
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 13 June 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.917588
 Yann Sévellec1
Yann Sévellec1 Eliette Ascencio2
Eliette Ascencio2 Pierre-Emmanuel Douarre1
Pierre-Emmanuel Douarre1 Benjamin Félix1Laurent Gal2Dominique Garmyn2
Benjamin Félix1Laurent Gal2Dominique Garmyn2 Laurent Guillier3
Laurent Guillier3 Pascal Piveteau4†
Pascal Piveteau4† Sophie Roussel1*†
Sophie Roussel1*†Listeria monocytogenes (Lm) is a ubiquitous bacterium that causes the serious foodborne illness listeriosis. Although soil is a primary reservoir and a central habitat for Lm, little information is available on the genetic features underlying the fitness of Lm strains in this complex habitat. The aim of this study was to identify (i) correlations between the strains fitness in soil, their origin and their phylogenetic position (ii) identify genetic markers allowing Lm to survive in the soil. To this end, we assembled a balanced panel of 216 Lm strains isolated from three major ecological compartments (outdoor environment, animal hosts, and food) and from 33 clonal complexes occurring worldwide. The ability of the 216 strains to survive in soil was tested phenotypically. Hierarchical clustering identified three phenotypic groups according to the survival rate (SR): phenotype 1 “poor survivors” (SR < 2%), phenotype 2 “moderate survivors” (2% < SR < 5%) and phenotype 3 “good survivors” (SR > 5%). Survival in soil depended neither on strains’ origin nor on their phylogenetic position. Genome-wide-association studies demonstrated that a greater number of genes specifically associated with a good survival in soil was found in lineage II strains (57 genes) than in lineage I strains (28 genes). Soil fitness was mainly associated with variations in genes (i) coding membrane proteins, transcription regulators, and stress resistance genes in both lineages (ii) coding proteins related to motility and (iii) of the category “phage-related genes.” The cumulative effect of these small genomic variations resulted in significant increase of soil fitness.
Listeria monocytogenes (Lm) is a facultative intracellular pathogen responsible for listeriosis, a serious foodborne disease affecting both humans and animals. Lm is a ubiquitous bacterium, which can be found in many habitats including soil, water, plants, animals, foodstuffs and humans. Farm animals are important reservoirs of Lm and they contribute to the circulation of Lm in the farm environment and to its transfer in soil through fecal shedding (Hurtado et al., 2017). Increasing amounts of data on the prevalence of Lm in wildlife suggest that various species of wild animals can act as reservoirs for Lm and may participate in its transfer to soil (Yoshida et al., 2000; Hellstrom et al., 2008; Gismervik et al., 2015; Weindl et al., 2016; Hydeskov et al., 2019; Parsons et al., 2019). Other routes of transfer of Lm to soil include organic fertilization and decaying plant material (McLaughlin et al., 2011).
Since the first discovery of occurrence of Lm in soil samples collected from farms in 1960 (Welshimer, 1960), Lm has been considered as a telluric bacterial species. Follow-up studies, listed in Vivant et al. (2013) have confirmed the presence of Lm in different soil types including uncultivated and cultivated soils and meadows. Soil may play a pivotal role in transmission of Lm to cultivated plants and farm animals, eventually leading to contamination of foodstuffs (Piveteau et al., 2011; Vivant et al., 2013; Kallipolitis et al., 2020). However, a deep understanding of the ecology of Lm in soil and of the contribution of intrinsic factors and intraspecific diversity to its fitness in this habitat is required for the management of Lm in food systems from the natural environment to the food chain.
Lm is a genetically heterogeneous species divided into 13 serotypes and three major phylogenetic lineages, of which lineages I and II are the most frequently encountered (Orsi et al., 2011). These two lineages group the serotypes most commonly associated with human listeriosis, including serotypes 1/2b and 4b (lineage I) and serotype 1/2a (lineage II). Most strains are grouped into major clonal complexes (CCs) that have descended from a common ancestor and accumulated differences among themselves by a predominantly mutational process (Ragon et al., 2008; Chenal-Francisque et al., 2011; Cantinelli et al., 2013; Haase et al., 2014). CCs evolve slowly over large temporal and geographic scales (Chenal-Francisque et al., 2011; Cantinelli et al., 2013).
Hypervirulent and hypovirulent CCs have been identified (Maury et al., 2016). Hyper virulent CCs such as for instance CC1, CC2, CC6, are those most likely to cause disease, in particular central nervous system or maternal-neonatal listeriosis (Maury et al., 2016) and account for the majority of human listeriosis outbreaks, sporadic human cases (Painset et al., 2019), and animals listeriosis (Dreyer et al., 2016; Papić et al., 2019). On the contrary, hypo virulent CCs, such as for instance CC9 and CC121, merely cause disease in highly immuno-compromised patients and show limited virulence in humanized mouse models (Maury et al., 2016). Many typing studies have indicated that the relative prevalence of CCs in animals, human clinical cases and in food is strikingly different (Henri et al., 2016; Felix et al., 2018; Painset et al., 2019). This suggests that some Lm strains thrive better in specific ecological compartments. To date, the link between the genetic diversity of Lm (lineage, serotype and CCs), the strain origin (food, animal, and environment) and Lm ability to survive in soil has yet to be investigated. From the point of view of food safety, investigating the fate of hyper virulent strains in agri-food systems once transferred in soil is of particular interest. This could lead to innovative measures of control and risk assessment.
Genome-wide association studies (GWAS) represent today powerful tools for the identification of associations between genomic elements and phenotypic properties. GWAS is a top-down approach that involves testing large numbers of genetic variants in a population of individual organisms with a given phenotype. GWAS dealing with food-related phenotypes was recently applied to Lm traits associated to cold, salt, acid, desiccation stresses (Hingston et al., 2017; Fritsch et al., 2019) adhesion to polystyrene (Mahoney et al., 2022) and persistence in dairy farms (Castro et al., 2021).
In this study, GWAS was performed to elucidate the phenotype-genotype relationships in Lm soil survival. We focused on a dataset of 216 strains selected to be representative of the major CCs or sequence types (STs) circulating between three major compartments of Lm: natural and farming environment, wild/farm animals and food. This panel was implemented to investigate genomic determinants of soil survival following three working hypotheses (i) soil fitness depends on the strain origin (ii) soil fitness depends on the phylogenetic position of the strains and (iii) soil fitness can be associated with specific genes.
The panel was composed of 216 Lm strains belonging to 33 clonal complexes from lineage I (n = 95) and lineage II (n = 121; Table 1 and Supplementary Table 1). The strains were collected over more than 30 years with most of them isolated after 2010, across 15 different European countries.
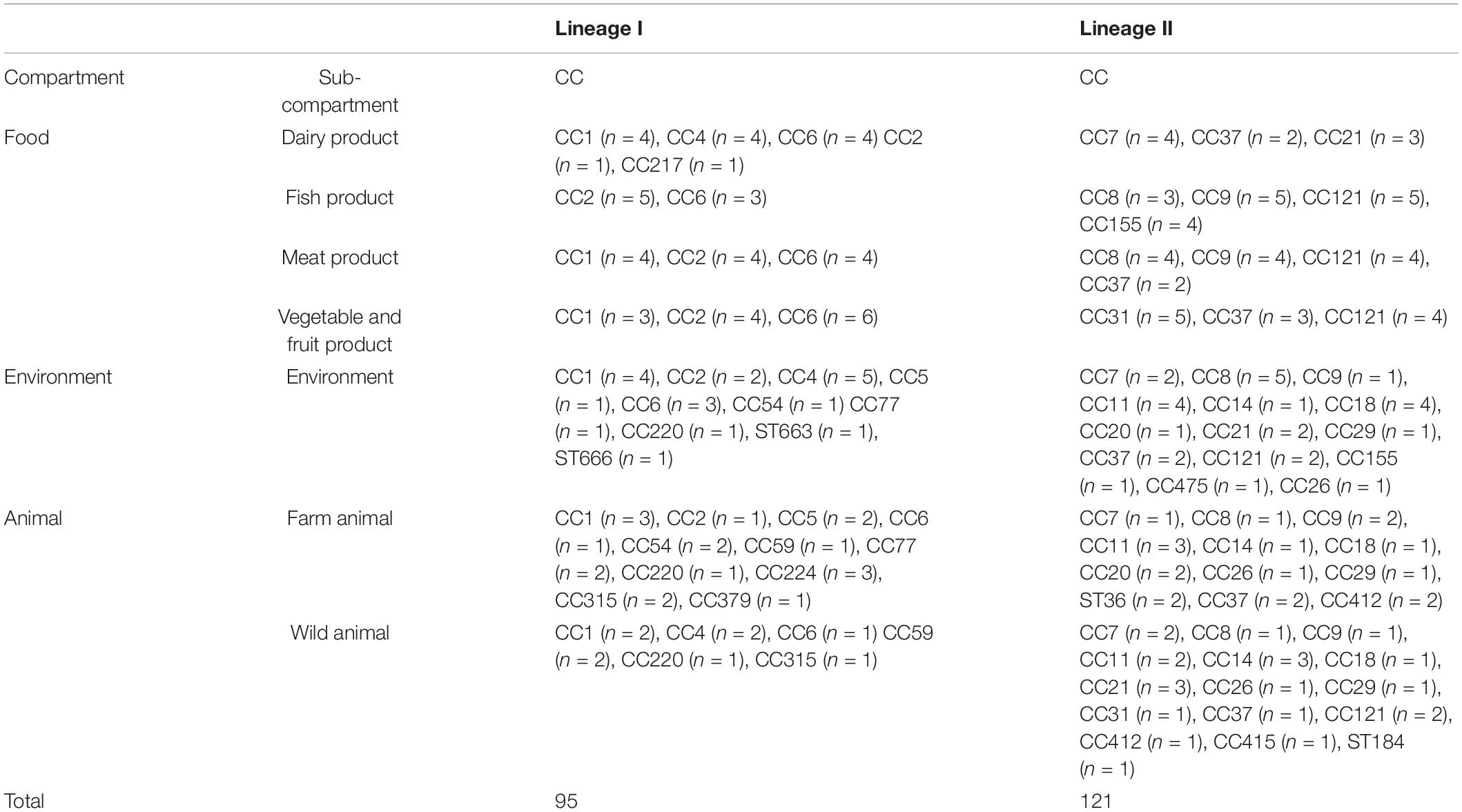
Table 1. Distribution of the 216 fully sequenced Listeria monocytogenes strains according to their origin, Lineage and clonal complex.
The 97 strains isolated from the environment and primary farm production were part of the strain collection (Félix et al., 2022) built in the frame of the European project ‘‘LISTADAPT’’ (Adaptive traits of Listeria monocytogenes to its diverse ecological niches).1 In all, strains were selected from farm animals (bovine, ovine, caprine, and pigs), wild animals (deer, badger, slugs, dolphin, wild boar, fox, and roe deer) and soil. They were selected to be representative of the major CCs or STs found in the LISTADAPT collection (Félix et al., 2022). In order to maximize the genetic diversity, at least two to four strains were selected per CC and ST with the greatest geographical breadth and the highest degree of ST diversity within each CC (Supplementary Table 1).
Additionally, 99 strains were selected from four ready-to-eat (RTE) food categories according to the risk food matrixes given by Painset et al. (2019). Of these, 11 strains were provided by the European LISEQ collection (Painset et al., 2019) and the rest of food strains were extracted from the ANSES Maisons-Alfort Laboratory for Food Safety collection of food isolates. The 10 major CCs circulating in secondary and tertiary food production were previously determined from two previous French studies (Maury et al., 2016; Felix et al., 2018) and one conducted in Europe (Painset et al., 2019). At least four strains collected from four different European countries and from as many STs as possible were selected within CC in order to maximize the genetic diversity (Supplementary Table 1).
Soil was collected from the upper superficial tillage layer at a depth of 0–10 cm in a field located in an experimental farm unit of INRAE (Bretenière, France). It is a silty clay soil containing 38% clay, 56% silt and 6% sand, with a pH of 6.9. Soil was sieved to 4 mm within 24 h after sampling then stored at –20°C for less than 1 month until use. Soil microcosms were prepared in 24-well microtiter plates by adding 0.5 g (equivalent dry weight) of soil per well. Plates were incubated 24 h at 25°C prior inoculation. Inocula were prepared from frozen stocks, by culturing in 10 mL of Trypton Soy Broth for 24 h at 25°C, then by sub-culturing (1%v/v) for 16 h under the same incubation conditions. Cells were washed twice in sterile distilled water then the final cell density was adjusted in sterile distilled water in order to inoculate microcosms at 106 CFU/g dry soil. The total volume of inoculum was calculated in order to adjust the soil water content to 60% of its Water Holding Capacity. Inoculated microcosms were incubated 36 h at 25°C. After incubation, 5 mL of Tryptone salt were added per soil microcosms. Soil slurries were serially diluted and Lm populations were numerated on Rapid’L mono (incubation 24–48 h at 37°C). The absence of indigenous Lm in soil was checked by direct plating on Rapid’L mono before inoculation. Experiments were performed in triplicates.
The strains were clustered based on their survival rate using the hclust R function (version 3.6.1) with the complete linkage method for hierarchical clustering. Ordinal logistic regression was used to explore the link between the strain ability to survive in soil and its origin. The polr R function was used to carry out the ordinal logistic regression.
The sequenced genomes were produced using paired-end sequencing (2 × 150 bp) on different Illumina instruments (NovaSeq 6000/NextSeq 500/Illumina HiSeq 2500). The raw reads were processed using the harmonized in-house workflow ARTWork.2 This pipeline performs various analyses (de novo assembly, scaffolding, annotation, quality control, inter-and intra-species contamination) that have been described in detail in previous studies (Vila Nova et al., 2019; Palma et al., 2020). Genome with a mean coverage < 30× and scaffolds with a length <200 bp were excluded. A cut-off for assembly was set at 200 contigs and an assembly length outside 2.7–3.2 Mbp. Sequence type (ST) and CC were also predicted based on the Listeria Multilocus ST (MLST) scheme (Moura et al., 2016). The accession numbers, the associated metadata and the metrics of the genome assemblies are provided in Supplementary Table 1.
Prediction of SNPs-Indels was performed using the Ivarcall2 pipeline (Felten et al., 2017) for alignment of the reads against the reference genome of Lm EGD-e (NC_003210.1). The phylogenetic tree was calculated on the pseudogenome based on genomic distance using iqtree V.1.6.9 (Schmidt et al., 2014). The best-fitted model for this dataset was determined to be a three-substitution type model with equal base parameter, empirical base frequencies (Kimura, 1981) and allowed proportion of invariant site (K3P + F + I). The tree was corrected for homologous recombination events using ClonalFrame (Didelot and Wilson, 2015). The genome of the L. innocua strain FR-FAR-WT-170 was used as outgroup to root the tree. The final recombination aware tree was visualized using iTOL (Letunic and Bork, 2019).
First, variable length K-mers were counted on all genomes using fsm-lite software with default parameters.3 The GWAS was then performed on the strain survival rate treated as a continuous trait using pyseer software (V.1.3.4) K-mer association study was applied on the full dataset (216 genomes) and three subsets including genomes from lineage I (n = 95), lineage II (n = 121) and genomes belonging to CC6 (n = 22).
Briefly, ClonalFrame inference (phylogeny tree) was used to generate a strain similarity matrix, based on the random effect model (phylogeny_distance.py script). The significance of the associations was determined according to their Bonferroni corrected p-value using the script count_pattern.py provided in the pyseer package: (i) full dataset p-value < 3.09–08 (ii) Lineage I p-value < 1.92–07 (iii) Lineage II p-value < 9.84–08) (iv) CC6 p-value < 9.00–03. K-mers significantly associated with soil fitness were then annotated on the reference genomes presenting the highest number of k-mers: LV-BOV_CP_29 for the whole collection (22/24) and the lineage II (2,148/2,242), DE-RDE-FE-17 for the lineage I (74/162) and FR-FI-U-UN-418 for the CC6 (2,243/3,056) (annotate_hits_pyseer script). The average maximum allele frequency (MAF) was determined by pyseer for each genes identified (Supplementary Table 2). The gene function was confirmed by BLAST analysis on the NCBI database.4 The PHASTER web server was used for the rapid identification of prophage sequences5 (Arndt et al., 2016, 2019).
In order to assess the association of soil phenotypes along the branches of the phylogenetic tree, the Bayesian inference method (Ansari and Didelot, 2016) was applied. The R package TreeBreaker was used in order to infer the evolution of a discrete phenotype distribution across a phylogenetic tree and to divide the tree into segments where their distributions are constant (Ansari and Didelot, 2016). Therefore, the core genome excluding recombination events and the discrete phenotypic traits (“fast” = 0, “slow” = 1) were supplied as input to this program.
The fate of the 216 strains was investigated during incubation in soil. The results were strain-dependent. Survival ranged from zero to 22% (Figure 1 and Supplementary Table 1). Hierarchical clustering grouped the strains into three discrete phenotypes, i.e., “poor,” “moderate,” and “good” according to their percent of survival in soil (Figure 1). For 54.4% of the strains, less than 2% of the initial population was detected after 36 h of incubation in soil (“poor survivors” = phenotype 1) and 36 strains had a survival rate of zero. Of these 36 strains, 12 were isolated from animal, 15 from food and 9 from environment. Thirty percent of the strains had survival rates between 2 and 5%. (“moderate survivors” = phenotype 2). A minority of strains (15.6%) had survival rates over 5% (“good survivors” = phenotype 3). The strain LV-BOV-CP-29 (CC11, Lineage II) isolated from animal presented the highest survival rate in soil.
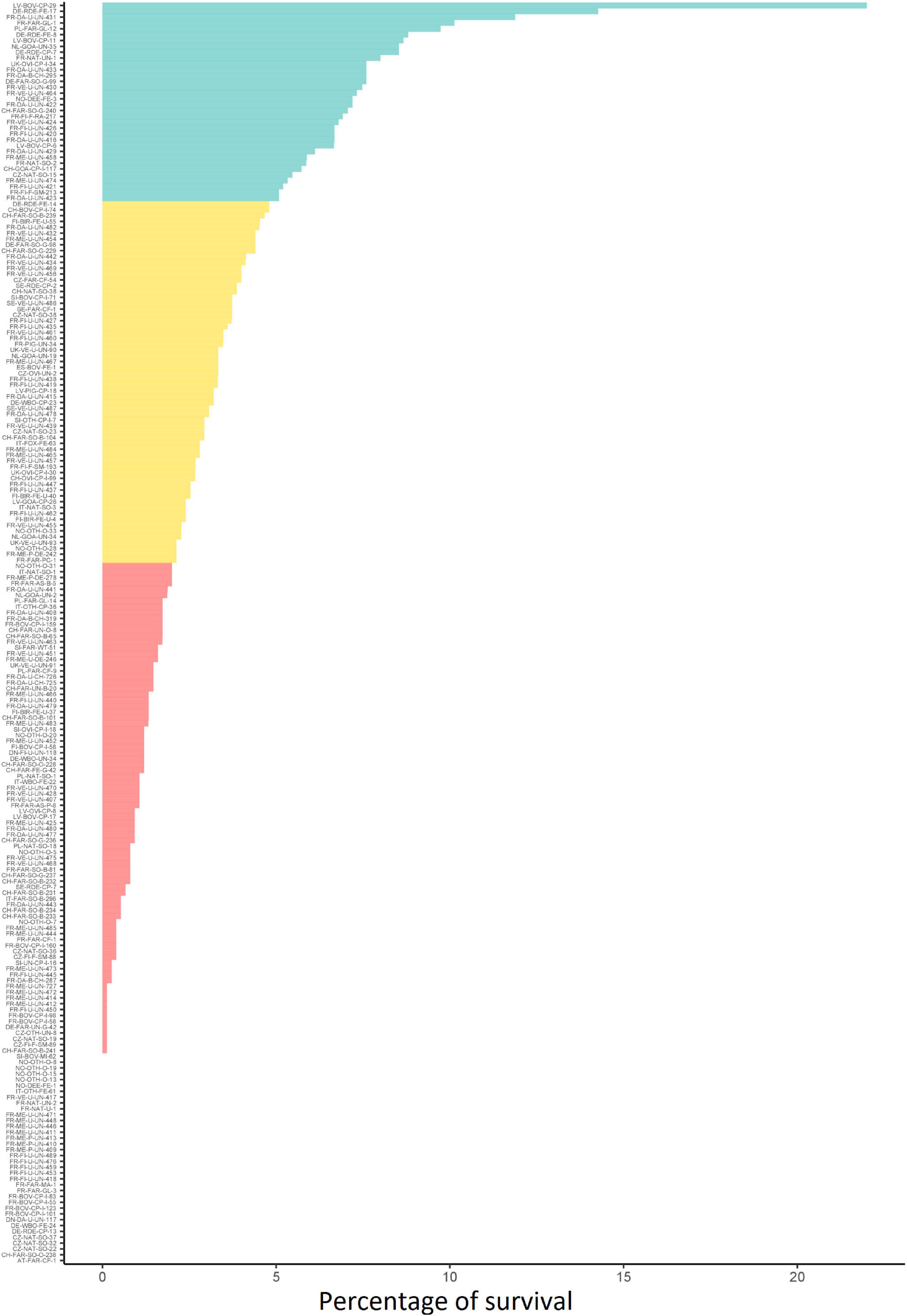
Figure 1. Soil survival phenotype of 216 strains tested. The three phenotypic classes of strain ability to survive in soil (blue = “good,” orange = “moderate” and red = “poor”) were determined by ascending hierarchical clustering.
Ordinal logistic regression of the phenotypes observed of the 216 strains was performed in order to investigate possible correlations between the phenotype and the origin of the strains. No significant correlation between the three ordered phenotypes characterizing strain’s potential to survive in soil and strain’s origin could be evidenced (Figure 2). Survival in soil does not depend of strain’s origin.
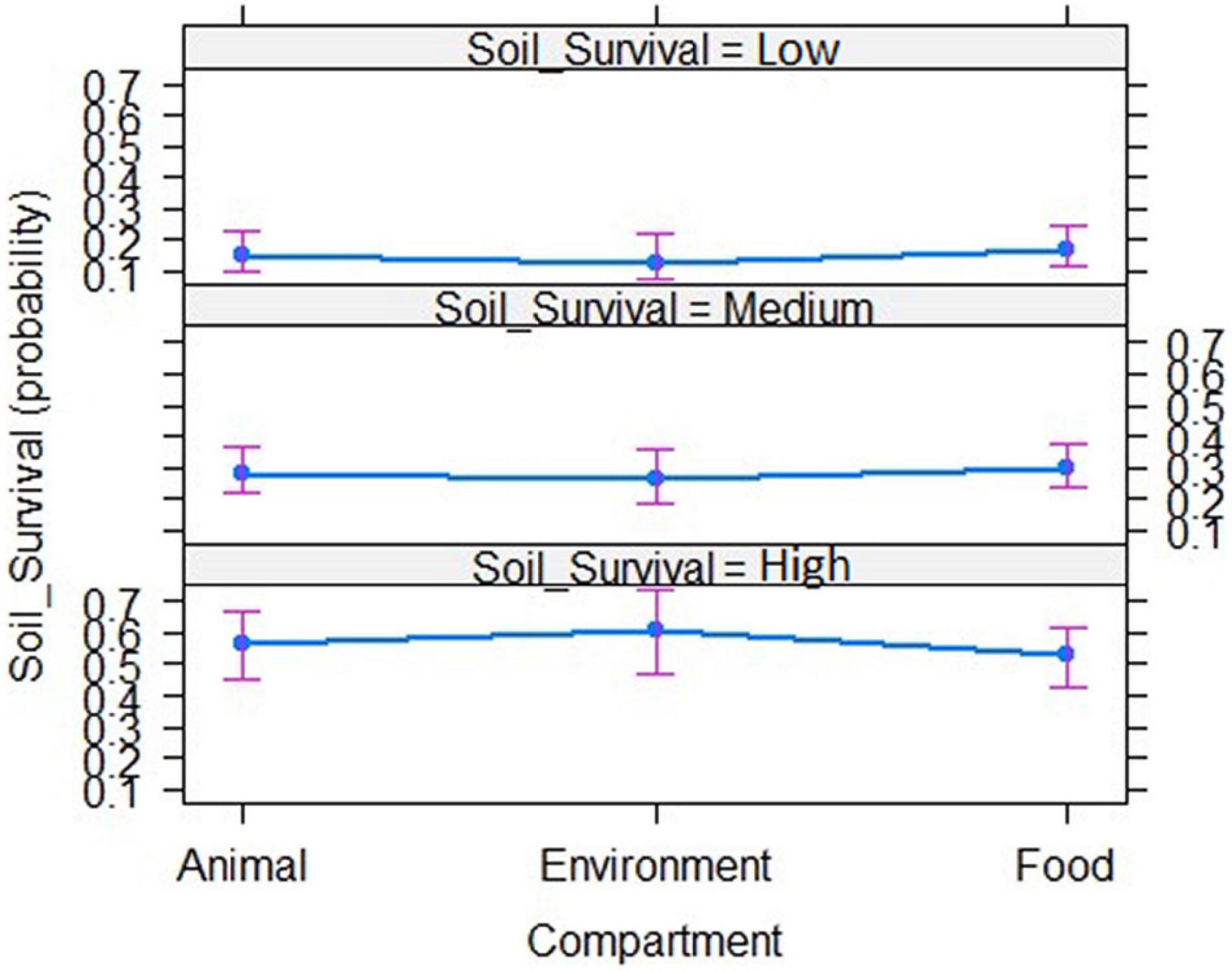
Figure 2. Ordinal logistic regression to assess correlations between the strain’s origin and its soil fitness from the phenotypic data collected from the 216 strains.
The distribution of the observed phenotypes within the phylogenetic tree based on the core genomes is presented in Figure 3. The three classes of phenotypes are randomly distributed on the tree leaves and analysis using Bayesian inference showed no stead. In addition, the posterior probabilities of phenotype changes on given branches (corresponding to Lineages, CC or any other clades) are not significant (Supplementary Figure 1). Survival in soil dos not depend of the phylogenetic position of strains.
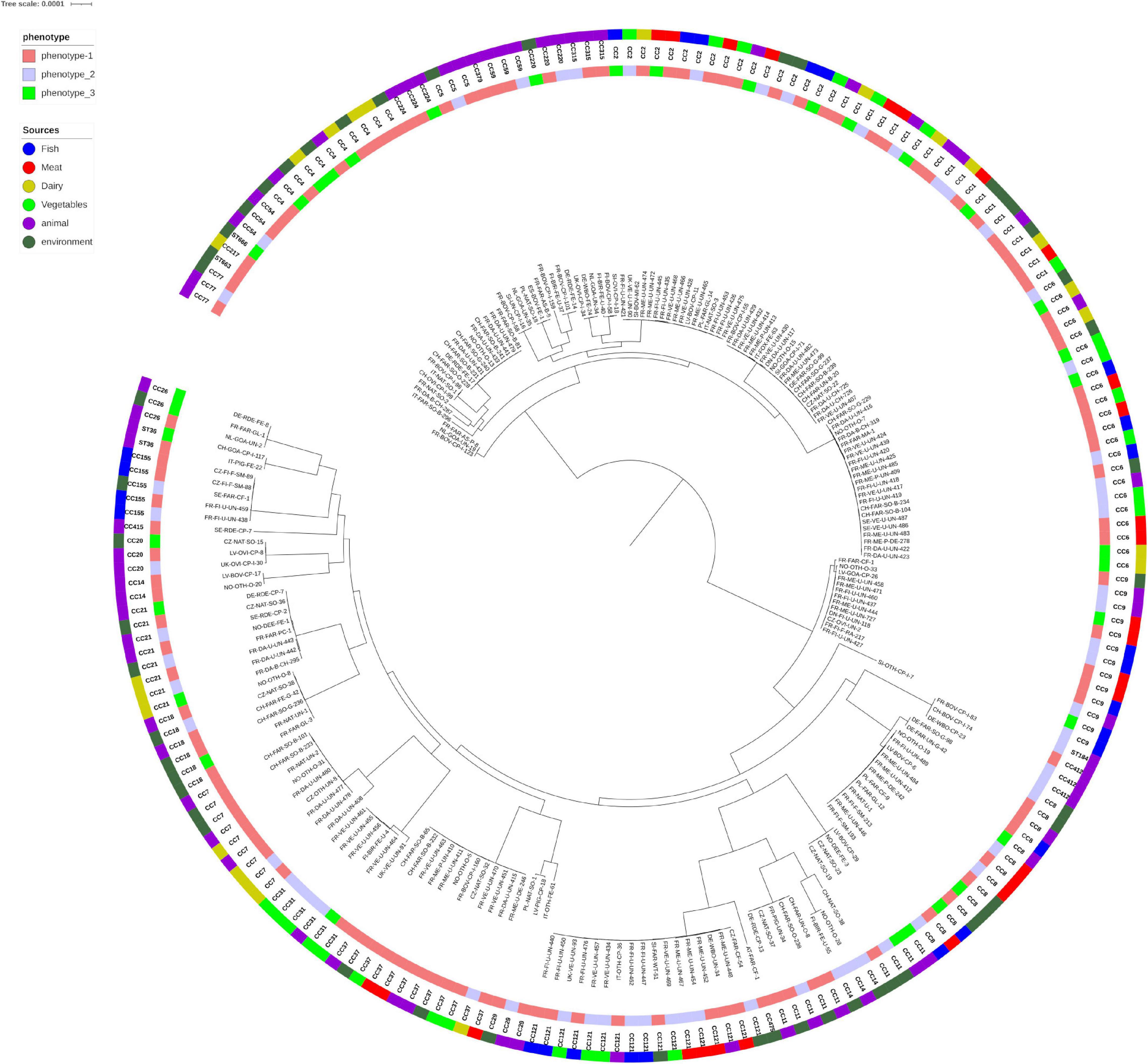
Figure 3. Phylogeny of the 216 Listeria monocytogenes genomes. The inner band indicates the phenotype of the strains and the outer band the different compartments and sub-compartments. The genomes clustered in two main branches corresponding to the lineage I and II. Inside each lineage, the strains were clustered according to their CC except for strains from CC14 (6 strains) and CC11 (9 strains) which present a polyphyletic structure between ST14 and ST91 and between ST 451 and ST11, respectively, (Figure 1). The pan-genome consisted of 2,384 core genes (plus a soft core of 126 genes present in more than 95% of the genomes).
The details of each gene and the number of significant k-mers associated with soil fitness are available in Supplementary Table 2.
Twenty-four k-mers, located in 11 genes, were associated with soil fitness (Figure 4 and Supplementary Table 2) with twenty two k-mers detected in the genome of strain LV-BOV-CP-29 (Strain presenting the highest survival rate 22% survival rate, lineage II, CC11). These 11 genes were present in few genomes (between three and six depending on the k-mer) with an average maximum allele frequency (MAF) between 0.014 and 0.0279. Interestingly, these genes were significantly associated with soil fitness specifically in Lineage II strains (Figure 4 and Supplementary Table 2).
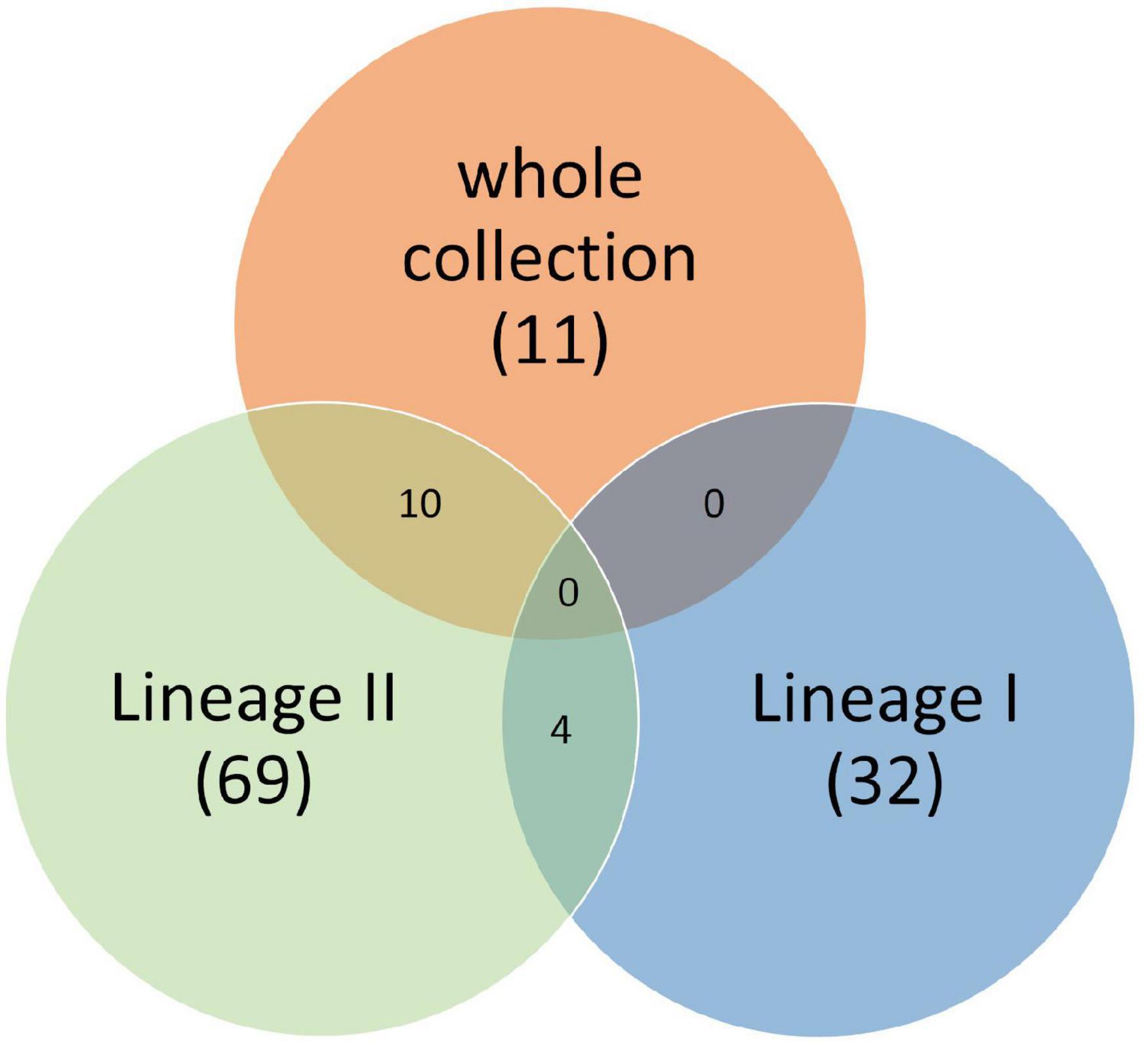
Figure 4. Venn diagram of genes significantly associated with soil survival extracted from the whole set of genomes, from the subset of Lineage I and from the subset of Lineage II.
Among the 11 genes identified, nine were significantly associated with high soil survival: the whiB gene, detected only in the best soil survivor strain (LV-BOV-CP-29) coding for a transcriptional regulator (Zheng et al., 2012) and eight genes annotated as prophage elements (Supplementary Table 2). Blast analysis of the LV-BOV-CP-29 genome revealed that these eight genes belong to a 38 Kb prophage integrated in the tRNA-Arg locus which shared 91% identity and 79% coverage with the listeria phage LP-101 (NC_024387.1) (Denes et al., 2014).
Additionally, two genes were significantly associated with poor soil survival: The rpoC gene codes for the beta subunit of the DNA-directed RNA polymerase and has been linked to resistance to cephalosporin and the HCFLCJGM_00555 gene encodes a hypothetical protein (Supplementary Table 2).
In lineage I, 162 k-mers covering 32 genes and presenting a low average MAF (between 0.02 and 0.07) were significantly associated with soil fitness (Figure 5 and Supplementary Table 2). Out of the 32 genes, 11 encode hypothetical proteins, four are transcription regulators including two regulators of stress response (PerR and SrlR), four genes are linked to Listeria monocytogenes motility (Figure 5 and Supplementary Table 2). Five genes coding membrane proteins were also identified, including four internalins (inlA, inlJ and two putative internalines) and two ABC transporters. Five genes belong to metabolism: Two of them are linked to resistance to oxidation (msrA and yqhD), two to cyanoamino acid metabolism (gmuD_1 and gmuD_2) and one related to fructose metabolism (malL_2). Finally, significant variation was identified in a putative anti-bacterial toxin gene. These 32 genes were not identified in the full dataset of 216 genomes (Figure 5).
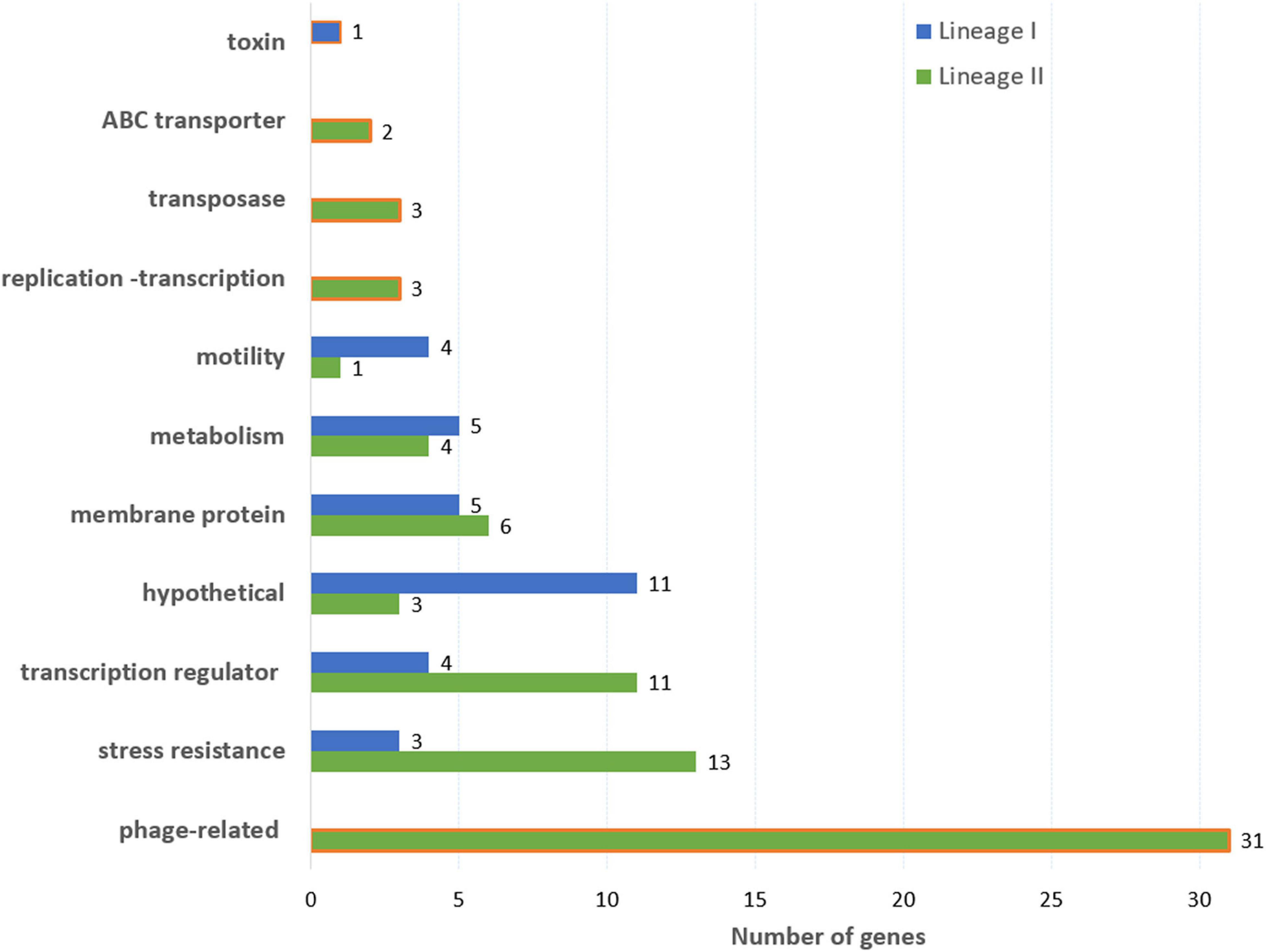
Figure 5. Function of genes associated with soil fitness in lineage I (95 genomes) and lineage II (121 genomes). The orange frame indicates that the gene function is only found in one lineage.
For four genes (msrA, IEOCHNDG_01235, gpx1 and mall_2) 88 k-mers detected were found in the genome of strains presenting poor soil survival. The 28 remaining genes were associated with high soil survival (Supplementary Table 2).
In lineage II, 2,243 k-mers covering 63 genes and 6 intergenic regions (Figure 5) were significantly associated with soil fitness. Most of these genes presented a low MAF (between 0.017 and 0.5 with only six genes presenting a MAF value above 0.1). 29 genes coding for phage elements (38 kb phage of LV-BOV-CP-29), 13 regulation factors (including three linked to stress resistance: perR, srlR, and ctsR), 12 stress resistance genes, 4 membrane proteins and the ackA gene associated with motility were detected. Notably, four of these genes (perR, cwlK, lytC, nagA) code for proteins involved in membrane metabolism or organization, especially in connection with growth promotion and stress induced membrane rearrangement.
The 69 genes detected for the lineage II include all the genes identified on the full dataset except HCFLCJGM_01110 (Figure 5) and four genes, srlR (coding a gluticol operon repressor), perR (coding a protein associated with peroxide resistance), iap (coding a secreted endopeptidase) and ycaD (coding a putative MFS transporter) also detected in lineage I.
For 57 genes, 95% of the k-mers identified were linked to high soil survival. For 12 genes, 94 k-mers were associated with poor soil survival. Among them, 43 k-mers were found in the gene HCFLCJGM_00555 and the intergenic region upstream this gene as well as 20 k-mers located in the msmE gene coding for an ABC transporter (Supplementary Table 2).
3,056 k-mers were identified as significantly associated with soil fitness in strains from CC6. These k-mers were located in 41 genes and 1 intergenic region (Supplementary Table 2). The majority of k-mers (2,310/3,056) were located in seven genes. Prokka_01766 is annotated as a pseudo gene corresponding to an HTH domain containing protein (706 k-mers). The ycaD gene is a putative MFS-transporter and a major salt resistance facilitator (457 k-mers). Prokka_01773 was annotated as a hypothetical protein (449 k-mers). ActA (189 k-mers) is coding a virulence factor. YlxH codes a flagellum site determining protein (172 k-mers). NemR codes a transcriptional regulator of the bile resistance efflux pump (170 k-mers). Finally, three genes (Prokka_01769, Prokka_01773 and Prokka_01774 encodes putative internalinprotein (81, 420, and 167 k-mers, respectively).
Most of the k-mers (2,242/3,056) were linked to poor soil survival. Seventeen prophage related genes were associated with poor soil fitness, of which five were related to phage LP-030-3 (NC_024384.1) (Supplementary Table 2). These five genes were detected in seven CC6 strains with poor soil survival and were also identified in other CCs that presented poor soil fitness such as CC7 strains (6/7 strains) and CC121 strains (11/18 strains).
Because little information is currently available on the fate of the various Clonal Complexes under conditions relevant to outdoor environments, we designed experiments on a panel of 216 strains representative of the overall genetic diversity and sources of Lm (natural environments, farm environments, wildlife, farm animals, food). The most prevalent CCs detected in an aggregated collection of outdoor environment strains were included in the panel of strains (Felix et al., 2018; Painset et al., 2019; Papić et al., 2019). Similarly, strains representative of the genetic diversity of Lm found in food were selected among the 10 most prevalent CCs found in 4 different food categories (Painset et al., 2019). To our knowledge, this is the first in-depth comparative investigation of a comprehensive collection of Lm strains grouping a balanced number of food and outdoor environment strains.
Survival in soil was used as proxy for the fitness of Lm in outdoor environments. Soil is a complex and heterogeneous environment in which bacteria may encounter a range of harsh conditions. Upon its arrival in soil, Lm has to face competition for resources and antibiosis depending on the characteristics and diversity of the soil microbiome (McLaughlin et al., 2011; Vivant et al., 2013; Bruce et al., 2017; Smith et al., 2018; Spor et al., 2020) and prophage infection (Williamson et al., 2007; Srinivasiah et al., 2008). Similarly, abiotic stress (nutrient starvation, temperature, osmotic variations and heavy metal exposure) may occur in soil (Oliver, 2010; Smith et al., 2018). Soil fitness is likely to require the contribution of a large array of genomic factors as suggested by deep transcriptome variations upon arrival of Lm in soil (Piveteau et al., 2011; Vivant et al., 2015).
In this strain panel, “poor” soil survivors were present in every CC, and no CC was strongly associated with “good” soil fitness. Information on the variability of Lm fitness in soil is scarce. However, a previous study suggested that survival in soil is strain-dependent (McLaughlin et al., 2011). In a longitudinal survey of farms and natural environments in North America, one genotype (Lineage II-Ribotype DUP-1039C) was significantly associated with both natural environments and farms (Sauders et al., 2006). This ribotype actually groups CC9, CC14, CC204, CC368, and CC436. However, in our study, the strains from CC9 and CC14 did not present higher survival rates in soil than strains from the other CCs of the dataset.
The panel of 216 genomes representative of Lm strains found in soil, animals and food was analyzed in order to obtain insight into the genetic determinants of Lm fitness in soil. We used genome-wide-association-studies in order to identify genomic markers linked to a phenotype. These studies using linear mixed models are a powerful tool to capture small variations underlying phenotypic differences (San et al., 2020). In the present work, genomic features significantly associated with soil fitness were identified through association studies. Only 11 genes were associated to the soil fitness of the full dataset of 216 genomes Our results confirm that soil fitness requires the contribution of several functions, and suggest that minor gene variations confer extra advantage when facing biotic and abiotic edaphic challenges. Strikingly, the number of gene variants found to be associated with survival in soil was greater in Lineage II strains (69) than in Lineage I strains (32). This is intriguing and calls for further investigations taking into account the evolutionary history of both Lineages.
While not detected in the dataset of 216 genomes, three transcription regulators associated with stress response were detected in both Lineages and were associated mostly with high soil fitness with only 6 hits out of 119 associated with poor soil fitness. The perR gene codes a putative peroxidase stress response regulator involved in H2O2 resistance and cell growth (Rea et al., 2005). The closely located ycaD gene, also linked to soil fitness in CC6 strains, codes a putative MFS transporter. This gene is known to be a major facilitator of salt resistance (Duché et al., 2002). Finally, srlR encodes a putative glucitol operon regulator involved in stress response as previously demonstrated for Lactobacillus paracasei (Palud et al., 2018). The variants of these genes were, however, different between Lineage I and II. This emphasizes how different genomic variations in key genes can induce similar effects.
In Lineage I and II alike, most of the variants connected to coping with environmental stresses were associated with transcription regulation, stress response, membrane protein and motility. As a feature of a ubiquitous bacterial species, a large percentage of the genome of Lm codes regulators (Glaser et al., 2001; den Bakker et al., 2013; Vivant et al., 2013). Response of Lm to the edaphic environmental factors requires transcription regulation and fine-tuning of gene expression (Piveteau et al., 2011; Vivant et al., 2015). In Lineage II strains, “good” soil fitness was linked to variations in the sequence of 11 transcription factors coding genes. Among these, ctsR encodes the class 3 stress gene repressor of Lm (Nair et al., 2000; Karatzas et al., 2003) and ackA is involved in chemotaxis and motility (Gueriri et al., 2008).
In Lineage I, flaA coding a flagelin and cheV involved in chemotaxis (Karatan et al., 2001) were associated with soil fitness. Likewise, in CC6 strains, multiple variants of ylxH, coding FlhF the protein involved in flagelin localization, were associated with high (46 k-mer) and poor (126 k-mer) soil fitness. The ctsR gene involved in flagellar transcription was associated with soil fitness in lineage II and in CC6 strains. Motility and chemotaxis associated genes are known to contribute to stress resistance in Listeria (Casey et al., 2014; Vivant et al., 2017) and are up-regulated in soil as a strategy to cope with harsh environments (Vivant et al., 2017).
Stress response is another important feature of the saprophytic lifestyle of Lm and the capacity to trigger the general stress response is required for survival in soil (Brunhede et al., 2020; Marinho et al., 2020). Our findings further support the importance of stress response in soil fitness. Interestingly, 13 stress resistance-coding genes in lineage II and five in lineage I were predicted to affect soil fitness. These genes include resistance to oxidative stress (cysK, uvrC in Lineage II; msrA and yqhD in Lineage I) (Duché et al., 2002; Hain et al., 2012; Liu et al., 2019; Patange et al., 2019) and resistance to antimicrobial agents (folP or rpoC) (Braschi et al., 2018; Wang et al., 2019). GWAS also highlighted significant associations with genes related to cell division and shape of the bacterial envelop. Altogether, these results show that the characteristics of the surface of the bacterium are a key feature of fitness in the edaphic environment, as differential transcriptome analysis suggested previously (Piveteau et al., 2011; Vivant et al., 2017).
GWAS underlined variations in genes coding internalins and uncharacterized membrane proteins as well. Two Internalin genes, inlJ and inlA were associated to high soil survival in lineages I,II and CC6 while three putative internalins were linked to poor soil fitness in the CC6. Some internalins such as inlL and inlA are involved in adherence and biofilm formation (Popowska et al., 2017) while the capacity to form biofilms improves soil survival (Salazar et al., 2013). Therefore, biofilm formation could be a decisive factor in soil survivability.
This study highlighted several phage-related genomic regions. In Lineage II strains, variations in the region of bacteriophage LP-101 correlated to soil fitness. Several genes from this phage region were detected, in particular a variant of yqbO. In Bacillus subtilis this gene codes a component of a defective prophage known to provide protection against other prophages (Klumpp and Loessner, 2013; Suzuki et al., 2020). It is likely that this prophage plays an important role in soil survival of Lineage II strains. On the contrary, 17 prophage-related genes presented variations specifically associated with “poor” soil fitness in CC6 strains. Most of them were associated with LP-030-3, a prophage integrated in comK. In Bacillus subtilis comK codes the master regulator required for expression of late competence genes. In Listeria monocytogenes, functional ComK is involved in stress regulation and phagosomal escape (Chatterjee et al., 2006; Rabinovich et al., 2012). This region is considered a rapid adaptation island because the mosaic structure of the comK prophage region enables rapid recombination (Verghese et al., 2011). In particular, excision of prophage A118 from the comK region restores expression of ComK, without entering the lytic phase (Rabinovich et al., 2012). Unfortunately, to date investigations on the role of LP-030-3-like prophages at this locus are lacking.
In this study, a panel of strains was built to cover the genomic diversity of Lm found in natural environments, animals and food. GWAS was performed on this panel in order to investigate the genomic features associated with the fitness of Lm in soil. Not surprisingly, fitness in this complex heterogeneous environment depends on the performance of a combination of several cellular processes. The synergy between several genomic variations could enhance the overall fitness of Lm clones among genetically related strains as well as between strains that are more distant. Several genomic variants of genes related to cell surface, transcription regulation and stress response were identified as significantly associated with soil fitness in our genetically diverse panel of Lm strains. This investigation also highlighted the importance of prophages in soil fitness but effects were either beneficial or detrimental according to the nature of the phage.
To gain a better understanding of the contributing effects of each of the genetic factors identified in this study, it would be of interest to investigate a larger genomic panel. It would then be possible to test the predictive power of these factors possibly by applying machine learning. The more promising factors could then be investigated by transcriptome analysis and reverse genetics.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YS contributed substantially to the study design, acquisition of strains and the corresponding genomes, data analysis, quality control of the genomic dataset as well as writing and editing the manuscript. EA was in charge of all to the phenotypic experiments and contributed to writing and revision of the manuscript. P-ED contributed substantially to the data analysis, as well as writing and editing the manuscript. BF contributed to the acquisition and selection of strains. LGa and DG contributed to the design and analysis of the phenotypic experiments. LGu was in charge of statistical analysis, selection of strains for sequencing and helped in drafting the manuscript. PP participated in the study design, provided strains from soil and designed and coordinated the study, and contributed to writing and editing of the manuscript. SR designed and coordinated the study and contributed to writing and editing of the manuscript. All authors read and approved the final manuscript.
This work was supported by the One Health European Joint Programme-European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement No. 773830).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Anna Oeverman (Department of Clinical Research and Veterinary Public Health, Vetsuisse Faculty, University of Bern, Switzerland) for providing 34 strains, isolated from a ruminant farm environment. We also thank Dr. Bojan Papic (Institute of Microbiology and Parasitology, Veterinary Faculty, University of Ljubljana, Slovenia) for providing 12 strains, isolated from farm animal autopsies and soil and six strains, isolated from farm or wild animal autopsies. We also thank Karol Romero and Karine Capitaine from ANSES, Laboratory for Food Safety, SEL Unit, responsible for sequencing of a substantial part of the strains, in collaboration with the Brain and Spine Institute sequencing platform.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.917588/full#supplementary-material
Supplementary Figure 1 | Evolution of a discrete soil phenotype distribution blue dot = “good,” orange dot = “moderate” and red dot = “poor”) on a phylogenetic tree including the genomes of 216 Listeria monocytogenes strains. The thickness of the branches is proportional to the posterior probability of having a change point. No change point was identified by treeBreaker. This shows that the three classes of phenotypes are randomly distributed on the tree leaves. The Bayesian inference method showed no steadiness distribution of phenotypes.
Supplementary Table 1 | Characteristics of the strain dataset.
Ansari, M. A., and Didelot, X. (2016). Bayesian Inference of the Evolution of a Phenotype Distribution on a Phylogenetic Tree. Genetics 204, 89–98. doi: 10.1534/genetics.116.190496
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arndt, D., Marcu, A., Liang, Y., and Wishart, D. S. (2019). PHAST, PHASTER and PHASTEST: tools for finding prophage in bacterial genomes. Brief. Bioinform. 20, 1560–1567. doi: 10.1093/bib/bbx121
Braschi, G., Serrazanetti, D. I., Siroli, L., Patrignani, F., De Angelis, M., and Lanciotti, R. (2018). Gene expression responses of Listeria monocytogenes Scott A exposed to sub-lethal concentrations of natural antimicrobials. Int. J. Food Microbiol. 286, 170–178. doi: 10.1016/j.ijfoodmicro.2018.07.026
Bruce, J. B., West, S. A., and Griffin, A. S. (2017). Bacteriocins and the assembly of natural Pseudomonas fluorescens populations. J. Evol. Biol. 30, 352–360. doi: 10.1111/jeb.13010
Brunhede, M. Z., Santos, P. T. D., Gal, L., Garmyn, D., Kallipolitis, B. H., and Piveteau, P. (2020). LisRK is required for optimal fitness of Listeria monocytogenes in soil. FEMS Microbiol. Lett. 367:fnaa188. doi: 10.1093/femsle/fnaa188
Cantinelli, T., Chenal-Francisque, V., Diancourt, L., Frezal, L., Leclercq, A., Wirth, T., et al. (2013). “Epidemic clones” of Listeria monocytogenes are widespread and ancient clonal groups. J. Clin. Microbiol. 51, 3770–3779. doi: 10.1128/JCM.01874-13
Casey, A., Fox, E. M., Schmitz-Esser, S., Coffey, A., McAuliffe, O., and Jordan, K. (2014). Transcriptome analysis of Listeria monocytogenes exposed to biocide stress reveals a multi-system response involving cell wall synthesis, sugar uptake, and motility. Front. Microbiol. 5:68. doi: 10.3389/fmicb.2014.00068
Castro, H., Douillard, F. P., Korkeala, H., and Lindstrom, M. (2021). Mobile Elements Harboring Heavy Metal and Bacitracin Resistance Genes Are Common among Listeria monocytogenes Strains Persisting on Dairy Farms. mSphere 6:e0038321. doi: 10.1128/mSphere.00383-21
Chatterjee, S. S., Hossain, H., Otten, S., Kuenne, C., Kuchmina, K., Machata, S., et al. (2006). Intracellular gene expression profile of Listeria monocytogenes. Infect. Immun. 74, 1323–1338. doi: 10.1128/IAI.74.2.1323-1338.2006
Chenal-Francisque, V., Lopez, J., Cantinelli, T., Caro, V., Tran, C., Leclercq, A., et al. (2011). Worldwide distribution of major clones of Listeria monocytogenes. Emerg. Infect. Dis. 17, 1110–1112. doi: 10.3201/eid/1706.101778
den Bakker, H. C., Desjardins, C. A., Griggs, A. D., Peters, J. E., Zeng, Q., Young, S. K., et al. (2013). Evolutionary Dynamics of the Accessory Genome of Listeria monocytogenes. PLoS One 8:e67511. doi: 10.1371/journal.pone.0067511
Denes, T., Vongkamjan, K., Ackermann, H. W., Moreno Switt, A. I., Wiedmann, M., and den Bakker, H. C. (2014). Comparative genomic and morphological analyses of Listeria phages isolated from farm environments. Appl. Environ. Microbiol. 80, 4616–4625. doi: 10.1128/AEM.00720-14
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient Inference of Recombination in Whole Bacterial Genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Dreyer, M., Aguilar-Bultet, L., Rupp, S., Guldimann, C., Stephan, R., Schock, A., et al. (2016). Listeria monocytogenes sequence type 1 is predominant in ruminant rhombencephalitis. Sci. Rep. 6:36419. doi: 10.1038/srep36419
Duché, O., Trémoulet, F., Glaser, P., and Labadie, J. (2002). Salt stress proteins induced in Listeria monocytogenes. Appl. Environ. Microbiol. 68, 1491–1498. doi: 10.1128/aem.68.4.1491-1498.2002
Felix, B., Feurer, C., Maillet, A., Guillier, L., Boscher, E., Kerouanton, A., et al. (2018). Population Genetic Structure of Listeria monocytogenes Strains Isolated From the Pig and Pork Production Chain in France. Front. Microbiol. 9:684. doi: 10.3389/fmicb.2018.00684
Félix, B., Sevellec, Y., Palma, F., Douarre, P.-E., Felten, A., Radomski, N., et al. (2022). A European-wide dataset to decipher adaptation mechanisms of Listeria monocytogenes to diverse ecological niches. Sci. Data 9:190. doi: 10.1038/s41597-022-01278-6
Felten, A., Vila Nova, M., Durimel, K., Guillier, L., Mistou, M.-Y., and Radomski, N. (2017). First gene-ontology enrichment analysis based on bacterial coregenome variants: insights into adaptations of Salmonella serovars to mammalian- and avian-hosts. BMC Microbiol. 17:222. doi: 10.1186/s12866-017-1132-1
Fritsch, L., Felten, A., Palma, F., Mariet, J.-F., Radomski, N., Mistou, M.-Y., et al. (2019). Insights from genome-wide approaches to identify variants associated to phenotypes at pan-genome scale: application to L. monocytogenes’ ability to grow in cold conditions. Int. J. Food Microbiol. 291, 181–188. doi: 10.1016/j.ijfoodmicro.2018.11.028
Gismervik, K., Aspholm, M., Rorvik, L. M., Bruheim, T., Andersen, A., and Skaar, I. (2015). Invading slugs (Arion vulgaris) can be vectors for Listeria monocytogenes. J. Appl. Microbiol. 118, 809–816. doi: 10.1111/jam.12750
Glaser, P., Frangeul, L., Buchrieser, C., Rusniok, C., Amend, A., Baquero, F., et al. (2001). Comparative genomics of Listeria species. Science 294, 849–852. doi: 10.1126/science.1063447
Gueriri, I., Bay, S., Dubrac, S., Cyncynatus, C., and Msadek, T. (2008). The Pta-AckA pathway controlling acetyl phosphate levels and the phosphorylation state of the DegU orphan response regulator both play a role in regulating Listeria monocytogenes motility and chemotaxis. Mol. Microbiol. 70, 1342–1357. doi: 10.1111/j.1365-2958.2008.06496.x
Haase, J. K., Didelot, X., Lecuit, M., Korkeala, H., and Achtman, M. (2014). The ubiquitous nature of Listeria monocytogenes clones: a large-scale Multilocus Sequence Typing study. Environ. Microbiol. 16, 405–416. doi: 10.1111/1462-2920.12342
Hain, T., Ghai, R., Billion, A., Kuenne, C. T., Steinweg, C., Izar, B., et al. (2012). Comparative genomics and transcriptomics of lineages I, II, and III strains of Listeria monocytogenes. BMC Genom. 13:144. doi: 10.1186/1471-2164-13-144
Hellstrom, S., Kiviniemi, K., Autio, T., and Korkeala, H. (2008). Listeria monocytogenes is common in wild birds in Helsinki region and genotypes are frequently similar with those found along the food chain. J. Appl. Microbiol. 104, 883–888. doi: 10.1111/j.1365-2672.2007.03604.x
Henri, C., Felix, B., Guillier, L., Leekitcharoenphon, P., Michelon, D., Mariet, J. F., et al. (2016). Population Genetic Structure of Listeria monocytogenes Strains as Determined by Pulsed-Field Gel Electrophoresis and Multilocus Sequence Typing. Appl. Environ. Microbiol. 82, 5720–5728. doi: 10.1128/aem.00583-16
Hingston, P., Chen, J., Dhillon, B. K., Laing, C., Bertelli, C., Gannon, V., et al. (2017). Genotypes Associated with Listeria monocytogenes Isolates Displaying Impaired or Enhanced Tolerances to Cold, Salt, Acid, or Desiccation Stress. Front. Microbiol. 8:369. doi: 10.3389/fmicb.2017.00369
Hurtado, A., Ocejo, M., and Oporto, B. (2017). Salmonella spp. and Listeria monocytogenes shedding in domestic ruminants and characterization of potentially pathogenic strains. Vet. Microbiol. 210, 71–76. doi: 10.1016/j.vetmic.2017.09.003
Hydeskov, H. B., Amar, C. F. L., Fernandez, J. R., John, S. K., Macgregor, S. K., Cunningham, A. A., et al. (2019). Listeria Monocytogenes Infection of Free-Living Western European Hedgehogs (Erinaceus Europaeus). J. Zoo Wildlife Med. 50, 183–189. doi: 10.1638/2018-0093
Kallipolitis, B., Gahan, C. G. M., and Piveteau, P. (2020). Factors contributing to Listeria monocytogenes transmission and impact on food safety. Curr. Opin. Food Sci. 36, 9–17. doi: 10.1016/j.cofs.2020.09.009
Karatan, E., Saulmon, M. M., Bunn, M. W., and Ordal, G. W. (2001). Phosphorylation of the response regulator CheV is required for adaptation to attractants during Bacillus subtilis chemotaxis. J. Biol. Chem. 276, 43618–43626. doi: 10.1074/jbc.m104955200
Karatzas, K. A., Wouters, J. A., Gahan, C. G., Hill, C., Abee, T., and Bennik, M. H. (2003). The CtsR regulator of Listeria monocytogenes contains a variant glycine repeat region that affects piezotolerance, stress resistance, motility and virulence. Mol. Microbiol. 49, 1227–1238. doi: 10.1046/j.1365-2958.2003.03636.x
Kimura, M. (1981). Estimation of evolutionary distances between homologous nucleotide sequences. Proc. Natl. Acad. Sci. U.S.A. 78, 454–458. doi: 10.1073/pnas.78.1.454
Klumpp, J., and Loessner, M. J. (2013). Listeria phages: genomes, evolution, and application. Bacteriophage 3:e26861. doi: 10.4161/bact.26861
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Liu, Y., Orsi, R. H., Gaballa, A., Wiedmann, M., Boor, K. J., and Guariglia-Oropeza, V. (2019). Systematic review of the Listeria monocytogenes σB regulon supports a role in stress response, virulence and metabolism. Future Microbiol. 14, 801–828. doi: 10.2217/fmb-2019-0072
Mahoney, D. B. J., Falardeau, J., Hingston, P., Chmielowska, C., Carroll, L. M., Wiedmann, M., et al. (2022). Associations between Listeria monocytogenes genomic characteristics and adhesion to polystyrene at 8 degrees C. Food Microbiol. 102:103915. doi: 10.1016/j.fm.2021.103915
Marinho, C. M., Garmyn, D., Gal, L., Brunhede, M. Z., O’Byrne, C., and Piveteau, P. (2020). Investigation of the roles of AgrA and σB regulators in Listeria monocytogenes adaptation to roots and soil. FEMS Microbiol. Lett. 367:fnaa036. doi: 10.1093/femsle/fnaa036
Maury, M. M., Tsai, Y. H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., et al. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 48, 308–313. doi: 10.1038/ng.3501
McLaughlin, H. P., Casey, P. G., Cotter, J., Gahan, C. G., and Hill, C. (2011). Factors affecting survival of Listeria monocytogenes and Listeria innocua in soil samples. Arch. Microbiol. 193, 775–785. doi: 10.1007/s00203-011-0716-7
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2016). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2:16185. doi: 10.1038/nmicrobiol.2016.185
Nair, S., Derré, I., Msadek, T., Gaillot, O., and Berche, P. (2000). CtsR controls class III heat shock gene expression in the human pathogen Listeria monocytogenes. Mol. Microbiol. 35, 800–811. doi: 10.1046/j.1365-2958.2000.01752.x
Oliver, J. D. (2010). Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol. Rev. 34, 415–425. doi: 10.1111/j.1574-6976.2009.00200.x
Orsi, R. H., Bakker, H. C., and Wiedmann, M. (2011). Listeria monocytogenes lineages: genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 301, 79–96. doi: 10.1016/j.ijmm.2010.05.002
Painset, A., Björkman, J. T., Kiil, K., Guillier, L., Mariet, J.-F., Félix, B., et al. (2019). LiSEQ – whole-genome sequencing of a cross-sectional survey of Listeria monocytogenes in ready-to-eat foods and human clinical cases in Europe. Microb. Genom. 5:e000257. doi: 10.1099/mgen.0.000257
Palma, F., Brauge, T., Radomski, N., Mallet, L., Felten, A., Mistou, M.-Y., et al. (2020). Dynamics of mobile genetic elements of Listeria monocytogenes persisting in ready-to-eat seafood processing plants in France. BMC Genom. 21:130. doi: 10.1186/s12864-020-6544-x
Palud, A., Salem, K., Cavin, J.-F., Beney, L., and Licandro, H. (2018). Identification and expression of Lactobacillus paracasei genes for adaptation to desiccation and rehydration. bioRxiv [preprint]. doi: 10.1101/475830
Papić, B., Pate, M., Félix, B., and Kušar, D. (2019). Genetic diversity of Listeria monocytogenes strains in ruminant abortion and rhombencephalitis cases in comparison with the natural environment. BMC Microbiol. 19:299. doi: 10.1186/s12866-019-1676-3
Parsons, C., Niedermeyer, T., Gould, N., Brown, P., Strules, J., Parsons, A. W., et al. (2019). Listeria monocytogenes at the human–wildlife interface: black bears (Ursus americanus) as potential vehicles for Listeria. Microb. Biotechnol. 13, 706–721. doi: 10.1111/1751-7915.13509
Patange, A., O’Byrne, C., Boehm, D., Cullen, P. J., Keener, K., and Bourke, P. (2019). The Effect of Atmospheric Cold Plasma on Bacterial Stress Responses and Virulence Using Listeria monocytogenes Knockout Mutants. Front. Microbiol. 10:2841. doi: 10.3389/fmicb.2019.02841
Piveteau, P., Depret, G., Pivato, B., Garmyn, D., and Hartmann, A. (2011). Changes in gene expression during adaptation of Listeria monocytogenes to the soil environment. PLoS One 6:e24881. doi: 10.1371/journal.pone.0024881
Popowska, M., Krawczyk-Balska, A., Ostrowski, R., and Desvaux, M. (2017). InlL from Listeria monocytogenes Is Involved in Biofilm Formation and Adhesion to Mucin. Front. Microbiol. 8:660. doi: 10.3389/fmicb.2017.00660
Rabinovich, L., Sigal, N., Borovok, I., Nir-Paz, R., and Herskovits, A. A. (2012). Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150, 792–802. doi: 10.1016/j.cell.2012.06.036
Ragon, M., Wirth, T., Hollandt, F., Lavenir, R., Lecuit, M., Le Monnier, A., et al. (2008). A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 4:e1000146. doi: 10.1371/journal.ppat.1000146
Rea, R., Hill, C., and Gahan, C. G. M. (2005). Listeria monocytogenes PerR mutants display a small-colony phenotype, increased sensitivity to hydrogen peroxide, and significantly reduced murine virulence. Appl. Environ. Microbiol. 71, 8314–8322. doi: 10.1128/AEM.71.12.8314-8322.2005
Salazar, J. K., Wu, Z., Yang, W., Freitag, N. E., Tortorello, M. L., Wang, H., et al. (2013). Roles of a Novel Crp/Fnr Family Transcription Factor Lmo0753 in Soil Survival, Biofilm Production and Surface Attachment to Fresh Produce of Listeria monocytogenes. PLoS One 8:e75736. doi: 10.1371/journal.pone.0075736
San, J. E., Baichoo, S., Kanzi, A., Moosa, Y., Lessells, R., Fonseca, V., et al. (2020). Current Affairs of Microbial Genome-Wide Association Studies: approaches, Bottlenecks and Analytical Pitfalls. Front. Microbiol. 10:3119. doi: 10.3389/fmicb.2019.03119
Sauders, B. D., Durak, M. Z., Fortes, E., Windham, K., Schukken, Y., and Lembo, A. J. Jr., et al. (2006). Molecular characterization of Listeria monocytogenes from natural and urban environments. J. Food Protect. 69, 93–105. doi: 10.4315/0362-028x-69.1.93
Schmidt, H. A., Minh, B. Q., von Haeseler, A., and Nguyen, L.-T. (2014). IQ-TREE: a Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Smith, A., Moorhouse, E., Monaghan, J., Taylor, C., and Singleton, I. (2018). Sources and survival of Listeria monocytogenes on fresh, leafy produce. J. Appl. Microbiol. 125, 930–942. doi: 10.1111/jam.14025
Spor, A., Camargo, A. R. O., Bru, D., Gaba, S., Garmyn, D., Gal, L., et al. (2020). Habitat Disturbances Modulate the Barrier Effect of Resident Soil Microbiota on Listeria monocytogenes Invasion Success. Front. Microbiol. 11:927. doi: 10.3389/fmicb.2020.00927
Srinivasiah, S., Bhavsar, J., Thapar, K., Liles, M., Schoenfeld, T., and Wommack, K. E. (2008). Phages across the biosphere: contrasts of viruses in soil and aquatic environments. Res. Microbiol. 159, 349–357. doi: 10.1016/j.resmic.2008.04.010
Suzuki, S., Yoshikawa, M., Imamura, D., Abe, K., Eichenberger, P., and Sato, T. (2020). Compatibility of Site-Specific Recombination Units between Mobile Genetic Elements. iScience 23:100805. doi: 10.1016/j.isci.2019.100805
Verghese, B., Lok, M., Wen, J., Alessandria, V., Chen, Y., Kathariou, S., et al. (2011). comK prophage junction fragments as markers for Listeria monocytogenes genotypes unique to individual meat and poultry processing plants and a model for rapid niche-specific adaptation, biofilm formation, and persistence. Appl. Environ. Microbiol. 77, 3279–3292. doi: 10.1128/AEM.00546-11
Vila Nova, M., Durimel, K., La, K., Felten, A., Bessières, P., Mistou, M.-Y., et al. (2019). Genetic and metabolic signatures of Salmonella enterica subsp. enterica associated with animal sources at the pangenomic scale. BMC Genom. 20:814. doi: 10.1186/s12864-019-6188-x
Vivant, A. L., Desneux, J., Pourcher, A. M., and Piveteau, P. (2017). Transcriptomic Analysis of the Adaptation of Listeria monocytogenes to Lagoon and Soil Matrices Associated with a Piggery Environment: comparison of Expression Profiles. Front. Microbiol. 8:1811. doi: 10.3389/fmicb.2017.01811
Vivant, A. L., Garmyn, D., Gal, L., Hartmann, A., and Piveteau, P. (2015). Survival of Listeria monocytogenes in Soil Requires AgrA-Mediated Regulation. Appl. Environ. Microbiol. 81, 5073–5084. doi: 10.1128/AEM.04134-14
Vivant, A. L., Garmyn, D., and Piveteau, P. (2013). Listeria monocytogenes, a down-to-earth pathogen. Front. Cell Infect. Microbiol. 3:87. doi: 10.3389/fcimb.2013.00087
Wang, Y., Li, X., Osmundson, T., Shi, L., and Yan, H. (2019). Comparative Genomic Analysis of a Multidrug-Resistant Listeria monocytogenes ST477 Isolate. Foodborne Pathog. Dis. 16, 604–615. doi: 10.1089/fpd.2018.2611
Weindl, L., Frank, E., Ullrich, U., Heurich, M., Kleta, S., Ellerbroek, L., et al. (2016). Listeria monocytogenes in Different Specimens from Healthy Red Deer and Wild Boars. Foodborne Pathog. Dis. 13, 391–397. doi: 10.1089/fpd.2015.2061
Welshimer, H. J. (1960). Survival of Listeria monocytogenes in soil. J. Bacteriol. 80, 316–320. doi: 10.1128/JB.80.3.316-320.1960
Williamson, K. E., Radosevich, M., Smith, D. W., and Wommack, K. E. (2007). Incidence of lysogeny within temperate and extreme soil environments. Environ. Microbiol. 9, 2563–2574. doi: 10.1111/j.1462-2920.2007.01374.x
Yoshida, T., Sugimoto, T., Sato, M., and Hirai, K. (2000). Incidence of Listeria monocytogenes in wild animals in Japan. J. Vet. Med. Sci. 62, 673–675. doi: 10.1292/jvms.62.673
Keywords: Listeria monocytogenes, soil, fitness, survival, ecotype, strain origin, clonal complex, genome-wide-association study
Citation: Sévellec Y, Ascencio E, Douarre P-E, Félix B, Gal L, Garmyn D, Guillier L, Piveteau P and Roussel S (2022) Listeria monocytogenes: Investigation of Fitness in Soil Does Not Support the Relevance of Ecotypes. Front. Microbiol. 13:917588. doi: 10.3389/fmicb.2022.917588
Received: 11 April 2022; Accepted: 17 May 2022;
Published: 13 June 2022.
Edited by:
Lin Lin, Jiangsu University, ChinaReviewed by:
Renato Hohl Orsi, Cornell University, United StatesCopyright © 2022 Sévellec, Ascencio, Douarre, Félix, Gal, Garmyn, Guillier, Piveteau and Roussel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Roussel, c29waGllLnJvdXNzZWxAYW5zZXMuZnI=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.