 Gang Zhang1‡
Gang Zhang1‡ Qinna Cui1,2,†,‡Jianjuan Li1,2Ruiliang Guo1,3Sébastien Olivier Leclercq4Lifeng Du5
Qinna Cui1,2,†,‡Jianjuan Li1,2Ruiliang Guo1,3Sébastien Olivier Leclercq4Lifeng Du5 Na Tang1,2Yuqin Song1Chao Wang1
Na Tang1,2Yuqin Song1Chao Wang1 Fangqing Zhao5
Fangqing Zhao5 Jie Feng1*
Jie Feng1*- 1State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 2College of Life Science, University of Chinese Academy of Sciences, Beijing, China
- 3School of Life Sciences, Zhengzhou University, Zhengzhou, China
- 4UMR ISP, INRAE, Université François Rabelais de Tours, Nouzilly, France
- 5Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing, China
In the worldwide health threat posed by antibiotic-resistant bacterial pathogens, mobile genetic elements (MGEs) play a critical role in favoring the dissemination of resistance genes. Among them, the genomic island GIsul2 and the ISCR-related element CR2-sul2 unit are believed to participate in this dissemination. However, the mobility of the two elements has not yet been demonstrated. Here, we found that the GIsul2 and CR2-sul2 units can excise from the host chromosomal attachment site (attB) in Shigella flexneri. Through establishing a two-plasmid mobilization system composed of a donor plasmid bearing the GIsul2 and a trap plasmid harboring the attB in recA-deficient Escherichia coli, we reveal that the integrase of GIsul2 can perform the excision and integration of GIsul2 and CR2-sul2 unit by site-specific recombination between att core sites. Furthermore, we demonstrate that the integrase and the att sites are required for mobility through knockout experiments. Our findings provide the first experimental characterization of the mobility of GIsul2 and CR2-sul2 units mediated by integrase. They also suggest a potential and unappreciated role of the GIsul2 integrase family in the dissemination of CR2-sul2 units carrying various resistance determinants in between.
Introduction
Mobile genetic elements (MGEs), including genomic islands (GIs), play a major role in the evolution of bacteria, and more specifically in the wide dissemination of antibiotic resistance genes (ARGs) (Juhas et al., 2009; Hall, 2010; Carraro and Burrus, 2014; Partridge et al., 2018; He et al., 2019; Halder et al., 2022; Li et al., 2022). Among them, GIsul2 was proposed to participate in the dissemination of sulphonamide resistance gene sul2 (Nigro and Hall, 2011; Harmer et al., 2017). GIsul2 was first detected in the chromosome of Enterobacter cloacae subspecies cloacae type strain ATCC 13047, in the chromosome of Shigella flexneri ATCC 700930 (also known as 2457T), and in the large conjugative plasmid pAB3 of Acinetobacter baumannii ATCC 17978 (Nigro and Hall, 2011). GIsul2 was then also reported in the chromosome of A. baumannii ATCC 19606 and plasmids of several γ-proteobacteria (Hamidian and Hall, 2016; Harmer et al., 2017). GIsul2 is usually inserted into the 3′ end of chromosomally-encoded guaA genes (GMP synthase) at the 5′-GAGTGGGA-3′ integration site (Harmer et al., 2017). GIsul2 and its derivatives are also frequently found as part of antibiotic resistance island B (ARI-B) of IncC plasmids (Anantham et al., 2015; Harmer and Hall, 2015), but in this case, the integration site is limited to 5′-GGGA-3′, which may correspond to a secondary integration site (Harmer et al., 2017). According to the recent genomic description (Harmer et al., 2017), the element consists of genes putatively involved in mobilization (excision/integration, replication, and transfer), followed by a toxin–antitoxin system, an arsenic resistance operon, a CR2 element, and the sul2 resistance gene (Figure 1).
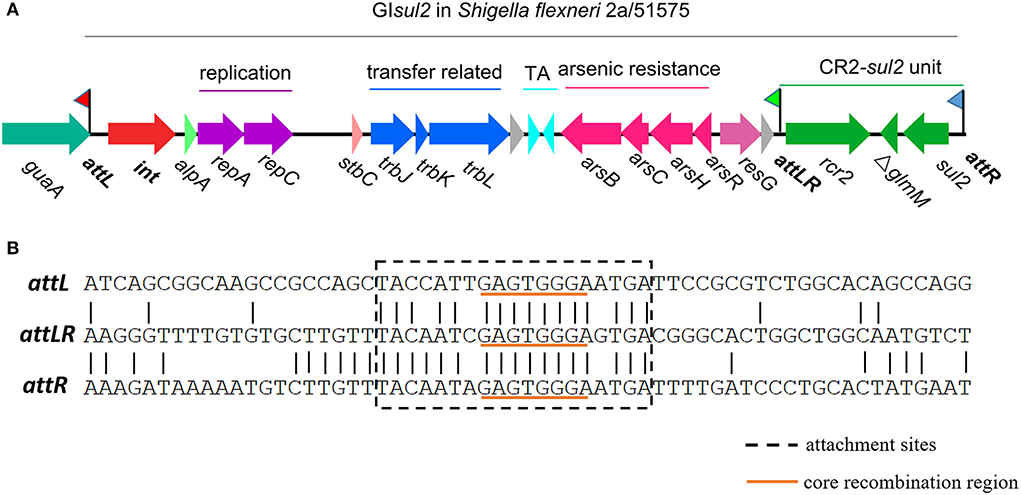
Figure 1. Genetic organization of genomic island GIsul2 in S. flexneri 2a str. 2457T (accession AE014073) or S. flexneri 51575 (accession AZPD01000036, position 2108-17567). (A) Organization of the GIsul2 element. The thick central line represents the backbone of the GIsul2 with flags representing the three attachment sites (attL/attLR/attR). Horizontal arrows indicate the position, size, and orientation of ORFs with the names below. The guaA gene encoding a guanosine monophosphate synthetase (GMP) is shown in teal. The int gene encoding the integrase is represented in red. Bright green, light red, and pink ORFs denote alpA, stbC, and resG genes, respectively. Genes putatively involved in replication, transfer-related, toxin-antitoxin system, arsenic resistance, and the CR2-sul2 unit are shown in purple, blue, cyan, pink and green, respectively. Gray ORFs encode hypothetical proteins. (B) Nucleotide alignment of att sites of GIsul2. The dotted box represents the expected att sites recognized by the integrase. The orange underlined motif indicates the putative core recombination region.
The first common region (CR) element, CR1, was discovered in the 3′-conserved segment (3′-CS) of some class 1 integrons, where it is associated with antibiotic resistance genes not embedded in gene cassettes and is usually found between partial duplications of the 3′-CS (Stokes et al., 1993; Partridge and Hall, 2003). Other CR elements were later identified in numerous bacteria, especially in Gram-negative pathogens, and are frequently linked to diverse ARGs (Partridge and Hall, 2003; Toleman et al., 2006a; Schleinitz et al., 2010; Fang et al., 2019). CR2 is found downstream of the sul2 gene in a head-to-head organization with a truncated glmM gene in between, a structure found at the 3′ end of GIsul2, and hereafter referred to as the CR2-sul2 unit (Figure 1). More commonly, CR2-sul2 units carry various other ARGs in between and locate on other MGEs. Recently, CR2 was also reported to likely contribute to the mobilization of the high-level tigecycline resistance gene tet(X) in Escherichia coli (Fang et al., 2019; He et al., 2019; Sun et al., 2019). CR elements are proposed to move by a process called rolling-circle replication because of the distant similarity of their encoded gene to the transposases of the IS91 family insertion sequences, known to promote their own spread through this mechanism (Toleman et al., 2006b). Accordingly, the term ISCR was proposed for these elements (Toleman et al., 2006a). CR element boundaries, called terIS and oriIS for 5′ and 3′ boundaries, respectively, were inferred from their genetic sequence for most elements, but not all (Toleman et al., 2006a; Lallement, 2018). The lack of experimental demonstration for a rcr (coding region of CR)-mediated rolling circle mechanism and the yet unclear boundaries of some CR elements make the hypothesis of CRs being IS elements still debatable. We therefore conservatively keep the CR denomination in this study to define these genetic elements.
Despite the numerous occurrences of GIsul2 and CR2-sul2 units in various bacteria, suggesting horizontal gene transfer, their mobility has not yet been demonstrated. GIsul2 mobilization was tested in an E. coli genetic context, but no excised circular intermediate could be detected (Harmer et al., 2017). In the present study, we demonstrate that GIsul2-encoded integrase mediates (i) the excision of GIsul2 and CR2-sul2 from their original chromosomal location, (ii) the integration of GIsul2 and CR2-sul2 in two-plasmid mobilization assay system in E. coli recA-deficient mutant. All these reactions are site-specific and rely on the presence of GIsul2 attachment (att) sites.
Results
GIsul2 and CR2-sul2 unit can excise from the chromosome of S. flexneri
By analyzing in detail the structure of GIsul2 reported in the reference sequence of S. flexneri 2a 2457T (GenBank accession AE014073), we found that three putative att sites were present in this island. In addition, to the already described attL and attR located at the left and right end of the GI (Harmer et al., 2017), a third att site, termed attLR, lies 112–130 bp upstream of the rcr2 gene (Figure 1A). The three sites are 19 bp-long and they all share the common central nucleotide motif 5′-GAGTGGGA-3′ (Figure 1B), previously proposed to be their core recombination site (Harmer et al., 2017). Interestingly, the putative terIS proposed for CR2 (5′-GGGAGTGACGGGCACTGGC-3′), which is the putative 5′ boundary of the element (Toleman et al., 2006a), starts in the middle of the core recombination region of attLR (Figure 1B). According to the possible recombination events between the putative att sites, Figure 2A shows the three predicted circular intermediates that could be produced, namely, excised GIsul2 (site-specific recombination between attL and attR), excised CR2-sul2 (site-specific recombination between attLR and attR), and an excised 12-kb segment named GI12K (site-specific recombination between attL and attLR). We first tested whether any of these circular intermediates could be naturally excised from the chromosome of S. flexneri 51,575. This strain carries a single copy of GIsul2 almost identical to that of S. flexneri 2a 2457T (GenBank accession number AZPD01000036, position 2,108–17,567), except for a 140-bp region in the trbL gene with several differences. GIsul2 in S. flexneri 51,575 is also inserted in the 3′ end of guaA gene, resulting in the same three att sites as in Figure 1.
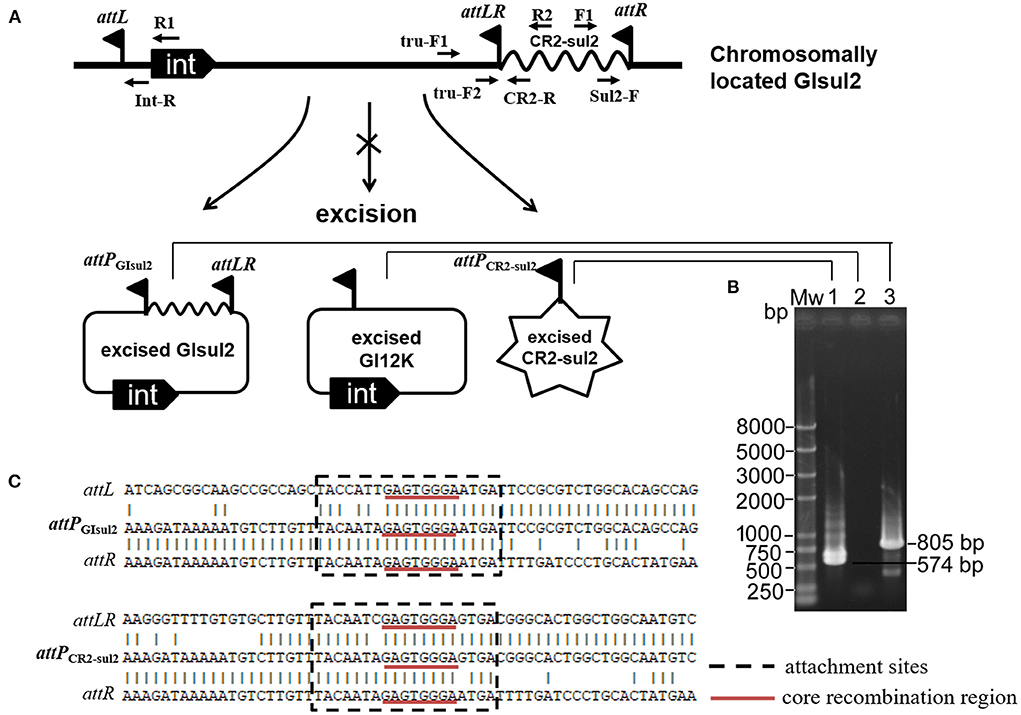
Figure 2. Excision of GIsul2 and of CR2-sul2 unit from the chromosome of S. flexneri 51575. (A) The predicted circular intermediates resulting from recombination between the three att sites of GIsul2. The crossed arrow indicates that the event could not be observed. Solid flags denote the various att sites. (B) Nested PCR assays for the detection of the three potential circular intermediates: Mw, M5 DL2000 plus DNA Marker (Mei5 Biotechnology, Beijing); lane 1, CR2-sul2 circular intermediate; lane 2, GI12K circular intermediate; lane 3, GIsul2 circular intermediate. (C) Alignment of the sequenced junctions resulting from the circularization of GIsul2 (attPGIsul2) and of CR2-sul2 unit (attPCR2−sul2) with their corresponding initial att sites.
The presence of the junction region of each predicted circular intermediate was tested through PCR amplification on crude DNA extracts from overnight culture in standard conditions using a nested procedure because of the very low amplification yield after the first round of PCR. Two pairs of nested primers F1/R1 and Sul2-F/Int-R were used for the detection of the GIsul2 circular form (805 bp), two pairs of nested primers F1/R2 and Sul2-F/CR2-R were used for the detection of the CR2-sul2 unit circular form (574 bp), and two pairs of nested primers tru-F1/R1 and tru-F2/Int-R were used for the detection of the GI12K circular form (767 bp; Figure 2A, Supplementary Table 1). These various PCR reactions resulted in only two target-size products (Figure 2B). The first corresponded to the junction in the circular form of GIsul2, resulting from the recombination between attL and attR sites. The second corresponded to the junction in the circular form of CR2-sul2 unit, resulting from the recombination between attLR and attR sites. Sequencing of the amplified junctions confirmed that they consisted of attL/attR and attLR/attR hybrids, hereafter named attPGIsul2 and attPCR2−sul2, respectively (Figure 2C, Supplementary Figure 1), indicating that GIsul2 and CR2-sul2 units are naturally excised from the chromosome of S. flexneri 51575.
A donor plasmid bearing GIsul2 can integrate into the chromosomal attB site downstream of guaA gene in recA-deficient E. coli
To avoid excision mediated by RecA-dependent homologous recombination and the difficulty of genetic operation in Shigella, pKDGItetWsul2 (24,157 bp) carrying a modified version of GIsul2, as a donor plasmid, was established and introduced into the recA-deficient strain, E. coli DH5α. pKDGItetWsul2 was constructed from pKD46, a temperature-sensitive plasmid that can be eliminated at 37 or 42°C (Datsenko and Wanner, 2000). pKDGItetWsul2 carries an ampicillin resistance marker and GIsul2 in which ΔglmM between rcr2 and sul2 genes was replaced by a constitutively expressed tetW gene (tetracycline resistance; Figure 3A, Supplementary Table 2). PCR assays targeting attPGIsul2 and attPCR2−sul2 were then performed on E. coli DH5α (pKDGItetWsul2) as in Section Introduction, and the amplified products (validated by sequencing) confirmed that the excision of GItetWsul2 and CR2-sul2 from donor plasmid can still occur in a recA− genetic context (Figure 3B).
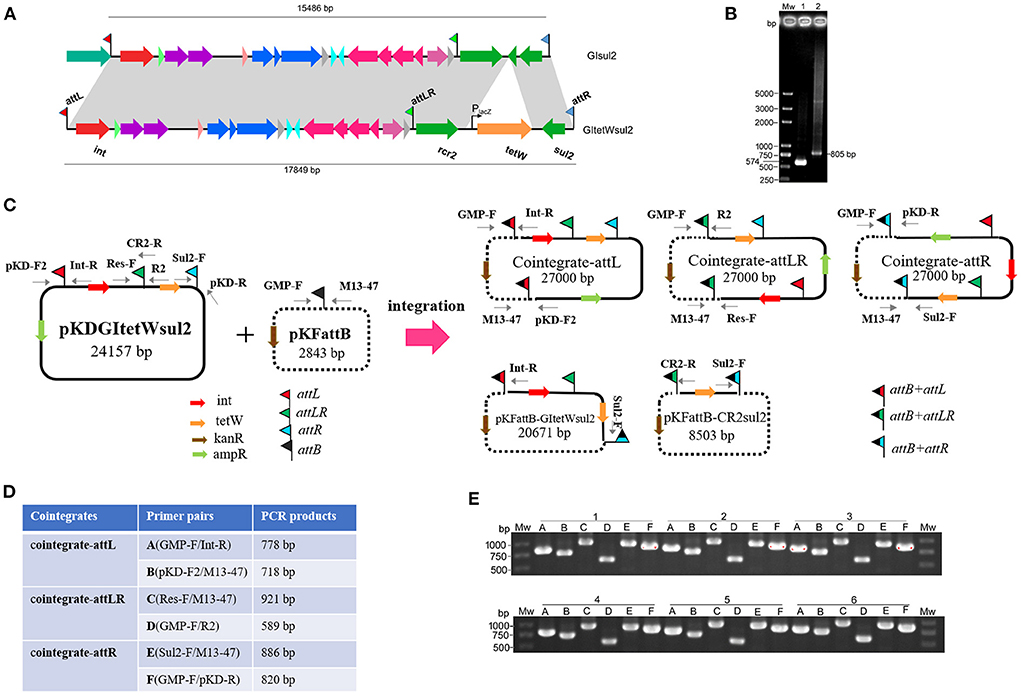
Figure 3. Co-integrate formation in E. coli DH5α through site-specific recombination at the att sites. (A) Construction of GItetWsul2. The gray shadings denote 100% identity between GItetWsul2 and the original GIsul2. ORFs (horizontal arrows) are colored as in Figure 1. (B) Detection of circular intermediates excised from the donor plasmid pKDGItetWsul2 in E. coli DH5α. Mw, DNA size marker; lane 1, CR2-sul2 unit circular intermediate; lane 2, GItetWsul2 circular intermediate. (C) Organization of the donor (pKDGItetWsul2) and trap (pFKattB) plasmids used in the two-plasmid integration experiment, and expected recombinant structures. Intervening ORFs are depicted with filled arrows while red, green, blue, and black flags denote attL, attLR, attR, and attB, respectively. The putative hybrid attachment sites resulting from site-specific recombination are represented by two-color flags. Primers used for the detection of the various structures are indicated. (D) Couples of primer pairs used to detect co-integrate structures in recombinant bacteria, and their expected amplification size. (E) PCR amplification using the six primer pairs from (D) on the plasmid extracts of six independent recombinant colonies. Colonies are labeled 1–6, with the six primer pairs amplification (A–E) displayed below each of them.
Since E. coli DH5α carries a GIsul2-empty attB site at the 3′ end of its guaA gene, we tested the integration potential of the excised elements into this chromosomal position. E. coli DH5α (pKDGItetWsul2) culture was grown overnight at 42°C to discard pKDGItetWsul2 and then transferred at 42°C on fresh LB plates containing tetracycline to select for colonies bearing chromosomally integrated circular intermediates. Twelve randomly selected resistant colonies were tested using the same six pairs of primers as in Figure 3D, except that the primer GMP-R replaced the vector-specific primer M13-47 (Supplementary Figure 2). Only Sul2-F/GMP-R and GMP-F/pKD-R PCR reaction provided an amplification (Supplementary Figure 2). Successful amplification was obtained in all colonies for the primer pairs, corresponding to the recombination between attR of pKDGItetWsul2 and attB. Sequencing of the PCR products confirmed the expected junction. The observations indicate that the chromosomal attB site can recombine with a plasmidic attR site in a recA− genetic context, leading to donor plasmid carrying GIsul2 integration into the chromosome.
The donor plasmid, GIsul2, and CR2-sul2 unit can integrate into the attB site of a trap plasmid in recA-deficient E. coli
To clearly demonstrate the mobilization mechanism for GIsul2 and CR2-sul2 units, we applied a two-plasmid assay system consisting of the donor plasmid and trap plasmid. A similar strategy was also used for studying integration mechanisms of pathogenicity islands in E. coli 536 (Wilde et al., 2008). The trap plasmid, pKFattB (2843 bp), was constructed by cloning an uninterrupted attB region (811 bp) into the high copy vector pKF18k-2 (Supplementary Table 2). This plasmid bears a kanamycin resistance gene (kanR) as the selective marker. The two plasmids pKDGItetWsul2 and pKFattB were co-transformed into E. coli DH5α. The correct clones were performed using the two-plasmid mobilization assay. Based on the GIsul2 and CR2-sul2 excisions observed above, we speculated that there could be five types of integrations: integration of the GItetWsul2 circular intermediate into the trap plasmid (pKFattB-GItetWsul2 in Figure 3C), integration of the CR2-sul2 intermediate (pKFattB-CR2sul2 in Figure 3C), and three possible integrations of the whole donor plasmid, one for each att site (cointegrate-attL/-attLR/-attR, Figure 3C).
First, we tested for the integration of the whole donor plasmid. There are three att sites in the donor plasmid, and we, therefore, designed six specific primer pairs (A to F) targeting each of the possible recombined sites (junctions; Figure 3D). Gel bands corresponding to the PCR product of every junction were detected in all 12 randomly selected recombinant plasmid extracts used as templates (six shown, Figure 3E). Sequencing of the PCR products confirmed that the amplification indeed corresponded to the recombination between attB and each of the three att sites (attL, attLR, and attR). To understand which type of integration dominated in each colony, we used BamHI to digest the recombinant plasmids. Two bands of 15.7 and 11.2 kb, respectively, corresponding to the digestion of the donor plasmid integration through attR-attB recombination, were observed for all colonies (Supplementary Figure 3). These results indicate that the integration of the donor plasmid into pKFattB mainly occurred in the attR site.
Second, we tested for the presence of structures matching the integration of the circular intermediates in the same 12 selected plasmid extracts. Primer pair Sul2-F/Int-R was used to detect pKFattB-GItetWsul2 structure, expected to produce an amplification size of 3,646 bp (Figure 3C). However, we only obtained bands around 10 kb which represented amplification of the donor plasmid from the cointegrates. We therefore digested the recombinant plasmids with NcoI restriction enzyme, which cuts only the backbone of the donor plasmid, resulting in digestion of the cointegrates. Using the NcoI digested products as a template, we obtain the expected 3,646 bp band for all six tested colonies (Figure 4A). Similarly, the primer pair Sul2-F/CR2-R was used for the detection of pKFattB–CR2sul2 structure after digestion of the cointegrates using NcoI. This PCR generated a product of the expected size (3,415 bp; Figure 4B), indicating that pKFattB-GItetWsul2 and pKFattB-CR2sul2 structures concurrently exist with cointegrate structures in the recombinant colonies.
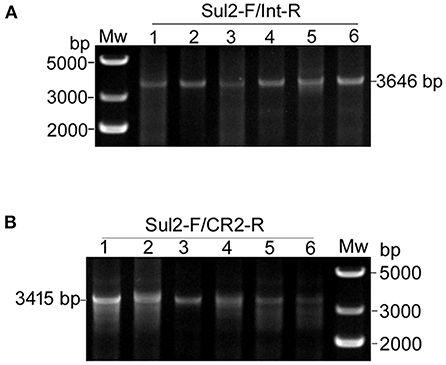
Figure 4. Detection of pKFattB-GItetWsul2 and pKFattB-CR2sul2 structures in recombinant colonies. PCR amplification using the primer pairs Sul2-F/Int-R and Sul2-F/CR2-R for the detection of GItetWsu2-pKFattB (A) and pKFattB-CR2sul2 (B) structures, respectively. PCR reactions were conducted on the same six recombinant colonies as in Figure 3E.
We finally applied nanopore sequencing on the recombinant plasmids extracted from five colonies and digested them with XbaI, which cuts only the trap plasmid pKFattB, and exactly once. The total number of reads ranged from 1,382,608 to 5,033,742 (Table 1, Supplementary Table 3) for all five samples, with most of them corresponding to the original trap plasmid pKFattB (80–88%), or to regions shared by two or more recombinant structures (ambiguous regions, 4–8% of the reads), or they did not properly map on any of the structures (~10%). Nonetheless, 1,741–34,155 of the reads mapped unambiguously to the cointegrate-attR structure while only 1–225 and 5–471 reads mapped unambiguously to the cointegrate-attL and cointegrate-attLR structures, respectively. An additional 3–147 and 386–776 reads mapped unambiguously on the pKFattB-GItetWsul2 and pKFattB-CR2sul2 structures, respectively. The higher number of reads mapping to the pKFattB-CR2sul2 structure compared to other structures (except cointegrate-attR) should be treated with caution since unambiguous matches require reads long enough to map both junctions with pKFattB, which is easier to achieve for pKFattB-CR2sul2 (reads of at least 5.7-kb) than for pKFattB-GItetWsul2 (reads of at least 18-kb) or other cointegrates (reads of at least 24.2-kb). Despite this limitation, these results provided further evidence that all the five predicted structures are present, with high dominance of the cointegrate-attR structure.
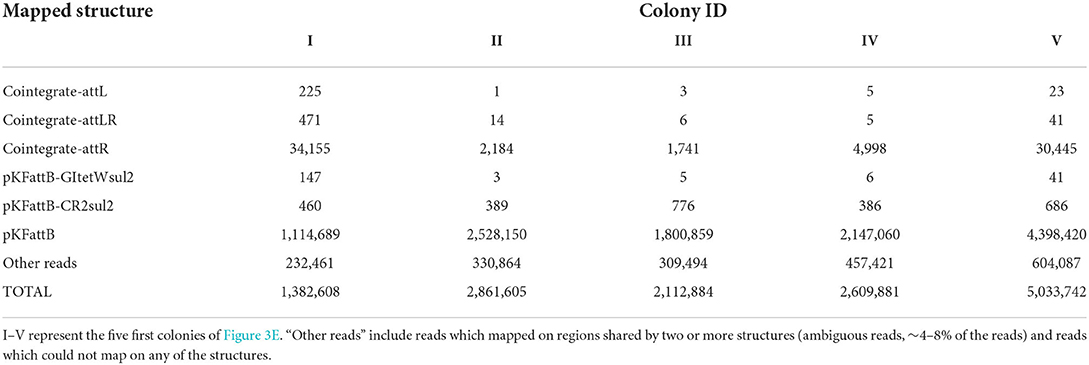
Table 1. Number of nanopore reads unambiguously mapping on each possible recombinant structure described in Figure 3C, in five of the 12 randomly selected recombinant colonies.
The integrase and att sites of GIsul2 are required for mobilization
To find out which genes or regions located in GIsul2 are required for integration, several knockout plasmids were constructed from the donor plasmid pKDGItetWsul2. The deleted regions are int, alpA/repA/C, resG, rcr2, or the att sites (Supplementary Table 2). Integration of these constructs into pKFattB was tested using the same PCR detection strategy as for pKDGItetWsul2, except that the primers CRUp-R and IntUp-R replaced the primers CR2-R and Int-R, respectively, when testing for the pKFattB-CR2sul2 and pKFattB-GItetWsul2 structures because of the deletion of rcr2 or int in one of the constructs. The deletion of the int gene resulted in the complete loss of integration ability including cointegrations, GIsul2, and CR2-sul2 integrations (Table 2). When the three att sites (attL, attLR, and attR) of pKDGItetWsul2 were deleted, only four colonies displayed the integration, but none of them carried any of the five expected recombinant structures (Table 2). A single type of integration was observed in all four clones involving the attB of pKFattB and a region 166 bp downstream of the deleted attR of pKDGItetWsul2, with the insertion site reduced to “TGGG” instead of “GAGTGGGA.” Deletion of rep, resG, and rcr2 regions had no effect on the recombination efficiency and the detection of the various structures. These results indicate that the integrase of GIsul2 and its att sites are required for the integration of donor plasmid, GIsul2, and CR2-sul2 unit into the trap plasmid, while rcr2-encoded putative transposase seemingly does not have any impact on the formation of recombination structures.

Table 2. Effect of deleting GIsul2-associated regions on integrations.
To confirm that no other factors encoded by GIsul2 are required for integration, we constructed a minimal donor plasmid containing only the int gene, attL, and a chloramphenicol resistance determinant (catR) on the pKD46 backbone, designated pKDmini (6,498 bp; Supplementary Figure 4A) and tested its integration into pKFattB. After co-transformation of pKDmini and pKFattB into E. coli DH5α and temperature shift, transformants were plated on a medium supplemented with chloramphenicol and kanamycin, and six colonies were randomly selected. The XbaI restriction enzyme analysis revealed that all colonies harbored the expected 9.3-kb band corresponding to the cointegrate of pKDmini with pKFattB, and 2.8-kb band corresponding to residual pKFattB (Supplementary Figure 4B). PCR and sequencing also confirmed the result, which demonstrated that Int is sufficient for the integration through the site-specific recombination when relevant att sites are present.
Unambiguous observation of GIsul2 and CR2-sul2 mobilization through restriction analysis by the introduction of sacB into the donor plasmid and overexpression of alpA
Due to the dominance of attR-based cointegrate during two-plasmid mobilization experiments, the potential integration of circularized CR2-sul2 or GItetWsul2 into the trap plasmid could not be observed distinctly through restriction enzyme digestions. The sacB gene, lethal for E. coli when expressed in the presence of sucrose, was therefore introduced into the backbone of pKDGItetWsul2, resulting in a new donor plasmid pKDGItetWsul2sacB (Supplementary Table 2). Cointegrates containing sacB are inhibited in the presence of sucrose, while colonies carrying only GItetWsul2 or CR2-sul2 unit will be able to grow normally. The two-plasmid mobilization experiment was repeated using pKDGItetWsul2sacB as the donor plasmid and with the addition of 20% sucrose during the integration step, resulting in the successful growth of recombinant colonies from which 36 were randomly selected for plasmid extraction. XbaI digestion (cutting only pKFattB) resulted in an 8.5-kb band for all the tested colonies (6 are represented in Figure 5A), suggesting that all recombinant colonies carried the pKFattB-CR2sul2 structure. Additional digestion using the BamHI enzyme (cutting CR2-sul2 unit once) plus XbaI produced two bands of 6.5-kb and 2-kb, consistent with the presence of pKFattB-CR2sul2 structure (Figure 5B). These results indicate that integration of the circularized CR2-sul2 unit into the trap plasmid can happen through site-specific recombination between the attPCR2−sul2 site and the attB site. No colony carrying the pKFattB-GItetWsul2 structure could have been detected using the sucrose negative selection.
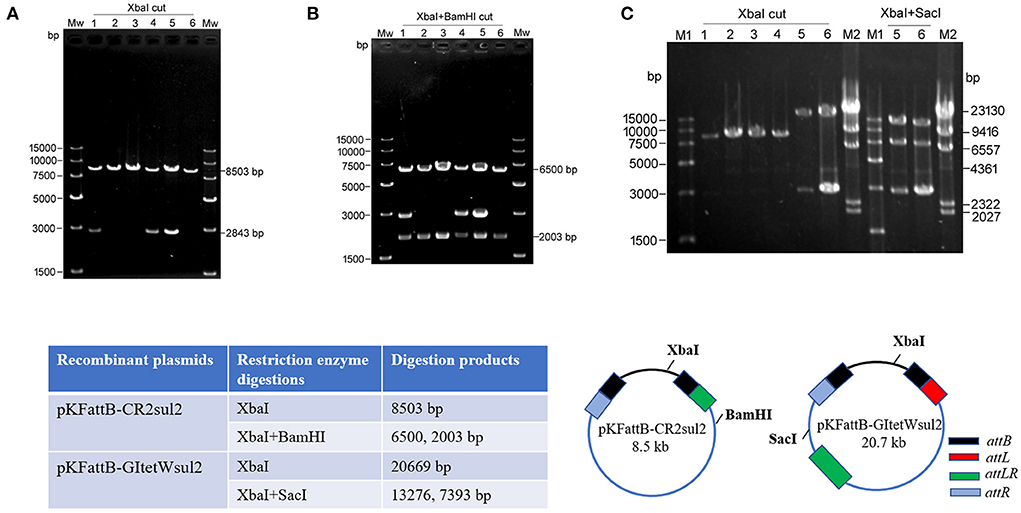
Figure 5. Integration of circular intermediates into the trap plasmid pKFattB under sucrose negative selection. (A) XbaI digestion of the plasmid extract from six independent recombinant colonies. Bands at 8,503 bp correspond to the recombinant plasmid pKFattB-CR2sul2 (CR2-sul2 inserted into pKFattB) and bands at 2,843 bp correspond to original pKFattB copies. (B) XbaI and BamHI double restriction of the same plasmid extracts. BamHI cuts CR2-sul2 unit once and not pKFattB, leading to the digestion of pKFattB-CR2sul2 into 6,500 and 2,003 bp bands. (C) Detection of GItetWsu2-pKFattB structure. Left panel, XbaI digestion of the plasmid extract from six independent recombinant colonies. M1, M2, different DNA markers. Right panel, XbaI and SacI double restriction of plasmid extracts (number 5 and 6). SacI cuts GItetWsu2 once and not pKFattB, leading to the digestion of GItetWsu2-pKFattB into 13,276 and 739 bp bands. The band at 2.8 kb is the residual pKFattB plasmid. The bottom schematics exhibit the locus of restriction enzymes and expected digestion sizes for restriction analysis.
AlpA is deemed to improve the excision of circular intermediates during site-specific recombination (Kirby et al., 1994; Lesic et al., 2004), and thus can increase the integration frequency of genomic islands. However, alpA gene deletion in our experiment did not result in significant changes for integration (Table 2). We speculated that alpA gene may not be expressed or maybe expressed moderately due to the lack of an efficient promoter in GIsul2. In view of this point, we used the expression vector pCOLADGm and constructed the resultant plasmid pCOLADalpAGm (see Section Materials and methods), under the control of a strong T7 promoter induced by IPTG. pCOLADalpAGm, pKDGItetWsul2, and pKFattB were simultaneously transformed into the recA-deficient strain. The integration experiment was performed on the correct strain carrying the three plasmids. Then 20 randomly selected colonies were analyzed and six samples were exhibited in agarose gel. Their extracted plasmid DNA was cut using XbaI and the two recombinant plasmids displayed the significant 20.7-kb band (lanes 5, 6 of left panel in Figure 5C), corresponding to the size of pKFattB-GItetWsul2. We further cut the two recombinant plasmids using double restriction enzymes XbaI and SacI and showed the correct expected bands, 13.3 and 7.4-kb (lanes 5 and 6 of right panel in Figure 5C). SacI only exists in pKFattB-GItetWsul2 structure and can cut to different sizes of fragments with XbaI (the bottom in Figure 5C). The weak 4,023 bp band was residual pCOLADalpAGm that was only cut by XbaI, and the 2,843-bp band still existed, which was the remnant pKFattB. The restriction enzyme analysis strongly indicates that GItetWsul2 movement occurred. Enzyme digestions of other recombinant plasmids also displayed the 8.5-kb band, indicative of CR2-sul2 movement. These integration experiments clearly validated that GIsul2 and CR2-sul2 units can move into the trap plasmid.
GIsul2 and CR2-sul2 units are widely distributed in proteobacteria
To get a better insight into the distribution of GIsul2, CR2-sul2 unit, and GI12K in bacterial species, we screened for their presence in the 169,991 complete bacterial genomes available in the NCBI GenBank database at the time of the study (see methods). The distribution of GIsul2 shows that this element has disseminated in proteobacteria, essentially β- and γ-proteobacteria, although one instance has been detected in the α-proteobacteria Sphingopyxis granuli (Figure 6, Supplementary Table 4). Within γ-proteobacteria, it is restricted to two species of Acinetobacter (A. baumannii and A. calcoaceticus), in which eight instances were detected (6.6% of the total number of genomes for these two species), and to the Enterobacterales clade, in which it was sporadically found in six common pathogens (one to four instances per species, representing 0.3–33% of the species screened genomes). Within β-proteobacteria, GIsul2 is mostly detected in species of the Alcaliganecae family, with eight instances distributed among five species. However, the total number of available β-proteobacterial genomes was much lower than those of γ-proteobacteria, leading to a comparatively higher prevalence of GIsul2 in this cluster (15–100% of the species screened genomes, Supplementary Table 4). Only three of the 31 instances of GIsul2 were detected on plasmid replicons of the complete genomes (Figure 6), consistent with its primary integration site at the 3′ end of the guaA gene.
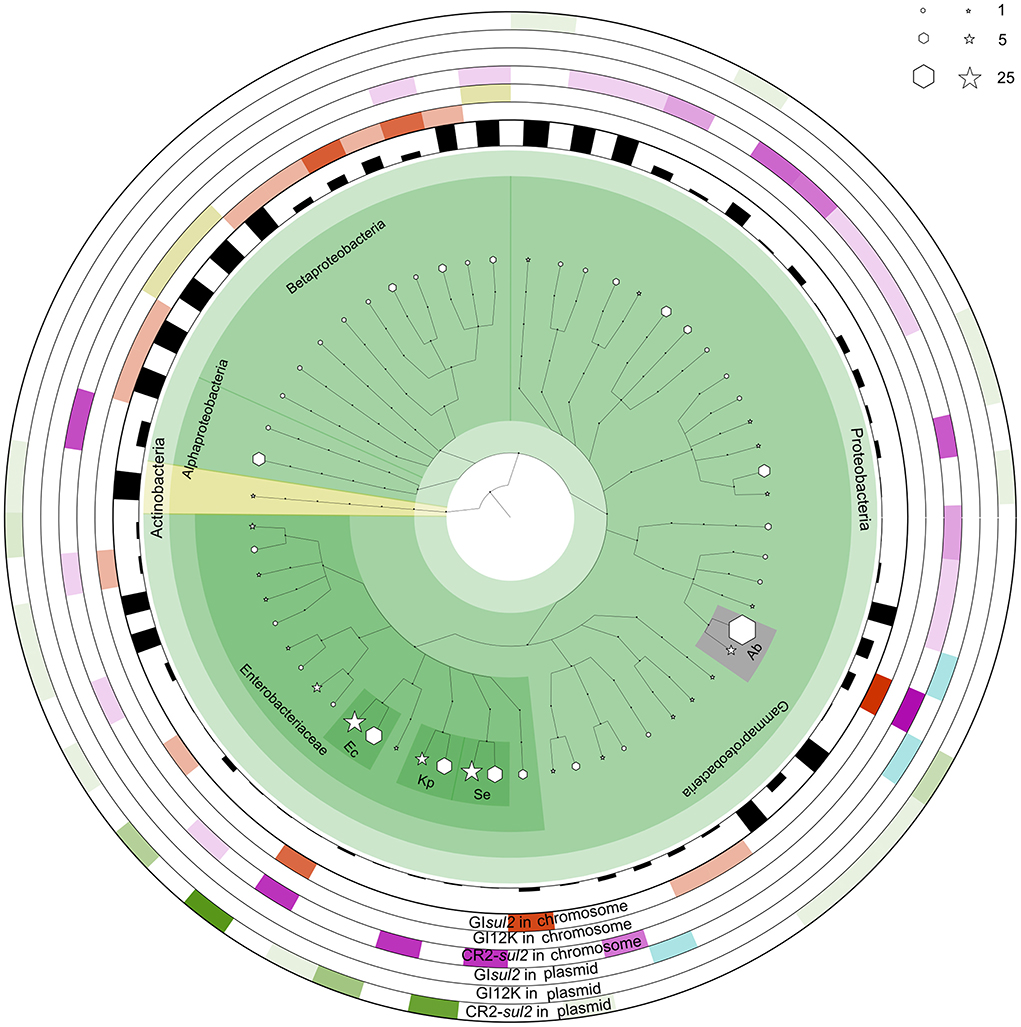
Figure 6. The distribution of GIsul2, CR2-sul2 unit, and GI12K in chromosomes and plasmids of NCBI complete genome database. The internal species tree represent the host distribution of the three elements. The size of the hexagon and star markers denotes the number of genomes of each host carrying the corresponding elements in chromosomes or plasmids, respectively. The bar height in the inner circle represents the proportion of genomes carrying the elements among the total genome of the species in the database. The six outermost heatmaps represent each element's abundance, separated by chromosomal or plasmid localization. The color intensity corresponds to the proportion of the host in the total number of genomes carrying the given element. Orange, olive green, and purple heatmaps represent the distribution of GIsul2, GI12K, and the CR2-sul2 unit in chromosomes, respectively. Blue and green heatmaps represent the dissemination of GIsul2 and CR2-sul2 units in the plasmids, respectively. Hosts with a total number of genomes >10 are highlighted and abbreviated as follows: Kp, Klebsiella pneumoniae; Ec, Escherichia coli; Se, Salmonella enterica; Ab, Acinetobacter baumannii.
CR2-sul2 unit was detected in one Actinobacteria, one α-proteobacteria, and two β-proteobacteria, but it is widespread in γ-proteobacteria with 180 instances detected in 30 species (Figure 6). These species belong to 6 different taxonomic orders (Supplementary Table 4), including the Aeromonadales, Oceanospirillales, Pasteurellales, and Vibrionales in which no GIsul2 was detected. The prevalence of CR2-sul2 unit within species is also substantially higher than the prevalence of GIsul2; for instance, CR2-sul2 unit was detected in 30.8, 17.8, 7.7, 6.6, and 5.4% of the A. baumannii, Vibrio cholerae, K. pneumoniae, S. enterica, and E. coli complete genomes, respectively, while GIsul2 was detected in only 5.8, 0, 0, 0, and 0.3% of the same genomes. Contrary to GIsul2, CR2-sul2 unit is also widely present on plasmids with 42% of the detections, although these plasmid-borne instances are mostly found in Enterobacteriaceae and A. baumannii (Figure 6). The last investigated element, GI12K, was found only in three complete genomes, belonging to three different species from three different orders of β-proteobacteria, and always in a chromosomal location (Figure 6).
When the NCBI draft genomes database was investigated, similar results were obtained (Supplementary Table 4). GIsul2 was found mostly in β-proteobacteria and γ-proteobacteria with more than 10 instances detected, respectively, in Bordetella bronchiseptica (24% of the total genomes of the species), and E. coli and A. baumannii (0.41 and 0.97% of the total genomes). The CR2-sul2 unit showed a wider distribution in γ-proteobacteria than GIsul2, but very few instances in β-proteobacteria. Its prevalence in draft genomes was similar to those in complete genomes with, e.g., 31 and 21% of the A. baumannii and V. cholerae genomes carrying it, respectively. The draft genome analysis revealed that GI12K was not restricted to β-proteobacteria since it was also detected in 19 γ-proteobacteria and one α-proteobacteria. However, its prevalence is usually extremely low in species with a sufficient number of available genomes (Supplementary Table 4). suggesting that these instances may be in fact GIsul2 structures for which the CR2-sul2 unit was incompletely sequenced or assembled.
Discussion
Although several studies showed that GIsul2 is distributed in various γ- and β-proteobacterial species (Nigro and Hall, 2011; Hamidian and Hall, 2016; Harmer et al., 2017), its mobility has never been demonstrated. In this report, we investigated the mobilization capacity of GIsul2 integrase to perform site-specific recombination at its cognate att sites. Contrary to most GIs studied to date, three related att sites were identified in GIsul2: the two expected attL and attR sites at the boundaries of the element and a third, termed attLR, lying upstream of the rcr2 putative transposase. This atypical GI structure carries an important mobile element, the CR2-sul2 unit, which can also be considered as an ISCR2-related element that plays an important role in transferring resistance determinants (Toleman et al., 2006a). But, the mobilization mechanism of ISCR2 is still not revealed through biological experiments. In the present study, excisions involving either attL-attR or attLR-attR complexes were both detected in RecA+ as well as in RecA− genetic backgrounds, indicating that the integrase can excise independently the GIsul2 and CR2-sul2 units (bounded by attLR and attR) from their respective insertion sites. However, detection of the circular intermediates required the use of nested PCR assays, suggesting that the excision process may be rare in standard culture conditions. This phenomenon was consistent with a previous report on the excision of high-pathogenicity island in Yersinia pseudotuberculosis (Lesic et al., 2004).
For most GI tyrosine integrases, the recombination process requires recombination directionality factors (RDFs). Notably, excision is usually controlled by excisionase proteins such as Xis in the phage λ model (Landy, 2014). The integrase gene of GIsul2 is followed by a gene annotated as alpA, which was initially proposed to act as a transcriptional regulator of CP4-57 prophage integrase, leading to the efficient excision of the prophage (Kirby et al., 1994). Later studies demonstrated that Hef, a protein homologous to AlpA, induces the excision of the high-pathogenicity island (HPI) of Y. pseudotuberculosis through excisionase activity (Lesic et al., 2004). Therefore, AlpA of GIsul2 likely also acts as an excisionase as previously suggested (Harmer et al., 2017), which may not be expressed or may be expressed slightly under standard culture conditions as is the case for excisionases of other integrative elements (Guédon et al., 2017). This speculation was verified by the introduction of alpA gene into expression vector pCOLADGm harboring T7 promoter during IPTG induction (Figure 6). Hence, we considered that the few observed circular intermediates could have arisen from expression leakage of the alpA gene or occasional AlpA-empty recombinations since excisionases are mostly helper proteins (Landy, 2014; Guédon et al., 2017). While the expression of Alp increased significantly, sufficient circular intermediates were supplied, and then GIsul2 and CR2-sul2 mobilization can be observed easily through restriction enzyme digestions.
Our findings are not consistent with the results of Harmer et al. (2017), who did not detect GIsul2 circular intermediates in their E. coli experimental strain despite a nested PCR procedure similar to ours. In their experiment, GIsul2 was not located in its typical integration site downstream of the guaA gene, but in an IncC-type plasmid in which the attL and attR sites were not conserved. We suspect that this unusual insertion site hampers the integrase-mediated excision because we showed that att sites are essential for proper integrase-mediated recombination. Despite its preferential chromosomal integration at the 3′-end of guaA, GIsul2 has been sporadically detected on plasmids at unrelated insertion sites (8, 9, 12, Figure 6). Here, we showed that elements devoid of att sites could still integrate into the donor plasmid, but at a very low rate and in a recombination site with reduced specificity (“TGGG” instead of “GAGTGGGA”). This suggests that Int behaves like any other integrase, for which target specificity may be loosened when no proper integration site is present in the cell (Guédon et al., 2017). However, non-specific integrations are likely a dead-end for the dissemination of GIsul2 because of the lack of proper excision when full-length attL/R sites are not present. GIsul2 is more probably an integrative and mobilizable element (IME), which relies on a helper conjugative element to transfer between bacteria (Guédon et al., 2017). The presence of plasmid-related transfer proteins into the element strongly supports this hypothesis, and the reported amino-acid similarity of these proteins to those found in IncP plasmids suggests that members of this plasmid family would be the helper elements (Harmer et al., 2017).
Finally, we showed that when the dominant cointegrate recombinants are eliminated by sucrose negative selection, integration of circularized intermediates into attB can be detected. Notably, integration of GIsul2 circular intermediate into the attB site of trap plasmid was observed through restriction enzyme analysis in recA− genetic context, indicating that the integrase can perform the final step of GIsul2 mobilization when molecular substrates are available. Even more interesting is our detection of CR2-sul2 integration in the attB site of the trap plasmid. CR2 transposase is very often located in the vicinity of sul2 with various ARGs in between, and several genomic evidences suggest that they represent independent mobile or mobilizable units (Leclercq et al., 2016; Xu et al., 2017). These CR2-sul2 units are therefore recognized as vectors of dissemination of antibiotic resistance in environmental bacteria and important clinical pathogens such as Vibrio spp., Pseudoalteromonas spp., Salmonella enterica, S. flexneri, and A. baumannii (Beaber et al., 2002; Toleman et al., 2006a; Yau et al., 2010; Leclercq et al., 2016; Harmer et al., 2017; Xu et al., 2017; Shi et al., 2019; Wüthrich et al., 2019). Especially, plasmid-borne ISCR2 carrying the high-level tigecycline resistance gene tet(X) has emerged in E. coli (Fang et al., 2019; He et al., 2019; Sun et al., 2019), which hints that we should pay close attention to the rise of this element. Interestingly, the boundaries of these transposons have been predicted to lie 119 bp upstream of the rcr gene and 304 bp upstream of sul2 (Leclercq et al., 2016), which correspond to the middle of the core recombination region of attLR, and the 3′-end of attR, respectively. In another study conducted on Aeromonas caviae, two independent ARG-carrying CR2-sul2 units are inserted in the attR core recombination region of a GI inserted in guaA. The GI harbors an integrase related to that of GIsul2 (Shi et al., 2019). Both integrations recreated perfect att core recombination regions (Shi et al., 2019), and very likely functional att sites. These observations, together with the results presented here, suggest that the integrase of GIsul2, or other integrases of the same family, may be the main drivers of the wide dissemination of CR2-sul2 units among proteobacteria. Future studies are need further to deeply investigate the influence of AlpA and possible host factors on integration or excision, providing a comprehensive understanding of mobilization.
Materials and methods
Bacterial strains and culture conditions
The genomic DNA of S. flexneri 51575 (kindly provided by Prof. Jianguo Xu from Chinese CDC) was extracted with the TIANamp Bacteria DNA Kit (TIANGEN BIOTECH, Beijing) and used to amplify the complete sequence region of GIsul2. E. coli DH5α deficient in recA was used for plasmid cloning and to perform all the integration experiments. IN-5 (KU736870) and IN-11 (KU736876) are, respectively tetW-containing and CR2-containing genomic regions cloned from pig manure's uncultured bacteria (Leclercq et al., 2016) and used for plasmid constructions. Unless otherwise stated, all strains were cultured at 37°C in a liquid LB medium. All plasmid constructions mentioned in this study are detailed in Supplementary Table 2.
PCR detection of circular intermediates
Detection of circular intermediates was performed using a nested PCR strategy on crude DNA from boiled overnight cultures (Lesic et al., 2004). To determine GIsul2 circular intermediates, the primer pair F1/R1 was first used to amplify the junction. Then the Sul2-F/Int-R primer pair was used for the second round of PCR amplification using the first-round PCR product as the template. Similarly, to detect CR2-sul2 unit and GI12K circular intermediates, a couple of primer pairs F1/R2 then Sul2-F/CR2-R, and tru-F1/R1 then tru-F2/Int-R were used, respectively. PCR conditions were as follows for both rounds: pre-denaturation at 95°C for 5 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 1 min; and then a final extension at 72°C for 5 min. All PCR amplifications were performed using the ploymerase TransStart® FastPfu Fly DNA Polymerase, which is a hot start, ultra-high fidelity, and high-processivity DNA polymerase (TRANS, Beijing, China). These PCR amplicons could also have theoretically arisen from chromosomal tandem duplications of each of the elements, but the fact that a nested PCR procedure was necessary to produce visible bands, together with the absence of such duplication in the genome sequence of the used strain, makes this possibility highly unlikely.
Construction of a two-plasmid assay system
The donor plasmid pKDGItetWsul2 was constructed in a two-step process. The first step consisted of the construction of a CR2′-tetW-sul′ fragment containing a constitutively expressed tetW gene. The tetW gene (2,070 bp) was amplified from the DNA of the IN-05 clone by using tetW-F and tetW-R primers. The PCR product was digested together with plasmid pUC19 by PstI and EcoRI enzymes and ligated using a T4 DNA ligase (New England Biolabs, Ipswich, MA, USA). The plasmid was termed pUC-tetW. The 3′ end of CR2 (1,156 bp) harboring the putative ori end was amplified from the IN-11 clone using G-UNIT-F/Rev-Pstv28cr2 primers. The tetW plus the PlacZ promoter sequences (2,403 bp) were amplified from pUC-tetW using G-tetWF/Rev-tetW primers. The partial sul2 sequence (789 bp) was amplified from IN-11 using Fwd-Pstv28cr2/G-UNIT-R primers. The above three fragments were assembled using the NEBuilder kit to obtain the CR2′-tetW-sul′ fragment (4,313 bp). The second step consisted of the aggregation of the CR2′-tetW-sul′ fragment with GIsul2 in a pKD46 backbone, lacking gam, bet, and exo regions, from four genomic fragments. First, the pKD46 backbone segment (4,199 bp) was obtained by PCR amplification using G-PKD-F and G-PKD-R primers from an intact pKD46 template. Second, the G-UP-floRF11 and G-UP-R primers were used to amplify a 2,505 bp fragment including the florfenicol resistance gene floR and the 5′ end of CR2 bearing attLR using the genomic DNA of the IN-11 clone as the template. The third fragment was the CR2′-tetW-sul′ constructed above. The fourth fragment included the 5′ end of sul2 and its upstream sequence (1,005 bp), obtained using G-DOWN-F and G-DOWN-R11 primers with DNA of the IN-11 clone as the template. The above four fragments were assembled using NEBuilder HiFi DNA Assembly Cloning Kit, resulting in the plasmid pKD-tetW-CR2. The 12-kb sequence of GIsul2 upstream of the CR2-sul2 unit was amplified from the genomic DNA of S. flexneri 51575 using 15K-F/CRUp-R primers, resulting in a 12,256 bp fragment. The recombinant plasmid pKD-tetW-CR2 was digested by BbvC1 restriction enzyme, producing overlap sequences with the corresponding two terminal regions of the GIsul2 12-kb fragment. The two fragments were ligated using the NEBuilder HiFi DNA Assembly Cloning Kit, resulting in the donor plasmid pKDGItetWsul2.
To construct the trap plasmid pKFattB, the attB region (811 bp), including the attB site (19 bp) and its upstream (319 bp) and downstream (473 bp) regions, was amplified from the chromosome of E. coli DH5α with primers G-GMP-SF and G-GMP-SR. These attB upstream and downstream regions corresponded to the 3′ end of guaA (336 bp) and the 5′ end of a hypothetical protein (381 bp), respectively. The high copy plasmid pKF18k-2 (Takara Bio Inc., Kasatsu, Japan) was digested using XbaI/HindIII enzymes and the digestion product was assembled together with the amplified attB region using NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs, Ipswich, MA, USA), resulting in the trap plasmid pKFattB.
Chromosomal integration and two-plasmid mobilization experiment in recA-deficient E. coli
pKDGItetWsul2 was transformed into E. coli DH5α, which was cultured on LB agar plates containing ampicillin (100 μg/ml) at 30°C. Single colonies (transformants) were selected and cultured at 30°C for 24 h in liquid medium containing tetracycline (20 μg/ml), then the temperature was increased to 42°C for 16 h to stop pKDGItetWsul2 replication. The resulting cultures were plated on LB medium containing tetracycline (20 μg/ml) at 42°C and then 12 colonies were selected and cultured in liquid LB at 42°C overnight. Crude DNA from these recombinant colonies was used as the template for PCR amplification.
The two-plasmid mobilization assay was performed according to a previous report (Wilde et al., 2008). pKDGItetWsul2 and pKFattB were co-transformed into E. coli DH5α, which was cultured on LB agar plates containing ampicillin (100 μg/ml) and kanamycin (50 μg/ml) at 30°C. The integration experiment was performed as in the chromosomal integration experiment, except that 50 μg/ml kanamycin was added to each medium. A total of 12 recombinant colonies were selected and their plasmids were extracted using TIANprep Mini Plasmid Kit (TIANGEN BIOTECH, Beijing). These recombinant plasmids were used as a template for PCR amplification and enzyme restriction analysis.
Nanopore sequencing and analysis
Five of the twelve recombinant plasmid extracts were digested with the XbaI restriction enzyme, cutting specifically the trap plasmid pKFattB 324 bp upstream of the attB site. The digestion products were sequenced using the PromethION platform (NextOmics, Wuhan, China). The raw reads data were demultiplexed and filtered with the Porechop v0.2.4 software (Wick, 2018). The quality control values were set as follows: (mean_qscore_template) ≥7 and sequence length ≥1,000 bp. Residual barcoding adapters were removed by trimming 100 bp on both sides of each read using cutadapt v2.9 (Martin, 2011). The filtered high-quality reads in fasta format were mapped against the pKFattB sequence and the sequence of each of the five expected recombinant structures using graphmap2 v0.6.3 (Sović et al., 2016) with the end-to-end owler parameter. The number of input structures mapped by each read was evaluated, and reads were categorized as unambiguous when they were properly mapped on only one structure and ambiguous when they were properly mapped on two or more structures.
Deletion of the int gene, att sites, and other mobility-associated genes or regions in GIsul2
Using pKDGItetWsul2 as a template, an int-deficient fragment of GIsul2 (int completely deleted) was amplified using the delINT-F/R primers. The backbone of pKDGItetWsul2 segment including the temperature-sensitive replicon and the ampicillin resistance marker was obtained using primers V15K-F/R. The two fragments were assembled using the NEBuilder Assembly Tool to produce the pKDGItetWsul2-Δint donor. Using the same strategy, the pKDGItetWsul2-Δrcr2, pKDGItetWsul2-ΔresG, and pKDGItetWsul2-Δrep donor plasmids were constructed using the delCR2-F/R and V15K-F/R, del-ser-F/R and Res-F/R, del-repF/R and V15K-F/V-D-repR primer pairs, respectively. The att-depleted plasmid donor was constructed as follows: attL-/attR-deficient fragment and the vector backbone fragment were amplified by PCR using del-21bp-F/R and VPKD-F/R primers, respectively, with pKDGItetWsul2 as the template. The two fragments were assembled using NEBuilder Assembly Tool, resulting in the attL/R-deficient plasmid. D-all21bpF/R and V-D-all21F/R primer pairs were then used to amplify two fragments devoid of attLR from the attL/R-deficient plasmid. The latter two fragments were assembled using NEBuilder Assembly Tool to construct the final attL/LR/R-deficient plasmid, named pKDGItetWsul2-Δatt. All primer sequences were designed according to the manufacturer's instructions (http://nebuilder.neb.com/), as shown in Supplementary Table 1. The constructed deletion plasmids served as donor plasmids for integration experiments using the two-plasmid mobilization system as described above.
Construction of pKDGItetWsul2sacB and pCOLADGm, and the mobilization assay
The MluI restriction enzyme (NEB) was used to cut pKDGItetWsul2 (24157 bp) and the sacB original vector pDM4 (7,104 bp). MluI cuts the araBAD promoter within pKDGItetWsul2 and cuts pDM4 twice, creating a 3.8-kb fragment that contains a chloramphenicol resistance gene and the sacB gene plus its promoter. The two fragments were purified and then ligated using a T4 DNA ligase (NEB). The ligation product was transformed into E. coli DH5α and cultured in LB containing tetracycline and chloramphenicol. Transformants were verified by MluI restriction analysis and sequencing. The new donor plasmid containing sacB was designated pKDGItetWsul2sacB and used for integration experiments with the trap plasmid pKFattB, as well as for the chromosomal integration experiment. Sucrose (20%) was used in addition to tetracycline and kanamycin in the LB medium used for the selection of the recombinant colonies.
To introduce alpA encoding region into the new two-plasmid system composed of pKDGItetWsul2sacB and pKFattB, pCOLADuet-1 (ColA origin, different from the origins of two-plasmid system) was used for overexpression of alpA gene under the control of IPTG. pCOLADuet-1 carries the kanamycin resistance determinant, similar to pKFattB. We used the gentamycin resistance gene derived from pEX18Gm to replace the kanamycin resistance gene. BspHI cut pCOLADuet-1 and pEX18Gm, respectively. Then using NEB T4 ligase ligated the two parts and then formed pCOLADGm. Amplification of alpA with primers alpANcoI-F and alpASalI-R was performed using the donor plasmid as the template. The PCR product was digested with NcoI and SalI restriction enzymes. The same double enzymes cut pCOLADGm. The two fragments were ligated by using T4 ligase. The resultant plasmid pCOLADalpAGm was constructed successfully through enzyme digestion and sequencing verification. pCOLADalpAGm, pKDGItetWsul2, and pKFattB were simultaneously transformed into the recA-deficient strain DH5α, and the LB agar plate containing tetracycline (10 μg/ml), kanamycin (50 μg/ml), and gentamycin (10 μg/ml) was screened at 30°C for 48 h. The correct strain was incubated in LB liquid culture carrying 20% sucrose, tetracycline (10 μg/ml), kanamycin (50 μg/ml), gentamycin (10 μg/ml), and IPTG (0.5 mM) at 30°C for 24 h. The culture dilution was plated LB harboring the same components as the liquid culture at 37°C for 48 h. Plasmids of the colonies extracted were the template, which was used for restriction enzyme analysis.
Bioinformatics methods and the distribution analysis of GI12K, GIsul2, and CR2-sul2
A total of 169,991 bacterial genomes were downloaded from the NCBI genome database [ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/] on 15 November 2018. The taxonomic lineage of each genome was retrieved from the NCBI taxonomy database. First, a search of regions matching the Int sequence with 95% amino acid identity and 98% coverage was performed. Preliminary results on the complete genomes database showed that the GI12K sequence coverage systematically exceeded 70% in genomes where a match on Int was detected with these parameters (Supplementary Figure 5). Hence, genomes returning a match to Int with 95% amino acid identity and 98% coverage were considered to carry GI12K. A search of the rcr2 and sul2 genes with a BLASTN cutoff of 95% nucleotide similarity was performed independently on all genomes, and genomes including both genes were considered as carrying the CR2-sul2 element. Preliminary results on complete genomes indicated that it is indeed the case since all the CR2-sul2 regions detected this way were surrounded by the typical 119 and 304 bp conserved regions ending at the core recombination sites of the att loci (Leclercq et al., 2016), despite inner structural variation. The distribution of the GI12K, GIsul2, and the CR2-sul2 unit was then estimated as follows: when only the match to the integrase was satisfied, the genome was considered to carry GI12K; when only the match to rcr2 and sul2 regions was satisfied, the genome was considered to carry a CR2-sul2 unit; finally, when all matches were satisfied, the genome was considered to carry GIsul2. This analysis was performed on the complete genome database and the draft genome database independently. The chromosomal or plasmid localization of the detected elements was returned only for the complete genome database, where the replicon type is known. Then, to illustrate the distribution of GI12K, GIsul2, and CR2-sul2, the bacterial taxonomic framework of the 169,991 bacterial genomes was built by using the software GraPhlAn based on the taxonomic lineage information (Asnicar et al., 2015), with the quantities for each GI being mapped to the taxonomic framework.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA608816.
Author contributions
GZ and QC designed and executed the work. GZ wrote the manuscript. JF and SL revised substantially the manuscript. JL and RG performed some detailed experiments. LD, NT, and FZ performed the distribution and drawn the relevant figure. YS and SL performed the bioinformatic analysis. CW did some figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grants 31870134, 32070075, 81861138053, and 31872632).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.905865/full#supplementary-material
References
Anantham, S., Harmer, C. J., and Hall, R. M. (2015). p39R861-4, A type 2 A/C2 plasmid carrying a segment from the A/C1 plasmid RA1. Microb. Drug Resist. 21, 571–576. doi: 10.1089/mdr.2015.0133
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C., and Segata, N. (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029
Beaber, J. W., Hochhut, B., and Waldor, M. K. (2002). Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J. Bacteriol. 184:4259–4269. doi: 10.1128/JB.184.15.4259-4269.2002
Carraro, N., and Burrus, V. (2014). Biology of three ICE families: SXT/R391, ICEBs1, and ICESt1/ICESt3. Microbiol. Spectr. 2, 1–20. doi: 10.1128/microbiolspec.MDNA3-0008-2014
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Fang, L. X., Chen, C., Yu, D. L., Sun, R. Y., Cui, C. Y., Chen, L., et al. (2019). Complete nucleotide sequence of a novel plasmid bearing the high-level tigecycline resistance gene tet(X4). Antimicrob. Agents Chemother. 63, e01373–e01319. doi: 10.1128/AAC.01373-19
Guédon, G., Libante, V., Coluzzi, C., Payot, S., and Leblond-Bourget, N. (2017). The obscure world of integrative and mobilizable elements, highly widespread elements that pirate bacterial conjugative systems. Genes 8:337. doi: 10.3390/genes8110337
Halder, U., Biswas, R., Kabiraj, A., Deora, R., Let, M., Roy, R. K., et al. (2022). Genomic, morphological, and biochemical analyses of a multi-metal resistant but multi-drug susceptible strain of Bordetella petrii from hospital soil. Sci. Rep. 12:8439. doi: 10.1038/s41598-022-12435-7
Hall, R. M. (2010). Salmonella genomic islands and antibiotic resistance in Salmonella enterica. Fut. Microbiol. 5, 1525–1538. doi: 10.2217/fmb.10.122
Hamidian, M., and Hall, R. M. (2016). Acinetobacter baumannii ATCC 19606 carries GIsul2 in a genomic island located in the chromosome. Antimicrob. Agents Chemother. 61, e01991–e01916. doi: 10.1128/AAC.01991-16
Harmer, C. J., and Hall, R. M. (2015). The A to Z of A/C plasmids. Plasmid 80, 63–82. doi: 10.1016/j.plasmid.2015.04.003
Harmer, C. J., Hamidian, M., and Hall, R. M. (2017). pIP40a, a type 1 IncC plasmid from 1969 carries the integrative element GIsul2 and a novel class II mercury resistance transposon. Plasmid 92, 17–25. doi: 10.1016/j.plasmid.2017.05.004
He, T., Wang, R., Liu, D., Walsh, T. R., Zhang, R., Lv, Y., et al. (2019). Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat Microbiol 4, 1450–1456. doi: 10.1038/s41564-019-0445-2
Hem, S., Wyrsch, E. R., Drigo, B., Baker, D. J., Charles, I. G., Donner, E., et al. (2022). Genomic analysis of carbapenem-resistant comamonas in water matrices: implications for public health and wastewater treatments. Appl. Environ. Microbiol. 16:e0064622. doi: 10.1128/aem.00646-22
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kirby, J. E., Trempy, J. E., and Gottesman, S. (1994). Excision of a P4-like cryptic prophage leads to Alp protease expression in Escherichia coli. J. Bacteriol. 176, 2068–2081. doi: 10.1128/jb.176.7.2068-2081.1994
Lallement, C. (2018). Caractérisation des séquences d'insertions ISCR bactériennes impliquées dans la résistance aux antibiotiques (Thesis). Université de Limoges, Limoges, France.
Landy, A. (2014). The λ integrase site-specific recombination pathway. Microbiol. Spectr. 3:MDNA3-0051-2014. doi: 10.1128/microbiolspec.MDNA3-0051-2014
Leclercq, S. O., Wang, C., Zhu, Y., Wu, H., Du, X., Liu, Z., et al. (2016). Diversity of the tetracycline mobilome within a chinese pig manure sample. Appl. Environ. Microbiol. 82, 6454–6462. doi: 10.1128/AEM.01754-16
Lesic, B., Bach, S., Ghigo, J. M., Dobrindt, U., Hacker, J., and Carniel, E. (2004). Excision of the high-pathogenicity island of Yersinia pseudotuberculosis requires the combined actions of its cognate integrase and Hef, a new recombination directionality factor. Mol. Microbiol. 52, 1337–1348. doi: 10.1111/j.1365-2958.2004.04073.x
Li, B., Chen, D., Lin, F., Wu, C., Cao, L., Chen, H., et al. (2022). Genomic Island-mediated horizontal transfer of the erythromycin resistance gene erm(X) among bifidobacteria. Appl. Environ. Microbiol. 88:e0041022. doi: 10.1128/aem.00410-22
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Nigro, S. J., and Hall, R. M. (2011). GIsul2, a genomic island carrying the sul2 sulphonamide resistance gene and the small mobile element CR2 found in the Enterobacter cloacae subspecies cloacae type strain ATCC 13047 from 1890, Shigella flexneri ATCC 700930 from 1954 and Acinetobacter baumannii ATCC 17978 from 1951. J. Antimicrob. Chemother. 66, 2175–2176. doi: 10.1093/jac/dkr230
Partridge, S. R., and Hall, R. M. (2003). In34, a complex In5 family class 1 integron containing orf513 and dfrA10. Antimicrob. Agents Chemother. 47, 342–349. doi: 10.1128/AAC.47.1.342-349.2003
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, e00088–e00017. doi: 10.1128/CMR.00088-17
Schleinitz, K. M., Vallaeys, T., and Kleinsteuber, S. (2010). Structural characterization of ISCR8, ISCR22, and ISCR23, subgroups of IS91-like insertion elements. Antimicrob. Agents Chemother. 54, 4321–4328. doi: 10.1128/AAC.00006-10
Shi, Y., Tian, Z., Leclercq, S. O., Zhang, H., Yang, M., and Zhang, Y. (2019). Genetic characterization and potential molecular dissemination mechanism of tet(31) gene in Aeromonas caviae from an oxytetracycline wastewater treatment system. J. Environ. Sci. 76, 259–266. doi: 10.1016/j.jes.2018.05.008
Sović, I., Šikić, M., Wilm, A., Fenlon, S. N., Chen, S., and Naragaran, N. (2016). Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 7:11307. doi: 10.1038/ncomms11307
Stokes, H. W., Tomaras, C., Parsons, Y., and Hall, R. M. (1993). The partial 3'-conserved segment duplications in the integrons In6 from pSa and In7 from pDGO100 have a common origin. Plasmid 30, 39–50 doi: 10.1006/plas.1993.1032
Sun, J., Chen, C., Cui, C. Y., Zhang, Y., Liu, X., Cui, Z. H., et al. (2019). Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 4, 1457–1464. doi: 10.1038/s41564-019-0496-4
Toleman, M. A., Bennett, P. M., and Walsh, T. R. (2006a). ISCR elements: novel gene-capturing systems of the 21st century? Microbiol. Mol. Biol. Rev. 70, 296–316. doi: 10.1128/MMBR.00048-05
Toleman, M. A., Bennett, P. M., and Walsh, T. R. (2006b). Common regions e.g. orf513 and antibiotic resistance: IS91-like elements evolving complex class 1 integrons. J. Antimicrob. Chemother. 58, 1–6. doi: 10.1093/jac/dkl204
Wick, R. (2018). Available online at: https://github.com/rrwick/Porechop
Wilde, C., Mazel, D., Hochhut, B., Middendorf, B., Le Roux, F., Carniel, E., et al. (2008). Delineation of the recombination sites necessary for integration of pathogenicity islands II and III into the Escherichia coli 536 chromosome. Mol. Microbiol. 68, 139–151. doi: 10.1111/j.1365-2958.2008.06145.x
Wüthrich, D., Brilhante, M., Hausherr, A., Becker, J., Meylan, M., and Perreten, V. (2019). A novel trimethoprim resistance gene, dfrA36, characterized from Escherichia coli from calves. mSphere 4, e00255–e00219. doi: 10.1128/mSphere.00255-19
Xu, Y., Wang, C., Zhang, G., Tian, J., Liu, Y., Shen, X., et al. (2017). ISCR2 is associated with the dissemination of multiple resistance genes among Vibrio spp. and Pseudoalteromonas spp. isolated from farmed fish. Arch. Microbiol. 199, 891–896. doi: 10.1007/s00203-017-1365-2
Keywords: integrase, genomic island (GI), mobilization, ISCR elements, site-specific recombination
Citation: Zhang G, Cui Q, Li J, Guo R, Leclercq SO, Du L, Tang N, Song Y, Wang C, Zhao F and Feng J (2022) The integrase of genomic island GIsul2 mediates the mobilization of GIsul2 and ISCR-related element CR2-sul2 unit through site-specific recombination. Front. Microbiol. 13:905865. doi: 10.3389/fmicb.2022.905865
Received: 28 March 2022; Accepted: 04 July 2022;
Published: 01 August 2022.
Edited by:
Yongfei Hu, China Agricultural University, ChinaReviewed by:
Shiwei Wang, Northwest University, ChinaHenan Li, Peking University People's Hospital, China
Copyright © 2022 Zhang, Cui, Li, Guo, Leclercq, Du, Tang, Song, Wang, Zhao and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Feng, fengj@im.ac.cn
†Present Address: Qinna Cui, BioQuant Center of the University of Heidelberg, Heidelberg, Germany; Max-Planck-Institute for Terrestrial Microbiology, Marburg, Germany
‡These authors have contributed equally to this work