 Sanqi Tang1,2†
Sanqi Tang1,2† Yuhui Li
Yuhui Li Zening Chen
Zening Chen Zhengjun Wu
Zhengjun Wu- 1Key Laboratory of Ecology of Rare and Endangered Species and Environmental Protection (Guangxi Normal University), Ministry of Education, Guilin, China
- 2Guangxi Key Laboratory of Rare and Endangered Animal Ecology, Guangxi Normal University, Guilin, China
- 3Key Laboratory of Animal Ecology and Conservation, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
Captive animals and wild animals may exhibit different characteristics due to the heterogeneity of their living environments. The gut microbiota play an important role in the digestion and absorption, energy metabolism, immune regulation, and physiological health of the host. However, information about the gut microbiota of captive and wild Gekko gecko is currently limited. To determine the difference in gut microbiota community composition, diversity, and structure between captive and wild geckos, we used the Illumina miseq platform to conduct high-throughput sequencing and bioinformatics analysis of the v3–v4 hypervariable region of 16S rRNA in 54 gecko samples. Our results showed that Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria were the dominant gut microbiota phyla of the gecko. The dominant genera comprised mainly Pseudomonas, Burkholderia-caballeronia-paraburkholderia, Ralstonia, Romboutsia, and Bacteroides. Captive geckos had significantly higher alpha diversity and potential pathogenic bacteria than wild populations. Moreover, significant differences in beta diversity of gut microbiota were observed between two populations. Functional prediction analysis showed that the relative abundance of functional pathways of wild geckos was more higher in metabolism, genetic information processing and organismal system function than those in captive geckos. Total length significantly affected gut microbial community (R2 = 0.4527, p = 0.001) and explained 10.45% of the total variation for gut microbial community variance between two groups. These results may be related to differences in diet and living environment between two populations, suggesting that the management of captive populations should mimic wild environments to the greatest extent possible to reduce the impact on their gut microbiota.
Introduction
The gut microbiota has symbiosis and coordinated evolution with the host, forming an overall system of interaction (Nicholson et al., 2012; Tremaroli and Bäckhed, 2012; Flint et al., 2015). The gut microbiota plays important roles in host growth and development (Egert et al., 2006; Sommer and Bäckhed, 2013), impacting energy budget (Semova et al., 2012), nutritional metabolism (Cani and Everard, 2016; Greer et al., 2016), vitamin synthesis (LeBlanc et al., 2013), digestion and absorption, colonization resistance and immune homeostasis (Hooper et al., 2012; Bengmark, 2013), and behavior and emotion (Heijtz et al., 2011; Ezenwa et al., 2012). At the same time, various factors also affect the gut microbiota, including host diet (Ley et al., 2008; De Filippo et al., 2010; Muegge et al., 2011; Wu et al., 2011), genetic and environmental history (Moeller et al., 2013), physiological status (Amato et al., 2013; McCord et al., 2014), and external environmental factors. Studies on human gut microbiota have identified the relationship between gut microbiota disorders and host infections with opportunistic pathogens (Shin et al., 2015), allergies (Bisgaard et al., 2011), and diseases (Ley et al., 2006; Sobhani et al., 2011; Vaarala, 2011).
Currently, the gut microbiota has been widely studied in invertebrates (Clark and Walker, 2018), amphibians (Bletz et al., 2016) and reptiles (Trevelline et al., 2019; Zhang et al., 2019Eliades et al., 2021), mammals (Hu et al., 2017; Song et al., 2017; Li et al., 2021), and birds (Ryu et al., 2012; Waite et al., 2012; Grond et al., 2018). In recent years, research into the gut microbiota in captive animals, such as Takydromus septentrionalis (Zhou et al., 2020), Neotoma albigula (Martínez-Mota et al., 2020), and Macaca mulatta (Chen et al., 2019) has also attracted extensive attention. The gut microbiota of captive animals may differ from those of wild animals due to dietary differences, antibiotic treatment, human activities, and exposure to other species in captivity (Alfano et al., 2015; Clayton et al., 2016; Chen et al., 2019; Eliades et al., 2021). Studies on mammals and reptiles—such as Hydrorga leptonyx (Nelson et al., 2013), Peromyscus maniculatus (Schmidt et al., 2019), Macaca mulatta (Chen et al., 2019) and Shinisaurus crocodilurus (Jiang et al., 2017), Crocodylus siamensis (Lin et al., 2019), and Brachylophus vitiensis (Eliades et al., 2021)—have confirmed that captivity may change the composition and abundance of the gut microbiota. Moreover, captivity may lead to an increase in potential pathogens in the animal gut microbiota, leading to an increased incidence of disease (Berry et al., 2012; Amato et al., 2016; Kohl et al., 2016; Xie et al., 2016). Conversely, a few studies reported that captivity may improve host immune status (Montalban-Arques et al., 2015), and even have beneficial effects on the development and behavior of the host (Heijtz et al., 2011). Therefore, an evaluation of the effects of captivity on the gut microbiota of wild animals in conservation and rescue breeding is necessary (Eliades et al., 2021).
In China, Tokay geckos are distributed mainly in Guangdong province, Guangxi Zhuang autonomous region, Yunnan province, Fujian province, and Taiwan (Huang et al., 1995; Tang et al., 1997). Geckos are used in traditional Chinese medicine (Li et al., 1996; Bauer, 2009). While medical demand for geckos is increasing, the captive breeding population cannot meet the market demand (Li et al., 1996). In addition, the natural habitat of geckos has been destroyed and gradually reduced, affecting their survival (Zhang et al., 2015). These factors have caused a decline in wild gecko populations, rendering ex-situ conservation the effective conservation approach for this species. Due to the heterogeneity of captive and natural environments, however, captive and wild geckos may differ greatly in diet composition. Moreover, captive populations are exposed to a greater extent to human activity than wild populations. The effect of different living environments on the gut microbiota of geckos remains unclear. We therefore analyzed the gut microbiota community of captive and wild geckos by 16S rRNA gene sequencing technology, and compared the microbial community diversity, richness, structure, and function of the two groups. Considering the differences in living environment between wild animals and captive animals, we propose the following predictions: (1) wild geckos have a higher diversity of gut microbiota than captive geckos and (2) the gut microbiota of captive geckos include more opportunistic pathogens.
Materials and Methods
Sample Collection
We collected a total of 54 samples, including 24 from wild geckos (18 males and 6 females) and 30 from captive geckos (22 males and 8 females). We captured the wild geckos in Jiangzhou District, Chongzuo City, Guangxi, and acquired the captive geckos from Nanning Junhao Wildlife Science and Technology Development Co., Ltd. Wild geckos eat mainly insects and moths from their natural environment (Liu et al., 1981; Tang et al., 1997). Captive individuals are fed on Zophobas morio and Tenebrio molitor and kept in temperature conditions of about 20°C. Average total length (TL) of wild and captive geckos is 233.92 ± 38.93 mm and 266.38 ± 32.82 mm, respectively. All Tokay geckos were healthy during the sampling period (Supplementary Table 1).
We used cloacal swabs—an acceptable source for non-destructive sampling of the gut microbiota of reptiles (Colston et al., 2015; Jiang et al., 2017)—to collect the gut microbiota of geckos. After sampling, we released all individuals back into the site of capture. We stored samples in a thermal insulation bucket with ice bags and transported them to a refrigerator at –20°C for storage.
DNA Extraction, PCR Amplification, and Sequencing
We extracted the total gut microbiota community genomic DNA from all samples using the FastDNA® Spin Kit for Soil (MP Biomedicals, United States) based on the manufacturer’s instructions. We detected the extraction quality of DNA by 1% agarose gel electrophoresis and determined the concentration and purity of DNA with a NanoDrop 2000 UV-vis spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States). We amplified the hypervariable region v3–v4 of the bacterial 16S rRNA gene with primer pairs 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) by an ABI GeneAmp® 9700 polymerase chain reaction (PCR) thermocycler (ABI, CA, United States) (Mori et al., 2014; Chen et al., 2019). Our PCR reaction parameters included initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 45 s, and ended with a final extension at 72°C for 10 min. For the PCR test we used TransGen AP221-02: TransStart Fastpfu DNA Polymerase with 20 μL of reaction system containing 5 × FastPfu Buffer 4 μL, 2.5 mM deoxy-ribonucleoside triphosphates (dNTPs) 2 μL, Forward Primer (5 μM) 0.8 μL, Reverse Primer (5 μM) 0.8 μL, FastPfu Polymerase 0.4 μL, bovine serum albumin (BSA) 0.2 μL, template DNA 10 ng, and finally ddH2O up to 20 μL. We performed PCR reactions in triplicate. We extracted the PCR product from 2% agarose gel, purified it using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to manufacturer’s instructions, and quantified it using Quantus™ Fluorometer (Promega, United States). We pooled purified amplicons in equimolar and paired-end sequenced them (2 × 300) on an Illumina miseq platform (Illumina, San Diego, CA, United States) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Processing of Sequencing Data and Quality Evaluation
We demultiplexed the raw 16S rRNA gene sequencing reads, quality-filtered them by Trimmomatic, and merged them by FLASH with the following criteria: (i) we truncated the 300 bp reads at any site receiving an average quality score of <20 over a 50 bp sliding window, and discarded both the truncated reads shorter than 50 bp and reads containing ambiguous characters; (ii) we assembled only overlapping sequences longer than 10 bp according to their overlapped sequence; we adopted a maximum mismatch ratio of overlap region of 0.2; we discarded reads that could not be assembled; (iii) we distinguished samples according to the barcode and primers, and adjusted the sequence direction, exact barcode matching, and 2 nucleotide mismatches in primer matching.
We obtained a total of 2,603,017 raw sequences from the v3–v4 region of the hypervariable region of 16S rRNA gene in 54 samples. After quality control, 2,504,574 sequences were effective (average ± SD = 48,204.02 ± 9,796.15 for each sample), and the average length of the sequences was 420.66 ± 7.13 bp (Supplementary Table 2). Our rarefaction curve tended to be flat, indicating that the amount of sequencing was reasonable, and the sequencing depth was sufficient (Supplementary Figure 1). The Good’s coverage estimates of the 54 samples ranged from 98.89% to 99.91%, indicating that we had identified almost all bacterial communities in the samples (Table 1).
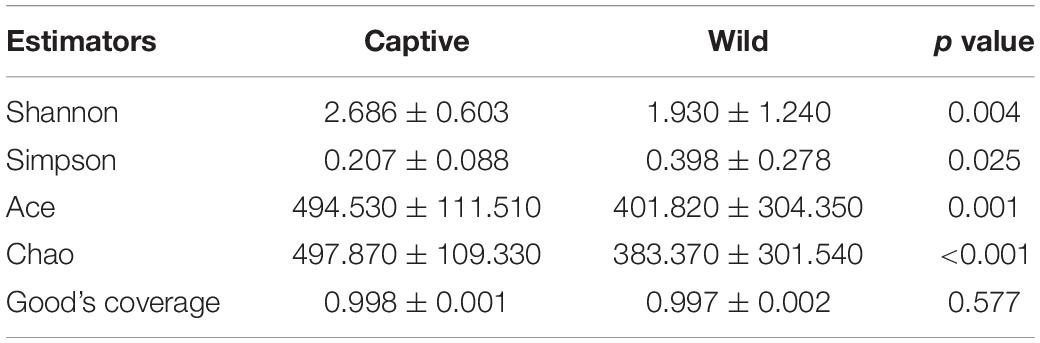
Table 1. Alpha diversity index of the gut microbiota in Tokay gecko.
Bioinformatics Analysis
We clustered operational taxonomic units (OTUs) with 97% similarity cutoff (Edgar, 2013) using UPARSE (version 7.0.10901) and identified and removed chimeric sequences using UCHIME. We analyzed the taxonomy of each OTU representative sequence by RDP Classifier (version 2.112) against the 16S rRNA database (for instance, Silva Release1383) using a confidence threshold of 70%. We used rarefaction curves to reflect whether sequencing data was reasonable, and the coverage index to indicate the real situation of sequencing results. We used the Mothur program to calculate the alpha diversity index to reflect community diversity (Shannon index and Simpson index) and community richness (Abundance-based Coverage Estimator (Ace) and Chao index) (version v.1.30.24). We used the Bray–Curtis distance algorithm for sample hierarchical cluster analysis of gut microbiota of captive and wild geckos and assessed beta diversity by principal coordinate analysis (PCoA) based on weighted and unweighted UniFrac distance metrics using QIIME (version 1.9.15). Adonis permutational multivariate analysis (Adonis/PERMANOVA) was performed to evaluate the dissimilarity among samples with permutation set at 999. We used the Wilcoxon rank-sum test to analyze gut microbiota diversity differences between wild and captive groups and adjusted the p values under the control of FDR level at 0.05. We performed linear discriminant analysis (LDA) effect size (LEfSe) analyses to identify potential microbial biomarkers between groups.6 For the LDA interpretation, we considered differences as significant for a p < 0.05 and an LDA score >4. We predicted functional profiles of microbial communities using PICRUSt2 (phylogenetic investigation of communities by reconstruction of unobserved states 2; Langille et al., 2013) (version 2.2.07). We performed the Wilcoxon rank-sum test to test the differences of functional pathways between the two groups in the Kyoto Encyclopedia of Genes and Genomes (KEGG) using IBM SPSS (version 23.0), and the threshold on the p value was set at 0.05. Detrended correspondence analysis (DCA) was conducted to obtain the length of first axis (R version 3.3.1, vegan package), RDA (Redundancy analysis), or CCA (Canonical correlation analysis) was chosen based on the value of DCA 1 (>4, CCA; <3 RDA; 3–4 RDA/CCA). The DCA analysis showed that the length of DCA 1 was 6.05, indicating a unimodal-model-based CCA analysis was more suitable than RDA analysis to test the effect of geckos’ total length and sex on microbial community at OTU level. A variation partitioning analysis (VPA) was conducted to examine the contribution of total length and sex factors in influencing microbial community structure as determined by CCA analysis (R version 3.3.1, vegan package). Further, the partial Mantel test was used to detect the Bray–Curtis distance matrix correlation between total length and sex and microbial community with 999 permutations [QIIME version 1.9.1 (see footnote 5)]. We used the non-parametric Spearman correlation test to analyze the correlation between total length and sex of geckos and the relative abundance of the top 50 microbial genera (R version 3.3.1, pheatmap package).
Results
Differences in Alpha and Beta Diversity of Gut Microbiota
Alpha diversity analysis showed that there were significant differences in community richness and diversity (p < 0.05; Table 1); captive geckos had higher Ace, Chao, and Shannon indices and a lower Simpson index than those of wild geckos (Table 1 and Figure 1).
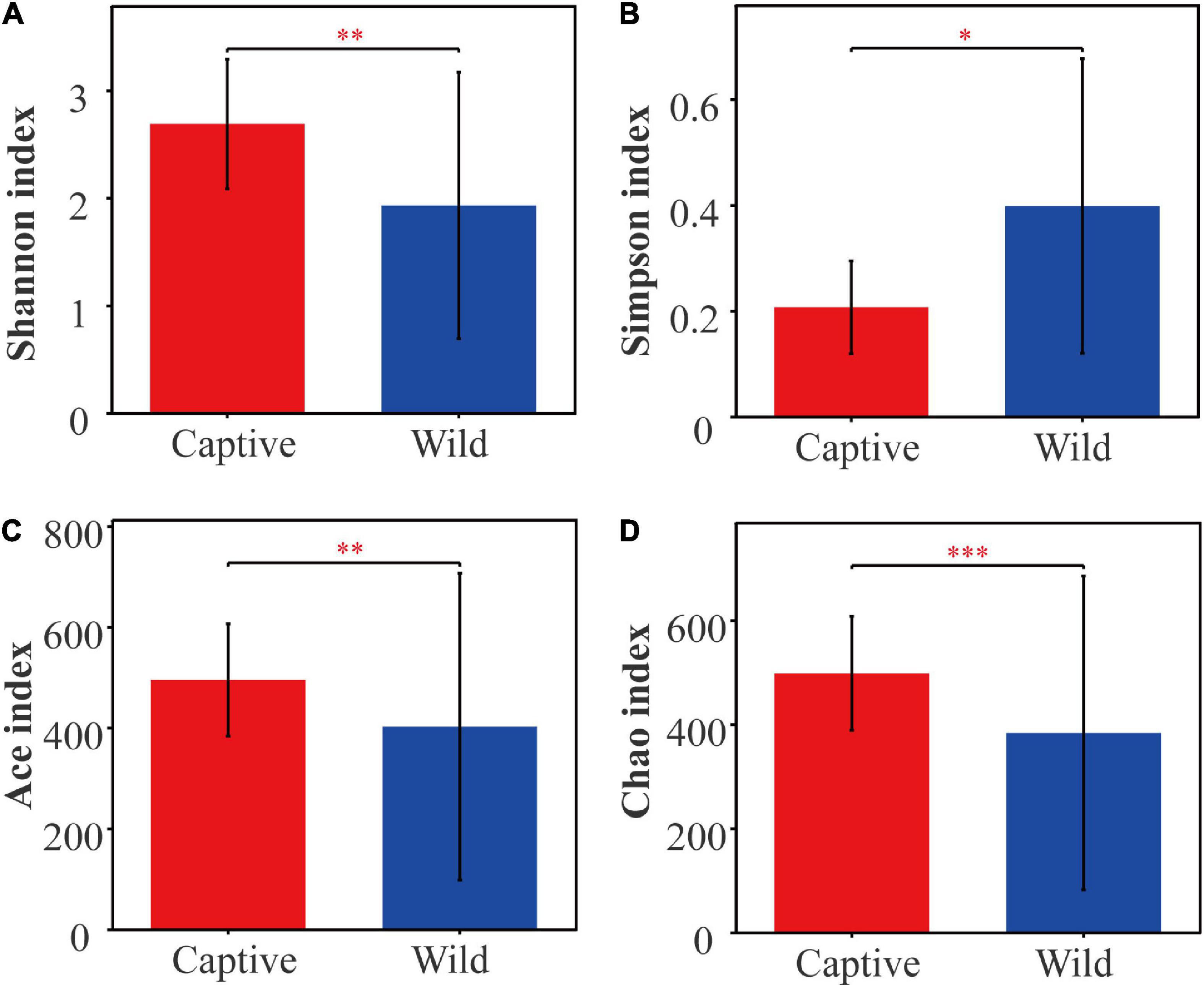
Figure 1. Alpha diversity of gut microbiota between captive and wild geckos. (A) Shannon index; (B) Simpson index; (C) Ace index; (D) Chao index. Significant difference 0.01 < p ≤ 0.05 was marked as “*”, 0.001 < p ≤ 0.01 was marked as “**”, and p ≤ 0.001 was marked as “***”.
Hierarchical cluster analysis based on Bray–Curtis distance algorithm showed that the wild and captive geckos divided into five small branches in total, without making two specific clusters according to groups (Figure 2). Our analysis clustered captive geckos in two branches, and wild geckos in three smaller branches. PCoA based on unweighted and weighted UniFrac distances revealed high clustering of the gut microbiota according to group, highlighting a significant separation between captive and wild geckos (Unweighted UniFrac: R2 = 0.2023, p = 0.001; Weighted UniFrac: R2 = 0.2177, p = 0.001; Figure 3).
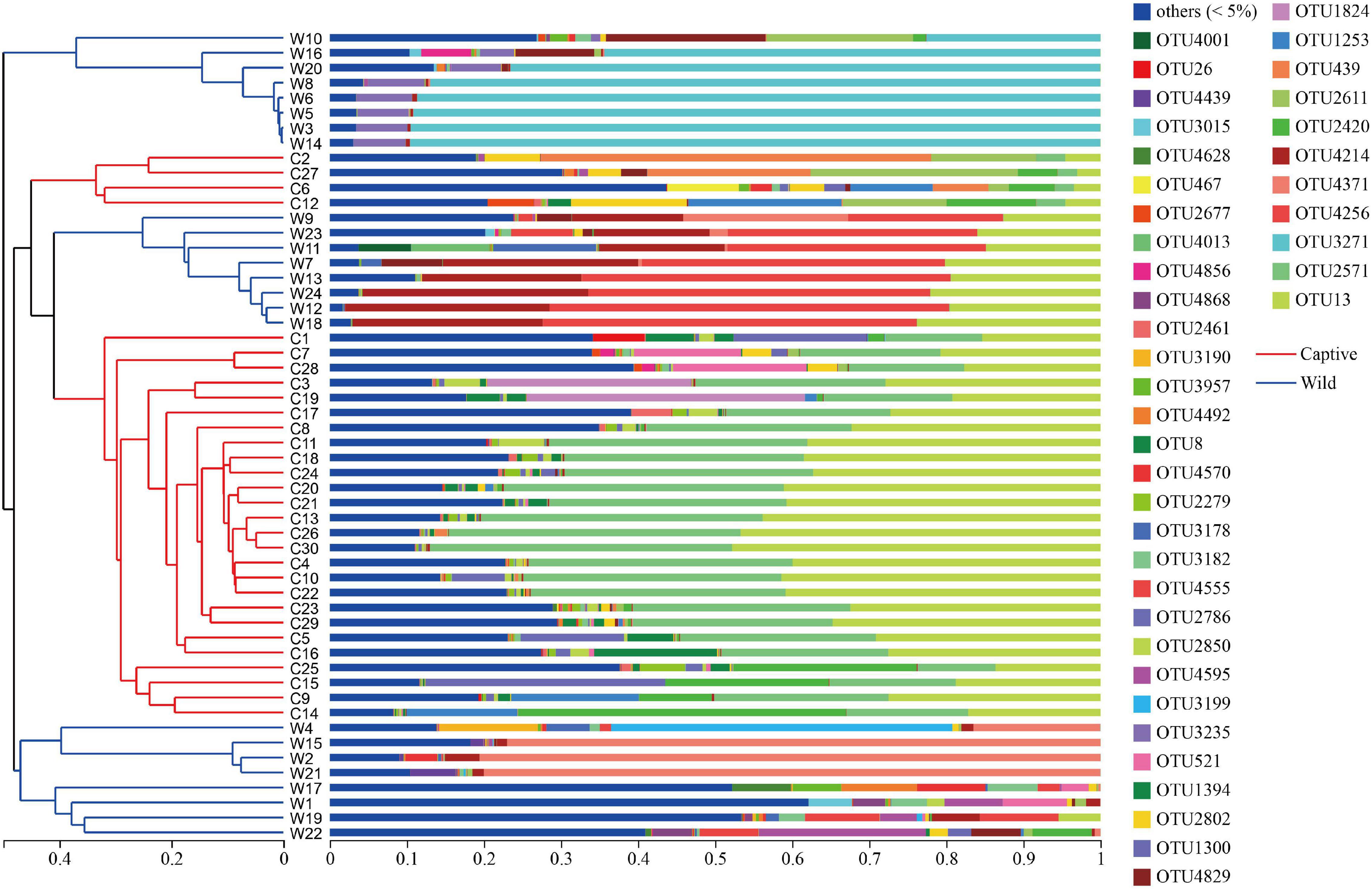
Figure 2. Hierarchical cluster analysis based on Bray–Curtis distance algorithm at the OTU level.
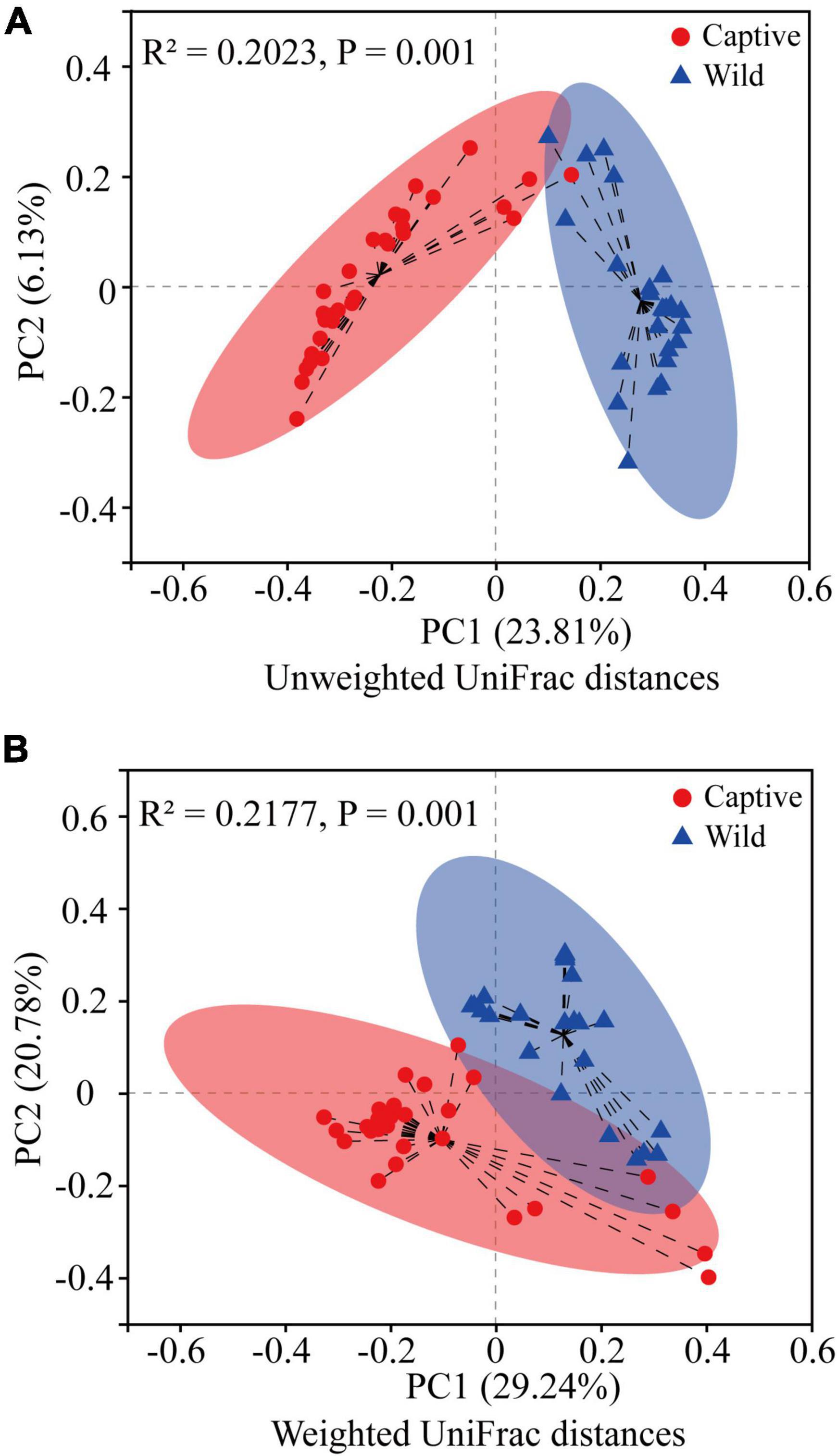
Figure 3. Difference in gut microbiota community structure between captive and wild geckos using Adonis test (permutation = 999). (A) A principal coordinate analysis of unweighted UniFrac distances; (B) A principal coordinate analysis of weighted UniFrac distances.
Differences in the Composition and Abundance of Gut Microbiota
We obtained 4,851 OTUs at 97% sequence similarity and classified them into 52 phyla, 153 classes, 353 orders, 586 families, and 1,320 genera. Of the 4,851 OTUs, 1,290 OTUs were shared by captive and wild geckos, whereas 2,130 OTUs and 1,431 OTUs were unique to captive geckos and wild geckos, respectively (Figure 4A). At the genus level, a total of 685 genera were shared by captive and wild geckos, with 358 genera specific to the captive group and 277 genera specific to the wild group (Figure 4B).
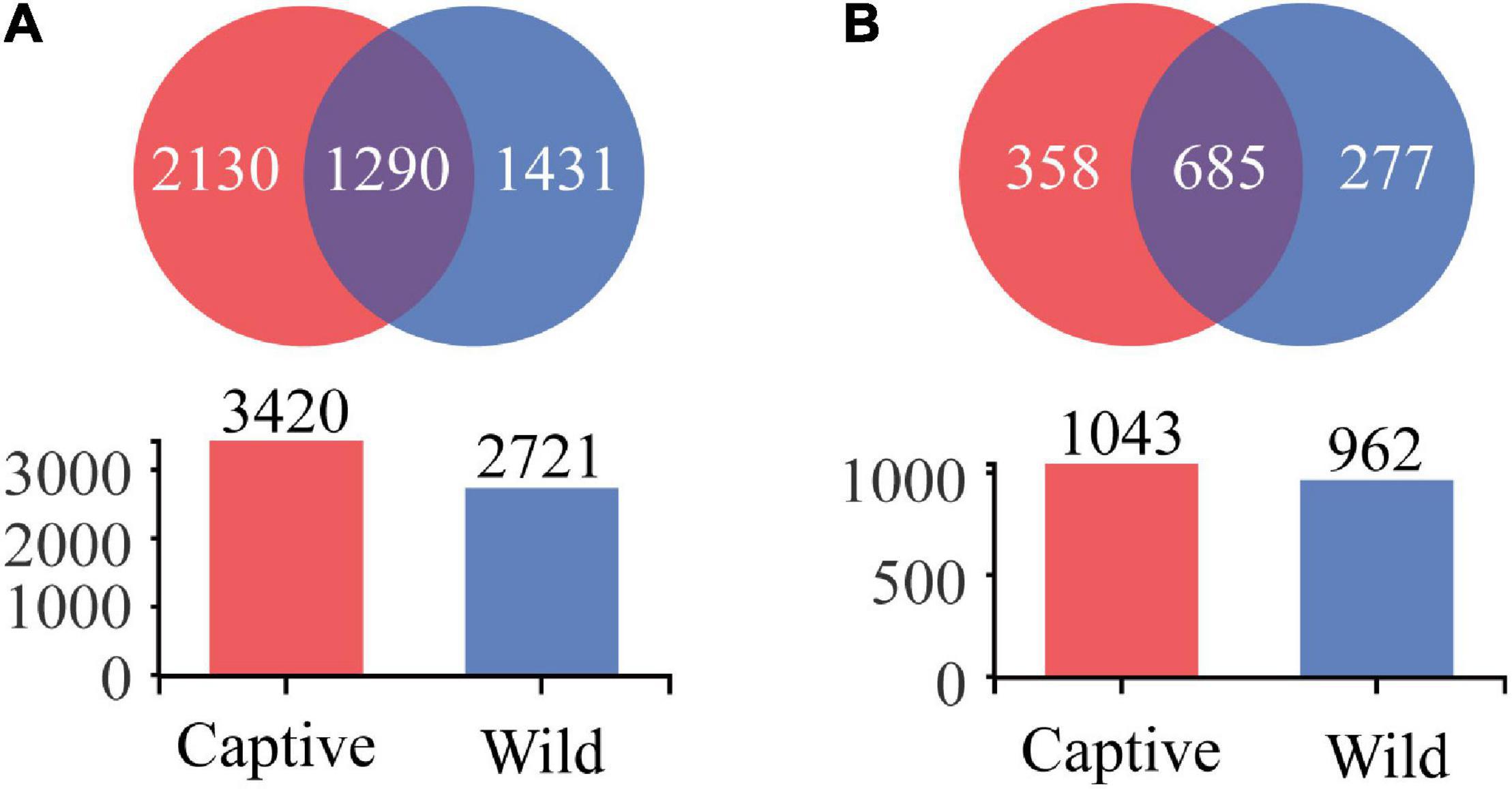
Figure 4. Venn diagram of gut microbiota in Tokay gecko at the OTU (A) and genus (B) level.
At the phylum level, Proteobacteria were the predominant phylum in two groups (captive: 63.88% ± 22.99%; wild: 50.67% ± 36.90%), followed by Firmicutes (captive: 15.29% ± 11.18%; wild: 31.45% ± 33.94%), Bacteroidetes (captive: 14.55% ± 22.11%; wild: 6.90% ± 9.90%), and Actinobacteria (captive: 3.51% ± 2.96%; wild: 8.87% ± 10.41%), respectively; Wilcoxon rank-sum test showed that the relative abundance of Acidobacteriota, Chloroflexi, Deferribacterota, and Patescibacteria was higher in the captive geckos than those in wild geckos (Figure 5A; Supplementary Table 3).
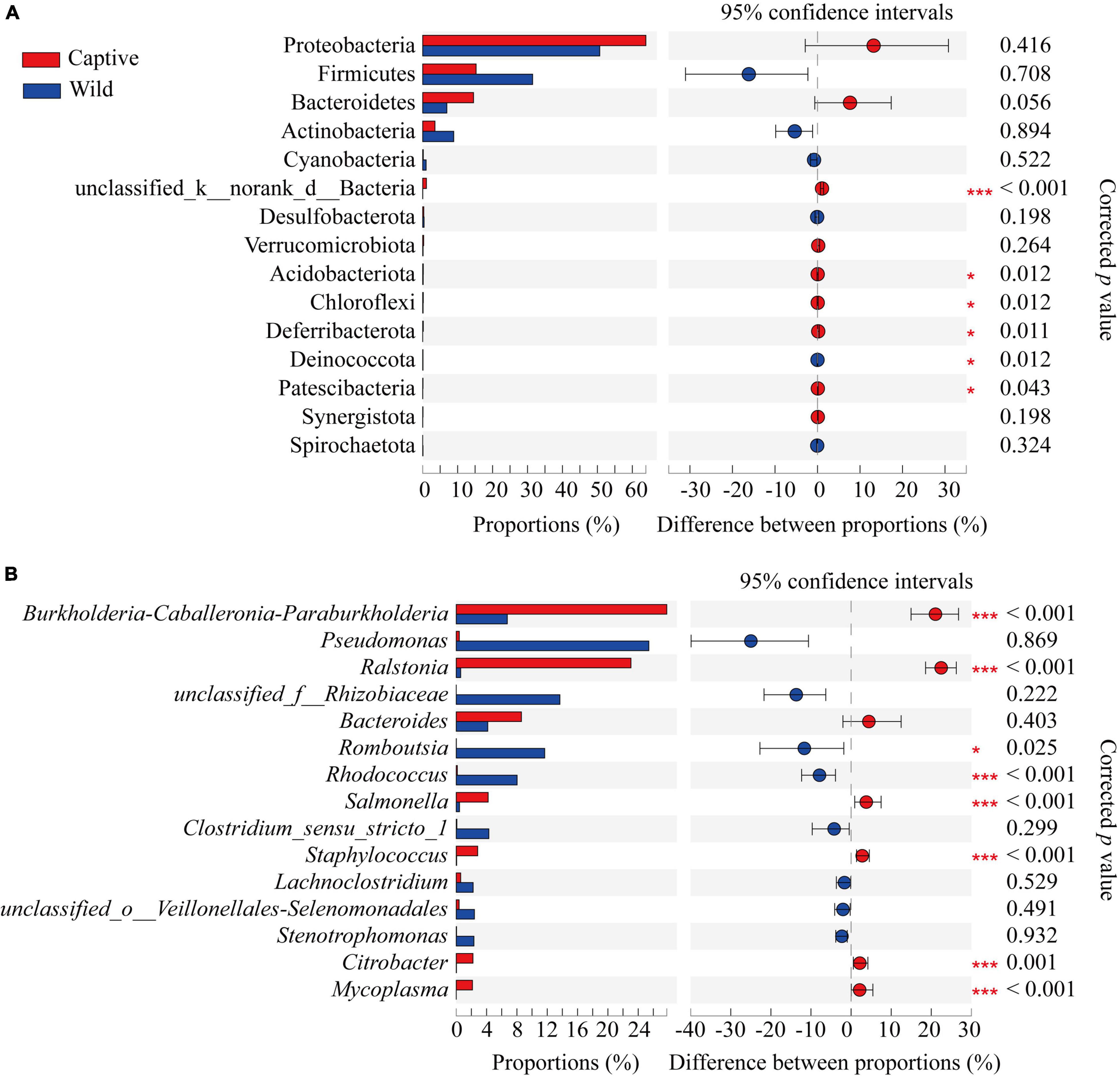
Figure 5. Differences in gut microbiota composition between captive and wild geckos at the phylum level (A) and genus level (B) of the top 15 taxa. Significant difference 0.01 < p ≤ 0.05 was marked as “*”, and p ≤ 0.001 was marked as “****”.
At the genus level, the main gut microbiota included Burkholderia-caballeronia-paraburkholderia (captive: 27.77% ± 13.56%; wild: 6.73% ± 8.94%), Ralstonia (captive: 23.05% ± 11.21%; wild: 0.59% ± 0.47%), Pseudomonas (captive: 0.41% ± 0.76%; wild: 25.40% ± 38.72%), and Bacteroides (captive 8.59% ± 18.71%; wild: 4.16% ± 6.38%); Wilcoxon rank-sum test showed that the relative abundance of Romboutsia and Rhodococcus was higher in wild geckos, and the relative proportion of Burkholderia-caballeronia-paraburkholderia, Ralstonia, Salmonella, Staphylococcus, Citrobacter, and Mycoplasma was higher in captive geckos (Figure 5B and Supplementary Table 4).
LEfSe analysis (LDA score > 4.0) showed that the order Burkholderiales, the family Burkholderiaceae, and the class Gammaproteobacteria were the most important taxa contributing to gut microbiota differences between captive and wild geckos (Figure 6).
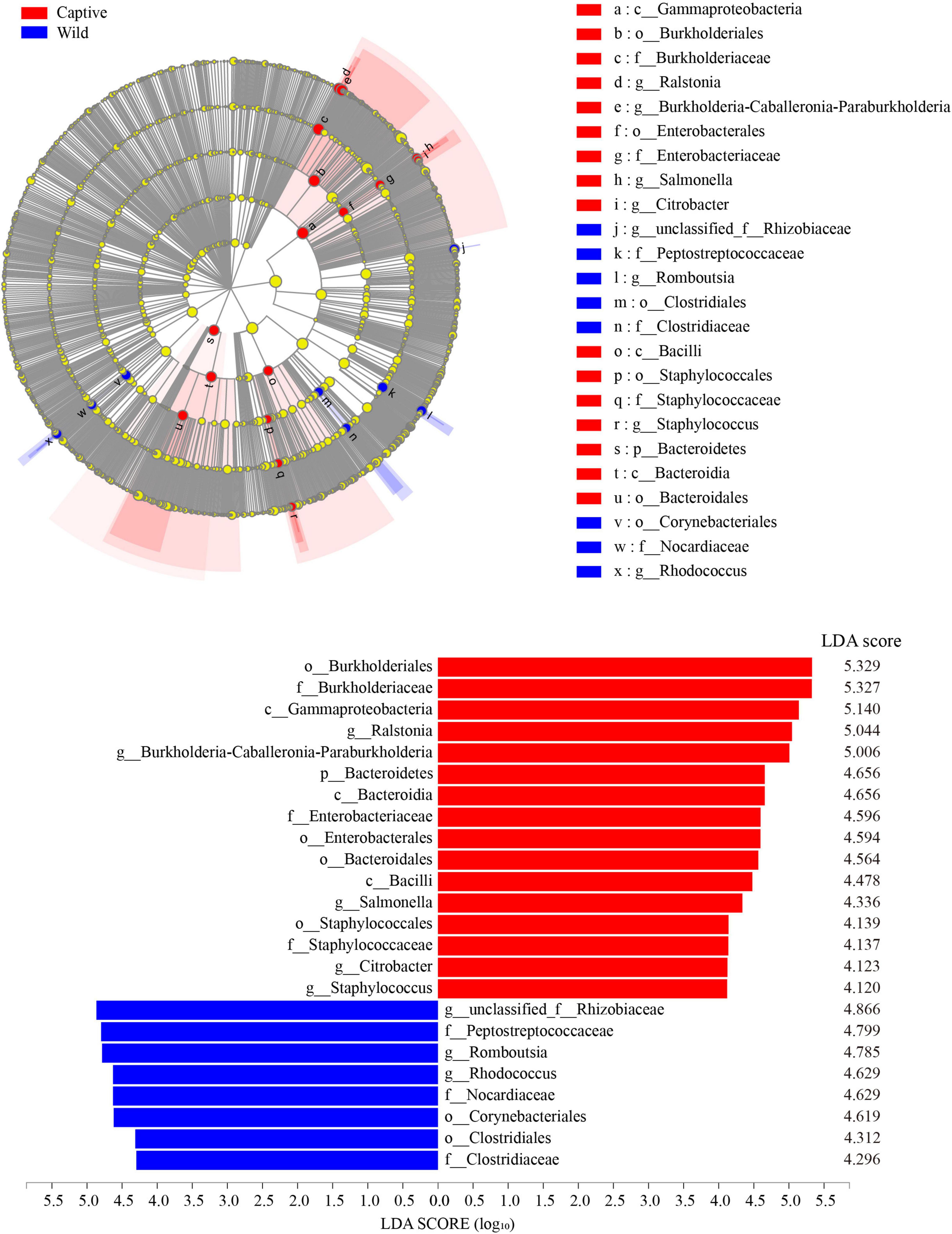
Figure 6. LEfSe (LDA Effect Size) analysis of the gut microbiota in Tokay gecko (LDA score > 4.0).
Gut Microbiota Functional Profile Prediction
Our results identified significant differences between two groups in three pathways at the KEGG pathway level 1 (Wilcoxon rank-sum test, p < 0.05), including those in metabolism, genetic information processing, and organismal systems, with a greater relative abundance in wild geckos than in captive geckos (Figure 7A). We detected 46 functional pathways at the KEGG pathway level 2, among which we identified significant differences in 27 pathways between captive and wild samples. The relative abundance of amino acid metabolism, energy metabolism and lipid metabolism in the gut microbiota of wild samples were higher than those of the captive population. However, we detected significant enrichment in global and overview maps, carbohydrate metabolism, and membrane transport in captive samples (Figure 7B).
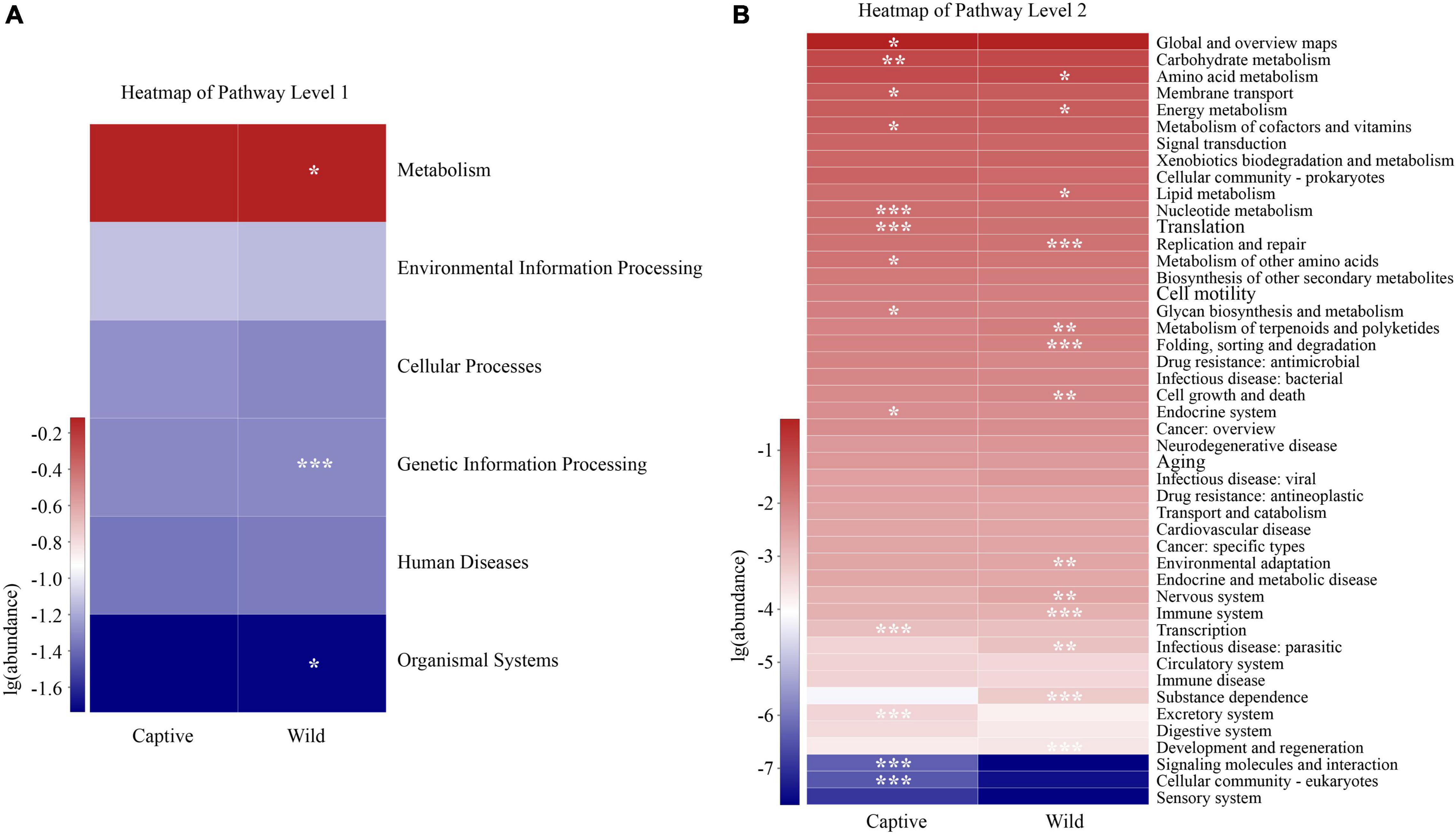
Figure 7. Differences in the functional profiles in level 1 (A) and level 2 (B) pathways of the gut microbiota in Tokay gecko. The p value was represented by asterisks. Significant difference 0.01 < p < 0.05 was marked as “*”, 0.001 < p < 0.01 was marked as “**”, and p < 0.001 was marked as “***”.
Effects of Total Length and Sex on Gut Microbiota of Tokay Gecko
Canonical correlation analysis results showed the first two axes explained 5.42% of total variance (CCA1: 4.04%; CCA2: 1.38%). Total length (R2 = 0.4527, p = 0.001) and sex (R2 = 0.3705, p = 0.001) significantly affected gut microbial community (Figure 8A). VPA demonstrated that total length of Tokay geckos explained 10.45% of the total variation for gut microbial community variance (Figure 8B). A partial Mantel test showed that total length was weakly and positively correlated with gut microbial community (r = 0.147, p = 0.019, conditioning on sex), whereas no significant correlation was observed between sex and gut microbial community (r = 0.004, p = 0.421, conditioning on total length). Spearman correlation showed that total length was significantly positively correlated with Burkholderia-Caballeronia-Paraburkholderia (r = 0.558, p < 0.001), Ralstonia (r = 0.478, p < 0.001), and Staphylococcus (r = 0.309, p = 0.023) (Supplementary Table 5).
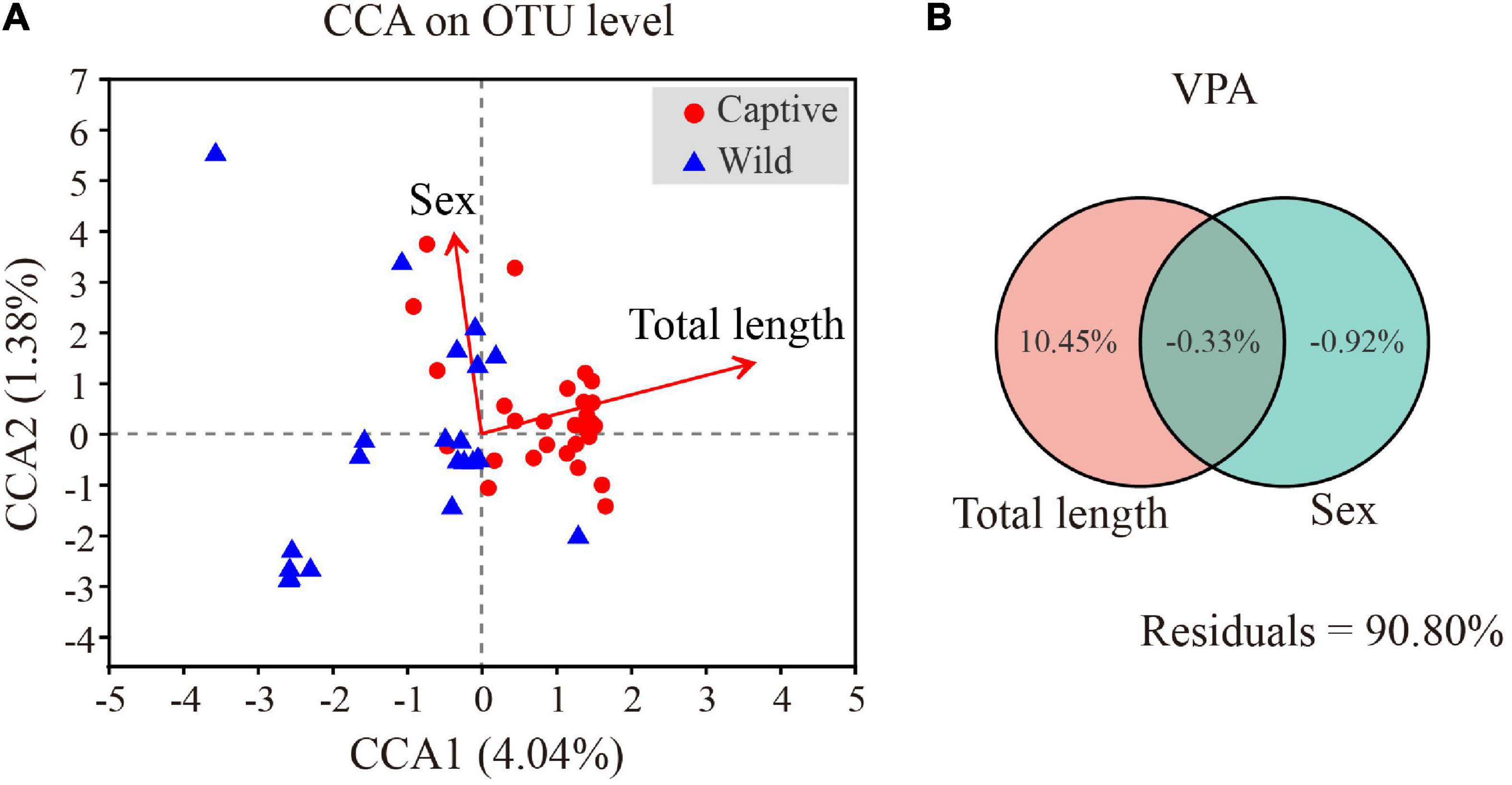
Figure 8. Effects of total length and sex on gut microbiota of Tokay gecko (A). Canonical correlation analysis of the microbial community on OTU level and physical parameters of Tokay gecko (B).
Discussion
In terms of microbial composition, Proteobacteria, Firmicutes and Bacteroidetes were the most dominant phyla in both captive and wild geckos, suggesting these microbes may play an important role in maintaining relative stability of gut microbiome. Other studies have also shown these microbes to be the main dominant phyla among vertebrate gut microbiota, including Moschus berezovskii and Moschus chrysogaster (Hu et al., 2017), Ciconia boyciana (Wu et al., 2021), Naja atra, Ptyas mucosus, Elaphe carinata, and Deinagkistrodon acutus (Zhang et al., 2019). However, we observed significant differences in microbial community structure between the captive and the wild geckos (Figures 2, 3). We also discovered a higher alpha diversity of gut microbiota in captive geckos than that in wild geckos, which was inconsistent with our prediction 1. Similar results have been found in several species, including Takifugu bimaculatus (Lei et al., 2020), Shinisaurus crocodilurus (Tang et al., 2020), Psittaciformes (Xenoulis et al., 2010), and Macaca mulatta (Chen et al., 2019). Moreover, captive geckos contained a larger total number of OTUs and a greater abundance of unique OTUs. The captive environment increased human contact, individual interaction, and drug use (McKenzie et al., 2017), which might allow a greater microbial diversity and more distinct taxa to colonize the intestinal tract of captive geckos. In addition, differences in dietary composition and quantities may also play an important role (De Filippo et al., 2010; Jiang et al., 2017). Captive geckos feed mainly on locusts, woodlouse worms and barley worms, whereas wild geckos eat mainly insects and moths (Liu et al., 1981; Tang et al., 1997). Moreover, captive geckos are fed adequate food at regular intervals, but wild geckos have erratic diets because of their need to search for natural food and the fluctuations in weather or habitat conditions. Therefore, although the gut microbiota of geckos inhabiting different living environments did not differ in their most dominant phyla, the diversity and structure of the microbial community differed significantly, indicating that the captive environment had an obvious influence on gecko gut microbiota.
At the genus level, we detected a higher relative abundance of Burkholderia-caballeronia-paraburkholderia, Ralstonia, and Staphylococcus in captive geckos. Many of the species in these genera have been confirmed to be pathogenic in humans and animals; for instance, Burkholderia pseudomallei was known to cause melioidoisis (Price et al., 2010); Ralstonia was proinflammatory during Parkinson’s disease (Keshavarzian et al., 2015). The higher abundance of opportunistic pathogens in captive geckos was consistent with our prediction 2. In captivity, both dietary shift, constant cohabitation with other congeners, limited range of activity, increased exposure to human-related microbes, and medicine intervention provide pathways for transmission of opportunistic pathogens, leading to differences in the gut microbial communities of captive and wild populations (Jiang et al., 2017; McKenzie et al., 2017). Nonetheless, the causes of differences in the gut microbiota between wild and captive geckos were unknown, since environmental microbes and dietary data were not collected. In addition, our result found total length was significantly positively correlated with these taxa (Supplementary Table 5). Thus, it is difficult to determine whether these bacteria, which are pathogenic to other animals, have an adverse effect on the geckos. More data are needed for further study.
Firmicutes and Bacteroidetes have an impact on the host’s metabolism and immune function mechanisms (Thomas et al., 2011; Zhang et al., 2019). Firmicutes can help to digest and absorb proteins and other nutrients (Kaakoush, 2015; Bernini et al., 2016; Berry, 2016). Most species of Bacteroidetes contribute to the degradation of carbohydrates and proteins (Fernando et al., 2010; Jami et al., 2014). The Firmicutes to Bacteroidetes (F/B) ratio has been studied in both humans and animals and appears to be related to host obesity (Ley et al., 2006; Turnbaugh et al., 2006; Vebo et al., 2016). An increased F/B ratio in gut microbiota indicates a greater energy harvesting capacity for hosts (Turnbaugh et al., 2006; Murphy et al., 2010). In our study, the F/B ratio in the gut microbiota of wild geckos was higher than that of captive geckos. While the diet of captive geckos includes Z. morio and T. molitor, with high protein and fat content, the diet of wild geckos remains unclear due to limiting conditions in the field. The higher F/B ratio in the wild geckos indicates gut microbiota more efficient at digesting food to help hosts obtain energy in wild populations—a favorable adaptive strategy for wild geckos that survive in harsh natural environments (Jami et al., 2014; Wu et al., 2021). Furthermore, the KEGG pathway analysis revealed that metabolism, genetic information processing, and organismal systems pathways were significantly enriched in wild geckos, providing further evidence that wild geckos may improve adaptability through the strong metabolic potential provided by their gut microbiota.
In summary, there were significant differences between captive and wild geckos in their respective gut microbiota community structures and diversity. Captive geckos had a higher diversity of gut microbiota, but more pathogenic bacteria, while wild geckos had a higher F/B ratio and more metabolism, genetic information processing, and organismal systems pathways. These differences are probably related to differences in living environments and diets of the two gecko populations. Our results could inform researchers in their efforts to further understand the relationship between the gut microbiota of geckos and their living environments and contribute to the comprehensive protection and management of this species in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject - PRJNA817415 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA817415).
Ethics Statement
The animal study was reviewed and approved by Laboratory Animal Care and Animal Ethics Committee of Guangxi Normal University.
Author Contributions
ZC and ZW contributed to conception and design of the study. SY and YTL organized the database. CH performed the statistical analysis. ST and YHL wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was financially supported by the National Natural Science Foundation of China (31760623 and 32160131), the Financial Project of State Forestry Administration (V2130211), Biodiversity Survey, Monitoring and Assessment Project of Ministry of Ecology and Environment, China (2019HB2096001006), and the Guangxi Natural Science Foundation, China (AD21220058).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are especially grateful to Huaxing Tang from Guangxi Chongzuo White-headed Langur Nature Reserve and Jiaxin Zhao and Rong Huang from Guangxi Encheng Nature Reserve for helping us to carry out the work in the study site.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.897923/full#supplementary-material
Footnotes
- ^ http://www.drive5.com/uparse/
- ^ http://sourceforge.net/projects/rdp-classifier/
- ^ https://www.arb-silva.de/
- ^ https://www.mothur.org/wiki/Download_mothur
- ^ http://qiime.org/install/index.html
- ^ http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload
- ^ https://github.com/picrust/picrust2/
References
Alfano, N., Courtiol, A., Vielgrader, H., Timms, P., Roca, A. L., and Greenwood, A. D. (2015). Variation in koala microbiomes within and between individuals: effect of body region and captivity status. Sci. Rep. 5:10189. doi: 10.1038/srep10189
Amato, K. R., Metcalf, J. L., Song, S. J., Hale, V. L., Clayton, J., Ackermann, G., et al. (2016). Using the gut microbiota as a novel tool for examining colobine primate GI health. Glob. Ecol. Conserv. 7, 225–237. doi: 10.1016/j.gecco.2016.06.004
Amato, K. R., Yeoman, C. J., Kent, A., Righini, N., Carbonero, F., Estrada, A., et al. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 7, 1344–1353. doi: 10.1038/ismej.2013.16
Bauer, A. M. (2009). Geckos in traditional medicine: forensic implications. Appl. Herpetol. 6, 81–96. doi: 10.1163/157075408x397509
Bengmark, S. (2013). Gut microbiota, immune development and function. Pharmacol. Res. 69, 87–113. doi: 10.1016/j.phrs.2012.09.002
Bernini, L. J., Simão, A. N. C., Alfieri, D. F., Lozovoy, M. A. B., Mari, N. L., de Souza, C. H., et al. (2016). Beneficial effects of Bifidobacterium lactis on lipid profile and cytokines in patients with metabolic syndrome: a randomized trial. Effects of probiotics on metabolic syndrome. Nutrition 32, 716–719. doi: 10.1016/j.nut.2015.11.001
Berry, D. (2016). The emerging view of Firmicutes as key fibre degraders in the human gut. Environ. Microbiol. 18, 2081–2083. doi: 10.1111/1462-2920.13225
Berry, D., Schwab, C., Milinovich, G., Reichert, J., Ben Mahfoudh, K., Decker, T., et al. (2012). Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 6, 2091–2106. doi: 10.1038/ismej.2012.39
Bisgaard, H., Li, N., Bonnelykke, K., Chawes, B. L. K., Skov, T., Paludan-Müller, G., et al. (2011). Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy. Clin. Immunol. 128, 646.e–652.e. doi: 10.1016/j.jaci.2011.04.060
Bletz, M. C., Goedbloed, D. J., Sanchez, E., Reinhardt, T., Tebbe, C. C., Bhuju, S., et al. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nat. Commun. 7:13699. doi: 10.1038/ncomms13699
Cani, P. D., and Everard, A. (2016). Talking microbes: when gut bacteria interact with diet and host organs. Mol. Nutr. Food. Res. 60, 58–66. doi: 10.1002/mnfr.201500406
Chen, T., Li, Y., Liang, J., Li, Y., and Huang, Z. (2019). Gut microbiota of provisioned and wild rhesus macaques (Macaca mulatta) living in a limestone forest in southwest Guangxi, China. MicrobiologyOpen 9:e981. doi: 10.1002/mbo3.981
Clark, R. I., and Walker, D. W. (2018). Role of gut microbiota in aging-related health decline: insights from invertebrate models. Cell. Mol. Life Sci. 75, 93–101. doi: 10.1007/s00018-017-2671-1
Clayton, J. B., Vangay, P., Huang, H., Ward, T., Hillmann, B. M., Al-Ghalith, G. A., et al. (2016). Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. U.S.A. 113, 10376–10381. doi: 10.1073/pnas.1521835113
Colston, T. J., Noonan, B. P., and Jackson, C. R. (2015). Phylogenetic Analysis of bacterial communities in different regions of the gastrointestinal tract of Agkistrodon piscivorus, the cottonmouth snake. PLoS One 10:e0128793. doi: 10.1371/journal.pone.0128793
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Edgar, R. C. (2013). Uparse: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Egert, M., de Graaf, A. A., Smidt, H., de Vos, W. M., and Venema, K. (2006). Beyond diversity: functional microbiomics of the human colon. Trends Microbiol. 14, 86–91. doi: 10.1016/j.tim.2005.12.007
Eliades, S. J., Brown, J. C., Colston, T. J., Fisher, R. N., Niukula, J. B., Gray, K., et al. (2021). Gut microbial ecology of the Critically Endangered Fijian crested iguana (Brachylophus vitiensis): effects of captivity status and host reintroduction on endogenous microbiomes. Ecol. Evol. 11, 4731–4743. doi: 10.1002/ece3.7373
Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science 338, 198–199. doi: 10.1126/science.1227412
Fernando, S. C., Purvis, H. T. I. I., Najar, F. Z., Sukharnikov, L. O., Krehbiel, C. R., Nagaraja, T. G., et al. (2010). Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microb. 76, 7482–7490. doi: 10.1128/AEM.00388-10
Flint, H. J., Duncan, S. H., Scott, K. P., and Louis, P. (2015). Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 74, 13–22. doi: 10.1017/s0029665114001463
Greer, R., Dong, X., Morgun, A., and Shulzhenko, N. (2016). Investigating a holobiont: microbiota perturbations and transkingdom networks. Gut Microb. 7, 126–135. doi: 10.1080/19490976.2015.1128625
Grond, K., Sandercock, B. K., Jumpponen, A., and Zeglin, L. H. (2018). The avian gut microbiota: community, physiology and function in wild birds. J. Avian. Biol. 49:e01788. doi: 10.1111/jav.01788
Heijtz, R. D., Wang, S., Anuar, F., Qian, Y., Björkholm, B., Samuelsson, A., et al. (2011). Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U.S.A. 108, 3047–3052. doi: 10.1073/pnas.1010529108
Hooper, L. V., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
Hu, X., Liu, G., Shafer, A. B. A., Wei, Y., Zhou, J., Lin, S., et al. (2017). Comparative analysis of the gut microbial communities in forest and alpine musk deer using high-throughput sequencing. Front. Microbiol. 8:572. doi: 10.3389/fmicb.2017.00572
Huang, C., Tang, Z., Li, H., and Yu, T. (1995). The preliminary research on statistic and ecological habits of Gekko gecko. J. Guangxi Norm. Univ. Nat. Sci. Ed. 13, 88–92.
Jami, E., White, B. A., and Mizrahi, I. (2014). Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS One 9:e85423. doi: 10.1371/journal.pone.0085423
Jiang, H., Ma, J., Li, J., Zhang, X., Li, L., He, N., et al. (2017). Diets alter the gut microbiome of crocodile lizards. Front. Microbiol. 8:2073. doi: 10.3389/fmicb.2017.02073
Kaakoush, N. O. (2015). Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 5:84. doi: 10.3389/fcimb.2015.00084
Keshavarzian, A., Green, S. J., Engen, P. A., Voigt, R. M., Naqib, A., Forsyth, C. B., et al. (2015). Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 30, 1351–1360. doi: 10.1002/mds.26307
Kohl, K. D., Brun, A., Magallanes, M., Brinkerhoff, J., Laspiur, A., Acosta, J. C., et al. (2016). Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 26, 1175–1189. doi: 10.1111/mec.13921
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
LeBlanc, J. G., Milani, C., de Giori, G. S., Sesma, F., van Sinderen, D., and Ventura, M. (2013). Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr. Opin. Biotech. 24, 160–168. doi: 10.1016/j.copbio.2012.08.005
Lei, Y., Wang, S., Chen, Y., Jiang, Y., and Zheng, Y. (2020). Microbial diversity in intestinal tract of wild and cultured puffer Takifugubimaculatus based on 16S rRNA gene sequence. Fish. Sci. 39, 579–584. doi: 10.16378/j.cnki.1003-1111.2020.04.016
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Li, H., Tang, Z., Yu, T., Lin, C., and Huang, C. (1996). Resources and protect of Gekko gecko in Guangxi. J. Guangxi Norm. Univ. Nat. Sci. Ed. 2, 64–68.
Li, Y., Chen, T., Li, Y., Tang, Y., and Huang, Z. (2021). Gut microbiota are associated with sex and age of host: evidence from semi-provisioned rhesus macaques in southwest Guangxi, China. Ecol. Evol. 11, 8096–8122. doi: 10.1002/ece3.7643
Lin, M., Zeng, C., Li, Z., Ma, Y., and Jia, X. (2019). Comparative analysis of the composition and function of fecal-gut bacteria in captive juvenile Crocodylus siamensis between healthy and anorexic individuals. MicrobiologyOpen 8:e929. doi: 10.1002/mbo3.929
Liu, S., Tang, D., and Liang, C. (1981). Feeding habits of large Gecko, Gekko gecko (Linnaeus). J. Jinan Univ. 4, 77–83.
Martínez-Mota, R., Kohl, K. D., Orr, T. J., and Dearing, M. D. (2020). Natural diets promote retention of the native gut microbiota in captive rodents. ISME J. 14, 67–78. doi: 10.1038/s41396-019-0497-6
McCord, A. I., Chapman, C. A., Weny, G., Tumukunde, A., Hyeroba, D., Klotz, K., et al. (2014). Fecal microbiomes of non-human primates in Western Uganda reveal species-specific communities largely resistant to habitat perturbation. Am. J. Primatol. 76, 347–354. doi: 10.1002/ajp.22238
McKenzie, V. J., Song, S. J., Delsuc, F., Prest, T. L., Oliverio, A. M., Korpita, T. M., et al. (2017). The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 57, 690–704. doi: 10.1093/icb/icx090
Moeller, A. H., Peeters, M., Ndjango, J.-B., Li, Y., Hahn, B. H., and Ochman, H. (2013). Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genom. Res. 23, 1715–1720. doi: 10.1101/gr.154773.113
Montalban-Arques, A., De Schryver, P., Bossier, P., Gorkiewicz, G., Mulero, V., Gatlin, D. M. I. I. I., et al. (2015). Selective manipulation of the gut microbiota improves immune status in vertebrates. Front. Immunol. 6:512. doi: 10.3389/fimmu.2015.00512
Mori, H., Maruyama, F., Kato, H., Toyoda, A., Dozono, A., Ohtsubo, Y., et al. (2014). Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 21, 217–227. doi: 10.1093/dnares/dst052
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
Murphy, E. F., Cotter, P. D., Healy, S., Marques, T. M., O’Sullivan, O., Fouhy, F., et al. (2010). Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59, 1635–1642. doi: 10.1136/gut.2010.215665
Nelson, T. M., Rogers, T. L., Carlini, A. R., and Brown, M. V. (2013). Diet and phylogeny shape the gut microbiota of Antarctic seals: a comparison of wild and captive animals. Environ. Microbiol. 15, 1132–1145. doi: 10.1111/1462-2920.12022
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Wei, J., et al. (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Price, E. P., Hornstra, H. M., Limmathurotsakul, D., Max, T. L., Sarovich, D. S., Vogler, A. J., et al. (2010). Within-host evolution of Burkholderia pseudomallei in four cases of acute melioidosis. PLoS Pathog. 6:e1000725. doi: 10.1371/journal.ppat.1000725
Ryu, H., Lu, J., Vogel, J., Elk, M., Chávez-Ramírez, F., Ashbolt, N., et al. (2012). Development and evaluation of a quantitative PCR assay targeting sandhill crane (Grus canadensis) fecal pollution. Appl. Environ. Microb. 78, 4338–4345. doi: 10.1128/AEM.07923-11
Schmidt, E., Mykytczuk, N., and Schulte-Hostedde, A. I. (2019). Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 13, 1293–1305. doi: 10.1038/s41396-019-0345-8
Semova, I., Carten, J. D., Stombaugh, J., Mackey, L. C., Knight, B., Farber, S. A., et al. (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microb. 12, 277–288. doi: 10.1016/j.chom.2012.08.003
Shin, N.-R., Whon, T. W., and Bae, J.-W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Sobhani, I., Tap, J., Roudot-Thoraval, F., Roperch, J. P., Letulle, S., Langella, P., et al. (2011). Microbial dysbiosis in Colorectal Cancer (CRC) patients. PLoS One 6:e16393. doi: 10.1371/journal.pone.0016393
Sommer, F., and Bäckhed, F. (2013). The gut microbiota — masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Song, C., Wang, B., Tan, J., Zhu, L., Lou, D., and Cen, X. (2017). Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol. Genet. Genom. 292, 407–414. doi: 10.1007/s00438-016-1282-0
Tang, G., Liang, X., Yang, M., Wang, T., Chen, J., Du, W., et al. (2020). Captivity influences gut microbiota in crocodile lizards (Shinisaurus crocodilurus). Front. Microbiol. 11:550. doi: 10.3389/fmicb.2020.00550
Tang, Z., Li, H., Chen, M., Huang, C., and Yu, T. (1997). An investigation on the ecology and geographic distribution of Gekko gecko in Guangxi. Guangxi Sci. 4, 259–263.
Thomas, F., Hehemann, J.-H., Rebuffet, E., Czjzek, M., and Michel, G. (2011). Environmental and gut Bacteroidetes: the food connection. Front. Microbiol. 2:93. doi: 10.3389/fmicb.2011.00093
Tremaroli, V., and Bäckhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Trevelline, B. K., MacLeod, K. J., Langkilde, T., and Kohl, K. D. (2019). Gestation alters the gut microbiota of an oviparous lizard. FEMS Microbiol. Ecol. 95:fiz086. doi: 10.1093/femsec/fiz086
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1131. doi: 10.1038/nature05414
Vaarala, O. (2011). The gut as a regulator of early inflammation in type 1 diabetes. Curr. Opin. Endocrinol. 18, 241–247. doi: 10.1097/MED.0b013e3283488218
Vebo, H. C., Karlsson, M. K., Avershina, E., Finnby, L., and Rudi, K. (2016). Bead-beating artefacts in the Bacteroidetes to Firmicutes ratio of the human stool metagenome. J. Microbiol. Meth. 129, 78–80. doi: 10.1016/j.mimet.2016.08.005
Waite, D. W., Deines, P., and Taylor, M. W. (2012). Gut microbiome of the critically endangered New Zealand parrot, the kakapo (Strigops habroptilus). PLoS One 7:e35803. doi: 10.1371/journal.pone.0035803
Wu, G. D., Chen, J., Hoffmann, C., Bittinger, K., Chen, Y., Keilbaugh, S. A., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. doi: 10.1126/science.1208344
Wu, H., Wu, F., Zhou, Q., and Zhao, D. (2021). Comparative analysis of gut microbiota in captive and wild Oriental White storks: implications for conservation biology. Front. Microbiol. 12:649466. doi: 10.3389/fmicb.2021.649466
Xenoulis, P. G., Gray, P. L., Brightsmith, D., Palculict, B., Hoppes, S., Steiner, J. M., et al. (2010). Molecular characterization of the cloacal microbiota of wild and captive parrots. Vet. Microbiol. 146, 320–325. doi: 10.1016/j.vetmic.2010.05.024
Xie, Y., Xia, P., Wang, H., Yu, H., Giesy, J. P., Zhang, Y., et al. (2016). Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 6:33350. doi: 10.1038/srep33350
Zhang, B., Ren, J., Yang, D., Liu, S., and Gong, X. (2019). Comparative analysis and characterization of the gut microbiota of four farmed snakes from southern China. Peer J. 7:e6658. doi: 10.7717/peerj.6658
Zhang, Y., Li, L., Li, N., Xu, Y., Zhao, C., Gu, Y., et al. (2015). Geographical variation color spots and tubercles of tokay gecko (Gekko gecko). Ecol. Sci. 34, 53–58.
Keywords: Gekko gecko, captive population, 16S rRNA, gut microbiota, wildlife, conservation, microbial community diversity
Citation: Tang S, Li Y, Huang C, Yan S, Li Y, Chen Z and Wu Z (2022) Comparison of Gut Microbiota Diversity Between Captive and Wild Tokay Gecko (Gekko gecko). Front. Microbiol. 13:897923. doi: 10.3389/fmicb.2022.897923
Received: 16 March 2022; Accepted: 11 May 2022;
Published: 17 June 2022.
Edited by:
Lifeng Zhu, Nanjing Normal University, ChinaReviewed by:
Wei Zhu, Chengdu Institute of Biology, ChinaHonghai Zhang, Qufu Normal University, China
Copyright © 2022 Tang, Li, Huang, Yan, Li, Chen and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengjun Wu, d3VfemhlbmdqdW5AYWxpeXVuLmNvbQ==; Zening Chen, Y2hlbnpuQGd4bnUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship