 Domonkos Sváb
Domonkos Sváb Linda Falgenhauer
Linda Falgenhauer Tünde Mag3
Tünde Mag3 Trinad Chakraborty
Trinad Chakraborty István Tóth
István Tóth- 1Veterinary Medical Research Institute, Budapest, Hungary
- 2Institute of Hygiene and Environmental Medicine and German Center for Infection Research (DZIF), Partner Site Giessen-Marburg-Langen, Justus Liebig University Giessen, Giessen, Germany
- 3National Public Health Center, Budapest, Hungary
- 4Institute of Medical Microbiology, German Center for Infection Research (DZIF), Partner Site Giessen-Marburg-Langen, Justus Liebig University Giessen, Giessen, Germany
Escherichia coli belonging to the enterohemorrhagic (EHEC), Shiga toxin-producing (STEC) and atypical enteropathogenic (aEPEC) pathotypes are significant foodborne zoonotic pathogens posing serious health risks, with healthy cattle as their main reservoir. A representative sampling of Hungarian cattle farms during 2017–2018 yielded a prevalence of 6.5 and 5.8% for STEC and aEPEC out of 309 samples. The draft genomes of twelve STEC (of them 9 EHEC) and four aEPEC of bovine origin were determined. For comparative purposes, we also included 3 EHEC and 2 aEPEC strains of human origin, as well four commensal isolates and one extraintestinal pathogenic E. coli (ExPEC) obtained from animals in a final set of 26 strains for a WGS-based analysis. Apart from key virulence genes, these isolates harbored several additional virulence genes with arrays characteristic for the site of isolation. The most frequent insertion site of Shiga toxin (stx) encoding prophages was yehV for the Stx1 prophage and wrbA and sbcB for Stx2. For O157:H7 strains, the locus of enterocyte effacement pathogenicity island was present at the selC site, with integration at pheV for other serotypes, and pheU in the case of O26:H11 strains. Several LEE-negative STEC and aEPEC as well as commensal isolates carried additional prophages, with an average of ten prophage regions per isolate. Comparative phylogenomic analysis showed no clear separation between bovine and human lineages among the isolates characterized in the current study. Similarities in virulence gene arrays and close phylogenetic relations of bovine and human isolates underline the zoonotic potential of bovine aEPEC and STEC and emphasize the need for frequent monitoring of these pathogens in livestock.
Introduction
Shiga toxin-producing Escherichia coli (STEC) are an intestinal pathotype of E. coli, currently comprising more than 400 serotypes (Blanco et al., 2004) their common feature being the production of Shiga toxin (also called Vero-cytotoxin, Stx), a potent cytotoxin inhibiting protein synthesis in the affected cell lines. A highly virulent subset of STEC strains termed enterohemorrhagic E. coli (EHEC) produce intimin apart from Stx, an adhesin that is responsible for attaching/effacing lesion on intestinal epithelial cells (Kaper et al., 2004). In addition to hemorrhagic colitis, EHEC cause a severe systemic complication called hemolytic-uremic syndrome (HUS), which results in acute renal failure in affected patients (Bielaszewska and Karch, 2005).
The primary reservoir of EHEC isolates is cattle, with isolates being present in the feces of the animals (Newell and Ragione, 2018). Human infection can result from direct contact with the animals or with their feces but occurs more frequently as a result of the consumption of undercooked meat, or of fresh vegetables, drinking water or meat contaminated with cattle feces (Chekabab et al., 2013; Terajima et al., 2017). The domestic animal reservoir, the severity of the disease, and low infective dose (<100 cells/person, Pulz et al., 2003), make EHEC serious and deadly zoonotic and foodborne pathogens. Enteropathogenic E. coli (EPEC) is the first described pathotype of E. coli and closely related to EHEC, responsible for a high proportion of children’s diarrhea mainly in countries with poor infrastructures (Ochoa and Contreras, 2011). Its key virulence factor is intimin, but unlike EHEC they do not produce Stx (Croxen et al., 2013). A subset of EPEC termed atypical EPEC (aEPEC) which are isolated from both humans and animals, do not carry the gene encoding the bundle-forming pilus (bfp). These isolates are genetically more closely related to EHEC, and it was recently suggested that subsets of aEPEC strains are possible progenitors of EHEC strains (Ferdous et al., 2015; Eichhorn et al., 2018).
An important feature of STEC, EHEC and EPEC is that their major virulence factors are carried on mobile genetic elements (MGE). Genes encoding stx are generally carried by lambdoid prophages, which are frequently inducible and transmissible (Ohnishi et al., 2002; Asadulghani et al., 2009; Rahman et al., 2018). In the case of both EHEC and EPEC, the eae gene encoding intimin is part of a 35–43 kb long pathogenicity island (PAI) termed locus of enterocyte effacement (LEE; reviewed by Jores et al., 2004), which carries genes encoding the type III secretion system, as well as effector proteins including translocated intimin receptor (tir) gene.
Highly virulent EHEC strains comprise members of the O157:H7 serotype (Hayashi, 2001; Perna et al., 2001; Paletta et al., 2020). Monitoring cattle for the presence of these strains has become a necessity, in order to understand the factors and risks associated with the transmission of EHEC O157 strains from animals to humans, as well as for epidemiological tracing.
The availability of affordable whole-genome sequencing (WGS) enables rapid assessment of genetic information encoded by a large set of strains, shedding light on of yet unknown genetic variants, additional virulence genes, and associated MGEs (Kulasekara et al., 2009). Genomic studies characterizing E. coli O157 strains revealed a remarkable genetic diversity of the serogroup (Ogura et al., 2006; Eppinger et al., 2011) especially regarding their prophage content (Shaaban et al., 2016; Sharma et al., 2019). A WGS-based study revealed that a specific subset of strains belonging to the serogroup, are frequently carried by cattle and responsible for the serious human outbreaks (Lupolova et al., 2016). It is therefore necessary to investigate cattle herds for the presence of these highly virulent strains and use the genome-based information to explore their genetic variability and assess their zoonotic potential. As the example of the recent outbreak caused by O80:H2 strains representing hybrid pathotype of STEC and ExPEC has shown (Nüesch-Inderbinen et al., 2018), the emergence of new, highly virulent pathotypes or clones is an ever-present possibility.
A previous explorative sampling of Hungarian cattle farms, which provided the first information on the prevalence of pathogenic strains of the O157 serogroup among cattle herds in Hungary used comprehensive PCR-based virulence profiling, and a classification based on pulsed-field gel electrophoresis (PFGE) together with multi-locus sequence typing (MLST) profiling of the isolated strains (Tóth et al., 2009). In addition to EHEC O157:H7, EPEC isolates of the same serotype, as well as those with other H types and atypical virulence gene repertoires are also present in cattle (Sváb et al., 2016).
The main aim in this study was to obtain further information regarding the prevalence of potentially zoonotic E. coli on Hungarian cattle farms, with a focus on the O157 serogroup and the STEC, EHEC and EPEC pathotypes. Using a WGS-based approach, we aimed to reveal potentially new genetic variants, virulence gene sets and MGEs, as well as to compare these data obtained from cattle to those of strains isolated in the earlier study together with those originating from human patients. In addition to assessing the occurrence of strains with high zoonotic potential, we also aimed to reveal potentially new genotypes, comprehensively map their MGEs, as well as their integration sites in the respective genomes.
Materials and Methods
Sample Collection and Bacterial Strains
Samples were collected from cattle farms throughout Hungary, both from the feces and milk of animals, as well as samples representing the environment, collected from the barn floor at selected farms. A total of 309 samples, comprising of fecal samples (n = 215), and milk samples (n = 81) from 215 healthy cattle, along with 13 samples from the farm environment were taken. Sampling was performed on 18 cattle farms representing different regions in Hungary involved in both dairy and meat production between 2017 and 2018. Samples were either processed immediately or stored at −70°C until required.
Strains were isolated using the ISO protocol 16,654:2001 recommended for the isolation of E. coli serotype O157 strains (Standard British and BSEN ISO, 2001). Briefly, following pre-enrichment in tryptone-soy broth (TSB) supplemented with bile salts, samples underwent immunomagnetic separation (IMS) with DynaBeads anti-E. coli O157 kit (Applied Biosystems) according to the manufacturer’s instruction. Samples were spread on sorbitol MacConkey (SMAC) agar plates, with cefixime as an additive selective agent.
From each sample, four sorbitol non fermenting (SNF) and one sorbitol-fermenting (SF) colony were picked from the SMAC agar plates and cultured in non-selective Luria–Bertani (LB) broth in 96-well plates. Every colony, which was positive for either of the key virulence genes, was stored and handled as a separate isolate in LB broth supplemented with 30% glycerol at −70°C.
Additionally, for comparative genomic purposes, we also included five STEC and EPEC strains isolated from human patients in 2018. One bovine EHEC and three bovine EPEC strains isolated by Tóth et al. (2009) were included in the sequencing as well.
Strains selected for further studies were grown overnight in LB broth, then the culture was mixed with 30% volume of sterile glycerol and stored at −70°C.
Detection of Key Virulence Genes
To detect the presence of STEC and EPEC among isolates, PCR screening of stx (Scheutz et al., 2012) and eae (China et al., 1996) genes was performed for all isolates, from which those for sequencing based were selected based on detected virulence profile.
Serotyping of Selected Isolates
Preliminary detection of the O157 antigen was performed with Oxoid latex agglutination test according to the manufacturer’s instruction. In all other cases of strains selected for genome sequencing serotyping was performed with O-specific and H-specific antisera following standard protocols (Ørskov and Ørskov, 1984). After obtaining whole-genome sequences, in silico serotyping was performed using SerotypeFinder 2.0 (Joensen et al., 2015).
Genome Sequencing
Seventeen bovine strains isolated in the current study were chosen for whole-genome sequencing (WGS). For comparative purposes, we also sequenced one EHEC O157:H7 as well as three EPEC strains isolated in the earlier study of Tóth et al. (2009), as well as two EPEC and three STEC strains of human origin, thus altogether 26 WGS were obtained. Isolates were chosen for WGS to represent not only different samples and sampling sites, but also distinctive arrays of key virulence genes. Strains containing either only one stx gene, or both stx1 and stx2 were selected as well. Genomic DNA of the strains was isolated with the ZymoResearch Quick-DNA Fungal/Bacterial Miniprep Kit according to the manufacturer’s instructions. Sequencing was performed on an Illumina NextSeq system as follows. Sequencing libraries were generated using Illumina Nextera XT Kit (Cat.Num.: FC-131-1096, Illumina, Eindhoven, Netherlands) as per manufacturer’s instructions. DNA sequencing was performed on an Illumina NextSeq 500 machine (Illumina, Eindhoven, Netherlands) with 2x150nt read length chemistry (mid-output/high output flow cells). Sequencing quality assessment and assembly was performed using ASA3P (Schwengers et al., 2020), including the SPAdes Software for assembly (Bankevich et al., 2012). The average read length was 122 nt and the coverage ranged from 62.14x to 171. 88x.The genomes were assembled into contigs ranging from 50 to 1947 in number (Table 1). All sequence data are available in GenBank through Bioproject no. PRJNA764596, in BioSample IDs SAMN21509413-SAMN21509438 and Sequence Read Archive nos. SRR15970245-SRR15970246.
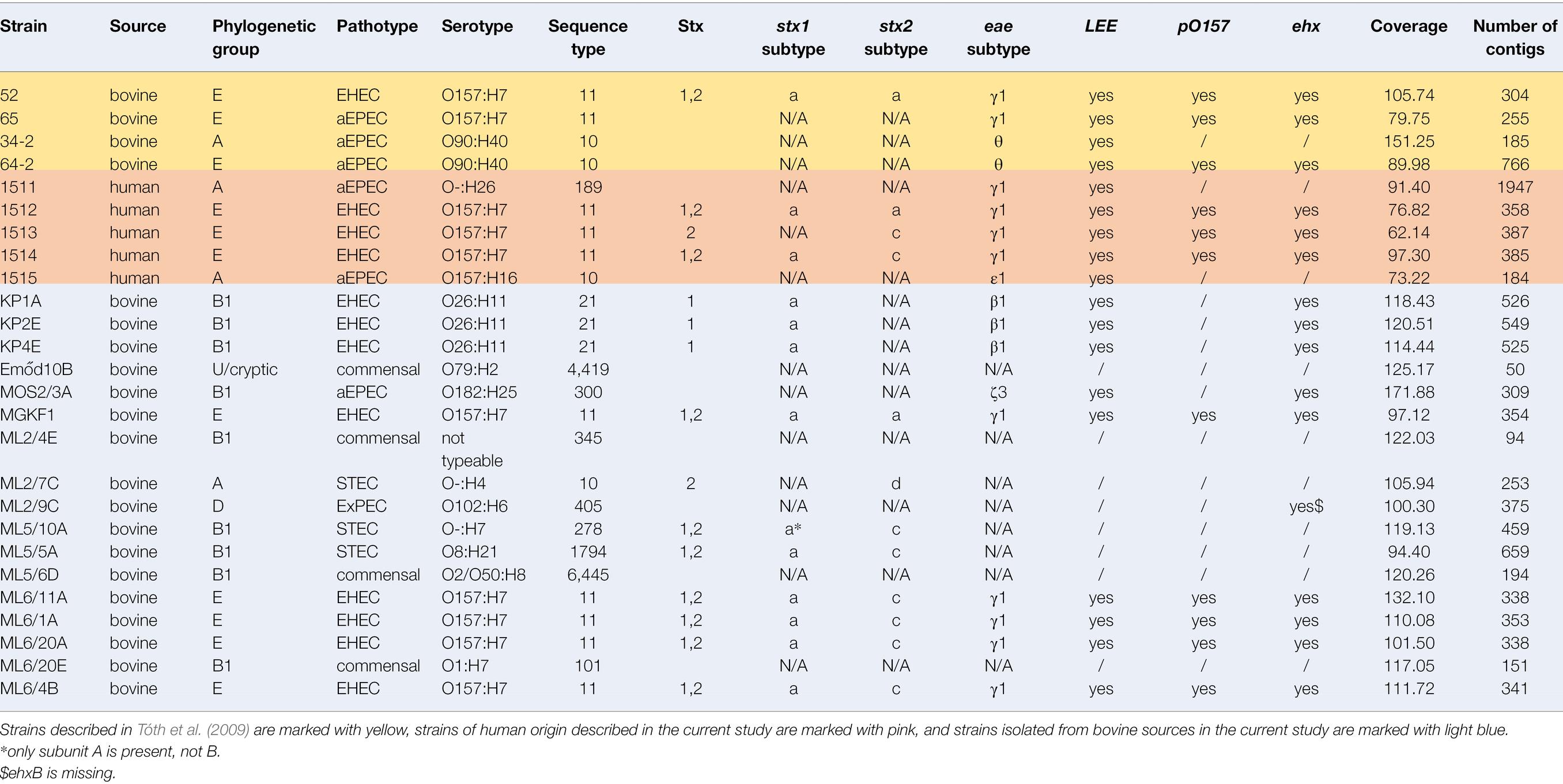
Table 1. Summary of sequencing results for strains with their whole-genome sequenced in this study.
Virulence, Antibiotic Resistance Gene and Prophage Searches
Virulence gene search was performed using VirulenceFinder (Joensen et al., 2015) and Virulence Factor Database (VFDB, Liu et al., 2022) antimicrobial resistance genes were searched for using ResFinder (Zankari et al., 2012), and prophage regions were searched by PHASTER (Arndt et al., 2016). Subtypes of intimin genes (eae) were determined using tblastN and the references depicted by Yang et al. (2020). The presence of the pO157 plasmid (accession number AB011549.2) was determined using blastN.
The intact or disrupted state of the characteristic stx prophage and LEE integration sites listed by Bonanno et al. (2015) and Saile et al. (2018), respectively, was checked by blastN of the respective empty integration sites. The flanking regions of occupied integration sites were analyzed using PHASTER to determine the nature of the respective phage or LEE island (e.g., whether this was a putative stx-encoding prophage or not).
Phylogenetic Grouping and MLST Analysis
Determination of the phylogenetic group was performed in silico using the online software tool “In Silico Clermont Phylotyper”1 based on the protocol of Clermont et al. (2013) that targets several housekeeping genes and additional regions. MLST was performed according to the Wirth scheme utilizing the nucleotide sequence of seven housekeeping genes (Wirth et al., 2006). A core-genome-based maximum likelihood phylogenetic tree was constructed with ParSNP implemented in the HarvestSuite program package (Treangen et al., 2014) using the genome set published by Arimizu et al. (2019). The full dataset is listed in Supplementary Table 1.
Results
Prevalence of Pathotypes Among the Isolates
To reveal the incidence of STEC and EPEC in Hungarian cattle herds, we sampled 215 animals from 18 cattle farms throughout Hungary, taking fecal samples from all animals, and milk samples from a total of 81 animals, as well as 13 environmental samples. A collection of 184 E. coli strains isolated from bovine sources during 2013–2017 was also examined.
The stx genes were detected in 20 out of 309 samples (6.5%) and from the stx-positive samples a total of 47 STEC isolates were identified, as multiple colonies were stored from each sample. Out of these, 18 also carried the eae gene, and they were classified as EHEC; these EHEC isolates represented nine samples altogether. A further 18 samples (5.8%) contained isolates carrying eae without stx, and altogether 44 such genotype isolates, classified as EPEC, were identified.
Draft genomes from a total of 12 bovine STEC, (out of them 9 EHEC), 4 bovine EPEC, 3 human EHEC, and 2 human EPEC strains, as well as 5 stx-, eae- E. coli strains labeled as “commensal” were analyzed. The bovine strains represented strains isolated in the current study as well as those from the earlier collection by Tóth et al. (2009). Sequencing data revealed that in addition to the O157:H7 strains 34 and 64 characterized in that study, the original samples contained further E. coli strains, named 34-2 and 64-2, respectively. In the present study, these novel isolates were characterized. Two human and one bovine isolate was negative for stx genes, that had previously been detected by PCR, but as they were eae-positive, they were reclassified as EPEC. In light of its sequence type and virulence gene array detailed below, one of the commensal strains was reclassified as ExPEC (Table 1).
The sizes of the genomes ranged from 4,595,164 to 5,962,783 bp, their GC content ranged from 50 to 51%.
Sequence Types, Phylogenetic Relations and Serotypes
To reveal the phylogenetic relationship of the isolates the main phylogenetic groups and the sequence types (STs) were determined (Table 1). Twelve different STs were detected. The most common ST was ST11 (n = 10), followed by ST10 (n = 4) and ST21 (n = 3). The remaining nine STs (Table 1) were represented by single isolates.
The core-genome-based phylogenetic relations were compared to a previously compiled global set of EPEC and STEC strains and is visualized in Figure 1.
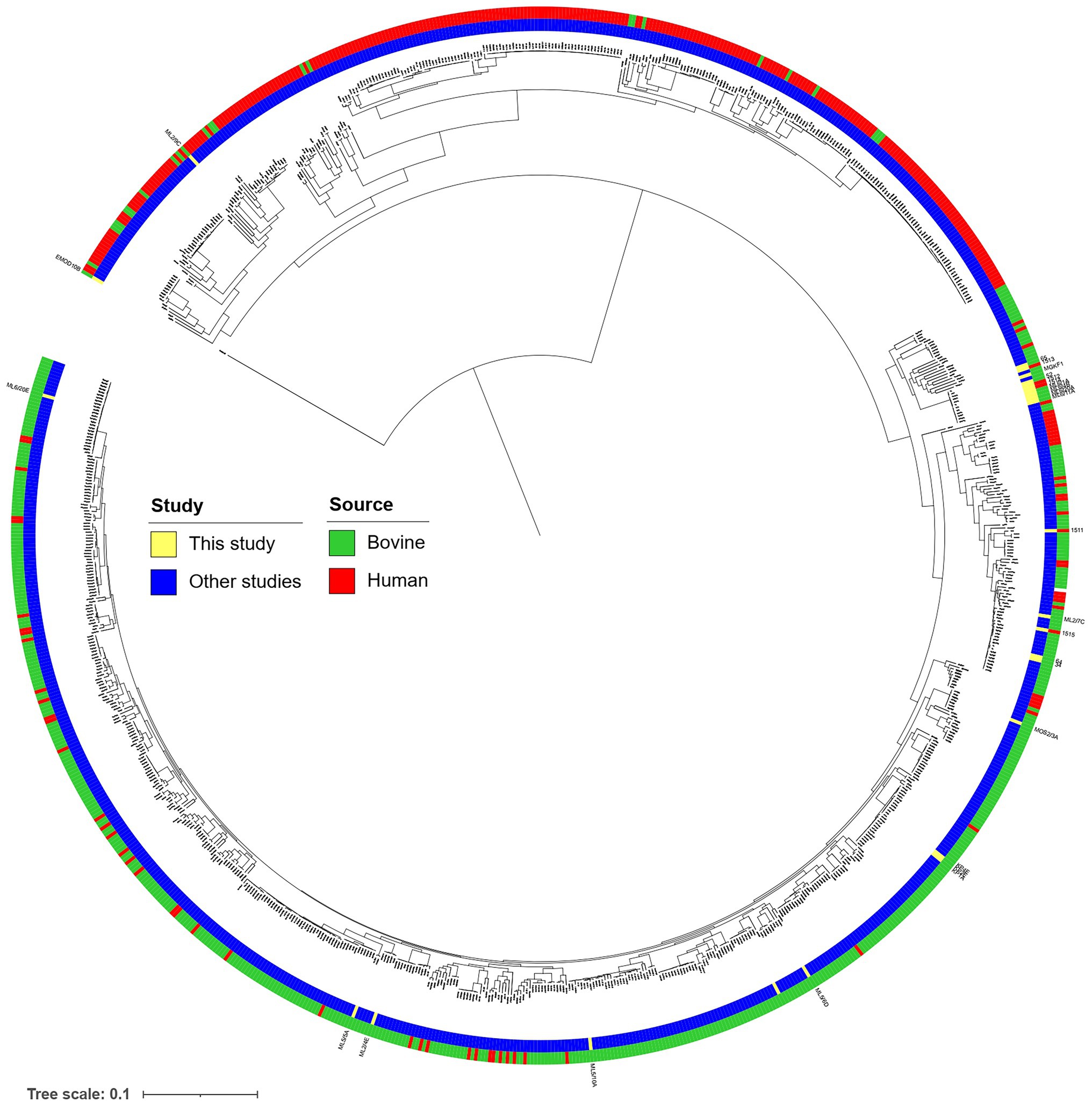
Figure 1. Core gene-based neighbor-joining tree of the 937 bovine and human commensal isolates with 197 human clinical isolates described in Arimizu et al. (2019), complemented with strains with WGS determined in the current study, marked with yellow.
All strains were serotyped using in silico tools (Table 1). The O157:H7 serotype was represented by EHEC strain 52 and EPEC strain 65 of earlier isolation (Tóth et al., 2009), as well by three strains of human origin (1,512, 1,513, 1,514), and by strains with the “ML6” designation, which were isolated from a single farm, but from different animals (Table 1), except for the commensal strain ML6/20E (Table 1). The STs of the O157 serogroup strains were remarkably uniform independently of their serotypes, as all were of ST11.
The “KP” strains (originating from one farm) were of the O26:H11 serotype and belonged all to ST21.
None of the remaining isolates represented any of the “big six” serogroups. This was true for ST10 isolates as well, it was represented by three different serotypes (O90:H40, O157:H4/H16). ST10 was represented by two bovine strains of earlier isolation (34-2, 64-2), one newly isolated bovine strain (ML2/7C) and one human (1515) strain. The serogroup of STEC strains ML2/7C and ML5/10A was O-:H4 and O-:H7, respectively. The human strain 1,511 depicted the O-:H26 serogroup. The commensal isolates exhibited various serotypes, the ExPEC ML2/9C represented serotype O102:H6.
Key Virulence Genes and Their Genetic Background
Characteristic Stx prophage, lambdoid phages and LEE integration sites in various strains were observed (Table 2).
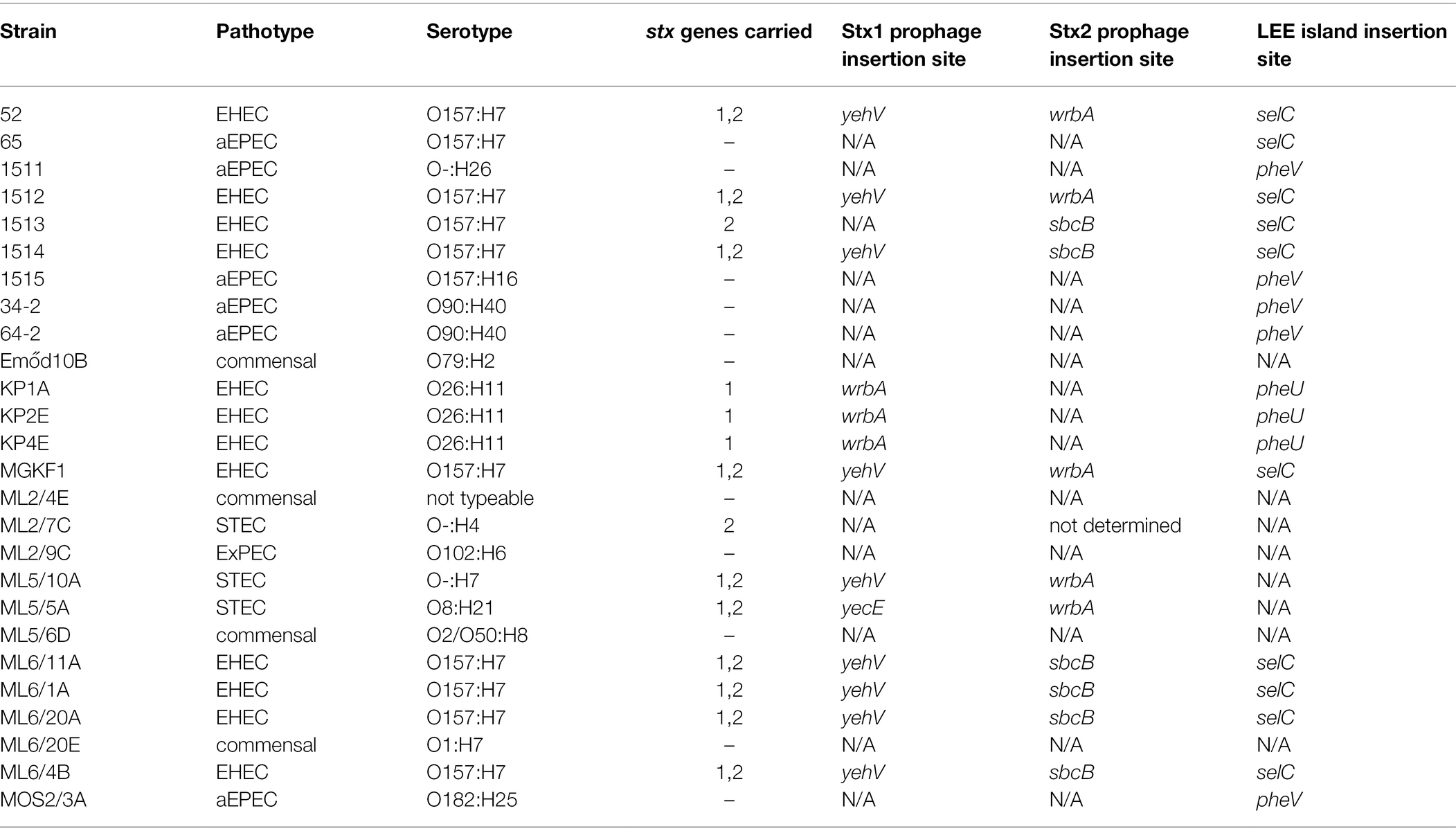
Table 2. Integration sites used by the Stx prophages and locus of enterocyte effacement (LEE) pathogenicity island in the strains with their whole-genome sequences determined in this study.
Thirteen isolates harbored a stx1, and twelve isolates carried the stx2 gene. Ten isolates including two human isolates carried both stx genes; eight were of the O157:H7 serotype and the MLST type ST11. Three different Stx1-prophage (yecE, yehV, wrbA) and two different Stx2-prophage integration sites were detected (sbcB, wrbA, Table 2).
A LEE island was detected in 18/26 whole-genome sequenced isolates. It was integrated at three different sites (selC, pheV, pheU, Table 2). The summary of the integration sites of Stx prophages and LEE islands in the investigated strains is shown in Table 2. In the case of human strains, the Stx1 prophage and the LEE tended to be integrated into the same, frequently used sites (yehV and selC, respectively), while the bovine strains were more diverse in this regard (yehV, wrbA and yecE for the Stx1 prophage, selC, pheU and pheV for LEE sites), with the tendency that strains from the same farm and with the same serotype showed similar integration patterns for these two MGEs. The detailed structure of the LEE of all WGS strains is shown in Figure 2 in a circularized form. The LEE island differed in the strains in several intergenic regions and in the two genes eae and tir, in which for some isolates no homology could be detected at the end of the gene (Figure 2).
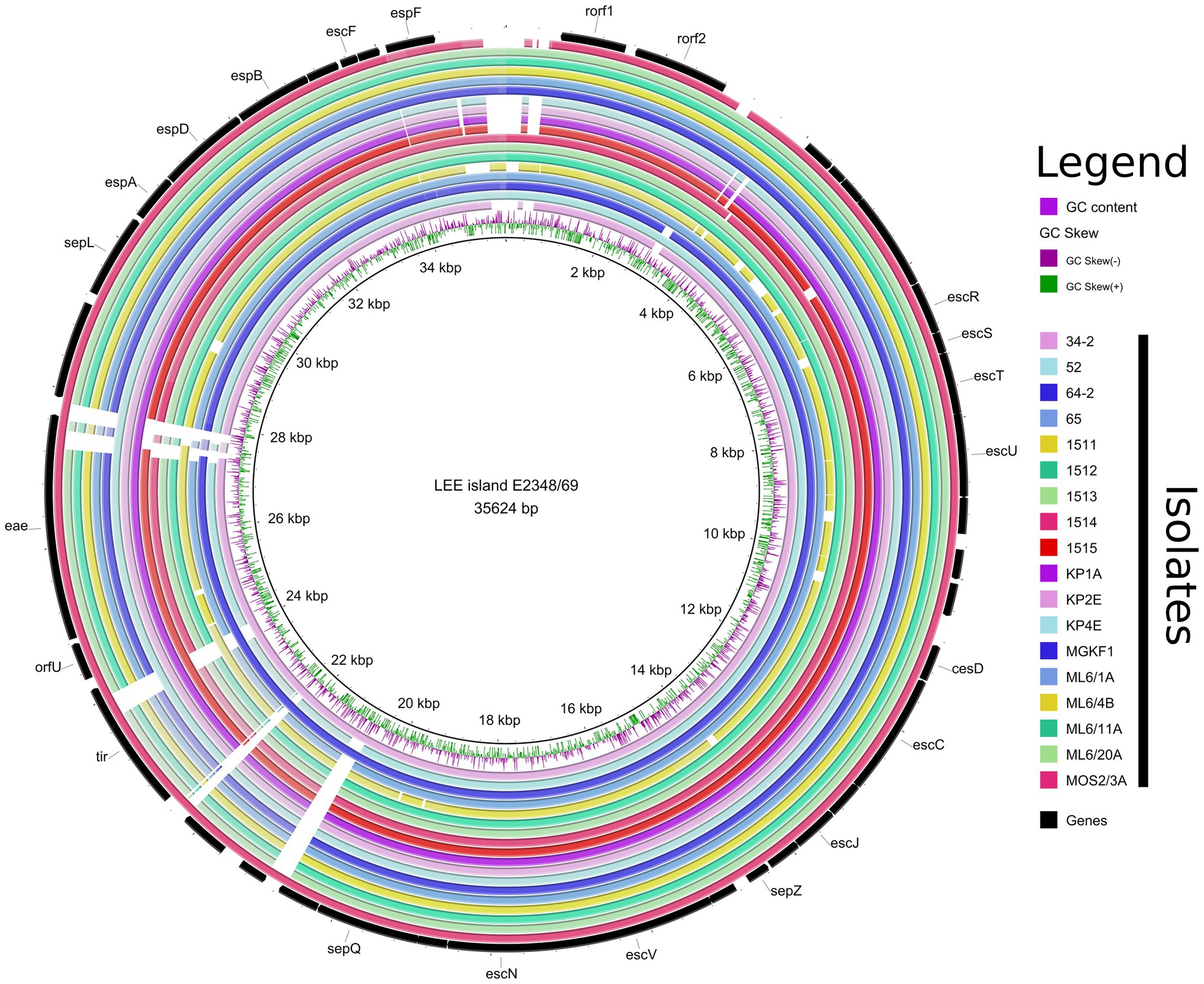
Figure 2. Representation of the LEE pathogenicity islands of all EPEC and EHEC strains with WGS determined in this study. The regions are shown circularized for a more compact view.
Additional Virulence Genes
Using VirulenceFinder and VFDB, we identified a total of 41 additional virulence-related genes in the genomes apart from the stx1 and stx2 genes (Table 3; Supplementary Table 2) as well as of the LEE-associated effectors. All eae-positive strains carried the tir gene, but only six strains harbored the tccP. Subtyping of the intimin gene (eae) revealed the presence of five different intimin subtypes (β1, γ1, ε1, ζ3, θ).
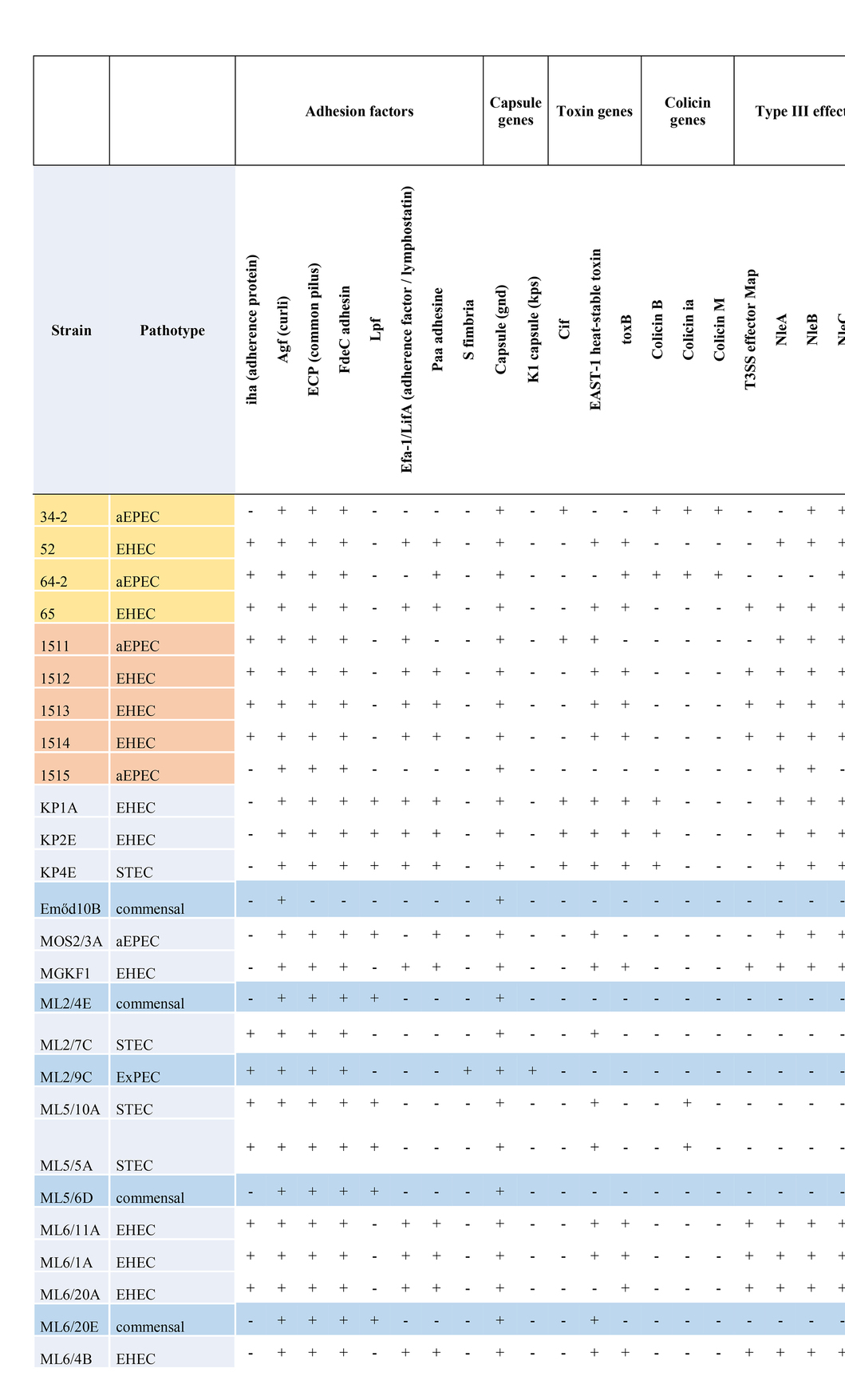
Table 3. Summary of additional virulence factors encoded by strains with WGS determined in this study.
All strains of the serotype O157:H7 also carried the pO157 plasmid that included several classical pO157 plasmid effectors, e.g., the enterohemolysin-encoding operon (ehxABCD). The bovine ExPEC strain ML2/9C carried pO157 that also included katP.
Other widely occurring virulence-related genes of note included the astA encoding the enteroaggregative E. coli heat-stable enterotoxin 1 (EAST1), the iha and lpfA encoding adhesins, the iss gene that encodes a serum survival factor as well as the gene encoding the iron siderophore enterobactin.
Based on the virulence gene content, four different pathotypes could be assigned to the whole-genome sequenced isolates (EHEC, EPEC, STEC and ExPEC). No strain harbored the genes encoding bundle-forming pilus (bfp), therefore all the EPEC strains with their WGS determined represented the atypical EPEC (aEPEC) strains. The most common pathotype was EHEC (n = 12), followed by aEPEC (n = 6) STEC (n = 3) and ExPEC (n = 1). The remaining four isolates were classified as commensal E. coli. A summary of additive virulence genes is shown in Table 3, while the detailed virulence-related gene content identified in each strain is given in Supplementary Table 2. The detailed comparative map of LEE islands highlighting the effector genes is shown in Figure 2.
Antimicrobial Resistance Genes
Five of 26 isolates sequenced carried antibiotic resistance genes (Table 4). The highest number of antibiotic resistance genes were found in an isolate lacking stx and eae, ML2/9C. It was classified as multidrug-resistant (MDR), because it harbored a total of 10 genes encoding resistance toward eight different groups of antimicrobials, including fluoroquinolones (aac(6’)Ib-cr), aminoglycosides (aac(3)-IIa, aadA5), macrolides (mph(A)), sulfonamides (sul1), tetracyclines (tet(B)), phenicols (catB3), and beta-lactams (blaCTX-M-15, blaOXA-1, Table 4). The blaCTX-M-15 gene represents an extended-spectrum beta-lactamase (ESBL). The other four isolates carrying antibiotic resistance genes (34-2, 64-2, 1,515, ML2/7C) harbored between one and three antibiotic resistance genes encoding resistance to aminoglycosides (strA/strB), sulfonamides (sul2) and/or beta-lactams (blaTEM-1B).
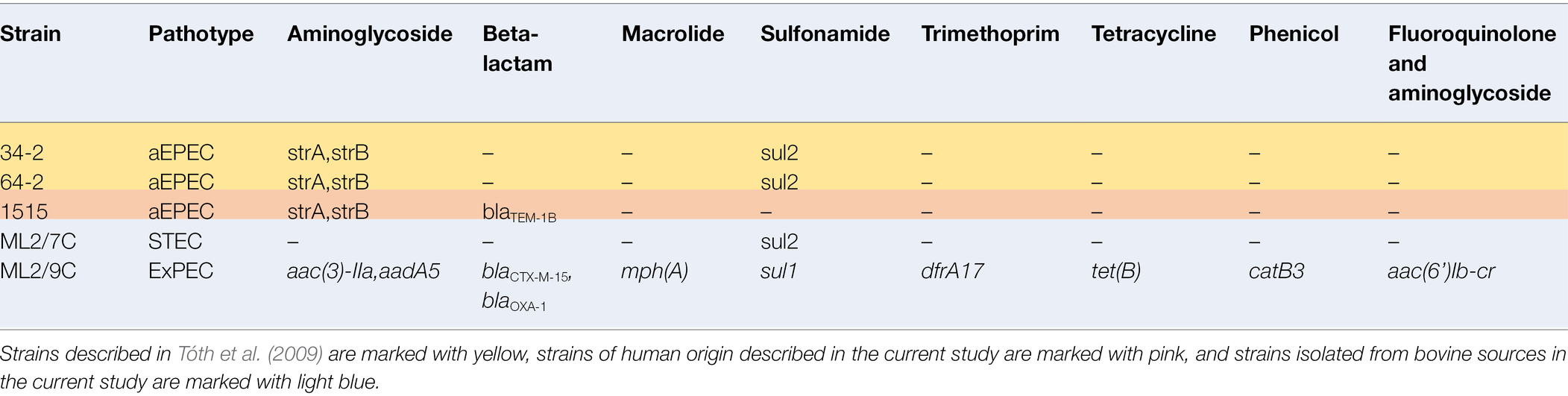
Table 4. Summary of identified antibiotic resistance genes in strains with their whole-genome sequences determined in this study.
Prophage Pool
Following a PHASTER search, in each genome of the investigated strains we found up to 5 intact prophages, between 2 and 12 incomplete prophage sequences, as well as up to 2 probable prophage-like sequences (Supplementary Table 3). We could not identify intact prophages in strains 65 and 1,513. A limitation of the results was that gapped genomes were used for the analysis; therefore, it is possible that the real number of prophages was higher in each strain than what was detected. The most prophage-rich genome was that of strain MOS2/3A with 4 complete, 10 incomplete and 2 phage-like regions. The length of the complete prophage regions ranged from 6.1 to 222 kb, and the percentage of complete prophages of the strains ranged from 43.80 to 56.15%. A summary of all detected prophages with accession numbers of their closest homologue sequences available in GenBank are listed in Supplementary Table 3.
Discussion
In this study, representative sampling of Hungarian cattle farms was carried out to detect STEC, EHEC and EPEC. The prevalence of aEPEC isolates was 8.3% (19/228), while that of STEC it was 8.7% (20/228), and that of EHEC alone 3.9% (9/228). These values are congruent with earlier findings and suggest that Hungary has an occurrence of STEC, EHEC and EPEC isolates that is comparable to other countries of the developed world (Hussein, 2007; Tóth et al., 2009; Dong et al., 2017). It must be noted however, that our method for isolation was optimized for the isolation of strains of the O157 serogroup, therefore strains of all pathotypes representing other serogroups could be underrepresented in our collection. In the earlier study of Tóth et al. (2009), twice as many EPEC isolates were detected as compared to EHEC among the O157:H7 serotype. Two such aEPEC isolates (64-2 and 65) from the earlier study had highly similar virulence gene arrays to EHEC isolates in the current study as well as from humans suggesting that there are several aEPEC ancestral isolates of EHEC, essentially only lacking the Stx prophage (Ferdous et al., 2015; Eichhorn et al., 2018). At the same time, it cannot be excluded that these aEPEC strains might have lost their Stx prophage(s) (Bielaszewska et al., 2008). As none of the EPEC in our current study carried the bfp gene, all of them fall into the category of aEPEC. It is also apparent that such strains are present in similar prevalence among Hungarian cattle like the STEC strains. There was a tendency of strains originating from the same farms to harbor key virulence gene carrying MGEs in the same integration sites, as well as similar sets of additive virulence genes and prophages.
The genomic variability of the investigated pathotypes is most significant with respect to virulence gene repertoires. Regarding the stx genes, only the stx1a subtype was found in all stx1+ strains, while stx2, where present, showed greater variability (stx2a, n = 3; stx2c, n = 8; stc2d, n = 1; Table 1). Almost all stx2 subtype a and c were detected in O157:H7 strains (2/3 and 6/8, respectively), whereas the single subtype d was detected in an O157:H4 serotype isolate (ML2/7C, ST1794). In the case of the strain ML2/7C the integration site for the Stx2 prophage could not be identified, as all the other characteristic integration sites were either intact or occupied by other prophages. This finding suggests the presence of an alternative, hitherto unknown integration site for the Stx2 prophage in this strain, a possibility that has been recently raised for STEC strains (Henderson et al., 2021). It is also noteworthy that the aEPEC strains had at least two of the typical Stx prophage integration sites unoccupied, therefore they can be considered as possible recipients of this MGE and have potential of becoming EHEC (Tables 3, 4).
The eae genes represented five types (β1, γ1, ε1, ζ3, θ). The most common subtype was γ1 (n = 11) and was detected almost exclusively in O157:H7 strains (exception: 1511,: H26) irrespective of their source (human/animal). The second most common subtype was β1, which was detected only in O26:H11 strains with the “KP” designation, originating from the same farm. These findings confirm the notion that the carriage of intimin subtypes is associated to serotypes of EHEC and EPEC (Oswald et al., 2000; Tarr and Whittam, 2002).
A notable difference could be observed in the integration sites used by the LEE when comparing aEPEC and EHEC strains. In EPEC strains, LEE was almost exclusively (5/6) integrated in pheV (exception: selC), while for EHEC strains, LEE was integrated either in selC (n = 9) or pheU (n = 3). This difference could be attributed to the different clonal lineages of strains representing the two pathotypes (Ingle et al., 2016).
Besides the stx genes and LEE-encoded effectors, additional virulence-related genes were identified all the strains. Except for ML2/7C, ML5/5A and ML5/10A, all EHEC strains as well as the aEPEC strain 65 carried the ehxA encoding enterohemolysin, which suggests to the presence of the pO157 plasmid in their case. This distribution is also congruent with the results of Arimizu et al. (2019), who found that STEC strains retained ehxA more frequently than stx-negative ones. The presence of at least one pO157-associated effector gene suggests the carriage of this virulence plasmid (Lim et al., 2010) by all the WGS strains in this study, which belonged to the O157:H7 serogroup. The majority of the aEPEC and EHEC strains harbored effectors of the OI-122 genomic island (efa1 and the non-LEE effectors nleABCD) widespread in these pathotypes (Karmali et al., 2003; Konczy et al., 2008) as well as additional adhesines, siderophores and other fitness factors (Table 3). Out of these, the LEE-negative strains (both STEC, commensal and the ExPEC ML2/9C) tended to carry alternative adhesin encoding genes: the iha, genes (Tarr and Whittam, 2002), and lpfA, the latter being widespread in various intestinal E. coli pathotypes (Zhou et al., 2019). Genes encoding the iron siderophore enterobactin (Raymond et al., 2003) were universally present, and the prevalence of the iss gene encoding an increased serum survival (Johnson et al., 2008) was also high.
Based on its virulence gene array containing S fimbria (Dobrindt et al., 2001) and on being a member of ST405, strain ML2/9C was categorized as ExPEC, with strains of this ST being reported as emerging uroseptic pathogens (Chowdhury et al., 2019). Strains of this pathotype are rarely reported from cattle (Bélanger et al., 2015; Schmidt et al., 2015).
The high number of prophages harbored by the sequenced genomes support the notion that these MGEs play a significant role in the evolution and genomic variability of intestinal E. coli pathotypes (Ogura et al., 2009; Shaaban et al., 2016). It must be noted however, that as we worked with draft genomes, probably not all prophage regions were identified in the genomes. This might be due to the fact that some phages are highly homologous in certain regions and assembling programs cannot distinguish between these regions but assemble these different regions into one single contig.
The distribution of antimicrobial resistance genes among the strains underlines the notion that these determinants are in circulation among bovine zoonotic E. coli, (Kennedy et al., 2017; Um et al., 2018) which can probably be attributed to the pressure of antimicrobials applied against animal pathogens (Hammerum and Heuer, 2009). The number of strains harboring antibiotic resistance genes was comparably low (5/26 of sequenced strains), probably owing to the banning of their use as growth-promoters in 2006 by the European Union. Only the ExPEC strain of bovine origin, ML2/9C showed a multidrug-resistance genotype, harboring genes encoding resistance toward eight different classes of antibiotics, including the ESBL gene blaCTX-M-15.
MLST analysis showed that all O157:H7 strains, regardless of pathotype, belonged to ST11, which is in line with earlier findings (González-Escalona and Kase, 2017; Galarce et al., 2021), while the EHEC O26:H11 strains represented ST21, which is also typical for strains of these sero- and pathotype (Abdalhamid et al., 2019; Galarce et al., 2021). A remarkable finding was that one bovine STEC, as well as two bovine and one human aEPEC strain belonged to ST10, which contains strains of various sero- and pathotypes, and is frequently isolated from human and animal sources (Manges et al., 2015).
We also attempted to place our strains on a global phylogenetic relationship of STEC and aEPEC strains, using a core-genome-based approach and the sequence set published by Arimizu et al. (2019). We found that the placement of bovine strains on the tree was generally aligned with their isolation source, and strains isolated from the same farm (i.e., the “KP” or “ML6” strains) tended to cluster together (Figure 1). The strains of human origin, however, were placed on clades with predominantly bovine isolates. This notion can be indicative of their close geographic origin, the genomic variability of the pathotype in general, as well as the zoonotic risk posed by the bovine strains isolated in our study.
In conclusion, we determined the WGS of 22 E. coli strains representing the STEC, EHEC, aEPEC and ExPEC pathotypes, both of human and bovine origin from Hungary, together with four commensal E. coli from bovine sources. Our results underline the pathogenic potential of bovine strains to humans, as EHEC and aEPEC strains of human origin as well as several bovine strains of the same pathotype harbored very similar virulence gene sets. The phylogenetic position of several human strains within the “bovine-like” lineages corroborates this notion. Several strains harbored Stx prophages with unusual combinations of integration sites. Comparative investigation of stx-negative, LEE-negative strains revealed the carriage of a small number of virulence genes, as well as a potential to carry virulence-related MGEs. The prophage content of the strains showed various profiles, with several widespread prophages with functions yet to be identified. Investigating the inducibility of prophages could be important toward understanding their role in the dissemination of virulence and fitness genes, as well their role in determining architecture of various STEC and aEPEC lineages within cattle populations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA764596, SAMN21509413-SAMN21509438, and SRR15970245-SRR15970246.
Author Contributions
IT and TC conceived and outlined the study. DS performed strain isolation and preliminary genetic characterization and participated in the genomic analysis as well. TM provided strains and performed serotyping. LF performed sequencing and bioinformatical analysis. DS, LF, and IT wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research, Development and Innovation Office (NKFIH), grant number K 124335 and the Bundesministerium fuer Bildung und Forschung (BMBF, Germany) within the German Center for Infection Research (DZIF; grant nos. 8032808818 and 8032808820 to TC) and the State Ministry of Higher Education, Research and Arts of the state Hessen (HMWK) through the HuKKH project (Hessisches universitaeres Kompetenzzentrum Krankenhaushygiene). DS was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank László Makrai and Viktor Jurkovich (University of Veterinary Medicine, Budapest, Hungary) for their invaluable help in the sample collection. We also thank Orsolya Saller for her technical assistance in the strain isolation. We also thank Christina Gerstmann for excellent technical assistance in genome sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.896296/full#supplementary-material
Supplementary Table 1 | Genome sequences used for the phylogenetic analysis of the strains with WGS determined in the current study.
Supplementary Table 2 | List of virulence genes identified by VirulenceFinder and VFDB in the WGS of strains determined in the current study.
Supplementary Table 3 | Prophages or prophage-like sequences identified by PHASTER in the WGS of strains determined in the current study.
Footnotes
References
Abdalhamid, B., McCutchen, E. L., Bouska, A. C., Weiwei, Z., Loeck, B., Hinrichs, S. H., et al. (2019). Whole genome sequencing to characterize Shiga toxin-producing Escherichia coli O26 in a public health setting. J. Infect. Public Heal. 12, 884–889. doi: 10.1016/j.jiph.2019.06.008
Arimizu, Y., Kirino, Y., Sato, M. P., Uno, K., Sato, T., Gotoh, Y., et al. (2019). Large-scale genome analysis of bovine commensal Escherichia coli reveals that bovine-adapted E. coli lineages are serving as evolutionary sources of the emergence of human intestinal pathogenic strains. Genome Res. 29, 1495–1505. doi: 10.1101/gr.249268.119
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Asadulghani, M., Ogura, Y., Ooka, T., Itoh, T., Sawaguchi, A., Iguchi, A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5:e1000408. doi: 10.1371/journal.ppat.1000408
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bélanger, L., Garenaux, A., Harel, J., Bouilanne, M., Nadeau, E., and Dozois, C. M. (2015). Escherichia coli from animal reservoirs as a potential source of human extraintestinal pathogenic E. coli. Pathog. Dis. 62, 1–10. doi: 10.1111/j.1574-695X.2011.00797.x
Bielaszewska, M., and Karch, H. (2005). Consequences of enterohaemorrhagic Escherichia coli infection for the vascular endothelium. Thromb. Haemost. 94, 312–318. doi: 10.1160/TH05-04-0265
Bielaszewska, M., Middendorf, B., Köck, R., Friedrich, A. W., Fruth, A., Karch, H., et al. (2008). Shiga toxin-negative attaching and effacing Escherichia coli: distinct clinical associations with bacterial phylogeny and virulence traits and inferred in-host pathogen evolution. Clin. Infect. Dis. 47, 208–217. doi: 10.1086/589245
Blanco, M., Blanco, J. E., Mora, A., Dahbi, G., Alonso, M. P., González, E. A., et al. (2004). Serotypes, virulence genes, and Intimin types of Shiga toxin (Verotoxin)-producing Escherichia coli isolates from cattle in Spain and identification of a new Intimin variant gene (eae-ξ). J. Clin. Microbiol. 42, 645–651. doi: 10.1128/JCM.42.2.645-651.2004
Bonanno, L., Loukiadis, E., Mariani-Kurkdjian, P., Oswald, E., Garnier, L., Michel, V., et al. (2015). Diversity of Shiga toxin-producing Escherichia coli (STEC) O26:H11 strains examined via stx subtypes and insertion sites of Stx and EspK bacteriophages. Appl. Environ. Microbiol. 81, 3712–3721. doi: 10.1128/AEM.00077-15
Chekabab, S. M., Paquin-Veillette, J., Dozois, C. M., and Harel, J. (2013). The ecological habitat and transmission of Escherichia coli O157:H7. FEMS Microbiol. Lett. 341, 1–12. doi: 10.1111/1574-6968.12078
China, B., Pirson, V., and Mainil, J. (1996). Typing of bovine attaching and effacing Escherichia coli by multiplex in vitro amplification of virulence-associated genes. Appl. Environ. Microbiol. 62, 3462–3465. doi: 10.1128/aem.62.9.3462-3465.1996
Chowdhury, P. R., McKinnon, J., Liu, M., and Djordjevic, S. P. (2019). Multidrug resistant Uropathogenic Escherichia coli ST405 With a novel, composite IS26 transposon in a unique chromosomal location. Front. Microbiol. 9:3212. doi: 10.3389/fmicb.2018.03212
Clermont, O., Christenson, J. K., Denamur, E., and Gordon, D. M. (2013). The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 5, 58–65. doi: 10.1111/1758-2229.12019
Croxen, M. A., Law, R. J., Scholz, R., Keeney, K. M., Wlodarska, M., and Finlay, B. B. (2013). Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26, 822–880. doi: 10.1128/CMR.00022-13
Dobrindt, U., Blum-Oehler, G., Hartsch, T., Gottschalk, G., Ron, E. Z., Fünfstück, R., et al. (2001). S-Fimbria-encoding determinant sfaI is located on Pathogenicity Island III536 of Uropathogenic Escherichia coli strain 536. Infect. Immun. 69, 4248–4256. doi: 10.1128/IAI.69.7.4248-4256.2001
Dong, H.-J., Lee, S., Kim, W., An, J.-U., Kim, J., Kim, D., et al. (2017). Prevalence, virulence potential, and pulsed-field gel electrophoresis profiling of Shiga toxin-producing Escherichia coli strains from cattle. Gut Pathog. 9:22. doi: 10.1186/s13099-017-0169-x
Eichhorn, I., Heidemanns, K., Ulrich, R. G., Schmidt, H., Semmler, T., Fruth, A., et al. (2018). Lysogenic conversion of atypical enteropathogenic Escherichia coli (aEPEC) from human, murine, and bovine origin with bacteriophage Φ3538 Δstx2::cat proves their enterohemorrhagic E. coli (EHEC) progeny. Int. J. Med. Microbiol. 308, 890–898. doi: 10.1016/j.ijmm.2018.06.005
Eppinger, M., Mammel, M. K., LeClerc, J. E., Ravel, J., and Cebula, T. A. (2011). Genome signatures of Escherichia coli O157:H7 isolates from the bovine host reservoir. Appl. Environ. Microbiol. 77, 2916–2925. doi: 10.1128/AEM.02554-10
Ferdous, M., Zhou, K., Mellmann, A., Morabito, S., Croughs, P. D., de Boer, R. F., et al. (2015). Is Shiga toxin-negative Escherichia coli O157:H7 Enteropathogenic or Enterohemorrhagic Escherichia coli? Comprehensive molecular analysis using whole-genome sequencing. J. Clin. Microbiol. 53, 3530–3538. doi: 10.1128/JCM.01899-15
Galarce, N., Sánchez, F., Escobar, B., Lapierre, L., Cornejo, J., Alegría-Morán, R., et al. (2021). Genomic epidemiology of Shiga toxin producing Escherichia coli isolated from the livestock-food-human Interface in South America. Animals 11:1845. doi: 10.3390/ani11071845
González-Escalona, N., and Kase, J. A. (2017). Virulence gene profiles and phylogeny of Shiga toxin-positive Escherichia coli strains isolated from FDA regulated foods during 2010-2017. PLoS One 14:e0214620. doi: 10.1371/journal.pone.0214620
Hammerum, A. M., and Heuer, O. E. (2009). Human health hazards from antimicrobial-resistant Escherichia coli of animal origin. Clin. Infect. Dis. 48, 916–921. doi: 10.1086/597292
Hayashi, T. (2001). Complete genome sequence of Enterohemorrhagic Eschelichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. doi: 10.1093/dnares/8.1.11
Henderson, S. T., Singh, P., Knupp, D., Lacher, D. W., Abu-Ali, G. S., Rudrik, J. T., et al. (2021). Variability in the occupancy of Escherichia coli O157 integration sites by Shiga toxin-encoding Prophages. Toxins 13:433. doi: 10.3390/toxins13070433
Hussein, H. S. (2007). Prevalence and pathogenicity of Shiga toxin-producing Escherichia coli in beef cattle and their products. J. Anim. Sci. 85, E63–E72. doi: 10.2527/jas.2006-421
Ingle, D. J., Tauschek, M., Edwards, D. J., Hocking, D. M., Pickard, D. J., Azzopardi, K. I., et al. (2016). Evolution of atypical enteropathogenic E. coli by repeated acquisition of LEE pathogenicity island variants. Nat. Microbiol. 1:15010. doi: 10.1038/nmicrobiol.2015.10
Joensen, K. G., Tetzschner, A. M. M., Iguchi, A., Aarestrup, F. M., and Scheutz, F. (2015). Rapid and easy In Silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
Johnson, T. J., Wannemuehler, Y. M., and Nolan, L. K. (2008). Evolution of the iss gene in Escherichia coli. Appl. Environ. Microbiol. 74, 2360–2369. doi: 10.1128/AEM.02634-07
Jores, J., Rumer, L., and Wieler, L. H. (2004). Impact of the locus of enterocyte effacement pathogenicity island on the evolution of pathogenic Escherichia coli. Int. J. Med. Microbiol. 294, 103–113. doi: 10.1016/j.ijmm.2004.06.024
Kaper, J. B., Nataro, J. P., and Mobley, H. L. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/nrmicro818
Karmali, M., Mascarenhas, M., Shen, S., Ziebell, K., Johnson, S., Reid-Smith, R., et al. (2003). Association of Genomic O Island 122 of Escherichia coli EDL 933 with Verocytotoxin-producing Escherichia coli Seropathotypes that are linked to epidemic and/or serious disease. J. Clin. Microbiol. 41, 4930–4940. doi: 10.1128/JCM.41.11.4930-4940.2003
Kennedy, C.-A., Fanning, S., Karczmarczyk, M., Byrne, B., Monaghan, Á., Bolton, D., et al. (2017). Characterizing the multidrug resistance of non-O157 Shiga toxin-producing Escherichia coli isolates from cattle farms and abattoirs. Microb. Drug Resist. 23, 781–790. doi: 10.1089/mdr.2016.0082
Konczy, P., Ziebell, K., Mascarenhas, M., Choi, A., Michaud, C., Kropinski, A. M., et al. (2008). Genomic O Island 122, locus for enterocyte effacement, and the evolution of virulent Verocytotoxin-producing Escherichia coli. J. Bacteriol. 190, 5832–5840. doi: 10.1128/JB.00480-08
Kulasekara, B. R., Jacobs, M., Zhou, Y., Wu, Z., Sims, E., Saenphimmachak, C., et al. (2009). Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes That may enhance virulence. Infect. Immun. 77, 3713–3721. doi: 10.1128/IAI.00198-09
Lim, J. Y., Yoon, J. W., and Hovde, C. J. (2010). A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 20, 5–14. doi: 10.4014/jmb.0908.08007
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Lupolova, N., Dallman, T. J., Matthews, L., Bono, J. L., and Gally, D. L. (2016). Support vector machine applied to predict the zoonotic potential of E. coli O157 cattle isolates. PNAS 113, 11312–11317. doi: 10.1073/pnas.1606567113
Manges, A. R., Harel, J., Masson, L., Edens, T. J., Portt, A., Reid-Smith, R. J., et al. (2015). Multilocus sequence typing and virulence gene profiles associated with Escherichia coli from human and animal sources. Foodborne Pathog. Dis. 12, 302–310. doi: 10.1089/fpd.2014.1860
Newell, D. G., and Ragione, R. M. L. (2018). Enterohaemorrhagic and other Shiga toxin-producing Escherichia coli (STEC): where are we now regarding diagnostics and control strategies? Transbound. Emerg. Dis. 65, 49–71. doi: 10.1111/tbed.12789
Nüesch-Inderbinen, M., Cerna, N., Wüthrich, D., Egli, A., and Stephan, R. (2018). Genetic characterization of Shiga toxin producing Escherichia coli belonging to the emerging hybrid pathotype O80:H2 isolated from humans 2010-2017 in Switzerland. Int. J. Med. Microbiol. 308, 534–538. doi: 10.1016/j.ijmm.2018.05.007
Ochoa, T. J., and Contreras, C. A. (2011). Enteropathogenic E. coli (EPEC) infection in children. Curr. Opin. Infect. Dis. 24, 478–483. doi: 10.1097/QCO.0b013e32834a8b8b
Ogura, Y., Kurokawa, K., Ooka, T., Tashiro, K., Tobe, T., Ohnishi, M., et al. (2006). Complexity of the genomic diversity in Enterohemorrhagic Escherichia coli O157 revealed by the combinational use of the O157 Sakai Oligo DNA microarray and the whole genome PCR scanning. DNA Res. 13, 3–14. doi: 10.1093/dnares/dsi026
Ogura, Y., Ooka, T., Iguchi, A., Toh, H., Asadulghani, M., Oshima, K., et al. (2009). Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. PNAS 106, 17939–17944. doi: 10.1073/pnas.0903585106
Ohnishi, M., Terajima, J., Kurokawa, K., Nakayama, K., Murata, T., Tamura, K., et al. (2002). Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. PNAS 99, 17043–17048. doi: 10.1073/pnas.262441699
Ørskov, F., and Ørskov, I. (1984). “2 serotyping of Escherichia coli,” in Methods in Microbiology. ed. T. Bergan (United States: Academic Press).
Oswald, E., Schmidt, H., Morabito, S., Karch, H., Marchès, O., and Caprioli, A. (2000). Typing of Intimin genes in human and animal Enterohemorrhagic and Enteropathogenic Escherichia coli: characterization of a new Intimin variant. Infect. Immun. 68, 64–71. doi: 10.1128/IAI.68.1.64-71.2000
Paletta, A. C. C., Castro, V. S., and Conte-Junior, C. A. (2020). Shiga toxin-producing and Enteroaggregative Escherichia coli in animal, foods, and humans: pathogenicity mechanisms, detection methods, and epidemiology. Curr. Microbiol. 77, 612–620. doi: 10.1007/s00284-019-01842-1
Perna, N. T., Plunkett, G., Burland, V., Mau, B., Glasner, J. D., Rose, D. J., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533. doi: 10.1038/35054089
Pulz, M., Matussek, A., Monazahian, M., Tittel, A., Nikolic, E., Hartmann, M., et al. (2003). Comparison of a Shiga toxin enzyme-linked Immunosorbent assay and two types of PCR for detection of Shiga toxin-producing Escherichia coli in human stool specimens. J. Clin. Microbiol. 41, 4671–4675. doi: 10.1128/JCM.41.10.4671-4675.2003
Rahman, M., Nabi, A., Asadulghani, M., Faruque, S. M., and Islam, M. A. (2018). Toxigenic properties and stx phage characterization of Escherichia coli O157 isolated from animal sources in a developing country setting. BMC Microbiol. 18:98. doi: 10.1186/s12866-018-1235-3
Raymond, K. N., Dertz, E. A., and Kim, S. S. (2003). Enterobactin: an archetype for microbial iron transport. PNAS 100, 3584–3588. doi: 10.1073/pnas.0630018100
Saile, N., Schuh, E., Semmler, T., Eichhorn, I., Wieler, L. H., Bauwens, A., et al. (2018). Determination of virulence and fitness genes associated with the pheU, pheV and selC integration sites of LEE-negative food-borne Shiga toxin-producing Escherichia coli strains. Gut Pathog. 10:43. doi: 10.1186/s13099-018-0271-8
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Schmidt, J. W., Agga, G. E., Bosilevac, J. M., Brichta-Harhay, D. M., Shackelford, S. D., Wang, R., et al. (2015). Occurrence of antimicrobial-resistant Escherichia coli and Salmonella enterica in the beef cattle production and processing continuum. Appl. Environ. Microbiol. 81, 713–725. doi: 10.1128/AEM.03079-14
Schwengers, O., Hoek, A., Fritzenwanker, M., Falgenhauer, L., Hain, T., Chakraborty, T., et al. (2020). ASA3P: An automatic and scalable pipeline for the assembly, annotation and higher level analysis of closely related bacterial isolates. PLoS Comput. Biol. 16:e1007134. doi: 10.1371/journal.pcbi.1007134
Shaaban, S., Cowley, L. A., McAteer, S. P., Jenkins, C., Dallman, T. J., Bono, J. L., et al. (2016). Evolution of a zoonotic pathogen: investigating prophage diversity in enterohaemorrhagic Escherichia coli O157 by long-read sequencing. Microb. Genom. 2:e000096. doi: 10.1099/mgen.0.000096
Sharma, V. K., Akavaram, S., Schaut, R. G., and Bayles, D. O. (2019). Comparative genomics reveals structural and functional features specific to He genome of a foodborne Escherichia coli O157:H7. BMC Genomics 20:196. doi: 10.1186/s12864-019-5568-6
Standard British and BSEN ISO (2001). ISO 16654/2001. Microbiology of food and animal feeding stuffs—Horizontal method for the detection of Escherichia coli O157.
Sváb, D., Bálint, B., Maróti, G., and Tóth, I. (2016). Cytolethal distending toxin producing Escherichia coli O157:H43 strain T22 represents a novel evolutionary lineage within the O157 serogroup. Infect. Genet. Evol. 46, 110–117. doi: 10.1016/j.meegid.2016.11.003
Tarr, C. L., and Whittam, T. (2002). Molecular evolution of the Intimin gene in O111 clones of pathogenic Escherichia coli. J. Bacteriol. 184, 479–487. doi: 10.1128/JB.184.2.479-487.2002
Terajima, J., Izumiya, H., Hara-Kudo, Y., and Ohnishi, M. (2017). Shiga toxin (Verotoxin)-producing Escherichia coli and foodborne disease: a review. Food Safety 5, 35–53. doi: 10.14252/foodsafetyfscj.2016029
Tóth, I., Schmidt, H., Kardos, G., Lancz, Z., Creuzburg, K., Damjanova, I., et al. (2009). Virulence genes and molecular typing of different groups of Escherichia coli O157 strains in cattle. Appl. Environ. Microbiol. 75, 6282–6291. doi: 10.1128/AEM.00873-09
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Um, M. M., Brugère, H., Kérourédan, M., Oswald, E., and Bibbal, D. (2018). Antimicrobial resistance profiles of Enterohemorrhagic and Enteropathogenic Escherichia coli of serotypes O157:H7, O26:H11, O103:H2, O111:H8, O145:H28 compared to Escherichia coli isolated from the same adult cattle. Microb. Drug Resist. 24, 852–859. doi: 10.1089/mdr.2017.0106
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
Yang, X., Sun, H., Fan, R., Fu, S., Zhang, J., Matussek, A., et al. (2020). Genetic diversity of the intimin gene (eae) in non-O157 Shiga toxin-producing Escherichia coli strains in China. Sci. Rep. 10:3275. doi: 10.1038/s41598-020-60225-w
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: EHEC, STEC, aEPEC, cattle, WGS, prophage, comparative genomics, virulence gene array
Citation: Sváb D, Falgenhauer L, Mag T, Chakraborty T and Tóth I (2022) Genomic Diversity, Virulence Gene, and Prophage Arrays of Bovine and Human Shiga Toxigenic and Enteropathogenic Escherichia coli Strains Isolated in Hungary. Front. Microbiol. 13:896296. doi: 10.3389/fmicb.2022.896296
Edited by:
James L. Bono, United States Department of Agriculture, United StatesReviewed by:
Sabine Delannoy, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), FranceIsha R. Patel, United States Food and Drug Administration, United States
Copyright © 2022 Sváb, Falgenhauer, Mag, Chakraborty and Tóth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domonkos Sváb, c3ZhYi5kb21vbmtvc0B2bXJpLmh1