 Lilian Lopez Leyva
Lilian Lopez Leyva Emmanuel Gonzalez
Emmanuel Gonzalez Noel W. Solomons4
Noel W. Solomons4 Kristine G. Koski
Kristine G. Koski
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 September 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.885588
This article is part of the Research Topic Risk of Dietary Hazardous Substances and Impact on Human Microbiota: Possible Role in Several Dysbiosis Phenotypes, Volume II View all 7 articles
There is evidence that breastfeeding practices may impact the milk microbiota diversity and differential abundance at the genera level; however, the possibility that distinct feeding practices, such as exclusive (EBF) and non-exclusive breastfeeding (non-EBF), might alter the milk microbiome at the species level has not been explored. This cross-sectional study analyzed the milk microbiome of 64 Mam-Mayan indigenous mothers from San Juan Ostuncalco in Guatemala. Two breastfeeding practices [exclusive (EBF) vs non-exclusive (non-EBF)] were analyzed at two stages of lactation [early (5–46 days post-partum) vs late (109–184 days post-partum)]. EBF was defined as offering only human milk and non-EBF was defined as feeding the infant herbal teas (agüitas) and/or complementary foods while continuing to breastfeed. Results identified four clusters with distinct microbial communities that segregated bacterial species by both breastfeeding practices and stage of lactation. Comparison among these clusters identified several notable patterns. First, during EBF, the microbiome differed by stage of lactation where there was a shift in differential abundance from Actinobacteria and Firmicutes in early to Bacteroidetes and Proteobacteria species in late lactation. Second, a similar comparison between non-EBF mothers by stage of lactation also identified a higher differential abundance of Actinobacteria and Firmicutes species in early lactation, but only Proteobacteria and not Bacteroidetes in late lactation, indicating a further shift in the milk microbial ecosystem with fewer oral bacteria present in late lactation. Third, comparisons between EBF and non-EBF mothers at both early and late lactation showed that mothers who exclusively breastfed had more differentially abundant species in early (11 vs 1) and late (13 vs 2) lactation. Fourth, EBF at early and late lactation had more commensal and lactic acid bacteria, including Lactobacillus gasseri, Granulicatella elegans, Streptococcus mitis, and Streptococcus parasanguinis, compared to those who did not exclusively breastfeed. Collectively, these results show that EBF has more differentially abundant bacteria, including commensal and lactic acid bacteria, and that the addition of agüitas (herbal teas) and/or complementary foods modify the milk microbiome composition by reducing the oral bacteria and introducing more environmentally sourced bacteria to the ecosystem.
Research on human milk microbiota has increased recently due to growing interest in understanding factors shaping the milk microbiome (Fitzstevens et al., 2017; Zimmermann and Curtis, 2020; Lopez Leyva et al., 2021a). Several studies in developed countries, where exclusive breastfeeding to 6 months is limited (UNICEF, 2019), have analyzed the human milk microbiota of breastfed and formula-fed infants (Bokulich et al., 2016; Martin et al., 2016; Carvalho-Ramos et al., 2018), noting that exclusive breastfeeding for 2 months was associated with healthier gut bacterial communities (Ho et al., 2018). In contrast, developing countries often report higher rates of exclusive breastfeeding for up to 6 months than developed countries (UNICEF, 2019). In fact, in Guatemala, exclusive breastfeeding rates exceed 53% for the first 5 months, and in rural communities, the prevalence increases to 61% compared to 41% worldwide (UNICEF, 2019). However, few studies have investigated breastfeeding practices in developing countries (Lopez Leyva et al., 2021a). In LMIC (low- and middle-income countries), cultural practices in early feeding may include ritual fluids. This is a common practice in Mexico (Deming et al., 2015; Rodríguez-Ramírez et al., 2016; Afeiche et al., 2018), Bangladesh, Brazil, India, Nepal, Pakistan, Peru, South Africa, and Tanzania (Patil et al., 2015). These ritual fluids or herbal teas are usually culturally ‘prescribed’ to maintain infant health (Wren et al., 2015) and to treat gastrointestinal infections, colic, stomach pain, constipation, sore throat, or fever (Doak et al., 2013), and for irritability and crying (Chomat et al., 2015).
The possibility that these ritual fluids and the introduction of complementary food might further impact the human milk microbiome has not been widely considered. Interestingly, there is emerging evidence to suggest that environmental bacteria in soil and water may function as a superorganism that can replenish human microbial communities as inoculants and provide beneficial microorganisms which could positively impact human health (Blum et al., 2019). Some of the benefits assigned to the interaction of bacteria present in the environment and the human microbiome include less propensity for allergies (Hanski et al., 2012), suppression of soil-borne pathogens, exposure to immunoregulation-inducing soil microorganisms, immune tolerance, and an increase in microbial diversity (Wall et al., 2015). Soil or water genera identified in human milk include Acinetobacter (Kumar et al., 2016; Sakwinska et al., 2016; Urbaniak et al., 2016; Drell et al., 2017; Patel et al., 2017), Bradyrhizobiaceae (Hunt et al., 2011), Novosphingobium (Jiménez et al., 2015), Pseudomonas (Hunt et al., 2011; Jiménez et al., 2015; Sakwinska et al., 2016; Pannaraj et al., 2017; Patel et al., 2017; Moossavi et al., 2019), Ralstonia (Hunt et al., 2011; Kumar et al., 2016; Vaidya et al., 2017; Williams et al., 2017; Moossavi et al., 2019), Sphingobium (Jiménez et al., 2015; Mueller et al., 2015), Sphingomonas (Hunt et al., 2011; Urbaniak et al., 2014; Jiménez et al., 2015; Davé et al., 2016; Kumar et al., 2016; Li et al., 2017; Ding et al., 2019; Hermansson et al., 2019), and Stenotrophomonas (Urbaniak et al., 2014; Davé et al., 2016). Although still debated (Salter et al., 2014; Jiménez et al., 2015; Sakwinska et al., 2016; Douglas et al., 2020), some of these environmental genera, such as Bradyrhizobiaceae, Pseudomonas, Sphingomonas, and Ralstonia, have been identified as part of the “core” human milk microbiome (Hunt et al., 2011; Jiménez et al., 2015). It has been suggested that human milk acquired these environmental bacteria through a maternal diet based on legumes (Drago et al., 2017), proximity to soil environments (Blum et al., 2019), and/or agrarian lifestyles (Yatsunenko et al., 2012; Clemente et al., 2015; Meehan et al., 2018).
Recently, we conducted a study in the Mam-Mayan indigenous community in Guatemala to identify maternal factors involved in modifying the human milk microbiome at both the genera (Lopez Leyva et al., 2021b) and species levels (Gonzalez et al., 2021) and identified distinct clustering of microbial communities associated with both stages of lactation and breastfeeding practices. The study conducted at the species level reported a shift from Staphylococcus and Streptococcus species in early lactation to Sphingobium and Pseudomonas species in late lactation (Gonzalez et al., 2021). However, in this later study, breastfeeding practices were not considered.
To date, no studies have investigated the relationships between the stage of lactation and breastfeeding practices in the shaping of the human milk microbiome at the species level. This is important because reporting at the species level provides more information about the functionality and facilitates biological interpretation by having an improved resolution of the data (Lopez Leyva et al., 2021a). The purpose of this study was to explore how two breastfeeding practices [exclusive (EBF) vs non-exclusive breastfeeding (non-EBF)] at two stages of lactation [early (5–46 days) vs late (109–184 days)] might modify the human milk microbiome. Our specific objectives were (1) to characterize the milk microbiome of EBF in early and late lactation in mothers living in eight rural Mam-speaking indigenous communities in Guatemala and (2) to compare shifts between mothers who exclusively breastfed vs those who did not exclusively breastfeed (EBF and non-EBF) at each stage of lactation.
This cross-sectional study was part of a collaboration between McGill University and the Center for Studies of Sensory Impairment, Aging, and Metabolism (CeSSIAM) in the Republic of Guatemala. Field studies were conducted from June 2012 through January 2013 in eight rural Mam-speaking communities of the San Juan Ostuncalco region in Guatemala (Chomat et al., 2015). The inclusion criteria were indigenous lactating women with infants at (1) early stage of lactation (5–46 days) or (2) late stage of lactation (109–184 days) and who had a vaginal delivery. The exclusion criteria were: (1) mothers with colostrum (milk < 4 days post-partum) and (2) mothers treated with antibiotics during the post-partum period. Ethical approval was obtained from the Institutional Review Boards of both institutions, and permission was obtained from community leaders and the local authorities of the Ministry of Health. All mothers provided written informed consent for participation in the study.
Two breastfeeding practices [exclusive (EBF) vs non-exclusive (non-EBF)] were analyzed at two stages of lactation [early (5–46 days post-partum) vs late (109–184 days post-partum)]. Exclusive breastfeeding (EBF) was defined as providing only human milk to the infant and non-exclusive breastfeeding (non-EBF) at the early stage was defined as providing water or agüitas in addition to human milk. Agüitas are ritual fluids; the infusions more commonly used are boiled water, sugar water, chamomile tea, corn paste water, anise water, orange leaf water, mint water, or sage water. Non-EBF at the late stage was defined as providing agüitas and/or complementary foods to the infant while they continued to breastfeed. From the sample of 64 women, the four groups created were early EBF (n = 15), early non-EBF (n = 14), late EBF (n = 18), and late non-EBF (n = 17).
The classification of the type of breastfeeding practice into groups was done based on a structured, in-depth questionnaire administered to mothers about independent factors (maternal, infant, and cultural practices and beliefs) that may influence breastfeeding initiation, exclusivity, and frequency (Wren et al., 2015). Trained local health care workers administered the questionnaire orally in either Spanish or Mam to participants during a 30- to 40-min interview (Wren et al., 2015). Feeding patterns were defined as EBF or non-EBF based on cumulative recall since birth. Mothers were asked if they ever fed their infant agüitas since birth (yes/no). If yes, the timing of the agüitas initiation was queried (hours, days, and weeks post-partum). Likewise, mothers were asked to identify from a list the type of agüitas given to the infant and provide an open-ended reason (Wren et al., 2015).
Milk samples from early lactation (5–46 days post-partum) and late lactation (109–184 days post-partum) were collected during the period from 2012 to 2013. These ranges were chosen to be consistent with our previous studies that measured infant growth (Li et al., 2016, 2019; Wren-Atilola et al., 2018). Prior to collection, the nipple and areola of the breast were cleaned with 70% ethyl alcohol. Human milk samples were collected during a 3-h time window in the morning from the breast not recently used for breastfeeding via full manual expression by a trained midwife, who used hand sanitizer before and after collection. This is important since other studies have shown differences in milk microbiome diversity with the use of breast pumps (Moossavi et al., 2019). Milk was collected into 60 ml plastic vials and immediately stored on ice. Samples were partitioned into 15 ml tubes at the field laboratory (−30°C) prior to transfer on dry ice to McGill University where they were stored at −80°C, which is known to preserve the milk microbiome integrity (Lyons et al., 2021), until DNA extraction for microbiome analysis was performed in 2018.
Methodology (sequencing and microbiome characterization) is as described in Gonzalez et al., 2021. Briefly, DNA extraction was done using 1 ml of milk with DNeasy Blood and Tissue mini kit from Qiagen according to the manufacturer’s protocol. For PCR, a region of ∼526 bp in the 16S rRNA gene, covering the V1–V3 region, was amplified with the universal eubacterial primers 27F: AGAGTTTGATCCTGGCTCAG and 533R: TTACCGCGGCTGCTGGCAC (Cabrera-Rubio et al., 2012). The subsequent 16S rRNA gene sequencing was performed using the Illumina MiSeq platform at McGill University and Génome Quebec Innovation Centre. Amplicons were assembled from 300 paired-end reads. Reagent controls were below the detection limit used by Génome Quebec Innovation Centre for quality assurance. Microbial data processing was performed using ANCHOR, a 16S rRNA gene amplicon pipeline for microbial analysis (Gonzalez et al., 2019). Briefly, Mothur (Schloss et al., 2009) was used to align and dereplicate sequences before high-count OTU selection at a count threshold of 36 across all samples. NCBI 16S rRNA RefSeq, NCBI non-redundant nucleotide, SILVA, and the Ribosomal Database Project (RDP) databases were used to annotate OTUs using BLASTn with criteria of > 99% for identity and coverage. When a BLASTn return had 100% identity and coverage hits across multiple databases, priority was given to NCBI 16S rRNA RefSeq due to the high standard of curation. Low counting amplicons (<36 counts) were binned to high-count OTUs at a lower threshold of >98% identity/coverage with multiple, equally good (highest identity/coverage); all the annotation was retained and reported. However, all annotation, and in particular species calls, should be considered putative even when sharing 100% sequence identity to a single species due to database errors. Bacteria with errors in the data repositories which qualified as unknown bacteria were identified as “DBinconsistency_MS” for multiple species or “DBinconsistency_MG” for multiple genera. Contamination was controlled at all stages of this analysis. This included the use of aseptic sampling protocols monitored by trained specialists as well as PCR blanks performed at Génome Quebec in Canada (no samples were sequenced if any bands were visible in negative controls). Contamination control continued at the bioinformatics stage where the Canadian Centre in Computational Genomics of McGill University carried out contemporary investigation and control via sample pre-processing including ordination analysis, control for sparsity and prevalence, and identifying putative contamination (decontam, R package; Supplementary File 1). Finally, the biostatistics analysis involved a differential abundance test (DESeq2) which contained a Cook’s distance analysis step prior to the test.
Alpha diversity of human milk samples was measured using Chao 1, Shannon, and Observed indices within the Phyloseq R package (McMurdie and Holmes, 2013). Significant differences between alpha-diversity indices were estimated using a t-test (for normally distributed values) or a non-parametric method (Mann–Whitney test). P-values were corrected for multiple comparisons using the Benjamini-Hochberg procedure. Beta diversity was assessed using the constrained correspondence analysis (CCA) ordination method. Dispersion ellipses were drawn using the veganCovEllipse function from the Vegan package in R (Oksanen et al., 2020). The significance of the different constraints in the CCA analysis was evaluated using an ANOVA-like permutation test (vegan package).
To characterize differentially abundant taxonomic units between groups of samples, parametric models developed in transcriptomics perform well when applied to microbiome biomarker data (uneven library sizes, sparsity, and sample representativity) (Gonzalez et al., 2015; Jonsson et al., 2016; Weiss et al., 2017; Minerbi et al., 2019; Brereton et al., 2020). DESeq2 procedure (Love et al., 2014) was used to calculate differentially abundant taxonomic units. Taxonomic units tested with a false discovery rate (FDR or the expected proportion of false-positive findings) <0.1 were considered significant (Anders et al., 2013; Love et al., 2014, 2016; Gonzalez et al., 2019).
Population characteristics are summarized in Table 1. The participating mothers were categorized into four groups depending on the stage of lactation (early or late) and breastfeeding practices (EBF or non-EBF). In early lactation, 51.7% of infants were EBF and 48.3% were non-EBF (added agüitas), whereas in late lactation, 51.4% were EBF and 48.6% were non-EBF (22.9% provided agüitas and human milk and 25.7% provided agüitas, complementary foods, and human milk). With regard to other breastfeeding practices, 59% breastfed within the first hour, and the average frequency of breastfeeding was 11 ± 3.5 feeds per day.
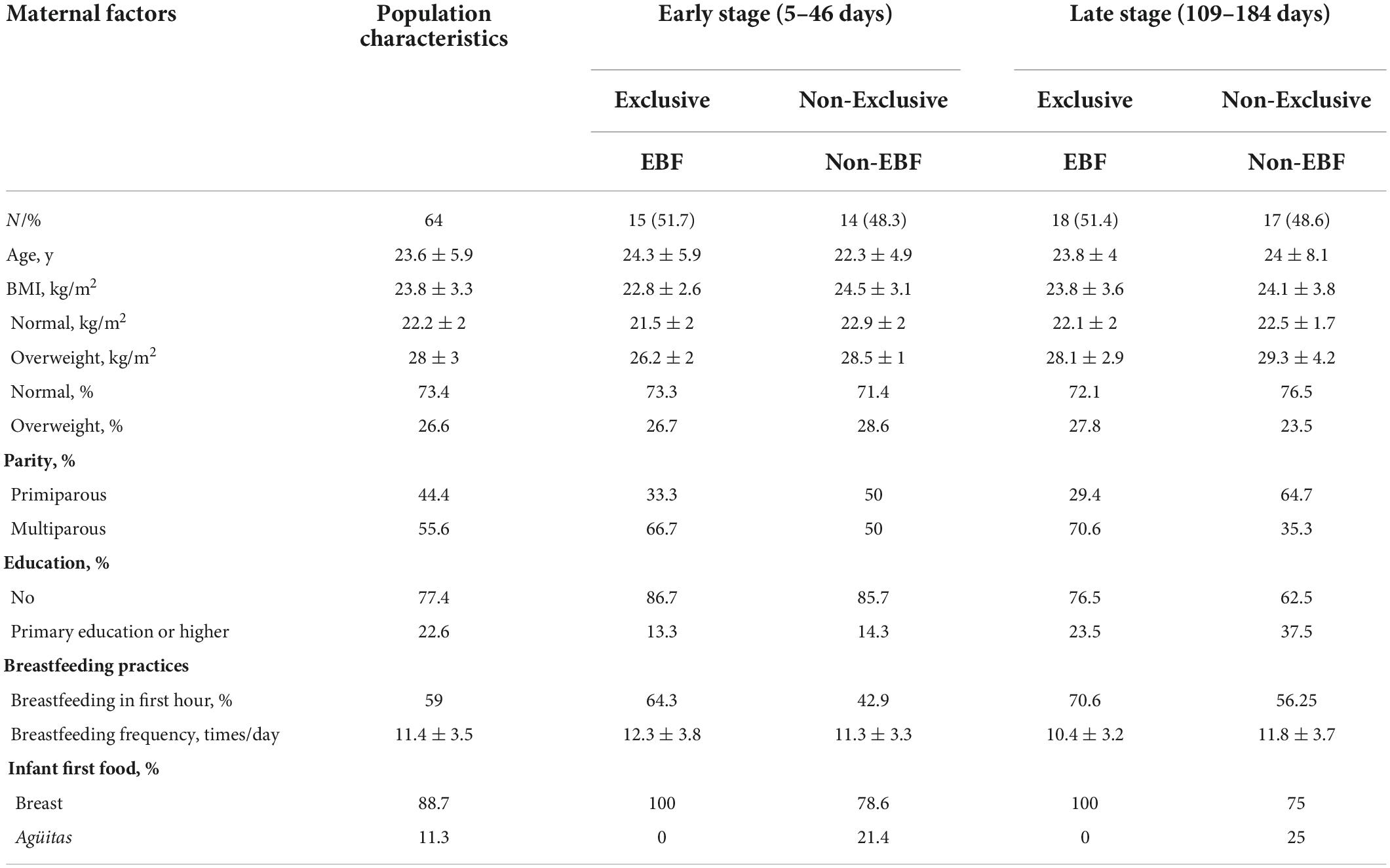
Table 1. Maternal characteristics and breastfeeding practices.
A total of 503 OTUs were assembled and captured 77,827 sequence reads across all 64 human milk samples (Supplementary File 2). These could be annotated as 287 OTUs, 134 genera, and 76 family or higher taxa, as well as 109 which could not be recognized as 99% similar (in both identity and coverage) to any known taxa and were labeled as unknown. Of the 287 OTUs annotated as putative species, the average BLASTn return identity was 99.8% including 173 perfect hits (100% identity). Main factors were projected onto an unconstrained ordination diagram (NMDS), and each variable regression was independently tested by Monte Carlo permutation (envfit function from R package Vegan). The goodness of fit obtained is described in Supplementary File 3.
Figure 1 shows the estimation of alpha diversity (A) and beta diversity (B). The indices Chao 1, Shannon, and Observed used to estimate alpha diversity identified differences between early and late lactation for EBF and non-EBF mothers. Differences in alpha diversity using Chao 1 were significant between early non-EBF and late EBF (FDR = 0.01) and between early EBF and late EBF (FDR = 0.04); using Observed, there were significant differences between early EBF and late EBF (FDR = 0.002), early non-EBF and late EBF (FDR = 0.005), and late EBF and late non-EBF (FDR = 0.08; Supplementary File 4). In beta-diversity analysis, CCA ordination significantly segregated (p = 0.021) the four groups: early EBF, early non-EBF, late EBF, and late non-EBF.
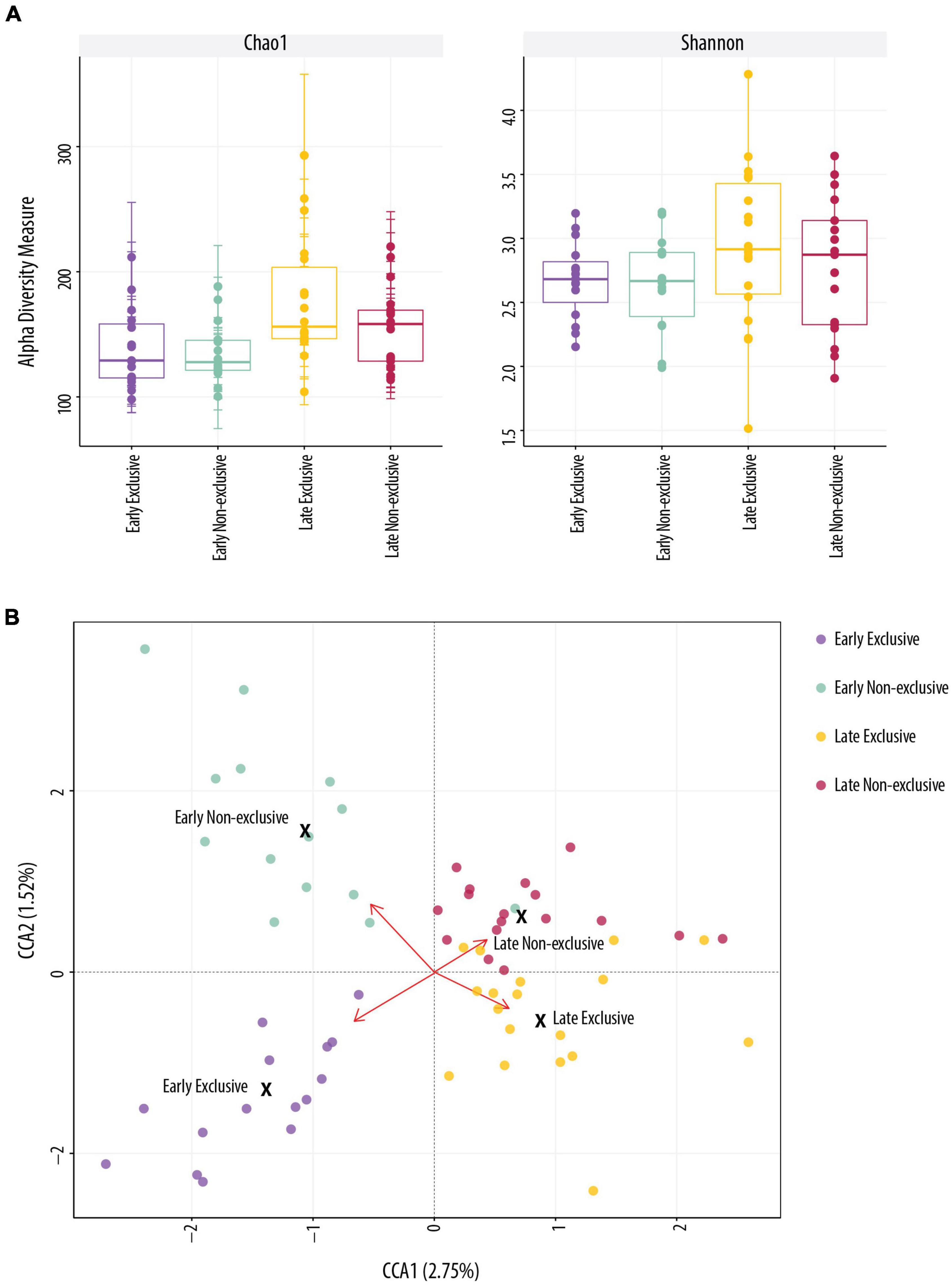
Figure 1. Alpha and beta diversity. (A) Alpha diversity indices were not significantly different in Shannon (t-test, p > 0.05) index across the four groups. Chao 1 was significant (t-test, p < 0.05) between early EBF (n = 15) and late EBF (n = 18). (B) Beta-diversity analysis using constrained correspondence analysis (CCA) ordination representation for each breastfeeding practice (Exclusive and Non-Exclusive) at early and late lactation stages (=0.021).
The differential abundance (DA) analysis between EBF at early and late stage identified 52 significant differentially abundant OTUs (FDR < 0.1), from which 24 OTUs were more abundant in the early stage and 28 OTUs in the late stage (Figure 2A). OTUs from Actinobacteria and Firmicutes were more abundant at the early stage, whereas at the late stage, Bacteroidetes and Proteobacteria were more abundant. The OTUs with the highest fold change (log2 FC ≤ −5) at early stage were the commensal and lactic acid bacteria: Lactobacillus gasseri_1 [false discovery rate (FDR) = 2.16 × 10–8; log2 FC = −11], the oral bacteria Streptococcus mitis_14 (FDR = 2.16 × 10–8; log2 FC = −7), Streptococcus parasanguinis_3 (FDR = 4.5 × 10–4; log2 FC = −7), and Corynebacterium xerosis_1, bacteria commonly found on the skin (FDR = 0.05; log2 FC = −5).
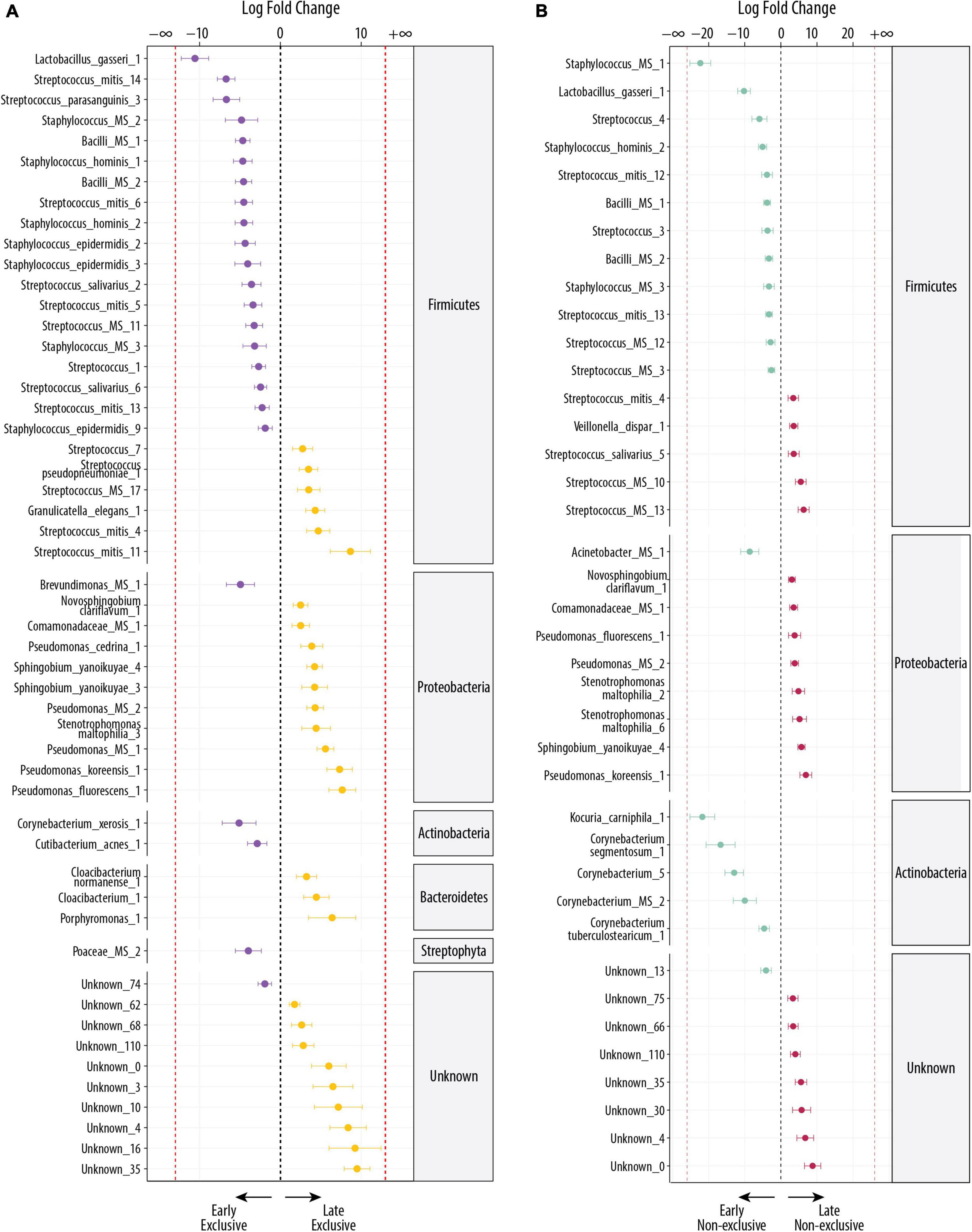
Figure 2. Differentially abundant bacteria associated with the lactation stage. Significantly different OTUs between groups were estimated using DESeq (FDR < 0.1). Species are grouped by phylum and ordered by logFC in each group. The dashed red line indicates “infinite” log fold change, where an OTU had detectable counts in samples from only a single group. (A) Differentially abundant OTUs between the early EBF (n = 15) and late EBF (n = 18) groups. Fifty-two OTUs were differentially abundant, of which 24 were more abundant at early EBF and 28 at late EBF. (B) Differentially abundant OTUs between the early non-EBF (n = 14) and late non-EBF (n = 17) groups. Thirty-nine OTUs were differentially abundant, of which 20 were more abundant at early non-EBF and 19 at late non-EBF.
At late stage, the OTUs with the highest log2 FC (log2 FC ≥ 5), excluding the ones labeled as unknown were Streptococcus mitis_11 (FDR = 0.003; log2 FC = 9), a species commonly found in the oral cavity, followed by OTUs commonly found in the environment, including Pseudomonas fluorescens_1 (FDR = 6 × 10–5; log2 FC = 8), Pseudomonas koreensis_1 (FDR = 4.35 × 10–5; log2 FC = 7), Porphyromonas_1 (FDR = 0.03; log2 FC = 6), and Pseudomonas MS_1 (FDR = 1.82 × 10–6; log2 FC = 6).
The DA analysis between non-EBF at the early and late stage identified 39 significant differential abundant OTUs (FDR < 0.1), from which 20 OTUs were more abundant in the early stage and 19 in the late stage (Figure 2B). The shift observed in EBF groups across early and late stages repeats in the non-EBF groups, where OTUs from Actinobacteria and Firmicutes dominated at the early stage and Proteobacteria at the late stage. In contrast to late EBF, OTUs from Bacteroidetes were not differentially abundant in non-EBF groups. The OTUs with the highest log2 FC (log2 FC ≤ −5) in early non-EBF were a mix of oral and environmental bacteria, but there was still a high presence of bacteria commonly found in oral cavity and human tissues: Staphylococcus MS_1, which shows 100% match with Staphylococcus haemolyticus, a species commonly found in human tissues (FDR = 4.9 × 10–13; log2 FC = −22), Kocuria carniphila_1 (FDR = 5.38 × 10–9; log2 FC = −22), Corynebacterium segmentosum_1 (FDR = 2.84 × 10–4; log2 FC = −17), the OTU Corynebacterium_5 (FDR = 4.61 × 10–6; log2 FC = −13), Lactobacillus gasseri_1 (FDR = 1.01 × 10–7; log2 FC = −10), Corynebacterium MS_2, which could be C. jeikeium, C. amycolatum, or C. xerosis (FDR = 0.01; log2 FC = −10), Acinetobacter MS_1 which could be A. guillouiae or A. lwoffii (FDR = 0.004 × 10–5; log2 FC = −9), the OTU Streptococcus_4 (FDR = 0.02; log2 FC = −6), and Staphylococcus hominis_2 (FDR = 6.88 × 10–5; log2 FC = −5).
The OTUs with the highest log2 FC (log2 FC ≥ 5) in late non-EBF, excluding the OTUs labeled as unknown, were predominantly environmental bacteria: Pseudomonas koreensis_1 (FDR = 8.17 × 10–4; log2 FC = 7), Streptococcus MS_13 (FDR = 2.23 × 10–4; log2 FC = 6), Sphingobium yanoikuyae_4 (FDR = 1.12 × 10–7; log2 FC = 6), Streptococcus MS_10 (FDR = 0.002; log2 FC = 5), and Stenotrophomona maltophilia_6 (FDR = 0.03; log2 FC = 5).
During early lactation, EBF mothers had more DA OTUs than non-EBF mothers. Figure 3A shows the 12 DA in EBF vs non-EBF mothers (FDR < 0.1), where 11 had higher relative abundance in early EBF and 1 had higher relative abundance in early non-EBF. Early EBF was the only group where two OTUs were identified as unique to the group. These were Corynebacterium jeikeium_1 (FDR = 7.08 × 10–4; log2 FC = −16) and Lactobacillales MS_1 (FDR = 0.02; log2 FC = −7). The first one is a species commonly found in the environment, and the second one could be Abiotrophia paraadiacens or Granulicatella adiacens. The rest of the OTUs more abundant in early EBF were a mix of oral, human tissue, and environmental bacteria: Staphylococcus epidermis_8 (FDR = 2.04 × 10–11; log2 FC = −24), Streptococcus mitis_14 (FDR = 2.04 × 10–11; log2 FC = −8), Veillonella_1 (FDR = 0.05; log2 FC = −8), Stenotrophomonas maltophilia_6 (FDR = 0.002; log2 FC = −7), Streptococcus parasanguinis_3 (FDR = 0.002; log2 FC = −7), Stenotrophomonas maltophilia_2 (FDR = 0.004; log2 FC = −6), Streptococcus mitis_5 (FDR = 0.005; log2 FC = −4), Streptococcus_MS_8 (FDR = 0.003; log2 FC = −4), and Pseudomonas_MS_2 (FDR = 0.1; log2 FC = −3), The only OTU DA in early non-EBF was unknown_160 (FDR = 0.1; log2 FC = 4).
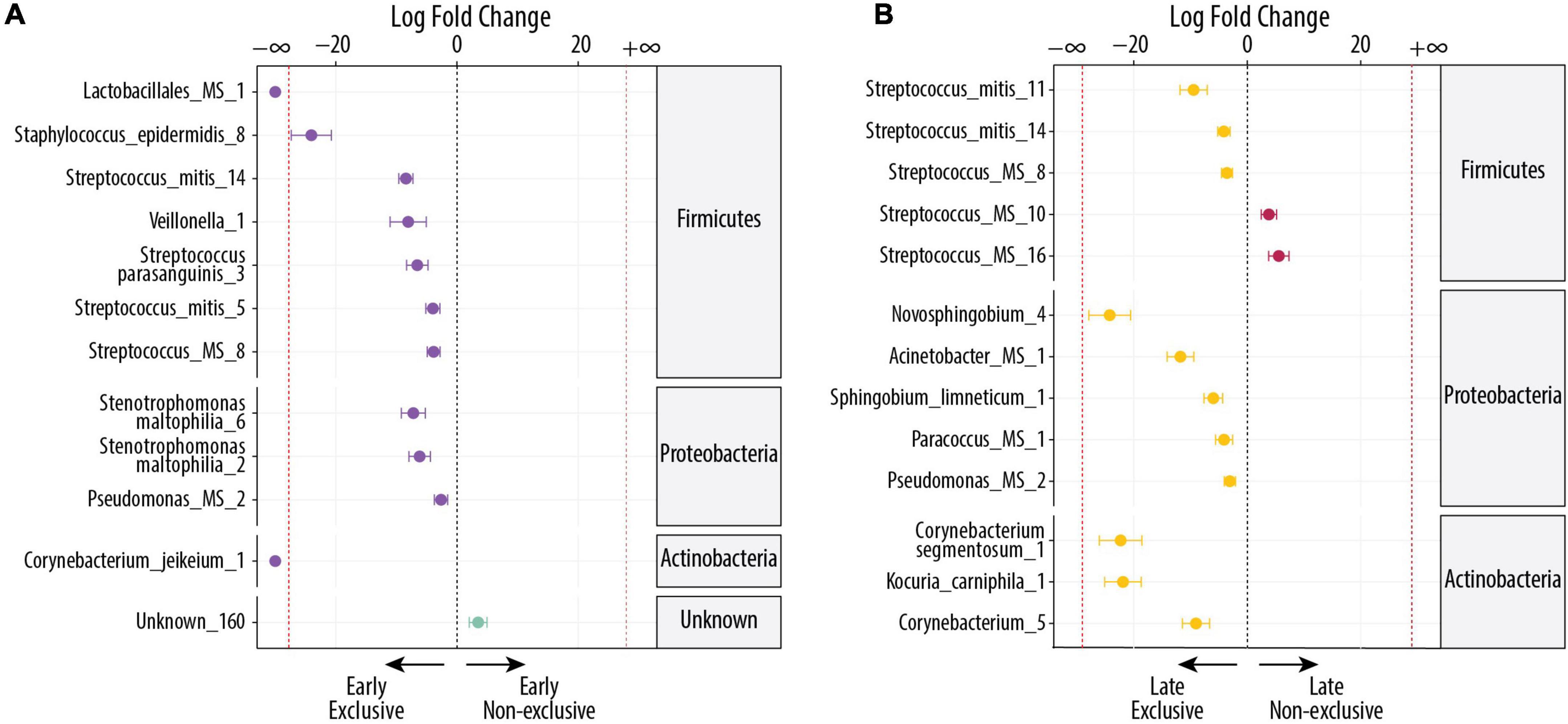
Figure 3. Differentially abundant bacteria associated with EBF and non-EBF. Significantly different OTUs between groups were estimated using DESeq (FDR < 0.1). Species are grouped by phylum and ordered by logFC in each group. The dashed red line indicates “infinite” fold change, where an OTU had detectable counts in samples from only a single group. (A) Differentially abundant OTUs between the early EBF (n = 15) and early non-EBF (n = 14) groups. Twelve OTUs were differentially abundant, of which 20 were more abundant in early EBF and 1 in early non-EBF. (B) Differentially abundant OTUs between the late EBF (n = 18) and late non-EBF (n = 17) groups. Thirteen OTUs were differentially abundant, of which 11 were more abundant in late EBF and 2 in late non-EBF.
During late lactation, the differences in DA OTUs between EBF and non-EBF mothers were similar to early lactation where EBF had more OTUs than non-EBF (Figure 3B). Of the 13 OTUs that were DA (FDR < 0.1), 11 OTUs belonged to late EBF and 2 OTUs to late non-EBF. In relation to the source, six of the 13 OTUs were identified as commonly found in the environment, and the rest (seven) were identified as commensal and commonly found in the oral cavity or human tissues. The OTUs in EBF with the highest log2 FC (log2 FC ≤ −5) were as follows: Novosphingobium_4 (FDR = 2.78 × 10–9; log2 FC = −24), Corynebacterium segmentosum_1 (FDR = 9.96 × 10–8; log2 FC = −22), Kocuria carniphila_1 (FDR = 9.11 × 10–10; log2 FC = −22), Acinetobacter_MS_1 (FDR = 1.97 × 10–5; log2 FC = −12), Streptococcus mitis_11 (FDR = 0.002; log2 FC = −9), Corynebacterium_5 (FDR = 0.004; log2 FC = −9), and Sphingobium limneticum_1 (FDR = 0.006; log2 FC = −6).
The only more abundant OTUs in late non-EBF were Streptococcus MS_16 (FDR = 0.03 × 10–4; log2 FC = 6), which could be S. mitis or S. pneumoniae and Streptococcus MS_10 (FDR = 0.08; log2 FC = 4), which could be S. mitis, S. oralis, S. pneumoniae, or S. pseudopneumoniae; all were OTUs commonly found in the oral cavity or human tissues (Figure 3B).
To our knowledge, this is the first study to analyze the impact of exclusive and non-exclusive breastfeeding on the human milk microbiome at the OTU level and to determine whether different breastfeeding practices further modified the milk microbiome during early and late lactation. Additional novel findings reported in this study include the following. When comparing the lactation stages in EBF, early lactation was associated with a higher relative abundance of commensal, oral, and lactic acid bacteria, whereas at late lactation, we observed several species commonly found in the environment. In non-EBF at early lactation, although there was a mix of bacteria of the oral cavity, bacteria of human tissues origin, and environmental bacteria, bacteria commonly found in the oral cavity and human tissues showed a higher presence than the environmental bacteria. Finally, milk of non-EBF at late lactation had predominantly environmental bacteria.
Streptococcus emerged as a dominant genus among all breastfeeding practices, but the OTUs differed by stage of lactation and by breastfeeding practice. S. mitis was observed in all breastfeeding practices, S. salivarius in early EBF and late non-EBF, and S. parasanguinis in early and late EBF. Streptococcus has been considered part of the “core” microbiota in human milk (Hunt et al., 2011; Cabrera-Rubio et al., 2012; Lackey et al., 2019; Moossavi et al., 2019), and in fact, the few studies that have reported results at the species level have also reported Streptococcus salivarius and Streptococcus mitis as part of the dominant species in human milk (Heikkilä and Saris, 2003; González et al., 2013; Jost et al., 2013). They are known prevalent species in the oral cavity of infants (Könönen, 2000). Of importance, Streptococcus salivarius is capable of suppressing the growth of pathogenic bacteria, such as Staphylococcus aureus, and it has also been reported as a putative probiotic (Damaceno et al., 2017).
In addition to Streptococcus (Hunt et al., 2011; Boix-Amorós et al., 2016), other dominant bacteria observed in the present study aligned with previous findings at the genera level. These included the presence of bacteria from the genera Pseudomonas (Hunt et al., 2011; Jeurink et al., 2013; Boix-Amorós et al., 2016), Sphingobium, and Novosphingobium (Jeurink et al., 2013). However, there is still a debate as to whether Novosphingobium and Pseudomonas are part of the “core” microbiota of human milk or contaminants (Salter et al., 2014; Jiménez et al., 2015). Novosphingobium has been considered a potential laboratory contaminant in sequence-based microbiome analyses (Salter et al., 2014), but it has also been considered part of the “core” microbiome of human milk (Jiménez et al., 2015). Since it was one of the dominant OTUs in our study and it was more abundant in EBF in late lactation, it would suggest that Novosphingobium may be acquired in late lactation through retrograde flow (Ramsay et al., 2004). In relation to Pseudomonas, we observed the OTUs Pseudomonas_MS_2 and P. cedrina in early EBF, Pseudomonas_MS_2 was also more abundant in late EBF, and Pseudomonas koreensis and P. fluorescens were more abundant in early non-EBF. Pseudomonas is a genus that has been reported as dominant in other studies (Hunt et al., 2011; Jeurink et al., 2013; Boix-Amorós et al., 2016), and according to the latest systematic review in human milk, the microbiome Pseudomonas has been found in 50% of the studies analyzed with a relative abundance ranging from <1 to 17% (Zimmermann and Curtis, 2020).
In our study, we observed similarities in the shift of microbial phyla, genera, and species between early and late lactation. In EBF, the bacterial composition shifted from a higher abundance of the phyla Actinobacteria and Firmicutes in early lactation to a higher abundance of Bacteroidetes, Proteobacteria, and unknown bacteria in late lactation. This same shift also occurred in mothers who did not exclusively breastfeed. At the genera level, we observed a shift from Corynebacterium, Staphylococcus, and Streptococcus in early lactation to Novosphingobium, Pseudomonas, Sphingobium, and Stenotrophomonas in late lactation. These findings align with previous results that analyzed the impact of the stage of lactation and confirmed a shift from early to late stage (Gueimonde et al., 2007; Collado et al., 2008; Cabrera-Rubio et al., 2012; Khodayar-Pardo et al., 2014; Latuga et al., 2014; Chen et al., 2018; Gonzalez et al., 2021; Lopez Leyva et al., 2021b). In a study at the species level (Gonzalez et al., 2021), we also observed a shift from Corynebacterium jeikeium, Lactobacillus gasseri, Staphylococcus hominis, Staphylococcus epidermidis, Streptococcus mitis, and Streptococcus parasanguinis species in early lactation to Sphingobium yanoikuyae, Pseudomonas putida, and Stenotrophomonas maltophilia species in late lactation, but this previous study only analyzed the stage of lactation and did not consider the type of breastfeeding practice (Gonzalez et al., 2021). In the present study, we confirmed the presence of distinct species in those who breastfed at early and late lactation, indicating that the stage of lactation is an important modifier of the human milk microbiome. Our conclusion is that this general shift remains regardless of the breastfeeding practices (EBF or non-EBF).
In EBF, more differentially abundant OTUs were observed at both stages of lactation, including lactic acid bacteria and commensal species with potential health benefits and specific functions. EBF had more abundance of lactic acid bacteria, including Granulicatella elegans, Lactobacillales_MS_1, Lactobacillus gasseri, Streptococcus mitis, and Streptococcus parasanguinis. The presence of these species is important due to the known benefits of LAB which include their ability to protect from harmful microorganisms and improve the nutrient acquisition of the host through their enzymatic functions (Vieco-Saiz et al., 2019). LAB can also counteract gastrointestinal infections, enhance lactose metabolism, minimize Helicobacter pylori infections, and strengthen immune responses (Ashraf and Shah, 2014). In fact, Lactobacillus gasseri has been recognized as a putative probiotic (Damaceno et al., 2017). Of note, both Lactobacillus gasseri and Granulicatella elegans were also present in the milk of mothers who did not exclusively breastfeed at early lactation. In contrast, when we compared EBF vs non-EBF at both stages of lactation, the addition of agüitas and/or complementary foods was associated with an overall lower abundance of LAB and commensal bacteria. Collectively, these results support the importance of WHO breastfeeding recommendations to continue exclusive breastfeeding for at least 6 months (World Health Organization [WHO], 2008), by showing a higher differential abundance of commensal and LAB associated with EBF.
The presence of environmental bacteria in human milk remains a controversial area with regard to their source (Salter et al., 2014; Jiménez et al., 2015; Sakwinska et al., 2016; Douglas et al., 2020). A recent review that considered environmental sources of bacteria in human milk found that only 40% of human milk bacteria were isolated from human tissue, while the other 60% were first observed in association with the environment (Togo et al., 2019). In our study, we found environmental bacteria in mothers who exclusively breastfed, including Brevundimonas_MS_1 and Poaceae_MS_2 in early lactation and Novosphingobium clariflavum_1, Comamonadaceae_MS_1, Pseudomonas cedrina_1, Pseudomonas_MS_2, Stenotrophomonas maltophilia_3, Pseudomonas_MS_1, Pseudomonas koreensis_1, Pseudomonas fluroescens_1, Cloacibacterium normanense, and the OTU Cloacibacterium_1 in late lactation, suggesting inoculation of human milk microbiome via either the entero-mammary pathway (Martín et al., 2004) or via retrograde flow (Ramsay et al., 2004). Alternatively, environmental bacteria could be introduced through water, or the herbs used for “agüitas”, the method of administration (spoon, bottle, or cup), or through the complementary foods provided to the infant. In our study, at early and late lactation, mothers who added agüitas and/or complementary foods also had a high abundance of environmental bacteria, including Acinetobacter MS_1, the OTUs Corynebacterium_5, Corynebacterium_MS_2, and Corynebacterium tuberculostearicum_1 in early lactation and Novosphingobium clariflavum_1, Comamonadaceae_MS_1, Pseudomonas fluorescens_1, Pseudomonas_MS_2, Pseudomonas koreensis_1, Stenotrophomona maltophilia_2, and Stenotrophomona maltophilia_ 6 in late lactation. Other studies have reported Pseudomonas and Sphingomonas as part of the “core” human milk microbiome (Hunt et al., 2011; Jiménez et al., 2015). The species Novosphingobium clariflavum, Stenotrophomona maltophilia, Pseudomonas fluroescens, and Pseudomona koreensis have been previously identified as species with extensive hydrocarbon degradation activity (Babalola and Ayangbenro, 2019; Gonzalez et al., 2021). Overall, there were fewer differentially abundant bacteria in the milk of mothers who did not exclusively breastfeed, but the associations and consequences of this on infant health will require further investigation.
This is the first study to characterize the DA of microbial species associated with EBF and to contrast it with shifts in the milk microbiome in mothers who did not exclusively breastfeed in the first 6 months of lactation. Our high-resolution analysis at the species level expands our understanding of the differences in milk microbiome composition, which may not be identifiable at the genera level. Finally, our milk collection methodology involved manual collection rather than the use of breast pumps, which has been shown to affect the milk microbiome analysis (Moossavi et al., 2019). Collectively, the aforementioned factors allowed us to characterize the human milk microbiome composition in a developing country and observe the impact of different breastfeeding practices at early and late lactation on the milk microbiome.
However, we recognize several limitations, including the cross-sectional nature of the study. Self-reported data, which were used to classify the mothers into the defined groups, may have introduced recall bias and could be a limitation. Moreover, we used the selected primers (27F/533R targeting the V1–V3 regions), which have high coverage for amplification of the genus Cutibacterium but not for species within the genus Bifodibacterium (Klindworth et al., 2012; Lopez Leyva et al., 2021a). Also, we are limited by the presence of several unknown or unidentified bacteria in the current libraries. Similarly, the lack of bacteriological studies of the water, soil, agüitas, and complementary foods limits the possibility of fully associating human milk microbiota with these factors.
Understanding the impact of breastfeeding practices and the lactation stage on the milk microbiome is critical for advancing our comprehension of an optimal milk microbiome, and for supporting the current breastfeeding recommendations. Our findings highlight the importance of exclusive breastfeeding in promoting milk microbiota with more commensal bacteria during the first 6 months of lactation compared to non-exclusive breastfeeding. In contrast, non-exclusive breastfeeding before 6 months was associated with lower abundance of commensal and lactic acid bacteria. Collectively, these findings advance our understanding of the factors that affect the milk microbiome, which could provide preliminary evidence to further strengthen the recommendation by health practitioners for “exclusive breastfeeding on demand for 6 months” (World Health Organization [WHO], 2008) in order to establish a healthier infant microbiome.
Finally, this study addressed several informational gaps. These included the impact of feeding agüitas and complementary foods to the infant on the milk microbiome profile of rural indigenous mothers and the recognition of environmental bacteria as part of the milk microbiome. However, there is a need to close other gaps in the scientific literature that still exist, and future studies are required. This need includes uncovering associations of the human milk microbiome with sources of environmental bacteria, with functional properties and with early infant growth and development. Application and investment in more advanced technologies for human milk microbiome analyses will advance our current understanding of the human milk microbiome and its impact on infant health.
Publicly available datasets were analyzed in this study. This data can be found here: raw sequence data has been deposited at the European Genome-Phenome Archive (EGAD00001004160).
The studies involving human participants were reviewed and approved by the Institutional Review Board (IRB) of McGill University and the Center for Studies of Sensory Impairment, Aging and Metabolism (CeSSIAM). The patients/participants provided their written informed consent to participate in this study.
KK, NS, EG, and LL: conceptualization, writing, reviewing, and editing of the manuscript. NS and KK: funding. All authors contributed to the article and approved the submitted version.
This study was supported by the Natural Sciences and Engineering Research Council of Canada Discovery (Grant #RGPIN-2016-0496) to KK and Consejo Mexiquense de Ciencia y Tecnología to LL.
Special thanks to McGill University and Genome Quebec Innovation Centre for technical assistance with 16S rRNA sequencing. We thank H. Wren, A. M. Chomat, and the CeSSIAM field team for sample collection and the participating mothers for providing valuable milk samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.885588/full#supplementary-material
Supplementary File 1 | Decontam ran on this dataset identified 1 OTU as putative contamination out of 1,505 OTUs. As expected, this OTU was not selected by DESeq2 as a candidate for differential abundance, due to low prevalence and high sparsity.
Supplementary File 2 | A total of 503 OTUs were assembled and captured 77,827 sequence reads across all 64 human milk samples. These could be annotated as 287 OTUs, 134 genera, and 76 family or higher taxa, as well as 109 which could not be recognized as 99% similar (in both identity and coverage) to any known taxa and were labeled as unknown.
Supplementary File 3 | Main factors were projected onto an unconstrained ordination diagram (NMDS), and each variable regression was independently tested by Monte Carlo permutation (envfit function from R package Vegan). The goodness of fit obtained is described in the table.
Supplementary File 4 | The indices Chao 1, Shannon, and Observed used to estimate alpha diversity identified differences between early and late lactation for EBF and non-EBF mothers. Alpha-diversity indices were not significantly different in Shannon (t-test, p > 0.05) across the four groups. Differences in alpha diversity using Chao 1 were significant between early non-EBF and late EBF (FDR = 0.01) and between early EBF and late EBF (FDR = 0.04) groups, and using Observed, there were significant differences between early EBF and late EBF (FDR = 0.002), early non-EBF and late EBF (FDR = 0.005), and late EBF and late non-EBF (FDR = 0.08).
BLASTn, basic local alignment search tool; CAP, constrained analysis of principal coordinates; CeSSIAM, Center for Studies of Sensory Impairment, Aging, and Metabolism; DA, differential abundance; DNA, deoxyribonucleic acid; ebf, exclusive breastfeeding; FC, fold change; FDR, false discovery rate; LMIC, low- and middle-income countries; NCBI, National Center for Biotechnology Information; OTU, operational taxonomic unit; PCR, polymerase chain reaction; WHO, World Health Organization.
Afeiche, M. C., Villalpando-Carrión, S., Reidy, K. C., Fries, L. R., and Eldridge, A. L. (2018). Many Infants and Young Children Are Not Compliant with Mexican and International Complementary Feeding Recommendations for Milk and Other Beverages. Nutrients 10:466. doi: 10.3390/nu10040466
Anders, S., McCarthy, D. J., Chen, Y., Okoniewski, M., Smyth, G. K., Huber, W., et al. (2013). Count-Based Differential Expression Analysis of RNA Sequencing Data Using R and Bioconductor. Nat. Protocols 8, 1765–1786. doi: 10.1038/nprot.2013.099
Ashraf, R., and Shah, N. P. (2014). Immune System Stimulation by Probiotic Microorganisms. Crit. Rev. Food Sci. Nutr. 54, 938–956. doi: 10.1080/10408398.2011.619671
Babalola, O. O., and Ayangbenro, A. S. (2019). Draft Genome Sequence of Pseudomonas Koreensis Strain AB36, Isolated from Gold Mining Soil. Microbiol. Reso. Announce. 8:e175–e119. doi: 10.1128/MRA.00175-19
Blum, W. E. H., Zechmeister-Boltenstern, S., and Keiblinger, K. M. (2019). Does Soil Contribute to the Human Gut Microbiome? Microorganisms 7:287. doi: 10.3390/microorganisms7090287
Boix-Amorós, A., Collado, M. C., and Mira, A. (2016). Relationship between Milk Microbiota, Bacterial Load, Macronutrients, and Human Cells during Lactation. Front. Microbiol. 7:492. doi: 10.3389/fmicb.2016.00492
Bokulich, N. A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H. D., et al. (2016). Antibiotics, Birth Mode, and Diet Shape Microbiome Maturation during Early Life. Sci. Trans. Med. 8:ra82–ra343. doi: 10.1126/scitranslmed.aad7121
Brereton, N. J. B., Gonzalez, E., Desjardins, D., Labrecque, M., and Pitre, F. E. (2020). Co-Cropping with Three Phytoremediation Crops Influences Rhizosphere Microbiome Community in Contaminated Soil. Sci. Total Environ. 711:135067. doi: 10.1016/j.scitotenv.2019.135067
Cabrera-Rubio, R., Collado, M. C., Laitinen, K., Salminen, S., Isolauri, E., and Mira, A. (2012). The Human Milk Microbiome Changes over Lactation and Is Shaped by Maternal Weight and Mode of Delivery. Am. J. Clin. Nutr. 96, 544–551. doi: 10.3945/ajcn.112.037382
Carvalho-Ramos, I. I., Duarte, R. T. D., Brandt, K. G., Martinez, M. B., and Taddei, C. R. (2018). Breastfeeding Increases Microbial Community Resilience. J. De Pediatria 94, 258–267. doi: 10.1016/j.jped.2017.05.013
Chen, P. W., Lin, Y. L., and Huang, M. S. (2018). Profiles of Commensal and Opportunistic Bacteria in Human Milk from Healthy Donors in Taiwan. J. Food Drug Anal. 26, 1235–1244. doi: 10.1016/j.jfda.2018.03.004
Chomat, A. M., Solomons, N. W., Koski, K. G., Wren, H. M., Vossenaar, M., and Scott, M. E. (2015). Quantitative Methodologies Reveal a Diversity of Nutrition, Infection/Illness, and Psychosocial Stressors During Pregnancy and Lactation in Rural Mam -Mayan Mother–Infant Dyads From the Western Highlands of Guatemala. Food Nutr. Bull. 36, 415–440. doi: 10.1177/0379572115610944
Clemente, J. C., Pehrsson, E. C., Blaser, M. J., Sandhu, K., Gao, Z., Wang, B., et al. (2015). The Microbiome of Uncontacted Amerindians. Sci. Adv. 1:e1500183. doi: 10.1126/sciadv.1500183
Collado, M. C., Isolauri, E., Laitinen, K., and Salminen, S. (2008). Distinct Composition of Gut Microbiota during Pregnancy in Overweight and Normal-Weight Women. Am. J. Clin. Nutr. 88, 894–899. doi: 10.1093/ajcn/88.4.894
Damaceno, Q. S., Souza, J. P., Nicoli, J. R., Paula, R. L., Assis, G. B., Figueiredo, H. C., et al. (2017). Evaluation of Potential Probiotics Isolated from Human Milk and Colostrum. Probiotics and Antimicrob. Proteins 9, 371–379. doi: 10.1007/s12602-017-9270-1
Davé, V., Street, K., Francis, S., Bradman, A., Riley, L., Eskenazi, B., et al. (2016). Bacterial Microbiome of Breast Milk and Child Saliva from Low-Income Mexican-American Women and Children. Pediatric Res. 79, 846–854. doi: 10.1038/pr.2016.9
Deming, D. M., Afeiche, M. C., Reidy, K. C., Alison, L. E., and Salvador, V.-C. (2015). Early Feeding Patterns among Mexican Babies: Findings from the 2012 National Health and Nutrition Survey and Implications for Health and Obesity Prevention. BMC Nutr. 1:40. doi: 10.1186/s40795-015-0035-5
Ding, M., Qi, C., Yang, Z., Jiang, S., Bi, Y., Lai, J., et al. (2019). Geographical Location Specific Composition of Cultured Microbiota and Lactobacillus Occurrence in Human Breast Milk in China. Food Function 10, 554–564. doi: 10.1039/C8FO02182A
Doak, C. M., van der Starre, R. E., van Beusekom, I., Campos Ponce, M., Vossenaar, M., and Solomons, N. W. (2013). Earlier Introduction of Aguitas Is Associated with Higher Risk of Stunting in Infants and Toddlers in the Western Highlands of Guatemala. Am. J. Clin. Nutr. 97, 631–636. doi: 10.3945/ajcn.112.047621
Douglas, C. A., Ivey, K. L., Papanicolas, L. E., Best, K. P., Muhlhausler, B. S., and Rogers, G. B. (2020). DNA Extraction Approaches Substantially Influence the Assessment of the Human Breast Milk Microbiome. Sci. Rep. 10:123. doi: 10.1038/s41598-019-55568-y
Drago, L., Toscano, M., De Grandi, R., Grossi, E., Padovani, E. M., and Peroni, D. G. (2017). Microbiota Network and Mathematic Microbe Mutualism in Colostrum and Mature Milk Collected in Two Different Geographic Areas: Italy versus Burundi. ISME J. 11, 875–884. doi: 10.1038/ismej.2016.183
Drell, T., Štšepetova, J., Simm, J., Rull, K., Aleksejeva, A., Antson, A., et al. (2017). The Influence of Different Maternal Microbial Communities on the Development of Infant Gut and Oral Microbiota. Sci. Rep. 7:9940. doi: 10.1038/s41598-017-09278-y
Fitzstevens, J. L., Smith, K. C., Hagadorn, J. I., Caimano, M. J., Matson, A. P., and Brownell, E. A. (2017). Systematic Review of the Human Milk Microbiota. Nutr. Clin. Practice 32, 354–364. doi: 10.1177/0884533616670150
Gonzalez, E., Brereton, N. J., Marleau, J., Guidi Nissim, W., Labrecque, M., Pitre, F. E., et al. (2015). Meta-Transcriptomics Indicates Biotic Cross-Tolerance in Willow Trees Cultivated on Petroleum Hydrocarbon Contaminated Soil. BMC Plant Biol. 15:246. doi: 10.1186/s12870-015-0636-9
Gonzalez, E., Brereton, N. J. B., Li, C., Lopez Leyva, L., Solomons, N. W., Agellon, L. B., et al. (2021). Distinct Changes Occur in the Human Breast Milk Microbiome Between Early and Established Lactation in Breastfeeding Guatemalan Mothers. Front. Microbiol. 12:557180. doi: 10.3389/fmicb.2021.557180
Gonzalez, E., Pitre, F. E., and Brereton, N. J. B. (2019). ANCHOR: A 16S RRNA Gene Amplicon Pipeline for Microbial Analysis of Multiple Environmental Samples. Environ. Microbiol. 21, 2440–2468. doi: 10.1111/1462-2920.14632
González, R., Maldonado, A., Martín, V., Mandomando, I., Fumadó, V., Metzner, K. J., et al. (2013). Breast Milk and Gut Microbiota in African Mothers and Infants from an Area of High HIV Prevalence. PLoS One 8:e80299. doi: 10.1371/journal.pone.0080299
Gueimonde, M., Laitinen, K., Salminen, S., and Isolauri, E. (2007). Breast Milk: A Source of Bifidobacteria for Infant Gut Development and Maturation? Neonatology 92, 64–66. doi: 10.1159/000100088
Hanski, I., von Hertzen, L., Fyhrquist, N., Koskinen, K., Torppa, K., Laatikainen, T., et al. (2012). Environmental Biodiversity, Human Microbiota, and Allergy Are Interrelated. Proc. Natl. Acad. Sci. 109, 8334–8339. doi: 10.1073/pnas.1205624109
Heikkilä, M. P., and Saris, P. E. (2003). Inhibition of Staphylococcus Aureus by the Commensal Bacteria of Human Milk. J. Appl. Microbiol. 95, 471–478. doi: 10.1046/j.1365-2672.2003.02002.x
Hermansson, H., Kumar, H., Collado, M. C., Salminen, S., Isolauri, E., and Rautava, S. (2019). Breast Milk Microbiota Is Shaped by Mode of Delivery and Intrapartum Antibiotic Exposure. Front. Nutr. 6:4. doi: 10.3389/fnut.2019.00004
Ho, N. T., Li, F., Lee-Sarwar, K. A., Tun, H. M., Brown, B. P., Pannaraj, P. S., et al. (2018). Meta-Analysis of Effects of Exclusive Breastfeeding on Infant Gut Microbiota across Populations. Nat. Commun. 9:4169. doi: 10.1038/s41467-018-06473-x
Hunt, K. M., Foster, J. A., Forney, L. J., Schütte, U. M., Beck, D. L., Abdo, Z., et al. (2011). Characterization of the Diversity and Temporal Stability of Bacterial Communities in Human Milk. PLoS One 6:e21313. doi: 10.1371/journal.pone.0021313
Jeurink, P. V., van Bergenhenegouwen, J., Jiménez, E., Knippels, L. M., Fernández, L., Garssen, J., et al. (2013). Human Milk: A Source of More Life than We Imagine. Benef. Microbes 4, 17–30. doi: 10.3920/BM2012.0040
Jiménez, E., de Andrés, J., Manrique, M., Pareja-Tobes, P., Tobes, R., Martínez-Blanch, J. F., et al. (2015). Metagenomic Analysis of Milk of Healthy and Mastitis-Suffering Women. J. Hum. Lactation 31, 406–415. doi: 10.1177/0890334415585078
Jonsson, V., Österlund, T., Nerman, O., and Kristiansson, E. (2016). Statistical Evaluation of Methods for Identification of Differentially Abundant Genes in Comparative Metagenomics. BMC Genomics 17:78. doi: 10.1186/s12864-016-2386-y
Jost, T., Lacroix, C., Braegger, C., and Chassard, C. (2013). Assessment of Bacterial Diversity in Breast Milk Using Culture-Dependent and Culture-Independent Approaches. Br. J. Nutr. 110, 1253–1262. doi: 10.1017/S0007114513000597
Khodayar-Pardo, P., Mira-Pascual, L., Collado, M. C., and Martínez-Costa, C. (2014). Impact of Lactation Stage, Gestational Age and Mode of Delivery on Breast Milk Microbiota. J. Perinatol. 34, 599–605. doi: 10.1038/jp.2014.47
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Könönen, E. (2000). Development of Oral Bacterial Flora in Young Children. Ann. Med. 32, 107–112. doi: 10.3109/07853890009011759
Kumar, H., du Toit, E., Kulkarni, A., Aakko, J., Linderborg, K. M., Zhang, Y., et al. (2016). Distinct Patterns in Human Milk Microbiota and Fatty Acid Profiles Across Specific Geographic Locations. Front. Microbiol. 7:1619. doi: 10.3389/fmicb.2016.01619
Lackey, K. A., Williams, J. E., Meehan, C. L., Zachek, J. A., Benda, E. D., Price, W. J., et al. (2019). What’s Normal? Microbiomes in Human Milk and Infant Feces Are Related to Each Other but Vary Geographically: The INSPIRE Study. Front. Nutr. 6:45. doi: 10.3389/fnut.2019.00045
Latuga, M. S., Stuebe, A., and Seed, P. C. (2014). A Review of the Source and Function of Microbiota in Breast Milk. Semin. Reproductive Med. 32, 068–073. doi: 10.1055/s-0033-1361824
Li, C., Solomons, N. W., Scott, M. E., and Koski, K. G. (2016). Minerals and Trace Elements in Human Breast Milk Are Associated with Guatemalan Infant Anthropometric Outcomes within the First 6 Months. J. Nutr. 146, 2067–2074. doi: 10.3945/jn.116.232223
Li, C., Solomons, N. W., Scott, M. E., and Koski, K. G. (2019). Anthropometry before Day 46 and Growth Velocity before 6 Months of Guatemalan Breastfed Infants Are Associated with Subclinical Mastitis and Milk Cytokines, Minerals, and Trace Elements. J. Nutr. 149, 1651–1659. doi: 10.1093/jn/nxz109
Li, S. W., Watanabe, K., Hsu, C. C., Chao, S. H., Yang, Z. H., Lin, Y. J., et al. (2017). Bacterial Composition and Diversity in Breast Milk Samples from Mothers Living in Taiwan and Mainland China. Front. Microbiol. 8:965. doi: 10.3389/fmicb.2017.00965
Lopez Leyva, L., Brereton, N. J. B., and Koski, K. G. (2021a). Emerging Frontiers in Human Milk Microbiome Research and Suggested Primers for 16S RRNA Gene Analysis. Comput. Struct. Biotechnol. J. 19, 121–133. doi: 10.1016/j.csbj.2020.11.057
Lopez Leyva, L., Gonzalez, E., Li, C., Ajeeb, T., Solomons, N. W., Agellon, L. B., et al. (2021b). Human Milk Microbiota in an Indigenous Population Is Associated with Maternal Factors, Stage of Lactation, and Breastfeeding Practices. Curr.Dev. Nutr. 5:nzab013. doi: 10.1093/cdn/nzab013
Love, M. I., Anders, S., Kim, V., and Huber, W. (2016). RNA-Seq Workflow: Gene-Level Exploratory Analysis and Differential Expression. F1000 Res. 4:1070. doi: 10.12688/f1000research.7035.2
Love, M. I., Huber, W., and Anders, S. (2014). Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lyons, K. E., Fouhy, F., O’ Shea, C. A., Ryan, C. A., Dempsey, E. M., Ross, R. P., et al. (2021). Effect of Storage, Temperature, and Extraction Kit on the Phylogenetic Composition Detected in the Human Milk Microbiota. Microbiol. 10:e1127. doi: 10.1002/mbo3.1127
Martin, R., Makino, H., Cetinyurek Yavuz, A., Ben-Amor, K., Roelofs, M., Ishikawa, E., et al. (2016). Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS One 11:e0158498. doi: 10.1371/journal.pone.0158498
Martín, R., Susana, L., Carlota, R., Esther, J., Marìa, L. M., Mónica, O., et al. (2004). The Commensal Microflora of Human Milk: New Perspectives for Food Bacteriotherapy and Probiotics. Trends Food Sci. Technol. 15, 121–127. doi: 10.1016/j.tifs.2003.09.010
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meehan, C. L., Lackey, K. A., Hagen, E. H., Williams, J. E., Roulette, J., Helfrecht, C., et al. (2018). Social Networks, Cooperative Breeding, and the Human Milk Microbiome. Am. J. of Hum. Biol. 30:e23131. doi: 10.1002/ajhb.23131
Minerbi, A., Gonzalez, E., Brereton, N. J. B., Anjarkouchian, A., Dewar, K., Fitzcharles, M. A., et al. (2019). Altered Microbiome Composition in Individuals with Fibromyalgia. PAIN 160, 2589–2602. doi: 10.1097/j.pain.0000000000001640
Moossavi, S., Sepehri, S., Robertson, B., Bode, L., Goruk, S., Field, C. J., et al. (2019). Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 25, 324e–335e. doi: 10.1016/j.chom.2019.01.011
Mueller, N. T., Bakacs, E., Combellick, J., Grigoryan, Z., and Dominguez-Bello, M. G. (2015). The Infant Microbiome Development: Mom Matters. Trends Mol. Med. 21, 109–117. doi: 10.1016/j.molmed.2014.12.002
Oksanen, J. F., Guillaume, B., Michael, F., Roeland, K., Pierre, L., and Peter, R. M. (2020). Vegan: Community Ecology Package (version 2.5-7). Available online at: https://CRAN.R-project.org/package=vegan. (accessed March 9, 2021).
Pannaraj, P. S., Li, F., Cerini, C., Bender, J. M., Yang, S., Rollie, A., et al. (2017). Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatrics 171:647. doi: 10.1001/jamapediatrics.2017.0378
Patel, S. H., Vaidya, Y. H., Patel, R. J., Pandit, R. J., Joshi, C. G., and Kunjadiya, A. P. (2017). Culture Independent Assessment of Human Milk Microbial Community in Lactational Mastitis. Sci. Rep. 7:7804. doi: 10.1038/s41598-017-08451-7
Patil, C. L., Turab, A., Ambikapathi, R., Nesamvuni, C., Chandyo, R. K., Bose, A., et al. (2015). Early Interruption of Exclusive Breastfeeding: Results from the Eight-Country MAL-ED Study. J. Health Populat. Nutr. 34:10. doi: 10.1186/s41043-015-0004-2
Ramsay, D. T., Kent, J. C., Owens, R. A., and Hartmann, P. E. (2004). Ultrasound Imaging of Milk Ejection in the Breast of Lactating Women. Pediatrics 113, 361–367. doi: 10.1542/peds.113.2.361
Rodríguez-Ramírez, S., Muñoz-Espinosa, A., Rivera, J. A., González-Castell, D., and González de Cosío, T. (2016). Mexican Children under 2 Years of Age Consume Food Groups High in Energy and Low in Micronutrients. J. Nutr. 146, 1916S–1923S. doi: 10.3945/jn.115.220145
Sakwinska, O., Moine, D., Delley, M., Combremont, S., Rezzonico, E., Descombes, P., et al. (2016). Microbiota in Breast Milk of Chinese Lactating Mothers. PLoS One 11:e0160856. doi: 10.1371/journal.pone.0160856
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and Laboratory Contamination Can Critically Impact Sequence-Based Microbiome Analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., and Hollister, E. B. (2009). Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Togo, A., Dufour, J. C., Lagier, J. C., Dubourg, G., Raoult, D., and Million, M. (2019). Repertoire of Human Breast and Milk Microbiota: A Systematic Review. Future Microbiol. 14, 623–641. doi: 10.2217/fmb-2018-0317
UNICEF. (2019). Infant and Young Child Feeding.” UNICEF DATA. 2019. Availabe online at: https://data.unicef.org/topic/nutrition/infant-and-young-child-feeding/. (accessed January 25, 2020).
Urbaniak, C., Angelini, M., Gloor, G. B., and Reid, G. (2016). Human Milk Microbiota Profiles in Relation to Birthing Method, Gestation and Infant Gender. Microbiome 4:1. doi: 10.1186/s40168-015-0145-y
Urbaniak, C., Cummins, J., Brackstone, M., Macklaim, J. M., Gloor, G. B., Baban, C. K., et al. (2014). Microbiota of Human Breast Tissue. Appl. Environ. Microbiol. 80, 3007–3014. doi: 10.1128/AEM.00242-14
Vaidya, Y. H., Patel, S. H., Patel, R. J., Pandit, R. J., Joshi, C. G., and Kunjadia, A. P. (2017). Human Milk Microbiome in Urban and Rural Populations of India. Meta Gene 13, 13–22. doi: 10.1016/j.mgene.2017.04.001
Vieco-Saiz, N., Belguesmia, Y., Raspoet, R., Auclair, E., Padgett, C., Bailey, C., et al. (2019). Benefits and Inputs From Lactic Acid Bacteria and Their Bacteriocins as Alternatives to Antibiotic Growth Promoters During Food-Animal Production. Front. Microbiol. 10:57. doi: 10.3389/fmicb.2019.00057
Wall, D. H., Nielsen, U. N., and Six, J. (2015). Soil Biodiversity and Human Health. Nature 528, 69–76. doi: 10.1038/nature15744
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Williams, J. E., Carrothers, J. M., Lackey, K. A., Beatty, N. F., York, M. A., Brooker, S. L., et al. (2017). “Human Milk Microbial Community Structure Is Relatively Stable and Related to Variations in Macronutrient and Micronutrient Intakes in Healthy Lactating Women. J. Nutr. 147, 1739–1748. doi: 10.3945/jn.117.248864
World Health Organization [WHO]. (2008). Indicators for Assessing Infant and Young Child Feeding Practices: Conclusions of a Consensus Meeting Held 6-8 November 2007 in Washington D.C., USA. Washington, DC: World Health Organization (WHO).
Wren, H. M., Solomons, N. W., Chomat, A. M., Scott, M. E., and Koski, K. G. (2015). Cultural Determinants of Optimal Breastfeeding Practices among Indigenous Mam -Mayan Women in the Western Highlands of Guatemala. J. Hum. Lactation 31, 172–184. doi: 10.1177/0890334414560194
Wren-Atilola, H. M., Solomons, N. W., Scott, M. E., and Koski, K. G. (2018). Infant Growth Faltering Linked to Subclinical Mastitis, Maternal Faecal-Oral Contamination, and Breastfeeding. Maternal Child Nutr. 15:e12756. doi: 10.1111/mcn.12756
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human Gut Microbiome Viewed across Age and Geography. Nature 486, 222–227. doi: 10.1038/nature11053
Keywords: human milk microbiome, exclusive breastfeeding, non-exclusive breastfeeding, environmental bacteria, 16S rRNA sequencing
Citation: Lopez Leyva L, Gonzalez E, Solomons NW and Koski KG (2022) Human milk microbiome is shaped by breastfeeding practices. Front. Microbiol. 13:885588. doi: 10.3389/fmicb.2022.885588
Received: 28 February 2022; Accepted: 08 August 2022;
Published: 08 September 2022.
Edited by:
Margarita Aguilera, University of Granada, SpainReviewed by:
Fiona Fouhy, Munster Technological University, IrelandCopyright © 2022 Lopez Leyva, Gonzalez, Solomons and Koski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristine G. Koski, a3Jpc3RpbmUua29za2lAbWNnaWxsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.