
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 23 June 2022
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.914764
This article is part of the Research Topic"One Health" Approach For Revealing Reservoirs And Transmission Of Antimicrobial Resistance, Volume IIView all 22 articles
 Xiaoshen Li1,2,3Longfei Xie1,2,3Honghao Huang1,2,3Zhi Li1,2,3Guihua Li1,2,3Peng Liu4
Xiaoshen Li1,2,3Longfei Xie1,2,3Honghao Huang1,2,3Zhi Li1,2,3Guihua Li1,2,3Peng Liu4 Danyu Xiao1,2,3Xucai Zhang1,2,3
Danyu Xiao1,2,3Xucai Zhang1,2,3 Wenguang Xiong1,2,3*
Wenguang Xiong1,2,3* Zhenling Zeng1,2,3*
Zhenling Zeng1,2,3*Livestock-associated methicillin-resistant Staphylococcus aureus (LA-MRSA) is an important zoonotic microorganism that is increasingly causing public health concern worldwide. The objective of this study was to determine the transmission and occurrence of MRSA in a slaughterhouse environment and evaluate its antimicrobial resistance and genetic characterization. In this study, we conducted a comprehensive epidemiological survey of S. aureus by spa typing and whole-genome sequencing (WGS) of samples obtained from the pork production chain, the environment, and community residents. To clarify the evolutionary relationships of MRSA sequence type (ST) 398 in this study and global isolates, 197 published whole-genome sequences data of MRSA ST398 strains were downloaded from the GenBank database and included in the phylogenetic analysis. A total of 585 porcine samples (snout and carcass swabs), 78 human nasal samples, and 136 environmental samples were collected. The MRSA isolates were detected at higher frequencies in samples from swine (15.0%) than carcasses (10.0%), slaughterhouse workers (8.0%), community residents (0%), and environment samples (5.9%). The spa typing results showed that t571 accounted for a higher proportion than other spa types. Closely related isolates from the samples of swine, slaughterhouse workers, carcasses, carrier vehicle, and surrounding fishpond water indicate that MRSA ST398 strains may spread among swine, humans, and the environment. MRSA ST398-t571 isolates were genetically different from global strains, except for two Korean isolates, which showed genetic closeness with it. In addition, a MRSA ST398 isolate recovered from an infected patient in Europe differed by only 31 SNPs from the airborne dust-associated strain isolated in this study, thereby suggesting potential transmission among different countries. Antimicrobial susceptibility testing results demonstrated that 99.0% (96/97) of MRSA and 95.1% (231/243) of methicillin-sensitive S. aureus (MSSA) showed multidrug-resistant (MDR) phenotypes. According to WGS analysis, the poxtA-carrying segment (IS431mec-optrA-IS1216-fexB-IS431mec) was reported in MRSA ST398 isolates for the first time. The coexistence of cfr and optrA in a plasmid was first detected in MRSA ST398. The potential transmission of MRSA among humans, animals, and the environment is a cause for concern. The emergence and transmission of LA-MRSA ST398 with high levels of resistance profiles highlight the urgent need for LA-MRSA surveillance.
Methicillin-resistant Staphylococcus aureus (MRSA) is an important pathogen that causes community- and hospital-acquired infections (Larsen et al., 2015; Nakaminami et al., 2020). Postoperative secondary infections caused by multidrug-resistant (MDR) MRSA make clinical treatment difficult (Van Duin and Paterson, 2016; Bassetti et al., 2020). Swine is an important host for the MRSA strain. Previous studies highlighted the potential zoonotic risks associated with MRSA carriage in swine farmers. Such carriage may be a consequence of frequent contact with infected animals, causing public health concern worldwide (Armand-Lefevre et al., 2005; Kinross et al., 2017).
In 2005, the Netherlands reported a case of livestock-associated MRSA (LA-MRSA) infection in a 6-month-old infant residing on a swine farm with sequence type (ST) 398 (Voss et al., 2005). According to the data from the GenBank database, we found that MRSA ST398 has increasingly spread worldwide and is widespread in Europe. It is currently found in more than 20 countries, including Germany, Denmark, Switzerland, Belgium, Netherlands, France, Italy, United Kingdom, Greece, Spain, Czech Republic, Poland, United States, and Canada. The sources are varied, but the predominant ones are swine and humans. In particular, the application of whole-genome sequencing (WGS) has revealed potential drivers for LA-MRSA ST398 dissemination, such as trading of colonized swine, contaminated transport vehicles, and colonized human (Grøntvedt et al., 2016; da Silva et al., 2017; Sieber et al., 2018). Notably, S. aureus protein A (spa) t571 is more strongly connected with human infections than animal colonization strains (Bonnet et al., 2018; Wardyn et al., 2018). Methicillin-sensitive S. aureus (MSSA) ST398-t571 was detected in nine families from the Dominican Republic living in New York; these families had no contact with livestock (Bhat et al., 2009). Furthermore, it was disseminated among detainees and patients in America (Davies et al., 2011; David et al., 2013). ST398-t571 MSSA isolates caused fatal necrotizing pneumonia and bloodstream infections in the United States and Europe (Rasigade et al., 2010; Bonnet et al., 2018; Wardyn et al., 2018).
The prevalence of MDR phenomenon in MRSA strains also attracted global attention. Oxazolidinones have never been approved for veterinary use in any country worldwide, but linezolid-resistant MRSA isolate has been reported in animal hosts. Linezolid is a last resort antimicrobial agent used to treat serious infections in humans caused by MDR Gram-positive bacteria, such as vancomycin-resistant enterococci, MRSA, and penicillin-resistant pneumococci (Bozdogan and Appelbaum, 2004). To date, three acquired genes, namely, cfr (Li S.-M. et al., 2018; Sun et al., 2018), optrA (Sun et al., 2018), and poxtA (Chen et al., 2021) conferring linezolid resistance have been mainly described in staphylococci of livestock origin. Aside from mediating linezolid resistance, these genes could confer reduced susceptibility or resistance to other antimicrobial agents (Shen et al., 2013; Wang Y. et al., 2015; Antonelli et al., 2018). Considering that they are frequently located in plasmids or other mobile genetic elements, they could be transferred between species and genera (Shen et al., 2013; Wang J. et al., 2015; Sun et al., 2018; Chen et al., 2021).
Unlike the widespread use of ST398 type of LA-MRSA strains in European and North American countries, LA-MRSA-ST9 predominates in China (Chuang and Huang, 2015; Wang J. et al., 2015; Li S.-M. et al., 2018). Previous studies showed that ST398 type of MSSA strains was frequently colonized in slaughter pigs in northeast China (Yan et al., 2014), while LA-MRSA ST398 strains were mainly isolated from humans in China (Song et al., 2017; Chen et al., 2020). In the last several years, MRSA ST398 has been identified sporadically in swine farms (Li W. et al., 2018; Zou et al., 2021), pork (Li et al., 2021), milk (Cui et al., 2020), and monkey feces (Tang et al., 2021) in China. Continued monitoring for MRSA in food-producing animals is urgently required. Comprehensive studies on the transmission of LA-MRSA among swine, humans, and the environment in swine slaughterhouses in China are few. To gain insight into the prevalence and transmission of LA-MRSA and MSSA in a typical swine slaughterhouse, spa typing, WGS, and bioinformatic analysis were conducted. Samples were collected from swine, carcass, environment, slaughterhouse workers, and neighboring community residents in Guangzhou, China.
A slaughterhouse with a capacity of 3,000 swine in Guangzhou, Guangdong, China, was selected for sampling. A total of 799 samples from swine, carcasses, and humans, as well as samples from their surrounding environmental samples were collected from November 2020 to April 2021. The environment samples included airborne dusk (800–130 cm off the ground; n = 60), effluent (∼30 ml; n = 5), sludge (∼30 g; n = 5), workstations (surface swabs; n = 5), vehicles carrying swine (surface swabs; n = 5), river water (∼30 ml; n = 5), fishpond water (∼30 ml; n = 5), farmland soil, and vegetables (∼30 g; n = 46). Air samples were collected using a stationary method by placing the plates with an open lid inside the slaughterhouse area. Swine snout (n = 535), human nasal (slaughterhouse workers, n = 50; community residents, n = 28), and carcass swabs (n = 50) were collected (Supplementary Table 1). The swine samples were mainly from Guangdong, Hunan, Guangxi, and Jiangxi Provinces. Samples from individual pigs were processed separately. The slaughterhouse workers and neighboring residents signed an informed consent form and were asked to agree to nose swabbing. Information details of the volunteers are summarized in Supplementary Table 2.
The samples were soaked in 7.5% sodium chloride broth (Land Bridge, Beijing, China) and cultured at 37°C for 12 h. Then, a loopful of the broth was streaked onto chromogenic S. aureus agar (Huankai Microbial, Guangdong, China). One colony with typical S. aureus morphology was selected from each sample and streaked onto Mannitol salt agar (Huankai Microbial, Guangdong, China) to obtain a purified isolate colony. Each isolate colony was identified by polymerase chain reaction (PCR) using the nuc gene to detect S. aureus and the nuc and mecA (or mecC) genes to detect the MRSA isolate (Zhang et al., 2004; Harrison et al., 2013).
Antimicrobial susceptibility was tested using the agar dilution method according to the guidelines of Clinical and Laboratory Standards Institute guidelines (CLSI, 2018). Tigecycline were assessed by broth microdilution as recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST Version 6.0) (EUCAST, 2016). S. aureus ATCC 29213 was set as the quality control strain. The antimicrobials were amoxicillin (AMO), oxacillin (OXA), penicillin (PEN), ceftiofur (CEF), cefoxitin (FOX), gentamicin (GEN), amikacin (AMI), doxycycline (DOX), tigecycline (TIG), florfenicol (FFC), erythromycin (ERY), tilmicosin (TMI), rifampicin (RIF), vancomycin (VAN), clindamycin (CLI), tiamulin (TIA), ciprofloxacin (CIP), enrofloxacin (ENR), fosfomycin (FOS), linezolid (LZD), tedizolid (TZD), and sulfamethoxazole-trimethoprim (SXT).
All identified MSSA and MRSA isolates were subjected to spa typing by PCR amplification and sequencing of the polymorphic region of the spa gene (Harmsen et al., 2003). Spa type was assigned using spa-plugin in BioNumerics v7.6 (Applied Maths, Sint-Martens-Latem, Belgium). Minimum spanning trees based on spa types were constructed in BioNumerics v7.6 (Applied Maths, Sint-Martens-Latem, Belgium). Based on spa type, source, and antimicrobial resistance (AMR), 50 MRSA isolates were selected for WGS (Supplementary Tables 4, 6). The bacterial DNA was extracted using the HiPure Bacterial DNA Kit (Magen, Guangzhou, China). Illumina NOVAseq 6000 sequencing platform (Novogene Company, Beijing, China) and PacBio RS II sequencing platform (Biochip Company, Tianjin, China) were used for WGS. SMRTbell library (10–20 kb in size) was prepared by the ligation of hairpin adaptors at both ends, and the resulting library was selected according to size using Blue Pippin with a 7–10 kb cutoff for Pacbio sequencing. Illumina Novaseq sequences were assembled using CLC Genomics Workbench 10 (CLC Bio, Aarhus, Denmark). Pacbio sequences were assembled using a hierarchical genome assembly process (Chin et al., 2013). The assembled Pacbio sequences were corrected by using Burrows-Wheeler Aligner’s Smith-Waterman Alignment software to ensure that their integrity was in accordance with those of Novaseq sequences (Li and Durbin, 2010). The genome assemblies of MRSA ST398 isolates were deposited in the GenBank and were registered with BioProject number PRJNA793117. The plasmid pSA159 carrying cfr and optrA in GD4SA159 and poxtA-positive strain GD4SA108 were annotated by RAST annotation server1 (Overbeek et al., 2014). Multi-locus sequence type (MLST), acquired resistance genes, and Staphylococcal cassette chromosome mec (SCCmec) were identified against MLST (Aanensen and Spratt, 2005), ResFinder (Zankari et al., 2012), and SCCmecFinder (Kaya et al., 2018) databases (accessed on 16 June 2020) using a mapping approach implemented in SRST2 (Inouye et al., 2014).
To clarify the genetic relatedness of MRSA ST398 in this study and global isolates, 197 published whole-genome sequences data of MRSA ST398 strains were downloaded from the GenBank database and included for analysis (Supplementary Table 3). Based on the draft genome sequences, a core-genome single nucleotide polymorphism (SNP)-based maximum-likelihood (ML) phylogenetic tree was constructed. The genome of MRSA ST398 isolate (GenBank accession number: JAASKV000000000) was used as a reference. The tree was constructed using Parsnp in the Harvest package (Treangen et al., 2014) with default parameter settings and was annotated on the EvolView website https://evolgenius.info. A comparison of the genetic context was generated using Easyfig 2.1 (Sullivan et al., 2011) and BLAST Ring Image Generator (BRIG) (Alikhan et al., 2011).
Descriptive and comparative analyses were performed in Graphpad Prism 8.0 (Graphpad Software, La Jolla, CA, United States), and statistical analyses were performed in SPSS 20.0 (IBM Corp., Armonk, NY, United States). Fisher’s exact test was used to test whether differences in drug resistance phenotypes among human-, environment-, and swine-associated isolates were significant, and also evaluated between MRSA and MSSA isolates.
Ninety-seven MRSA and 243 MSSA isolates were collected from 799 samples (Supplementary Table 1). MRSA isolates were detected in swine (15.0%, 80/535), carcass (10.0%, 5/50), slaughterhouse workers (8.0%, 4/50), and environmental samples (5.9%, 8/136), whereas no MRSA-positive isolate was found in the samples from community (Figure 1A). MSSA-positive isolates were detected in swine (35.5%, 190/535), carcasses (18.0%, 9/50), slaughterhouse workers (26.0%, 13/50), community residents (7.1%, 2/28), and environmental samples (21.3%, 29/136) (Figure 1A).
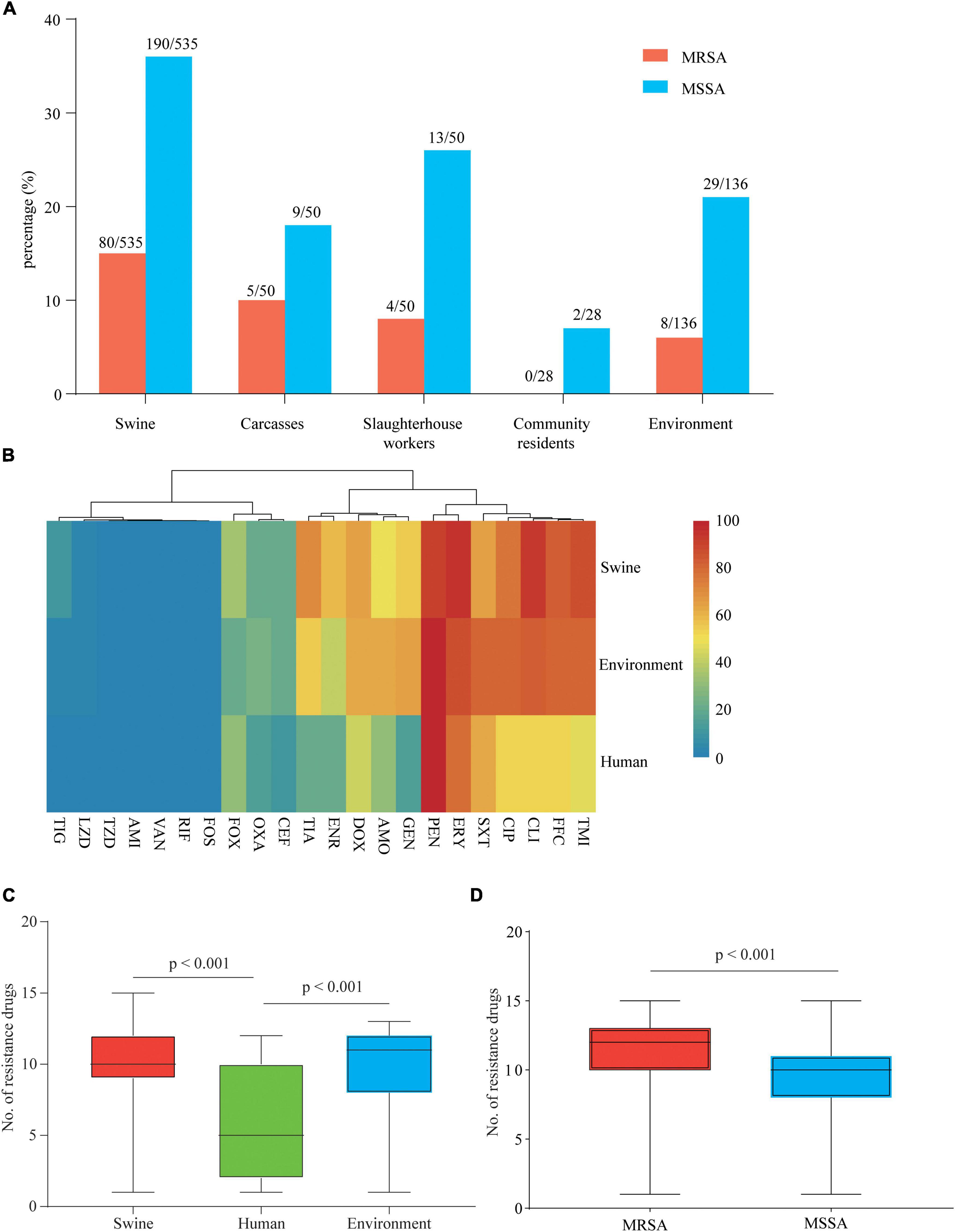
Figure 1. Prevalence and drug resistance of isolates recovered from samples from various origins. (A) Prevalence of methicillin-susceptible Staphylococcus aureus (MSSA) and methicillin-resistant S. aureus (MRSA) in samples from various origins along and around a slaughterhouse in Guangzhou, China. (B) Each cell in the heat map indicates the percentage of strains with resistance to a particular drug. (C) The bar graph indicates the total number of drugs with resistance within each swine, human, and environment-associated isolate. (D) The bar graph indicates the total number of drugs with resistance within each MSSA and MRSA isolate. The numbers of swine, human, and environment-associated strains were 284, 19, and 37, respectively. The numbers of MSSA and MRSA isolates were 243 and 97, respectively.
Antimicrobial susceptibility tests of 340 isolates were performed using 22 antimicrobials (Supplementary Table 4). According to the results, 99.0% (96/97) of MRSA and 95.1% (231/243) of MSSA showed MDR phenotypes. Swine- and environment-associated isolates were resistant to a larger number of antimicrobial agents compared with human-associated isolates (median 10, 11 vs. 5; p < 0.001) (Figure 1C). The swine- and environment-associated isolates showed high resistance levels to florfenicol, erythromycin, tilmicosin, and clindamycin (> 80%). The trimethoprim/sulfonamide resistance rate of environment-associated isolates was significantly higher than that of isolates from swine or human samples (p < 0.05). All isolates from different sources showed high resistance to penicillin (> 90%) but low resistance to tigecycline, linezolid, fosfomycin, and rifampicin (< 11.47%). All isolates were susceptible to amikacin, vancomycin, and Acetazolamide (Figure 1B and Supplementary Table 5). Compared with MSSA isolates, MRSA isolates were resistant to most of the antimicrobial agents (median 12 vs. 10; p < 0.001) (Figure 1D). The majority of isolates recovered in the study showed a MDR phenotype.
In total, 26 spa types were identified among the 340 S. aureus isolates, with MSSA isolates (25 spa types) showing greater diversity than MRSA (six spa types) and three isolates were not typed (Figure 2A and Supplementary Table 4). Besides t571 (64.9%, 63/97), t034, t899, t114, and t1793 were also detected in MRSA isolates. In addition to t571 (72.8%, 177/243), t034, t899, t114, t1793, and t011 were also detected in MSSA isolates (Figure 2A).
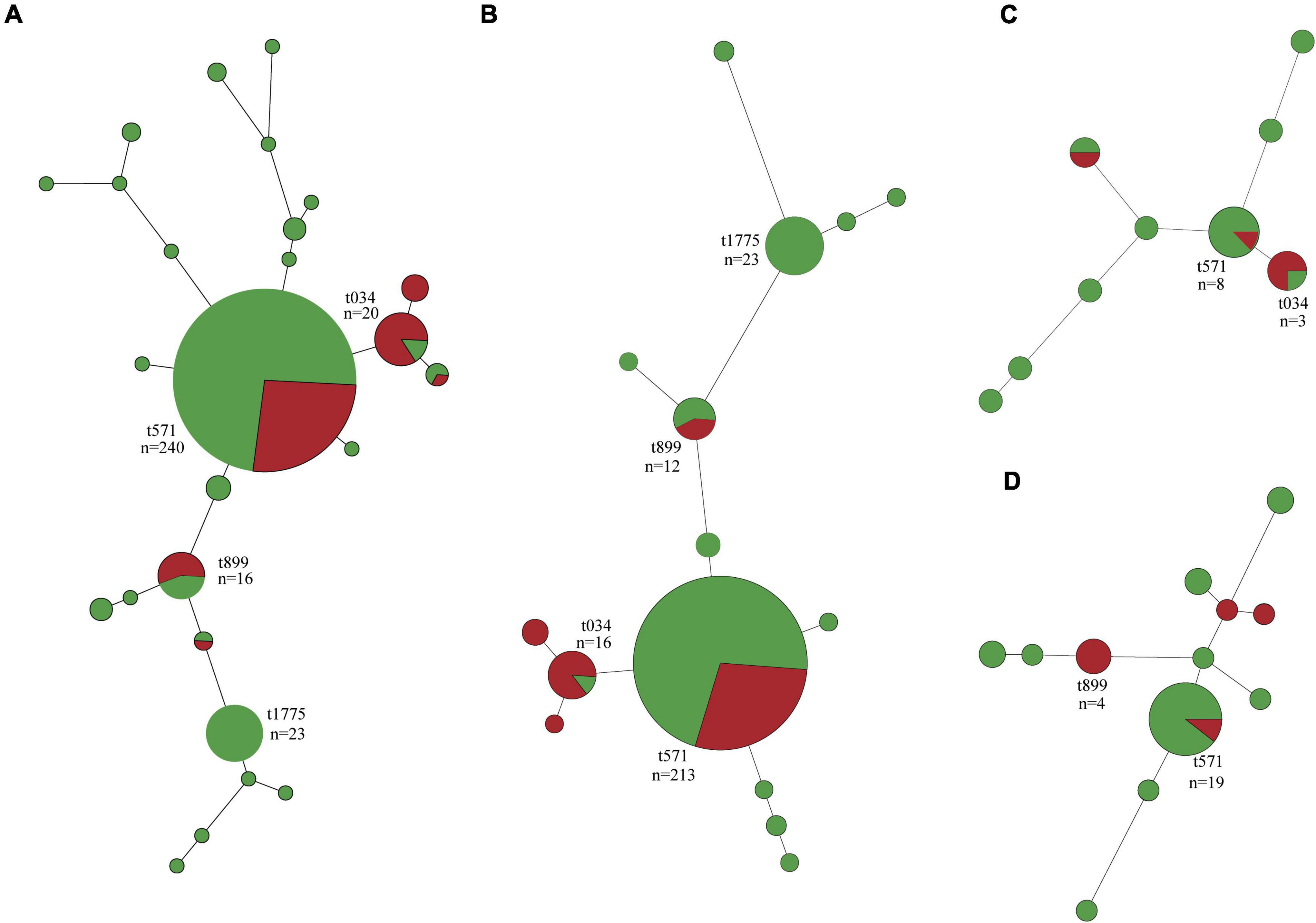
Figure 2. Minimum spanning tree of methicillin-susceptible Staphylococcus aureus (MSSA, n = 243) and methicillin-resistant S. aureus (MRSA, n = 97) isolates by spa type. (A) All isolates. (B) Isolates of swine origin. (C) Isolates of human origin. (D) Isolates of environmental origin. Each node represents a single spa type. The size of the node is proportional to the number of isolates represented by a said node. Branch lengths between nodes are proportional to the number of alleles that differ between the two linked nodes. Selected nodes are labeled with corresponding spa type and number of isolates represented. Red nodes, MRSA; green nodes, MSSA.
Swine-associated MRSA isolates were dominated by t571 (71.4%, 60/84); spa type t034 and t899 accounted for a small proportion (Figure 2B). The human- and environment-associated MRSA isolates primarily had t034 (50%, 2/4) and t899 (50%, 4/8), respectively (Figures 2C,D). MSSA t571 clonal lineage predominated in swine, human, and environment-associated isolates (Figure 2).
Among 50 MRSA isolates sequenced, ST398 was the dominant clonal lineage (84.0%, 42/50). Most MRSA isolates belonged to ST398-t571 (42.0%, 21/50), followed by ST398-t034 (30%, 15/50), ST398-t011 (10%, 5/50), ST9-t899 (14.0%, 7/50), and one ST1-t114 isolate associated with human.
Phylogenetic analyses revealed that MRSA ST398 was mainly clustered into three clades, corresponding to clonal lineage identified as SCCmec V-t034, SCCmec V-t011, and SCCmec IV-t011 (Figure 3). Notably, SCCmec were diversified in MRSA ST398-t571 clonal lineage, such as SCCmec V, IX, and XII subtypes, which were predicted with a template coverage of less than 60%. These isolates differed in 6,893 core genome SNPs. Interestingly, the isolates (block 1) found in this study were genetically different from global strains, except for the Korean strains (block 2) that showed close genetic relationship with the abovementioned isolates. Swine-associated Korean isolate PJFA-521M (GenBank accession number: SRKD00000000) was closely related to human-associated Korean isolate PJFH-522M (GenBank accession number: RKRI00000000) (2 SNPs difference) and swine-associated Chinese isolate (HN2SA18) (35 SNPs difference) (Figure 3). Furthermore, a strain OP17 (GenBank accession number: VKBM00000000) isolated from an infected patient in Europe differed by only 31 SNPs from the airborne dust-associated isolate (GDK3SA10) isolated in this study. The potential transmission of MRSA among swine, human, carcass, carrier vehicle, and fishpond water was observed around the slaughterhouse. Swine-associated isolate GDA3SA01 was closely related to GD4SA98 recovered from carrier vehicle (swine) (17 SNPs difference). Meanwhile, GD4SA98 was closely related to the GD4SA24 recovered from a slaughterhouse worker (1 SNP difference). GD4SA24 was closely related to GD4SA93 recovered from the surrounding fishpond water (with a difference of 8 SNPs). GD4SA24 was closely related to swine-associated isolate GDA3SA01 (with a difference of 16 SNPs). Swine-associated isolate GD4SA116 was closely related to GD4SA159 recovered from carcass (with a difference of 1 SNP). This was also observed among slaughterhouse workers, as GD3SAM4 was closely related to GD3SAT1 (with a difference of 1 SNP).
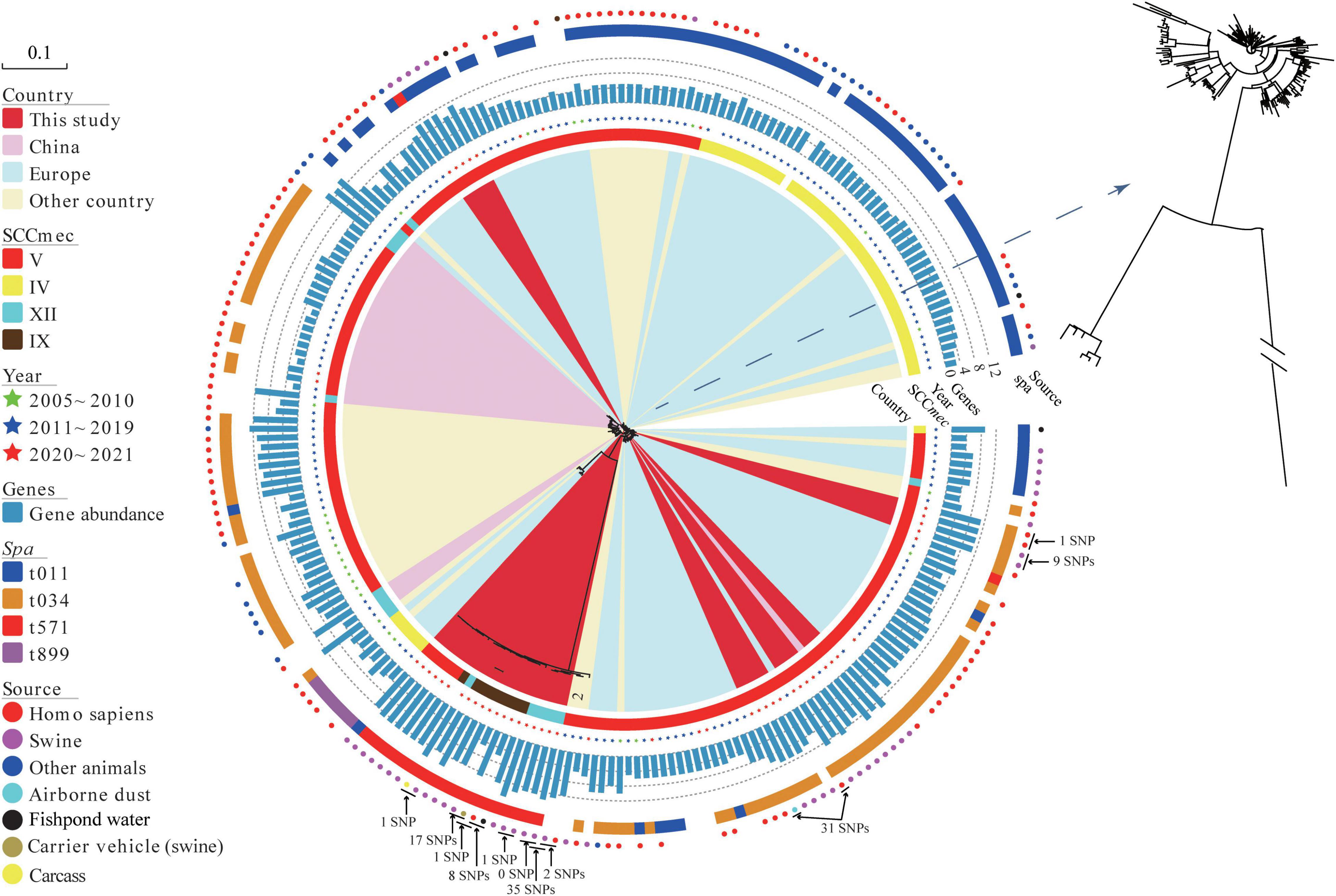
Figure 3. The maximum likelihood (ML) phylogenetic tree of 42 methicillin-resistant Staphylococcus aureus (MRSA) ST398 isolates from China based on single nucleotide polymorphisms in the core genomes. A total of 197 global MRSA ST398 isolates were included for comparison. Regions were indicated by leaf color. Sources and years of the isolates were indicated by circles and stars in different colors, respectively. SCCmec and spa types were indicated by strips in different colors. The branch of Block 2 represents Korean isolates. The total number of antibiotic resistance genes in each isolate was represented by the length of a blue bar chart. Details of the genes of each isolate are given in Supplementary Table 7. The core of phylogenetic tree was amplified for a clearer display.
Antibiotic resistance genes (ARGs) in MRSA ST398 isolates (42 sequenced isolates in this study and 197 records retrieved in the GenBank database) are displayed in Supplementary Figure 1 (Supplementary Table 7). A total of thirty-three ARGs grouped into nine types were found in samples from swine, human, and environment. Figure 3 shows that the abundance of resistance genes of sequenced MRSA ST398 isolates in this study was higher than that of global isolates. Notably, the prevalence rates of penicillin/methicillin resistance gene blaZ and tetracycline resistance gene tetM in MRSA ST398 were 95.82% and 88.28%, respectively. In addition, the detection rates of tetracycline resistance gene tet(K), trimethoprim resistance gene dfrG, lincomycin resistance gene lnu(B), macrolides-lincosamides-streptogramin B (MLSB) resistance gene erm(C), lincosamides-pleuromutilins-streptogramins A resistance gene lsa(E), aminoglycoside resistance genes aac(6′)-aph(2″), ant(9)-Ia, and aadD, and florfenicol resistance gene fexA ranged from 20% to 50%. The multiresistance genes cfr, optrA, and poxtA were detected in MRSA ST398. The coexistence of the cfr and optrA genes was detected in a plasmid of GD4SA159 with oxazolidinone sensitivity that was isolated from carcass. In addition, the swine-associated isolate GD4SA108 carried poxtA along with the resistance genes aac(6′)-aph(2″), aadD, ant(6)-Ia, blaZ, dfrG, erm(C), erm(T), fexA, fexB, lnu(B), lsa(E), mecA, tet(L), tet(M), and qacG. Alarmingly, two slaughterhouse worker-associated isolates (GD3SAM4 and GD3SAT1) carried the cfr gene, as well as the ant(9)-Ia, blaZ, dfrG, erm(A), erm(C), fexA, mecA, tet(K), tet(M), and vga(E). The two isolates showed an MDR phenotype, which indicated resistance to beta-lactams, tetracyclines, florfenicol, erythromycin, tilmicosin, clindamycin, tiamulin, and ciprofloxacin.
To characterize the genetic environment of the cfr and optrA genes, plasmid pSA159 (GenBank accession no. CP090408) from MRSA ST398 isolate GD4SA159 was completely sequenced. It was 52,881 bp in length and had an average GC content of 38.0%. A total of 53 open reading frames (ORFs) coding for proteins of > 50 amino acids were identified. Except for the 20 ORFs encoding hypothetical proteins with no defined function, the products of the remaining 36 ORFs exhibited identities ranging from 33.0% to 100.0% to proteins with known functions, such as AMR, plasmid replication, and conjugative transfer or transposition. The plasmid pSA159 with the optrA-fexA-aacA-aphD-aadD-ble-cfr resistance gene cluster was subjected to a comparative analysis with other plasmids (Figure 4). The 16,956 bp segment between optrA and tnpA showed 99.68% identity with the cfr- and optrA-carrying region of plasmid pwo28-3 from pig-associated S. sciuri (KT601170). The segment truncated optrA displayed 99.8% identity with the corresponding region of plasmid pk8D6P-cfr from duck-associated S. sciuri (CP065793). The segment between tnpA and IS257 showed 99.96% identity with the corresponding region of plasmid pWo27-9 from S. sciuri (KX982169).
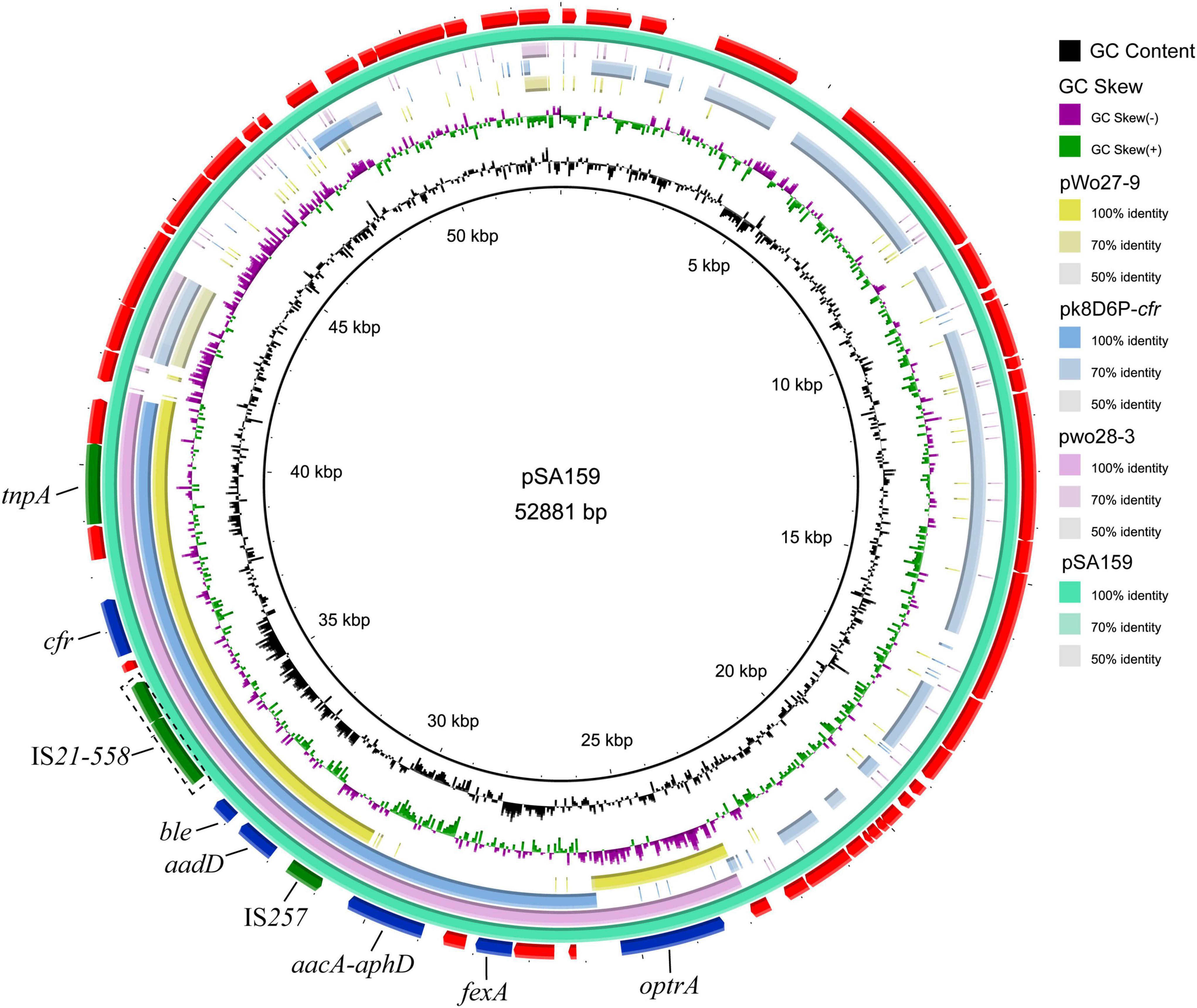
Figure 4. Ring comparison of plasmid pSA159 (GD4SA159) using BRIG. Arrows indicate the positions and directions of gene transcription. Blue and green arrows indicate resistance genes and mobile genetic elements (MGEs), respectively. Genes encoding hypothetical proteins and proteins for plasmid stability are depicted in red.
The genome of MRSA-ST398 GD4SA108 (GenBank accession no. CP090375) containing the poxtA gene was completely sequenced and was 2,864,441 bp in length. It had an average GC content of 32.0%. As shown in Figure 5, the genetic context of poxtA from plasmids pY80, pC10, pfas4-1, and pDY32-poxtA was commonly flanked by two copies of IS1216 elements in the same orientation. By contrast, the poxtA gene from chromosomal genome of GD4SA108 was flanked by two copies of IS431mec elements in the same orientation. It could form a circular intermediate (7,270 bp, region A) that contained IS1216 and fexB genes (Figure 5). Plasmids pC10, pfas4-1, and pDY32-poxtA from Enterococcus faecalis, E. hirae, and E. faecium carried the poxtA gene and tetracycline resistance genes tetM and tetL. Plasmid pY80 from S. haemolyticus contained tetL and aadD1 aside from the poxtA gene. The results suggested that poxtA gene may be transmitted with other drug resistance genes, especially tetracycline-resistant genes.
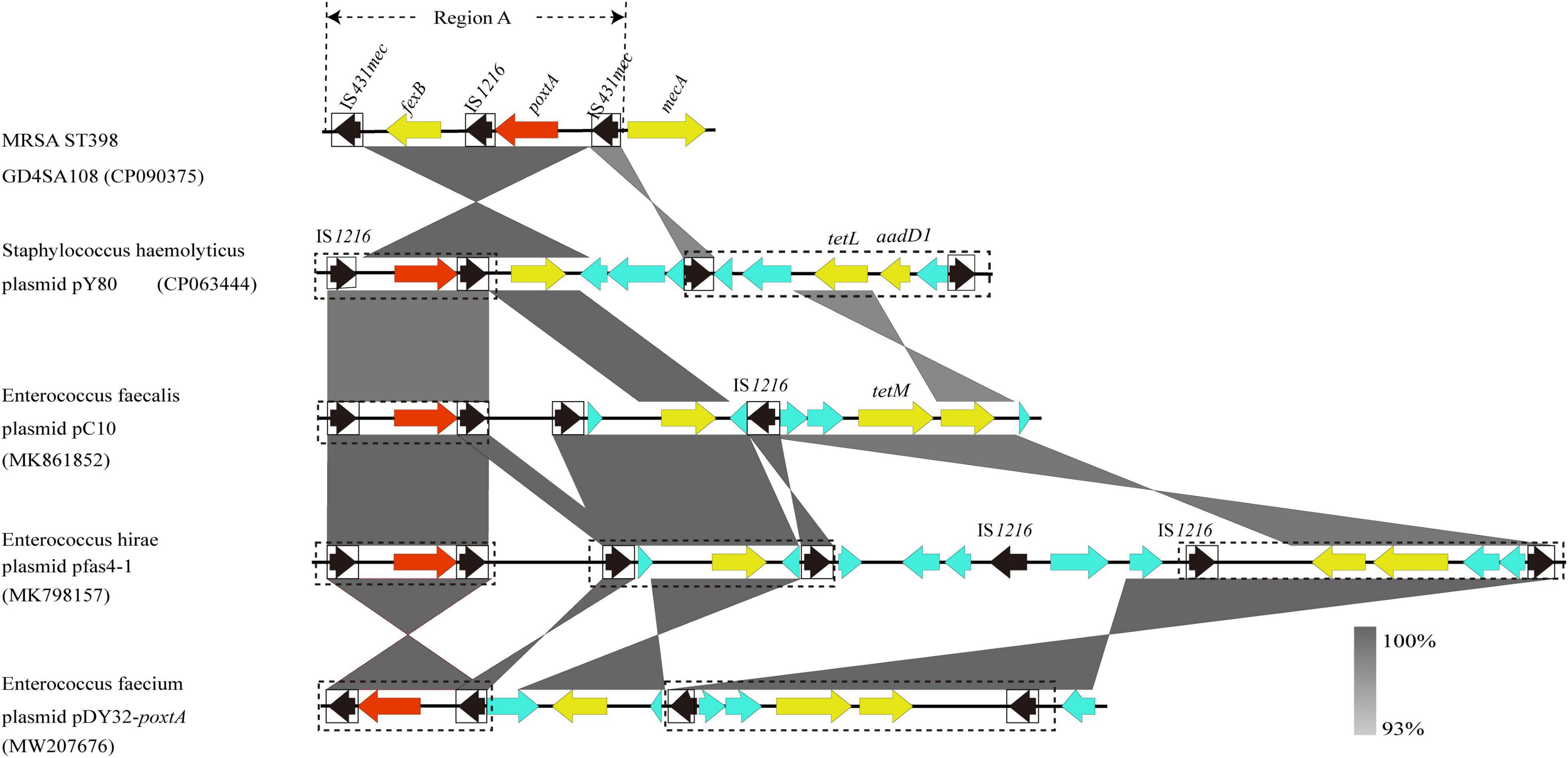
Figure 5. Schematic presentation of the genetic context of poxtA-flanking regions in the chromosomal DNAs of the isolate GD4SA108 compared with the corresponding regions of other strains in the NCBI database. Regions of >93% homologies are shaded gray. Differently colored arrows indicate the direction of transcription of the different genes. The poxtA gene is shown as a red arrow, and yellow arrows indicate other resistance genes. Genes with unknown or other functions are depicted in light blue. MGEs are indicated as black dotted boxes. Insertion sequences are indicated as black boxes, with the black arrow inside the box showing the transposase gene. Region A could form a circular intermediate.
MRSA ST9 is a common clonal lineage among swine-associated MRSA in China (Zhou et al., 2018). In this study, fewer MRSA ST9 isolates were found in swine. In contrast, the prevalence of MRSA ST398, which is typically associated with swine and farmers in Europe, was detected in the slaughterhouse. Phylogenetic analyses revealed that MRSA ST398 was predominantly associated with the t571 circulating in the slaughterhouse. This finding differed from those of previous studies, in which t011 and t034 were commonly associated with MRSA ST398 globally (Hansen et al., 2019; Pirolo et al., 2019; Schnitt et al., 2020). The swine contaminated with MRSA ST398 was probably raised in a farm setting before being transported to the slaughterhouse, and this assumption was supported by the occurrence of MRSA ST398-t034/t571 in a pig farm in southern China (Li W. et al., 2018). Additionally, the potential spread was predicted among swine, workers, carcass, carrier vehicle, and surrounding fishpond water, as supported by the analysis of SNP difference (Bartels et al., 2015; Schürch et al., 2018; Bouchami et al., 2020). A previous study on the drivers of LA-MRSA CC398 demonstrated that swine movements were crucial for the spread of MRSA CC398 (Sieber et al., 2018). As a means of transporting for swine, vehicles that test positive for MRSA ST398 were highly likely to contaminate the transport route, leading to widespread infection. In addition, the carcasses transported to the pork market were positive for MRSA ST398, thereby indicating a potential food safety risk. Slaughterhouse workers had a higher detection rate of MRSA ST398 compared with community residents, perhaps due to the prolonged exposure to slaughterhouse conditions. Although it was not detected in the samples from residents, MRSA ST398 was detected in the nearby fishpond and airborne dust, suggesting that residents exposed to the environment were still at risk of infection. The suggestion was corroborated by a previous report (Bos et al., 2016). The infected slaughterhouse workers, carrier vehicles, fishponds, and carcasses could serve as a source for the transmission of MRSA ST398 in the community. However, large-scale sampling and further studies should be conducted to confirm this.
Phylogenetic analysis of MRSA ST398 suggested that infected human or swine may drive the spread of MRSA ST398 among different countries. The diversity of colonizing sources suggested their potential role as reservoirs for transmission. Half of the sequenced MRSA ST398 isolates showed difference in evolutionary relationship (block 1, spa type t571), indicating the development of diversity in the population of MRSA ST398 isolates. MSSA ST398-t571 was relatively rare among livestock isolates (Lienen et al., 2020) and was commonly associated with human infection. Amazingly, spa type t571 was the dominant clonal lineage in MRSA and MSSA isolates in the slaughterhouse (Bonnet et al., 2018; Wardyn et al., 2018). The virulence levels of MRSA- and MSSA-t571 isolates in this study were unclear. The frequent reports of invasive infections caused by S. aureus ST398-t571 isolates suggested that persistent genotypic surveillance of S. aureus strains in humans and animals should be practiced (Song et al., 2017; Chen et al., 2020). The prevalence of MRSA ST398 in the slaughterhouse may be closely related to foreign pig trading and traveling. The details of pig trading and history of human travel abroad could not be identified exactly. Thus, further studies are needed to clarify the reason for the prevalence of MRSA ST398 lineage.
The MRSA carriage among swine, carcass, slaughterhouse workers, and environmental samples in this study was higher than that in samples from slaughterhouses from Shandong (fatteners, 8.2%, 7/85; carcasses, 1.0%, 1/91; slaughterhouse workers, 0%, 0/16; environmental samples, 1.9%, 1/52) (Sun et al., 2019). The difference in MRSA carriage could be due to differences in the location and size of the slaughterhouse. The high prevalence of MRSA carriage in this slaughterhouse might result from the beta-lactam antibiotic usage pressure in animal production (Qiao et al., 2018). Antibiotic resistance is a serious threat to global public health. The resistance phenotypes of swine-associated isolates were generally more severe than those in human-associated isolates (e.g., resistance to florfenicol, gentamicin, doxycycline, clindamycin, tilmicosin, tiamulin, enrofloxacin, and ciprofloxacin) (Figures 1B,C). The differences in AMR phenotype between swine- and human-associated isolates could be due to differences in the type and amount of antimicrobial used. The analyses of AMR genotypes of MRSA ST398 showed that the extremely high detection of tetM (88.28%) was consistent with the characteristic of tetracycline resistance of CC398 clonal lineage (Ward et al., 2014), suggesting that heavy tetracycline use in the swine industry may have promoted the dissemination of this clonal lineage (Moodley et al., 2011).
Oxazolidinones are essential drugs used to address the MDR of Gram-positive bacteria. The linezolid resistance genes cfr, optrA, and poxtA are of concern, because they are often located in mobile genetic elements (MGEs), which could transmit among bacteria by horizontal gene transfer. The coexistence of cfr and optrA in a plasmid was first detected in MRSA ST398, although it has been reported in S. sciuri and E. gallinarum (Li et al., 2016; Fan et al., 2017; Coccitto et al., 2022). The poxtA gene is commonly flanked by IS1216 element (Antonelli et al., 2018; Chen et al., 2021). However, the novel genetic context of poxtA gene flanked by two copies of IS431mec elements was identified, thereby increasing the awareness of the spread of poxtA gene. These cfr, optrA, and poxtA-carrying plasmids or fragments may be disseminated among MRSA in the slaughterhouse and possibly spread to MRSA of human origin. The cfr, optrA, and poxtA genes lead to resistance to chloramphenicol and florfenicol aside from linezolid (Long et al., 2006; Wang Y. et al., 2015; Antonelli et al., 2018). As the use of chloramphenicol in food animals was banned in China in 2002, florfenicol appears to be the only antimicrobial agent to select cfr, optrA, and poxtA from isolates of food animal origin. Thus, rational use of florfenicol in food-producing animals is urgently needed. In addition, SCCmec elements play a crucial role in the spread of methicillin resistance and evolution of MRSA. SCCmec V and SCCmec IV types among CC398-MRSA were prevalent worldwide (Wulf et al., 2008; Kadlec et al., 2009; Lienen et al., 2020). Compared with global isolates, SCCmec IV lineage isolate was absent, and the SCCmec V lineage dominated in MRSA ST398 isolates sequenced in this study. Interestingly, the types of SCCmec XII and IX were predicted in MRSA ST398-t571 isolates. These results indicated a rapid evolution of SCCmec elements in MRSA ST398-t571, which was similar to the findings from a previous study (Ji et al., 2020).
This study provided a longitudinal investigation of the presence of MRSA in a swine slaughterhouse in China. MRSA ST398 may spread among swine, humans, and the environment in the slaughterhouse, and it showed high levels of resistance profiles. This is the first report of the coexistence of cfr and optrA genes in a plasmid in MRSA ST398 isolates. The poxtA-carrying segment (IS431mec-optrA-IS1216-fexB-IS431mec) was reported in MRSA ST398 isolates for the first time. The spa typing results confirmed that t571 clonal linage dominated in MRSA or MSSA strains. This study underscores the importance of surveillance for MRSA ST398 in swine and indicates a high likelihood for the spread of MRSA ST398 from the swine production chain to human.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The study was approved by the South China Agriculture University (SCAU) Animal Ethics Committee. All animals were sampled under authorization from the Institutional Animal Care and Use Committees (IACUCs) of SCAU. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XL wrote the first draft of the manuscript. XL, WX, and ZZ contributed to the conception and design of the study. LX, HH, ZL, GL, PL, DX, and XZ performed the sampling and statistical analysis. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program [2019BT02N054] and Guangdong Basic and Applied Basic Research Foundation [2020A1515010917].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.914764/full#supplementary-material
Aanensen, D. M., and Spratt, B. G. (2005). The multilocus sequence typing network: mlst. net. Nucleic Acids Res. 33(Suppl._2), W728–W733. doi: 10.1093/nar/gki415
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Antonelli, A., D’Andrea, M. M., Brenciani, A., Galeotti, C. L., Morroni, G., Pollini, S., et al. (2018). Characterization of poxtA, a novel phenicol–oxazolidinone–tetracycline resistance gene from an MRSA of clinical origin. J. Antimicrob. Chemother. 73, 1763–1769. doi: 10.1093/jac/dky088
Armand-Lefevre, L., Ruimy, R., and Andremont, A. (2005). Clonal comparison of Staphylococcus aureus isolates from healthy pig farmers, human controls, and pigs. Emerg. Infect. Dis. 11, 711–714.
Bartels, M., Larner-Svensson, H., Meiniche, H., Kristoffersen, K., Schønning, K., Nielsen, J., et al. (2015). Monitoring meticillin resistant Staphylococcus aureus and its spread in Copenhagen, Denmark, 2013, through routine whole genome sequencing. Euro Surveill. 20:21112. doi: 10.2807/1560-7917.es2015.20.17.21112
Bassetti, M., Eckmann, C., Giacobbe, D. R., Sartelli, M., and Montravers, P. (2020). Post-operative abdominal infections: epidemiology, operational definitions, and outcomes. Intensive Care Med. 46, 163–172. doi: 10.1007/s00134-019-05841-5
Bhat, M., Dumortier, C., Taylor, B. S., Miller, M., Vasquez, G., Yunen, J., et al. (2009). Staphylococcus aureus ST398, New York City and Dominican Republic. Emerg. Infect. Dis. 15, 285–287.
Bonnet, I., Millon, B., Meugnier, H., Vandenesch, F., Maurin, M., Pavese, P., et al. (2018). High prevalence of spa type t571 among methicillin-susceptible Staphylococcus aureus from bacteremic patients in a French University Hospital. PLoS One 13:e0204977. doi: 10.1371/journal.pone.0204977
Bos, M. E., Verstappen, K. M., Van Cleef, B. A., Dohmen, W., Dorado-García, A., Graveland, H., et al. (2016). Transmission through air as a possible route of exposure for MRSA. J. Expo. Sci. Environ. Epidemiol. 26, 263–269. doi: 10.1038/jes.2014.85
Bouchami, O., Fraqueza, M. J., Faria, N. A., Alves, V., Lawal, O. U., de Lencastre, H., et al. (2020). Evidence for the dissemination to human of methicillin-resistant Staphylococcus aureus ST398 through the pork production chain: a study in a Portuguese slaughterhouse. Microorganisms 8:1892. doi: 10.3390/microorganisms8121892
Bozdogan, B., and Appelbaum, P. C. (2004). Oxazolidinones: activity, mode of action, and mechanism of resistance. Int. J. Antimicrob. Agents 23, 113–119. doi: 10.1016/j.ijantimicag.2003.11.003
Chen, H., Yin, Y., Li, X., Li, S., Gao, H., Wang, X., et al. (2020). Whole-genome analysis of livestock-associated methicillin-resistant Staphylococcus aureus sequence type 398 strains isolated from patients with bacteremia in China. J. Infect. Dis. 221, (Suppl. 2), S220–S228. doi: 10.1093/infdis/jiz575
Chen, L., Hu, J.-X., Liu, C., Liu, J., Ma, Z.-B., Tang, Z.-Y., et al. (2021). Identification of the Multiresistance Gene poxtA in Oxazolidinone-Susceptible Staphylococcus haemolyticus and Staphylococcus saprophyticus of Pig and Feed Origins. Pathogens 10:601. doi: 10.3390/pathogens10050601
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Chuang, Y.-Y., and Huang, Y.-C. (2015). Livestock-associated meticillin-resistant Staphylococcus aureus in Asia: an emerging issue? Int. J. Antimicrob. Agents 45, 334–340. doi: 10.1016/j.ijantimicag.2014.12.007
CLSI (2018). Performance Standards for Antimicrobial Susceptibility Testing, Twenty-Eighth Edn. Wayne, PA: CLSI.
Coccitto, S. N., Cinthi, M., Fioriti, S., Morroni, G., Simoni, S., Vignaroli, C., et al. (2022). Linezolid-resistant Enterococcus gallinarum isolate of swine origin carrying cfr, optrA and poxtA genes. J. Antimicrob. Chemother. 77, 331–337. doi: 10.1093/jac/dkab408
Cui, M., Li, J., Ali, T., Kalim, K., Wang, H., Song, L., et al. (2020). Emergence of livestock-associated MRSA ST398 from bulk tank milk, China. J. Antimicrob. Chemother. 75, 3471–3474. doi: 10.1093/jac/dkaa367
da Silva, A. G., Baines, S. L., Carter, G. P., Heffernan, H., French, N. P., Ren, X., et al. (2017). A phylogenomic framework for assessing the global emergence and evolution of clonal complex 398 methicillin-resistant Staphylococcus aureus. Microb. Genomics 3:e000105. doi: 10.1099/mgen.0.000105
David, M. Z., Siegel, J., Lowy, F. D., Zychowski, D., Taylor, A., Lee, C. J., et al. (2013). Asymptomatic Carriage of Sequence Type 398, spa Type t571 Methicillin-Susceptible Staphylococcus aureus in an Urban Jail: a Newly Emerging, Transmissible Pathogenic Strain. J. Clin. Microbiol. 51, 2443–2447. doi: 10.1128/jcm.01057-13
Davies, P. R., Wagstrom, E. A., and Bender, J. B. (2011). Lethal necrotizing pneumonia caused by an ST398 Staphylococcus aureus strain. Emerg. Infect. Dis. 17, 1152–1153. doi: 10.3201/eid/1706.101394
EUCAST (2016). Breakpoint Tables for Interpretation of MICs and Zone Diameters, Version 6.0. Available online at: https://www.eucast.org/ (accessed November 20, 2016).
Fan, R., Li, D., Feßler, A. T., Wu, C., Schwarz, S., and Wang, Y. (2017). Distribution of optrA and cfr in florfenicol-resistant Staphylococcus sciuri of pig origin. Vet. Microbiol. 210, 43–48. doi: 10.1016/j.vetmic.2017.07.030
Grøntvedt, C. A., Elstrøm, P., Stegger, M., Skov, R. L., Skytt Andersen, P., Larssen, K. W., et al. (2016). Methicillin-resistant Staphylococcus aureus CC398 in humans and pigs in Norway: a “One Health” perspective on introduction and transmission. Clin. Infect. Dis. 63, 1431–1438. doi: 10.1093/cid/ciw552
Hansen, J. E., Ronco, T., Stegger, M., Sieber, R. N., Fertner, M. E., Martin, H. L., et al. (2019). LA-MRSA CC398 in dairy cattle and veal calf farms indicates spillover from pig production. Front. Microbiol. 10:2733. doi: 10.3389/fmicb.2019.02733
Harmsen, D., Claus, H., Witte, W., Rothganger, J., Claus, H., Turnwald, D., et al. (2003). Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J. Clin. Microbiol. 41, 5442–5448. doi: 10.1128/jcm.41.12.5442-5448.2003
Harrison, E. M., Paterson, G. K., Holden, M. T., Morgan, F. J., Larsen, A. R., Petersen, A., et al. (2013). A Staphylococcus xylosus isolate with a new mecC allotype. Antimicrob. Agents Chemother. 57, 1524–1528. doi: 10.1128/AAC.01882-12
Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Ji, X., Krüger, H., Feßler, A. T., Liu, J., Zeng, Z., Wang, Y., et al. (2020). A novel SCCmec type V variant in porcine MRSA ST398 from China. J. Antimicrob. Chemother. 75, 484–486. doi: 10.1093/jac/dkz445
Kadlec, K., Ehricht, R., Monecke, S., Steinacker, U., Kaspar, H., Mankertz, J., et al. (2009). Diversity of antimicrobial resistance pheno- and genotypes of methicillin-resistant Staphylococcus aureus ST398 from diseased swine. J. Antimicrob. Chemother. 64, 1156–1164. doi: 10.1093/jac/dkp350
Kaya, H., Hasman, H., Larsen, J., Stegger, M., Johannesen, T. B., Allesøe, R. L., et al. (2018). SCC mec Finder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere 3:e00612-17. doi: 10.1128/msphere.00612-17
Kinross, P., Petersen, A., Skov, R., Van Hauwermeiren, E., Pantosti, A., Laurent, F., et al. (2017). Livestock-associated meticillin-resistant Staphylococcus aureus (MRSA) among human MRSA isolates, European Union/European Economic Area countries, 2013. Euro Surveill. 22, 16–00696. doi: 10.2807/1560-7917.es.2017.22.44.16-00696
Larsen, J., Petersen, A., Sørum, M., Stegger, M., van Alphen, L., Valentiner-Branth, P., et al. (2015). Meticillin-resistant Staphylococcus aureus CC398 is an increasing cause of disease in people with no livestock contact in Denmark, 1999 to 2011. Euro Surveill. 20:30021. doi: 10.2807/1560-7917.es.2015.20.37.30021
Li, D., Wang, Y., Schwarz, S., Cai, J., Fan, R., Li, J., et al. (2016). Co-location of the oxazolidinone resistance genes optrA and cfr on a multiresistance plasmid from Staphylococcus sciuri. J. Antimicrob. Chemother. 71, 1474–1478. doi: 10.1093/jac/dkw040
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Tang, T., Stegger, M., Dalsgaard, A., Liu, T., and Leisner, J. J. (2021). Characterization of antimicrobial-resistant Staphylococcus aureus from retail foods in Beijing, China. Food Microbiol. 93:103603. doi: 10.1016/j.fm.2020.103603
Li, S.-M., Zhou, Y.-F., Li, L., Fang, L.-X., Duan, J.-H., Liu, F.-R., et al. (2018). Characterization of the multi-drug resistance gene cfr in methicillin-resistant Staphylococcus aureus (MRSA) strains isolated from animals and humans in China. Front. Microbiol. 9:2925. doi: 10.3389/fmicb.2018.02925
Li, W., Liu, J.-H., Zhang, X.-F., Wang, J., Ma, Z.-B., Chen, L., et al. (2018). Emergence of methicillin-resistant Staphylococcus aureus ST398 in pigs in China. Int. J. Antimicrob. Agents 51, 275–276. doi: 10.1016/j.ijantimicag.2017.10.013
Lienen, T., Schnitt, A., Hammerl, J. A., Maurischat, S., and Tenhagen, B.-A. (2020). Genomic distinctions of LA-MRSA ST398 on dairy farms from different German federal states with a low risk of severe human infections. Front. Microbiol. 11:575321. doi: 10.3389/fmicb.2020.575321
Long, K. S., Poehlsgaard, J., Kehrenberg, C., Schwarz, S., and Vester, B. (2006). The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 50, 2500–2505. doi: 10.1128/AAC.00131-06
Moodley, A., Nielsen, S. S., and Guardabassi, L. (2011). Effects of tetracycline and zinc on selection of methicillin-resistant Staphylococcus aureus (MRSA) sequence type 398 in pigs. Vet. Microbiol. 152, 420–423. doi: 10.1016/j.vetmic.2011.05.025
Nakaminami, H., Hirai, Y., Nishimura, H., Takadama, S., and Noguchi, N. (2020). Arthritis caused by MRSA CC398 in a patient without animal contact, Japan. Emerg. Infect. Dis. 26, 795–797. doi: 10.3201/eid2612.203738
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214.
Pirolo, M., Gioffrè, A., Visaggio, D., Gherardi, M., Pavia, G., Samele, P., et al. (2019). Prevalence, molecular epidemiology, and antimicrobial resistance of methicillin-resistant Staphylococcus aureus from swine in southern Italy. BMC Microbiol. 19:51. doi: 10.1186/s12866-019-1422-x
Qiao, M., Ying, G.-G., Singer, A. C., and Zhu, Y.-G. (2018). Review of antibiotic resistance in China and its environment. Environ. Int. 110, 160–172. doi: 10.1016/j.envint.2017.10.016
Rasigade, J.-P., Laurent, F., Hubert, P., Vandenesch, F., and Etienne, J. (2010). Lethal necrotizing pneumonia caused by an ST398 Staphylococcus aureus strain. Emerg. Infect. Dis. 16:1330. doi: 10.3201/eid1608.100317
Schnitt, A., Lienen, T., Wichmann-Schauer, H., Cuny, C., and Tenhagen, B.-A. (2020). The occurrence and distribution of livestock-associated methicillin-resistant Staphylococcus aureus ST398 on German dairy farms. J. Dairy Sci. 103, 11806–11819. doi: 10.3168/jds.2020-18958
Schürch, A., Arredondo-Alonso, S., Willems, R., and Goering, R. (2018). Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin. Microbiol. Infect. 24, 350–354. doi: 10.1016/j.cmi.2017.12.016
Shen, J., Wang, Y., and Schwarz, S. (2013). Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 68, 1697–1706. doi: 10.1093/jac/dkt092
Sieber, R. N., Skov, R. L., Nielsen, J., Schulz, J., Price, L. B., Aarestrup, F. M., et al. (2018). Drivers and dynamics of methicillin-resistant livestock-associated Staphylococcus aureus CC398 in pigs and human in Denmark. mBio 9:e2142-18. doi: 10.1128/mbio.02142-18
Song, Z., Gu, F.-F., Guo, X.-K., Ni, Y.-X., He, P., and Han, L.-Z. (2017). Antimicrobial resistance and molecular characterization of Staphylococcus aureus causing childhood pneumonia in Shanghai. Front. Microbiol. 8:455. doi: 10.3389/fmicb.2017.00455
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Sun, C., Chen, B., Hulth, A., Schwarz, S., Ji, X., Nilsson, L. E., et al. (2019). Genomic analysis of Staphylococcus aureus along a pork production chain and in the community, Shandong Province, China. Int. J. Antimicrob. Agents 54, 8–15. doi: 10.1016/j.ijantimicag.2019.03.022
Sun, C., Zhang, P., Ji, X., Fan, R., Chen, B., Wang, Y., et al. (2018). Presence and molecular characteristics of oxazolidinone resistance in staphylococci from household animals in rural China. J. Antimicrob. Chemother. 73, 1194–1200. doi: 10.1093/jac/dky009
Tang, Y., Qiao, Z., Wang, Z., Li, Y., Ren, J., Wen, L., et al. (2021). The Prevalence of Staphylococcus aureus and the Occurrence of MRSA CC398 in Monkey Feces in a Zoo Park in Eastern China. Animals 11:732. doi: 10.3390/ani11030732
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Van Duin, D., and Paterson, D. L. (2016). Multidrug-resistant bacteria in the community: trends and lessons learned. Infect. Dis. Clin. 30, 377–390. doi: 10.1016/j.idc.2016.02.004
Voss, A., Loeffen, F., Bakker, J., Klaassen, C., and Wulf, M. (2005). Methicillin-resistant Staphylococcus aureus in pig farming. Emerg. Infect. Dis. 11, 1965–1966. doi: 10.3201/eid1112.050428
Wang, Y., Lv, Y., Cai, J., Schwarz, S., Cui, L., Hu, Z., et al. (2015). A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicrob. Chemother. 70, 2182–2190. doi: 10.1093/jac/dkv116
Wang, J., Lin, D.-C., Guo, X.-M., Wei, H.-K., Liu, X.-Q., Chen, X.-J., et al. (2015). Distribution of the multidrug resistance gene cfr in Staphylococcus isolates from pigs, workers, and the environment of a hog market and a slaughterhouse in Guangzhou, China. Foodborne Pathog. Dis. 12, 598–605. doi: 10.1089/fpd.2014.1891
Ward, M. J., Gibbons, C. L., McAdam, P. R., Bunnik, B. A. D. V., Girvan, E. K., Edwards, G. F., et al. (2014). Time-scaled evolutionary analysis of the transmission and antibiotic resistance dynamics of Staphylococcus aureus clonal complex 398. Appl. Environ. Microbiol. 80, 7275–7282.
Wardyn, S. E., Stegger, M., Price, L. B., and Smith, T. C. (2018). Whole-genome analysis of recurrent Staphylococcus aureus t571/ST398 infection in farmer, Iowa, USA. Emerg. Infect. Dis. 24, 153–154. doi: 10.3201/eid2401.161184
Wulf, M., Sørum, M., Van Nes, A., Skov, R., Melchers, W., Klaassen, C., et al. (2008). Prevalence of methicillin-resistant Staphylococcus aureus among veterinarians: an international study. Clin. Microbiol. Infect. 14, 29–34. doi: 10.1111/j.1469-0691.2007.01873.x
Yan, X., Yu, X., Tao, X., Zhang, J., Zhang, B., Dong, R., et al. (2014). Staphylococcus aureus ST398 from slaughter pigs in northeast China. Int. J. Med. Microbiol. 304, 379–383. doi: 10.1016/j.ijmm.2013.12.003
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, K., Sparling, J., Chow, B. L., Elsayed, S., Hussain, Z., Church, D. L., et al. (2004). New quadriplex PCR assay for detection of methicillin and mupirocin resistance and simultaneous discrimination of Staphylococcus aureus from coagulase-negative staphylococci. J. Clin. Microbiol. 42, 4947–4955. doi: 10.1128/jcm.42.11.4947-4955.2004
Zhou, W., Li, X., Osmundson, T., Shi, L., Ren, J., and Yan, H. (2018). WGS analysis of ST9-MRSA-XII isolates from live pigs in China provides insights into transmission among porcine, human and bovine hosts. J. Antimicrob. Chemother. 73, 2652–2661. doi: 10.1093/jac/dky245
Keywords: swine slaughterhouse, multi-drug resistance, MRSA ST398, spa typing, whole-genome sequencing, public health
Citation: Li X, Xie L, Huang H, Li Z, Li G, Liu P, Xiao D, Zhang X, Xiong W and Zeng Z (2022) Prevalence of Livestock-Associated MRSA ST398 in a Swine Slaughterhouse in Guangzhou, China. Front. Microbiol. 13:914764. doi: 10.3389/fmicb.2022.914764
Received: 07 April 2022; Accepted: 17 May 2022;
Published: 23 June 2022.
Edited by:
Ziad Daoud, Central Michigan University, United StatesReviewed by:
Chen Kaichao, City University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Li, Xie, Huang, Li, Li, Liu, Xiao, Zhang, Xiong and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenguang Xiong, eGlvbmd3Z0BzY2F1LmVkdS5jbg==; Zhenling Zeng, emx6ZW5nQHNjYXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.