
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 June 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.903024
 Niloofar Vaghefi1,2
Niloofar Vaghefi1,2 Stefan Kusch3
Stefan Kusch3 Márk Z. Németh4
Márk Z. Németh4 Diána Seress4
Diána Seress4 Uwe Braun5
Uwe Braun5 Susumu Takamatsu6
Susumu Takamatsu6 Ralph Panstruga3
Ralph Panstruga3 Levente Kiss1,4,7*
Levente Kiss1,4,7*Powdery mildew fungi (Erysiphaceae), common obligate biotrophic pathogens of many plants, including important agricultural and horticultural crops, represent a monophyletic lineage within the Ascomycota. Within the Erysiphaceae, molecular phylogenetic relationships and DNA-based species and genera delimitations were up to now mostly based on nuclear ribosomal DNA (nrDNA) phylogenies. This is the first comprehensive genome-scale phylogenetic analysis of this group using 751 single-copy orthologous sequences extracted from 24 selected powdery mildew genomes and 14 additional genomes from Helotiales, the fungal order that includes the Erysiphaceae. Representative genomes of all powdery mildew species with publicly available whole-genome sequencing (WGS) data that were of sufficient quality were included in the analyses. The 24 powdery mildew genomes included in the analysis represented 17 species belonging to eight out of 19 genera recognized within the Erysiphaceae. The epiphytic genera, all but one represented by multiple genomes, belonged each to distinct, well-supported lineages. Three hemiendophytic genera, each represented by a single genome, together formed the hemiendophytic lineage. Out of the 14 other taxa from the Helotiales, Arachnopeziza araneosa, a saprobic species, was the only taxon that grouped together with the 24 genome-sequenced powdery mildew fungi in a monophyletic clade. The close phylogenetic relationship between the Erysiphaceae and Arachnopeziza was revealed earlier by a phylogenomic study of the Leotiomycetes. Further analyses of powdery mildew and Arachnopeziza genomes may discover signatures of the evolutionary processes that have led to obligate biotrophy from a saprobic way of life. A separate phylogeny was produced using the 18S, 5.8S, and 28S nrDNA sequences of the same set of powdery mildew specimens and compared to the genome-scale phylogeny. The nrDNA phylogeny was largely congruent to the phylogeny produced using 751 orthologs. This part of the study has revealed multiple contamination and other quality issues in some powdery mildew genomes. We recommend that the presence of 28S, internal transcribed spacer (ITS), and 18S nrDNA sequences in powdery mildew WGS datasets that are identical to those determined by Sanger sequencing should be used to assess the quality of assemblies, in addition to the commonly used Benchmarking Universal Single-Copy Orthologs (BUSCO) values.
Nuclear ribosomal DNA (nrDNA) sequences, above all the internal transcribed spacer (ITS), the large subunit (LSU or 28S) and the small subunit (SSU or 18S) nrDNA sequences, have been at the epicenter of molecular identification and phylogenetic studies of all groups of fungi since the nascence of this research field in the late 1980s. According to Web of Science, the paper reporting the first universal primers to amplify the 18S and the ITS region of nrDNA in fungi (White et al., 1990) is probably the most cited publication in mycology, with over 40,000 citations to date. More than two decades later, a comprehensive, multi-laboratory comparison of different nuclear and mitochondrial DNA loci widely used in fungal phylogenetics concluded that the nrDNA ITS sequences are still the only reliable species DNA markers that can be used to infer phylogenies across all fungal groups (Schoch et al., 2012). In some intensively studied groups of fungi, multi-locus analyses based on specific sets of loci have already been established as the frameworks for phylogenetic analyses and molecular taxonomic studies (e.g., Marin-Felix et al., 2019; Vaghefi et al., 2020; Poudel et al., 2021); however, in most fungal groups, nrDNA sequences are still the sole basis for phylogenetic works (e.g., Crous et al., 2019, 2020, 2021).
An important group of ascomycetous plant pathogens, the Erysiphaceae, known as the powdery mildew fungi, is a good example for a large monophyletic lineage within which molecular phylogenetic relationships as well as DNA-based species and genera delimitations are mostly based on nrDNA phylogenies. The Erysiphaceae include more than 900 species belonging to 19 genera. All species are obligate biotrophic plant pathogens, i.e., they take up nutrients from living host plant tissues only (Hückelhoven and Panstruga, 2011) and cannot grow and produce spores for reproduction without being functionally connected to the infected and living host plant tissues. Altogether, powdery mildew fungi can colonize more than 10,000 dicot and monocot species in different parts of the world (Amano, 1986; Braun and Cook, 2012). Some powdery mildew species infect only a single host plant species or a few closely related hosts belonging to the same genus (e.g., Meeboon et al., 2017; Kiss et al., 2018). Others are known from many, only distantly related plants (e.g., Meeboon and Takamatsu, 2016; Braun et al., 2019; Kiss and Vaghefi, 2021; Young and Kiss, 2021). Finally, some host plants can be infected by more than one powdery mildew species, which sometimes belong to different genera (e.g., Takamatsu et al., 2007; Kiss et al., 2008; Desprez-Loustau et al., 2018; Kelly et al., 2021; Faticov et al., 2022). Some species have become invasive in different parts of the world (Kiss, 2005; Desprez-Loustau et al., 2010; Kiss et al., 2020). Important crops, including wheat, barley, grapevine, as well as fruit and vegetable species, are commonly colonized by diverse powdery mildew fungi (Glawe, 2008). Despite extensive research on their pathogenesis, epidemiology and control, these powdery mildew species remain amongst the economically most important plant pathogens in agriculture and horticulture worldwide due to the combined effect of costs of chemical crop protection measures and yield losses (Calonnec et al., 2004; Fondevilla and Rubiales, 2012; Fuller et al., 2014; Dunn and Gaynor, 2020). Others are well-known tree pathogens (Marçais and Desprez-Loustau, 2014; Demeter et al., 2021), and some have become model organisms in plant pathology research (Gadoury et al., 2012; Bindschedler et al., 2016; Kuhn et al., 2016) or in the study of wild plant pathosystems (Susi et al., 2015).
Before the era of DNA-based phylogenies, the generic phylogenetic concept in the Erysiphaceae was based on the morphological characteristics of the sexual morphs (teleomorphs), known as chasmothecia (formerly: cleistothecia; Braun, 1987, 1995). The complex geometries of the appendage tips of some chasmothecia have captured the attention and admiration of early mycologists and microscopists already in the 19th century (Hirata et al., 2000) and were useful in grouping powdery mildews in a few genera. Other characteristics of chasmothecia, such as the number and shape of asci and ascospores, have also been used to define the genera within the Erysiphaceae. Species within genera were mainly distinguished based on their host plants and the morphology of the sexual and asexual morphs (Braun, 1987, 1995). The early speculations on the evolution of powdery mildew fungi focused on chasmothecia, and presumed that species/genera with simple, mycelioid chasmothecial appendages were ancestral, and those with more complex appendages have appeared later during the evolution of the Erysiphaceae (Braun, 1987).
It was, therefore, surprising that the first phylogenetic analyses of powdery mildews based on nrDNA sequences did not support the classic, well-established generic concept of the Erysiphaceae (Takamatsu et al., 1998, 1999; Saenz and Taylor, 1999; Hirata et al., 2000; Mori et al., 2000a; Matsuda and Takamatsu, 2003). As one of the very first molecular phylogenetic analyses concluded, ‘appendage morphology does not always accurately reflect the phylogeny of the powdery mildews’ (Takamatsu et al., 1999). In fact, species were clearly grouped according to the morphological characteristics of their asexual morphs (anamorphs), irrespective of the morphology of their chasmothecia (Saenz and Taylor, 1999; Takamatsu, 2004). This has been confirmed by all nrDNA phylogenetic analyses conducted to date (e.g., Marmolejo et al., 2018; Bradshaw and Tobin, 2020; Kiss et al., 2020). The discovery that it is, in fact, the conidiogenesis and the morphology of the asexual morphs that mirror the phylogenetic relationships within the Erysiphaceae has triggered major changes in the taxonomy of powdery mildew fungi, especially at the genus level (Braun et al., 2002; Braun, 2011; Braun and Cook, 2012). The currently accepted genera are listed in Table 1.

Table 1. The current classification of the Erysiphaceae.
The nrDNA phylogenies have also led to the development of a new hypothesis about the evolution of the distinctive chasmothecial appendages of powdery mildews, and a better understanding of their life cycles. This was needed because, in contrast to the early speculations, all phylogenetic analyses have shown that species with complex appendages on their chasmothecia have appeared first, and those with simple, mycelioid appendages only later during evolution (for reviews, see Takamatsu, 2013a; Takamatsu, 2018). Most of the latter species infect herbaceous plants, while species with complex appendages are pathogens of trees or shrubs, and usually have more restricted host ranges than herb-pathogenic species (Mori et al., 2000a; Takamatsu et al., 2000; Takamatsu, 2013b). To explain these results, Takamatsu (2004) hypothesized that appendage morphology reflects the adaptation of the respective powdery mildew species to overwintering on deciduous woody hosts, mostly in regions with temperate climate. Appendages with complex geometries play a vital role in securing the attachment of many chasmothecia to the bark of their deciduous hosts at the end of the growing season, when those are washed off the canopy by autumn rains, or detached, e.g., by air movements, before leaf fall (for a review, see Takamatsu, 2013b). Some of the chasmothecia that become attached to the woody parts of their deciduous hosts survive the winter period, and release ascospores in spring in the close vicinity of the freshly emerged shoots and leaves, which are then infected by ascospores, re-starting the infection cycle. This was well documented for example in Erysiphe necator on grapevine (Pearson and Gadoury, 1987; Gadoury et al., 2012) and E. alphitoides on sessile oak (Quercus petraea) (Dantec et al., 2015). Takamatsu (2004) has also hypothesized that simple, mycelioid appendages of chasmothecia of diverse herb-parasitic powdery mildew fungi may be a result of convergent evolution that has repeatedly occurred in different lineages of the Erysiphaceae as an adaptation to their herbaceous host plants. It appears that evolutionary changes in the characteristics of the conidiogenesis (e.g., whether conidia are produced singly or in chains), reflected by the morphology of the conidiophores, have happened slower than changes in the morphology of chasmothecia, and nrDNA phylogenies of the Erysiphaceae are capturing their phenotypic evolution based on patterns of their conidiogenesis (Takamatsu, 2013b).
DNA regions other than nrDNA loci have also been tested for their use in delimiting species in some groups of the Erysiphaceae. The main objectives were to introduce new loci, including protein-coding regions, in phylogenetic analyses (Inuma et al., 2007; Liu et al., 2021); to develop new species-specific DNA barcodes (Ellingham et al., 2019; Shirouzu et al., 2020); and to test the hypothesis of interspecific hybridization in powdery mildew fungi (Seko et al., 2011). Intraspecific, host-driven differentiation of some powdery mildew species has also been tested with new DNA markers, e.g., in Podosphaera xanthii (Vela-Gorcía et al., 2014) and Erysiphe quercicola (Desprez-Loustau et al., 2017). Amongst the non-nrDNA loci, the MCM7 gene was the most useful in distinguishing closely related powdery mildew taxa (Ellingham et al., 2019; Shirouzu et al., 2020). This gene encodes one of the highly conserved mini-chromosome maintenance proteins (MCMs) that is required for eukaryotic genome replication (Raja et al., 2011).
Internal transcribed spacer sequences are still the most commonly used species-level DNA barcodes available for powdery mildew fungi, despite their limitations. These include intragenomic variations within some powdery mildew species (Kovács et al., 2011) and lack of resolution power to discriminate between some other species that can be distinguished based on morphology and host range (Braun et al., 2019; Qiu et al., 2020). ITS sequences as species barcodes have limitations in other fungal groups as well (Kiss, 2012; Stadler et al., 2020).
Traditionally, powdery mildew fungi were classified as members of the monotypic order Erysiphales (i.e., consisting of a single family, the Erysiphaceae) (Braun and Cook, 2012). However, comprehensive multi-gene analyses of the class Leotiomycetes have recently shown that powdery mildew fungi group together with a few Arachnopeziza species within the order Helotiales, and together form the ‘erysiphoid clade’ (Johnston et al., 2019). A taxonomic consequence of this phylogenetic analysis is that the family level classification of powdery mildew fungi, i.e., the status of the Erysiphaceae, remains unchanged, but the order Erysiphales is not retained and the Erysiphaceae is now regarded to be part of the Helotiales sensu Johnston et al. (2019).
Another outcome of the multi-gene analyses performed by Johnston et al. (2019) is that Arachnopeziza spp. were identified as the closest known saprobic relatives of powdery mildew fungi. Little is known about Arachnopeziza spp.; it appears that these fungi are decomposers of diverse organic substrates, such as decaying wood and leaf debris, mostly in forest ecosystems (Hirata et al., 2000; Baldrian et al., 2016; Kosonen et al., 2021). Earlier, when the available nrDNA and other sequences were much more limited, the Myxotrichaceae, including Oidiodendron, Byssoascus, and Myxotrichum species were considered as the closest known saprobic relatives of the Erysiphaceae based on nrDNA phylogenies and molecular clock calculations (Berbee and Taylor, 1993, 2001; Sugiyama et al., 1999; Mori et al., 2000b; Takamatsu, 2004). Fungi belonging to the Myxotrichaceae are also known as decomposers of dead plant materials and other organic materials. Further phylogenetic analyses indicated that Chlorociboria and Cyttaria (Wang et al., 2006a,b) or the Myxotrichaceae and Pleuroascus (Peterson and Pfister, 2010) are the closest known saprobic relatives of powdery mildew fungi. Deciphering the evolutionary origin of the Erysiphaceae may shed light on how a fungal lineage has evolved from a saprobic lifestyle to obligate biotrophy.
A part of the comprehensive phylogenetic analyses performed by Johnston et al. (2019) used over 3,000 concatenated orthologous single-copy genes extracted from the genomes of 49 strains belonging to the Leotiomycetes. Their analysis showed that Oidiodendron maius, representing the Myxotrichaceae, was not a close relative of the ‘erysiphoid clade’ consisting of powdery mildew fungi and Arachnopeziza. This was also revealed by other analyses of the same study (Johnston et al., 2019) based on 15 genes commonly used in recent fungal phylogenies. Another genome-scale study revealed the phylogeny of the entire phylum Ascomycota, using 815 single-copy orthologs from genomes of 1,107 ascomycetes (Shen et al., 2020). That study did not include Arachnopeziza genomes but confirmed that O. maius is not closely related to powdery mildew fungi.
So far, only one study has reconstructed evolutionary relationships within the Erysiphaceae based on genome-scale data (Frantzeskakis et al., 2019a). The aim of that analysis was to test the relationship between Parauncinula polyspora, an early-diverged powdery mildew fungus with a surprisingly small genome, and other, well-known powdery mildews based on non-nrDNA loci. The analysis used 1,964 single-copy orthologs identified in the genomes of seven powdery mildew and nine other fungi belonging to the Leotiomycetes. Arachnopeziza genomes were not included in that study (Frantzeskakis et al., 2019a).
Clearly, further comprehensive genome-scale phylogenies are needed to learn more about the evolution of powdery mildews and to reveal their closest saprobic relatives. Comparative genomics analyses have already contributed to a better understanding of the interactions between some powdery mildew fungi and their plant hosts, including rapid evolution of the host range patterns (Spanu et al., 2010; Wicker et al., 2013; Frantzeskakis et al., 2018, 2019b; Wu et al., 2018; Barsoum et al., 2019; Müller et al., 2019), but comprehensive analyses of host-parasite interactions in many species and genera of the Erysiphaceae have not been performed yet. Phylogenetic analyses of nrDNA loci have already revealed many aspects of their evolution that are consistent with morphological characteristics of different groups, and their host-pathogen interaction patterns (Takamatsu, 2013a,b). Multi-gene studies may be useful to enrich results based solely on nrDNA analyses, above all to disentangle complexes of closely allied species that cannot be properly resolved in phylogenetic analyses based on ITS sequences only, such as the Erysiphe aquilegiae complex (Shin et al., 2019; Bradshaw et al., 2020), and to identify the closest saprobic relatives of the Erysiphaceae.
More and more genome assemblies are reported for different powdery mildew species (e.g., Kusch et al., 2020, 2022a; Kim et al., 2021; Polonio et al., 2021), and these data offer new avenues to understand the powdery mildew lifestyle. However, most powdery mildew genomes published so far are highly fragmented (Bindschedler et al., 2016; Barsoum et al., 2019) as these are generally large, gene-poor, and contain a high proportion of repetitive elements compared to other ascomycetes (Spanu et al., 2010; Wicker et al., 2013; Frantzeskakis et al., 2018). These quality issues sometimes limit the use of powdery mildew genomes in further studies. The main goals of this study were to (i) perform genome-scale phylogenetic analyses using single-copy orthologs identified in the genomes of as many powdery mildew fungi and presumed saprobic relatives as possible; (ii) compare the results to nrDNA phylogenies of the same isolates; (iii) reveal quality issues associated with the use of the currently available powdery mildew whole-genome sequencing (WGS) datasets in phylogenetic studies; and (iv) update the taxonomy of the genome-sequenced powdery mildew species where needed.
To obtain a comprehensive set of genomes representative of powdery mildews sequenced to date, the European Nucleotide Archive (ENA), Joint Genome Institute (JGI), and National Center for Biotechnology Information (NCBI) were searched using the search term Erysiphaceae in January 2022. For Blumeria graminis, the most studied powdery mildew species infecting several cereal and wild grass species, and Erysiphe necator, the causal agent of grape powdery mildew, multiple genomes are available in public repositories; therefore, only four to five representative genomes were retrieved. For all other powdery mildew species that had a sequenced genome, all available genomes were obtained from the databases listed above, except for E. alphitoides specimen MS-42D (Dutech et al., 2020), for which only the raw data are available in ENA, and the genome was retrieved from http://arachne.pierroton.inra.fr/AlphiGeno/ (Table 2).
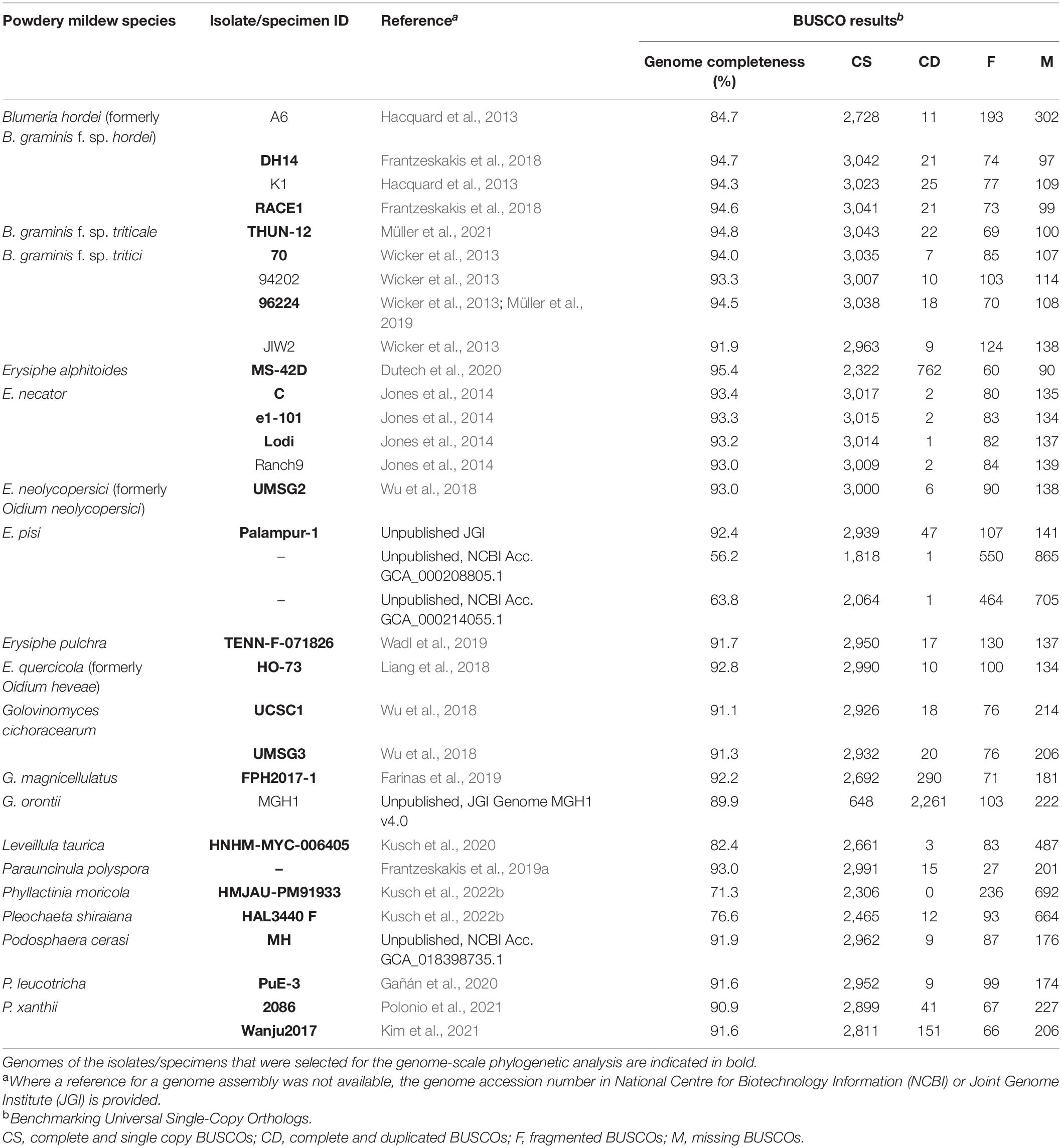
Table 2. List of powdery mildew genomes assessed for completeness through identification of Benchmarking Universal Single-Copy Orthologs (BUSCO) using BUSCO v.5.2.2 (Simão et al., 2015) and Leotiomycetes dataset Odb10.
To assess the quality of the accessed genome assemblies, we used the Benchmarking Universal Single-Copy Orthologs (BUSCO) v.2.5.5 (Simão et al., 2015) and the Leotiomycetes odb10 database, which includes a total of 3,234 single-copy BUSCOs. Where multiple genomes were available for a species, and to remove low-quality genomes and minimize missing data, only genomes with highest completeness were retained for the phylogenetic analyses. The final dataset for genome-scale phylogenetic analyses included 24 powdery mildew genomes (Table 2). A set of genomes selected to represent close relatives of powdery mildew fungi within the Leotiomycetes were also included in the genome-scale analysis (Table 3); these were selected based on previous phylum-level and class-level phylogenies (Johnston et al., 2019).
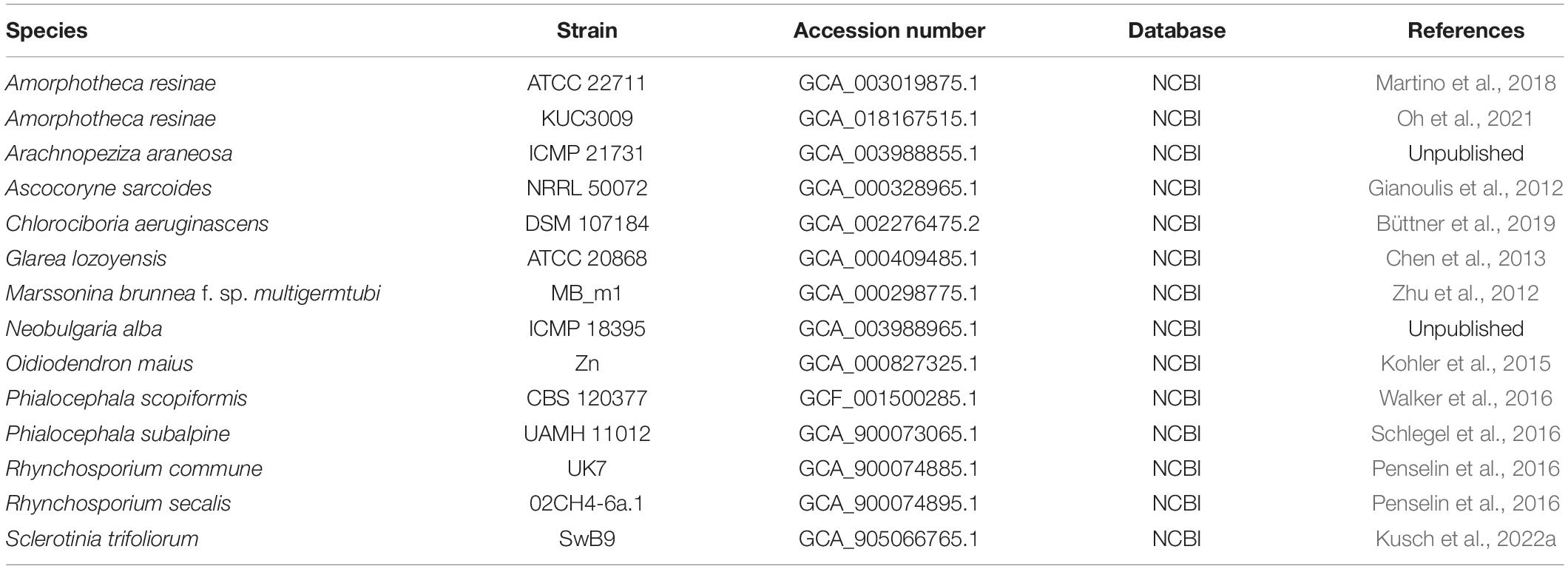
Table 3. List of non-powdery mildew Leotiomycete genomes included in the genome-scale phylogenetic analysis.
We used single-copy orthologous amino acid sequences obtained using BUSCO for phylogenomic inference. First, extracted protein sequences for all genomes were analyzed using OrthoFinder v.2.5.1 (Emms and Kelly, 2019) to identify single-copy orthologs shared across all genomes. OrthoFinder assigned a total of 113,072 proteins (99.9% of total) to 3,427 orthogroups and identified 751 orthogroups that existed in all target genomes in single copies. Amino acid sequences were aligned separately using MAFFT v.7.453 (Katoh and Standley, 2013) with the BLOSUM62 matrix of substitutions. Ambiguously aligned regions were removed using Gblocks v. 0.91b (Castresana, 2000; Talavera and Castresana, 2007) using default settings. A maximum likelihood (ML) phylogenetic tree based on the concatenated alignment of amino acid sequences was generated with 1,000 bootstrap replicates using RAxML-NG v.1.0.1 (Kozlov et al., 2019), under the JTT+I+G4+F amino acid substitution model identified by ModelTest-NG v.0.1.6 (Darriba et al., 2020). Sclerotinia trifoliorum strain SwB9 was used as the outgroup (Kusch et al., 2022b).
We aimed to produce a separate phylogeny based on nrDNA sequences of the same set of powdery mildew specimens to compare with the phylogeny based on the single-copy orthologs. For this, we searched the NCBI GenBank database for 5.8S, 18S, and 28S nrDNA sequences of each powdery mildew specimen included in our genome-scale study (Table 4). Out of the 24 specimens included in the genome-scale analyses, only 10 had their nrDNA loci sequenced by Sanger sequencing, and only one of them, originally recognized as Oidium heveae HO-73, had all the three loci sequenced prior to this study (Table 4). (HO-73 should be identified as an isolate of E. quercicola based on Wu et al., 2019; see below.) Subsequently, we attempted to extract the missing nrDNA loci from the published genome assemblies for inclusion in the nrDNA phylogenetic analysis. For this, we converted the genome assemblies of target species to BLAST databases in Geneious Prime1 (Kearse et al., 2012) and used 5.8S, 18S, and 28S sequences of reference specimens (Kiss et al., 2020) as queries in BLAST searches against the genomes to retrieve the respective sequences from each of the assemblies. The extracted nrDNA fragments were used in BLAST searches against the NCBI nrDNA database to ensure these belonged to the target powdery mildew species and are suitable for the phylogenetic analysis. Where nrDNA sequences of specimens were available in the NCBI GenBank database, we aligned these against the sequences obtained from the genomes for comparison (Table 5). To avoid misidentification of sequences as nrDNA, the Megablast function was used, and contigs/scaffolds were only reported to contain nrDNA sequence fragments if these had a query coverage of 100% and BLAST hit length of at least 300 bp. The identified fragments were subsequently used in BLAST searches against the NCBI nrDNA database to ensure these belonged to the nrDNA region.
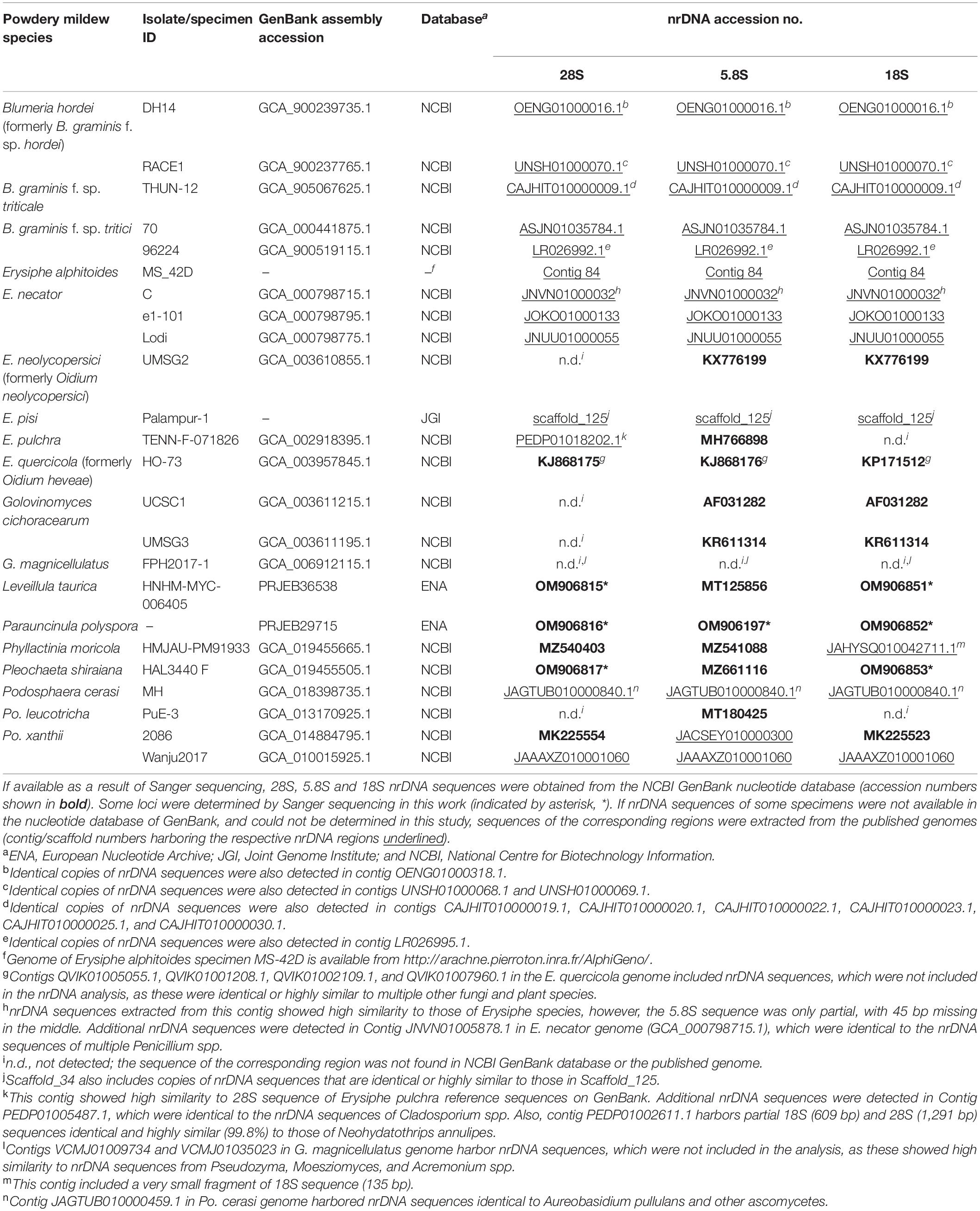
Table 4. List of powdery mildew specimens included in the genome-scale and nuclear ribosomal (nrDNA) phylogenetic analyses.
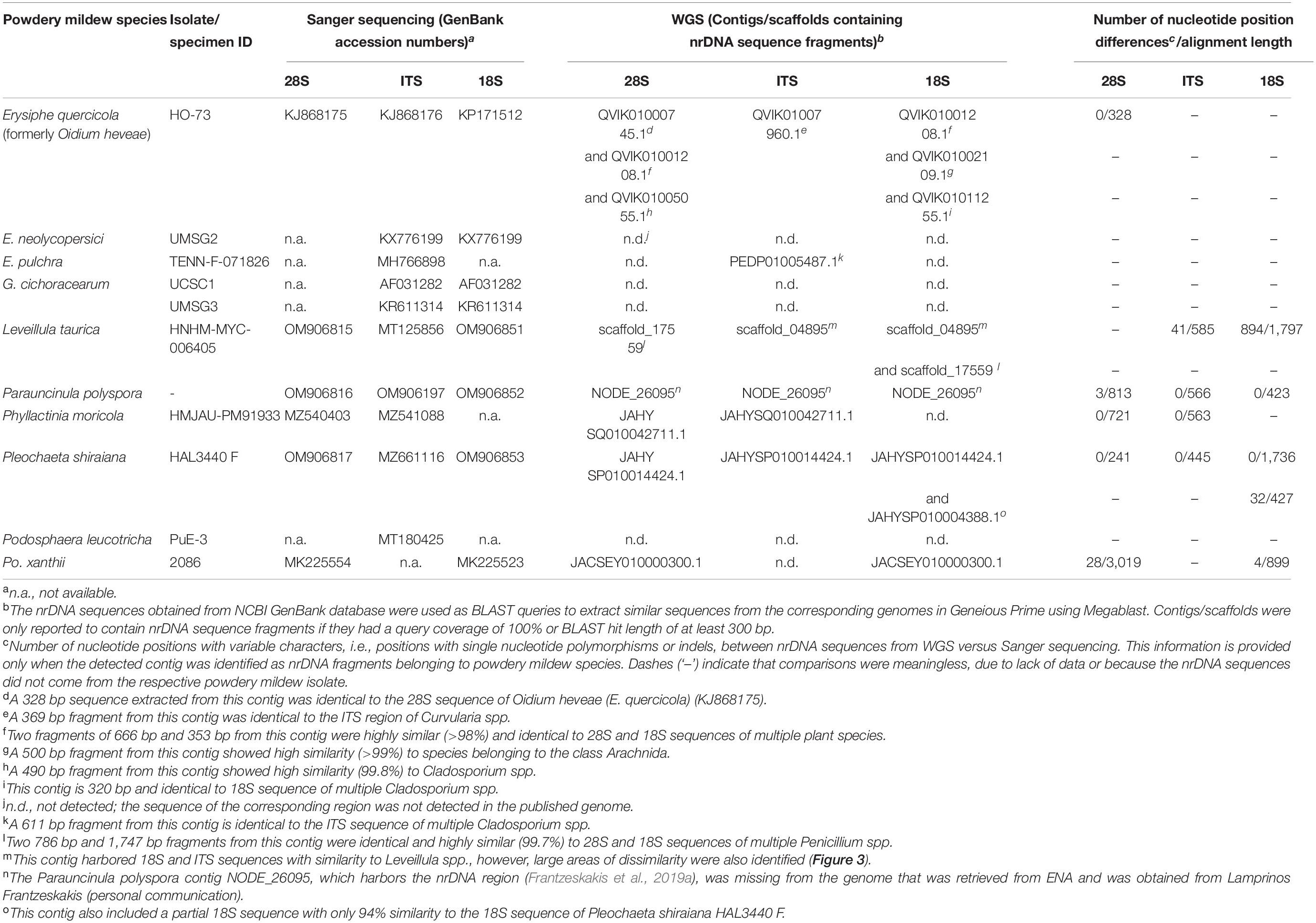
Table 5. Comparison of 28S, ITS and 18S sequences extracted from whole genome sequencing (WGS) datasets (genome assembly accession numbers shown in Table 4) to sequences determined by Sanger sequencing available at NCBI GenBank database.
In addition, we amplified and sequenced the 18S and 28S regions in Leveillula taurica HNHM-MYC-006405 and Pleochaeta shiraiana HAL3440 F, as well as the 18S, 28S and ITS regions of the P. polyspora specimen studied by Frantzeskakis et al. (2019a), according to the protocol described by Kiss et al. (2020). The newly obtained nrDNA sequences were deposited in GenBank (Table 4).
The 5.8S, 18S, and 28S sequences extracted from genome assemblies, retrieved from NCBI GenBank or determined in this work (Table 4), were included in the nrDNA analysis. When one or more of these loci were not available in either of the respective genomes or as separate GenBank entries for the respective specimens, and we did not have access to herbarium specimens or DNA from the respective specimens to amplify and sequence the loci, these were coded as missing. All powdery mildew specimens included in the genome-scale phylogenetic analysis were also used in the nrDNA-based phylogenetic analysis, except for Golovinomyces magnicellulatus FPH2017-1 as the nrDNA loci were missing from its genomes and were not available as separate entries in GenBank.
Sequences available for each region were aligned using MAFFT v.7.450 (Katoh and Standley, 2013) as implemented in Geneious Prime. Alignments were manually trimmed and concatenated into a supermatrix, with missing data represented as gaps. A Maximum Likelihood analysis of the concatenated alignment was run using RAxML v.8 (Stamatakis, 2018) in Geneious Prime with 1,000 bootstrap replicates based on the GTR substitution model with gamma-distribution rate variation for individual partitions. Parauncinula polyspora was used as the outgroup based on Kusch et al. (2022a).
Some of the published powdery mildew genome assemblies are available in GenBank under species names that need to be updated. The taxonomy of the genus Blumeria has recently changed (Liu et al., 2021); we followed the new nomenclature and renamed the powdery mildew isolates from barley as B. hordei in this work. The species first described as Oidium neolycopersici from tomato (Kiss et al., 2001) was re-classified as Pseudoidium neolycopersici (Braun and Cook, 2012) and recently as Erysiphe neolycopersici (Hsiao et al., 2022). Therefore, the latter name was used here as a synonym of O. neolycopersici. Powdery mildew on rubber tree (Hevea brasiliensis) has long been attributed to Oidium heveae; however, Wu et al. (2019) revealed that the causal agent of this disease is Erysiphe quercicola, a species known to infect diverse host plant species. As the ITS sequence of the genome-sequenced isolate known as O. heveae HO-73, available in GenBank under acc. no. KJ868176, is identical to several ITS sequences of E. quercicola analyzed by Wu et al. (2019), we propose to use the binomial E. quercicola for isolate HO-73, especially because of confusions concerning the precise identification of powdery mildew anamorphs listed as O. heveae in different works (Braun and Cook, 2012; Wu et al., 2019).
After assessment of publicly available powdery mildew genome assemblies, representative genomes of all powdery mildew species with published WGS data were included in the genome-scale phylogenetic analysis (Table 4) except for Golovinomyces orontii MGH1 (Micali et al., 2008) as its genome showed a high number of duplicated BUSCOs (2,261) and only 648 single-copy BUSCOs (Table 2). Therefore, the analysis was undertaken based on 751 single-copy orthologous sequences from 38 selected Leotiomycete genomes (24 powdery mildew genomes and 14 additional genomes from Helotiales) (Tables 3, 4). The final alignment included a total of 197,082 sites, with 0.74% gaps and 30.73% invariant sites. In the resulting phylogeny, the Erysiphaceae formed a monophyletic group with maximum bootstrap support as expected. Arachnopeziza araneosa was identified as the closest saprobic relative of powdery mildew fungi (Figure 1), which is in agreement with the analysis of Johnston et al. (2019). Alignments and trees produced in this study are available in the Supplementary Material.
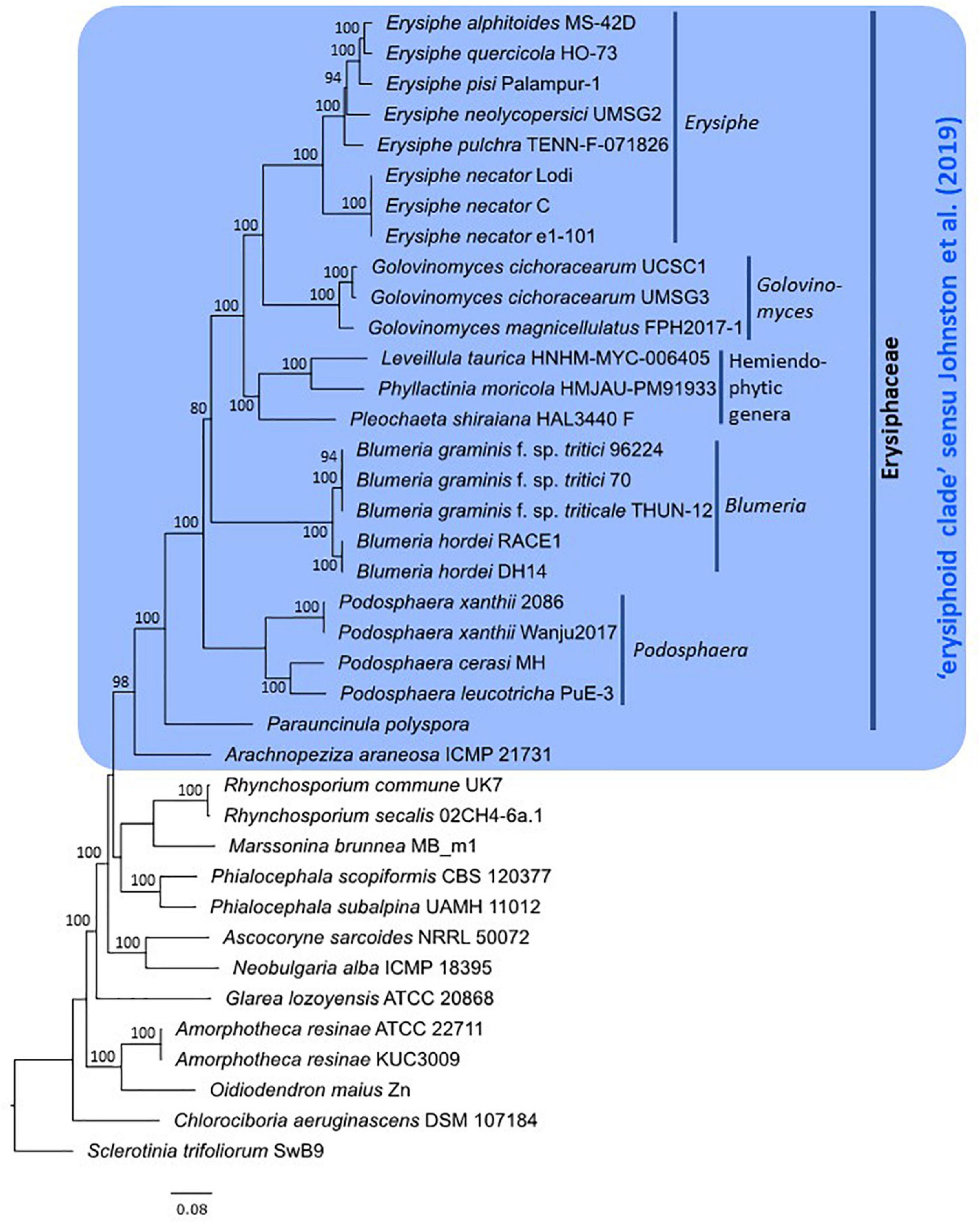
Figure 1. Phylogeny of powdery mildew species and closely related Leotiomycetes based on 751 orthologous protein sequences. Maximum likelihood phylogeny was inferred from a concatenated alignment of amino acid sequences using RAxML-NG v.1.0.1 (Kozlov et al., 2019) under the JTT+I+G4+F substitution model. Taxon labels include species names followed by the specimen/strain accession numbers, except for Parauncinula polyspora, for which a herbarium specimen is not available (Frantzeskakis et al., 2019a). Bootstrap support values greater than 70% are shown at the edges. The tree is rooted to Sclerotinia trifoliorum strain SwB9 (Kusch et al., 2022b). The scale bar represents nucleotide substitutions per site.
The 24 powdery mildew genomes included in the analysis represented eight genera out of 19 recognized within the Erysiphaceae (Table 1). The epiphytic genera Erysiphe, Golovinomyces, Blumeria, and Podosphaera, all represented by multiple genomes, belonged each to distinct clades with 100% bootstrap support (Figure 1). Within Erysiphe, E. alphitoides, E. quercicola, and E. pulchra representing sect. Microsphaera (Takamatsu et al., 2015a), together with E. pisi belonging to sect. Erysiphe, formed a lineage with maximum bootstrap support. Erysiphe necator, a representative of sect. Uncinula within the genus (Takamatsu et al., 2015b), belonged to another lineage. Within Podosphaera, P. leucotricha and P. cerasi, representing sect. Podosphaera of the genus, formed a fully supported clade, while P. xanthii, a representative of sect. Sphaerotheca, belonged to another clade. Similar to a previous study (Frantzeskakis et al., 2019a), the analysis identified Parauncinula polyspora, another epiphytic powdery mildew species, as belonging to an early diverged lineage of the Erysiphaceae. Three genera, Leveillula, Phyllactinia, and Pleochaeta, each represented by a single genome, formed a distinct lineage known as the hemiendophytic lineage within the Erysiphaceae (Takamatsu, 2013a,b; Kusch et al., 2022a).
Out of those 14 other taxa from the Helotiales included in this analysis, Arachnopeziza araneosa was the only one that belonged to the large monophyletic clade including all the 24 genome-sequenced powdery mildew fungi (Figure 1). The close phylogenetic relationship between the Erysiphaceae and Arachnopeziza has already been shown by Johnston et al. (2019) based on the analysis of the class Leotiomycetes that included three powdery mildew species and was built on a partly overlapping set of a total of 3,156 single-copy orthologs.
All powdery mildew specimens included in the genome-scale phylogenetic analysis were also used in the nrDNA analysis, except for G. magnicellulatus FPH2017-1 as the 5.8S, 18S, and 28S nrDNA sequences were missing from its published genome. These sequences were not available for this specimen as separate entries in GenBank either and we had no access to any materials of the G. magnicellulatus isolate FPH2017-1 to determine the missing nrDNA sequences in this study. Therefore, the nrDNA analysis consisted of 23 taxa and a total of 2,666 sites (5.8S: 153 bp, 18S: 1,695 bp, and 28S: 818 bp), 34.8% of which were variable. Whenever possible, the 5.8S, 18S, and/or 28S nrDNA sequences extracted from the respective genomic databases were used in the analysis. When some of those sequences were not found in the respective WGS contigs/scaffolds, sequences from the same specimens/isolates were retrieved from GenBank or PCR-amplified and determined in this study or coded as missing. The nrDNA phylogeny (Figure 2) was largely congruent to the phylogeny produced based on 751 orthologous proteins (Figure 1).
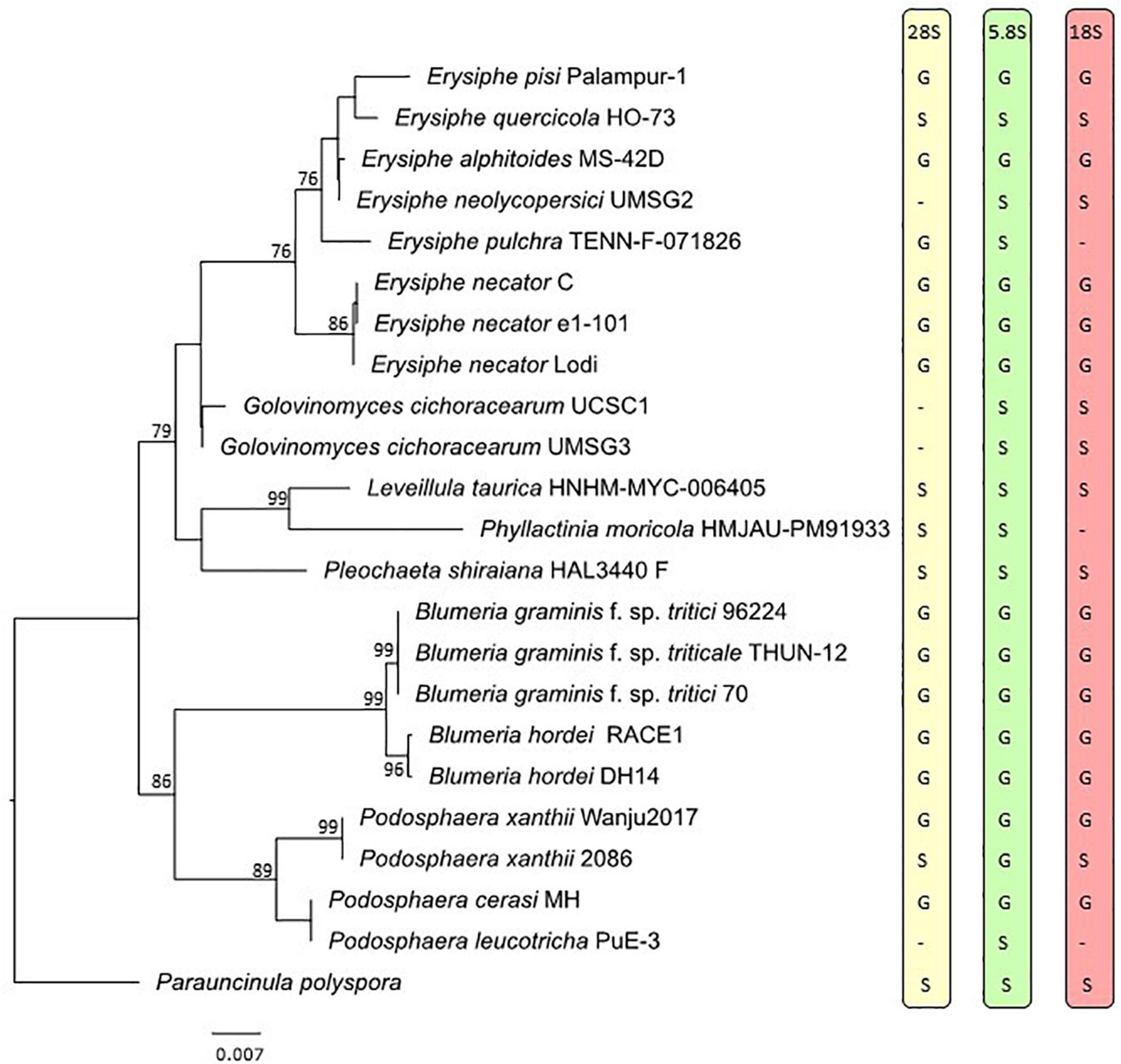
Figure 2. Maximum likelihood phylogeny based on the concatenated sequences of the 5.8S, 18S, and 28S regions of the nuclear ribosomal DNA of representative powdery mildew taxa. Bootstrap values greater than 70% are shown above or below the branches. The tree is rooted to Parauncinula polyspora specimen sequenced by Frantzeskakis et al. (2019a). Taxon labels include species names followed by the specimen accession numbers, except for Parauncinula polyspora, for which a herbarium specimen is not available (Frantzeskakis et al., 2019a). The letters on the right side of the tree indicate source of the 28S, 5.8S, and 18S nrDNA sequences as G (extracted from the genome) or S (produced through Sanger sequencing or obtained from NCBI GenBank database). Dashes indicate missing loci. The scale bar represents nucleotide substitutions per site.
Extracting the nrDNA sequences from published powdery mildew genomes resulted in detection of contaminating DNA in multiple genomes (Table 4). For example, multiple nrDNA sequences were detected in the genome of E. necator C genome (GCA_000798715.1), which were identical to those of Penicillium spp. strains. Likewise, additional nrDNA sequences were detected in E. pulchra TENN-F-071826 (GCA_002918395.1) and E. quercicola (formerly O. heveae) HO-73 (GCA_003957845.1) genomes, which were highly similar to the nrDNA sequences of Cladosporium spp. and various plant species, respectively. Although no nrDNA sequences with any similarity to powdery mildew species could be retrieved from the genome of G. magnicellulatus (GCA_006912115.1), two contigs (VCMJ01009734 and VCMJ01035023) were found to carry nrDNA sequences with high similarity to nrDNA sequences of Pseudozyma spp., Moesziomyces spp., and Acremonium spp. (Table 4).
The presence of multiple dissimilar copies of nrDNA sequences in some powdery mildew assemblies prompted us to identify powdery mildew nrDNA sequences within the analyzed genomes and compare those to the 18S, 28S and ITS sequences of the same specimens if they were available in NCBI GenBank database. In some powdery mildew genome assemblies, nrDNA sequences obtained from Sanger sequencing and WGS data were identical (for example Phyllactinia moricola HMJAU-PM91933 and Pleochaeta shiraiana HAL3440 F; Table 5). In others, some or all the available nrDNA sequences differed in a few, or many, nucleotide positions when extracted from WGS contigs and compared to the results of Sanger sequencing (Table 5). As an example, Figure 3 reveals the variation of nrDNA sequences obtained by the two methods in Leveillula taurica HNHM-MYC-006405.
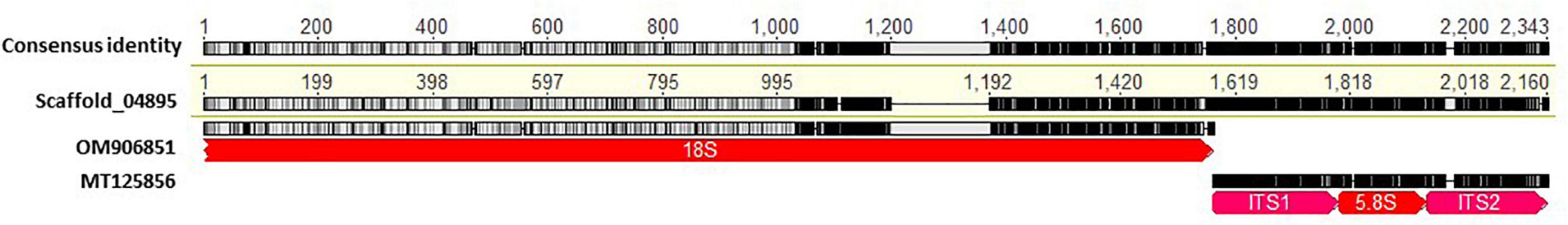
Figure 3. Alignment of 18S (GenBank accession no. OM906851) and ITS (GenBank accession no. MT125856) sequences of Leveillula taurica HNHM-MYC-006405 produced via Sanger sequencing against the sequence of Scaffold-04895 extracted from the genome assembly of the same strain (Assembly project PRJEB36538 from ENA database). nrDNA annotations are depicted by arrowed boxes. Black and gray colors indicate dissimilar and identical nucleotides, respectively, with gaps depicted by horizontal lines.
The nuclear ribosomal DNA region is an essential component of the genomes of all prokaryotes and eukaryotes as the genes included in this region encode ribosomal RNA (rRNA) molecules. These molecules are integral parts of cytoplasmic ribosomes, the major protein synthesis machinery of all living cells. All eukaryotic genomes contain multiple, tandemly repeated copies of a cluster consisting of 18S, 5.8S, and 28S rRNA genes, and the ITS1 and ITS2 regions flanking the 5.8S rRNA gene. This unit is sometimes designated as the 45S nrDNA cluster (Rosato et al., 2016) and it is first transcribed into a single precursor RNA, which is then further processed to produce the 18S, 5.8S and 28S rRNA molecules (Eickbush and Eickbush, 2007). The nuclear ribosomal 5S gene, which is another multi-copy nrDNA unit in all eukaryotic genomes, is transcribed independently of the 45S nrDNA cluster (Naidoo et al., 2013) and it may be localized in another part of the genome (Müller et al., 2019).
Multiple copies of both the 45S and the 5S nrDNA units are mostly needed in those stages of the cell cycles when the demand for protein synthesis is high. In fungi, a comprehensive genome-based analysis revealed that the copy number of the 45S nrDNA cluster varied considerably across phylogenetic lineages, ranging from an estimated 14 to 1,442 copies, with a mean value of 113 copies in the 91 taxa examined (Lofgren et al., 2019). Within the Erysiphaceae, B. graminis is the only species with an estimated copy number of the nrDNA units: Müller et al. (2019) detected approximately 800 copies of the 45S nrDNA cluster on chromosome 9 of B. graminis, and approximately 1,300 copies of the 5S nrDNA unit on chromosome 5.
There is usually low or no intragenomic variation amongst sequences of the nrDNA units, which has been attributed to the concerted evolution of these loci (Eickbush and Eickbush, 2007; Naidoo et al., 2013). In fungi, the 18S and 28S rRNA genes, and especially the ITS region that includes the 5.8S rRNA gene, have long been the most important and best-known DNA regions during phylogenetic and identification works (Schoch et al., 2012). In the present study it was, therefore, surprising to note how difficult is to work with the nrDNA region in some powdery mildew genomes. Some publicly available genome assemblies did not contain all rRNA genes of the nrDNA region (Figure 2 and Table 4). In two WGS assemblies, the 28S, ITS, and/or 18S nrDNA sequences differed in many nucleotide positions from those determined by Sanger sequencing in the same specimens (Figure 3 and Table 5). These differences may have been the result of mis-assembly issues that are common with short-read WGS. Conversely, all nrDNA loci were always reliably amplified through specific PCRs, and their sequences were accurately determined by Sanger sequencing in the Erysiphaceae (e.g., Marmolejo et al., 2018; Bradshaw and Tobin, 2020; Kiss et al., 2020). Therefore, we suggest that the presence of 28S, ITS, and 18S rDNA sequences in powdery mildew (and other) WGS datasets that are identical to those resulted from Sanger sequencing of the respective loci should be used to assess the quality of assemblies, in addition to the commonly used BUSCO values.
Our study has also revealed that a number of published powdery mildew genome assemblies are contaminated with nrDNA sequences from non-target organisms. Misclassification of sequences in reference databases and contamination of public genome assemblies with sequences from other organisms is a common problem that has been the subject of many studies (Kryukov and Imanishi, 2016; Breitwieser et al., 2019; Lupo et al., 2021). Our analyses indicated that contaminant sequences are common in some of the draft powdery mildew genomes, which are, in fact, metagenomes. This is partly linked to the obligate biotrophic nature of powdery mildews, i.e., the fact that isolates cannot be grown in the absence of their host plants. DNA has to be extracted from powdery mildew samples that almost inevitably contain host plant DNA and also DNA from multiple non-target organisms (predominantly microbes) that are associated with powdery mildew colonies (Panstruga and Kuhn, 2019). Contamination in genome assemblies can complicate downstream analyses and lead to misleading results; therefore, it is necessary to implement multiple methods and algorithms to assess and exclude contaminant sequences from draft powdery mildew and other genomes before making these public (Cornet et al., 2018; Kahlke and Ralph, 2018; Low et al., 2019; Wood et al., 2019; Kusch et al., 2020, 2022a).
Genome assemblies of other obligate biotrophic plant pathogens may also be contaminated with DNA regions coming from non-target organisms. For example, Zaccaron and Stergiopoulos (2021) have recently pointed out that the genome assembly of the oomycete Albugo laibachii infecting the leaves of experimental Arabidopsis thaliana plants (Kemen et al., 2011) contains the ITS region and many GC-rich regions of a powdery mildew fungus (Golovinomyces sp.), which may have infected the sampled leaves in addition to A. laibachii without being noticed.
The debate related to specimen-based versus environmental DNA (eDNA)-only research in biology, and particularly in fungal biology, is ongoing, especially in biodiversity and taxonomy studies (e.g., Truong et al., 2017; Lücking and Hawksworth, 2018). DNA samples of powdery mildew fungi used for WGS can be considered as eDNA because they are not extracted from pure cultures of the target isolates, as explained above. There is no consensus amongst diverse laboratories engaged in powdery mildew WGS to deposit herbarium specimens and/or other samples, such as DNA samples, of the powdery mildew-infected plant materials used for WGS in internationally recognized herbaria, fungaria or other fungal collections. This is the reason for the missing specimen or voucher accession numbers for some of the powdery mildew materials that were included in WGS projects and used in this study. Deposition of such materials in internationally accessible collections should be required by research journals before the publication of WGS results. The collections would preserve the specimens and other samples and would make them available for further studies similar to fungal biodiversity and taxonomy studies (Verkley et al., 2015).
This work included all powdery mildew species with publicly available genomes that were suitable for a genome-scale phylogenetic analysis using single-copy proteins. In total, 24 powdery mildew genome assemblies were used in the phylogenetic analyses, which represented eight out of the 19 genera that are currently recognized within the Erysiphaceae. Our analysis built on 751 single-copy orthologs resulted in a phylogeny that is largely congruent to nrDNA sequences-based phylogeny of the same set of specimens. These results indicated that phylogenetic analyses of nrDNA sequences are sufficient to delimit genera within the Erysiphaceae, which are also defined based on morphological characteristics (Braun and Cook, 2012; Marmolejo et al., 2018; Kiss et al., 2020). To test this presumption, further phylogenomic analyses should be conducted when genome assemblies become available for those powdery mildew genera that currently lack WGS data.
One of the main goals of this work was to reveal the closest known saprobic relatives of powdery mildew fungi. Recently, new genome assemblies were published for a number of taxa belonging to the order Helotiales (Table 3), which has enabled a more extensive taxon sampling and higher resolution of phylogenomic analyses within those fungal groups that were considered earlier as close saprobic relatives of the Erysiphaceae. Our genome-scale phylogenetic analysis identified the Arachnopezizaceae as a putative sister group of powdery mildew fungi. According to our study, the Myxotrichaceae (including Oidiodendron), which were previously considered as the closest saprobic relatives of powdery mildew fungi (Peterson and Pfister, 2010; Takamatsu, 2013a,2018), and Chlorociboria, another genus considered earlier as a close relative of the Erysiphaceae (Wang et al., 2006a,b), were only distantly related to powdery mildew fungi. Our results supported the finding of a comprehensive genome-scale analysis of 49 Leotiomycetes genomes that included the genomes of three powdery mildew species, and a genome of an Arachnopeziza, a Chlorociboria, and an Oidiodendron strain, and that grouped the three powdery mildew fungi and Arachnopeziza together in the newly defined ‘erysiphoid’ clade of the Helotiales (Johnston et al., 2019). Further analyses of powdery mildew and Arachnopeziza genomes may discover signatures of the evolutionary processes that have led to obligate biotrophy from a saprobic way of life.
DNA sequence data produced in this study were deposited in NCBI GenBank database and accession numbers are presented in Table 4. Alignments and phylogenetics trees are provided as Supplementary Material.
LK designed and coordinated the research. NV and LK wrote the manuscript. NV carried out all phylogenetic and phylogenomic analyses. SK contributed to genome analyses. MN and DS determined all 18S, ITS and 28S sequences newly reported in this study. UB provided taxonomic expertise, ST and RP phylogenetic and genomic expertise. UB, ST, RP, SK, and LK provided expertise on powdery mildew evolution and biology. All authors commented on early drafts of the manuscript, read, and approved the final version.
This work was partly supported by a grant of the Australia-Germany Joint Research Co-Operation Scheme, and the program PPP Australia 2019 funded by the German Academic Exchange Service (DAAD), awarded to LK and RP. It was further supported by the German Research Foundation (Deutsche Forschungsgemeinschaft; DFG) project number 274444799 [Grant 861/14-2 awarded to RP] in the context of the DFG-funded priority program SPP1819 “Rapid evolutionary adaptation – potential and constraints,” and also by the University of Southern Queensland. The support of a János Bolyai Research Scholarship of the Hungarian Academy of Sciences, awarded to MN, is also acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.903024/full#supplementary-material
Amano, K. (1986). Host Range and Geographical Distribution of the Powdery Mildew Fungi. Tokyo: Japan Scientific Societies Press.
Baldrian, P., Zrůstová, P., Tláskal, V., Davidová, A., Merhautová, V., and Vrška, T. (2016). Fungi associated with decomposing deadwood. Fungal Ecol. 23, 109–122. doi: 10.1016/j.funeco.2016.07.001
Barsoum, M., Sabelleck, B., Spanu, D., and Panstruga, R. (2019). Rumble in the effector jungle: candidate effector proteins in interactions of plants with powdery mildew and rust fungi. Crit. Rev. Plant Sci. 38, 255–279. doi: 10.1080/07352689.2019.1653514
Berbee, M. L., and Taylor, J. W. (1993). Dating the evolutionary radiation of the true fungi. Can. J. Bot. 71, 1114–1127.
Berbee, M. L., and Taylor, J. W. (2001). “Fungal molecular evolution: gene trees and geologic time,” in The Mycota VII. Part B, Systematics and Evolution, eds D. J. McLaughlin, E. G. Mclaughlin, and P. A. Lemke (Berlin: Springer-Verlag), 229–245.
Bindschedler, L. V., Panstruga, R., and Spanu, P. D. (2016). Mildew-Omics: how global analyses aid the understanding of life and evolution of powdery mildews. Front. Plant Sci. 7:123. doi: 10.3389/fpls.2016.00123
Bradshaw, M., Braun, U., Götz, M., Takamatsu, S., Brand, T., Cabrera, M. G., et al. (2020). Contributions to the phylogeny and taxonomy of the Erysiphaceae (powdery mildews) – part 1. Sydowia 73, 89–112.
Bradshaw, M., and Tobin, P. (2020). Sequencing herbarium specimens of a common detrimental plant disease (powdery mildew). Phytopathology 110, 1248–1254. doi: 10.1094/PHYTO-04-20-0139-PER
Braun, U. (2011). The current systematics and taxonomy of the powdery mildews (Erysiphales): an overview. Mycoscience 52, 210–212. doi: 10.1007/s10267-010-0092-1
Braun, U., and Cook, R. T. A. (2012). Taxonomic Manual of the Erysiphales (Powdery Mildews). Utrecht: CBS-KNAW Fungal Biodiversity Centre.
Braun, U., Cook, R. T. A., Inman, A. J., and Shin, H.-D. (2002). “The taxonomy of the powdery mildew fungi,” in The Powdery Mildews: A Comprehensive Treatise, eds R. R. Bélanger, W. R. Bushnell, A. J. Dik, and T. L. W. Carver (St. Paul, MN: American Phytopathogical Society), 13–55. doi: 10.1016/j.funbio.2010.08.008
Braun, U., Shin, H.-D., Takamatsu, S., Meeboon, J., Kiss, L., Lebeda, A., et al. (2019). Phylogeny and taxonomy of Golovinomyces orontii revisited. Mycol. Prog. 18, 335–357. doi: 10.1007/s11557-018-1453-y
Breitwieser, F. P., Pertea, M., Zimin, A. V., and Salzberg, S. L. (2019). Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res. 29, 954–960. doi: 10.1101/gr.245373.118
Büttner, E., Liers, C., Gebauer, A. M., Collemare, J., Navarro-Muñoz, J. C., Hofrichter, M., et al. (2019). Draft genome sequence of the wood-staining ascomycete Chlorociboria aeruginascens DSM 107184. Microbiol. Resour. Announc. 8, e249–e219. doi: 10.1128/MRA.00249-19
Calonnec, A., Cartolaro, P., Poupot, C., Dubourdieu, D., and Darriet, P. (2004). Effects of Uncinula necator on the yield and quality of grapes (Vitis vinifera) and wine. Plant Pathol. 53, 434–445.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, L., Yue, Q., Zhang, X., Xiang, M., Wang, C., Li, S., et al. (2013). Genomics-driven discovery of the pneumocandin biosynthetic gene cluster in the fungus Glarea lozoyensis. BMC Genom. 14:339. doi: 10.1186/1471-2164-14-339
Cornet, L., Meunier, L., Vlierberghe, M. V., Léonard, R. R., Durieu, B., Lara, Y., et al. (2018). Consensus assessment of the contamination level of publicly available cyanobacterial genomes. PLoS One 13:e0200323. doi: 10.1371/journal.pone.0200323
Crous, P. W., Cowan, D. A., Maggs-Kölling, G., Yilmaz, N., Thangavel, R., Wingfield, M. J., et al. (2021). Fungal planet description sheets: 1182–1283. Persoonia 46, 313–528. doi: 10.3767/persoonia.2021.46.11
Crous, P. W., Wingfield, M. J., Chooi, Y.-H., Gilchrist, C. L. M., Lacey, E., Pitt, J. I., et al. (2020). Fungal Planet description sheets: 1042-1111. Persoonia 44, 301–459. doi: 10.3767/persoonia.2020.44.11
Crous, P. W., Wingfield, M. J., Lombard, L., Roets, F., Swart, W. J., Alvarado, P., et al. (2019). Fungal planet description sheets: 951–1041. Persoonia 43, 223–425. doi: 10.3767/persoonia.2019.43.06
Dantec, C. F., Ducasse, H., Capdevielle, X., Fabreguettes, O., Delzon, S., and Desprez-Loustau, M. L. (2015). Escape of spring frost and disease through phenological variations in oak populations along elevation gradients. J. Ecol. 103, 1044–1056. doi: 10.1111/1365-2745.12403
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Demeter, L., Molnár, ÁP., Öllerer, K., Csóka, G., Kiš, A., Vadász, C., et al. (2021). Rethinking the natural regeneration failure of pedunculate oak: the pathogen mildew hypothesis. Biol. Cons. 253:108928. doi: 10.1016/j.biocon.2020.108928
Desprez-Loustau, M. L., Courtecuisse, R., Robin, C., Husson, C., Moreau, P. A., Blancard, D., et al. (2010). Species diversity and drivers of spread of alien fungi (sensu lato) in Europe with a particular focus on France. Biol. Inv. 12, 157–172. doi: 10.1007/s10530-009-9439-y
Desprez-Loustau, M.-L., Massot, M., Feau, N., Fort, T., De Vicente, A., Torés, J. A., et al. (2017). Further support of conspecificity of oak and mango powdery mildew and first report of Erysiphe quercicola and Erysiphe alphitoides on mango in mainland Europe. Plant Dis. 101, 1086–1093. doi: 10.1094/PDIS-01-17-0116-RE
Desprez-Loustau, M.-L., Massot, M., Toïgo, M., Fort, T., Aday Kaya, A. G., Boberg, J., et al. (2018). From leaf to continent: the multi-scale distribution of an invasive cryptic pathogen complex on oak. Fungal Ecol. 36, 39–50. doi: 10.1016/j.funeco.2018.08.001
Dunn, M. W., and Gaynor, L. G. (2020). Impact and control of powdery mildew on irrigated soybean varieties grown in Southeast Australia. Agronomy 10:514. doi: 10.3390/agronomy10040514
Dutech, C., Feau, N., Lesur, I., Ehrenmann, F., Letellier, T., Li, B., et al. (2020). An easy and robust method for isolation and validation of single-nucleotide polymorphic markers from a first Erysiphe alphitoides draft genome. Mycol. Prog. 19, 615–628. doi: 10.1007/s11557-020-01580-w
Eickbush, T. H., and Eickbush, D. G. (2007). Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175, 477–485. doi: 10.1534/genetics.107.071399
Ellingham, O., David, J., and Culham, A. (2019). Enhancing identification accuracy for powdery mildews using previously underexploited DNA loci. Mycologia 111, 798–812. doi: 10.1080/00275514.2019.1643644
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Farinas, C., Gluck-Thaler, E., Slot, J. C., and Peduto Hand, F. (2019). Whole-genome sequence of the phlox powdery mildew pathogen Golovinomyces magnicellulatus strain FPH2017-1. Microbiol. Resour. Announc. 8, e852–e819. doi: 10.1128/MRA.00852-19
Faticov, M., Desprez-Loustau, M.-L., Kiss, L., Massot, M., D’Arcier, J. F., Mutz, J., et al. (2022). Niche differentiation within a cryptic pathogen complex: climatic drivers and hyperparasitism at multiple spatial scales. Ecography 2022:e06062. doi: 10.1111/ecog.06062
Fondevilla, S., and Rubiales, D. (2012). Powdery mildew control in pea. A review. Agron. Sustain. Dev. 32, 401–409.
Frantzeskakis, L., Kracher, B., Kusch, S., Yoshikawa-Maekawa, M., Bauer, S., Pedersen, C., et al. (2018). Signatures of host specialization and a recent transposable element burst in the dynamic one-speed genome of the fungal barley powdery mildew pathogen. BMC Genom. 19:381. doi: 10.1186/s12864-018-4750-6
Frantzeskakis, L., Németh, M. Z., Barsoum, M., Kusch, S., Kiss, L., Takamatsu, S., et al. (2019a). The Parauncinula polyspora draft genome provides insights into patterns of gene erosion and genome expansion in powdery mildew fungi. Mbio 10:e01692–19. doi: 10.1128/mBio.01692-19
Frantzeskakis, L., Kusch, S., and Panstruga, R. (2019b). The need for speed: compartmentalized genome evolution in filamentous phytopathogens. Mol. Plant Pathol. 20, 3–7. doi: 10.1111/mpp.12738
Fuller, K. B., Alston, J. M., and Sambucci, O. S. (2014). The value of powdery mildew resistance in grapes: evidence from California. Wine Econ. Policy 3, 90–107.
Gadoury, D. M., Cadle-Davidson, L., Wilcox, W. F., Dry, I. B., Seem, R. C., and Milgroom, M. G. (2012). Grapevine powdery mildew (Erysiphe necator): a fascinating system for the study of the biology, ecology and epidemiology of an obligate biotroph. Mol. Plant Pathol. 13, 1–16. doi: 10.1111/j.1364-3703.2011.00728.x
Gañán, L., White, R. A. III, Friesen, M. L., Peever, T. L., and Amiri, A. (2020). A genome resource for the apple powdery mildew pathogen Podosphaera leucotricha. Phytopathology 110, 1756–1758. doi: 10.1094/PHYTO-05-20-0158-A
Gianoulis, T. A., Griffin, M. A., Spakowicz, D. J., Dunican, B. F., Alpha, C. J., Sboner, A., et al. (2012). Genomic analysis of the hydrocarbon-producing, cellulolytic, endophytic fungus Ascocoryne sarcoides. PLoS Genet. 8:e1002558. doi: 10.1371/journal.pgen.1002558
Glawe, D. A. (2008). The powdery mildews: a review of the world’s most familiar (yet poorly known) plant pathogens. Annu. Rev. Phytopathol. 46, 27–51. doi: 10.1146/annurev.phyto.46.081407.104740
Hacquard, S., Kracher, B., Maekawa, T., Vernaldi, S., Schulze-Lefert, P., and van Themaat, E. V. L. (2013). Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc. Natl. Acad. Sci. U.S.A. 110, E2219–E2228. doi: 10.1073/pnas.1306807110
Haeckel, E. (1899-1904). Kunstformen der Natur. [Art Forms in Nature.]. Leipzig: Bibliographisches Institut.
Han, J. G., Sung, G. H., and Shin, H. D. (2014). Proliferodiscus inspersus var. magniascus and Rodwayella citrinula, two unrecorded taxa of Hyaloscyphaceae (tribe Arachnopezizeae) in Korea. Mycobiology 42, 86–91. doi: 10.5941/MYCO.2014.42.1.86
Hirata, T., Cunnington, J. H., Paksiri, U., Limkaisang, S., Shishkoff, N., Grigaliunaite, B., et al. (2000). Evolutionary analysis of subsection Magnicellulatae of Podosphaera section Sphaerotheca (Erysiphales) based on the rDNA internal transcribed spacer sequences with special reference to host plants. Can. J. Bot. 78, 1521–1530. doi: 10.1139/cjb-78-12-1521
Hsiao, H.-Y., Ariyawansa, H. A., Hsu, C.-C., Wang, C.-J., and Shen, Y.-M. (2022). New records of Powdery mildews from Taiwan: Erysiphe ipomoeae comb. nov., E. aff. betae on buckwheat, and E. neolycopersici comb. nov. on Cardiospermum halicacabum. Diversity 14:204. doi: 10.3390/d14030204
Hückelhoven, R., and Panstruga, R. (2011). Cell biology of the plant–powdery mildew interaction. Curr. Opin. Plant Biol. 14, 738–746. doi: 10.1016/j.pbi.2011.08.002
Inuma, T., Khodaparast, S. A., and Takamatsu, S. (2007). Multilocus phylogenetic analyses within Blumeria graminis, a powdery mildew fungus of cereals. Mol. Phylogenet. Evol. 44, 741–751. doi: 10.1016/j.ympev.2007.01.007
Johnston, P. R., Quijada, L., Smith, C. A., Baral, H. O., Hosoya, T., Baschien, C., et al. (2019). A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA Fungus 10, 1. doi: 10.1186/s43008-019-0002-x
Jones, L., Riaz, S., Morales-Cruz, A., Amrine, K. C., McGuire, B., Gubler, W. D., et al. (2014). Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator. BMC Genom. 15:1081. doi: 10.1186/1471-2164-15-1081
Kahlke, T., and Ralph, P. J. (2018). BASTA – Taxonomic classification of sequences and sequence bins using last common ancestor estimations. Methods Ecol. Evol. 10, 100–103. doi: 10.1111/2041-210X.13095
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelly, L. A., Vaghefi, N., Bransgrove, K., Fechner, N. A., Stuart, K., Pandey, A. K., et al. (2021). One crop disease, how many pathogens? Podosphaera xanthii and Erysiphe vignae sp. nov. identified as the two species that cause powdery mildew of mungbean (Vigna radiata) and black gram (V. mungo) in Australia. Phytopathology 111, 1193–1206. doi: 10.1094/PHYTO-12-20-0554-R
Kemen, E., Gardiner, A., Schultz-Larsen, T., Kemen, A. C., Balmuth, A. L., Robert-Seilaniantz, A., et al. (2011). Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol. 9:e1001094. doi: 10.1371/journal.pbio.1001094
Kim, S., Subramaniyam, S., Jung, M., Oh, E. A., Kim, T. H., and Kim, J. G. (2021). Genome resource of Podosphaera xanthii, the host-specific fungal pathogen that causes cucurbit powdery mildew. Mol. Plant-Microbe Interac. 34, 457–459. doi: 10.1094/MPMI-11-20-0307-A
Kiss, L. (2005). Powdery mildews as invasive plant pathogens: new epidemics caused by two North American species in Europe. Mycol. Res. 109, 259–260. doi: 10.1017/s0953756205232793
Kiss, L. (2012). Limits of nuclear ribosomal DNA internal transcribed spacer (ITS) sequences as species barcodes for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109:E1811. doi: 10.1073/pnas.1207143109
Kiss, L., Cook, R. T. A., Saenz, G. S., Cunnington, J. H., Pascoe, I., Bardin, M., et al. (2001). Identification of two powdery mildew fungi, Oidium neolycopersici sp. nov. and O. lycopersici, infecting tomato in different parts of the world. Mycol. Res. 105, 684–697.
Kiss, L., Jankovics, T., Kovács, G. M., and Daughtrey, M. L. (2008). Oidium longipes, a new powdery mildew fungus on petunia in the USA: a potential threat to ornamental and vegetable solanaceous crops. Plant Dis. 92, 818–825.
Kiss, L., Stuart, K., Grigg, J., Calvert, J., and Dearnaley, J. D. W. (2018). First report of powdery mildew on goji berry (Lycium barbarum) caused by Arthrocladiella mougeotii in Queensland. Australia. Plant Dis. 102:446. doi: 10.1094/PDIS-07-17-1055-PDN
Kiss, L., and Vaghefi, N. (2021). First report of powdery mildew of rainforest spinach (Elatostema reticulatum), native to Australia, caused by Podosphaera xanthii. Australas. Plant Dis. Notes 16:8. doi: 10.1007/s13314-021-00424-0
Kiss, L., Vaghefi, N., Bransgrove, K., Dearnaley, J. D. W., Takamatsu, S., Tan, Y. P., et al. (2020). Australia: a continent without native powdery mildews? The first comprehensive catalogue indicates recent introductions and multiple host range expansion events, and leads to the re-discovery of Salmonomyces as a new lineage of the Erysiphales. Front. Microbiol. 11:1571. doi: 10.3389/fmicb.2020.01571
Kohler, A., Kuo, A., Nagy, L. G., Morin, E., Barry, K. W., Buscot, F., et al. (2015). Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 47, 410–415. doi: 10.1038/ng.3223
Kosonen, T., Huhtinen, S., and Hansen, K. (2021). Taxonomy and systematics of Hyaloscyphaceae and Arachnopezizaceae. Persoonia 46, 26–62. doi: 10.3767/persoonia.2021.46.02
Kovács, G. M., Jankovics, T., and Kiss, L. (2011). Variation in the nrDNA ITS sequences of some powdery mildew species: do routine molecular identification procedures hide valuable information? Eur. J. Plant Pathol. 131, 135–141. doi: 10.1007/s10658-011-9793-3
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kryukov, K., and Imanishi, T. (2016). Human contamination in public genome assemblies. PLoS One 11:e0162424. doi: 10.1371/journal.pone.0162424
Kuhn, H., Kwaaitaal, M., Kusch, S., Acevedo-Garcia, J., Wu, H., and Panstruga, R. (2016). Biotrophy at its best: Novel findings and unsolved mysteries of the Arabidopsis-powdery mildew pathosystem. Arabidopsis Book 14:e0184. doi: 10.1199/tab.0184
Kusch, S., Vaghefi, N., Takamatsu, S., Liu, S. H.-Y., Németh, M. Z., Seress, D., et al. (2022a). First draft genome assemblies of Pleochaeta shiraiana and Phyllactinia moricola, two tree-parasitic powdery mildew fungi with hemiendophytic mycelia. Phytopathology 112, 961–967. doi: 10.1094/PHYTO-08-21-0337-A
Kusch, S., Larrouy, J., Ibrahim, H. M., Mounichetty, S., Gasset, N., Navaud, O., et al. (2022b). Transcriptional response to host chemical cues underpins the expansion of host range in a fungal plant pathogen lineage. ISME J. 16, 138–148. doi: 10.1038/s41396-021-01058-x
Kusch, S., Németh, M. Z., Vaghefi, N., Ibrahim, H. M., Panstruga, R., and Kiss, L. (2020). A short-read genome assembly resource for Leveillula taurica causing powdery mildew disease of sweet pepper (Capsicum annuum). Mol. Plant-Microbe Interac. 33, 782–786. doi: 10.1094/MPMI-02-20-0029-A
Liang, P., Liu, S., Xu, F., Jiang, S., Yan, J., He, Q., et al. (2018). Powdery mildews are characterized by contracted carbohydrate metabolism and diverse effectors to adapt to obligate biotrophic lifestyle. Front. Microbiol. 9:3160. doi: 10.3389/fmicb.2018.03160
Liu, M., Braun, U., Takamatsu, S., Hambleton, S., Shoukouhi, P., Bisson, K. R., et al. (2021). Taxonomic revision of Blumeria based on multi-gene DNA sequences, host preferences and morphology. Mycoscience 62, 143–165. doi: 10.47371/mycosci.2020.12.003
Lofgren, L. A., Uehling, J. K., Branco, S., Bruns, T. D., Martin, F., and Kennedy, P. G. (2019). Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol. Ecol. 28, 721–730. doi: 10.1111/mec.14995
Low, A. J., Koziol, A. G., Manninger, P. A., Blais, B., and Carrillo, C. D. (2019). ConFindr: rapid detection of intraspecies and cross-species contamination in bacterial whole-genome sequence data. PeerJ 7:e6995. doi: 10.7717/peerj.6995
Lücking, R., and Hawksworth, D. L. (2018). Formal description of sequence-based voucherless Fungi: promises and pitfalls, and how to resolve them. IMA Fungus 9, 143–165. doi: 10.5598/imafungus.2018.09.01.09
Lupo, V., Van Vlierberghe, M., Vanderschuren, H., Kerff, F., Baurain, D., and Cornet, L. (2021). Contamination in reference sequence databases: time for divide-and-rule tactics. Front. Microbiol. 12:755101. doi: 10.3389/fmicb.2021.755101
Marçais, B., and Desprez-Loustau, M. L. (2014). European oak powdery mildew: impact on trees, effects of environmental factors, and potential effects of climate change. Ann. Forest Sci. 71, 633–642. doi: 10.1007/s13595-012-0252-x
Marin-Felix, Y., Hernández-Restrepo, M., Iturrieta-González, I., García, D., Gené, J., Groenewald, J. Z., et al. (2019). Genera of phytopathogenic fungi: GOPHY 3. Stud. Mycol. 94, 1–124. doi: 10.1016/j.simyco.2019.05.001
Marmolejo, J., Siahaan, S. A., Takamatsu, S., and Braun, U. (2018). Three new records of powdery mildews found in Mexico with one genus and one new species proposed. Mycoscience 59, 1–7. doi: 10.1016/j.myc.2017.06.010
Martino, E., Morin, E., Grelet, G. A., Kuo, A., Kohler, A., Daghino, S., et al. (2018). Comparative genomics and transcriptomics depict ericoid mycorrhizal fungi as versatile saprotrophs and plant mutualists. New Phytol. 217, 1213–1229. doi: 10.1111/nph.14974
Matsuda, S., and Takamatsu, S. (2003). Evolution of host–parasite relationships of Golovinomyces (Ascomycete: Erysiphaceae) inferred from nuclear rDNA sequences. Mol. Phylogenet. Evol. 27, 314–327. doi: 10.1016/S1055-7903(02)00401-3
Meeboon, J., Siahaan, S. A., Fujioka, K., and Takamatsu, S. (2017). Molecular phylogeny and taxonomy of Parauncinula (Erysiphales) and two new species P. polyspora and P. uncinata. Mycoscience 58, 361–368. doi: 10.1016/j.myc.2017.04.007
Meeboon, J., and Takamatsu, S. (2016). Notes on powdery mildews (Erysiphales) in Thailand II. Erysiphe species on Adoxaceae, Anacardiaceae, Apocynaceae, Araliaceae, Aristolochiaceae, Bixaceae, Brassicaceae, Cleomaceae, Convolvulaceae, Cucurbitaceae and Euphorbiacea. Trop. Plant Pathol. 41, 357–369. doi: 10.1007/s40858-016-0111-7
Micali, C., Gollner, K., Humphry, M., Consonni, C., and Panstruga, R. (2008). The powdery mildew disease of Arabidopsis: a paradigm for the interaction between plants and biotrophic fungi. Arabidopsis Book 6:e0115. doi: 10.1199/tab.0115
Mori, Y., Sato, Y., and Takamatsu, S. (2000a). Evolutionary analysis of the powdery mildew fungi using nucleotide sequences of the nuclear ribosomal DNA. Mycologia 92, 74–93. doi: 10.2307/3761452
Mori, Y., Sato, Y., and Takamatsu, S. (2000b). Molecular phylogeny and radiation time of Erysiphales inferred from the nuclear ribosomal DNA sequences. Mycoscience 41, 437–447.
Müller, M. C., Kunz, L., Graf, J., Schudel, S., and Keller, B. (2021). Host adaptation through hybridization: genome analysis of triticale powdery mildew reveals unique combination of lineage-specific effectors. Mol. Plant-Microbe Interac. 34, 1350–1357. doi: 10.1094/MPMI-05-21-0111-SC
Müller, M. C., Praz, C. R., Sotiropoulos, A. G., Menardo, F., Kunz, L., Schudel, S., et al. (2019). A chromosome-scale genome assembly reveals a highly dynamic effector repertoire of wheat powdery mildew. New Phytol. 221, 2176–2189. doi: 10.1111/nph.15529
Naidoo, K., Steenkamp, E. T., Coetzee, M. P. A., Wingfield, M. J., and Wingfield, B. D. (2013). Concerted evolution in the ribosomal RNA cistron. PLoS One 8:e59355. doi: 10.1371/journal.pone.0059355
Oh, J. J., Kim, Y. J., Kim, J. Y., Kwon, S. L., Lee, C., Lee, M. E., et al. (2021). Genomic analysis and assessment of melanin synthesis in Amorphotheca resinae KUC3009. J. Fungi 7:289. doi: 10.3390/jof7040289
Panstruga, R., and Kuhn, H. (2019). Mutual interplay between phytopathogenic powdery mildew fungi and other microorganisms. Mol. Plant Pathol. 20, 463–470. doi: 10.1111/mpp.12771
Pearson, R. C., and Gadoury, D. M. (1987). Cleistothecia, the source of primary inoculum for grape powdery mildew in New York. Phytopathology 77, 1509–1514. doi: 10.1094/PDIS.1997.81.8.922
Penselin, D., Münsterkötter, M., Kirsten, S., Felder, M., Taudien, S., Platzer, M., et al. (2016). Comparative genomics to explore phylogenetic relationship, cryptic sexual potential and host specificity of Rhynchosporium species on grasses. BMC Genom. 17:953. doi: 10.1186/s12864-016-3299-5
Peterson, K. R., and Pfister, D. H. (2010). Phylogeny of Cyttaria inferred from nuclear and mitochondrial sequence and morphological data. Mycologia 102, 1398–1416. doi: 10.3852/10-046
Polonio, Á, Díaz-Martínez, L., Fernández-Ortuño, D., de Vicente, A., Romero, D., López-Ruiz, F. J., et al. (2021). A hybrid genome assembly resource for Podosphaera xanthii, the main causal agent of powdery mildew disease in cucurbits. Mol. Plant-Microbe Interac. 34, 319–324. doi: 10.1094/MPMI-08-20-0237-A
Poudel, B., Shivas, R. G., Adorada, D. L., Barbetti, M. J., Bithell, S. L., Kelly, L. A., et al. (2021). Hidden diversity of Macrophomina associated with broadacre and horticultural crops in Australia. Eur. J. Plant Pathol. 161, 1–23. doi: 10.1007/s10658-021-02300-0
Qiu, P. L., Liu, S. Y., Bradshaw, M., Rooney-Latham, S., Takamatsu, S., Bulgakov, T. S., et al. (2020). Multi-locus phylogeny and taxonomy of an unresolved, heterogeneous species complex within the genus Golovinomyces (Ascomycota, Erysiphales), including G. ambrosiae, G. circumfusus and G. spadiceus. BMC Microbiol. 20:51. doi: 10.1186/s12866-020-01731-9
Raja, H., Schoch, C. L., Hustad, V., Shearer, C., and Miller, A. (2011). Testing the phylogenetic utility of MCM7 in the Ascomycota. MycoKeys 1, 63–94. doi: 10.3897/mycokeys.1.1966
Rosato, M., Kovařík, A., Garilleti, R., and Rosselló, J. A. (2016). Conserved organisation of 45S rDNA sites and rDNA gene copy number among major clades of early land plants. PLoS One 11:e0162544. doi: 10.1371/journal.pone.0162544
Saenz, G. S., and Taylor, J. W. (1999). Phylogeny of the Erysiphales (powdery mildews) inferred from internal transcribed spacer (ITS) ribosomal DNA sequences. Can. J. Bot. 77, 150–169.
Schlegel, M., Münsterkötter, M., Güldener, U., Bruggmann, R., Duò, A., Hainaut, M., et al. (2016). Globally distributed root endophyte Phialocephala subalpina links pathogenic and saprophytic lifestyles. BMC Genom. 17:1015. doi: 10.1186/s12864-016-3369-8
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Seko, Y., Heluta, V., Grigaliunaite, B., and Takamatsu, S. (2011). Morphological and molecular characterization of two ITS groups of Erysiphe (Erysiphales) occurring on Syringa and Ligustrum (Oleaceae). Mycoscience 52, 174–182. doi: 10.1007/S10267-010-0088-X
Shen, X. X., Steenwyk, J. L., LaBella, A. L., Opulente, D. A., Zhou, X., Kominek, J., et al. (2020). Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci. Adv. 6:eabd0079. doi: 10.1126/sciadv.abd0079
Shin, H. D., Meeboon, J., Takamatsu, S., Adhikari, M. K., and Braun, U. (2019). Phylogeny and taxonomy of Pseudoidium pedaliacearum. Mycol. Prog. 18, 237–246. doi: 10.1007/s11557-018-1429-y
Shirouzu, T., Takamatsu, S., Hashimoto, A., Meeboon, J., and Ohkuma, M. (2020). Phylogenetic overview of Erysiphaceae based on nrDNA and MCM7 sequences. Mycoscience 61, 249–258. doi: 10.1016/j.myc.2020.03.006
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Spanu, P. D., Abbott, J. C., Amselem, J., Burgis, T. A., Soanes, D. M., Stüber, K., et al. (2010). Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330, 1543–1546. doi: 10.1126/science.1194573
Stadler, M., Lambert, C., Wibberg, D., Kalinowski, J., Cox, R. J., Kolařík, M., et al. (2020). Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales. Ascomycota). Mycol. Prog. 19, 235–245. doi: 10.1007/s11557-019-01552-9
Stamatakis, A. (2018). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sugiyama, M., Ohara, A., and Mikawa, T. (1999). Molecular phylogeny of onygenalean fungi based on small subunit ribosomal DNA (SSU rDNA) sequences. Mycoscience 40, 251–258.
Susi, H., Barrès, B., Vale, P. F., and Laine, A.-L. (2015). Co-infection alters population dynamics of infectious disease. Nat. Commun. 6:5975. doi: 10.1038/ncomms6975
Takamatsu, S. (2004). Phylogeny and evolution of the powdery mildew fungi (Erysiphales, Ascomycota) inferred from nuclear ribosomal DNA sequences. Mycoscience 45, 147–157. doi: 10.1007/s10267-003-0159-3
Takamatsu, S. (2013a). Origin and evolution of the powdery mildews (Ascomycota, Erysiphales). Mycoscience 54, 75–86. doi: 10.1016/j.myc.2012.08.004
Takamatsu, S. (2013b). Molecular phylogeny reveals phenotypic evolution of powdery mildews (Erysiphales, Ascomycota). J. Gen. Plant Pathol. 79, 218–226. doi: 10.1007/s10327-013-0447-5
Takamatsu, S. (2018). Studies on the evolution and systematics of powdery mildew fungi. J. Gen. Plant Pathol. 84, 422–426. doi: 10.1007/s10327-018-0805-4
Takamatsu, S., Braun, U., Limkaisang, S., Kom-Un, S., Sato, Y., and Cunnington, J. H. (2007). Phylogeny and taxonomy of the oak powdery mildew Erysiphe alphitoides sensu lato. Mycol. Res. 111, 809–826. doi: 10.1016/j.mycres.2007.05.013
Takamatsu, S., Hirata, T., and Sato, Y. (1998). Phylogenetic analysis and predicted secondary structures of the rDNA internal transcribed spacers of the powdery mildew fungi (Erysiphaceae). Mycoscience 39, 441–453. doi: 10.1007/BF02460905
Takamatsu, S., Hirata, T., and Sato, Y. (2000). A parasitic transition from trees to herbs occurred at least twice in tribe Cystotheceae (Erysiphaceae): evidence from nuclear ribosomal DNA. Mycol. Res. 104, 1304–1311.
Takamatsu, S., Hirata, T., Sato, Y., and Nomura, Y. (1999). Phylogenetic relationships of Microsphaera and Erysiphe section Erysiphe (powdery mildews) inferred from the rDNA ITS sequences. Mycoscience 40, 259–268.
Takamatsu, S., Ito, H., Shiroya, Y., Kiss, L., and Heluta, V. (2015a). First comprehensive phylogenetic analysis of the genus Erysiphe (Erysiphales, Erysiphaceae) I. The Microsphaera lineage. Mycologia 107, 475–489. doi: 10.3852/15-007
Takamatsu, S., Ito, H., Shiroya, Y., Kiss, L., and Heluta, V. (2015b). First comprehensive phylogenetic analysis of the genus Erysiphe (Erysiphales, Erysiphaceae) II: the Uncinula lineage. Mycologia 107, 903–914. doi: 10.3852/15-062
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Truong, C., Mujic, A. B., Healy, R., Kuhar, F., Furci, G., Torres, D., et al. (2017). How to know the fungi: combining field inventories and DNA-barcoding to document fungal diversity. New Phytol. 214, 913–919. doi: 10.1111/nph.14509
Vaghefi, N., Thompson, S. M., Kimber, R. B. E., Thomas, G. J., Kant, P., Barbetti, M. J., et al. (2020). Multi-locus phylogeny and pathogenicity of Stemphylium species associated with legumes in Australia. Mycol. Prog. 19, 381–396. doi: 10.1007/s11557-020-01566-8
Vela-Gorcía, D., Bellon-Gomez, D., Lopez-Ruiz, F., Torés, J. A., and Perez-Garcia, A. (2014). The Podosphaera fusca TUB2 gene, a molecular “Swiss Army knife” with multiple applications in powdery mildew research. Fungal Biol. 118, 228–241. doi: 10.1016/j.funbio.2013.12.001
Verkley, G. J. M., Rossman, A., and Crouch, J. A. (2015). “The Role of Herbaria and Culture Collections,” in Systematics and Evolution. The Mycota (A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research), Vol. 7B, eds D. McLaughlin and J. Spatafora (Berlin: Springer), doi: 10.1007/978-3-662-46011-5_8
Wadl, P. A., Mack, B. M., Beltz, S. B., Moore, G. G., Baird, R. E., Rinehart, T. A., et al. (2019). Development of genomic resources for the powdery mildew, Erysiphe pulchra. Plant Dis. 103, 804–807. doi: 10.1094/PDIS-05-18-0719-A
Walker, A. K., Frasz, S. L., Seifert, K. A., Miller, J. D., Mondo, S. J., LaButti, K., et al. (2016). Full genome of Phialocephala scopiformis DAOMC 229536, a fungal endophyte of spruce producing the potent anti-insectan compound rugulosin. Genome Announc. 4:e01768–15. doi: 10.1128/genomeA.01768-15
Wang, Z., Binder, M., Schoch, C. L., Johnston, P. R., Spatafora, J. W., and Hibbett, D. S. (2006a). Evolution of helotialean fungi (Leotiomycetes, Pezizomycotina): a nuclear rDNA phylogeny. Mol. Phylog. Evol. 41, 295–312. doi: 10.1016/j.ympev.2006.05.031
Wang, Z., Johnston, P. R., Takamatsu, S., Spatafora, J. W., and Hibbett, D. S. (2006b). Toward a phylogenetic classification of the Leotiomycetes based on rDNA data. Mycologia 98, 1065–1075. doi: 10.3852/mycologia.98.6.1065
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: a Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press), 315–322.
Wicker, T., Oberhaensli, S., Parlange, F., Buchmann, J. P., Shatalina, M., Roffler, S., et al. (2013). The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat. Genet. 45, 1092–1096. doi: 10.1038/ng.2704
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wu, H., Pan, Y., Di, R., He, Q., Rajaofera, M. J. N., Liu, W., et al. (2019). Molecular identification of the powdery mildew fungus infecting rubber trees in China. For. Pathol. 49:e12519. doi: 10.1111/efp.12519
Wu, Y., Ma, X., Pan, Z., Kale, S. D., Song, Y., King, H., et al. (2018). Comparative genome analyses reveal sequence features reflecting distinct modes of host-adaptation between dicot and monocot powdery mildew. BMC Genom. 19:705. doi: 10.1186/s12864-018-5069-z
Young, A., and Kiss, L. (2021). First report of powdery mildew of coastal wattle (Acacia sophorae) caused by Erysiphe quercicola. Australas. Plant Dis. Notes 16:9. doi: 10.1007/s13314-021-00421-3
Zaccaron, A. Z., and Stergiopoulos, I. (2021). Characterization of the mitochondrial genomes of three powdery mildew pathogens reveals remarkable variation in size and nucleotide composition. Microb. Genom. 7:000720. doi: 10.1099/mgen.0.000720
Keywords: contaminated genomes, contaminating sequences, metagenomes, obligate biotrophs, phylogenomics, plant–microbe interactions, single-copy orthologs, whole-genome sequencing
Citation: Vaghefi N, Kusch S, Németh MZ, Seress D, Braun U, Takamatsu S, Panstruga R and Kiss L (2022) Beyond Nuclear Ribosomal DNA Sequences: Evolution, Taxonomy, and Closest Known Saprobic Relatives of Powdery Mildew Fungi (Erysiphaceae) Inferred From Their First Comprehensive Genome-Scale Phylogenetic Analyses. Front. Microbiol. 13:903024. doi: 10.3389/fmicb.2022.903024
Received: 23 March 2022; Accepted: 10 May 2022;
Published: 09 June 2022.
Edited by:
Christian Koch, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanyReviewed by:
Peter Johnston, Manaaki Whenua Landcare Research, New ZealandCopyright © 2022 Vaghefi, Kusch, Németh, Seress, Braun, Takamatsu, Panstruga and Kiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Levente Kiss, TGV2ZW50ZS5LaXNzQHVzcS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.