 Hailan Zhao1,2†
Hailan Zhao1,2† Jiaqi Wang1,2†
Jiaqi Wang1,2† Yao Peng3
Yao Peng3 Xunchao Cai3
Xunchao Cai3 Yandi Liu1,2
Yandi Liu1,2 Wenqi Huang1,2
Wenqi Huang1,2 Hongli Huang1,2*
Hongli Huang1,2* Yuqiang Nie1,2*
Yuqiang Nie1,2*- 1Department of Gastroenterology and Hepatology, The Second Affiliated Hospital, School of Medicine, South China University of Technology, Guangzhou, Guangdong, China
- 2Department of Gastroenterology and Hepatology, Guangzhou First People's Hospital, School of Medicine, South China University of Technology, Guangzhou, Guangdong, China
- 3Department of Gastroenterology and Hepatology, Shenzhen University General Hospital, Shenzhen, Guangdong, China
Background: Paraclostridium bifermentans is the most diverse distributed species of Paraclostridium and can cause fatal human infections under rare conditions. However, its pathogenic mechanisms and adaptation ability behind infections remain unclear. Herein, we reported the complete genome sequence of P. bifermentans HD0315_2 isolated from the feces of a patient with Crohn's disease. Then, we performed genomic analyses to understand its pathogenic mechanisms and adaptation ability.
Results: The de novo assembly revealed that the HD0315_2 strain carried a circular chromosome of 3.27 Mb and six circular plasmids (19.41 to 139.50 kb). The phylogenomic analysis assigned the HD0315_2 strain as P. bifermentans and reclassified some previously non-P. bifermentans strains into this clade. The general genomic features showed that this species harbored a flexible genomic pool characterized by variable genome length and multiple plasmids. Then, the HD0315_2 strain was predicted as a human pathogen with high probability, and Listeria LIPI-1 virulence proteins were identified on its genome. Besides, abundant antibiotics/metal/stress resistant genes, such as asrABCH, cat, mccF, macB, entS, albA, bcrA, and tetB, were carried by either the genome or the plasmids. Furthermore, we proposed that transposase-directed horizontal gene transfer was responsible for the distribution of multiple copies of the hin gene in the plasmids.
Conclusion: The flexible genomic pool of P. bifermentans encodes abundant functions for antimicrobial or oxidative stress resistance, helping it successfully inhabit and adapt to diverse environments. Moreover, P. bifermentans HD0315_2 might infect hosts via a Listeria LIPI-1-like cycle, with the help of a plasmid expressing the Hin DNA invertase to evade host immune responses.
Introduction
Paraclostridium spp. belongs to the class Clostridia of Gram-positive, rod-shaped, and obligate anaerobe bacteria that reside in various mesophilic conditions, such as soil, marine habitats, polluted waters, and clinical specimens (Jyothsna et al., 2016). To date, only two species have been formally published under this genus (i.e., P. bifermentans and P. benzoelyticum) (Jyothsna et al., 2016). Among them, the most frequently isolated and best-characterized species is P. bifermentans (Hale et al., 2016). Additionally, in 2016, P. bifermentans was reclassified from Clostridium bifermentans (Jyothsna et al., 2016). This species has been previously recognized as nonpathogenic unless co-inhabited with Clostridium perfringens, producing putrid lesions in guinea pigs (Weinberg and Séguin, 1918). However, 15 cases of P. bifermentans infections in humans have been recently summarized and included brain abscess, lymphadenitis, necrotizing endometritis, a prosthetic knee joint infection, empyema, and endocarditis (Kolander et al., 1989; Edagiz et al., 2015; Hale et al., 2016; Biswas et al., 2018; Barrett et al., 2020). Kutsuna et al. have shown that P. bifermentans PAGU1678T exacerbates mouse's pathological conditions in a dextran sulfate sodium-induced colitis model (Kutsuna et al., 2018). Although P. bifermentans has emerged as a rare pathogen in humans, it thrives as a human pathogen under specific conditions and rises to major health hazards. Nevertheless, no report has determined any suggestive genes causing its pathogenicity, although various draft genomes have been published. On the other hand, the high similarity of 16S rRNA sequences within the genus (e.g., P. bifermentans and P. benzoelyticum share a 16S rRNA sequence similarity > 99%) is difficult for their taxonomic identification and characterization of biological traits. Herein, we isolated P. bifermentans strains from the feces of patients with Crohn's disease (CD) for the first time. Additionally, we showed that P. bifermentans strains were only isolated from patients with CD but not from patients with ulcerative colitis (UC), underlying the potential involvement of this species in CD. Then, the complete genome of P. bifermentans HD0315_2, isolated from the feces of a young male patient with CD, was assembled de novo using reads from Illumina and Nanopore sequencing. Furthermore, whole-genome sequence-based taxonomy identification and comparative genome analyses were performed to clarify the general genome function and its specificities regarding virulence, adaptation, and pathogenic effects.
Materials and methods
Isolation and characterization of strains
Anaerobic conditions (90% N2, 5% CO2, and 5% H2) were used during the isolation and culture of P. bifermentans strains. First, fresh fecal samples were collected from a 24-year-old Chinese man with active CD from Guangdong (China) who suffered from recurrent abdominal pain, changes in stool traits, and perianal exudate. Briefly, 10 ml of sterile PBS was added per gram of fresh feces to a 50-ml conical tube, then vortexed and left to settle. Next, the feces suspension was transferred to anaerobic blood culture bottles (BD, BACTECTM Lytic/10 Anaerobic/F Culture Vials, America) supplemented with sterile sheep blood and rumen fluid. Bottles were incubated under anaerobic conditions at 37°C for 30 d as described by Lagier's culturomics strategy (Lagier et al., 2018). Then, 1 ml of suspension was sterilely aspirated from the incubated culture and serially diluted (10 to 1012). Finally, 100 μl of each dilution was evenly plated on Brain-Heart Infusion (BHI) or reinforced clostridia agar plates to harvest colonies. Purification was further conducted by streaking. The harvested colonies were enriched in BHI medium at 37°C for 3 d and identified by MALDI Biotyper RTC (Bruker Daltonics, Germany). Single and sufficiently grown colonies were directly transferred to the MALDI Biotyper RTC 96 target spot, and 1 μl of Bruker bacterial test standard (BTS) and matrix solution were sequentially added to prepare the detection target. A spectrum score of 2.3 was used as the threshold for high-confident taxa identification at the species level. Then, the 16S rRNA sequence of one strain (HD0315_2), identified as Paraclostridium sp., was obtained by PCR using the 8f/1492r primer pair and sent to Beijing Genomics institution (BGI) for Sanger sequencing. For taxa identification, the 16S rRNA sequence was aligned using the NCBI nucleotide (nt) collection database. Finally, species were determined with 100% sequence coverage and > 97% sequence identity.
Genome sequencing and assembly
Genomic DNA extraction was performed using the TaKaRa MiniBEST Bacteria Genomic DNA Extraction Kit (Takara, Japan) following the manufacturer's instructions. The DNA quality was checked using the Synergy HTX Multi-Mode Reader (BioTek, USA). Genome sequencing was separately performed under two platforms to generate long and short reads by the Nanopore PromethION platform (MAGIGENE, Guangzhou, China) and the Illumina NovaSeq platform (Novogene, Nanjing, China), respectively. The long-read sequencing library was constructed using the SQK-LSK109 kit (Oxford Nanopore Technologies, UK) according to the manufacturer's instructions. Sequencing and base calling were performed using MinKNOW v1.15.4 with the FLO-MINSP6 flow cell (Oxford Nanopore Technologies, UK) and low-quality reads (i.e., scores <7) were removed. The short-read sequencing library was constructed with a 350 bp insert size and sequenced using the PE150 strategy. Adapter trimming and low-quality reads (Phred score ≤ 20) were removed for quality control. The de novo genome assembly was conducted using the Unicycler v0.4.9b assembler (Wick et al., 2017), with the default hybrid assembly pipeline.
Phylogenomic characterization and plasmid detection
Taxonomy assignment was further curated using the gtdbtk_wf workflow implemented in GTDT-Tk (Parks et al., 2018). The average nucleotide identity (ANI) between the HD0315_2 strain and the phylogenomic close genomes were calculated using fastANI (Jain et al., 2018). A phylogenomic tree based on the whole genome protein sequences was constructed using CVTree3 with default parameters (K-tuple length 3, 4, 5, 6, 7) (Zuo and Hao, 2015). Plasmids were predicted using PlasForest v1.2 (https://github.com/leaemiliepradier/PlasForest) and mlplasmids v2.1.0 (https://gitlab.com/sirarredondo/analysis_mlplasmids) based on machine learning from sequence homology and pentamer frequencies.
Comprehensive genome annotation
Comprehensive genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016). Functional genome annotation with multiple databases, including Carbohydrate-Active enZYmes (CAZy), Cluster of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG), was performed using eggNOGMapper v2.1.5 (Huerta-Cepas et al., 2017). Comprehensive genome analysis, including the annotation of subsystems, identification of specialty genes (transporters, virulence factors, drug targets, antibiotic resistance genes, and antimicrobial resistance genes), and phylogenetic analysis, was conducted using PATRIC v3.6.10 (https://www.patricbrc.org/). Pathogenicity prediction was performed using PathogenFinder v1.1 (https://cge.cbs.dtu.dk/services/PathogenFinder/). Genomic islands (GI) were identified using IslandViewer 4 (Bertelli et al., 2017).
Quality assurance
The HD0315_2 strain was repeatedly sub-cultured on broth agar plates to confirm the obtainment of a single colony before sequencing. Taxa assignments were cross-validated by both MALDI Biotyper RTC and full-length 16S rRNA sequencing, which determined that the HD0315_2 strain belongs to the Paraclostridium genus (Supplementary Tables S1, S2). Quality control was conducted by using CheckM v1.0.12 to evaluate the assembly quality and using samtools v1.9 and bcftools v1.10.2 to check the variants through mapping the Illumina reads to the contigs. Then, the assembled genome sequence was loaded onto GTDB-Tk for taxonomy assignments.
Results and discussion
Genomic features and phylogenomic tree
The hybrid assembly using Nanopore and Illumina reads generated seven complete circular contigs without Ns. The results showed that the (1) high-quality contigs were assembled using the Unicycler assembler (contamination = 0.70%, completeness = 99.30%, Strain heterogeneity = 0.00%), (2) the pair-end Illumina reads showed an overall 99.61% alignment rate to the assembled contigs, and (3) only three-point sites were determined as SNP sites, all located in the chromosome (i.e., contig HD0315_2) (Supplementary Table S3). The plasmid identification analysis showed that the biggest contig was the chromosome sequence, while the remaining six contigs were plasmids. The taxa identification workflow implemented in GTDB-Tk identified the HD0315_2 strain as P. bifermentans based on the sequence ANI (> 96) from the biggest contig (chromosome). The six smaller contigs did not return the bacteria marker genes when loaded into the workflow, which validated them as plasmids. The whole-genome sequencing of P. bifermentans HD0315_2 comprehends a circular chromosome with 3,265,124 bp, 3, 366 CDSs, 51 rRNA, 103 tRNAs, and a G + C content of 28.8%; and six plasmids with lengths, G + C contents, and CDSs of the six plasmids are between 19,414–139,499 bp, 24.6–27.6%, and 23–162, respectively (Table 1). Currently, only two complete genomes of the Paraclostridium genus have been released in the NCBI genome database, and both belong to P. bifermentans species. Similar to the HD0315_2 strain, these two genomes carry multiple plasmids and close genome G + C content (<0.5% variations). Moreover, the chromosome lengths in this species varied from 3.27 to 3.64 Mb, indicating a flexible genomic pool not only from plasmid sequences but also from genome accessory sequences. Although the chromosome length of the HD0315_2 strain is the shortest and has a lower G/G+P proportion, it carries more rRNAs/tRNAs and is associated with more plasmids and might represent a more efficient protein synthesis capability of HD0315_2, in contrast to the Cbm strain that is characterized by an opposite pattern. Detailed comparisons of the genomic features between these strains are listed in Table 1.
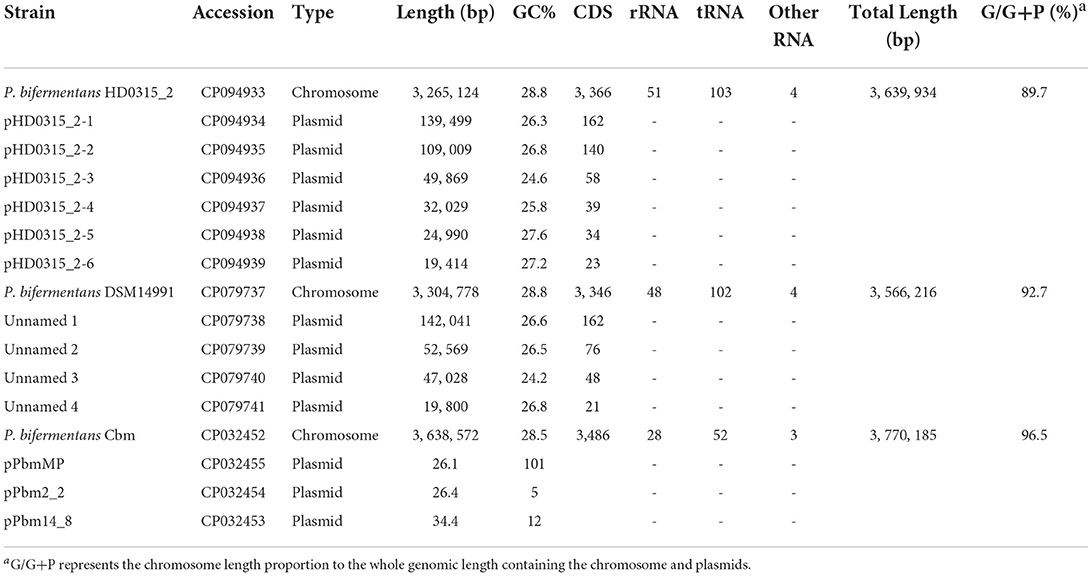
Table 1. Genomic features of the strains in the genus Paraclostridium with completed genome sequences.
The genome sequence alignment among HD0315_2, DSM14991, and Cbm strains indicated that large sequence rearrangement/transversion events happened in Cbm, close to the homologous replicon origin region of HD0315_2, which might result in the variations of rRNAs and tRNAs gain or loss (Figure 1A). Five GIs were predicted in the genome sequence, two were exclusive for HD0315_2 (located at 2,096,711–2,102,952 bp and 2,112,391–2,122,603 bp). However, none of them were assigned as pathogenic or antibiotic-resistant GIs by the IslandViewer tool. Then, we used PATRIC to collect and analyze the top 50 genomes close to HD0315_2 (indicated by the genome distance) (Supplementary Table S4). A phylogenomic tree was constructed using these genomes and showed that these strains can be divided into four major clades. The biggest clade (clade 1) consists of 28 strains that could be assigned as P. bifermentans. We found that some strains in clade 1 were previously assigned as non-Paraclostridium, such as Clostridium sp. NCR, Candidatus Dorea massiliensis AP6, Clostridium cuniculi BSD2780061688_150302_F12, Clostridium thiosulfatireducens src6, and Acinetobacter sp. RIT592, and reclassified as P. bifermentans in the present study. Moreover, SKVG24, previously proposed as the P. dentum type strain (Choksket et al., 2020), is proven to be an invalidly assigned species (https://lpsn.dsmz.de/search?word=Paraclostridium), was clustered into clade 1 and should be reclassified as P. bifermentans (Figure 1). Additionally, clade 2 was composed of P. benzoelyticum, which was the most phylogenomically close species to P. bifermentans. Clades 3 and 4 consisted of species distant from P. bifermentans, such as Eubacterium tenue and Clostridium sordellii (Figure 1B). A novel P. bifermentans subspecies, named Paraclostridium bifermentans subsp. muricolitidis subsp. nov., was recently proposed by Kutsuna et al. (2019). We found that the HD0315_2 strain was close to Paraclostridium bifermentans subsp. bifermentans (Figure 1B). The phylogenomic relationship between these strains was further validated by the ANI analysis (Figure 1C).
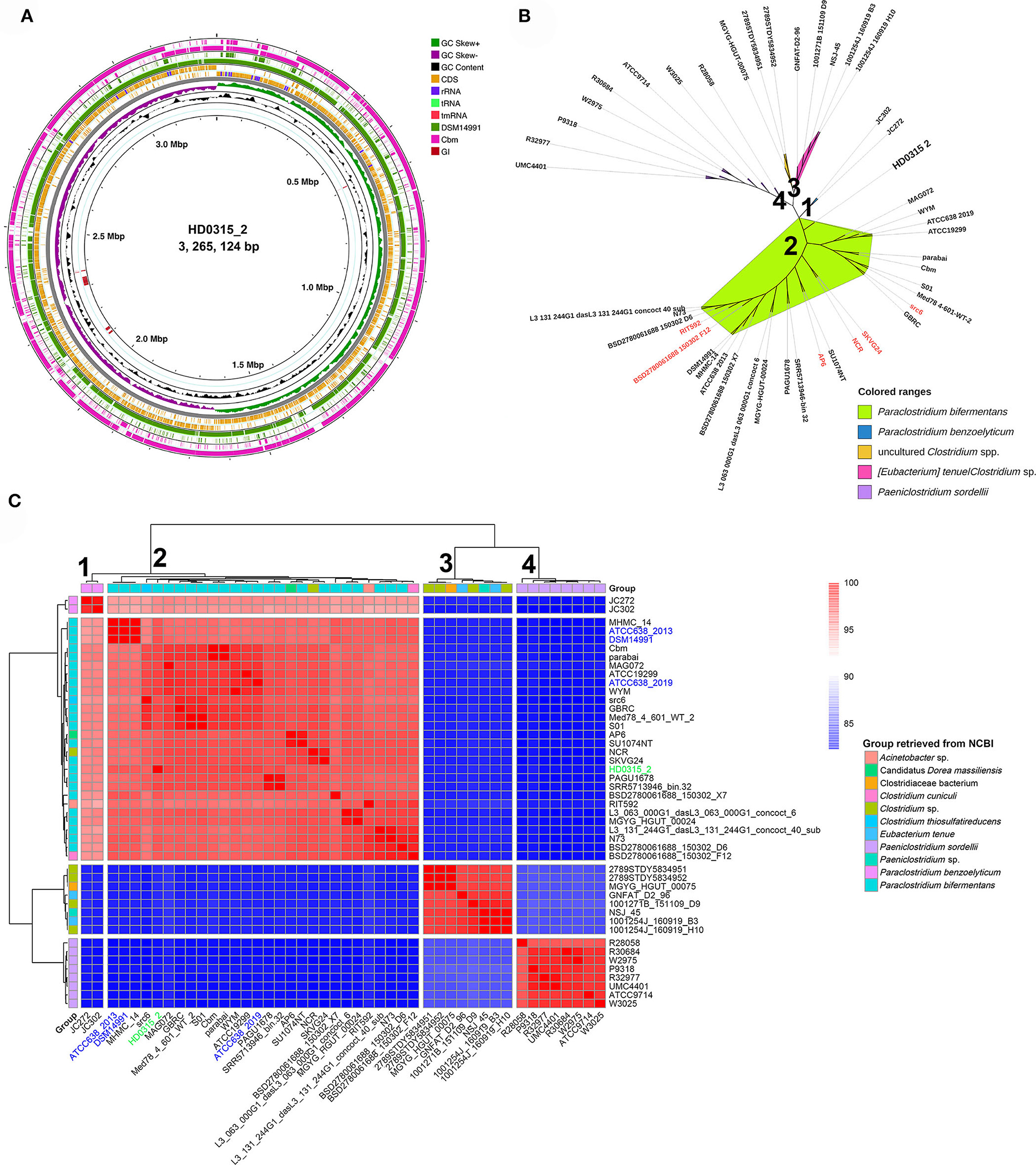
Figure 1. The circular display of the complete genome sequence feature and phylogenomic tree originated from P. bifermentans HD0315_2. (A) From outer to inner rings: genome sequence of P. bifermentans Cbm, the genome sequence of P. bifermentans DSM14991, CDSs on the forward/reverse strand, GC skew, GC content, and genome islands. RNAs and repeat sequence regions are displayed in the CDSs circles. GI represents genome islands. (B) Phylogenomic tree constructed from WGS of the phylogenetic close strains. Numbers marked on the tree branch represent four Clades divided by phylogenomic analyses. Leaf labels with red color are those reclassified as P. bifermentans members in this study. Previously documented taxonomic information of them are listed in Supplementary Table S1. (C) Heatmap displaying the ANI between the phylogenetically close Paraclostridium spp. The color bar on the right indicates the ANI value calculated using fastANI. Numbers marked alongside the dendrogram branch represent four Clades divided by clustering. Strains names marked with blue color are type strains of P. bifermentans those with different genome versions or from different institution collections (e.g., ATCC or DSM). Strain HD0315_2 is marked with green color.
General gene functions
A total of 1,188 subsystem features were revealed by RAST in the genome of P. bifermentans HD0315_2 (Supplementary Figure S2). Most features were assigned to “Protein Metabolism (237)” and “Amino Acids and Derivatives (179).” The wide distribution or habitats of Paraclostridium spp. have suggested that this genus has an excellent ability to survive in various environments (Rai et al., 2015), which was endowed by the genomic function. We found that 75 genes were tightly correlated to the adaptation of HD0315_2, including 42 assigned to “Virulence, Disease and Defense,” 7 assigned to “Phages, Prophages, Transposable elements, Plasmids,” and 26 assigned to “Stress Response” (Supplementary Figure S2). The main resistance coding genes were those sub-assigned to tetracycline (4), fluoroquinolones (2), cobalt-zinc-cadmium (11), copper (2), and multidrug-compounds (12). The virulence factor coding genes were mainly sub-assigned to Mycobacterium-involved virulence operon (10) and Listeria LIPI-1-extended proteins (3). Stress-response coding genes were mainly sub-assigned to osmotic stress (4) and oxidative stress (15) (Supplementary Table S5). However, the plasmid annotation using RAST presented poor results, and only 15 genes in the plasmids were assigned to the subsystem (data not shown). Among them, two genes were in pHD0315_2-1 code bile hydrolysis (1) and cobalt-zinc-cadmium resistance genes (1). Paraclostridium strains have been observed expressing multiple antibiotic-resistance genes (e.g., chloramphenicol, tetracycline, and gentamicin) (Liang et al., 2020), metal/metalloid (e.g., Se, Hg, As, and Zn) (Wang et al., 2021; Zhang et al., 2022) or toxic compounds (e.g., ciprofloxacin) (Fang et al., 2021) and were also deduced from the genomic coding functions in this study. Importantly, coding genes identified as Listeria LIPI-1-extended proteins in the HD0315_2 genome have been well-characterized for the infection cycle processes of “Internalization,” “Escape from the vacuole,” and “Reinfection” (see Supplementary Figure S3 for details). Overall, the functions encoded by the HD0315_2 genome contribute to survival in diverse environments. Meanwhile, for the multiple plasmids, the identification of functions was difficult due to the poor annotations.
Pathogenicity and survival features
Since various pathogenicity or virulence factor coding genes were annotated in the genome sequence, we further checked the possibility of the HD0315_2 strain being a human pathogen using PathogenFinder. The results have suggested that the HD0315_2 strain is a human pathogen with a high probability (0.77 and 0.98), besides 11 and one pathogenic protein family detected in the chromosome of HD0315_2 and the pHD0315_2-3 plasmid sequence, respectively (Supplementary Table S6). Interestingly, all pathogenic proteins detected in the chromosome sequence were homologs [e.g., putative malate-2H(+)/Na(+)-lactate antiporter, PTS-system-phospho carrier protein, and ribonuclease Ph] of the Clostridium difficile, and the pathogenic protein detected in the pHD0315_2-3 sequence was an amino acid/peptide transporter homolog of Clostridium botulinum (see Supplementary Table S4 for details). However, well-characterized toxins (e.g., enterotoxins and cytotoxins) that can cause diarrhea and inflammation by C. difficile (Butler et al., 2016) were not detected in the HD0315_2 strain. The detected pathogenic proteins were related to pathogen virulence and surviving regulation. For example, ribonuclease Ph is present in many pathogen ns and has been proven to help them withstand oxidative stress or oxidative bursts of neutrophils and/or macrophages from hosts (Lawal et al., 2011). Moreover, the Hin DNA invertase coding gene (i.e., hin) was carried by five out of the six plasmids (Figure 2). The Hin DNA invertase has been found to regulate flagellar phase variations in pathogens (Merickel and Johnson, 2001), which would help the HD0315_2 strain to evade host immune responses. Other features, not in the RAST subsystem but annotated by PGAP, included antimicrobial/metal resistance genes (e.g., asrABCH, cat, mccF, macB, entS, albA, bcrA, and tetB), transposase coding genes [e.g., tnXo19 (Tn3 family), and iSDku1 (IS256 family)] (Figures 2A–D,F). Moreover, four hin genes were linked with tnXo19, indicating a high transfer of the hin gene by transposase-directed horizontal gene transfer (HGT) events, which would also help the HD0315_2 strain to evade host immune responses. Meanwhile, mccF, macB, entS, alba, and bcrA (coding microcin C7 self-immunity protein, macrolide export ATP-binding/permease protein, enterobactin exporter, antilisterial bacteriocin subtilosin biosynthesis protein, and bacitracin transport ATP-binding protein, respectively) might help the HD0315_2 strain to resist microcin produced from Paraclostridium members or microbial toxins from other bacteria. Consequently, these plasmid features would make this strain not only resistant to host responses but also to harsh environments. Hence, P. bifermentans HD0315_2 might live under antimicrobial or oxidative stress and might infect the host cell via a Listeria LIPI-1-like cycle. Thus, the chromosome or plasmid-encoded functions would help P. bifermentans HD0315_2 survive during the whole infection cycle by evading host immune responses or resisting medical compounds.
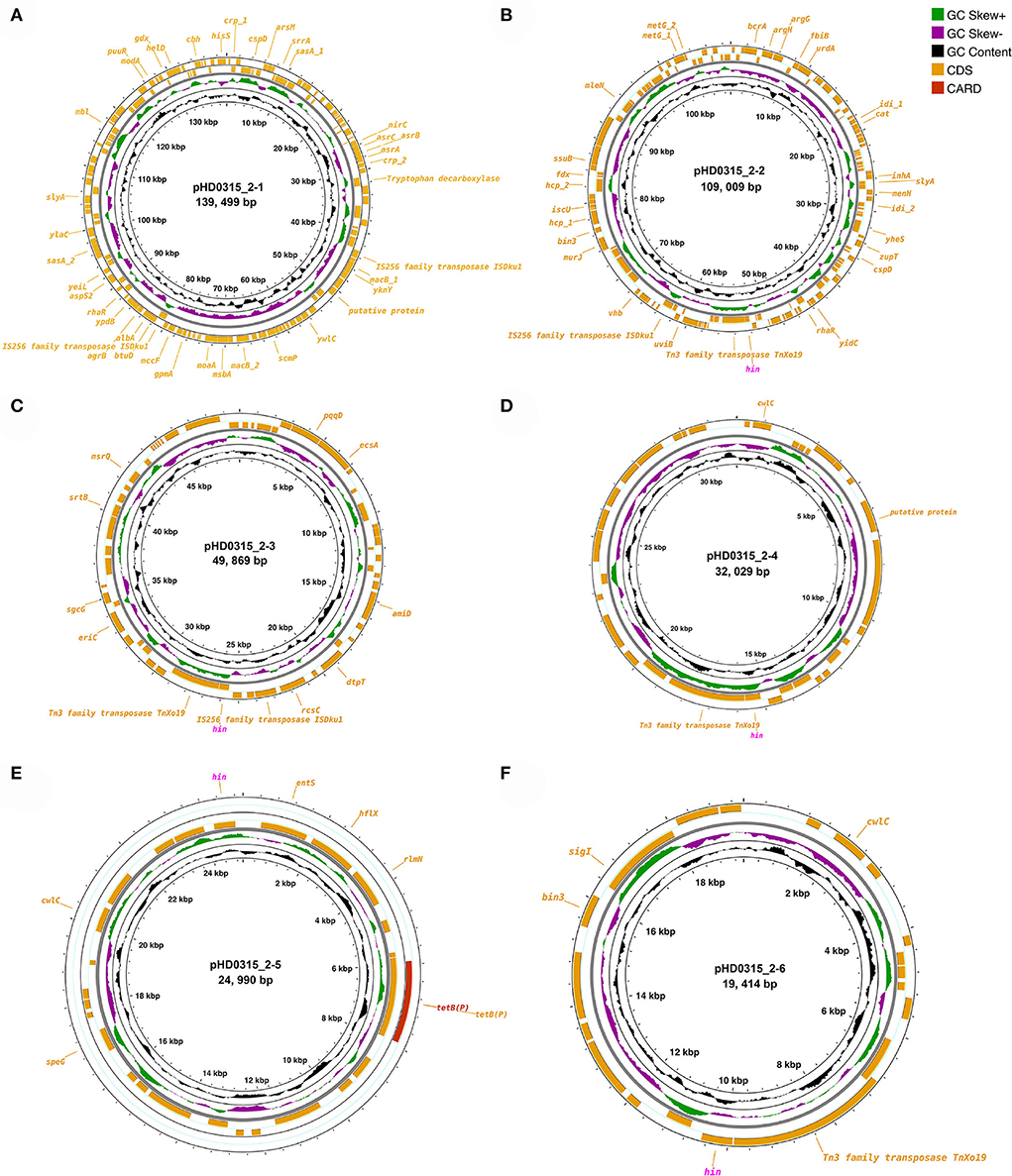
Figure 2. Circular display of the plasmids identified in P. bifermentans HD0315_2. (A–F) From outer to inner rings: antibiotic-resistant gene based on CARD annotation, CDSs on the forward/reverse strand, GC skew, and GC content. CDSs labeled with magenta color are genes (hin) coding Hin DNA invertase. CDSs without a label are genes coding hypothetical proteins.
Conclusion
In summary, we reported the complete genome sequence of P. bifermentans HD0315_2 and analyzed its general genomic features, phylogenomic relationship, and pathogenic potential. The results revealed that multiple plasmids are present in P. bifermentans strains. Additionally, some species previously identified as non-P. bifermentans were reclassified as P. bifermentans by the phylogenomic analysis. The pathogenicity proteins encoded by the HD0315_2 strain genome suggested that it can infect host cells via a Listeria LIPI-1-like cycle. Moreover, both the chromosome and plasmids were found coding abundant antimicrobial or oxidative stress resistance functions. Additionally, transposase-directed HGT generated the distribution of multiple copies of the hin gene in the plasmids, which might help the strain evade host immune responses. Therefore, in the present study, we expanded the knowledge regarding the general genomic features of P. bifermentans, revealed some of its environmental/host adaptation mechanisms, and proposed, for the first time, its pathogenicity model at the genomic level.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
This study was approved by our Institutional Review Board (July 24, 2019; reference No. K-2019-146-01) and was compliant with all relevant ethical regulations. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HZ performed the bacteria isolation and taxa identification. YP and XC performed the WGS-based analysis. HZ, JW, and XC drafted the manuscript. YP, YL, and WH reviewed and revised the manuscript. HH and YN designed the whole study. All authors made substantial and direct contributions to the work, and read and approved the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81871905 and 41907214); Natural Science Foundation of Guangdong Province (2020A1515011000); and Guangzhou Planned Project of Science and Technology (202002030288 and 202002020012).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.928153/full#supplementary-material
References
Barrett, L. F., Saragadam, S. D., DiMaria, C. N., and Delgado-Daza, A. (2020). Infection of a prosthetic knee joint with Clostridium bifermentans. Oxf. Med. Case Reports. 2020, omaa057. doi: 10.1093/omcr/omaa057
Bertelli, C., Laird, M. R., Williams, K. P., Group, S. F. U. R. C., Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Biswas, R., Raja, S., Sistla, S., Gopalakrishnan, M., and Saxena, S. K. (2018). Brain abscess and cervical lymphadenitis due to Paraclostridium bifermentans: a report of two cases. Anaerobe. 51, 8–11. doi: 10.1016/j.anaerobe.2018.03.006
Butler, M., Olson, A., Drekonja, D., Shaukat, A., Schwehr, N., Shippee, N., et al. (2016). Early diagnosis, prevention, and treatment of Clostridium difficile: Update [Internet]. Rockville, MD: Agency for Healthcare Research and Quality.
Choksket, S., Jain, A., Sharma, D., Grover, V., and Korpole, S. (2020). Paraclostridium dentum, a novel species with pathogenic features isolated from human dental plaque sample. Anaerobe. 65, 102239. doi: 10.1016/j.anaerobe.2020.102239
Edagiz, S., Lagace-Wiens, P., Embil, J., Karlowsky, J., and Walkty, A. (2015). Empyema caused by Clostridium bifermentans: a case report. Can. J. Infect. Dis. Med. Microbiol. 26, 105–107. doi: 10.1155/2015/481076
Fang, H., Oberoi, A. S., He, Z., Khanal, S. K., and Lu, H. (2021). Ciprofloxacin-degrading Paraclostridium sp. isolated from sulfate-reducing bacteria-enriched sludge: optimization and mechanism. Water Res. 191, 116808. doi: 10.1016/j.watres.2021.116808
Hale, A., Kirby, J. E., and Albrecht, M. (2016). Fatal spontaneous Clostridium bifermentans necrotizing endometritis: a case report and literature review of the pathogen. Open Forum Infect Dis. 3, ofw095. doi: 10.1093/ofid/ofw095
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-07641-9
Jyothsna, T. S., Tushar, L., Sasikala, C., and Ramana, C. V. (2016). Paraclostridium benzoelyticum gen. nov., sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii. Int. J. Syst. Evol. Microbiol. 66, 1268–1274. doi: 10.1099/ijsem.0.000874
Kolander, S. A., Cosgrove, E. M., and Molavi, A. (1989). Clostridial endocarditis: report of a case caused by Clostridium bifermentans and review of the literature. Arch. Intern. Med. 149, 455–456. doi: 10.1001/archinte.1989.00390020143032
Kutsuna, R., Miyoshi-Akiyama, T., Mori, K., Hayashi, M., Tomida, J., Morita, Y., et al. (2019). Description of Paraclostridium bifermentans subsp. muricolitidis subsp. nov., emended description of Paraclostridium bifermentans (Sasi Jyothsna et al. 2016), and creation of Paraclostridium bifermentans subsp. bifermentans subsp. nov. Microbiol. Immunol. 63, 1–10. doi: 10.1111/1348-0421.12663
Kutsuna, R., Tomida, J., Morita, Y., and Kawamura, Y. (2018). Paraclostridium bifermentans exacerbates pathosis in a mouse model of ulcerative colitis. Plos ONE. 13, e0197668. doi: 10.1371/journal.pone.0197668
Lagier, J-. C., Dubourg, G., Million, M., Cadoret, F., Bilen, M., et al. (2018). Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 16, 540–550. doi: 10.1038/s41579-018-0041-0
Lawal, A., Jejelowo, O., Chopra, A. K., and Rosenzweig, J. A. (2011). Ribonucleases and bacterial virulence. Microb. Biotechnol. 4, 558–571. doi: 10.1111/j.1751-7915.2010.00212.x
Liang, Z., Zhang, Y., He, T., Yu, Y., Liao, W., Li, G., et al. (2020). The formation mechanism of antibiotic-resistance genes associated with bacterial communities during biological decomposition of household garbage. J. Hazard. Mater. 398, 122973. doi: 10.1016/j.jhazmat.2020.122973
Merickel, S. K., and Johnson, R. C. (2001). “Hin/Gin-Mediated Site-Specific DNA Inversion,” in Encyclopedia of Genetics, eds S. Brenner, J. H. Miller (Academic Press: New York), 938–942. doi: 10.1006/rwgn.2001.0566
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Rai, A., Ramana, C. V., Uppada, J., and Sasikala, C. (2015). Paraclostridium. Bergey's Manual of Systematics of Archaea and Bacteria. 2015, 1–12. doi: 10.1002/9781118960608.gbm01635
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Wang, Y., Wei, D., Li, P., Jiang, Z., Liu, H., Qing, C., et al. (2021). Diversity and arsenic-metabolizing gene clusters of indigenous arsenate-reducing bacteria in high arsenic groundwater of the Hetao Plain, Inner Mongolia. Ecotoxicology 30, 1680–1688. doi: 10.1007/s10646-020-02305-1
Weinberg, M., and Séguin, P. (1918). La gangrene Gazeuse, Monographies de ITnstitut Pasteur. Paris, Masson and Cie.
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Zhang, X., Wang, L., Zeng, T., Liu, Y., Wang, G., Liu, J., et al. (2022). The removal of selenite and cadmium by immobilized biospheres: Efficiency, mechanisms and bacterial community. Environ. Res. 211, 113025. doi: 10.1016/j.envres.2022.113025
Keywords: Paraclostridium, P. bifermentans, antimicrobial resistance, pathogenicity, Listeria LIPI-1, multiple plasmids, phylogenomic tree
Citation: Zhao H, Wang J, Peng Y, Cai X, Liu Y, Huang W, Huang H and Nie Y (2022) Genomic insights from Paraclostridium bifermentans HD0315_2: General features and pathogenic potential. Front. Microbiol. 13:928153. doi: 10.3389/fmicb.2022.928153
Received: 25 April 2022; Accepted: 25 July 2022;
Published: 24 August 2022.
Edited by:
Chinyere Okoro, University of Surrey, United KingdomReviewed by:
Sonia Agrawal, AstraZeneca, United StatesPrashant P. Patil, National Institute of Allergy and Infectious Diseases (NIH), United States
Copyright © 2022 Zhao, Wang, Peng, Cai, Liu, Huang, Huang and Nie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuqiang Nie, ZXluaWV5dXFpYW5nQHNjdXQuZWR1LmNu; Hongli Huang, ZXlodWFuZ2hsQHNjdXQuZWR1LmNu
†These authors have contributed equally to this work