
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 July 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.917553
This article is part of the Research Topic Advanced Microbial Engineering and Biohybrid Technologies for Waste Biorefinery View all 7 articles
 Zivile Kondrotaite
Zivile Kondrotaite Laura C. Valk
Laura C. Valk Francesca Petriglieri
Francesca Petriglieri Caitlin Singleton
Caitlin Singleton Marta Nierychlo
Marta Nierychlo Morten K. D. Dueholm
Morten K. D. Dueholm Per H. Nielsen*
Per H. Nielsen*The Saprospiraceae family within the phylum Bacteroidota is commonly present and highly abundant in wastewater treatment plants (WWTPs) worldwide, but little is known about its role. In this study, we used MiDAS 4 global survey with samples from 30 countries to analyze the abundance and distribution of members of Saprospiraceae. Phylogenomics were used to delineate five new genera from a set of 31 high-quality metagenome-assembled genomes from Danish WWTPs. Newly designed probes for fluorescence in situ hybridization (FISH) revealed rod-shaped morphologies for all genera analyzed, including OLB8, present mostly inside the activated sludge flocs. The genomes revealed potential metabolic capabilities for the degradation of polysaccharides, proteins, and other complex molecules; partial denitrification; and storage of intracellular polymers (glycogen, polyphosphate, and polyhydroxyalkanoates). FISH in combination with Raman microspectroscopy confirmed the presence of intracellular glycogen in Candidatus Brachybacter, Candidatus Parvibacillus calidus (both from the former genus OLB8), and Candidatus Opimibacter, and the presence of polyhydroxyalkanoates in Candidatus Defluviibacterium haderslevense and Candidatus Vicinibacter. These results provide the first overview of the most abundant novel Saprospiraceae genera present in WWTPs across the world and their potential involvement in nutrient removal and the degradation of macromolecules.
Wastewater treatment is an essential process that protects humans and the environment from anthropogenic waste, and currently, it is shifting toward resource recovery and sustainability (Loosdrecht et al., 2016; Qasim, 2017). Microorganisms are the most important actors in wastewater treatment systems, with their capabilities to degrade diverse organic substances and to cycle elements such as nitrogen, phosphorus, and carbon (Daims et al., 2006b; Wen et al., 2010; Zaborowska et al., 2019). However, most of the microorganisms involved in these processes are still poorly described.
The phylum Bacteroidota (former Bacteroidetes) is one of the most abundant and diverse lineages in wastewater treatment plants (WWTPs), comprising bacteria with various metabolisms (Thomas et al., 2011; Kwon et al., 2019; Al Ali et al., 2020). Their main role in WWTPs has been described in relation to the degradation of complex organic substances, such as proteins, starch, cellulose, and fibers (Yang et al., 2017). In addition, members of this phylum are able to degrade dead cells and exopolysaccharides into simpler organic compounds, such as lactate and ethanol, which can then be utilized by other members of the community (Al Ali et al., 2020).
Saprospiraceae is the most abundant family in this phylum, with representatives in WWTPs across the world (Muszyński et al., 2015; Yang et al., 2015, 2017; Al Ali et al., 2020; Cao et al., 2020). Several isolates have been obtained from different water environments (Hosoya et al., 2006; Chen et al., 2014; McIlroy and Nielsen, 2014) and are reported to have variable morphology, spanning from filamentous to rods of different lengths, with some genera forming helical gliding filaments (McIlroy and Nielsen, 2014). Interestingly, filamentous Saprospiraceae such as Haliscomenobacter can occasionally be involved in bulking problems in WWTPs (Xu et al., 2018). However, the most abundant genera, such as OLB8, are widely found in different environments such as activated sludge (Al Ali et al., 2020; Aqeel and Liss, 2020; Seshan et al., 2021) and anammox reactors (Speth et al., 2016). The first metagenome-assembled genome (MAG) of the genus OLB8 was recovered from partial nitrification anammox bioreactors, where the bacterium was known to function as an aerobe heterotroph (Speth et al., 2016). Members of the genus OLB8 were also found in high abundance in reactors with polyphosphate-accumulating organism (PAO)-enriched biomass (Arumugam et al., 2019), bioreactors with a high ratio of food to microorganisms (Roy et al., 2020), reactors with increased concentration of ammonia (Aqeel and Liss, 2020), deammonification reactors (Chini et al., 2020), and also in reactors related to degradation of aromatic compounds (Seshan et al., 2021). Over the years, OLB8 bacteria were speculated to be involved in phosphorus and nitrogen removal in addition to the degradation of different carbon compounds in WWTPs, but their function was never fully resolved. Most of the Saprospiraceae genera are still uncharacterized, with unknown functions; thus, further analysis is needed to fully understand their role in WWTPs.
Since only a few Saprospiraceae isolates exist from the WWTP ecosystem, the function most of these microorganisms can only be inferred using metabolic models derived from annotated genomes MAGs, preferably high-quality (HQ) MAGs (Parks et al., 2017; McDaniel et al., 2021; Singleton et al., 2021). While in 2015, only five genomes were available for Saprospiraceae (McIlroy and Nielsen, 2014), recent effort has raised that number to the 134 representative public genomes currently available in the GTDB (R06-RS202) (Parks et al., 2020). However, extensive metabolic reconstruction is still lacking. In situ studies have shown a strong metabolic capacity to decompose proteins, lipids, and other macromolecules into simple compounds (Luo et al., 2020). For example, the genus Candidatus Epiflobacter plays an important role in protein degradation by the production of extracellular enzymes (Xia et al., 2008). Moreover, members of Saprospiraceae are often found in microbial communities involved in nitrogen removal (Sun et al., 2012; Cao et al., 2020), indicating their potential role as nitrogen-transforming bacteria, although this has to be confirmed experimentally.
The taxonomy of Bacteroidota has undergone several changes in the last decade, resulting in inconsistent classification, with major differences depending on the reference database used (Hahnke et al., 2016; Parks et al., 2020; Schoch et al., 2020). MiDAS, a comprehensive and ecosystem-specific full-length 16 rRNA gene reference database of microbes found in Danish and global WWTPs, was established 5 years ago and is still expanding (Karst et al., 2018; Nierychlo et al., 2020; Dueholm et al., 2022). It is a powerful tool that provides taxonomy information for all bacteria and is useful for both community composition analysis and for the design and re-evaluation of oligonucleotide probes for fluorescence in situ hybridization (FISH). The FISH technique is important for the visualization of microorganisms of interest and also essential for the validation of genomic information when combined with other in situ techniques (Nielsen et al., 2009).
In this study, we investigate the diversity of 5 highly abundant novel genera within the Saprospiraceae in activated sludge WWTPs worldwide. The morphology and spatial arrangement of the Saprospiraceae species were studied with newly designed genus- and family-level FISH probes. The FISH probes were also used to investigate the presence of intracellular storage polymers with FISH–Raman microspectroscopy. A detailed metabolic reconstruction was made using the recently published ecosystem-specific HQ MAGs (Singleton et al., 2021). By combining the global MiDAS 4 16S rRNA gene reference (Dueholm et al., 2022) and the HQ MAG database with FISH techniques, we obtained a comprehensive understanding of the diversity and function of abundant members of Saprospiraceae in the activated sludge ecosystem.
Sampling of full-scale plants across the world was coordinated by the MiDAS Global Consortium and carried out within the Global MiDAS project (Dueholm et al., 2022). Global activated sludge samples from the process tanks were preserved in RNA later and shipped to Aalborg University with cooling elements.
Sequencing of full-length 16S rRNA genes was performed as previously described by Karst et al. (2018). After sequencing, data for bioinformatics analysis was performed as explained in Dueholm et al. (2020, 2022). Raw and assembled sequencing data are available at the NCBI SRA database BioProject ID: PRJNA728873. Data were analyzed using R v4.1.1 (R Core Team, 2020) and R studio software v1.4.1717 (RStudio Team, 2020) and visualized with ampvis2 v2.7.10 (Andersen et al., 2018) and ggplot v3.2.1 (Wickham, 2016) packages. The Kruskal–Wallis test (Kruskal-Wallis Test, 2008) was used to determine statistically significant differences in genus abundance across different groupings.
Phylogenetic analysis of 16S rRNA gene sequences and design of FISH probes for Saprospiraceae were performed using ARB software v6.0.6 (Ludwig, 2004). The phylogenetic maximum likelihood tree was constructed based on comparative analysis of aligned 16S rRNA gene sequences, retrieved from MiDAS 4 (Dueholm et al., 2022), using the GTR method and a 1,000 replicates bootstrap analysis. Coverage and specificity of the FISH probes were evaluated and validated in silico with mathFISH software (Yilmaz et al., 2011) for hybridization efficiencies of target and potentially weak non-target matches. When needed, unlabeled competitor, and helper probes were designed. All probes were purchased from Biomers (Ulm, Germany), labeled with cyanine 3 (Cy3), cyanine 5 (Cy5), Atto 532, Atto 565, Atto 594, and Atto 488 fluorochromes.
The biomass collected from full-scale activated sludge WWTPs was fixed with 50% ethanol (final volume) or 4% PFA (final volume), and stored at −20°C for further analysis. FISH was performed as described by Nielsen et al. (2009). Optimal formamide concentrations for each new FISH probe (Supplementary Table S1) were determined after performing hybridization at different formamide concentrations in the range of 0–70% (with 5% increments). The intensity of 50 cells was measured using ImageJ software (Schneider et al., 2012). EUBmix (Amann et al., 1990; Daims et al., 1999) was used as a general probe to target all bacteria. Microscopic analysis was performed with either an Axioskop epifluorescence microscope (Car Zeiss, Germany) equipped with a LEICA DFC7000 T CCD camera or a white light laser confocal microscope (Leica TCS SP8 X, Leica Microsystems, Germany). qFISH was performed for each genus-specific probe, targeting bacteria of interest, labeled with Cy3 dye, and used together with EUBmix labeled with Cy5 as a universal probe. Analysis was performed on a set of 30 images captured using a 63 x microscope objective on a white light laser confocal microscope (Leica TCS SP8 X, Leica Microsystems, Germany) and analyzed with DAIME v2.2.2 software (Daims et al., 2006a). Raman microspectroscopy was applied in combination with FISH. This approach allows the detection of general cellular components, such as nucleic acids, membrane lipids or proteins, as well as intracellular storage polymers, such as polyphosphate (poly-P), glycogen, and polyhydroxyalkanoates (PHA), as previously described (Fernando et al., 2019).
Genomes from Saprospiraceae were identified in a set of 1,083 HQ MAGs recovered from Danish WWTPs (Singleton et al., 2021). Taxonomic classification at the phylogenetic level in relation to the genome taxonomy database and maximum likelihood placement was conducted using GTDB-Tk v1.4.1 (Chaumeil et al., 2019) (Refseq release 95) “classify.” Pyani v0.2.11 (Pritchard et al., 2016) was used to determine the species (using a 95% average nucleotide identity (ANIb) cutoff) (Singleton et al., 2021) and genus (using a 75–77% ANI cutoff). To determine MAG completeness and contamination statistics, CheckM v1.1.2 (Parks et al., 2015) “lineage_wf” was used. Multiple sequence alignments of 120 concatenated single copy proteins, trimmed to ~5,000 amino acids, produced by GTDB-Tk were used as input for IQ-TREE v2.0 (Nguyen et al., 2015) to create a maximum likelihood tree using the WAG+G model and 100-replicates bootstrap analysis. Saprospiraceae genomes were selected for inclusion in the bootstrapped tree based on the GTDB-Tk v1.5.0 tree and the paraphyletic clade incorporating all genomes of interest. The resulting bootstrapped tree of Saprospiraceae was further examined and rooted in ARB v6.0.3 (Ludwig, 2004), and ITOL v6 (Letunic and Bork, 2021) was used for tree visualization with final aesthetic changes made in Inkscape v1.0.1. Members of the genera Lewinella, Haliscomenobacter, and Saprospira were used as an outgroup.
MAGs were annotated using EnrichM v0.5.0 (github.com/geronimp/enrichM) “annotate” with default settings to blast the MAG proteins against the uniref100 database (Suzek et al., 2015) annotated with KEGG (Kanehisa, 2000). Orthology (KO) numbers (EnrichM database v10). In addition, the MAGs were uploaded to the “MicroScope Microbial Genome Annotation & Analysis Platform” (Vallenet et al., 2019) to examine the gene synteny and cross-validate KO annotations found using EnrichM. The presence of a metabolic pathway was assumed when the full set of genes, defined by the KEGG module and “MicroScope Microbial Genome Annotation & Analysis Platform,” were predicted. In this study, pathways are considered present if 100% of the genes in the KEGG or custom module were encoded.
In order to determine the most abundant genera of Saprospiraceae and to investigate their diversity and key factors correlating with their global presence, a detailed analysis was performed on the V1-V3 amplicon data obtained from the recent Global MiDAS survey (Dueholm et al., 2022), comprising 929 samples from 30 countries. Bacteroidota was the second most abundant phylum in the WWTPs (Supplementary Figure S1), with Saprospiraceae being the most abundant family (Figure 1A), in accordance with previous observations (Yang et al., 2015, 2017; Al Ali et al., 2020; Cao et al., 2020; Luo et al., 2020). Saprospiraceae was very diverse and comprised more than 162 different genera based on MiDAS 4, which included the known Ca. Epiflobacter (Xia et al., 2008), Phaeodactylibacter (Chen et al., 2014), and Haliscomenobacter (van Veen et al., 1973), but the majority of the genera were putative with assigned placeholder names by AutoTax (Dueholm et al., 2020) and undescribed. The high diversity within the family and the difference in taxonomic classification using different databases suggest that the classification and taxonomy of Saprospiraceae need revision. In this study, we decided to focus on the three most abundant and novel genera defined by MiDAS 4.7.1 with the provisional names OLB8, midas_g_17, and midas_g_65. In most of the countries, these three genera co-existed in the same plants, with the highest global abundances recorded mostly in European countries, such as Germany (2.6% relative read abundance) for OLB8, Italy (0.9%) for midas_g_17, and Portugal (0.6) for midas_g_65 (Figure 1B).
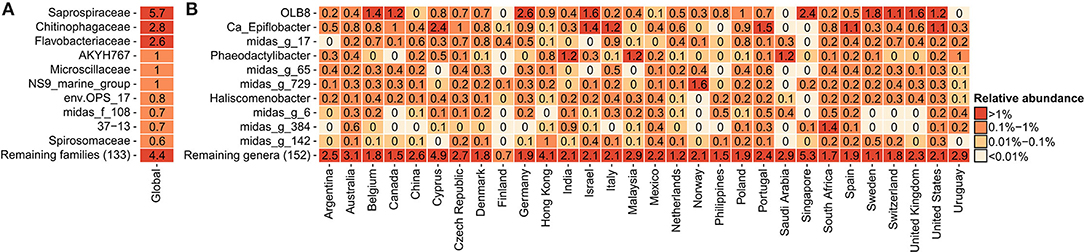
Figure 1. Global average relative read abundance in 929 activated sludge samples: (A) 10 most abundant families within Bacteroidota and (B) 10 most abundant genera within Saprospiraceae.
More in-depth analyses of the five most abundant species in genera OLB8 (midas_s_29, midas_s_3279), midas_g_17 (midas_s_17), and midas_g_65 (midas_s_65, midas_s_177) were carried out to give insights into factors affecting their occurrence across the world (Figure 2). Some studies have shown a seasonal distribution of members belonging to Saprospiraceae, with a potential preference for lower temperatures (Muszyński et al., 2015; Xu et al., 2018; Luo et al., 2020). We applied Köppen–Geiger climate classification (Kottek et al., 2006) to determine differences in the abundance distribution within different climate zones. While midas_s_3279 had higher abundance in tropical/megathermal, dry (desert and semi-arid), and temperate/mesothermal climates, midas_s_65, midas_s_17, and midas_s_29 seemed to be most abundant in temperate/mesothermal climate, continental/microthermal, and polar climates, featuring more humid and colder climates (Figure 2A and Supplementary Table S2).
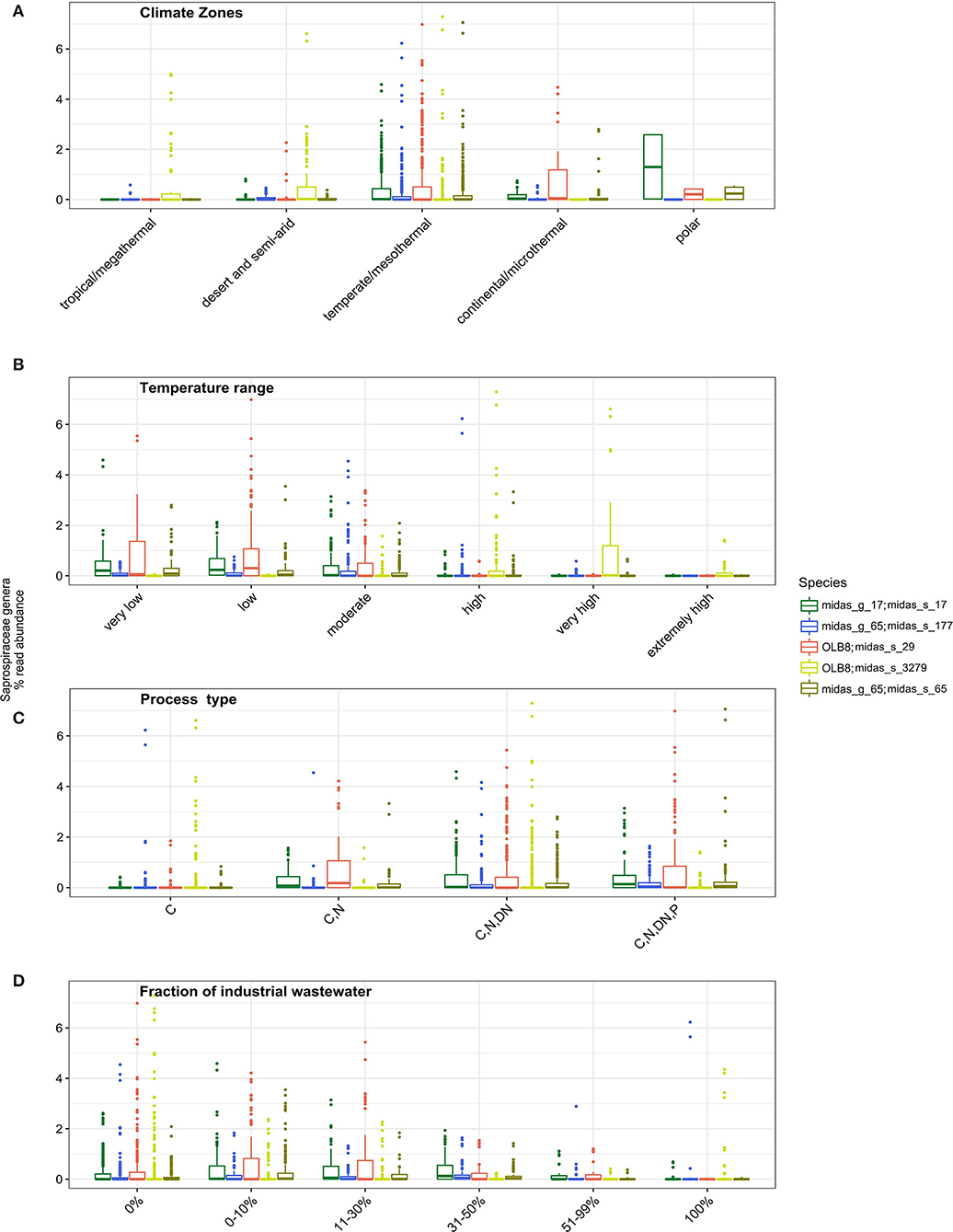
Figure 2. Occurrence of different Saprospiraceae species across the world in relation to (A) different climate zone groupings (tropical/megathermal climate, 29 plants; dry (desert and semi-arid) climate, 48 plants; temperate/mesothermal climate, 368 plants; continental/microthermal climate, 24 plants; polar climate, 2 plants) (Kruskal–Wallis for all species p < 0.05; Supplementary Table S3); (B) global average relative read abundance of highly abundant species of Saprospiraceae in different temperature ranges analyzed in process tanks [very low (1–10.0°C, 43 plants), low (10.1–15.0°C, 96 plants), moderate (15.1–20.0°C, 112 plants), high (20.1–25°C, 73 plants), very high (25.1–30.0°C, 48 plants), extremely high (30.1–38.0°C, 32 plants)] (Kruskal–Wallis for all species p < 0.05; Supplementary Table S3); (C) process type (C−113 plants; C, N, 48 plants; C, N, DN, 208 plants, C, N, DN, P, 111 plants), (C, carbon removal; N, nitrification; DN, denitrification; P, biological P removal), (Kruskal–Wallis for all species p < 0.05; Supplementary Table S3) and (D) In WWTPs with different fractions of industrial wastewater (0%, 169 plants; 0–10%, 105 plants; 11–30%, 67 plants; 31–50%, 41 plants; 51–99%, 20 plants; 100%, 40 plants) (Kruskal–Wallis for all species p < 0.05; Supplementary Table S3).
We observed a strong effect of temperature in process tanks of WWTPs on the abundance of the members of Saprospiraceae. The highest abundance was noticed at very low, low, and moderate temperatures for midas_s_29, midas_s_17, midas_s_65, and midas_s_177, while at the higher process tank temperature ranges, these members were present at a very low abundance (Figure 2B). Unlike all other members, midas_s_3279 was mostly abundant in WWTPs with very high temperatures (25.1–30°C) (Figure 2B). In addition, midas_s_3279 was observed in high relative abundance in deammonification reactors (up to 15%) (Supplementary Figure S2) with low oxygen and high temperatures.
The process design influenced the abundance of most species, with highest abundances occurring in more advanced plants with biological N and P removal (Figure 2C). This is supported by previous observations of highly abundant members of Saprospiraceae in plants performing N and P removal with anaerobic and anoxic stages (Sun et al., 2012; Yang et al., 2015). The fraction of industrial wastewater in the influent (here given as the COD fraction) (Figure 2D) was also shown as an important and statistically significant factor influencing the abundance of all the species, based on Kruskal–Wallis statistical analysis. All species were found mainly in municipal plants with low or no influx of industrial wastewater (<30%) (Figure 2D) in agreement with other studies (Yang et al., 2017; Chi et al., 2018; Luo et al., 2020; Schambeck et al., 2020).
16S rRNA gene-based phylogenetic analysis of Saprospiraceae was performed using full-length sequences obtained from MiDAS 4 (Dueholm et al., 2022) and from 32 HQ MAGs classified as the genera OLB8, midas_g_17, and midas_g_65, recovered from 23 Danish WWTPs (Singleton et al., 2021; Figure 3).
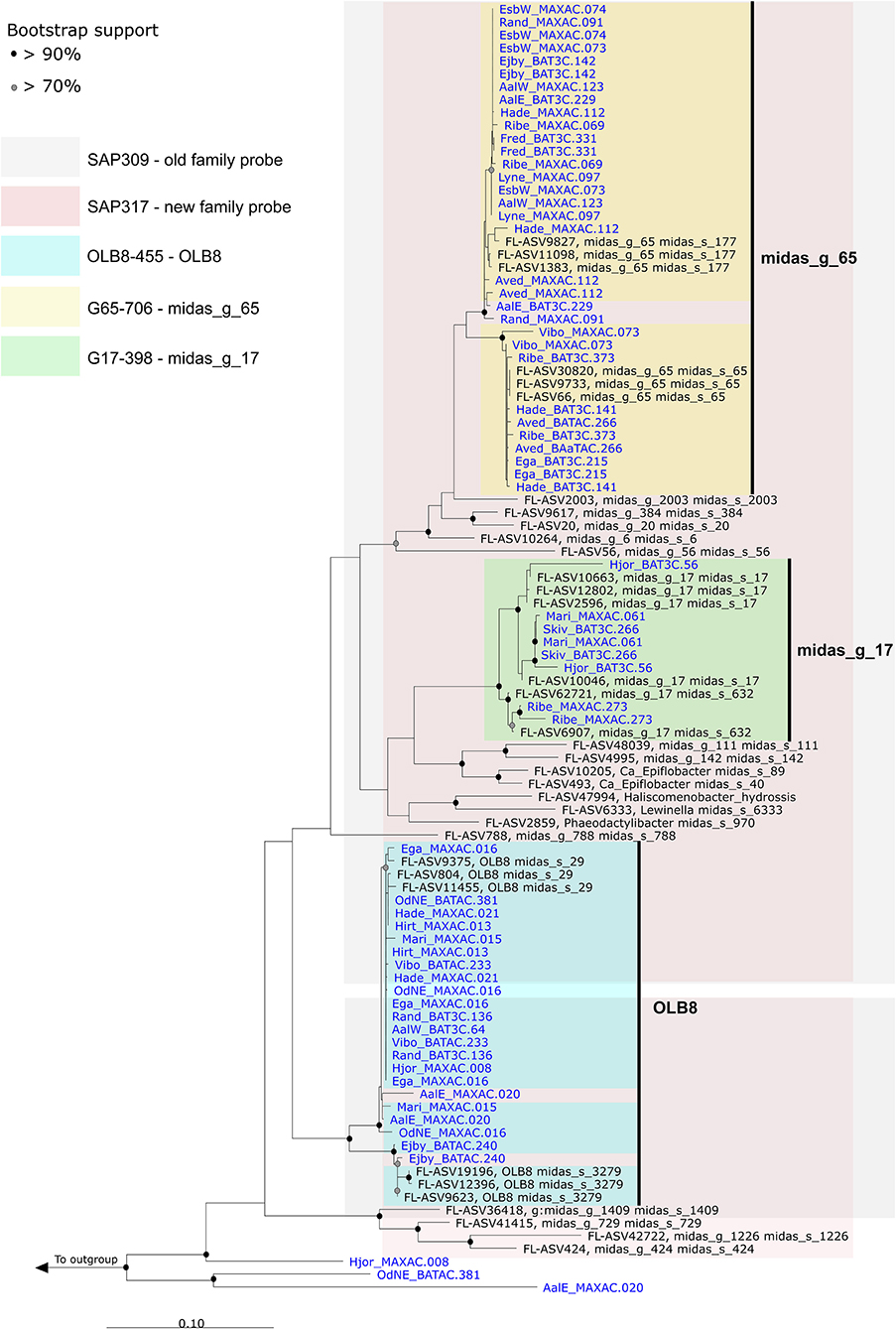
Figure 3. Maximum-likelihood (PhyML) phylogenetic tree based on 16S rRNA genes of the Saprospiraceae including full-length sequences from MiDAS 4 (black) and MAG (blue). Colors indicate the coverage of FISH probes designed in this study. Bootstrap values from 1000 re-samplings are indicated for branches with >70% (gray circles) and >90% (black circles) support. The scale bar represents substitutions per nucleotide base.
The first FISH probes targeting Saprospiraceae were designed over 27 years ago (Wagner et al., 1994), and to date, there are only 21 FISH probes available to target 11 specific genera, species, or clusters within this family (Ginige et al., 2004; McIlroy and Nielsen, 2014). Among these, the widely applied family-level probe SAP-309 (Schauer and Hahn, 2005) was tested in silico using the MiDAS 4 reference (Figure 3). Even though the test indicated a good specificity, the use of this probe as a general probe cannot be recommended due to insufficient coverage (Supplementary Table S1). A new family-level probe SAP-317 was designed and optimized for a better coverage of Saprospiraceae present in WWTPs (Figure 3 and Supplementary Table S1). Furthermore, three additional probes were designed using the MiDAS 4 taxonomy, with competitors and helper probes, targeting the genera OLB8, midas_g_17, and midas_g_65 (Figure 3 and Supplementary Table S1). When applied to full-scale activated sludge biomass, all three probes exclusively hybridized to short, thick rods found predominantly as single cells inside the flocs and rarely attached to other bacteria (Supplementary Figure S3).
Quantitative FISH analysis with all three new probes showed that the biovolume fractions of the biomass were lower than those of the relative read abundance by amplicon sequencing using V1-V3 primers (Supplementary Table S4). It could be due to the presence of 2–3 copies of the 16S rRNA gene within each MAG (Figure 3 and Supplementary Data File 1) or due to the different sizes of cells within the activated sludge biomass. Primers can have a considerable impact on the abundance recorded by the amplicon sequencing (Dueholm et al., 2022), so we compared the average global abundance obtained using the two commonly applied primer sets, V1–V3 and V4. The average abundance of members of Saprospiraceae (genus level) was similar between both primers sets (Supplementary Figure S4).
Overall, 32 HQ MAGs representing different genera in Saprospiraceae (Supplementary Table S5) from 23 Danish WWTPs (Singleton et al., 2021) were investigated. Completeness and contamination of all MAGs analyzed ranged between 91.88–99.01% and 0–4.95%, respectively, and they all contained 2–3 copies of the 16S rRNA gene (Supplementary Data File 1).
Phylogenomic analysis using the GTDB-Tk (Figure 4) showed that all OLB8 midas_s_29 MAGs grouped with the OLB8 genus as one novel species (>95% ANI) (Chaumeil et al., 2019), and one MAG representing midas_s_3279, which grouped with OLB8 sp001567405 (GCA_001567405.1) species. The analysis additionally showed that all MAGs from midas_g_65 clustered together with the UBA3362 genus (GCA_002360455.1), while the MAGs from midas_g_17 represented a novel genus and did not cluster with any of the known Saprospiraceae genera.
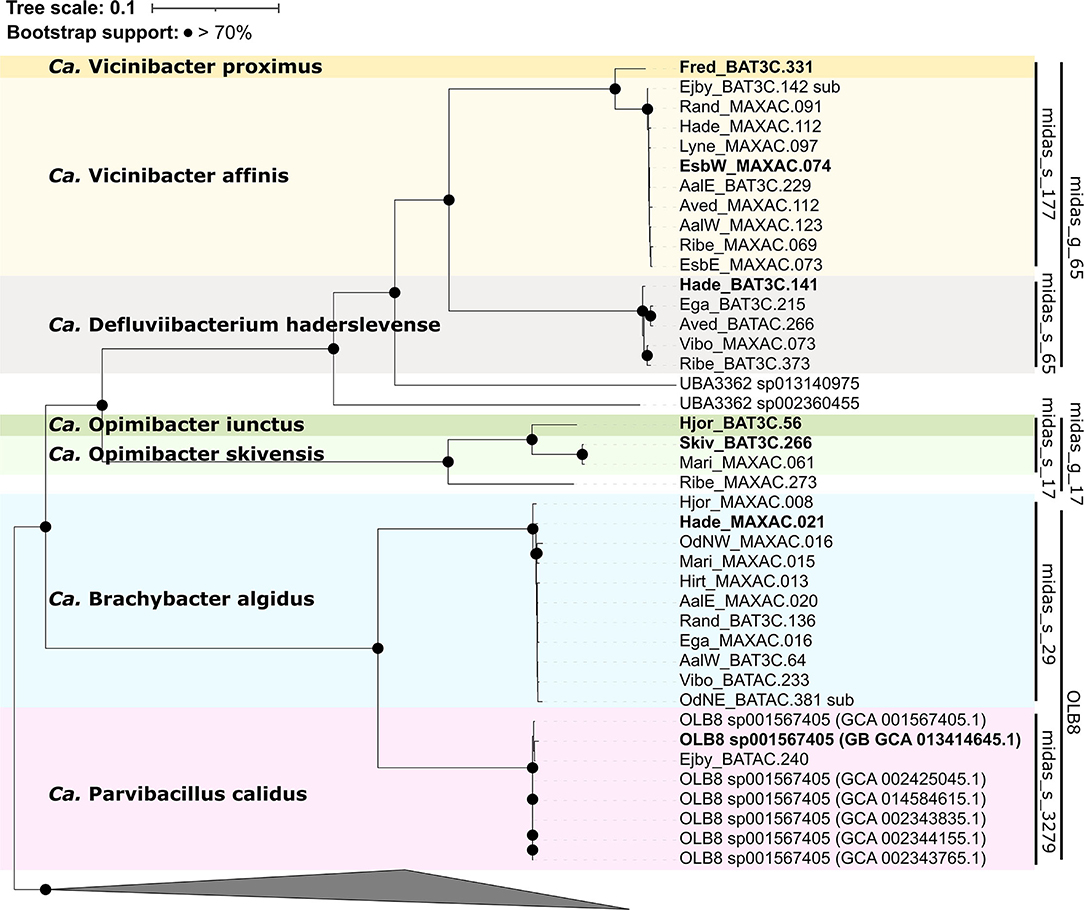
Figure 4. Maximum likelihood genome tree created from the concatenated alignment of 120 single-copy marker gene proteins trimmed to 5,000 amino acids using the GTDB-Tk v1.5.0. Branches with bootstraps support >70% are indicated by black dots. Species representatives are in bold. The genera Lewinella, Haliscomenobacter, and Saprospira were used as the outgroup.
To determine the taxonomic rank classification, analysis of the full genome average nucleotide identity (ANI) was performed with proposed boundaries for genus (75–77%) and species (>95%) levels (Barco et al., 2020; Parks et al., 2020). The analysis showed that the HQ MAGs represented six novel genera and seven novel species (Figure 4), contrary to previous 16S rRNA gene-based taxonomic analysis. Based on the phylogenomic analysis, the species midas_s_29 (>98% ANI) and midas_s_3279 (>99% ANI), both belonging to the genus OLB8 according to MiDAS and GTDB-Tk taxonomy, represent two different genera (<75% ANI clustering) (Supplementary Figure S5). In addition, based on ANI clustering, 16 MAGs representing midas_g_65 clustered in two different genera and three different species, grouping together next to the close relative UBA3362 sp002360455 (Parks et al., 2017) but representing a separate genus (<75% ANI) (Supplementary Figure S5). Furthermore, midas_s_17 and midas_s_632, both belonging to the MiDAS-defined genus midas_g_17 based on ANI clustering analysis, appeared to be two novel genera with three novel species (Supplementary Figure S5). Due to the very low abundance of midas_s_632 in activated sludge in both Danish and global WWTPs, we excluded it from further analysis. Overall, the clustering of analyzed members did not show any major differences between the 16S rRNA gene and the genome tree (Figures 3, 4).
For the two new genera encompassing midas_s_29 and midas_s_3279, respectively, we propose the names Candidatus Brachybacter algidus (Supplementary Table S6) and Candidatus Parvibacillus calidus (Supplementary Table S7). For further analysis, seven additional HQ MAGs from the public GTDB (OLB8 sp001567405) were included in the metabolic reconstruction for better representation of Ca. P. calidus. For midas_s_65, we propose the name Candidatus Defluciibacterium haderslevense (Supplementary Table S8), and for midas_s_177, which represents two different species based on ANI analysis, we propose the names Candidatus Vicinibacter proximus (Supplementary Table S9) and Candidatus Vicinibacter affinis (Supplementary Table S10). For midas_s_17, which includes two species, we propose the names Candidatus Opimibacter skivensis (Supplementary Table S11) and Candidatus Opimibacter iunctus (Supplementary Table S12).
In this section, we have investigated the metabolic potential of key wastewater treatment processes: carbon degradation, nitrogen transformation, polyphosphate accumulation, and fermentation (Figure 5 and Supplementary Data Files 3, 4). The five genera analyzed showed diverse metabolisms, with a general heterotrophic lifestyle.
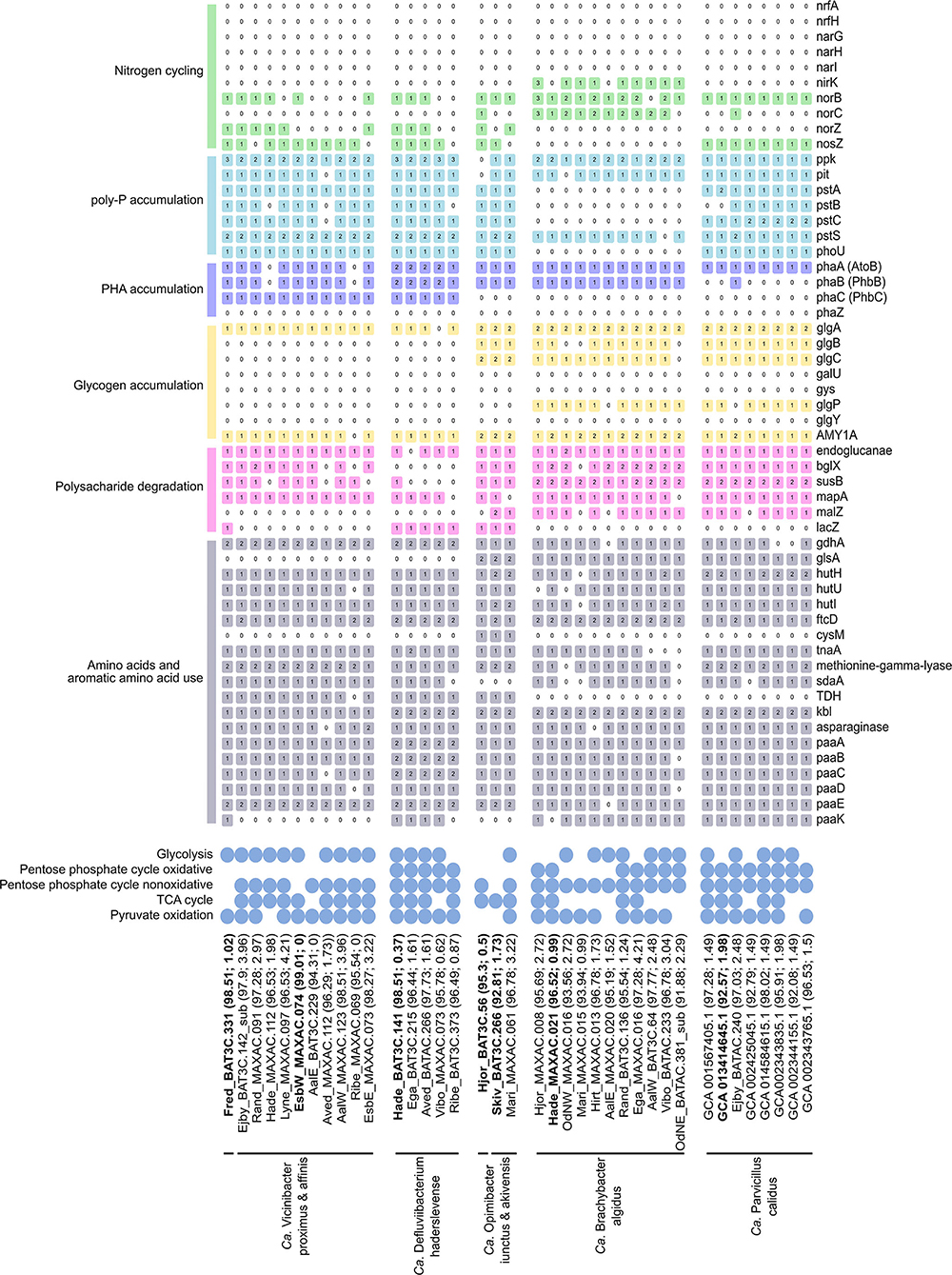
Figure 5. Basic functional potential of the Ca. Vicinibacter, Ca. Defluviibacterium haderslevense, Ca. Opimibacter, Ca. Brachybacter algidus, and Ca. Parvibacillus calidus. The number in colored boxes represents gene copy number. The gene list follows the progression in the text. For the full list of gene names and associated KO numbers, see Supplementary data 3. The genomes are ordered as in the genome tree in Figure 4, with their genome completeness and contamination indicated within the parentheses. Species representatives are in bold.
The potential for glucose utilization through the full Embden–Meyerhof–Parnas (EMP) pathway (KEGG module: M00001) was encoded in MAGs from Ca. B. algidus, Ca. P. calidus, and Ca. O. skivensis. Even though the non-oxidative pentose phosphate pathway (M00007) was predicted in all analyzed species, the full pentose phosphate pathway (M00004) was only predicted in Ca. B. algidus and Ca. P. calidus. A complete TCA cycle (M00009) was found in MAGs from all analyzed species (Figure 5). The cytochrome c oxidase (M00155) pathway was also identified in all species; in addition, the aerobic respiration (cytochrome c) pathway was present in MAGs from Ca. B. algidus (Supplementary Data File 4).
In previous studies of Bacteroidota, it was shown that some genera are able to utilize polysaccharides as the carbon source (Xia et al., 2008; Terrapon et al., 2018; Lapébie et al., 2019). The genes for endoglucanase and beta-glucosidase (blgX), responsible for cellulose degradation to glucose, were found in most of the MAGs (Figure 5). In addition, genes responsible for starch/glycan (susB) degradation to glucose (Kitamura et al., 2008) were present in all the MAGs, except those representing Ca. D. haderslevense and Ca. O. skivensis (Figure 5). Maltose phosphorylase (mapA), which is used for maltose degradation (Gopal et al., 2010), was present in all five genera. In 13 MAGs representing Ca. O. skivensis, Ca. B. algidus, and Ca. P. calidus, the gene malZ, responsible for sucrose degradation, was found. In addition, the lacZ gene, which divides lactose into glucose and galactose, was found in all MAGs from Ca. Opimibacter, Ca. D. haderslevense, and Ca. V. proximus (Figure 5). The capability to degrade polysaccharides was indicated by the presence of susC/D homologs, which predict polysaccharide utilization loci (PULs), present in many Bacteroidota genomes. In addition, the phylum-exclusive type IX secretion system (T9SS), which is highly associated with polysaccharide degradation when PUL-encoded enzymes are secreted by T9SS (McKee et al., 2021), was found in all five genera (Supplementary Data File 3). These findings agree with studies of many isolates of Bacteroidota having high numbers of extracellular carbohydrate active enzymes, indicating a potential to degrade various high-molecular weight polysaccharides (McKee et al., 2021).
In addition, it has been observed that Ca. Epiflobacter is able to utilize amino acids as a carbon source under oxic, anoxic, and short-term anaerobic conditions (Xia et al., 2008). In all MAGs from all five genera genes were found for the degradation of several amino acids, such as glutamate (glutamate dehydrogenase), glutamine (glutaminase), histidine (hutHUI, ftcD, fold), L-cysteine (cysM/tnaA/CTH), methionine (methionine-gamma-lyase), L-serine (sdaA), threonine (TDH, kbl), tryptophan (tnaA), alanine (ald), and asparagine (asparaginase) (Figure 5).
Fermentation is another important growth metabolism for bacteria present in WWTPs (Kong et al., 2008). According to genome annotation, all five newly described genera have, to some extent, the possibility to ferment (Supplementary Data File 3). All MAGs from Ca. Opimibacter, Ca. B. algidus, and Ca. P. calidus had all genes (ppC, gltA, acnA, icd) for degradation of phosphoenolpyruvate to 2-oxoglutarate, which is part of mixed acid fermentation (Friesen, 1988). In addition, the adh gene for acetaldehyde degradation to ethanol was present in all MAGs from Ca. Opimibacter, Ca. Vicinibacter, and Ca. D. haderslevense. Overall, 30 MAGs encoded alanine dehydrogenase (ald) for reduction of pyruvate to alanine. The reaction is reversible and has been reported to play an important role in redox balance maintenance during the fermentative growth and could additionally facilitate anaerobic growth or alanine use (Ward et al., 2000). Although all 5 genera have genes which, to some extent, may support anaerobic fermentation, it does not appear to be a key function.
Denitrification is important in wastewater treatment, converting nitrate (NO) to dinitrogen gas (N2) (von Sperling, 2007). However, none of the MAGs analyzed were predicted to be full denitrifiers as they all lacked genes for dissimilatory nitrate reductase (napAB, narGHI), and other genes involved in the denitrification pathway were sporadically encoded by the different species (Figure 5). Genes for nitrite (NO) reduction to nitric oxide (NO) (nirK or nirS) were predicted in nine MAGs from Ca. B. algidus, and NO reduction to nitrous oxide (N2O) (norBC) was predicted in 11 MAGs from Ca. O. iunctus (1/1), Ca. B. algidus (9/11), and Ca. P. calidus (1/8). Furthermore, the norB gene was annotated by EnrichM in 11 MAGs from Ca. D. haderslevense, Ca. Vicinibacter, and Ca. Opimibacter, while norC was present only in one MAG (Figure 5). Based on MicroScope annotation, in 10 of these 11 MAGs (Figure 5), the quinol-dependent nitric-oxide reductase gene (norZ) was predicted. NorZ can be found in many denitrifying organisms and various pathogenic species which lack the full denitrifying process. For those organisms, NorZ seems to be used for a defense, rather than energy conservation (Hendriks et al., 2000). The nosZ gene (K00376) was found in 23 MAGs representing Ca. P. calidus (8/8), Ca. Opimibacter (2/3), Ca. D. haderslevense (4/6), and Ca. Vicinibacter (9/10) (Figure 5), indicating a possible reduction of nitrous oxide (N2O) to N2. Potential usage of nitrous oxide as an electron acceptor was expected as the nosZ was previously predicted in five genomes from Bacteroidota (Bacteroidetes bacterium OLB8, OLB9, OLB10, OLB1, OLB12) (Speth et al., 2016). Overall, the use of oxidized nitrogen compounds in anaerobic respiration is hypothesized to be uncommon in Saprospiraceae, but perhaps some of them can be important as non-denitrifying nitrous oxide oxidizers (Ren et al., 2019). Nitrate/nitrite transporter, NarK gene (Noji et al., 1989), was not present in any of the genera, and multiple ABC transporter systems, which could be used for the translocation of many substrates across the membrane, including nitrogen-related compounds (Moir and Wood, 2001), were found in all five genera with a high copy number (Supplementary Data File 3).
Uptake and removal of phosphate are a key process among the PAOs and common in WWTPs with enhanced biological phosphorus removal (Nielsen et al., 2019). While no members of Saprospiraceae have previously been suggested to be PAOs, the full set of known genes encoding polyphosphate accumulation (pit, pstABCS, phoU, ppk) was predicted in all MAGs from Ca. D. haderslevense, Ca. Vicinibacter, Ca. Opimibacter, and Ca. P. calidus (Figure 5). In particular, the pit transporter gene (pit) for phosphate transport is suggested to be a main indicator for the PAO bacteria (Nielsen et al., 2019), suggesting these species could be potential PAOs.
Most PAOs accumulate PHA and/or glycogen as an intracellular carbon and energy source under dynamic oxic–anoxic conditions (Akbari et al., 2021; Petriglieri et al., 2021b). Previous studies of Saprospiraceae have not found PHA accumulation (Xia et al., 2008), but the genes (PhaABC) for PHA accumulation were predicted in 14 MAGs from the genera Ca. D. haderslevense and Ca. Vicinibacter (Figure 5), suggesting possible PHA accumulation and PHA usage. The full gene set for glycogen biosynthesis (glgABC) was encoded in 15 MAGs from Ca. Opimibacter, Ca. P. Validus, and Ca. B. algidus (Figure 5). Even though the well-known glycogen degradation pathway (M00855) was not present in Ca. Opimibacter, Ca. P. calidus, and Ca. B. algidus, all of them had the alpha-amylase (AMY1A) gene (Figure 5), which may be used for glycogen/starch degradation to dextrin and maltose (Janecek, 1997). Taken together, none of the MAGs investigated encoded both PHA and glycogen accumulation, suggesting they do not follow the conventional PAO metabolism.
New FISH probes were applied to full-scale activated sludge biomass to determine the morphology of the novel genera. Samples exclusively containing target species (based on amplicon sequencing) were selected to visualize individual species. All species were mostly present deep inside the flocs, usually as single cells with variations in size (Table 1). Ca. B. algidus was occasionally found as microcolonies. Although previous studies found some members of Saprospiraceae are epiphytic bacteria (Xia et al., 2008; McIlroy and Nielsen, 2014), only a few cells from Ca. Opimibacter, Ca. B. algidus, Ca. P. calidus, and Ca. D. haderslevense were attached to filamentous bacteria. Interestingly, members of Ca. Vicinibacter were attached to clusters of the PAO Ca. Accumulibacter aalborgensis and to a few filaments from the phylum Chloroflexota (Figure 6), indicating possible symbiotic relationships between these bacteria. This needs, however, to be verified by more detailed studies.

Table 1. Summary table of morphology and presence of intracellular storage polymers detected by FISH and FISH–Raman microspectroscopy in the Saprospiraceae species of interest.
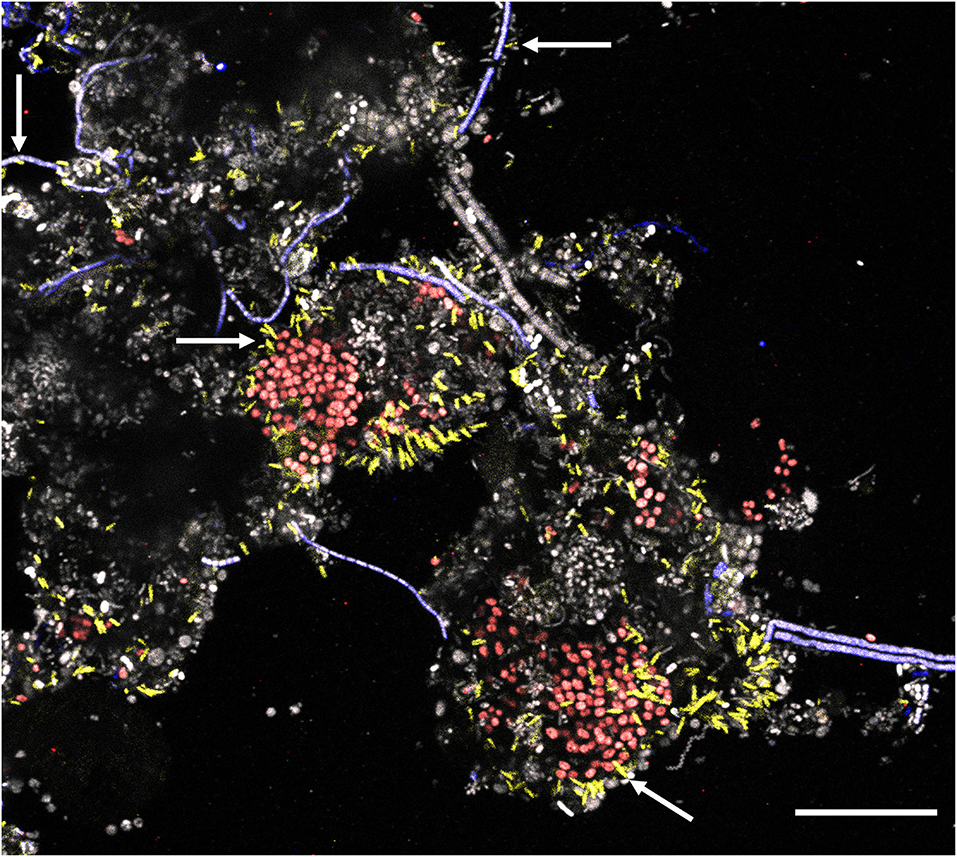
Figure 6. Multicolor FISH micrograph of Ca. Vicinibacter (yellow, G65-705) attached to Ca. Accumulibacter aalborgensis (red, Acc470 ref for that probe) and some to Chloroflexi filaments (blue, CFXmix ref for that probe). All other bacteria are gray. Scale bar is 20 μm.
To confirm key physiological features of the novel Saprospiraceae genera, Raman microspectroscopy was performed in combination with the FISH. The species from the five genera showed in situ presence of peaks associated with common biological components, such as phenylalanine, nucleic acids, and lipids (Supplementary Figure S6). We did not detect poly-P in any of the genera, despite their potential based on the MAG annotation. Glycogen was detected in Ca. B. algidus, Ca. P. calidus, and Ca. Opimibacter (Table 1 and Supplementary Figure S6), whereas Ca. D. haderslevense and Ca. Vicinibacter had a peak indicative of the storage polymer PHA (Table 1 and Supplementary Figure S6), agreeing with the metabolic annotation. These results indicate that they are not PAOs, but the storage polymers glycogen or PHA may be important under dynamic substrate conditions.
Saprospiraceae is a diverse family comprising 162 genera as assessed by our comprehensive global analysis. However, as only a few genera were present and abundant in most WWTPs globally, we have mainly focused on these, although many other genera and species could be abundant in a few plants across the world. The huge variation from plant to plant may be due to the high impact of immigration from different source communities to the WWTPs (Dottorini et al., 2021). The highly abundant former genus OLB8 was found in different WWTP designs, which comprises two different genera, Ca. Brachybacter and Ca. Parvibacillus, based on ANI. Ca. Brachybacter and Ca. Parvibacillus were mainly present in conventional nutrient removal plants and in deammonification plants, respectively.
The only abundant Saprospiraceae isolate from WWTPs is Haliscomenobacter, which is described as strictly aerobic heterotrophic filamentous bacteria. Similar to Ca. Epiflobacter, Haliscomenobacter has the ability to degrade polysaccharides and amino acids with the proposed ability to accumulate carbohydrate storage polymers but not PHA (van Veen et al., 1973; Xia et al., 2008). Our study showed that all analyzed genera have the potential to degrade different polysaccharides such as starch and many amino acids and may grow under both oxic and anoxic conditions. Some members from Saprospiraceae (Ca. Epiflobacter) are also known to be epiphytic bacteria attached to filamentous bacteria and may have a symbiotic relationship. However, only Ca. Vicinibacter was found to be attached in some WWTPs to Ca. Accumulibacter and some filamentous bacteria from the Chloroflexota phylum.
As bacteria from Saprospiraceae are often found in WWTPs designed for nutrient removal, it is assumed that they are involved in the removal of nitrogen and phosphorus. Specifically, Ca. Parvibacillus was abundantly present in deammonification plants, which are characterized by high temperature (20.1–30.0°C) and high ammonium concentration. However, none of the genera analyzed were denitrifiers as nitrate and nitrite reductases were not encoded or encoded in only a few of the MAGs, respectively. Only Ca. Brachybacter algidus had a gene set for reduction of NO to N2O. Most MAGs encoded nitrous oxide reductase, indicating that they might act as non-denitrifying nitrous oxide reducers, but further experiments are needed to confirm this.
Some genera had a potential to be PAOs, but none of them had the canonical pathway known from Ca. Accumulibacter and Ca. Dechloromonas with the dynamic accumulation of poly-P, PHA, and glycogen (Petriglieri et al., 2021a,b). Although the genera had the potential to accumulate poly-P, this was not observed in our in situ studies. Physiological experiments are required to find or confirm PAO metabolism as the genes are indicative but not conclusive. As predicted, Ca. D. haderslevense and Ca. Vicinibacter accumulated PHA, but not glycogen. Ca. Opimibacter, Ca. Brachybacter, and Ca. Parvibacillus calidus all stored glycogen, but not PHA, so they all seem well adapted to dynamic oxic–anoxic conditions but seemed not to be directly involved in the biological P removal.
Our study provides a deeper insight into a very diverse Saprospiraceae family through in situ studies and genomic information that could also be used for future research, for example, using metatranscriptomics, in order to fully understand and confirm this family's involvement in different processes of wastewater treatment.
Description of “Candidatus Brachybacter algidus” gen. nov. sp. nov. (OLB8 midas_s_29) “Candidatus Brachybacter algidus,” (bra.chi.bac'ter, G. m. adj. brachis, small; N.L. m. n. bacteri, rod-shaped bacterium; N.L. m. n. Brachybacter, indicating a small rod-shaped bacterium; al'gi.dus, L. m. adj. algidus, indicating the highest abundance of this microorganism in colder climates). This taxon was represented by the MAG Hade_18-Q3-R52-61_MAXAC.021. The complete protologue can be found in Supplementary Table S6.
Description of “Candidatus Parvibacillus calidus” gen. nov. sp. nov. (OLB8 midas_s_3279) “Candidatus Parvibacillus calidus,” (par.vi.ba.cil'lus, L. m. adj. parvus, small; N.L. m. n. bacillus, rod-shaped bacterium; N.L. m. n. parvibacillus, indicating a small rod-shaped bacterium; ca'li.dus, L. m. adj. calidus, indicating the highest abundance of this microorganism in warmer climates). This taxon was represented by GCA_013414645.1. The complete protologue can be found in Supplementary Table S7.
Description of “Candidatus Defluviibacterium haderslevense” gen. nov. sp. nov. (midas_g_65 midas_s_65) “Candidatus Defluviibacterium haderslevense,” (de.flu.vi.i.bac.te'rium, L. n. n. defluvium, sewage; N.L. m. n. bacterium, rod-shaped bacterium; N.L. m. n. defluviibacterium, indicating a rod-shaped bacterium found in sewage; ha.der.sle.ven'sis, N. L. fem. adj. haderslevense pertaining to the city of Haderslev, where the sample was obtained from which the MAG was produced). This taxon was represented by the MAG Hade_18-Q3-R52-61_BAT3C.141. The complete protologue can be found in Supplementary Table S8.
Description of “Candidatus Vicinibacter” gen. nov. (midas_g_65 midas_s_177) “Candidatus Vicinibacter,” (vi.ci.ni.bac'ter, L. m. adj. vicinus, close; N.L. m. n. bacter, rod-shaped bacterium; N.L. m. n. vicinibacter, indicating rod-shaped bacteria often attached to other bacterial clusters). Description of “Candidatus Vicinibacter affinis” sp. nov. “Candidatus Vicinibacter affinis,” (af.fi'nis L. m. adj. affinis, indicating the close phylogenetic relationship with the other Candidatus Vicinibacter species). This taxon was represented by the MAG EsbW_18-Q3-R4-48_MAXAC.074. The complete protologue can be found in Supplementary Table S10. Description of “Candidatus Vicinibacter proximus” sp. nov. “Candidatus Vicinibacter proximus,” (pro'xi.mus, L. m. adj. proximus, indicating the close phylogenetic relationship with the other Candidatus Vicinibacter species). This taxon was represented by the MAG Fred_18-Q3-R57-64_BAT3C.331. The complete protologue can be found in Supplementary Table S9.
Description of “Candidatus Opimibacter” gen. nov. (midas_g_17 midas_s_17) “Candidatus Opimibacter,” (o.pi.mi.bac'ter, L. m. adj. opimus, fat; N.L. m. n. bacter, rod-shaped bacterium; N.L. m. n. opimibacter, indicating a fat rod-shaped bacterium). Description of “Candidatus Opimibacter skivensis” sp. nov. “Candidatus Opimibacter skivensis,” (ski.ven'sis, N.L. m. adj. skivensis, indicating the location of Skive, from where the MAG was obtained). This taxon was represented by the MAG Skiv_18-Q3-R9-52_BAT3C.266. The complete protologue can be found in Supplementary Table S11. Description of “Candidatus Opimibacter iunctus” sp. nov. “Candidatus Opimibacter iunctus,” (iun'ctus, L. m. adj. iunctus, indicating the close phylogenetic relationship with Candidatus Opimibacter skivensis). This taxon was represented by the MAG Hjor_18-Q3-R7-51_BAT3C.56. The complete protologue can be found in Supplementary Table S12.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
ZK and PN designed the study and wrote the manuscript. FP and MN performed 16S rRNA gene phylogenetic analyses. MN designed the FISH probes. ZK performed the FISH probe optimization, imaging, and qFISH. FP performed the FISH–Raman analyses. ZK, LV, and CS performed the phylogenomic analysis and metabolic reconstruction. ZK and MD performed amplicon sequencing bioinformatic analysis. All authors reviewed and approved the final manuscript.
This project was funded by the Villum Foundation (Dark Matter grant13351) and Aalborg University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.917553/full#supplementary-material
Akbari, A., Wang, Z., He, P., Wang, D., Lee, J., Han, I., et al. (2021). Unrevealed roles of polyphosphate-accumulating microorganisms. Microb. Biotechnol. 14, 82–87. doi: 10.1111/1751-7915.13730
Al Ali, A. A., Naddeo, V., Hasan, S. W., and Yousef, A. F. (2020). Correlation between bacterial community structure and performance efficiency of a full-scale wastewater treatment plant. J. Water Process Eng. 37, 101472. doi: 10.1016/j.jwpe.2020.101472
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925. doi: 10.1128/aem.56.6.1919-1925.1990
Andersen, K.S., Kirkegaard, R.H., Karst, S.M., and Albertsen, M. (2018). ampvis2: an R package to analyse and visualise 16S rRNA amplicon data. bioRxiv [Preprint] 299537. doi: 10.1101/299537
Aqeel, H., and Liss, S. N. (2020). Autotrophic fixed-film systems treating high strength ammonia wastewater. Front. Microbiol. 11, 551925. doi: 10.3389/fmicb.2020.551925
Arumugam, K., Bagc,i, C., Bessarab, I., Beier, S., Buchfink, B., Górska, A., et al. (2019). Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data. Microbiome 7, 61. doi: 10.1186/s40168-019-0665-y
Barco, R. A., Garrity, G. M., Scott, J. J., Amend, J. P., Nealson, K. H., and Emerson, D. (2020). A genus definition for Bacteria and Archaea based on a standard genome relatedness index. MBio 11, e02475-19. doi: 10.1128/mBio.02475-19
Cao, J., Zhang, T., Wu, Y., Sun, Y., Zhang, Y., Huang, B., et al. (2020). Correlations of nitrogen removal and core functional genera in full-scale wastewater treatment plants: influences of different treatment processes and influent characteristics. Bioresour. Technol. 297, 122455. doi: 10.1016/j.biortech.2019.122455
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2019, btz848. doi: 10.1093/bioinformatics/btz848
Chen, Z., Lei, X., Lai, Q., Li, Y., Zhang, B., Zhang, J., et al. (2014). Phaeodactylibacter xiamenensis gen. nov., sp. nov., a member of the family Saprospiraceae isolated from the marine alga Phaeodactylum tricornutum. Int. J. Syst. Evol. Microbiol. 64, 3496–3502. doi: 10.1099/ijs.0.063909-0
Chi, X., Li, A., Li, M., Ma, L., Tang, Y., Hu, B., et al. (2018). Influent characteristics affect biodiesel production from waste sludge in biological wastewater treatment systems. Int. Biodeterior. Biodegrad. 132, 226–235. doi: 10.1016/j.ibiod.2018.04.010
Chini, A., Ester Hollas, C., Chiapetti Bolsan, A., Venturin, B., Bonassa, G., Egidio Cantão, M., et al. (2020). Process performance and anammox community diversity in a deammonification reactor under progressive nitrogen loading rates for swine wastewater treatment. Bioresour. Technol. 311, 123521. doi: 10.1016/j.biortech.2020.123521
Daims, H., Brühl, A., Amann, R., Schleifer, K.-H., and Wagner, M. (1999). The domain-specific probe eub338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/S0723-2020(99)80053-8
Daims, H., Lücker, S., and Wagner, M. (2006a). daime, a novel image analysis program for microbial ecology and biofilm research. Environ. Microbiol. 8, 200–213. doi: 10.1111/j.1462-2920.2005.00880.x
Daims, H., Taylor, M. W., and Wagner, M. (2006b). Wastewater treatment: a model system for microbial ecology. Trends Biotechnol. 24, 483–489. doi: 10.1016/j.tibtech.2006.09.002
Dottorini, G., Michaelsen, T. Y., Kucheryavskiy, S., Andersen, K. S., Kristensen, J. M., Peces, M., et al. (2021). Mass-immigration determines the assembly of activated sludge microbial communities. Proc. Natl. Acad. Sci. U.S.A. 118, e2021589118. doi: 10.1073/pnas.2021589118
Dueholm, M. K. D., Nierychlo, M., Andersen, K. S., Rudkjøbing, V., Knutsson, S., Arriaga, S., et al. (2022). MiDAS 4: A global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 13, 1908. doi: 10.1038/s41467-022-29438-7
Dueholm, M. S., Andersen, K. S., McIlroy, S. J., Kristensen, J. M., Yashiro, E., Karst, S. M., et al. (2020). Generation of comprehensive ecosystem-specific reference databases with species-level resolution by high-throughput full-length 16s rrna gene sequencing and automated taxonomy assignment (AutoTax). MBio 11:e01557-20. doi: 10.1128/mBio.01557-20
Fernando, E. Y., McIlroy, S. J., Nierychlo, M., Herbst, F.-A., Petriglieri, F., Schmid, M. C., et al. (2019). Resolving the individual contribution of key microbial populations to enhanced biological phosphorus removal with Raman–FISH. ISME J. 13, 1933–1946. doi: 10.1038/s41396-019-0399-7
Friesen, J. D. (1988). Escherichia coli and Salmonella typhimurium: cellular and molecular biology. Science 240, 1678.
Ginige, M. P., Hugenholtz, P., Daims, H., Wagner, M., Keller, J., and Blackall, L. L. (2004). Use of stable-isotope probing, full-cycle rrna analysis, and fluorescence in situ hybridization-microautoradiography to study a methanol-fed denitrifying microbial community. Appl. Environ. Microbiol. 70, 588–596. doi: 10.1128/AEM.70.1.588-596.2004
Gopal, S., Berg, D., Hagen, N., Schriefer, E.-M., Stoll, R., Goebel, W., et al. (2010). Maltose and maltodextrin utilization by listeria monocytogenes depend on an inducible ABC transporter which is repressed by glucose. PLoS ONE 5, e10349. doi: 10.1371/journal.pone.0010349
Hahnke, R. L., Meier-Kolthoff, J. P., García-López, M., Mukherjee, S., Huntemann, M., Ivanova, N. N., et al. (2016). Genome-based taxonomic classification of bacteroidetes. Front. Microbiol. 7, 2003. doi: 10.3389/fmicb.2016.02003
Hendriks, J., Oubrie, A., Castresana, J., Urbani, A., Gemeinhardt, S., and Saraste, M. (2000). Nitric oxide reductases in bacteria. Biochim. Biophys. Acta 1459, 266–273. doi: 10.1016/S0005-2728(00)00161-4
Hosoya, S., Arunpairojana, V., Suwannachart, C., Kanjana-Opas, A., and Yokota, A. (2006). Aureispira marina gen. nov., sp. nov., a gliding, arachidonic acid-containing bacterium isolated from the southern coastline of Thailand. Int. J. Syst. Evol. Microbiol. 56, 2931–2935. doi: 10.1099/ijs.0.64504-0
Janecek, S. (1997). α-amylase family: molecular biology and evolution. Prog. Biophys. Mol. Biol. 67, 67–97. doi: 10.1016/S0079-6107(97)00015-1
Kanehisa, M. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Karst, S. M., Dueholm, M. S., McIlroy, S. J., Kirkegaard, R. H., Nielsen, P. H., and Albertsen, M. (2018). Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat. Biotechnol. 36, 190–195. doi: 10.1038/nbt.4045
Kitamura, M., Okuyama, M., Tanzawa, F., Mori, H., Kitago, Y., Watanabe, N., et al. (2008). structural and functional analysis of a glycoside hydrolase family 97 enzyme from Bacteroides thetaiotaomicron. J. Biol. Chem. 283, 36328–36337. doi: 10.1074/jbc.M806115200
Kong, Y., Xia, Y., and Nielsen, P. H. (2008). Activity and identity of fermenting microorganisms in full-scale biological nutrient removing wastewater treatment plants. Environ. Microbiol. 10, 2008–2019. doi: 10.1111/j.1462-2920.2008.01617.x
Kottek, M., Grieser, J., Beck, C., Rudolf, B., and Rubel, F. (2006). World map of the Köppen-Geiger climate classification updated. Meteorol. Z. 15, 259–263. doi: 10.1127/0941-2948/2006/0130
Kruskal-Wallis Test (2008). The Concise Encyclopedia of Statistics. New York, NY: Springer New York.
Kwon, K., Kwon, Y. M., and Kim, S.-J. (2019). “Aerobic hydrocarbon-degrading bacteroidetes,” in Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes, ed T. J. McGenity (Cham: Springer International Publishing), 1–19.
Lapébie, P., Lombard, V., Drula, E., Terrapon, N., and Henrissat, B. (2019). Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 2043. doi: 10.1038/s41467-019-10068-5
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, gkab301. doi: 10.1093/nar/gkab301
Loosdrecht, M. C. M., van Nielsen, P. H., Lopez-Vazquez, C. M., and Brdjanovic, D. (Eds.). (2016). Experimental Methods in Wastewater Treatment. London: IWA Publishing.
Ludwig, W. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Luo, Y., Yao, J., Wang, X., Zheng, M., Guo, D., and Chen, Y. (2020). Efficient municipal wastewater treatment by oxidation ditch process at low temperature: bacterial community structure in activated sludge. Sci. Total Environ. 703, 135031. doi: 10.1016/j.scitotenv.2019.135031
McDaniel, E. A., Wahl, S. A., Ishii, S., Pinto, A., Ziels, R., Nielsen, P. H., et al. (2021). Prospects for multi-omics in the microbial ecology of water engineering. Water Res. 205, 117608. doi: 10.1016/j.watres.2021.117608
McIlroy, S. J., and Nielsen, P. H. (2014). “The family Saprospiraceae,” in The Prokaryotes. Springer Berlin Heidelberg, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg), 863–889.
McKee, L. S., La Rosa, S. L., Westereng, B., Eijsink, V. G., Pope, P. B., and Larsbrink, J. (2021). Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13, 559–581. doi: 10.1111/1758-2229.12980
Moir, J. W. B., and Wood, N. J. (2001). Nitrate and nitrite transport in bacteria. Cell Mol. Life Sci. 58, 215–224. doi: 10.1007/PL00000849
Muszyński, A., Tabernacka, A., and Miłobedzka, A. (2015). Long-term dynamics of the microbial community in a full-scale wastewater treatment plant. Int. Biodeterior. Biodegrad. 100, 44–51. doi: 10.1016/j.ibiod.2015.02.008
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nielsen, P. H., Daims, H., and Lemmer, H. (Eds.). (2009). FISH Handbook for Biological Wastewater Treatment: Identification and Quantification of Microorganisms in Activated Sludge and Biofilms by FISH. New York, NY; London : IWA Publishing.
Nielsen, P. H., McIlroy, S. J., Albertsen, M., and Nierychlo, M. (2019). Re-evaluating the microbiology of the enhanced biological phosphorus removal process. Curr. Opin. Biotechnol. 57, 111–118. doi: 10.1016/j.copbio.2019.03.008
Nierychlo, M., Andersen, K. S., Xu, Y., Green, N., Jiang, C., Albertsen, M., et al. (2020). MiDAS 3: an ecosystem-specific reference database, taxonomy and knowledge platform for activated sludge and anaerobic digesters reveals species-level microbiome composition of activated sludge. Water Res. 182, 115955. doi: 10.1016/j.watres.2020.115955
Noji, S., Nohno, T., Saito, T., and Taniguchi, S. (1989). The narK gene product participates in nitrate transport induced in Escherichia coli nitrate-respiring cells. FEBS Lett. 252, 139–143. doi: 10.1016/0014-5793(89)80906-8
Parks, D. H., Chuvochina, M., Chaumeil, P.-A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P.-A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Petriglieri, F., Singleton, C., Kondrotaite, Z., Dueholm, M. K. D., McDaniel, E. A., McMahon, K. D., et al. (2021a). Re-evaluation of the phylogenetic diversity and global distribution of the genus Candidatus Accumulibacter. mSystems. doi: 10.1101/2021.12.20.473458
Petriglieri, F., Singleton, C., Peces, M., Petersen, J. F., Nierychlo, M., and Nielsen, P. H. (2021b). “Candidatus Dechloromonas phosphoritropha” and “Ca. D. phosphorivorans”, novel polyphosphate accumulating organisms abundant in wastewater treatment systems. ISME J. 15, 3605–614. doi: 10.1101/2020.11.05.369777
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Qasim, S. R. (2017). Wastewater Treatment Plants: Planning, Design, and Operation, 2nd Edn. New York, NY: Routledge.
R Core Team (2020). R: A Language and Enviroment for Statistical Computing. Viena: R Foundation for Statistical Computing.
Ren, Y., Ngo, H. H., Guo, W., Ni, B.-J., and Liu, Y. (2019). Linking the nitrous oxide production and mitigation with the microbial community in wastewater treatment: a review. Bioresour. Technol. Rep. 7, 100191. doi: 10.1016/j.biteb.2019.100191
Roy, D., Lemay, J.-F., Drogui, P., Tyagi, R. D., Landry, D., and Rahni, M. (2020). Identifying the link between MBRs' key operating parameters and bacterial community: a step towards optimized leachate treatment. Water Res. 172, 115509. doi: 10.1016/j.watres.2020.115509
Schambeck, C. M., Magnus, B. S., de Souza, L. C. R., Leite, W. R. M., Derlon, N., Guimarães, L. B., et al. (2020). Biopolymers recovery: dynamics and characterization of alginate-like exopolymers in an aerobic granular sludge system treating municipal wastewater without sludge inoculum. J. Environ. Manag. 263, 110394. doi: 10.1016/j.jenvman.2020.110394
Schauer, M., and Hahn, M. W. (2005). Diversity and phylogenetic affiliations of morphologically conspicuous large filamentous bacteria occurring in the pelagic zones of a broad spectrum of freshwater habitats. Appl. Environ. Microbiol. 71, 1931–1940. doi: 10.1128/AEM.71.4.1931-1940.2005
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Schoch, C. L., Ciufo, S., Domrachev, M., Hotton, C. L., Kannan, S., Khovanskaya, R., et al. (2020). NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database J. Biol. Databases Curat 2020, baaa062. doi: 10.1093/database/baaa062
Seshan, H., Santillan, E., Constancias, F., Chandra Segaran, U.S., Williams, R.B.H., and Wuertz, S. (2021). Metagenomics and Metatranscriptomics Reveal Pathway of 3-Chloroaniline Degradation in Wastewater Reactors. bioRxiv 2021.05.02.442374. doi: 10.1101/2021.05.02.442374
Singleton, C. M., Petriglieri, F., Kristensen, J. M., Kirkegaard, R. H., Michaelsen, T. Y., Andersen, M. H., et al. (2021). Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 12, 2009. doi: 10.1038/s41467-021-22203-2
Speth, D. R., in't Zandt, M. H., Guerrero-Cruz, S., Dutilh, B. E., and Jetten, M. S. M. (2016). Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nat. Commun. 7, 11172. doi: 10.1038/ncomms11172
Sun, G., Zhu, Y., Saeed, T., Zhang, G., and Lu, X. (2012). Nitrogen removal and microbial community profiles in six wetland columns receiving high ammonia load. Chem. Eng. J. 203, 326–332. doi: 10.1016/j.cej.2012.07.052
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B., Wu, C. H., and the UniProt Consortium (2015). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. doi: 10.1093/bioinformatics/btu739
Terrapon, N., Lombard, V., Drula, É., Lapébie, P., Al-Masaudi, S., Gilbert, H. J., et al. (2018). PULDB: the expanded database of polysaccharide utilization loci. Nucleic Acids Res. 46, D677–D683. doi: 10.1093/nar/gkx1022
Thomas, F., Hehemann, J.-H., Rebuffet, E., Czjzek, M., and Michel, G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2, 93. doi: 10.3389/fmicb.2011.00093
Vallenet, D., Calteau, A., Dubois, M., Amours, P., Bazin, A., Beuvin, M., et al. (2019). MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 48, gkz926. doi: 10.1093/nar/gkz926
van Veen, W. L., van der Kooij, D., Geuze, E. C. W. A., and van der Vlies, A. W. (1973). Investigations on the sheathed bacterium Haliscomenobacter hydrossis gen.n., sp.n., isolated from activated sludge. Antonie Van Leeuwenhoek 39, 207–216. doi: 10.1007/BF02578853
von Sperling, M. (2007). Wastewater Characteristics, Treatment and Disposal, Biological Wastewater Treatment Series. London: IWA Publication.
Wagner, M., Amann, R., Kämpfer, P., Assmus, B., Hartmann, A., Hutzler, P., et al. (1994). Identification and in situ detection of gram-negative filamentous bacteria in activated sludge. Syst. Appl. Microbiol. 17, 405–417. doi: 10.1016/S0723-2020(11)80058-5
Ward, D. E., Kengen, S. W. M., van der Oost, J., and de Vos, W. M. (2000). Purification and Characterization of the alanine aminotransferase from the hyperthermophilic archaeon pyrococcus furiosus and its role in alanine production. J. Bacteriol. 182, 2559–2566. doi: 10.1128/JB.182.9.2559-2566.2000
Wen, Q., Chen, Z., Tian, T., and Chen, W. (2010). Effects of phosphorus and nitrogen limitation on PHA production in activated sludge. J. Environ. Sci. 22, 1602–1607. doi: 10.1016/S1001-0742(09)60295-3
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis, 2nd Edn. Cham: Use R! Springer International Publishing; Springer.
Xia, Y., Kong, Y., Thomsen, T. R., and Halkjær Nielsen, P. (2008). Identification and ecophysiological characterization of epiphytic protein-hydrolyzing Saprospiraceae (“Candidatus Epiflobacter” spp.) in activated sludge. Appl. Environ. Microbiol. 74, 2229–2238. doi: 10.1128/AEM.02502-07
Xu, S., Yao, J., Ainiwaer, M., Hong, Y., and Zhang, Y. (2018). Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. BioMed Res. Int. 2018. 1–8. doi: 10.1155/2018/8278970
Yang, Q., Xiong, P., Ding, P., Chu, L., and Wang, J. (2015). Treatment of petrochemical wastewater by microaerobic hydrolysis and anoxic/oxic processes and analysis of bacterial diversity. Bioresour. Technol. 196, 169–175. doi: 10.1016/j.biortech.2015.07.087
Yang, Q., Zhao, H., and Du, B. (2017). Bacteria and bacteriophage communities in bulking and non-bulking activated sludge in full-scale municipal wastewater treatment systems. Biochem. Eng. J. 119, 101–111. doi: 10.1016/j.bej.2016.12.017
Yilmaz, L. S., Parnerkar, S., and Noguera, D. R. (2011). mathFISH, a web tool that uses thermodynamics-based mathematical models for in silico evaluation of oligonucleotide probes for fluorescence in situ hybridization. Appl. Environ. Microbiol. 77, 1118–1122. doi: 10.1128/AEM.01733-10
Keywords: wastewater, activated sludge, Saprospiraceae, OLB8, high-quality MAGs, FISH–Raman, MiDAS
Citation: Kondrotaite Z, Valk LC, Petriglieri F, Singleton C, Nierychlo M, Dueholm MKD and Nielsen PH (2022) Diversity and Ecophysiology of the Genus OLB8 and Other Abundant Uncultured Saprospiraceae Genera in Global Wastewater Treatment Systems. Front. Microbiol. 13:917553. doi: 10.3389/fmicb.2022.917553
Received: 11 April 2022; Accepted: 30 May 2022;
Published: 08 July 2022.
Edited by:
Akihiko Terada, Tokyo University of Agriculture and Technology, JapanReviewed by:
Tomonori Kindaichi, Hiroshima University, JapanCopyright © 2022 Kondrotaite, Valk, Petriglieri, Singleton, Nierychlo, Dueholm and Nielsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Per H. Nielsen, cGhuQGJpby5hYXUuZGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.