.6.2126-2132.1998
Ravatn, R., Studer, S., Springael, D., Zehnder, A. J., and van der Meer, J. R. (1998a). Chromosomal integration, tandem amplification, and deamplification in Pseudomonas putida F1 of a 105-kilobase genetic element containing the chlorocatechol degradative genes from Pseudomonas sp. strain B13. J. Bacteriol. 180, 4360–4369. doi: 10.1128/JB.180.17.4360-4369.1998
Reva, O. N., and Tümmler, B. (2005). Differentiation of regions with atypical oligonucleotide composition in bacterial genomes. BMC Bioinformatics 6:251. doi: 10.1186/1471-2105-6-251
Rodríguez-Beltrán, J., Sørum, V., Toll-Riera, M., de la Vega, C., Peña-Miller, R., and San Millán, Á (2020). Genetic dominance governs the evolution and spread of mobile genetic elements in bacteria. Proc. Natl. Acad. Sci. U.S.A. 117, 15755–15762. doi: 10.1073/pnas.2001240117
Ryan, M. P., Pembroke, J. T., and Adley, C. C. (2009). Novel Tn4371-ICE like element in Ralstonia pickettii and genome mining for comparative elements. BMC Microbiol. 9:242. doi: 10.1186/1471-2180-9-242
Sentchilo, V., Czechowska, K., Pradervand, N., Minoia, M., Miyazaki, R., and van der Meer, J. R. (2009). Intracellular excision and reintegration dynamics of the ICEclc genomic island of Pseudomonas knackmussii sp. strain B13. Mol. Microbiol. 72, 1293–1306. doi: 10.1111/j.1365-2958.2009.06726.x
Singh, R., Trivedi, V. D., and Phale, P. S. (2013). Metabolic regulation and chromosomal localization of carbaryl degradation pathway in Pseudomonas sp. strains C4, C5 and C6. Arch. Microbiol. 195, 521–535. doi: 10.1007/s00203-013-0903-9
Strnad, H., Ridl, J., Paces, J., Kolar, M., Vlcek, C., and Paces, V. (2011). Complete genome sequence of the haloaromatic acid-degrading bacterium Achromobacter xylosoxidans A8. J. Bacteriol. 193, 791–792. doi: 10.1128/JB.01299-10
van der Meer, J. R., and Sentchilo, V. (2003). Genomic islands and the evolution of catabolic pathways in bacteria. Curr. Opin. Biotechnol. 14, 248–254.
Verma, H., Kumar, R., Oldach, P., Sangwan, N., Khurana, J. P., Gilbert, J. A., et al. (2014). Comparative genomic analysis of nine Sphingobium strains: insights into their evolution and hexachlorocyclohexane (HCH) degradation pathways. BMC Genomics 15:1014. doi: 10.1186/1471-2164-15-1014
Verma, N., Kantiwal, U., Yadav, Y. K., Teli, S., Goyal, D., and Pandey, J. (2019). “Catalytic promiscuity of aromatic ring-hydroxylating dioxygenases and their role in the plasticity of xenobiotic compound degradation,” in Microbial Metabolism of Xenobiotic Compounds, ed. P. Arora (Singapore: Springer), 123–143.
Wiegand, I., Hilpert, K., and Hancock, R. E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175.
Wozniak, R. A., Fouts, D. E., Spagnoletti, M., Colombo, M. M., Ceccarelli, D., Garriss, G., et al. (2009). Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5:e1000786. doi: 10.1371/journal.pgen.1000786
Wozniak, R. A., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563. doi: 10.1038/nrmicro2382
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Keywords: Pseudomonas, integrative conjugative element (ICE), comparative and functional genomics, phylogenomics, naphthalene metabolism, Co-Zn-Cd tolerance, niche adaptation, evolution-speciation
Citation: Mohapatra B, Malhotra H and Phale PS (2022) Life Within a Contaminated Niche: Comparative Genomic Analyses of an Integrative Conjugative Element ICEnahCSV86 and Two Genomic Islands From Pseudomonas bharatica CSV86T Suggest Probable Role in Colonization and Adaptation. Front. Microbiol. 13:928848. doi: 10.3389/fmicb.2022.928848
Received: 26 April 2022; Accepted: 08 June 2022;
Published: 06 July 2022.
Edited by:
Roshan Kumar, Magadh University, IndiaReviewed by:
François Delavat, Université de Nantes, FranceCopyright © 2022 Mohapatra, Malhotra and Phale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prashant S. Phale, cHBoYWxlQGlpdGIuYWMuaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.