 Effie C. Kisgeropoulos
Effie C. Kisgeropoulos Vivek S. Bharadwaj
Vivek S. Bharadwaj David W. Mulder
David W. Mulder Paul W. King
Paul W. King- National Renewable Energy Lab, Biosciences Center, Golden, CO, United States
The [FeFe]-hydrogenases are enzymes that catalyze the reversible activation of H2 coupled to the reduction–oxidation of electron carriers. Members of the different taxonomic groups of [FeFe]-hydrogenases display a wide range of preference, or bias, for H2 oxidation or H2 production reactions, despite sharing a common catalytic cofactor, or H-cluster. Identifying the properties that control reactivity remains an active area of investigation, and models have emerged that include diversity in the catalytic site coordination environments and compositions of electron transfer chains. The kinetics of proton-coupled electron transfer at the H-cluster might be expected to be a point of control of reactivity. To test this hypothesis, systematic changes were made to the conserved cysteine residue that functions in proton exchange with the H-cluster in the three model enzymes: CaI, CpII, and CrHydA1. CaI and CpII both employ electron transfer accessory clusters but differ in bias, whereas CrHydA1 lacks accessory clusters having only the H-cluster. Changing from cysteine to either serine (more basic) or aspartate (more acidic) modifies the sidechain pKa and thus the barrier for the proton exchange step. The reaction rates for H2 oxidation or H2 evolution were surveyed and measured for model [FeFe]-hydrogenases, and the results show that the initial proton-transfer step in [FeFe]-hydrogenase is tightly coupled to the control of reactivity; a change from cysteine to more basic serine favored H2 oxidation in all enzymes, whereas a change to more acidic aspartate caused a shift in preference toward H2 evolution. Overall, the changes in reactivity profiles were profound, spanning 105 in ratio of the H2 oxidation-to-H2 evolution rates. The fact that the change in reactivity follows a common trend implies that the effect of changing the proton-transfer residue pKa may also be framed as an effect on the scaling relationship between the H-cluster di(thiolmethyl)amine (DTMA) ligand pKa and Em values of the H-cluster. Experimental observations that support this relationship, and how it relates to catalytic function in [FeFe]-hydrogenases, are discussed.
Introduction
The [FeFe]-hydrogenase class of enzymes fulfill significant roles in H2 metabolism and energy transduction. They catalyze the reversible reaction, H2↔2H+ + 2e- by performing either H2 gas evolution (i.e., proton reduction) or H2 oxidation. This is mediated by a complex metallocofactor, or H-cluster, consisting of a diiron site ([2Fe]H) coordinated by a conserved cysteine to a [4Fe-4S] cubane ([4Fe-4S]H; Figure 1). To achieve this reversibility, the enzyme must couple dynamic fluxes in both PT and ET to catalytic H2 activation. As a result, the catalytic function of the H-cluster must be adapted to a changing energy landscape. [FeFe]-hydrogenases therefore represent an ideal model system for understanding the mechanisms that enzymes employ to control proton-coupled electron transfer (PCET) at metal sites to accomplish chemical transformation reactions. While most [FeFe]-hydrogenases possess neutral reactivity profiles, i.e., similar rates for both H2 evolution and H2 oxidation, there are instances where reactivity for either one or the other is favored. For example, the overall catalytic preference across the diversity of [FeFe]-hydrogenase from Clostridium pasteurianum spans an impressive seven orders of magnitude from CpI (neutral reactivity) to CpII (biased toward H2 oxidation) and CpIII (biased toward H2 evolution; Adams, 1990; Poudel et al., 2016; Artz et al., 2020b). The wide range in reactivity stands in contrast to the fact that all [FeFe]-hydrogenases studied so far share a common H-cluster cofactor.
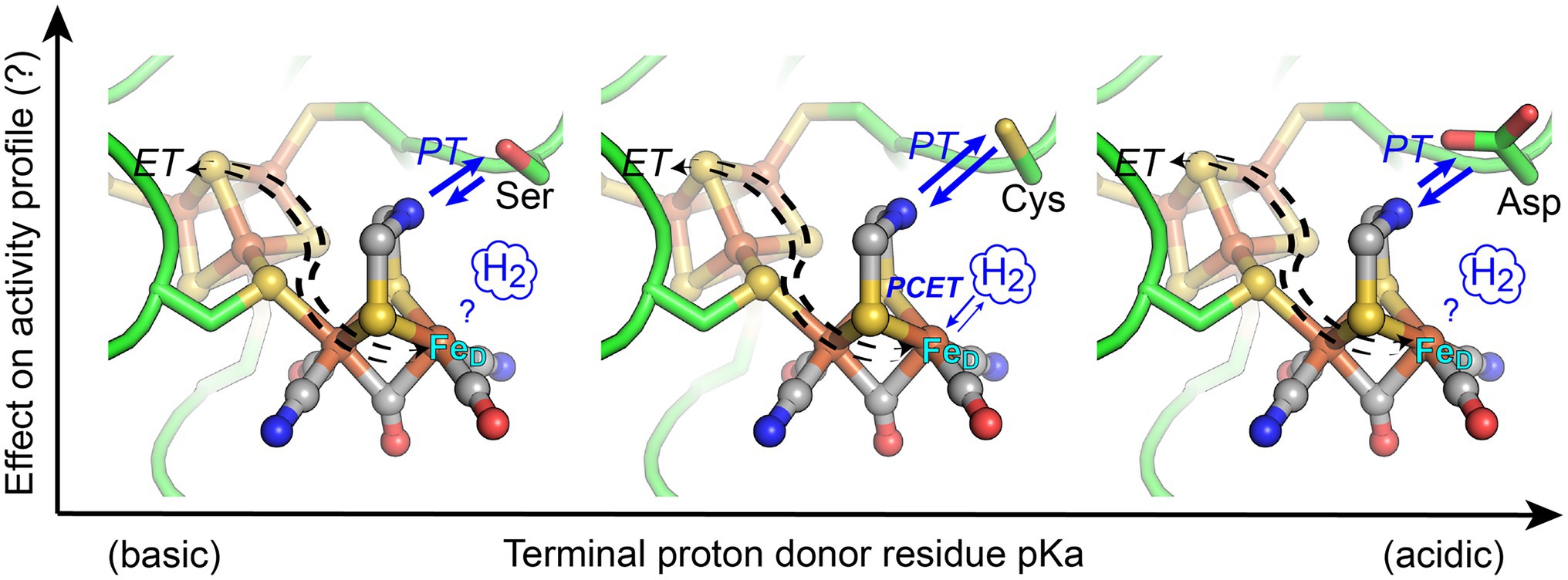
Figure 1. Structural depiction of the [FeFe]-hydrogenase H-cluster active-site where coordination of orthogonal electron transfer (ET) and proton transfer (PT) pathways by proton-coupled electron transfer (PCET) at the distal Fe atom (FeD) of [2Fe]H results in catalytic activation of H2. In this work, the conserved Cys residue (middle panel), which functions in proton exchange with the H-cluster di(thiolmethyl)amine (DTMA) ligand, was changed to either Ser (more basic pKa, left) or Asp (more acidic pKa, right). The activity profile of enzyme variants was measured to ascertain the extent that engineered variations to the pKa of the exchange step alter reactivity of [FeFe]-hydrogenases. Blue arrows indicate relative changes in the proton transfer direction bias for the variants. Structural representation from PDB 3C8Y, with substitution of Ser or Asp by alignment to PDB 6GLZ.
Diversity in the amino acids that comprise the catalytic site environments, differential stabilization of catalytic intermediates, amino acid composition of proton transfer pathways, and differences in thermodynamic profiles of electron transfer relays have all been proposed to account for the observed differences in reactivity profiles of [FeFe]-hydrogenases (Cornish et al., 2011; Hexter et al., 2012; Ginovska-Pangovska et al., 2014; Artz et al., 2017; Caserta et al., 2018; Duan et al., 2018; Gauquelin et al., 2018; Rodríguez-Maciá et al., 2019; Senger et al., 2019; Lampret et al., 2020). It is conceivable these properties converge to control PCET chemistry at the H-cluster, and changes in the coupling of protons and electrons at the H-cluster may also have a role in the control of reactivity. Computational and experimental studies have shown that PT flux to the H-cluster involves defined, well-conserved structural elements (Long et al., 2014; Duan et al., 2018; Lampret et al., 2020) including a strictly conserved cysteine residue that forms an H-bonding network with the di(thiolmethyl)amine ligand (DTMA) and distal Fe (FeD) of [2Fe]H in catalytically active enzymes (Figure 1). Cysteine is a relatively basic residue (free cysteine pKa ~8) and mediates the proton exchange step with the H-cluster during catalysis. Electron transfer can also be a control point of catalytic reactivity due to variability in midpoint-potentials of accessory iron–sulfur clusters (referred to as F-clusters) and/or their electronic interactions with the H-cluster (Adams et al., 1989; Mulder et al., 2013; Artz et al., 2017; Rodríguez-Maciá et al., 2017, 2019, 2020; Caserta et al., 2018). Together these mechanisms exert control over the PT and ET fluxes in the enzyme and contribute to the control of the PCET kinetics at the H-cluster. For example, the fine-tuning of the thermodynamic equilibria between reaction intermediates (Artz et al., 2017), H-bonding effects on the electronic structure of the H-cluster (Mulder et al., 2014; Pham et al., 2018), or matching of proton transfer pKa values to the H-cluster DTMA ligand pKa (Lampret et al., 2020; Birrell et al., 2021) are all mechanisms that have been demonstrated to influence the catalytic cycle and overall reactivity profile of the [FeFe]-hydrogenases.
To address understanding of the PCET mechanism in [FeFe]-hydrogenases, this work summarizes the results on testing the hypothesis that the pKa of the conserved residue that mediates the coupling of PT to ET (or PCET) at the H-cluster can be tuned to control reactivity in [FeFe]-hydrogenases. Using [FeFe]-hydrogenases that differ in their intrinsic reactivity, we applied reverse engineering to replace the conserved cysteine with either a more basic (serine) or acidic (aspartate) residue (Figure 1). The enzymes examined include CaI from Clostridium acetobutylicum (neutral reactivity) and CpII from C. pasteurianum (H2 oxidation bias), both of which have accessory F-clusters with different thermodynamic profiles (Adams et al., 1989), and HydA1 from Chlamydomonas reinhardtii (CrHydA1, neutral reactivity), which possesses only the catalytic H-cluster. Computational modeling was used to evaluate the pKa’s of the native and enzyme variants, and the H2 oxidation and evolution rates were measured to assess the reactivity profiles. The results of this work, in the context of previous biophysical studies of the reaction intermediates and redox profiles of related variants, are integrated into an overall thermodynamic model of reactivity control in [FeFe]-hydrogenase.
Materials and Methods
Expression, Purification, and Activity Measurements of [FeFe]-hydrogenases
CaI, CpII, and CrHydAI [FeFe]-hydrogenases were expressed and purified as previously described (King et al., 2006; Mulder et al., 2014; Ratzloff et al., 2018; Artz et al., 2020b) and assayed for both H2 evolution and H2 oxidation (uptake) using standard biochemical assays (Duan et al., 2018; Artz et al., 2020b).
H2 production was measured by gas chromatography (Agilent Technologies) using methyl viologen (MV) as the electron donor. The 2 ml reactions were set up in 13 ml anaerobic vials containing 1 μg to 1 mg of enzyme in 50 mM Tris (pH 8–8.3), 200–300 mM NaCl, 5% glycerol, and 10–100 mM sodium dithionite (DT); enzyme concentrations were varied as needed to measure kinetic parameters. Reactions were carried out at 37°C and initiated by the addition of MV to a final concentration of 5–80 mM. The reported rates were measured in the initial linear phase (varying from 5 to 40 min for the native enzymes and enzyme variants) of the reaction.
H2 oxidation was monitored by UV–Vis using either methylene blue (MB) monitored at 664 nm (ε = 95,000 M−1 cm−1) or benzyl viologen (BV) monitored at 600 nm (ε = 10,000 M−1 cm−1) as the electron acceptor (Cenens et al., 1988; Dörner and Boll, 2002; Duan et al., 2018). The 2 ml reactions were set up in 4 ml, septa-sealed cuvettes, with 1 μg to 1 mg of enzyme at various concentrations in 50 mM Tris pH 8–8.3, 300 mM NaCl, and 5% glycerol. The cuvettes were then either sparged continuously under H2 for 5 min, or subjected to 10 vacuum/refill cycles with H2 on a Schlenk line, and allowed to incubate under an overpressure of H2 at room temperature for 5–10 min. The reaction was then initiated with the addition of the redox dye, to a final concentration of 38 μM (MB) or 10 mM (BV). No reduction of either dye was observed when added to cuvettes containing only buffer. The Vmax values for CaI, CpI, and CrHydA1 are similar when either MV or MB is used as the acceptor (Adams and Mortenson, 1984; Artz et al., 2020b). The values for CpII are maximal using MB as the acceptor (Adams and Mortenson, 1984); therefore, MB was used for all CpII H2 oxidation assays. Maximal rates were calculated over the initial 1–5 min of the reaction.
Structural Models and pKa Calculations of [FeFe]-hydrogenase Variants
Since experimentally characterized and holo-structures of CrHydA1, CpII, and CaI are not yet available, the open-source neural-network-based protein prediction tool AlphaFold2 was used for their structure prediction (Jumper et al., 2021). For each protein, only the amino acid sequence was used as input to AlphaFold2 and five models were generated. Since Alphafold2 does not incorporate ligands into its structure predictions, the top-ranked AlphaFold2 predicted structure for each [FeFe]-hydrogenase was aligned with the experimental X-ray crystal structure for CpI (PDB ID: 3C8Y) for the incorporation of the H-cluster into the predicted structures. The H-cluster-bound top-ranked models for each [FeFe]-hydrogenase were then used for pKa estimations for its titratable residues using the Propka ver. 3.1 software package (Bas et al., 2008; Søndergaard et al., 2011). Of particular interest are the pKa predictions for the conserved cysteine residue (and aspartic acid for the C→D variant) that functions in proton exchange with the H-cluster. Since Propka does not consider residues with bulk pKa values >10 as titratable, values for the serine residue in the C→S variants were not predicted in this study, and experimental values from related model systems were used (Bruice et al., 1962; Liepinsh and Otting, 1996). It may be noted that these pKa calculations are intended as estimates and not exact determinations of the pKa values. This will require more computationally intensive techniques such as constant pH molecular dynamics simulations which are not undertaken in this study.
Results
pKa Calculations and Activity Profiles of Native [FeFe]-hydrogenases
The strictly conserved cysteine residue of catalytically active [FeFe]-hydrogenases (protein sequence number C298 CaI, C169 CpII, and C169 CrHydA1) is known to function in mediating the exchange of protons between the conserved proton transfer pathway and the H-cluster during catalysis (Cornish et al., 2011; Ginovska-Pangovska et al., 2014; Mulder et al., 2014; Lampret et al., 2020). The free-energy of proton transfer, ΔGPT, is related to the pKa of the exchange site by Equation 1;
To examine the extent that the proton-transfer residue pKa controls H-cluster protonation and enzyme reactivity (Figure 1), we first evaluated the cysteine pKa values using computational approaches and established a baseline of activity values for the model [FeFe]-hydrogenases, CaI, CpII, and CrHydA1 (Table 1). Proton-transfer residue pKa values were calculated using Propka (Olsson et al., 2011; Søndergaard et al., 2011), a computational treatment for the empirical prediction of pKa values which also takes into account the influences of the protein environment (vide infra). For the [FeFe]-hydrogenases listed in Table 1, the determined cysteine pKa values ranged 11.5–12. It may be noted that the calculated values are more basic compared to the pKa value for a cysteine residue in bulk solution. This can be attributed to the fact that Propka considers the contributions from desolvation energies, H-bonding energies, electrostatic reorganization energies, and coulombic interactions that are all cumulatively added to the bulk pKa value of a given titratable residue. A greater degree of H-bonding shifts the pKa to be more basic, while a greater degree of desolvation makes conjugate bases more basic and conjugate acids more acidic (Bas et al., 2008). For the conserved cysteine in [FeFe]-hydrogenases, these collective effects contribute to an overall more basic pKa value.
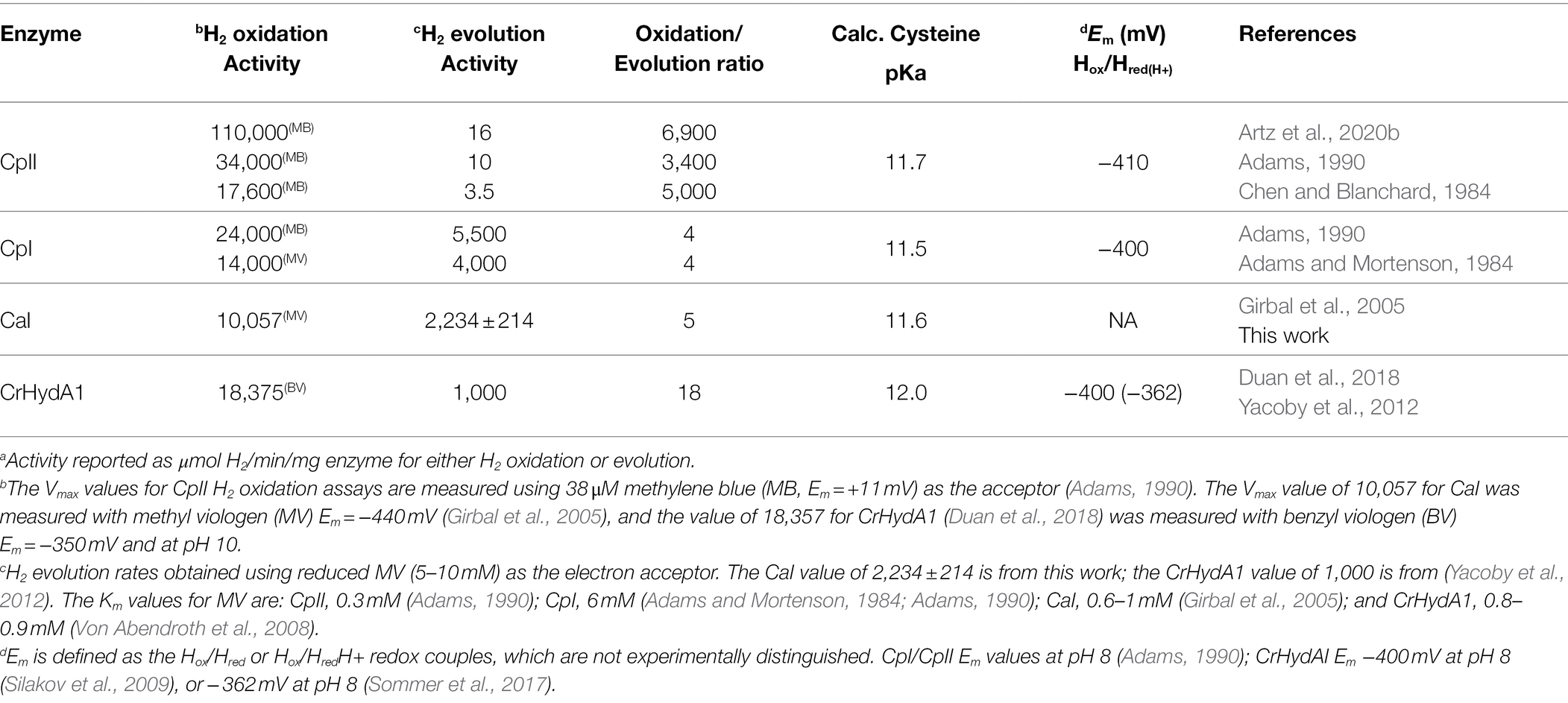
Table 1. Overview of WT [FeFe]-hydrogenase activitya profiles.
Table 1 also shows H2 evolution and H2 oxidation rates for the model [FeFe]-hydrogenases. The corresponding reactivity preference, also referred to as catalytic bias (Abou Hamdan et al., 2012; Artz et al., 2020b; Fourmond et al., 2021; Mulder et al., 2021), can be discerned from the ratio of H2 oxidation activity to H2 evolution activity. A ratio approaching 1 is representative of the energy landscapes of the enzyme being leveled so that it no longer catalytically favors one direction or another. In such a case, the enzyme is described as having a “neutral” catalytic bias. As shown in Table 1, the dye-assays show that CaI, CpI, and CrHydA1 each have an H2 oxidation-to-H2 evolution ratio of <18, with CaI and CpI being slightly more neutral in bias than CrHydA1.
In contrast, and as shown previously (Adams and Mortenson, 1984; Chen and Blanchard, 1984; Adams, 1990; Artz et al., 2020b), CpII has a large bias toward H2 oxidation, with an H2 oxidation-to-H2 evolution ratio that is two orders of magnitude greater than for CaI, CpI, or CrHydA1 (Table 1). It is noted that the calculated cysteine pKa values for all the enzymes examined here are generally basic and that all the catalytic [FeFe]-hydrogenases incorporate cysteine at the exchange site (an exception to cysteine occurs in the sensory [FeFe]-hydrogenases, see Poudel et al., 2016; Chongdar et al., 2018; Fasano et al., 2021). Thus, it does not appear that the pKa of the exchange step is used to control the reactivity or catalytic bias. The fact that the relative oxidation/evolution reaction profiles differ by as much as 103, with cysteines having similar pKa values, implies other factors, such as F-cluster reduction potentials, dynamic secondary interactions, and local electrostatics around the H-cluster (Adams et al., 1989; Caserta et al., 2018; Artz et al., 2020b), might be critical to the control of reactivity between enzyme types. It is also noted that the Em of the Hox/Hred transition is similar in value for CpI, CpII, and CrHydA1 (Adams et al., 1989; Silakov et al., 2009) and near the value of the H+/H2 couple (−413 mV vs. SHE at pH 7, 1 atm H2), which is consistent with the minimal overpotential requirement of these enzymes for catalysis.
Cys→Ser Variant pKa and Activity Profiles
To probe how differences in reactivity of CaI, CpII, and CrHydA1 are influenced by the pKa of the nearby proton donor residue, we examined enzyme variants where the cysteine residue is either altered to a more basic serine (C→S) or more acidic aspartate (C→D) residue. Previous site-saturation studies at the cysteine position have shown that these two mutations retain H-cluster cofactor incorporation with decreased H2 evolution activity (Knörzer et al., 2012; Morra et al., 2012; Mulder et al., 2014; Duan et al., 2018); however, the effects on H2 oxidation and full activity profiles in the variants are less understood.
The Propka method used to calculate cysteine pKa values is not configured to calculate serine pKa values (see the section Materials and Methods for a detailed discussion). However, experimental measurements of the pKa of -OH moieties in mimics of the catalytic triad in chymotrypsin assign the pKa of serine to be ~13.6 (Bruice et al., 1962). In this enzyme, the serine-OH group is deprotonated by the Nε atom of a nearby histidine during catalytic esterification of aromatic amino acids (Bruice et al., 1962; Frey, 2001). The basicity of the histidine Nε atom, with a pKa of 12, is within the pKa range of the H-cluster DTMA ligand that was determined from the modeling of FTIR spectro-electrochemical data from CrHydA1 collected at different pH values (Hred/HredH+ pKa = 7.2 and, Hsred/HsredH+ pKa = 11.6; Sommer et al., 2017; Birrell et al., 2021). Based on these similarities to [FeFe]-hydrogenase, we have tentatively assigned the serine residue as having a pKa of ≥13.6. Other experimental studies have measured the pKa of the hydroxyl group on a serine amino acid to be >16 using NMR (Liepinsh and Otting, 1996), further indicating its highly basic nature.
Using redox-dye mediated assays on the purified CrHydA1 C169S variant, H2 oxidation and evolution activities were determined and compared to CaI C298S (Cornish et al., 2011) and CpII C169S (Artz et al., 2020a; Table 2). The reactivity results show a collective shift toward H2 oxidation reactivity for all the enzymes. Whereas CpII C169S maintained the highest H2 oxidation preference among the C→S variants, the reactivity preference of CaI C298S likewise shifted over 100-fold toward H2 oxidation compared to WT CaI. It is also noted that while all C→S variants exhibit a decrease in the absolute reactivity rates compared to WT counterparts, this decrease is consistently more pronounced for the H2 evolution rates (Table 2), as has been observed for CpI (Cornish et al., 2011). This can be viewed as consistent with the basic pKa shift in relation to native Cys for the PT exchange step, and the transfer of protons from the H-cluster being more favored (discussed in more detail below).
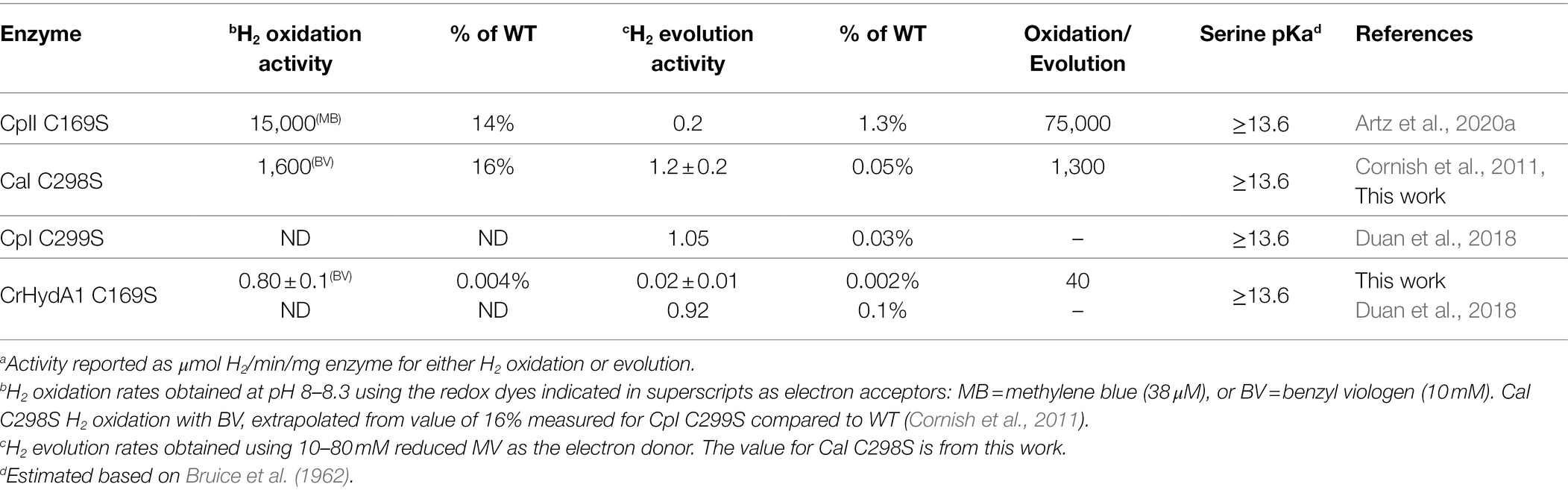
Table 2. Activitya profiles of [FeFe]-hydrogenase C→S variants.
Cys→Asp Variant pKa and Activity Profiles
The effect of introducing a more acidic proton donor residue near the H-cluster in CaI, CpII, and CrHydA1 was also examined using the C→D variant (Table 3). In contrast to the C→S variant, it could be expected that the more acidic pKa of the carboxyl group (pKa ≈ 4–6, Table 3) would cause a shift in the reactivity profile toward H2 evolution, resulting in oxidation/evolution ratios below 1. CrHydA1 C169D displayed a ratio <1 (oxidation/evolution ratio = 0.3) although this still falls within the definition for neutral bias given above, while CaI C298D also retains a neutral bias (Morra et al., 2012). Remarkably, CpII C169D had neutral reactivity, with an oxidation/evolution ratio of 11 (Table 3).
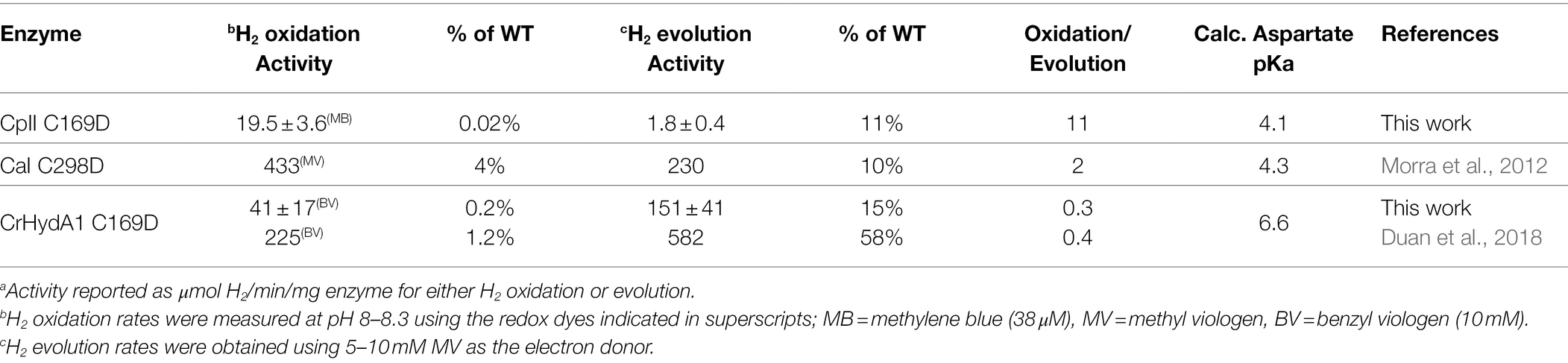
Table 3. Activitya profiles of [FeFe]-hydrogenase C→D variants.
Although the WT activity profiles of CaI and CrHydAI are also considered neutral, the H2 oxidation/evolution values for their C→D variants decreased relative to the WT, indicating a skewing toward H2 evolution that was not present before. This is much more striking in the case of CpII, where the H2 oxidation/evolution ratio for the C169D variant is shifted three orders of magnitude from WT in the direction of H2 evolution, effectively neutralizing the CpII bias toward H2 oxidation. As observed for the C→S variants, there is an overall decrease in the absolute H2 reactivity rates of the C→D variants compared to WT counterparts, with the decrease now consistently more pronounced for the H2 oxidation rates. From this, it appears that the pKa shift of the C→D substitution provides an overall leveling of the catalytic bias (oxidation/evolution ratio approaching 1).
Discussion
Here, we have tested the prediction that reactivity in [FeFe]-hydrogenases can be tuned through perturbing the pKa value of the amino acid in the secondary coordination sphere that mediates the proton exchange step with the H-cluster. Modulation of the proton exchange site pKa was achieved by mutation of Cys and led to pronounced shifts in the reactivity profiles of all the [FeFe]-hydrogenases that were tested, regardless of the presence of F-clusters or the intrinsic bias (Table 4; Figure 1). The native [FeFe]-hydrogenases establish a common baseline for comparison between the different enzymes as they exhibit similar properties, such as the pKa of the conserved cysteine residue (pKa = 11.5–12) and the Em for the Hox/Hred couple, which is relevant to the initial step for proton binding or H2 activation (CpII, −410 mV; CpI, −400 mV; and CrHydAI, −400 mV; Adams, 1990; Silakov et al., 2009). The observed trends can be summarized as follows: the C→S substitution resulted in a more basic pKa that shifted reactivity toward H2 oxidation, while the C→D substitution resulted in a more acidic pKa that shifted the reactivity toward that of H2 evolution, resulting in an overall neutralization of the catalytic bias.
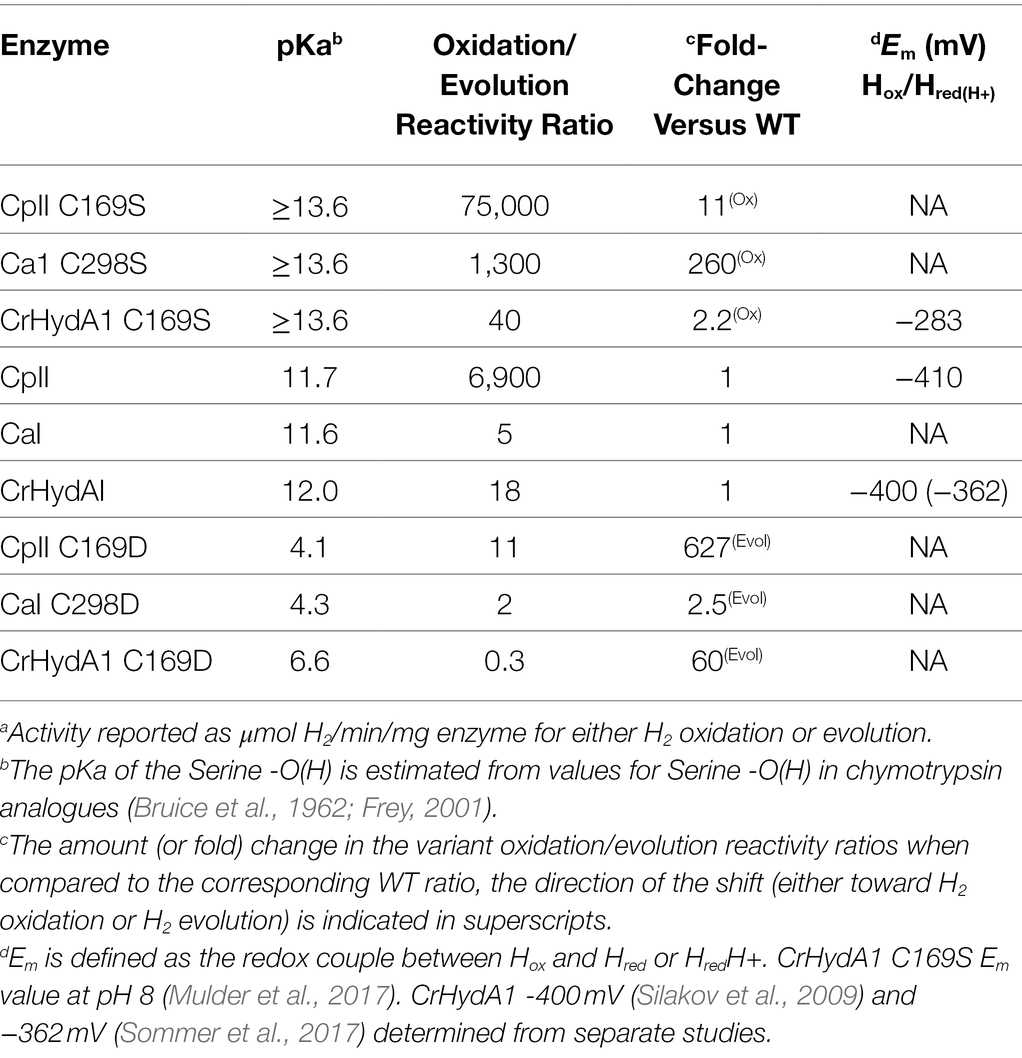
Table 4. Summary of exchange site pKa on [FeFe]-hydrogenase activitya profiles.
[FeFe]-hydrogenase Reactivity and the pKa, Em Scaling Relationship
The selection of either Cys, Ser, or Asp as the proton exchange site residue imparts a profound control over reactivity, which we propose in part results from perturbing the balance between exchange site pKa (Tables 1–3) and the pKa of the DTMA ligand of the H-cluster (Figure 2), and changes to H-bonding at the DTMA in the variants (Duan et al., 2018; Pham et al., 2018). The change from Cys to Ser also coincides with previous observations (Mulder et al., 2017) of a positive shift in the measured H-cluster reduction potentials, consistent with changes to the H-cluster electronic structure. The values for CrHydA1 C169S compared to WT are −283 and −400 mV vs. NHE for reduction of Hox to Hred, and −431 and −460 mV vs. NHE for reduction of Hred to Hhyd or Hsred, respectively (Silakov et al., 2009; Mulder et al., 2017; Table 4). Conversely, it has been shown that CaI C298D has a proportionately higher population of Hox under reduction with either H2 or sodium dithionite compared to WT CaI (Morra et al., 2016). This difference is consistent with a negative shift in the Em value, and presumably a shift toward a more basic pKa value of the H-cluster DTMA ligand.
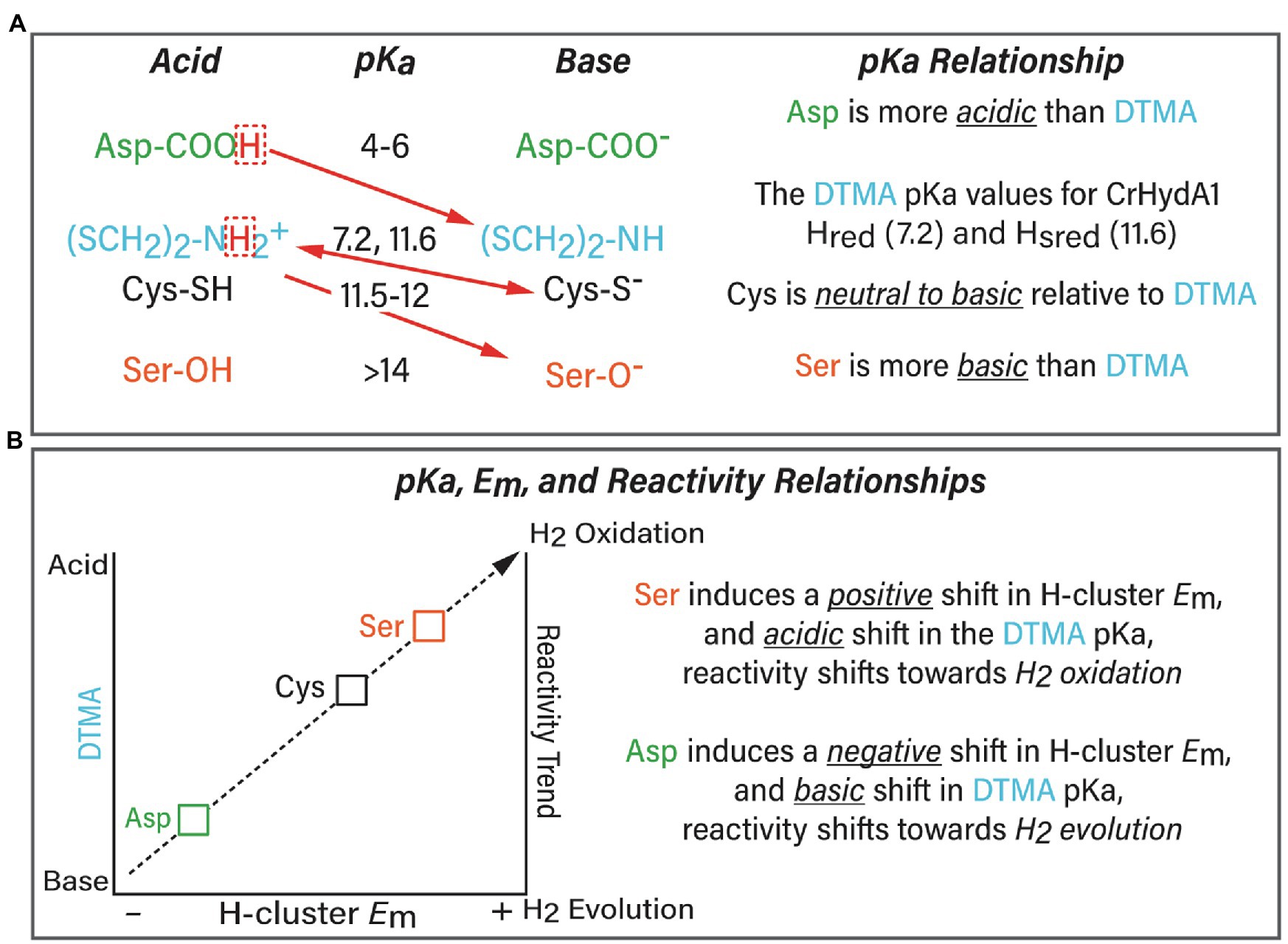
Figure 2. (A) Summary of the relationship between the exchange site pKa (Asp, green; Cys, black; and Ser, orange) and pKa of the H-cluster DTMA ligand (blue). The preferred proton transfer direction is indicated with red arrows. The pKa values for Asp, Cys, and Ser were calculated in this study, and the measured pKa values for the bridging ligand di(thiolmethyl)amine (DTMA, blue) were taken from (Sommer et al., 2017; Birrell et al., 2021) for CrHydA1. The scaling model predicts that Asp induces a basic shift whereas Ser induces an acidic shift in DTMA pKa, this would create an even more favorable direction bias than indicated in the Figure. (B) Summary of the model of the pKa, Em, and reactivity relationships based on the results of this work.
The differential stabilization of H-cluster oxidation states and Em shifts can be explained within the framework of the linear-scaling relationships between the pKa and Em of a transition metal catalyst (Figure 2; Bullock et al., 2014; Brereton et al., 2020; Puthenkalathil and Ensing, 2021). In this relationship, there is a direct correlation of the metal site pKa to the Em value of the reduction potential, where a more acidic pKa is matched by a more positive reduction potential. For the serine variant of CrHydA1, substitution of the more basic serine would be predicted to induce a relative acidic shift in the H-cluster DTMA pKa value, and the Em values of the H-cluster would be predicted to shift to more positive values. This is in fact the case (Table 4), which explains in part why the serine variants of CrHydA1 and CaI exhibit higher populations of the reduced states, predominantly Hsred and Hhyd, compared to the native enzyme under the same reducing conditions (Mulder et al., 2014, 2017; Pham et al., 2018; Ratzloff et al., 2018). The corresponding effect is also observed for the CaI aspartate variant, which maintains a high population of Hox under reducing conditions (Morra et al., 2016). Thus, the effect of the cysteine variants on [FeFe]-hydrogenase reactivity can be modeled as a manifestation of combined changes to the pKa relationship with DTMA, and to the Em values of the H-cluster, which are summarized in Figure 2. The collective effect leads to a change in reactivity of [FeFe]-hydrogenase for H2 oxidation (e.g., acidic H-cluster pKa and positive Em shift) or H2 evolution (e.g., basic H-cluster pKa and negative Em shift; Artz et al., 2020b). The relationship between the exchange site pKa and the H-cluster DTMA pKa modeled in Figure 2 predicts that there should be a corresponding negative shift in the value of Em for C→D variants. This prediction is supported by IR spectra of the H2, and dithionite reduced CaI C298D that clearly shows a high population of Hox, and only minor populations of reduced states compared to the spectra of WT CaI, prepared under the same conditions (Morra et al., 2016).
Expanding the Model for Exchange Site Influence on Reactivity and the Special Case of CpII
It is also possible that the differences in reactivity arise from contributions other than pKa effects. In agreement with this, there are changes in the H-cluster electronic structures of Cys variants evidenced by changes in the FTIR and EPR spectra of catalytic intermediates compared to native enzymes (Mulder et al., 2013, 2014, 2017; Morra et al., 2016), that may occur from secondary sphere effects in the variants. Examples of these effects include the hydrophobicity, charge, electrostatics, and H-bonding networks that can control FeS cluster electronic structures and reduction potentials (Zuris et al., 2010; Hosseinzadeh and Lu, 2016). Indeed, the Hhyd state of the CrHydA1 C→S variant has been modeled as having a more contracted H-bonding network between the H-cluster FeD and the serine O-atom, when compared to the distance from FeD and the S-atom of cysteine in the native enzyme (Pham et al., 2018). The shorter N–H-O bond lengths vs. N–H-S accounted for shifts in the Fe-H/D frequencies in the NRVS spectra of Hhyd, and likely account for frequency shifts in vCO modes of redox intermediates in the FTIR spectra of the Cys-to-Ser variant versus wild-type (see Mulder et al., 2014, 2017).
Furthermore, whereas the model we set forth in Figure 2 correlates trends in reactivity to changes in pKa, it does not completely account for differences in intrinsic reactivity between the different types of [FeFe]-hydrogenases we tested. Specifically, the fact that the cysteine variants of CpII have comparatively low H2 evolution rates indicates that control of reactivity is not entirely embodied within the pKa relationship. The fact that cysteine has a basic pKa in all [FeFe]-hydrogenases, yet CpII is strongly biased toward H2 oxidation, implies that the H-cluster may be tuned differently compared to CpI/CaI, or CrHydA1. This could include effects such as dynamic secondary interactions and local electrostatics around the H-cluster, or differential stabilization of catalytic intermediates (Artz et al., 2020b). Mechanisms such as long-range potential effects from F-clusters may also contribute to the differences in reactivity of CpII vs. CpI/CaI and CrHydA1. These details indicate there are additional layers of control of H-cluster reactivity, for example, from the surrounding dielectric (Artz et al., 2020b), spin (de)localization, and other aspects of the electronic structure, the effects of which are being currently investigated by our group.
Conclusion
The outcome of these results identifies that the exchange site residue in proton transfer at the H-cluster can be used to control the reaction equilibria of [FeFe]-hydrogenases for H2 activation. Together, the interplay of pKa and electronic effects integrate as a framework for rationalizing the overall free energy landscape surrounding the H-cluster in terms of a scaling relationship, useful for understanding how [FeFe]-hydrogenase control reactivity (Figure 2). Fine-tuning the H-cluster electronic structure by other outer non-coordinating residues is emerging from biophysical and structural studies of [FeFe]-hydrogenase diversity (Poudel et al., 2016; Caserta et al., 2018; Chongdar et al., 2018; Rodríguez-Maciá et al., 2019; Artz et al., 2020b; Lampret et al., 2020; Fasano et al., 2021) and informed by decades of studies on iron–sulfur clusters (Beinert et al., 1997; Bominaar et al., 1997; Brereton et al., 1998, 1999; Ye et al., 2019). Combining mutational studies with biochemical and biophysical analysis in the context of these findings will likely contribute to further insights into the exquisite control of catalysis and reactivity in [FeFe]-hydrogenases.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Author Contributions
All authors analyzed data respective to their experiments. EK, DM, and PK performed protein expression, purification, and biochemical assays. VB conducted computational analyses on [FeFe]-hydrogenases. All authors contributed to the writing and/or editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the U.S. Department of Energy Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, Photosynthetic Systems Program.
Licenses and Permissions
This work was authored in part by the Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308.The U.S. Government retains and the publisher, by accepting the article for publication, acknowledge that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.
Conflict of Interest
All authors are employees of Alliance for Sustainable Energy, LLC, an M&O contractor of the U.S. Government.
Author Disclaimer
The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PT, Proton transfer; ET, Electron transfer; H-bonding, hydrogen-bonding.
References
Abou Hamdan, A., Dementin, S., Liebgott, P.-P., Gutierrez-Sanz, O., Richaud, P., De Lacey, A. L., et al. (2012). Understanding and tuning the catalytic bias of hydrogenase. J. Am. Chem. Soc. 134, 8368–8371. doi: 10.1021/ja301802r
Adams, M. W. W. (1990). The structure and mechanism of iron-hydrogenases. Biochim. Biophys. Acta 1020, 115–145.
Adams, M. W., Eccleston, E., and Howard, J. B. (1989). Iron-sulfur clusters of hydrogenase I and hydrogenase II of Clostridium pasteurianum. Proc. Natl. Acad. Sci. 86, 4932–4936. doi: 10.1073/pnas.86.13.4932
Adams, M. W., and Mortenson, L. E. (1984). The physical and catalytic properties of hydrogenase II of Clostridium pasteurianum. A comparison with hydrogenase I. J. Biol. Chem. 259, 7045–7055. doi: 10.1016/S0021-9258(17)39835-6
Artz, J. H., Mulder, D. W., Ratzloff, M. W., Lubner, C. E., Zadvornyy, O. A., Levan, A. X., et al. (2017). Reduction potentials of [FeFe]-hydrogenase accessory iron–sulfur clusters provide insights into the energetics of proton reduction catalysis. J. Am. Chem. Soc. 139, 9544–9550. doi: 10.1021/jacs.7b02099
Artz, J., Mulder, D., Ratzloff, M., Peters, J., and King, P. (2020a). The hydricity and reactivity relationship in FeFe-hydrogenases [online]. [Preprint]. doi: 10.21203/rs.3.rs-77874/v1
Artz, J. H., Zadvornyy, O. A., Mulder, D. W., Keable, S. M., Cohen, A. E., Ratzloff, M. W., et al. (2020b). Tuning catalytic bias of hydrogen gas producing hydrogenases. J. Am. Chem. Soc. 142, 1227–1235. doi: 10.1021/jacs.9b08756
Bas, D. C., Rogers, D. M., and Jensen, J. H. (2008). Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73, 765–783. doi: 10.1002/prot.22102
Beinert, H., Holm, R. H., and Münck, E. (1997). Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277, 653–659. doi: 10.1126/science.277.5326.653
Birrell, J. A., Rodríguez-Maciá, P., and Hery-Barranco, A. (2021). A beginner’s guide to thermodynamic modelling of [FeFe] hydrogenase. Catalysts 11:238. doi: 10.3390/catal11020238
Bominaar, E. L., Achim, C., Borshch, S. A., Girerd, J. J., and Münck, E. (1997). Analysis of exchange interaction and electron delocalization as intramolecular determinants of intermolecular electron-transfer kinetics. Inorg. Chem. 36, 3689–3701. doi: 10.1021/ic961298q
Brereton, P. S., Duderstadt, R. E., Staples, C. R., Johnson, M. K., and Adams, M. W. W. (1999). Effect of serinate ligation at each of the iron sites of the [Fe4S4] cluster of Pyrococcus furiosus ferredoxin on the redox, spectroscopic, and biological properties. Biochemistry 38, 10594–10605. doi: 10.1021/bi990671d
Brereton, K. R., Smith, N. E., Hazari, N., and Miller, A. J. M. (2020). Thermodynamic and kinetic hydricity of transition metal hydrides. Chem. Soc. Rev. 49, 7929–7948. doi: 10.1039/D0CS00405G
Brereton, P. S., Verhagen, M. F. J. M., Zhou, Z. H., and Adams, M. W. W. (1998). Effect of iron-sulfur cluster environment in modulating the thermodynamic properties and biological function of ferredoxin from Pyrococcus furiosus. Biochemistry 37, 7351–7362. doi: 10.1021/bi972864b
Bruice, T. C., Fife, T. H., Bruno, J. J., and Brandon, N. E. (1962). Hydroxyl group catalysis. II. The reactivity of the hydroxyl group of serine. The nucleophilicity of alcohols and the ease of hydrolysis of their acetyl esters as related to their pKa'*. Biochemistry 1, 7–12. doi: 10.1021/bi00907a002
Bullock, R. M., Appel, A. M., and Helm, M. L. (2014). Production of hydrogen by electrocatalysis: making the H–H bond by combining protons and hydrides. Chem. Commun. 50, 3125–3143. doi: 10.1039/c3cc46135a
Caserta, G., Papini, C., Adamska-Venkatesh, A., Pecqueur, L., Sommer, C., Reijerse, E., et al. (2018). Engineering an [FeFe]-hydrogenase: do accessory clusters influence O2 resistance and catalytic bias? J. Am. Chem. Soc. 140, 5516–5526. doi: 10.1021/jacs.8b01689
Cenens, J., Schoonheydt, R. A., and Voor, L. (1988). Visible spectroscopy of methylene blue on hectorite, Laponite B and Barasym in aqueous uspension. Clay Clay Miner., 214–224. doi: 10.1346/CCMN.1988.0360302
Chen, J.-S., and Blanchard, D. K. (1984). Purification and properties of the H2-oxidizing (uptake) hydrogenase of the N2-fixing anaerobe Clostridium pasteurianum W5. Biochem. Biophys. Res. Commun. 122, 9–16. doi: 10.1016/0006-291X(84)90431-5
Chongdar, N., Birrell, J. A., Pawlak, K., Sommer, C., Reijerse, E. J., Rüdiger, O., et al. (2018). Unique spectroscopic properties of the H-cluster in a putative sensory [FeFe] hydrogenase. J. Am. Chem. Soc. 140, 1057–1068. doi: 10.1021/jacs.7b11287
Cornish, A. J., Gärtner, K., Yang, H., Peters, J. W., and Hegg, E. L. (2011). Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J. Biol. Chem. 286, 38341–38347. doi: 10.1074/jbc.M111.254664
Dörner, E., and Boll, M. (2002). Properties of 2-Oxoglutarate: ferredoxin oxidoreductase from Thauera aromatica and its role in enzymatic reduction of the aromatic ring. J. Bacteriol. 184, 3975–3983. doi: 10.1128/JB.184.14.3975-3983.2002
Duan, J., Senger, M., Esselborn, J., Engelbrecht, V., Wittkamp, F., Apfel, U.-P., et al. (2018). Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases. Nat. Commun. 9:4726. doi: 10.1038/s41467-018-07140-x
Fasano, A., Land, H., Fourmond, V., Berggren, G., and Léger, C. (2021). Reversible or irreversible catalysis of H+/H2 conversion by FeFe hydrogenases. J. Am. Chem. Soc. 143, 20320–20325. doi: 10.1021/jacs.1c09554
Fourmond, V., Plumeré, N., and Léger, C. (2021). Reversible catalysis. Nature reviews. Nat. Rev. Chem. 5, 348–360. doi: 10.1038/s41570-021-00268-3
Frey, P. A. (2001). Review: strong hydrogen bonding in molecules and enzymatic complexes. Magn. Reson. Chem. 39, S190–S198. doi: 10.1002/mrc.953
Gauquelin, C., Baffert, C., Richaud, P., Kamionka, E., Etienne, E., Guieysse, D., et al. (2018). Roles of the F-domain in [FeFe] hydrogenase. Biochim. Biophys. Acta 1859, 69–77. doi: 10.1016/j.bbabio.2017.08.010
Ginovska-Pangovska, B., Ho, M.-H., Linehan, J. C., Cheng, Y., Dupuis, M., Raugei, S., et al. (2014). Molecular dynamics study of the proposed proton transport pathways in [FeFe]-hydrogenase. Biochim. Biophys. Acta 1837, 131–138. doi: 10.1016/j.bbabio.2013.08.004
Girbal, L., Abendroth, G. V., Winkler, M., Benton, P. M. C., Meynial-Salles, I., Croux, C., et al. (2005). Homologous and heterologous overexpression in clostridium acetobutylicum and characterization of purified clostridial and algal Fe-only hydrogenases with high specific activities. Appl. Environ. Microbiol. 71, 2777–2781. doi: 10.1128/AEM.71.5.2777-2781.2005
Hexter, S. V., Grey, F., Happe, T., Climent, V., and Armstrong, F. A. (2012). Electrocatalytic mechanism of reversible hydrogen cycling by enzymes and distinctions between the major classes of hydrogenases. Proc. Natl. Acad. Sci. 109, 11516–11521. doi: 10.1073/pnas.1204770109
Hosseinzadeh, P., and Lu, Y. (2016). Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta 1857, 557–581. doi: 10.1016/j.bbabio.2015.08.006
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
King, P. W., Posewitz, M. C., Ghirardi, M. L., and Seibert, M. (2006). Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. J. Bacteriol. 188, 2163–2172. doi: 10.1128/JB.188.6.2163-2172.2006
Knörzer, P., Silakov, A., Foster, C. E., Armstrong, F. A., Lubitz, W., and Happe, T. (2012). Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J. Biol. Chem. 287, 1489–1499. doi: 10.1074/jbc.M111.305797
Lampret, O., Duan, J., Hofmann, E., Winkler, M., Armstrong, F. A., and Happe, T. (2020). The roles of long-range proton-coupled electron transfer in the directionality and efficiency of [FeFe]-hydrogenases. Proc. Natl. Acad. Sci. 117, 20520–20529. doi: 10.1073/pnas.2007090117
Liepinsh, E., and Otting, G. (1996). Proton exchange rates from amino acid side chains— implications for image contrast. Magn. Reson. Med. 35, 30–42. doi: 10.1002/mrm.1910350106
Long, H., King, P. W., and Chang, C. H. (2014). Proton transport in Clostridium pasteurianum [FeFe] hydrogenase I: a computational study. J. Phys. Chem. B 118, 890–900. doi: 10.1021/jp408621r
Morra, S., Giraudo, A., Di Nardo, G., King, P. W., Gilardi, G., and Valetti, F. (2012). Site saturation mutagenesis demonstrates a central role for cysteine 298 as proton donor to the catalytic site in CaHydA [FeFe]-hydrogenase. PLoS One 7:e48400. doi: 10.1371/journal.pone.0048400
Morra, S., Maurelli, S., Chiesa, M., Mulder, D. W., Ratzloff, M. W., Giamello, E., et al. (2016). The effect of a C298D mutation in CaHydA [FeFe]-hydrogenase: insights into the protein-metal cluster interaction by EPR and FTIR spectroscopic investigation. Biochim. Biophys. Acta 1857, 98–106. doi: 10.1016/j.bbabio.2015.10.005
Mulder, D. W., Guo, Y., Ratzloff, M. W., and King, P. W. (2017). Identification of a catalytic iron-hydride at the H-cluster of [FeFe]-hydrogenase. J. Am. Chem. Soc. 139, 83–86. doi: 10.1021/jacs.6b11409
Mulder, D. W., Peters, W. J., and Raugei, S. (2021). Catalytic bias in oxidation–reduction catalysis. Chem. Commun. 57, 713–720. doi: 10.1039/D0CC07062A
Mulder, D. W., Ratzloff, M. W., Bruschi, M., Greco, C., Koonce, E., Peters, J. W., et al. (2014). Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase. J. Am. Chem. Soc. 136, 15394–15402. doi: 10.1021/ja508629m
Mulder, D. W., Ratzloff, M. W., Shepard, E. M., Byer, A. S., Noone, S. M., Peters, J. W., et al. (2013). EPR and FTIR analysis of the mechanism of H2 activation by [FeFe]-hydrogenase HydA1 from Chlamydomonas reinhardtii. J. Am. Chem. Soc. 135, 6921–6929. doi: 10.1021/ja4000257
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: consistent treatment of internal and surface residues in empirical pka predictions. J. Chem. Theory Comput. 7, 525–537. doi: 10.1021/ct100578z
Pham, C. C., Mulder, D. W., Pelmenschikov, V., King, P. W., Ratzloff, M. W., Wang, H., et al. (2018). Terminal hydride species in [FeFe]-hydrogenases are vibrationally coupled to the active site environment. Angew. Chem. 130, 10765–10769. doi: 10.1002/ange.201805144
Poudel, S., Tokmina-Lukaszewska, M., Colman, D. R., Refai, M., Schut, G. J., King, P. W., et al. (2016). Unification of [FeFe]-hydrogenases into three structural and functional groups. Biochim. Biophys. Acta 1860, 1910–1921. doi: 10.1016/j.bbagen.2016.05.034
Puthenkalathil, R. C., and Ensing, B. (2021). Linear scaling relationships to predict pKa’s and reduction potentials for bioinspired hydrogenase catalysis. Inorg. Chem. 61, 113–120. doi: 10.1021/acs.inorgchem.1c02429
Ratzloff, M. W., Artz, J. H., Mulder, D. W., Collins, R. T., Furtak, T. E., and King, P. W. (2018). CO-bridged H-cluster intermediates in the catalytic mechanism of [FeFe]-hydrogenase CaI. J. Am. Chem. Soc. 140, 7623–7628. doi: 10.1021/jacs.8b03072
Rodríguez-Maciá, P., Breuer, N., Debeer, S., and Birrell, J. A. (2020). Insight into the redox behavior of the [4Fe–4S] subcluster in [FeFe] hydrogenases. ACS Catal. 10, 13084–13095. doi: 10.1021/acscatal.0c02771
Rodríguez-Maciá, P., Kertess, L., Burnik, J., Birrell, J. A., Hofmann, E., Lubitz, W., et al. (2019). His-ligation to the [4Fe–4S] subcluster tunes the catalytic bias of [FeFe] hydrogenase. J. Am. Chem. Soc. 141, 472–481. doi: 10.1021/jacs.8b11149
Rodríguez-Maciá, P., Pawlak, K., Rüdiger, O., Reijerse, E. J., Lubitz, W., and Birrell, J. A. (2017). Intercluster redox coupling influences protonation at the H-cluster in [FeFe] hydrogenases. J. Am. Chem. Soc. 139, 15122–15134. doi: 10.1021/jacs.7b08193
Senger, M., Eichmann, V., Laun, K., Duan, J., Wittkamp, F., Knör, G., et al. (2019). How [FeFe]-hydrogenase facilitates bidirectional proton transfer. J. Am. Chem. Soc. 141, 17394–17403. doi: 10.1021/jacs.9b09225
Silakov, A., Kamp, C., Reijerse, E., Happe, T., and Lubitz, W. (2009). Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii. Biochemistry 48, 7780–7786. doi: 10.1021/bi9009105
Sommer, C., Adamska-Venkatesh, A., Pawlak, K., Birrell, J. A., Rüdiger, O., Reijerse, E. J., et al. (2017). Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J. Am. Chem. Soc. 139, 1440–1443. doi: 10.1021/jacs.6b12636
Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M., and Jensen, J. H. (2011). Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295. doi: 10.1021/ct200133y
Von Abendroth, G., Stripp, S., Silakov, A., Croux, C., Soucaille, P., Girbal, L., et al. (2008). Optimized over-expression of [FeFe] hydrogenases with high specific activity in Clostridium acetobutylicum. Int. J. Hydrog. Energy 33, 6076–6081. doi: 10.1016/j.ijhydene.2008.07.122
Yacoby, I., Tegler, L. T., Pochekailov, S., Zhang, S., and King, P. W. (2012). Optimized expression and purification for high-activity preparations of algal [FeFe]-hydrogenase. PLoS One 7:e35886. doi: 10.1371/journal.pone.0035886
Ye, M., Thompson, N. B., Brown, A. C., and Suess, D. L. M. (2019). A synthetic model of enzymatic [Fe4S4]–alkyl intermediates. J. Am. Chem. Soc. 141, 13330–13335. doi: 10.1021/jacs.9b06975
Keywords: [FeFe]-hydrogenase, proton-coupled electron transfer, enzymatic reactivity, H-cluster, pKa and proton transfer, catalytic bias
Citation: Kisgeropoulos EC, Bharadwaj VS, Mulder DW and King PW (2022) The Contribution of Proton-Donor pKa on Reactivity Profiles of [FeFe]-hydrogenases. Front. Microbiol. 13:903951. doi: 10.3389/fmicb.2022.903951
Edited by:
Stefan Frielingsdorf, Technical University of Berlin, GermanyReviewed by:
James Birrell, Max Planck Institute for Chemical Energy Conversion, GermanyGustav Berggren, Uppsala University, Sweden
This work is authored by Effie C. Kisgeropoulos, Vivek S. Bharadwaj, David W. Mulder and Paul W. King, © 2022 Alliance for Sustainable Energy, LLC. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul W. King, cGF1bC5LaW5nQG5yZWwuZ292