 Zhengang Miao
Zhengang Miao Guangyuan Wang
Guangyuan Wang Heng Shen1
Heng Shen1 Dean W. Gabriel
Dean W. Gabriel Wenxing Liang
Wenxing Liang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 June 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.925868
This article is part of the Research Topic Interactions Between Filamentous Plant Pathogens with their Host Plants and Biocontrol Agents View all 16 articles
DNA adenine N6-methylation (6mA) plays a critical role in various biological functions, but its occurrence and functions in filamentous plant pathogens are largely unexplored. Botrytis cinerea is an important pathogenic fungus worldwide. A systematic analysis of 6mA in B. cinerea was performed in this study, revealing that 6mA is widely distributed in the genome of this fungus. The 2 kb regions flanking many genes, particularly the upstream promoter regions, were susceptible to methylation. The role of BcMettl4, a 6mA methyltransferase, in the virulence of B. cinerea was investigated. BcMETTL4 disruption and point mutations of its catalytic motif “DPPW” both resulted in significant 6mA reduction in the genomic DNA and in reduced virulence of B. cinerea. RNA-Seq analysis revealed a total of 13 downregulated genes in the disruption mutant ΔBcMettl4 in which methylation occurred at the promoter sites. These were involved in oxidoreduction, secretory pathways, autophagy and carbohydrate metabolism. Two of these genes, BcFDH and BcMFS2, were independently disrupted. Knockout of BcFDH led to reduced sclerotium formation, while disruption of BcMFS2 resulted in dramatically decreased conidium formation and pathogenicity. These observations indicated that 6mA provides potential epigenetic markers in B. cinerea and that BcMettl4 regulates virulence in this important plant pathogen.
DNA methylation is one of the basic epigenetic markers involved in regulation of various biological functions in both prokaryotes and eukaryotes (Bird, 2007; Vasu and Nagaraja, 2013). Among the methylation modifications, DNA adenine N6-methylation (6mA) and 5-methylcytosine (5mC) are two of the most important. The 5mC modification has been well studied as an epigenomic marker in eukaryotes (Zemach et al., 2010), while adenine N6-methylation has been found to be predominantly distributed in prokaryotic genomes (Blow et al., 2016) and eukaryotic RNA (Fu et al., 2014). In recent years, 6mA has also been found to be widespread in the genomic DNA of many eukaryotes, including fungi (Mondo et al., 2017), Chlamydomonas (Fu et al., 2015), Caenorhabditis elegans (Greer et al., 2015), Arabidopsis thaliana (Liang et al., 2018), rice (Zhou et al., 2018) and humans (Xiao et al., 2018). However, compared with those in prokaryotes, the characteristics of 6mA methylation in eukaryotes are still largely unknown.
Previous studies have shown that 6mA is dynamically controlled by methyltransferase and demethylase (Greer et al., 2015; Zhang et al., 2015). In prokaryotes, M.MunI is a classic 6mA methyltransferase (Iyer et al., 2016). It has been well documented that the orthologs of MT-A70 that evolved from M.MunI (Iyer et al., 2011) in eukaryotes are important 6mA methyltransferases (Bujnicki et al., 2002). Three MT-A70 methyltransferases, namely, Mettl4 in Bombyx mori (Wang et al., 2018) and Mettl3 and Mettl14 in humans (Wang et al., 2016), have been shown to play important roles in regulating the levels of 6mA. Mta1, an MT-A70 ortholog in Oxytricha, can form a dimer with Mta9 and has the function of a DNA methyltransferase (Woodcock et al., 2020). We found an MT-A70 homologous protein, BcMettl4, in Botrytis cinerea, but the function of this protein in DNA methylation was unclear.
Botrytis cinerea is an important necrotrophic fungal pathogen that infects over 1,000 plants and causes gray mold disease. Many economically important vegetables and fruits, including strawberry, tomato, cucumber and grape, are susceptible to B. cinerea. B. cinerea is ranked as the second most common plant pathogenic fungus in the world (Dean et al., 2012) and causes severe preharvest and postharvest economic losses worldwide (Mousavi-Derazmahalleh et al., 2019). However, the biological role of the 6mA DNA modification in B. cinerea is still unclear.
In this study, the 6mA DNA modification in B. cinerea was investigated and 6mA was found to be widely distributed in B. cinerea. The gene BcMETTL4 was disrupted in the wild-type strain B05.10. Methylated DNA immunoprecipitation sequencing (MeDIP-Seq) was further employed to analyze the methylome characteristics of the wild-type strain B05.10 and the gene disruption mutant ΔBcMettl4. Deletion of BcMETTL4 caused a global reduction in DNA 6mA. RNA-Seq analysis was subsequently used to investigate global gene expression patterns in the disruption mutant ΔBcMettl4 and its wild-type strain B05.10. Finally, the relationships between 6mA, gene expression and virulence were investigated. Expression of key genes involved in virulence and conidiation was strongly affected by 6mA modification in B. cinerea.
Strain B05.10 of B. cinerea used in this study was isolated from Vitis vinifera (Yang et al., 2018). The wild-type strain, gene deletion derivatives and complemented strains were grown on PDA plates (20% potato, 2% dextrose, and 1.5% agar). The growth ratios, conidial development, and sclerotium formation of different strains of B. cinerea were measured according to previously described methods (Yang et al., 2019). For mycelial growth assays, the different strains were cultured at 25°C for 3 day on PDA plates supplemented with 0.5 mM CR, 0.73 mM SDS, 20 mM H2O2, 1 M sorbitol, 1 M KCl, 1 M NaCl, and 1 M glycerine.
To construct the gene mutant cassette for targeted deletions, two flanking sequences of each gene were obtained from genomic DNA of strain B05.10 by PCR. The hygromycin B phosphotransferase (HPH) gene was derived from the plasmid pBS-HPH1 (Yang et al., 2018). The two flanking fragments and the HPH gene were fused by splice overlap PCR. The resulting deletion cassettes (Promoter:HPH:Terminator) were connected to the pMD19-T vector for sequencing. The gene deletion cassette was transformed into strain B05.10.
For complementation, full-length genes, including promoters and terminators, were amplified from the B05.10 genome. The resulting PCR products were cloned into plasmid pBS-neo (Yang et al., 2018). The resulting vectors were subsequently transformed into the deletion mutants.
To construct BcMettl4-GFP, BcDamt-GFP, and BcMettl4Δ NLS-GFP fusion cassettes, the open reading frames (ORFs) of BcMETTL4 and BcDAMT without the stop codons were amplified by PCR from strain B05.10. The NLS of BcMettl4 was deleted using overlap PCR (Heckman and Pease, 2007). The PCR products were ligated into plasmid pNAH-OGG-G418 that was derived by replacing the hygromycin B resistance gene of pNAH-OGG (Schumacher, 2012) with a geneticin G418 resistance gene from plasmid pBS-neo (Yang et al., 2018).
For generation of the BcMettl4 point mutant, we first designed primers that contained the mutated site. The sites upstream and downstream of the mutated site were obtained by PCR and the products were joined by overlap PCR. The resulting PCR products were cloned into the plasmid pBS-neo and transformed into the ΔBcMettl4 strain using standard protoplast formation procedures as previously described (Gronover et al., 2001; Yang et al., 2018).
All primers and their descriptions are shown in Supplementary Table 1. Positive transformants were selected on PDA plates with 0.01% hygromycin B or 0.01% geneticin G418 sulfate. PCR was used to confirm the positive transformants.
Genomic DNA from various strains was extracted using a Fungi Genomic DNA Purification Kit (Sangon Biotech, China). After denaturation at 98°C for 5 min, the extracted DNA was chilled on ice for 10 min and then transferred to a HybondTM-N + membrane. After drying at room temperature, the DNA was fixed to the membrane by UV cross-linking. The HybondTM-N + membrane was blocked in 5% skim milk powder for 1.5 h at room temperature, followed by incubation with 6mA antibody (1:2,000 dilution, Abcam, ab151230) for 10 h at 4°C. The membrane was then washed four times for 8 min using TBST [25 mM Tris–HCl (pH 7.4), 140 mM NaCl and 0.1% Tween-20]. After incubation with secondary antibody (1:5,000 dilution, Sigma A0545) at room temperature for 2 h, the membrane was washed 4 times for 8 min using TBST. Finally, an ECL Immunoblotting Detection Kit (Thermo Fisher) was used for signal visualization. The input DNA was normalized by using methylene blue.
MeDIP-Seq was performed according to previously described protocols (Fu et al., 2015; Chen et al., 2018; Zhou et al., 2018). In brief, the genomic DNAs of different strains were extracted from conidia grown in YEPD medium for 24 h at 25°C, followed by incubation with RNase A overnight. After sonication, the DNA fragments (200–400 bp) were repaired, followed by adaptor ligation. The resulting DNA was purified using a TIANquick Midi Purification Kit (DP204, TIANGEN BIOTECH, China). The purified DNA fragments were denatured at 94°C for 15 min and chilled on ice for 10 min. An aliquot of denatured DNA was used as input. The remaining DNA was incubated with anti-6mA antibody at 4°C for 12 h. After elution by 6mA salt competition, a 6mA-IP-Seq library was constructed by PCR amplification. The resulting DNA fragments were sequenced with a HiSeq 2500 (Illumina) (Zhou et al., 2018). Raw reads were trimmed to remove adaptors and low-quality bases, and the reads with 6mA-rich regions were called with MASC2 (Liu, 2014).
After conidial germination in YEPD medium at 25°C for 24 h, the mycelia of different strains were collected by centrifugation for RNA extraction. Total RNA was extracted using TRIzol Reagent (Invitrogen). A NanoDrop spectrophotometer (Thermo Scientific) was employed to determine the RNA concentration, quality and integrity. Poly-T oligo-attached magnetic beads were purified to enrich mRNA. First-strand cDNA was obtained using random oligonucleotides and SuperScript II. Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The resulting cDNA fragments were further adenylated at the 3’ ends and ligated with Illumina PE adaptor oligonucleotides. PCR was performed to enrich the DNA fragments with ligated adaptors on both ends. After purification using an AMPure XP system and quantification using an Agilent high-sensitivity DNA assay, the obtained sequencing library was sequenced on a NovaSeq 6000 platform (Illumina).
TopHat (v2.0.12) was employed to map the obtained sequences against the database of B. cinerea (Trapnell et al., 2009). The mapped genes were calculated using HTSeq (v0.6.1) (Anders et al., 2015). Gene expression was quantitatively estimated by using the reads per kilobase per million mapped reads (RPKM) value (Trapnell et al., 2010). DESeq was used to analyze the differentially expressed genes between the wild-type and the gene deletion mutants (Wang et al., 2010).
After growth on PDA plates at 25°C for 7 days, the conidia produced by different strains of B. cinerea were harvested and resuspended in buffer (6.7 mm K2HPO4 and 10 mm glucose). A total of 5 μl of conidia suspension (5.0 × 105 conidia/ml) was transferred to 8-week-old tomato (Solanum lycopersicum) leaves, incubated at 25°C for 72 h, and developing disease lesions caused by different strains were measured.
Additionally, infection-related morphogenesis was observed on onion epidermis. The conidial suspensions (3.0 × 103 conidia/ml) were inoculated onto the epidermis. After culturing for 12 h at 25°C in the dark, the onion skin was stained with aniline blue and observed.
The expression levels of BcMETTL4 and BcDAMT in wild-type B05.10 in interaction between B. cinerea and tomato leaves were investigated by real-time PCR. In brief, 45 ml of conidia suspension (5.0 × 106 conidia/ml) and 5 ml of YEPD medium were mixed and 3 g of sterile fresh intact tomato leaves (3 weeks old) was added, and then gently agitated in a shaker at 25°C. Samples were collected at 6 h intervals for 24 h. RNA was extracted according to a previously described protocol (Yang et al., 2018). cDNA was biosynthesized by reverse transcription using a PrimeScript RT Kit (Takara, Japan). The primers used are shown in Supplementary Table 1. Relative expression levels were calculated using the 2–ΔΔCt method (Livak and Schmittgen, 2001).
The mycelia grown in YEPD medium at 25°C for 24 h were collected. A nuclear protein extraction kit (R0050, Solarbio) was used to separate nuclear and cytoplasmic proteins. The proteins were separated by SDS-PAGE and immunoblotted using anti-GFP antibody (ab290, Abcam), anti-H3 antibody (ab1791, Abcam), and anti-TUBA4A antibody (D110022, BBI Life Sciences).
To investigate the presence of 6mA in B. cinerea, we searched the genome of B. cinerea B05.10 using BLAST and found two potential candidates for 6mA methyltransferase. One candidate was BCIN_12g06440 (also known as BC1G_13837), encoding a hypothetical protein (NCBI id: XP_024552389.1) which is a 462-amino-acid ortholog of MT-A70 in B. cinerea (Figure 1A). The other was BCIN_02g05970 (also known as BC1G_16157), encoding a hypothetical protein (255 amino acids) containing a N6-adenineMlase (probable N6-adenine methyltransferase) catalytic domain (NCBI id: XP_024547190.1) (Figure 1B). As shown in Figure 1A, the protein encoded by BCIN_12g06440 contained a conserved catalytic motif (“DPPW”) and a nuclear localization signal (NLS). However, the protein encoded by BCIN_02g05970 contained only one catalytic motif, “DPPF” (Figure 1B).
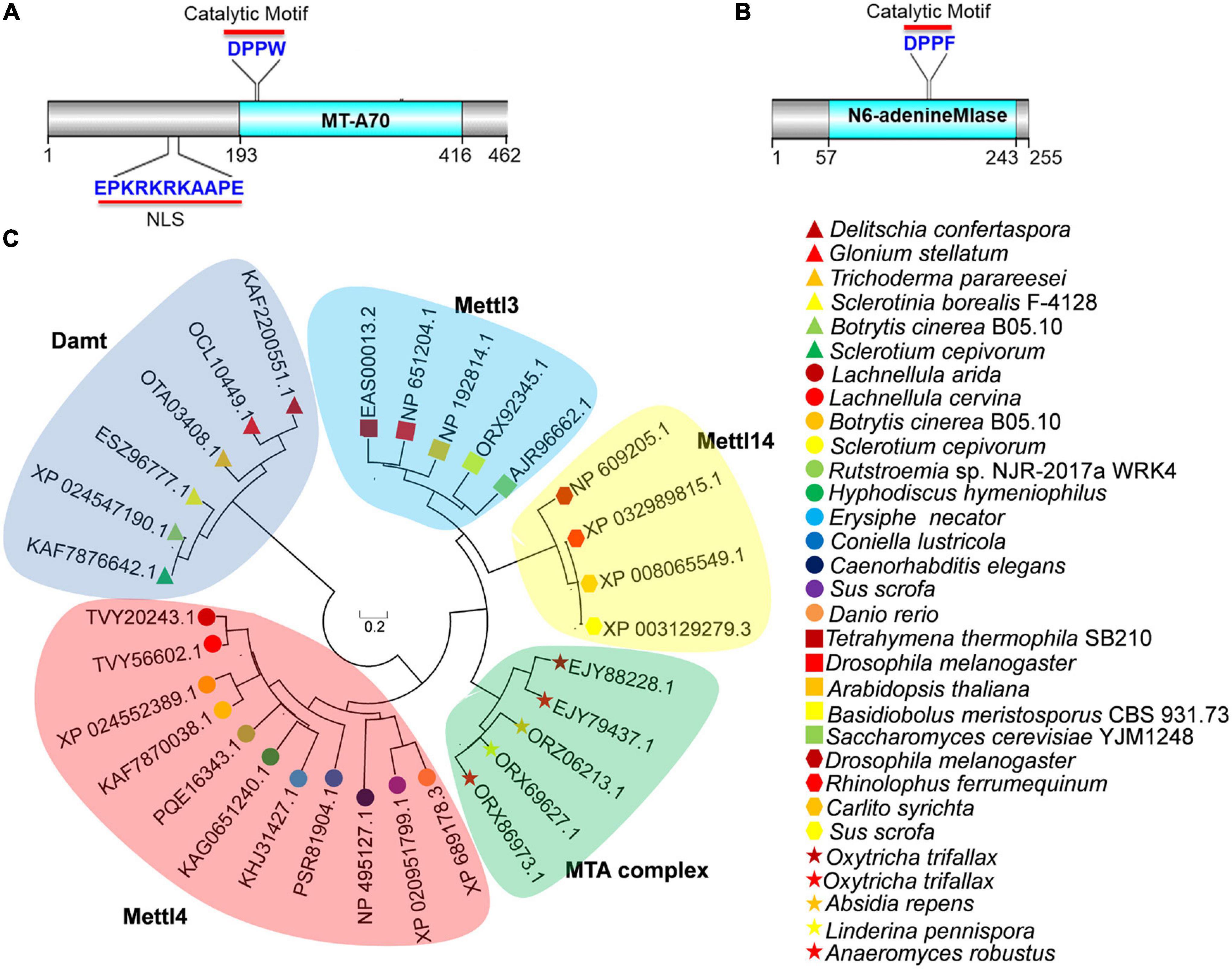
Figure 1. Bioinformatics analysis of BcMettl4 and BcDamt. (A) The structural domains encoded by BCIN_12g06440. (B) The structural domain encoded by BCIN_02g05970. (C) Phylogenetic tree of methyltransferase orthologs based on a neighbor-joining analysis.
A variety of methyltransferases, including Mta1, Mta9, Mettl3, Mettl4, Mettl14, and Damt, have previously been reported (Wang et al., 2016, 2018; Chen et al., 2018; Beh et al., 2019; Woodcock et al., 2020). A phylogenetic tree of methyltransferase orthologs was constructed using MEGA 7.0 software, revealed that the protein encoded by BCIN_12g06440 in B. cinerea was closely related to a branch representing the MT-A70 ortholog Mettl4, while the protein encoded by BCIN_02g05970 belonged to a clade of Damt (Figure 1C). The hypothetical proteins encoded by BCIN_12g06440 and BCIN_02g05970 were thus named BcMettl4 and BcDamt, respectively. The roles of BcMettl4 and BcDamt in the virulence of B. cinerea were subsequently studied in some detail.
Since BcMettl4 contains a predicted NLS signal (Figure 1A), this protein might localize in the nucleus. To test this hypothesis, two fusion proteins, BcMettl4-GFP and BcMettl4Δ NLS-GFP (without a NLS), were expressed in strain B05.10 of B. cinerea. After growth on PDA plates for 3 days, the transformants containing the fusion protein BcMettl4-GFP or BcMettl4Δ NLS-GFP were observed by fluorescence microscopy. The results showed that the GFP signal of BcMettl4-GFP overlapped with nuclei stained with DAPI fluorescent dye, while BcMettl4Δ NLS-GFP was present in both the cytoplasm and the nucleus (Figures 2A,B). A western blot analysis was performed using extracted total proteins, nuclear proteins and cytoplasmic proteins and revealed that BcMettl4-GFP was significantly enriched in the nucleus compared with BcMettl4Δ NLS-GFP (Figure 2C). These findings confirmed that BcMettl4 was located in the nucleus and that the NLS of BcMettl4 played an important role in controlling the subcellular localization of BcMettl4. By contrast, the fusion protein BcDamt-GFP was mainly located in the cytoplasm (Figures 2A–C).
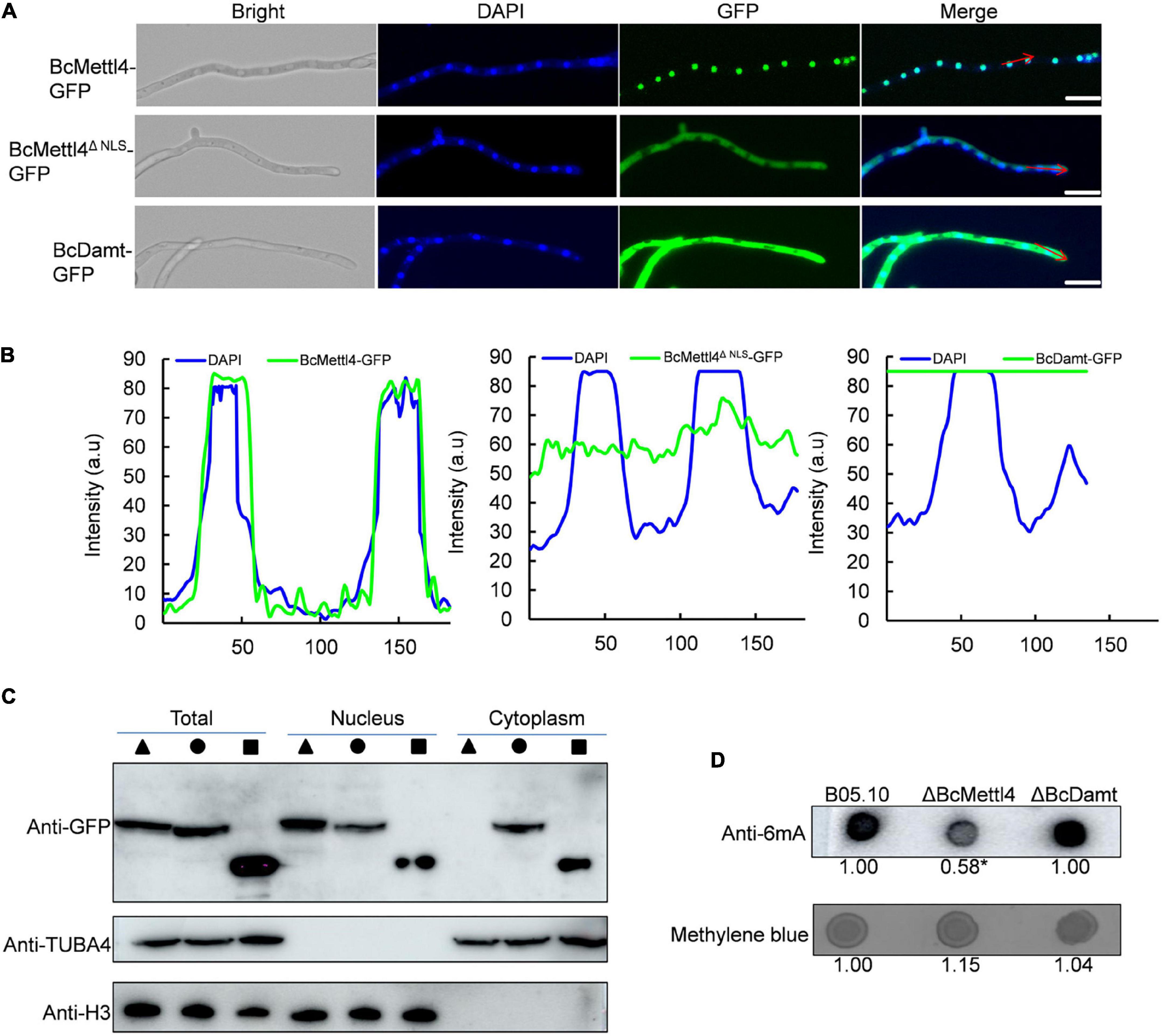
Figure 2. Subcellular localization of BcMettl4 and BcDamt, and effects of BcMETTL4 and BcDAMT deletion on genome methylation. (A) Mycelia images for subcellular localization of the BcMettl4-GFP, BcMettl4Δ NLS-GFP, and BcDamt-GFP. Scale bar = 20 μm. (B) Fluorescence intensity of BcMettl4-GFP, BcMettl4Δ NLS-GFP, and BcDamt-GFP. The blue line represents DAPI while the green line shows GFP. The signal areas were marked by red lines in Figure 2A and the signal quantification were calculated by ImageJ. (C) Western blot analysis of BcMettl4-GFP (triangle), BcMettl4Δ NLS-GFP (circle), and BcDamt-GFP (square). Histone H3 served as a reference for proteins in the nucleus. Alpha tubulin 4a was used as a control for cytoplasmic proteins. (D) Dot blot assay for verification the presence of 6mA in the genome DNA of B. cinerea. A total of 200 ng DNA was investigated by dot blot assay. Relative 6mA abundance in different strains was calculated by ImageJ. The relative ratio after normalization is to reflect the difference among various treatments. The asterisk means significant difference in values at p < 0.05.
The genes BcMETTL4 and BcDAMT were disrupted in the wild-type strain B05.10. PCR amplification revealed that one transformant, ΔBcMettl4, exhibited BcMETTL4 gene disruption (Supplementary Figure 1), and another transformant, ΔBcDamt, exhibited BcDAMT gene deletion (Supplementary Figure 2). To investigate the presence of 6mA DNA modification in B. cinerea, the genomic DNAs (gDNAs) of B05.10, ΔBcMettl4, and ΔBcDamt were extracted and a dot blot assay performed using a specific 6mA antibody. As shown in Figure 2D, a strong immune blot signal was detected in the gDNA of B05.10. Compared with those of B05.10 and ΔBcDamt, the signal in the gene deletion mutant ΔBcMettl4 was significantly weaker, based on ImageJ analysis (p < 0.05) (Figure 2D).
Taken together, these observations suggested that 6mA was a widespread DNA modification in the genome of B. cinerea and that BcMettl4 may play a role in modifying nuclear DNA.
The transcript levels of BcMETTL4 and BcDAMT at the point of germination in culture in the presence of tomato leaf tissue to 24 h later were assessed as a proxy for the early interaction stage between B. cinerea and tomato leaves. As shown in Figure 3A, the expression levels of BcMETTL4 and BcDAMT remained stable until 12 h and then increased approximately four-fold compared to those at 0 h. However, after 24 h of growth, the expression of BcMETTL4 increased approximately 110-fold, compared with a twofold increase in BcDAMT gene expression (Figure 3A). Highly induced expression of BcMETTL4 by presence of tomato leaf tissue indicated that BcMettl4 might be involved in the virulence of B. cinerea.
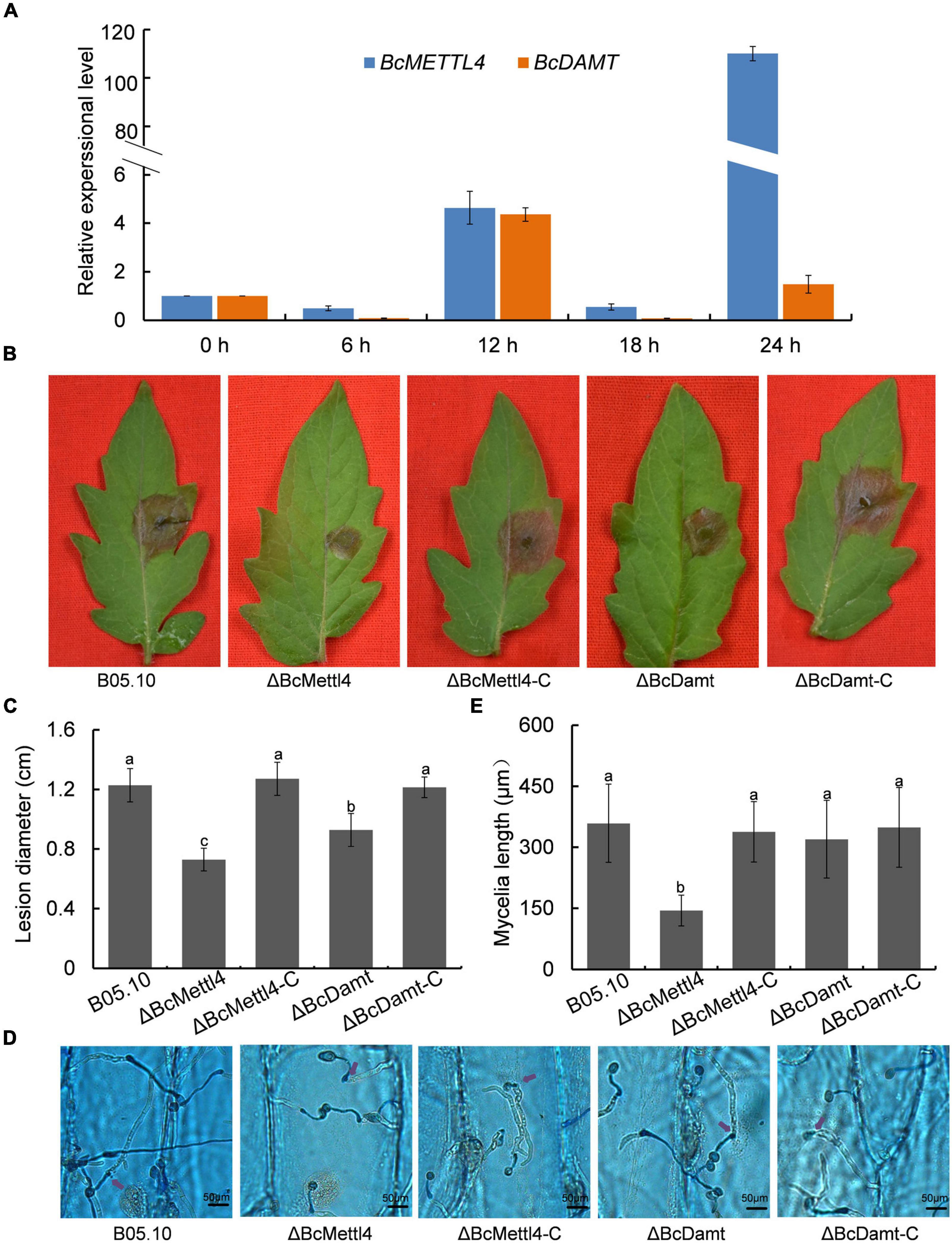
Figure 3. Effects of BcMETTL4 and BcDAMT disruption on virulence and conidia infection. (A) The expression levels of BcMETTL4 and BcDAMT at early infection stages evaluated by qRT-PCR. (B) Lesions on tomato leaves caused by different strains 72 h after inoculation. (C) Lesion diameter of the strains inoculated on tomato leaves. Three experiments were carried out and nine leaves were measured in one biological experiment. Statistical tests were carried out using Tukey’s test. The same letters marked on the bars indicate no significant difference in values at p < 0.05. (D) Infection of conidia from different strains after 12 h of incubation on onion epidermis. Mycelia growing outside onion cells were colored blue, while mycelia growing inside onion cells are not stained. Penetrated positions are marked by purple arrows. (E) Mycelia length of different strains at 12 h after conidia infection on onion epidermis. Data are given as the mean ± SD, n = 10. The different letters showed on the columns were significantly different at p < 0.05.
The impacts of BcMETTL4 and BcDAMT disruption on the virulence of B. cinerea were therefore investigated. First, the mutants ΔBcMettl4 and ΔBcDamt were complemented with the genes BcMETTL4 and BcDAMT, respectively. These two genes including their native promoters and terminators were amplified from wild-type strain B05.10 genomic DNA using the primers listed in Supplementary Table 1. The obtained fragments were inserted into the plasmid pBS-neo and subsequently transformed into the mutants according to the method described above. Finally, two complemented strains, ΔBcMettl4-C and ΔBcDamt-C, were obtained. Deletion of any of the genes, that is, BcMETTL4 or BcDAMT, reduced the pathogenicity of B. cinerea on tomato, especially deletion of BcMETTL4 (Figure 3B). As shown in Figure 3C, the spreading of the lesion induced by the disruption mutant ΔBcMettl4 was significantly reduced compared with that induced by the wild-type strain B05.10 or the disruption mutant ΔBcDamt.
We further examined the ability of these strains to penetrate onion epidermal cells. As shown in Figure 3D, deletion of BcMETTL4 weakened the infection of mycelia in onion epidermal cells. Mycelia length of different strains of B. cinerea at 12 h after conidia infection on onion epidermis were further investigated. It was found that the mycelia length of ΔBcMettl4 invaded onion cells were 144 μm, while those of other strains, B05.10, ΔBcMettl4-C, ΔBcDamt, and ΔBcDamt-C, were 359, 338, 320, and 349 μm (Figure 3E), respectively, revealing that BcMETTL4 interruption decreased virulence of B. cinerea on onion by reducing the conidia infectivity.
The reduction in genome-wide 6mA (Figure 2D) in the disruption mutant ΔBcMettl4 led to investigations of the biological phenotypes of the different mutations affecting 6mA. After 3 days of cultivation on PDA plates, there were no obvious differences in mycelial growth of B05.10, ΔBcMettl4, ΔBcDamt, ΔBcMettl4-C, and ΔBcDamt-C (Figure 4A). Disruption of BcMETTL4 or BcDAMT also did not affect conidial formation (Figure 4A). However, after growth on PDA plates for 30 days in darkness, ΔBcMettl4 exhibited significantly decreased sclerotial formation compared with B05.10, ΔBcMettl4-C, ΔBcDamt, and ΔBcDamt-C (Figure 4B). Our previous studies have shown that the reduction of sclerotia in B. cinerea is often accompanied by a decrease in pathogenicity (Yang et al., 2015; Wang et al., 2020). These observations suggested that the reduction in sclerotia in the BcMETTL4 mutant might also be correlated with a decrease in the pathogenicity of B. cinerea.
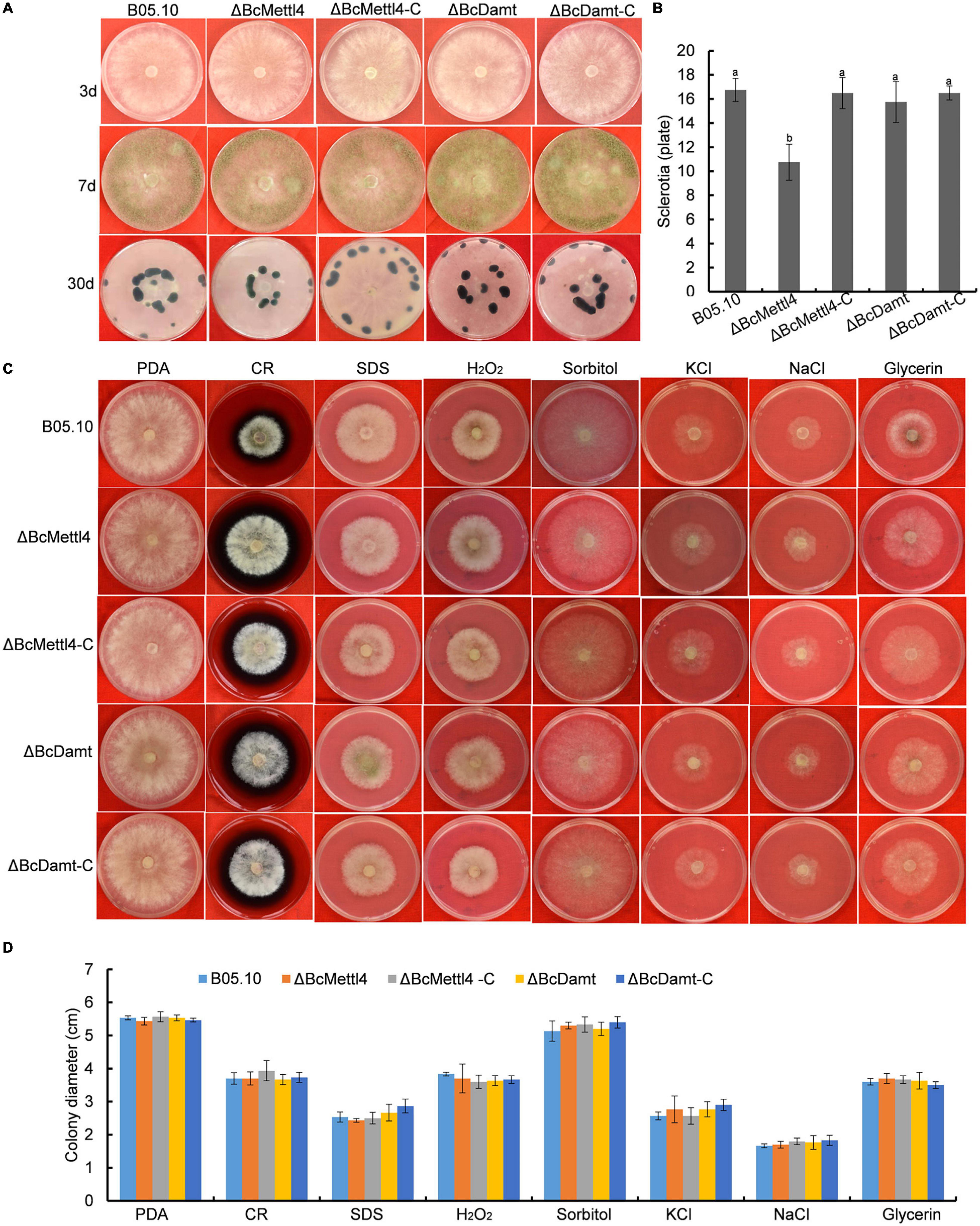
Figure 4. Mycelial growth, conidial and sclerotial formation and stress tolerance of the indicated strains. (A) Mycelial growth, conidial and sclerotial development of various strains grown on PDA plates. (B) Number of sclerotia produced by various strains grown on PDA plates for 30 days in darkness. Bars denote standard errors from three plates. Tukey’s test was employed to calculate significance. The same letters indicated on the bars mean no significant difference in values at p < 0.05. (C) The sensitivity of various strains on stress tolerance. (D) Colony diameter of the indicated strains grown on various stress conditions. Bars represent standard errors from three plates.
The sensitivity of different strains to stress tolerance was also measured. Disruption of BcMETTL4 or BcDAMT did not cause significant sensitivity to cell wall stress, osmotic stabilization, or oxidative stress (Figures 4C,D), indicating that BcMETTL4 and BcDAMT were not involved in the regulation of cell stress tolerance.
Methyltransferases contain a conserved catalytic motif, “DPPW.” A 3-dimensional structure of BcMettl4 was constructed, and the signature motif “DPPW” was found to be located in the catalytic domain (Figure 5A). Multiple sequence alignments of the catalytic motifs in 6mA methyltransferases were performed. The results showed that the conserved catalytic motif “DPPW” in BcMettl4 exhibited high similarity with Mettl4 orthologs (Figure 5B). To study the catalytic function of BcMettl4, the catalytic domain “DPPW” was mutated to “APPA” in BcMettl4. After the mutation to “APPA,” the methylation level of DNA in B. cinerea decreased (Figure 5C) and both virulence and sclerotium formation decreased (Figures 5D,E), which was consistent with the phenotype of the ΔBcMettl4 mutant. These data suggested that the catalytic motif “DPPW” plays a key role in the enzymatic function of BcMettl4 and that BcMettl4 is involved in the regulation of DNA methylation levels and virulence.
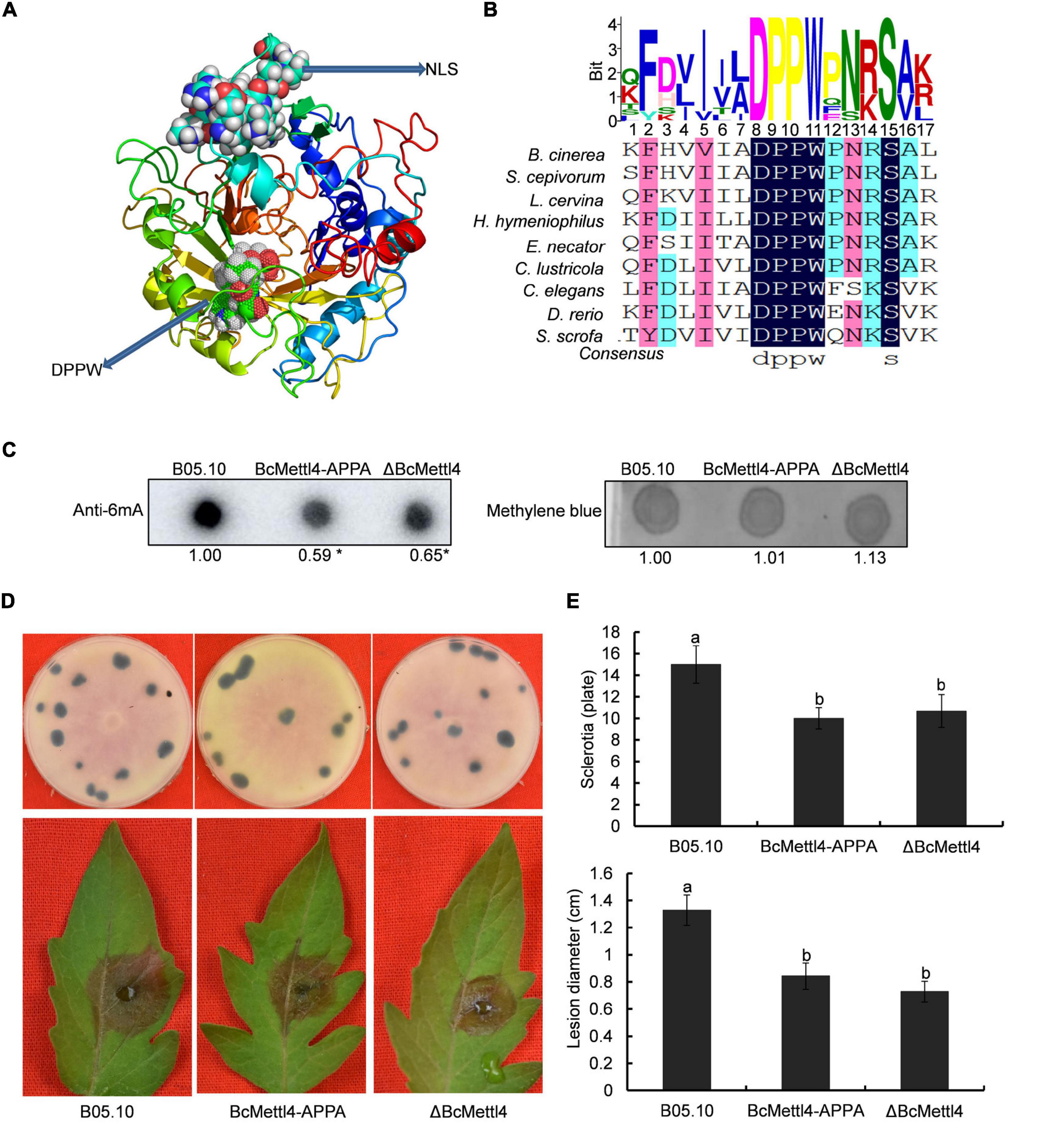
Figure 5. Impact of BcMettl4 loss-of-function on DNA modification, pathogenicity, and sclerotia formation of B. cinerea. (A) Three-dimensional structure of BcMettl4 modeled were predicted by I-TASSER. (B) Sequence alignment of motifs in methyltransferase form B. cinerea, Sclerotium cepivorum, Lachnellula cervine, Hyphodiscus hymeniophilus, Erysiphe necator, Coniella lustricola, Caenorhabditis elegans, Danio rerio, and Sus scrofa. (C) Dot blot assay of 6mA in the indicated strains. A total of 200 ng DNA was investigated on each dot. The relative ratio of 6mA in wild-type strain B05.10 and point mutant BcMettl4-APPA was calculated by ImageJ. The asterisk represents significant difference at p < 0.05. (D) The disease lesions caused by various strains following inoculation on tomato leaves for 3 days and the sclerotia formation of various strains after grown in darkness for 30 days. (E) Lesion diameter of the strains inoculated on tomato leaves and the number of sclerotia in various stains. Error-bars were from three experiments. For lesion diameter, nine leaves were investigated in one biological experiment. Three plates were used to count sclerotia. Tukey’s test was used to calculate significance. The same letters marked on the columns were not significantly different at p < 0.05.
To detect naturally occurring methylated genes globally regulated by BcMETTL4 in B. cinerea, we performed a MeDIP-Seq analysis of the 6mA methylomes of the disruption mutant ΔBcMettl4 and its wild-type strain B05.10. The expression of BcMETTL4 increased significantly after 24 h infection (Figure 3A). We therefore collected the samples of conidial germination at 24 h for MeDIP-seq. Three biological replicates for each strain of ΔBcMettl4 and B05.10 were investigated in MeDIP-Seq experiments. A total of 30,320 6mA-enriched regions (peaks) were captured from the three B05.10 biological replicates, while a total of 28,385 methylation peaks were obtained from the three ΔBcMettl4 biological replicates (Supplementary Table 2). These observations further confirmed that disruption of BcMETTL4 reduced the methylation level of B. cinerea. After assembling the attained sequences, it was found that most of the 6mA peaks (57%) were distributed in the upstream regions of genes, followed by 24% in the exon, and 15% in the downstream regions in wild-type B05.10 and the mutant ΔBcMettl4 (Figures 6A,B). The 6mA peaks were further investigated and wild-type B05.10 exhibited 1,297 unique 6mA peaks while 689 unique 6mA peaks were identified in the mutant ΔBcMettl4 (Figure 6C). There were 8,357 6mA peaks that were found in both the wild-type B05.10 and the mutant ΔBcMettl4 (Figure 6C). Among the unique 6mA peaks of wild-type B05.10, 2,396 genes were detected (Figure 6D and Supplementary Table 3). However, only 1,330 genes were detected in the unique 6mA peaks of the mutant ΔBcMettl4 (Figure 6D and Supplementary Table 4).
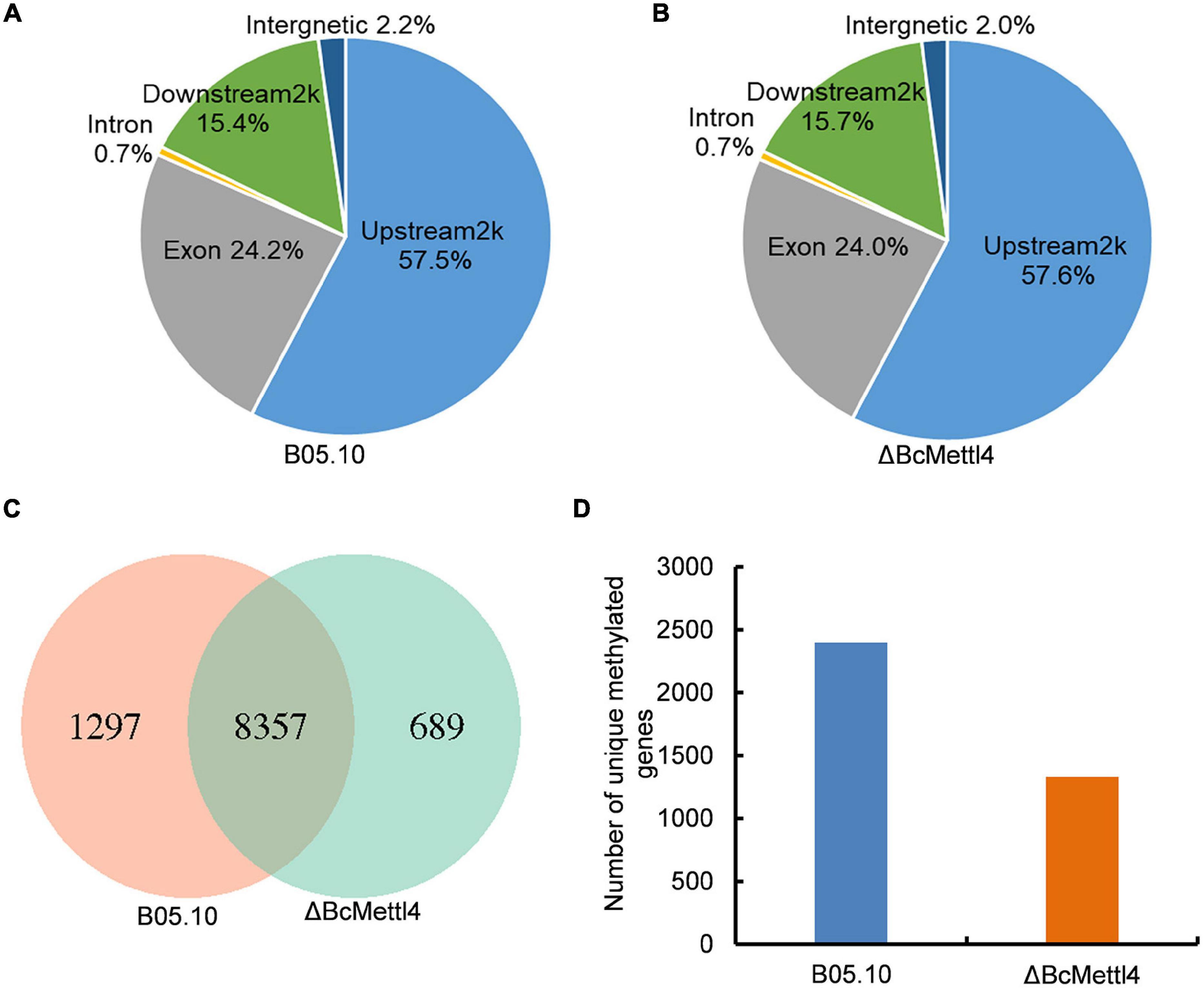
Figure 6. The 6mA methylated genes in B. cinerea. (A) Peak distribution in genomic elements of the wild-type strain B05.10. (B) Peak distribution in genomic elements of the disruption mutant ΔBcMettl4. (C) Venn diagram comparing unique 6mA peaks between the wild-type B05.10 and the mutant ΔBcMettl4. (D) The number of unique methylated genes in wild-type strain B05.10 and the disruption mutant ΔBcMettl4.
Since the majority of the 6mA peaks were identified in the regions upstream of the affected genes (Figures 6A,B), involvement of the promoter regions and regulation of gene expression was indicated. To identify all genes that might exhibit differential expression due to the disruption of BcMETTL4 in B. cinerea, we performed an RNA-Seq analysis of the full transcriptomic profiles of the wild-type strain B05.10 and the disruption mutantΔBcMettl4. The differentially expressed genes were further analyzed using DESeq software (Wang et al., 2010). A total of 792 genes, including 601 upregulated genes (Supplementary Table 5) and 191 downregulated genes (Supplementary Table 6), were found to be differentially expressed in the BcMETTL4 mutant compared with the wild-type strain B05.10, which accounted for 4.82% (792/16,448) of the annotated genes in B. cinerea.
To test the hypothesis that the unique genes modified at promoter sites might be directly regulated by BcMettl4, the transcription levels of the unique genes were further analyzed in the mutant ΔBcMettl4. After disruption of BcMETTL4, among the unique methylated genes modified at promoter sites in the wild-type strain B05.10, 32 genes were upregulated (Supplementary Table 7) and 13 genes were downregulated (Figures 7A,B). Since the virulence of the BcMETTL4 mutant was significantly reduced, we hypothesized that the 13 methylated genes modified in the promoter region with downregulated expression might be associated with pathogenicity. The genes shown in Figure 7A were therefore subsequently further analyzed.
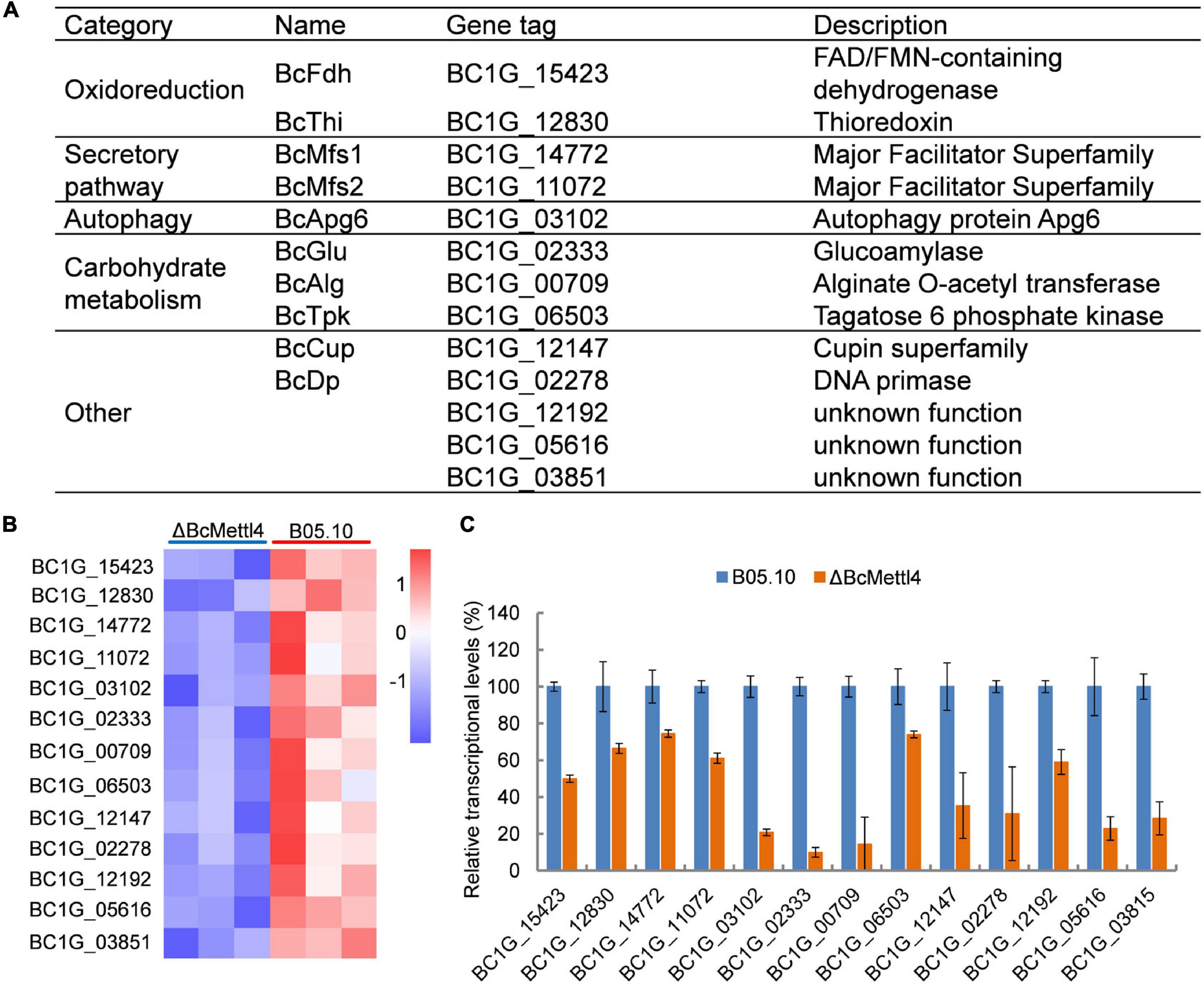
Figure 7. The downregulated methylated genes in which modification occurs in the promoter region. (A) Categories of the screened genes. (B) RNA-Seq analysis of 13 methylated genes. The colored squares show the level of gene expression referred to log2 FPKM value in each of the three replicas. (C) Validation of RNA-Seq data of selected genes by fluorescent real-time qRT-PCR. The reference gene used in this study was Beta-tubulin. Data are given as the mean ± SD, n = 3.
The reduced transcript levels of the 13 genes shown in Figure 7B were further validated by fluorescent real-time qRT-PCR (Figure 7C). As shown in Figure 7A, these methylated genes were divided into five categories, including oxidoreduction, secretory pathways, autophagy and carbohydrate metabolism, according to their biological functions. It has been well documented that proteins related to oxidative stress sensitivity (Yang et al., 2015), autophagy (Ren et al., 2018), and secretion (Zhang et al., 2016) are important factors affecting the virulence of B. cinerea. These observations indicated that the downregulated methylated genes related to oxidoreduction, secretory pathways and autophagy may cause a decrease in the pathogenicity of B. cinerea.
Deletion of BcMETTL4 both reduced the methylation and expression levels of 13 genes (Figure 7). We speculated that these genes might be involved in virulence of B. cinerea. Two of them were randomly selected for targeted disruption. BcMFS2 encoding a major facilitator superfamily (MFS) transporter and BcFDH encoding a FAD/FMN-containing dehydrogenase were independently deleted in the wild-type strain B05.10. Two mutants, ΔBcMfs2 and ΔBcFdh, were subsequently obtained. The genes BcMFS2 and BcFDH were also cloned and complemented both mutants, resulting in transformants, ΔBcMfs2-C and ΔBcFdh-C, respectively. Deletion of BcMFS2 reduced sclerotium formation (Figures 8A,B). Further infection assays of tomato leaves were performed, and the results showed that deletion of BcMFS2 did not reduce the virulence of B. cinerea (Figures 8A,B). These findings indicated that BcMFS2 was involved in sclerotium formation but not in the virulence of B. cinerea. Another interesting finding was that deletion of BcFDH severely hindered conidium formation in B. cinerea (Figure 8B). Infection of tomato leaves by ΔBcFdh was further performed, and the results showed that deletion of the BcFDH gene reduced the pathogenicity of B. cinerea by 20% (Figure 8B). These observations demonstrated that BcFDH was involved in both conidium formation and the pathogenicity of B. cinerea.
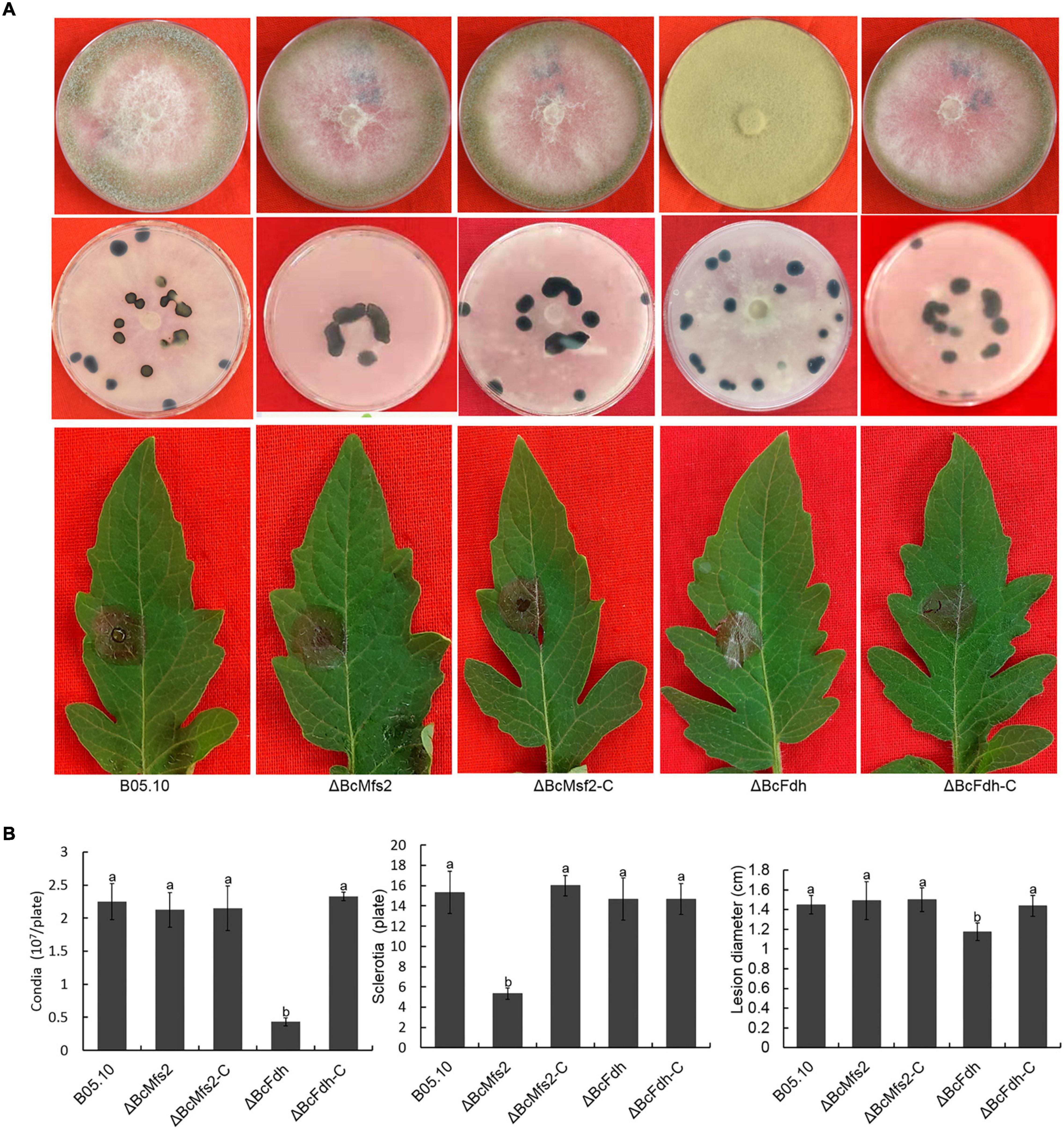
Figure 8. Impact of BcMFS2 and BcFDH deletion on pathogenicity of B. cinerea. (A) Effect of deletions BcMFS2 or BcFDH on conidia biosynthesis, sclerotia formation and virulence. (B) Quantification of conidia, sclerotia and pathogenicity of different strains. Bars represent standard errors from three experiments. In each biological experiment, lesion diameters of nine leaves were investigated. Three plates were used to count sclerotia and conidia. Statistical tests were carried out using Tukey’s test. The different letters marked on the columns indicate statistical significance p < 0.05.
As a basic epigenetic marker, DNA methylation has been widely documented in both eukaryotes and prokaryotes (Bird, 2007; Vasu and Nagaraja, 2013). However, its characteristics in B. cinerea are still elusive. In this study, we found that 6mA is a widespread DNA modification in the B. cinerea genome. Previous studies have shown that DNA methylation levels are dynamic and controlled by two enzymes, methyltransferase and demethylase (Greer et al., 2015; Zhang et al., 2015). MT-A70 is an important 6mA methyltransferase (Bujnicki et al., 2002). Gray mold contains two homologous methyltransferases, BcMettl4 (Figure 1A) and BcDamt (Figure 1B). As shown in Figure 2D, disruption of BcMETTL4 but not BcDAMT resulted in a decrease in the DNA methylation level of B. cinerea, suggesting that BcMettl4 is related to 6mA modification. Previous reports have shown that the signature motif “DNSH/PP/YFW” of 6mA methyltransferase is responsible for substrate binding and catalytic activity (Iyer et al., 2016; Wang et al., 2019). After the mutation of “DPPW” to “APPA,” the methylation level in B. cinerea decreased (Figure 5C), demonstrating a catalytic role of “DPPW” in BcMettl4. Consistent with these results, the AMT1 APPA mutant of Tetrahymena thermophila had a reduced 6mA level (Wang et al., 2019). Unlike the genome of B. cinerea, the genome of Phytophthora sojae contains three 6mA methyltransferase genes (DAMT1, DAMT2, and DAMT3), and single knockouts of any of these genes in the P. sojae strain P6497 resulted in a significant decrease in DNA methylation (Chen et al., 2018).
Using MeDIP-Seq analysis, we identified a large number of 6mA peaks that were distributed in the genome of B. cinerea. As shown in Figures 6A,B, 6mA was significantly enriched in upstream and downstream gene regions in both the wild-type strain B05.10 and the disruption mutant ΔBcMettl4. Consistent with our observations, almost all of the 6mA were found in the promoter regions, either at or slightly downstream of the transcription start sites in early-diverging fungi, Hesseltinella vesiculosa, Absidia repens, Lobosporangium transversale, and Syncephalastrum racemosum (Mondo et al., 2017). Several other studies also revealed that the transcription initiation sites of Chlamydomonas (Fu et al., 2015) and Phytophthora (Chen et al., 2018) were readily modified by 6mA. In Arabidopsis, the 6mA modification level in gene bodies was higher than that of intergenic regions (Liang et al., 2018). Similar to the result for Arabidopsis, DNA 6mA modification was preferentially distributed in the gene bodies rather than the intergenomic regions in Tetrahymena (Wang et al., 2017). However, 6mA was evenly distributed on chromosomes and correlated with 5mC distribution in rice (Zhou et al., 2018). Inconsistent with the above findings, DNA 6mA modification was significantly enriched in exon regions in the human genome (Xiao et al., 2018). Taken together, the results suggested that the genomic localization pattern of DNA 6mA modification in different species varies greatly, indicating that these modifications are hitherto unknown epigenetic markers.
Botrytis cinerea is an important necrotizing pathogen. It has been well documented that cellular accumulation of reactive oxygen species (ROS) is one of the immune responses of plants after infection by pathogens (Kaku et al., 2006; Cheval et al., 2013; Wada et al., 2019). Previous studies have documented that the virulence of pathogens is often related to resistance to oxidative stress (Yan et al., 2011; Yang et al., 2015, 2018). In a previous study, we found that knocking out the catalase BcCAT (BC1G_12146) reduced the toxicity of gray mold (Wang et al., 2020). In this study, BcCAT was downregulated in the disruption mutant ΔBcMettl4 (Supplementary Table 6). We also found that three oxidative stress-related genes, BcCATA (BC1G_01095), BcCAT3 (BC1G_02407), and BcCAT4 (BC1G_09386), were upregulated in ΔBcMettl4 (Supplementary Table 5), which might compensate for the effect of downregulation of BcCAT. After BcMETTL4 was inactivated, two methylated genes, BcFDH and BcTHI, which are related to oxidoreduction, were also downregulated (Figure 7A). Further experiments confirmed that knockout of BcFDH reduced the pathogenicity of B. cinerea (Figure 8B). The disruption of BcFDH severely hindered conidia formation (Figure 8B). The genome of B. cinerea contains 16 genes involved in conidiation (Amselem et al., 2011). We found that only one of them, BcPPOA80 (BC1G_14780), were downregulated when BcMETTL4 was deleted (Supplementary Table 6), indicating that the downregulation of only BcPPOA80 might have little effect on conidia formation. However, it is still completely unknown why the conidia formation was severely hindered when the BcFDH gene was deleted.
Two methylated genes, BcMFS1 and BcMFS2, which are associated with MFS transporters, were downregulated in the mutant ΔBcMettl4 (Figure 7A). After BcMFS2 was deleted, the mutant’s ability to form sclerotia was reduced (Figure 8A). BcSec14 and BcSec31 are involved in vesicle transport, and deletion of BcSec14 and BcSec31 resulted in a reduction in pathogenicity and protein secretion in gray mold (Zhang et al., 2016). It has been well documented that MFS is the largest group of membrane transporters (Law et al., 2008). MFS transporters are also involved in cell secretion and can transport some secondary metabolites and compounds (Vela-Corcía et al., 2019). The polysaccharide components of the cell wall are mainly hydrolyzed into monosaccharides and oligosaccharides by degrading enzymes, thereby reducing the protection of the cell wall against pathogens and increasing the ability of pathogens to infect plants (Cantarel et al., 2009; Kubicek et al., 2014). A cell polysaccharide-degrading enzyme-encoding gene, BcGLU, was found among the downregulated methylated genes (Figure 7A); this gene may be involved in the pathogenicity of B. cinerea. Taken together, these findings suggest that BcMettl4 may regulate pathogenicity via its effect on multiple pathways.
DNA adenine N6-methylation sites are widely distributed in the genome of B. cinerea. BcMettl4 is nuclear-localized and contains the conserved motif “DPPW,” which is likely related to its interaction with DNA for methylation modification. MeDIP-Seq showed that the regions 2,000 bp upstream and downstream of the genes were preferentially modified by methylation in B. cinerea. BcMETTL4 disruption and mutation of “DPPW” to “APPA” both resulted in a significant reduction in the genomic methylation and pathogenicity of B. cinerea. These observations indicate that 6mA provides potential epigenetic markers in B. cinerea and that BcMettl4 regulates the level of DNA methylation in this important plant pathogen. Our results extend the current understanding of the global regulatory role of 6mA in filamentous plant pathogens.
The data has been made available at NCBI with accession numbers PRJNA817858 and PRJNA817835.
ZM, GW, and WL contributed to the conception and design of the study. ZM, GW, and XW performed the experiments. ZM, GW, HS, and XW organized the database. ZM, HS, and XW performed the statistical analysis. ZM, GW, and WL wrote the first draft of the manuscript. GW, WL, and DG reviewed and edited the manuscripts. All authors provided comments on the manuscript and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31972213), the Shandong Provincial Natural Science Foundation (ZR2020KC003 and ZR2019MC052), the Key Research and Development Program of Shandong Province (2019YQ017), Taishan Scholar Construction Foundation of Shandong Province (tshw20130963), and Shandong Province “Double-Hundred Talent Plan” (WST2018008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.925868/full#supplementary-material
Supplementary Figure 1 | Disruption of BcMETTL4 and validation of three different mutants by PCR. (A) Marker exchange technique used for disruption of BcMETTL4. (B) PCR products with Mettl4-in-F/R (the first four lanes except Marker) or Mettl4-out-F/R (the last four lanes) from the genomic DNAs of wild-type (lanes 1 and 5) and ΔBcMettl4 (three different mutants).
Supplementary Figure 2 | Work procedure for BcDAMT deletion. (A) Marker exchange technique used for disruption of BcDAMT. (B) PCR products with DAMT-in-F/R (the first four lanes except Marker) or DAMT-out-F/R (the last four lanes) from the genomic DNAs of wild-type (lanes 1 and 5) and ΔBcDamt (three different mutants).
Amselem, J., Cuomo, C. A., van Kan, J. A., Viaud, M., Benito, E. P., Couloux, A., et al. (2011). Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 7:e1002230. doi: 10.1371/journal.pgen.1002230
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Beh, L. Y., Debelouchina, G. T., Clay, D. M., Thompson, R. E., Lindblad, K. A., Hutton, E. R., et al. (2019). Identification of a DNA N6-adenine methyltransferase complex and its impact on chromatin organization. Cell 177, 1781–1796.e25. doi: 10.1016/j.cell.2019.04.028
Blow, M. J., Clark, T. A., Daum, C. G., Deutschbauer, A. M., Fomenkov, A., Fries, R., et al. (2016). The epigenomic landscape of prokaryotes. PLoS Genet. 12:e1005854. doi: 10.1371/journal.pgen.1005854
Bujnicki, J. M., Feder, M., Radlinska, M., and Blumenthal, R. M. (2002). Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol. 55, 431–444. doi: 10.1007/s00239-002-2339-8
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Chen, H., Shu, H., Wang, L., Zhang, F., Li, X., Ochola, S. O., et al. (2018). Phytophthora methylomes are modulated by 6mA methyltransferases and associated with adaptive genome regions. Genome Biol. 19:181. doi: 10.1186/s13059-018-1564-4
Cheval, C., Aldon, D., Galaud, J. P., and Ranty, B. (2013). Calcium/calmodulin-mediated regulation of plant immunity. Biochim. Biophys. Acta 1833, 1766–1771. doi: 10.1016/j.bbamcr.2013.01.031
Dean, R., Van Kan, J. A., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Fu, Y., Dominissini, D., Rechavi, G., and He, C. (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306. doi: 10.1038/nrg3724
Fu, Y., Luo, G. Z., Chen, K., Deng, X., Yu, M., Han, D., et al. (2015). N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161, 879–892. doi: 10.1016/j.cell.2015.04.010
Greer, E. L., Blanco, M. A., Gu, L., Sendinc, E., Liu, J., Aristizabal-Corrales, D., et al. (2015). DNA methylation on N6-adenine in C. elegans. Cell 161, 868–878. doi: 10.1016/j.cell.2015.04.005
Gronover, C. S., Kasulke, D., Tudzynski, P., and Tudzynski, B. (2001). The role of G protein alpha subunits in the infection process of the gray mold fungus Botrytis cinerea. Mol. Plant Microbe Interact. 14, 1293–1302. doi: 10.1094/mpmi.2001.14.11.1293
Heckman, K. L., and Pease, L. R. (2007). Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932. doi: 10.1038/nprot.2007.132
Iyer, L. M., Abhiman, S., and Aravind, L. (2011). Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 101, 25–104. doi: 10.1016/b978-0-12-387685-0.00002-0
Iyer, L. M., Zhang, D., and Aravind, L. (2016). Adenine methylation in eukaryotes: apprehending the complex evolutionary history and functional potential of an epigenetic modification. BioEssays 38, 27–40. doi: 10.1002/bies.201500104
Kaku, H., Nishizawa, Y., Ishii-Minami, N., Akimoto-Tomiyama, C., Dohmae, N., Takio, K., et al. (2006). Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc. Natl. Acad. Sci. U. S. A. 103, 11086–11091. doi: 10.1073/pnas.0508882103
Kubicek, C. P., Starr, T. L., and Glass, N. L. (2014). Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451. doi: 10.1146/annurev-phyto-102313-045831
Law, C. J., Maloney, P. C., and Wang, D. N. (2008). Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 62, 289–305. doi: 10.1146/annurev.micro.61.080706.093329
Liang, Z., Shen, L., Cui, X., Bao, S., Geng, Y., Yu, G., et al. (2018). DNA N(6)-adenine methylation in Arabidopsis thaliana. Dev. cell 45, 406–416.e3. doi: 10.1016/j.devcel.2018.03.012
Liu, T. (2014). Use model-based analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol. Biol. 1150, 81–95. doi: 10.1007/978-1-4939-0512-6_4
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Mondo, S. J., Dannebaum, R. O., Kuo, R. C., Louie, K. B., Bewick, A. J., LaButti, K., et al. (2017). Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 49, 964–968. doi: 10.1038/ng.3859
Mousavi-Derazmahalleh, M., Chang, S., Thomas, G., Derbyshire, M., Bayer, P. E., Edwards, D., et al. (2019). Prediction of pathogenicity genes involved in adaptation to a lupin host in the fungal pathogens Botrytis cinerea and Sclerotinia sclerotiorum via comparative genomics. BMC Genom. 20:385. doi: 10.1186/s12864-019-5774-2
Ren, W., Liu, N., Sang, C., Shi, D., Zhou, M., Chen, C., et al. (2018). The autophagy gene BcATG8 regulates the vegetative differentiation and pathogenicity of Botrytis cinerea. Appl. Environ. Microbiol. 84, e2455–e2417. doi: 10.1128/aem.02455-17
Schumacher, J. (2012). Tools for Botrytis cinerea: new expression vectors make the gray mold fungus more accessible to cell biology approaches. Fungal Genet. Biol. 49, 483–497. doi: 10.1016/j.fgb.2012.03.005
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Vasu, K., and Nagaraja, V. (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rew. 77, 53–72. doi: 10.1128/mmbr.00044-12
Vela-Corcía, D., Aditya Srivastava, D., Dafa-Berger, A., Rotem, N., Barda, O., and Levy, M. (2019). MFS transporter from Botrytis cinerea provides tolerance to glucosinolate-breakdown products and is required for pathogenicity. Nat. Commun. 10:2886. doi: 10.1038/s41467-019-10860-3
Wada, S., Cui, S., and Yoshida, S. (2019). Reactive Oxygen Species (ROS) Generation Is Indispensable for Haustorium Formation of the Root Parasitic Plant Striga hermonthica. Front. Plant Sci. 10:328. doi: 10.3389/fpls.2019.00328
Wang, G., Song, L., Bai, T., and Liang, W. (2020). BcSas2-Mediated Histone H4K16 Acetylation Is Critical for Virulence and Oxidative Stress Response of Botrytis cinerea. Mol. Plant Microbe Interact. 33, 1242–1251. doi: 10.1094/mpmi-06-20-0149-r
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wang, P., Doxtader, K. A., and Nam, Y. (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 63, 306–317. doi: 10.1016/j.molcel.2016.05.041
Wang, X., Li, Z., Zhang, Q., Li, B., Lu, C., Li, W., et al. (2018). DNA methylation on N6-adenine in lepidopteran Bombyx mori. Biochim. Biophys. Acta Gene Regul. Mech. [Epub ahead of print]. doi: 10.1016/j.bbagrm.2018.07.013
Wang, Y., Chen, X., Sheng, Y., Liu, Y., and Gao, S. (2017). N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res. 45, 11594–11606. doi: 10.1093/nar/gkx883
Wang, Y., Sheng, Y., Liu, Y., Zhang, W., Cheng, T., Duan, L., et al. (2019). A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription. Nucleic Acids Res. 47, 11771–11789. doi: 10.1093/nar/gkz1053
Woodcock, C. B., Horton, J. R., Zhang, X., Blumenthal, R. M., and Cheng, X. (2020). Beta class amino methyltransferases from bacteria to humans: evolution and structural consequences. Nucleic Acids Res. 48, 10034–10044. doi: 10.1093/nar/gkaa446
Xiao, C. L., Zhu, S., He, M., Chen, D., Zhang, Q., Chen, Y., et al. (2018). N(6)-methyladenine DNA modification in the human genome. Mol. Cell 71, 306–318.e7. doi: 10.1016/j.molcel.2018.06.015
Yan, L., Yang, Q., Jiang, J., Michailides, T. J., and Ma, Z. (2011). Involvement of a putative response regulator Brrg-1 in the regulation of sporulation, sensitivity to fungicides, and osmotic stress in Botrytis cinerea. Appl. Microbiol. Biotechnol. 90, 215–226. doi: 10.1007/s00253-010-3027-z
Yang, Q., Song, L., Miao, Z., Su, M., Liang, W., and He, Y. (2019). Acetylation of BcHpt lysine 161 regulates Botrytis cinerea sensitivity to fungicides, multistress adaptation and virulence. Front. Microbiol. 10:2965. doi: 10.3389/fmicb.2019.02965
Yang, Q., Yin, D., Yin, Y., Cao, Y., and Ma, Z. (2015). The response regulator BcSkn7 is required for vegetative differentiation and adaptation to oxidative and osmotic stresses in Botrytis cinerea. Mol. Plant Pathol. 16, 276–287. doi: 10.1111/mpp.12181
Yang, Q., Zhang, J., Hu, J., Wang, X., Lv, B., and Liang, W. (2018). Involvement of BcYak1 in the regulation of vegetative differentiation and adaptation to oxidative stress of Botrytis cinerea. Front. Microbiol. 9:281. doi: 10.3389/fmicb.2018.00281
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zhang, G., Huang, H., Liu, D., Cheng, Y., Liu, X., Zhang, W., et al. (2015). N6-methyladenine DNA modification in Drosophila. Cell 161, 893–906. doi: 10.1016/j.cell.2015.04.018
Zhang, Z., Li, H., Qin, G., He, C., Li, B., and Tian, S. (2016). The MADS-Box transcription factor Bcmads1 is required for growth, sclerotia production and pathogenicity of Botrytis cinerea. Sci. Rep. 6:33901. doi: 10.1038/srep33901
Keywords: Botrytis cinerea, 6mA, methyltransferase, MeDIP-Seq, virulence
Citation: Miao Z, Wang G, Shen H, Wang X, Gabriel DW and Liang W (2022) BcMettl4-Mediated DNA Adenine N6-Methylation Is Critical for Virulence of Botrytis cinerea. Front. Microbiol. 13:925868. doi: 10.3389/fmicb.2022.925868
Received: 22 April 2022; Accepted: 23 May 2022;
Published: 30 June 2022.
Edited by:
Qunqing Wang, Shandong Agricultural University, ChinaReviewed by:
Qiya Yang, Jiangsu University, ChinaCopyright © 2022 Miao, Wang, Shen, Wang, Gabriel and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenxing Liang, d2xpYW5nMUBxYXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.