 Coco Koedooder1,2,3*
Coco Koedooder1,2,3* Etai Landou4
Etai Landou4 Futing Zhang1,2
Futing Zhang1,2 Siyuan Wang1,2
Siyuan Wang1,2 Subhajit Basu1,2,5
Subhajit Basu1,2,5 Ilana Berman-Frank6
Ilana Berman-Frank6 Yeala Shaked1,2
Yeala Shaked1,2 Maxim Rubin-Blum3
Maxim Rubin-Blum3- 1The Fredy and Nadine Herrmann Institute of Earth Sciences, Hebrew University of Jerusalem, Jerusalem, Israel
- 2The Interuniversity Institute for Marine Sciences in Eilat, Eilat, Israel
- 3Israel Oceanographic and Limnological Research, Haifa, Israel
- 4Mina and Everard Goodman Faculty of Life Sciences, Bar Ilan University, Ramat Gan, Israel
- 5Microsensor Research Group, Max Planck Institute for Marine Microbiology, Bremen, Germany
- 6Department of Marine Biology, Leon H. Charney School of Marine Sciences, University of Haifa, Haifa, Israel
Trichodesmium are filamentous cyanobacteria of key interest due to their ability to fix carbon and nitrogen within an oligotrophic marine environment. Their blooms consist of a dynamic assemblage of subpopulations and colony morphologies that are hypothesized to occupy unique niches. Here, we assessed the poorly studied diversity of Trichodesmium in the Red Sea, based on metagenome-assembled genomes (MAGs) and hetR gene-based phylotyping. We assembled four non-redundant MAGs from morphologically distinct Trichodesmium colonies (tufts, dense and thin puffs). Trichodesmium thiebautii (puffs) and Trichodesmium erythraeum (tufts) were the dominant species within these morphotypes. While subspecies diversity is present for both T. thiebautii and T. erythraeum, a single T. thiebautii genotype comprised both thin and dense puff morphotypes, and we hypothesize that this phenotypic variation is likely attributed to gene regulation. Additionally, we found the rare non-diazotrophic clade IV and V genotypes, related to Trichodesmium nobis and Trichodesmium miru, respectively that likely occurred as single filaments. The hetR gene phylogeny further indicated that the genotype in clade IV could represent the species Trichodesmium contortum. Importantly, we show the presence of hetR paralogs in Trichodesmium, where two copies of the hetR gene were present within T. thiebautii genomes. This may lead to the overestimation of Trichodesmium diversity as one of the copies misidentified T. thiebautii as Trichodesmium aureum. Taken together, our results highlight the importance of re-assessing Trichodesmium taxonomy while showing the ability of genomics to capture the complex diversity and distribution of Trichodesmium populations.
Introduction
Trichodesmium is a genus of filamentous cyanobacteria known for its ability to form large visible surface blooms in tropical and subtropical regions of the ocean. Having first been described in 1770 by James Cook (Transcription of National Library of Australia, 2004), Trichodesmium have been extensively studied due to their ability to form blooms of large biomass supporting marine food webs through their nitrogen and carbon fixing capabilities (Dugdale et al., 1961) and subsequent relevance to biogeochemical cycles within oligotrophic marine environments (Capone et al., 1997; McKinna, 2015; Capone, 2021).
Trichodesmium blooms are dynamic over space and time, typically consisting of a complex assemblage of several different subpopulations and colony morphologies that are predicted to exhibit unique ecological lifestyles (Hansel et al., 2016; Gradoville et al., 2017; Eichner et al., 2019; Delmont, 2021; Held et al., 2021; Wang et al., 2021). For example, the comparison of two distinct puff-shaped morphotypes termed “dense” and “thin” colonies, isolated from the Red Sea displayed a remarkable heterogeneity in their preference to capture and center dust (Wang et al., 2021). While colony morphotypes cannot always be linked to different genotypes or species (Rouco et al., 2016; Gradoville et al., 2017) genomic information regarding Trichodesmium colonies in this region is lacking in comparison to studies conducted in the Atlantic and Pacific Oceans.
The diversity of Trichodesmium bloom-forming populations was first assessed via colony morphology (Janson et al., 1995). With the advent of molecular techniques, the use of single-marker genes represented a more accurate and consistent phylogenetic technique (McManus and Katz, 2009). Marker genes that have been used to assess Trichodesmium diversity include the 16S rRNA, hetR, a regulatory gene for the development of heterocysts, and nifH, which encodes the nitrogenase iron protein. The hetR gene is considered a good marker to assess Trichodesmium diversity as it is more variable (10%) in comparison to genetic markers such as 16S ribosomal RNA (2–3%) or nifH (2%; Orcutt et al., 2002; Lundgren et al., 2005).
Phylogenetic analysis using hetR as a marker gene separates Trichodesmium into four distinct clades: clade I (Trichodesmium thiebautii, Trichodesmium hildebrandtii, Trichodesmium tenue, and Trichodesmium pelagicum), clade II (Trichodesmium aureum), clade III (Trichodesmium erythraeum, Trichodesmium havanum), and clade IV (Trichodesmium contortum, Trichodesmium tenue; Lundgren et al., 2005; Hynes et al., 2012). Recently, a fifth clade (clade V) consisting of the non-diazotrophic Trichodesmium miru was proposed by Delmont (2021). Currently, genomes of clades I, III, (and V) are available, including that of the culturable clade III lineage Trichodesmium erythraeum IMS101 (Walworth et al., 2015).
To better understand the taxonomic diversity of Trichodesmium in the Red Sea, we isolated colonies from the Gulf of Aqaba and compared the resulting metagenome-assembled genomes (MAGs) to those of other Trichodesmium populations from the Indian, Pacific and Atlantic Oceans. We confirmed the presence of these genomes in the Red Sea based on amplicon sequencing of the hetR gene, and, in light of our findings, evaluated the ability of this marker gene to capture Trichodesmium diversity.
Materials and Methods
Trichodesmium Sampling and Extraction
To assess the Trichodesmium population of the Red Sea, Trichodesmium colonies were handpicked and separated into three distinct morphotypes (~100–200 colonies each) throughout the winter bloom (November 2020) using a 100 μm phytoplankton net at 20 m depth in the Gulf of Aqaba (Eilat, Israel; 29.56°N, 34.95°E; Figure 1). Colonies were washed three times in 0.2 μm filtered seawater before being filtered on a 0.2 μm PCC filter using a vacuum pump. Filters were frozen in liquid N2 and kept at −80°C until analysis.
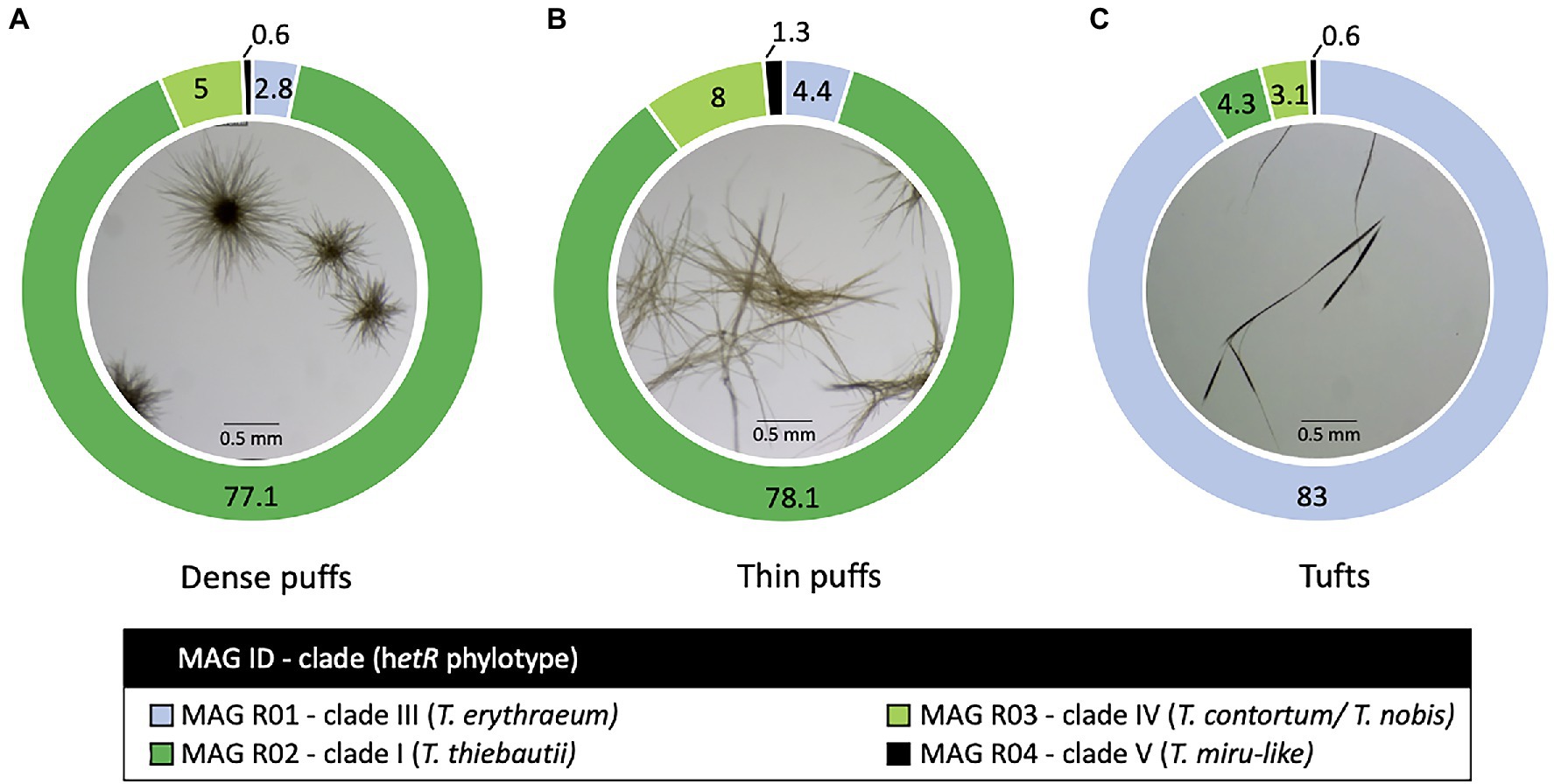
Figure 1. Trichodesmium colony morphotypes and their relative read abundance (%) in the Red Sea. Both (A) dense and (B) thin puff-shaped morphotypes, primarily consisted of Trichodesmium thiebautii genotype (dark green; MAG R02), while tuft-shaped morphotypes (C) were dominated by Trichodesmium erythraeum (light blue; MAG R01). All morphotypes contained low read abundance of non-diazotrophic MAG R03 (light green; MAG R03) and MAG R04 (black; MAG R04) genotypes, clustering within clade IV and V respectively.
DNA was extracted from three Trichodesmium samples using the DNAeasy Plant Mini Kit (Qiagen) following the manufacturer’s instructions. Metagenomic libraries were prepared at HyLabs (Rehovot, Israel) and sequenced with 30–60 million 2 × 150 bp reads on Illumina HiSeq 3000 at GENEWIZ (Leipzig, Germany). The metagenomic samples can be found under the accession numbers SRR17940154-6.
Metagenomic Analysis
Trichodesmium MAGs were binned from assemblies based on the raw reads from this study (PRJNA804487) and those from the previously published metagenomes of Trichodesmium from the Pacific (PRJNA435427; PRJNA358796) and Atlantic (PRJNA330990) Oceans (Frischkorn et al., 2017, 2018a; Gradoville et al., 2017). Metagenomes were assembled, annotated, and binned using the ATLAS (v2) pipeline (Kieser et al., 2020; Supplementary R Markdown File). Briefly, raw sequences underwent quality control through the BBTools suite (Bushnell, 2014; Bushnell et al., 2017) and were assembled using metaSPAdes (Nurk et al., 2017; k-mer lengths: 21, 33, 55, 99, and 121 bp). MAGs were binned from each sample using MetaBAT 2 (Kang et al., 2019), MaxBin 2.0 (Wu et al., 2016), and VAMB (Nissen et al., 2021). The completeness and redundancy of each bin were assessed using CheckM (Parks et al., 2015). A non-redundant set of bins was produced using DAS Tool (Sieber et al., 2018) and dRep (Olm et al., 2017) based on an average nucleotide identity (ANI) cutoff of 97.5%. MAGs were taxonomically characterized using the genome taxonomy database tool kit GTDB-tk (Parks et al., 2018). Genes were predicted using Prodigal (Hyatt et al., 2010). We refer to the Red-Sea MAGs from this study as MAG R and MAGs based on other studies as MAG T. The genome of the cultured strain Trichodesmium erythraeum IMS101 and five TARA Oceans MAGs, including two novel non-diazotrophic genomes (Delmont, 2021), were incorporated in the follow-up analysis. The ANI values for each MAG were analyzed using ANIb module in pyANI (Pritchard et al., 2015).
Amplicon Sequencing of the hetR Gene
Trichodesmium samples (tuft and puff morphotypes) for amplicon sequencing of the hetR were collected in the Gulf of Aqaba (same location as above) during several blooms (2013–2019). DNA was extracted using the phenol-chloroform method (Massana et al., 1997). Partial hetR gene sequences (~355 bp) were amplified using the forward primer (hetrR_50F), 5′-ATTGAACCYA AACGGGTT-3′ and reverse primer (hetR_381R), 5′-CGCTTAATATGTY CTGYCAAAGCTT-3′, which were deduced from conserved regions of a Trichodesmium hetR nucleotide sequence alignment. 2 × 250 bp reads were sequenced on Illumina MiSeq, following library preparation at HyLabs (Rehovot, Israel). The hetR amplicon samples can be found under the accession numbers SAMN25885796-803. Sequences were merged, denoised, and called into amplicon sequence variants (ASVs) using DADA2 in Qiime2 (Callahan et al., 2016; Bolyen et al., 2019). The ASVs were clustered into five OTUs at 98% similarity for further downstream analyses using the VSEARCH consensus taxonomy classifier (Rognes et al., 2016).
Trichodesmium Phylogeny
We constructed multi-locus and single marker gene (hetR) phylogenies of Trichodesmium. We used the following genomes: the four Trichodesmium MAGs from this study, five Trichodesmium MAGs from the TARA Oceans dataset (Delmont, 2021) and that of Trichodesmium erythraeum IMS101 PRJNA318 (Walworth et al., 2015). The phylogenomic tree was constructed from a concatenated gene-alignment of a 251 single-copy gene-set hidden Markov Models (HMMs) for Cyanobacteria using GToTree (v.1.16.12; default settings; Lee, 2019). The aligned protein sequences were refined using Gblocks (0.91b; default settings) to eliminate poorly aligned positions and divergent regions (Castresana, 2000). A tree was subsequently constructed from the cleaned alignment using IQtree2 (v2.1.3) which implements ModelFinder (v1) to estimate the best-fit model (Q.plant+F + I + G4; Kalyaanamoorthy et al., 2017; Minh et al., 2020). Shimodaira–Hasegawa approximate likelihood-ratio test (SH-aLRT) and ultrafast bootstrap approximation (UFBoot) branch support values were estimated from 1,000 bootstraps. The tree was visualized using FigTree (v1.4.4; Figure 2) and rooted with Okeania hirsuta (GCA_003838225; Moss et al., 2018) as an outgroup.
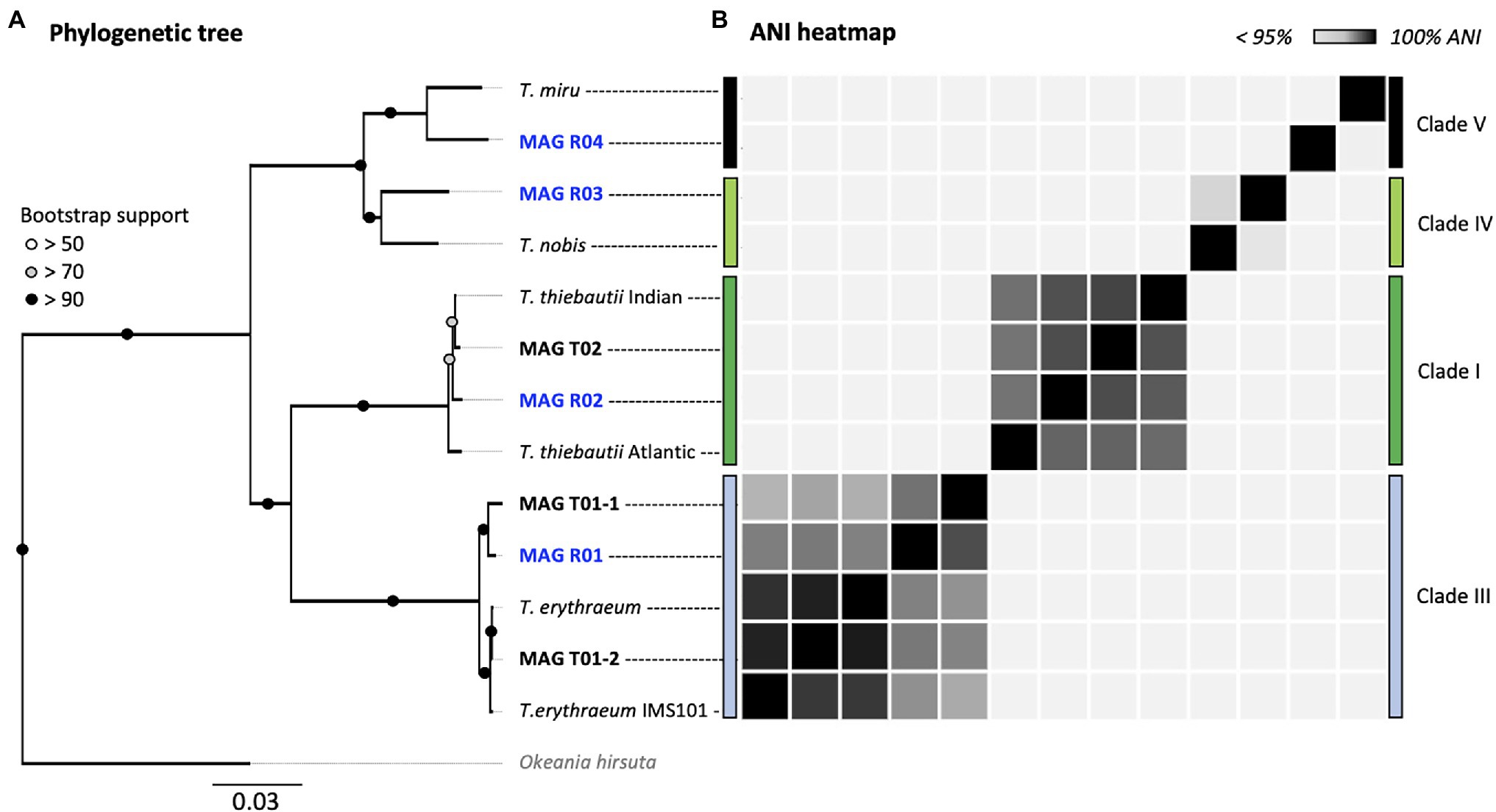
Figure 2. Multi-locus (251 HMMs) phylogenomic tree (A) and ANI heatmap (B) of Trichodesmium MAGs. All MAGs assembled in this study are marked in bold. Red Sea MAGs (our samples) are marked in blue. The phylogeny includes five MAGs from the TARA Oceans dataset and the laboratory culture Trichodesmium erythraeum IMS101. The tree was rooted at Okeania hirsuta (gray). The accession numbers for each MAG can be found in Supplementary Tables 1, 2, and their ANI values in Supplementary Tables 3a,b.
The hetR marker gene was identified in Trichodesmium MAGs using the Rapid Annotation using Subsystem Technology (RAST v2.0; Aziz et al., 2008). The hetR sequence and the neighboring genes cluster were identified by searching for the HetR amino acid sequence (Tery_1921; Q93CE9) using BLAST on the SEED (v2.0) server (Aziz et al., 2012; Overbeek et al., 2013). Coverage of the raw reads from the hetR-containing contig was further assessed in IGV (v2.12.3; Robinson et al., 2011). The marker gene sequences from each Trichodesmium MAG were aligned with previously published ones (Janson et al., 1999; Orcutt et al., 2002; Hynes et al., 2012) and those from our Red Sea hetR amplicon sequencing using the Multiple Alignment Fast Fourier Transformation (MAFFT v7.490; L-INS-i) software (Katoh et al., 2002, 2005, 2019). All the hetR sequences used in this study can be found in Supplementary Table 5. This alignment was further cleaned using GBlocks (v0.91b; default settings; Castresana, 2000). A phylogenetic tree rooted at the sequence of Okeania hirsuta, was constructed using IQtree2 (v2.1.3) for hetR (best-fit model TPM3 + F + I) and visualized using FigTree (v1.4.4; Figure 3). Similarly, the phylogeny of the rbcL gene from Trichodesmium MAGs was also assessed (best-fit model TN + F + G4) and visualized in IQtree2 (Supplementary Figure 1).
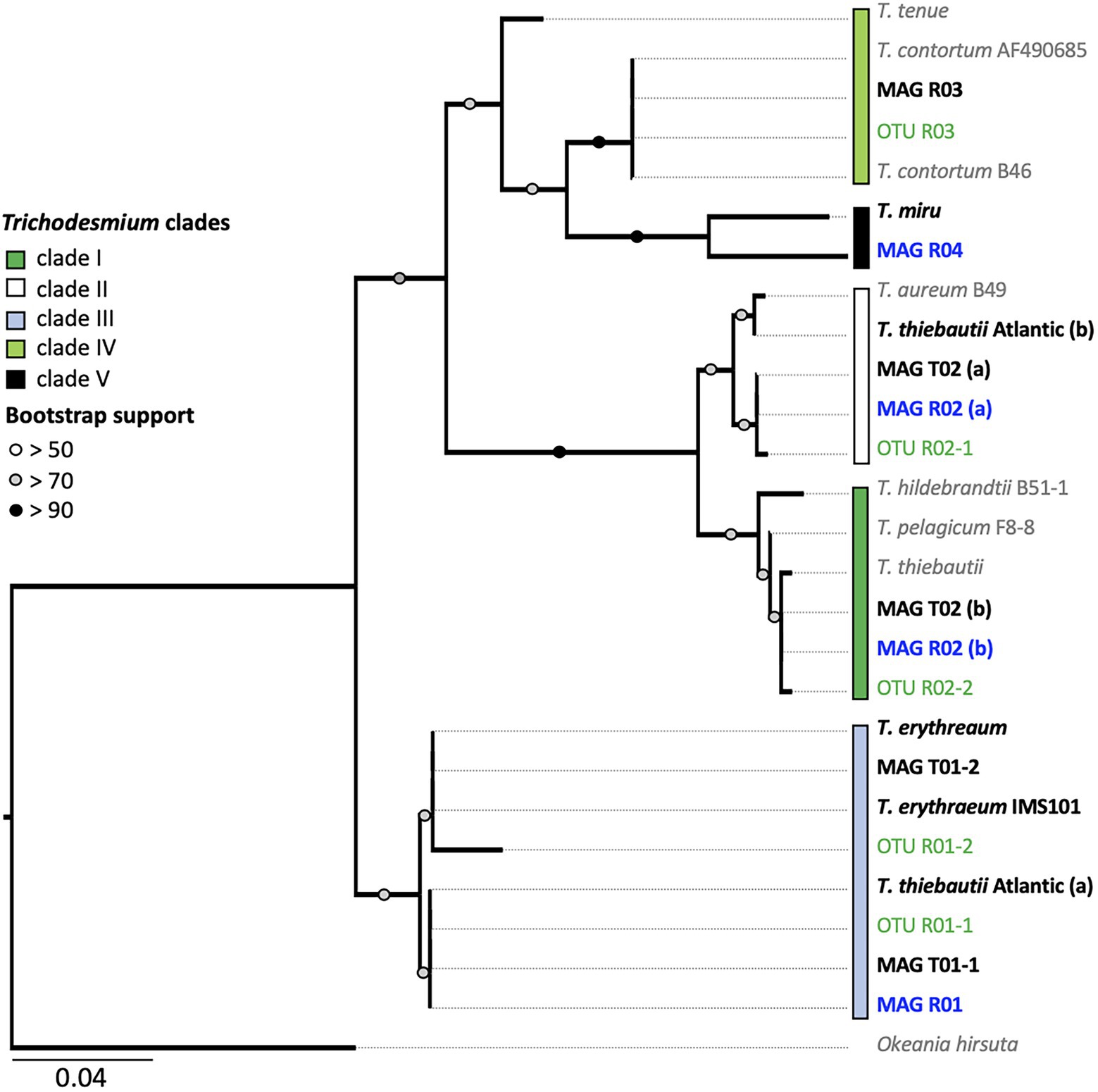
Figure 3. Trichodesmium hetR phylogeny. The tree is based on the alignment of 312 nucleotide sequences. Bold text indicates the hetR sequences from metagenome-assembled genomes (MAGs; Red Sea sequences are marked in blue). Amplicon hetR sequences are shown in green. The five proposed Trichodemium clades are shown with colored boxes. Note that clades IV and V are polyphyletic and are therefore not completely resolved. MAGs R02 and T03 contain two hetR sequences, clustering within clades I and II, respectively. The average percentage identity (ANI) matrix between the hetR gene sequences can be found in Supplementary Table 4.
Results and Discussion
Four non-redundant Trichodesmium MAGs were assembled from the Red Sea (Figure 1; Supplementary Table 1). Briefly, MAG R01 (T. erythraeum) and MAG R04 (T. miru-like) were assembled from the tuft sample. MAG R02 (T. thiebautii) was curated from dense-puff samples, while MAG R03 (T. nobis-like) was from long puffs. The MAGs were of high quality, being 90% complete and with less than 5% redundancy, apart from MAG R04 which was 86.29% complete. To provide a wider framework of Trichodesmium diversity, we assembled three non-redundant Trichodesmium MAGs from the Pacific and the Atlantic Ocean, based on previously published raw data (Frischkorn et al., 2017, 2018b; Gradoville et al., 2017). These included two high-quality MAGs T01-2 and T02, as well as the lower quality MAG T01-1 (Supplementary Table 2).
Our results suggest that T. erythraeum and T. thiebautii are the dominant Trichodesmium lineages in the Red Sea, where MAG R01 clustered with T. erythraeum (97.3% ANI), and MAG R02 with T. thiebautii (97.7–98.3% ANI; Figure 2; Supplementary Table 3). The dominance of these lineages in the Red Sea could be confirmed from hetR gene phylotyping of Trichodesmium populations across several seasons (Supplementary Figure 2). Tuft colonies were primarily composed of T. erythraeum (~83% read abundance; Figure 1C; Orcutt et al., 2002).
Both thin and dense puff-shaped colonies were dominated by T. thiebautii (~78% read abundance; Supplementary Table 1; Figures 1A,B). This finding is similar to another genomic study where radial (dense-like) and non-radial puff morphologies were linked to the same genotype of T. thiebautii (Gradoville et al., 2017). Puff-shaped Trichodesmium colonies have the unique capability to interact, capture, and concentrate dust at the colony’s core (Rubin et al., 2011) which can allow colonies to obtain limiting nutrients from the marine environment, such as iron or phosphorous (Basu and Shaked, 2018; Basu et al., 2019). In the field, thin puffs isolated from the Red Sea were shown to be more interactive in comparison to dense puffs in the capturing and concentrating of dust at the colony’s core, while exhibiting pronounced gliding motility (Wang et al., 2021). Our findings highlight that these morphological variations do not appear to be explained genomically and that other yet uncharacterized factors are at play. We hypothesize that the observed variations in puff morphologies concerning the centering of dust (Kessler et al., 2019; Wang et al., 2021) are due to differences in gene regulation in response to yet unknown environmental conditions which may include nutrient limitation or grazing. In a laboratory setting, single filaments of T. erythraeum IMS101 clustered into the puff and tuft-shaped colonies when subjected to iron or phosphorous limitation (Tzubari et al., 2018). Nonetheless, Trichodesmium displays functional variability and heterogeneity at a single-colony level that remains largely unexplained (Eichner et al., 2019; Held et al., 2021) and subsequently the ability to predict whether a colony will center dust still requires ongoing exploration.
Our data revealed subspecies diversity for both T. thiebautii and T. erythraeum (Figure 2). Trichodesmium thiebautii MAGs appeared to cluster according to broad geographical patterns as MAG R02 was more similar to that of T. thiebautii populations isolated from the Indian Ocean (98.3% ANI), than to the Atlantic T. thiebautii MAG (97.7% ANI). This concurs with the previous findings (Delmont, 2021). We also found genomic diversity within T. erythraeum as revealed from two distinct T. erythraeum MAGs from the Pacific Ocean (96.6% ANI). MAG T01-1 was highly similar to that of T. erythraeum isolated from the Red Sea (97.6% ANI; MAG R01), whereas MAG T01-2 was closely related to T. erythraeum IMS101 (99.4% ANI), which was isolated from the Atlantic Ocean (Prufert-Bebout et al., 1993). The hetR gene phylotyping of Trichodesmium populations in the Red Sea indicates the occurrence of an additional T. erythraeum subspecies similar to that of MAG T01-2 (Figure 3). We were, however, unable to assemble its genome or link it to a specific morphotype. It is still unclear if these two T. erythraeum subspecies within the Red Sea and the Pacific Ocean occupy distinct ecological niches.
We detected two potentially non-diazotrophic species within the Red Sea with minor read abundance in all three samples (Supplementary Table 1). Both genotypes could not be linked to a specific morphotype, and it is plausible that they either interleaved with T. thiebautii puffs or T. erythraeum tufts, or existed as single filaments. The low coverage reflected a previous estimate of the non-diazotrophic Trichodesmium of the Red Sea (Delmont, 2021). Whereas MAG R03 clustered within clade IV, which includes the non-diazotrophic T. nobis (95.6% ANI), and MAG R04 clustered within clade V containing the non-diazotrophic T. miru (94.6% ANI), both MAGs are on the threshold of being a distinct species within clade IV and V, respectively. We, therefore, turned to the single-marker gene hetR to further differentiate these MAGs, as hetR phylogeny captures a larger diversity of Trichodesmium than is possible through the limited number of Trichodesmium genomes currently available. The hetR sequences, but not the genomes, are available for T. pelagicum (AF490696.1), T. hildebrandtii (AF490679.1) in clade I, T. aureum (AF490680.1) in clade II, and T. contortum (AF013031.1), T. tenue (AF013033.1) in clade IV and T. miru in clade V.
We show that T. nobis and T. contortum are closely related, potentially representing the same non-diazotrophic Trichodesmium species. Given that T. nobis has not been described morphologically, and the genome of T. contortum has not been sequenced, both can be linked through the phylogeny of marker genes. Whereas the previously published genome of T. nobis did not contain the hetR gene (Delmont, 2021), the clade IV MAG R03 did, and its hetR gene sequence was 99.78% similar to that of T. contortum (Figure 3; Supplementary Table 4). Complementing our findings, morphological descriptions of T. contortum depict the species as single spiral-shaped trichomes, rather than colonies, that are sporadically present in low abundance (<1% of total Trichodesmium biomass) within samples (Janson et al., 1995; Letelier and Karl, 1996; Orcutt et al., 2002). MAG R03 lacked the nitrogenase gene cluster, confirming that this clade is non-diazotrophic.
Our results indicate that hetR phylotyping can overestimate the diversity within Trichodesmium populations, in particular regarding clade II. Intriguingly, both T. thiebautii MAGs contained two distinct hetR copies (906 bp), which had 28 (MAG R02) and 29 bp (MAG T02) single nucleotide polymorphisms within a sequence. Further inspection of the two copies showed that they were encoded in close vicinity of each other and that the synteny is highly conserved across all the Trichodesmium genomes (Figure 4). Metagenomic reads of MAG T02 mapped uniformly to both hetR copies in both the dense (50.53 and 49.47% hetR reads) and long-puff samples (49.21 and 50.21% hetR reads), indicating that they likely originate from a single population DNA. The putative pseudogenes between the two hetR genes did not match any known viral sequence, isolate spacer, or metagenomic spacer within the Integrated Microbial Genome/Virus Repository (IMG/VR v3; 10−5 cutoff value; Roux et al., 2021). Therefore, we suspect that these hetR genes are likely paralogs, although it is still unclear how T. thiebautii benefits from retaining both hetR sequences. Each hetR sequence was placed into a distinct phylogenetic clade, where one copy grouped with the hetR sequence of T. aureum (clade II), and the other one was placed within clade I, together with T. thiebautii, T. pelagicum, and T. hildebrandtii (Figure 3).
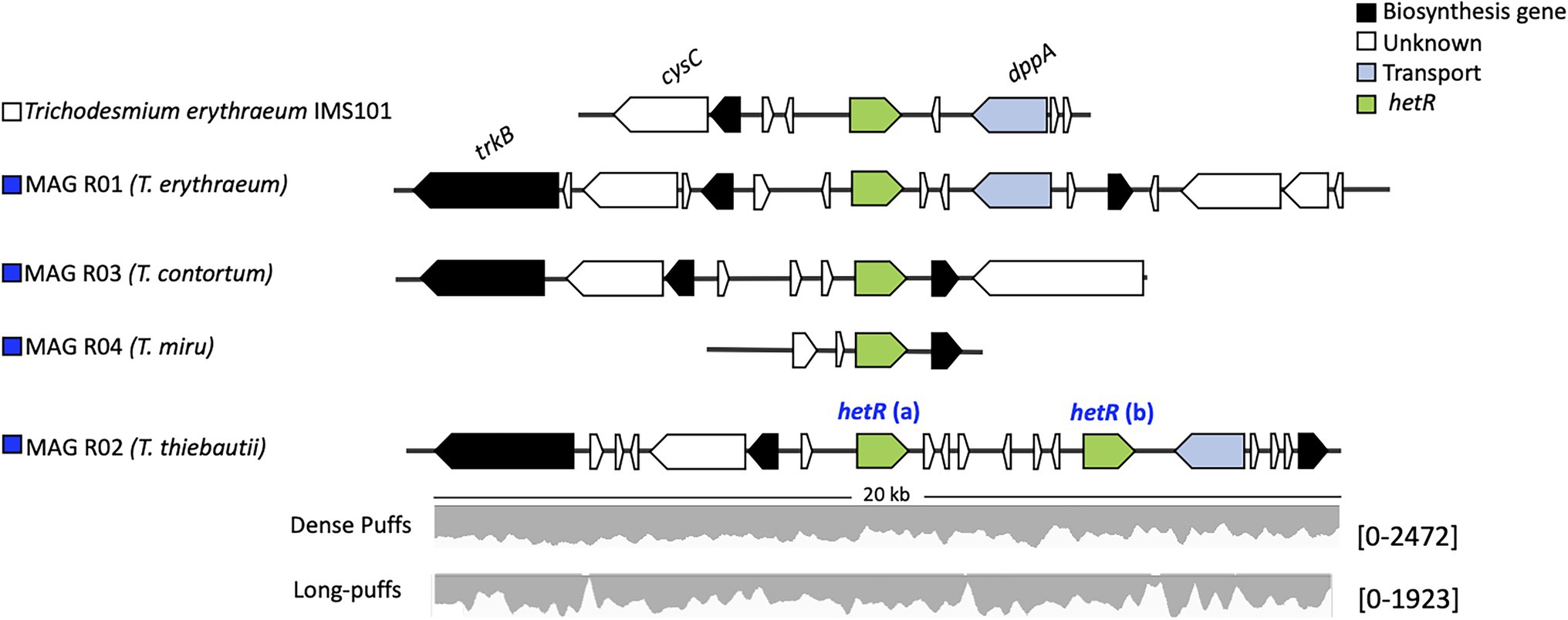
Figure 4. The hetR gene clusters in Trichodesmium sp. The synteny of the hetR genes (green) and the neighboring genes of the cluster is conserved between the Trichodesmium MAGs and Trichodesmium erythraeum IMS101. MAG R02 (Trichodesmium thiebautii) contains two hetR genes. Raw read counts, listed within the brackets, were mapped to the hetR gene cluster of MAG R02 from both the dense and long puff samples.
Amplicon sequencing of the hetR gene showed highly similar abundances of T. thiebautii and T. aureum during most sampling days. Some fluctuations in the relative read abundance were observed (Supplementary Figure 2). These findings can hint at the occurrence of T. aureum in the Red Sea. Yet, the presence of T. aureum-like hetR sequence as a paralog in a single population of T. thiebautii can lead to similar observations. Trichodesmium aureum has previously been described based on cloning and Sanger sequencing of the hetR gene whereas its morphology has been described poorly (Lundgren et al., 2005; Hynes et al., 2012). Therefore, past distinctions made between T. thiebautii and T. aureum may reflect the morphological diversity within the same genotype or species. We note that the Atlantic T. thiebautii contained one hetR paralog that clustered with the hetR sequence of clade IV (T. erythraeum), yet we cannot exclude a binning artifact in this case, as the sequences did not belong to a single scaffold.
Conclusion and Future Perspectives
Morphotype metagenomics enabled us to better capture the diversity that is present within Trichodesmium populations, while revisiting past attempts to differentiate between colonies (Scornavacca et al., 2020; Smith and Hahn, 2021). We were able to reconstruct the genomes of four different Trichodesmium species isolated from the Red Sea, that were subsequently assigned to four different Trichodesmium clades including the more elusive non-diazotrophic clades IV and V. Single-colony, rather than bulk population analysis, may help address whether these non-diazotrophic lineages occur as single free-floating filaments, from colonies of yet unknown morphology or are included within the single T. erythraeum puff or T. thiebautii puff colonies. We further show that hetR gene-based phylotyping could overestimate Trichodesmium diversity, highlighting the importance of re-assessing past attempts at phylogeny. Other marker genes such as full-length 16S rRNA as suggested by Delmont (2021) or the single-copy cyanobacterial marker rbcL (Singh et al., 2015) may assess the phylogenetic diversity of Trichodesmium (Supplementary Figure 1), although further verification will be required. Overall, the phylogenetic and functional diversity of Trichodesmium in the world’s oceans is still poorly understood to date. Thus, genomics in combination with gene expression and physiology, especially as single-colony approaches, will remain an important avenue for future exploration of these key species.
Data Availability Statement
The four Trichodesmium genomes, the raw metagenomic sequences and the hetR amplicon sequencing data presented in this study are deposited under the BioProject ID number PRJNA804487.
Author Contributions
CK, YS, and MR-B conceived the study. CK, FZ, and SW collected and extracted the metagenomic samples. Under guidance of MR-B, CK analyzed the metagenomic data. EL and SB collected samples for hetR amplicon sequencing. Under the guidance of IB-F, EL extracted and analyzed the amplicon sequencing data. The manuscript was compiled and written by CK with the help of MR-B. YS, MR-B, IB-F, and EL provided feedback and guidance during the writing process. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Israel Science Foundation (260/21) and the Israel-United States Bi-National Science Foundation (2020041) to YS. This work was also financially supported in part by the Schulich Marine Studies Initiative to IB-F. MR-B acknowledges the support of the Israel Science Foundation (913/19), the United States-Israel Binational Science Foundation (2019055), and the Israel Ministry of Science and Technology (1126). FZ thanks the PBC Fellowship Program for Outstanding Chinese and Indian Post-Doctoral Fellows.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Murielle Dray for technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.879970/full#supplementary-material
References
Aziz, R. K., Bartels, D., Best, A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 1–15. doi: 10.1186/1471-2164-9-75
Aziz, R. K., Devoid, S., Disz, T., Edwards, R. A., Henry, C. S., Olsen, G. J., et al. (2012). SEED servers: high-performance access to the SEED genomes, annotations, and metabolic models. PLoS One 7:e48053. doi: 10.1371/journal.pone.0048053
Basu, S., Gledhill, M., de Beer, D., Matondkar, S. G. P., and Shaked, Y. (2019). Colonies of marine cyanobacteria Trichodesmium interact with associated bacteria to acquire iron from dust. Commun. Biol. 2, 284–288. doi: 10.1038/s42003-019-0534-z
Basu, S., and Shaked, Y. (2018). Mineral iron utilization by natural and cultured Trichodesmium and associated bacteria. Limnol. Oceanogr. 63, 2307–2320. doi: 10.1002/lno.10939
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bushnell, B. (2014). BBTools software package. Available at: https://sourceforge.net/projects/bbmap/ (Accessed December 16, 2021).
Bushnell, B., Rood, J., and Singer, E. (2017). BBMerge—accurate paired shotgun read merging via overlap. PLoS One 12:e0185056. doi: 10.1371/journal.pone.0185056
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Capone, D. G. (2021). Coming full circle on diazotrophy in the marine cyanobacterium Trichodesmium. Proc. Natl. Acad. Sci. U. S. A. 118:e2117967118. doi: 10.1073/pnas.2117967118
Capone, D. G., Zehr, J. P., Paerl, H. W., Bergman, B., and Carpenter, E. J. (1997). Trichodesmium, a globally significant marine cyanobacterium. Science 276, 1221–1229. doi: 10.1126/science.276.5316.1221
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.A026334
Delmont, T. O. (2021). Discovery of nondiazotrophic Trichodesmium species abundant and widespread in the open ocean. Proc. Natl. Acad. Sci. U. S. A. 118:e2112355118. doi: 10.1073/PNAS.2112355118
Dugdale, R. C., Menzel, D. W., and Ryther, J. H. (1961). Nitrogen fixation in the Sargasso Sea. Deep-Sea Res. 7, 297–300. doi: 10.1016/0146-6313(61)90051-X
Eichner, M., Basu, S., Gledhill, M., de Beer, D., and Shaked, Y. (2019). Hydrogen dynamics in Trichodesmium colonies and their potential role in mineral iron acquisition. Front. Microbiol. 10:1565. doi: 10.3389/fmicb.2019.01565
Frischkorn, K. R., Haley, S. T., and Dyhrman, S. T. (2018a). Coordinated gene expression between Trichodesmium and its microbiome over day-night cycles in the north pacific subtropical gyre. ISME J. 12, 997–1007. doi: 10.1038/s41396-017-0041-5
Frischkorn, K. R., Krupke, A., Guieu, C., Louis, J., Rouco, M., Estrada, A. E. S., et al. (2018b). Trichodesmium physiological ecology and phosphate reduction in the western tropical South Pacific. Biogeosciences 15, 5761–5778. doi: 10.5194/BG-15-5761-2018
Frischkorn, K. R., Rouco, M., van Mooy, B. A. S., and Dyhrman, S. T. (2017). Epibionts dominate metabolic functional potential of Trichodesmium colonies from the oligotrophic ocean. ISME J. 11, 2090–2101. doi: 10.1038/ismej.2017.74
Gradoville, M. R., Crump, B. C., Letelier, R. M., Church, M. J., and White, A. E. (2017). Microbiome of Trichodesmium colonies from the north pacific subtropical gyre. Front. Microbiol. 8:1122. doi: 10.3389/fmicb.2017.01122
Hansel, C. M., Buchwald, C., Diaz, J. M., Ossolinski, J. E., Dyhrman, S. T., van Mooy, B. A. S., et al. (2016). Dynamics of extracellular superoxide production by Trichodesmium colonies from the Sargasso Sea. Limnol. Oceanogr. 61, 1188–1200. doi: 10.1002/lno.10266
Held, N. A., Sutherland, K. M., Webb, E. A., McIlvin, M. R., Cohen, N. R., Devaux, A. J., et al. (2021). Mechanisms and heterogeneity of in situ mineral processing by the marine nitrogen fixer Trichodesmium revealed by single-colony metaproteomics. ISME Commun. 1, 1–9. doi: 10.1038/s43705-021-00034-y
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 1–11. doi: 10.1186/1471-2105-11-119
Hynes, A. M., Webb, E. A., Doney, S. C., and Waterbury, J. B. (2012). Comparison of cultured Trichodesmium (Cyanophyceae) with species characterized from the field. J. Phycol. 48, 196–210. doi: 10.1111/J.1529-8817.2011.01096.X
Janson, S., Bergman, B., Carpenter, E. J., Giovannoni, S. J., and Vergin, K. (1999). Genetic analysis of natural populations of the marine diazotrophic cyanobacterium Trichodesmium. FEMS Microbiol. Ecol. 30, 57–65. doi: 10.1111/J.1574-6941.1999.TB00635.X
Janson, S., Siddiqui, P. J. A., Walsby, A. E., Romans, K. M., Carpenter, E. J., and Bergman, B. (1995). Cytomorphological of the planktonic diazotrophic cyanobacteria Trichodesmium spp. from the Indian Ocaen and Caribbean and Sargasso seas. J. Phycol. 31, 463–477. doi: 10.1111/J.0022-3646.1995.00463.X
Kalyaanamoorthy, S., Minh, B., Wong, T., von Haeseler, A., and Jermiin, L. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/PEERJ.7359
Katoh, K., Kuma, K. I., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kessler, N., Armoza-Zvuloni, R., Wang, S., Basu, S., Weber, P. K., Stuart, R. K., et al. (2019). Selective collection of iron-rich dust particles by natural Trichodesmium colonies. ISME J. 14, 91–103. doi: 10.1038/s41396-019-0505-x
Kieser, S., Brown, J., Zdobnov, E. M., Trajkovski, M., and McCue, L. A. (2020). ATLAS: a snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinformatics 21:257. doi: 10.1186/S12859-020-03585-4
Lee, M. D. (2019). GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164. doi: 10.1093/bioinformatics/btz188
Letelier, R. M., and Karl, D. M. (1996). Role of Trichodesmium spp. in the productivity of the subtropical north pacific ocean. Mar. Ecol. Prog. Ser. 133, 263–273. doi: 10.3354/MEPS133263
Lundgren, P., Janson, S., Jonasson, S., Singer, A., and Bergman, B. (2005). Unveiling of novel radiations within Trichodesmium cluster by hetR gene sequence analysis. Appl. Environ. Microbiol. 71, 190–196. doi: 10.1128/AEM.71.1.190-196.2005
Massana, R., Murray, A. E., Preston, C. M., and DeLong, E. F. (1997). Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara channel. Appl. Environ. Microbiol. 63, 50–56. doi: 10.1128/aem.63.1.50-56.1997
McKinna, L. I. W. (2015). Three decades of ocean-color remote-sensing Trichodesmium spp. in the world’s oceans: a review. Prog. Oceanogr. 131, 177–199. doi: 10.1016/j.pocean.2014.12.013
McManus, G. B., and Katz, L. A. (2009). Molecular and morphological methods for identifying plankton: what makes a successful marriage? J. Plankton Res. 31, 1119–1129. doi: 10.1093/plankt/fbp061
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Moss, N. A., Leão, T., Rankin, M. R., McCullough, T. M., Qu, P., Korobeynikov, A., et al. (2018). Ketoreductase domain dysfunction expands chemodiversity: malyngamide biosynthesis in the cyanobacterium Okeania hirsuta. ACS Chem. Biol. 13, 3385–3395. doi: 10.1021/ACSCHEMBIO.8B00910
Nissen, J. N., Johansen, J., Allesøe, R. L., Sønderby, C. K., Armenteros, J. J. A., Grønbech, C. H., et al. (2021). Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560. doi: 10.1038/S41587-020-00777-4
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/GR.213959.116
Olm, M. R., Brown, C. T., Brooks, B., and Banfield, J. F. (2017). dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868. doi: 10.1038/ismej.2017.126
Orcutt, K. M., Rasmussen, U., Webb, E. A., Waterbury, J. B., Gundersen, K., and Bergman, B. (2002). Characterization of Trichodesmium spp. by genetic techniques. Appl. Environ. Microbiol. 68, 2236–2245. doi: 10.1128/AEM.68.5.2236-2245.2002
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2013). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/GR.186072.114
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2015). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Prufert-Bebout, L., Paerl, H. W., and Lassen, C. (1993). Growth, nitrogen fixation, and spectral attenuation in cultivated Trichodesmium species. Appl. Environ. Microbiol. 59, 1367–1375. doi: 10.1128/AEM.59.5.1367-1375.1993
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/NBT.1754
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/PEERJ.2584
Rouco, M., Haley, S. T., and Dyhrman, S. T. (2016). Microbial diversity within the Trichodesmium holobiont. Environ. Microbiol. 18, 5151–5160. doi: 10.1111/1462-2920.13513
Roux, S., Páez-Espino, D., Chen, I. M. A., Palaniappan, K., Ratner, A., Chu, K., et al. (2021). IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 49, D764–D775. doi: 10.1093/NAR/GKAA946
Rubin, M., Berman-Frank, I., and Shaked, Y. (2011). Dust-and mineral-iron utilization by the marine dinitrogen-fixer Trichodesmium. Nat. Geosci. 4, 529–534. doi: 10.1038/ngeo1181
Scornavacca, C., Delsuc, F., and Galtier, N. (eds.) (2020). Phylogenetics in the Genomic Era. Available at: https://hal.inria.fr/PGE/ (Accessed December 14, 2021).
Sieber, C. M. K., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843. doi: 10.1038/s41564-018-0171-1
Singh, P., Fatma, A., and Mishra, A. K. (2015). Molecular phylogeny and evogenomics of heterocystous cyanobacteria using rbcl gene sequence data. Ann. Microbiol. 65, 799–807. doi: 10.1007/S13213-014-0920-1
Smith, M. L., and Hahn, M. W. (2021). New approaches for inferring phylogenies in the presence of paralogs. Trends Genet. 37, 174–187. doi: 10.1016/J.TIG.2020.08.012
Transcription of National Library of Australia (2004). Cook’s Journal: Daily Entries, 22 August 1770. South Seas, 287. Available at: http://nla.gov.au/nla.cs-ss-jrnl-cook-17700822 (Accessed February 10, 2022).
Tzubari, Y., Magnezi, L., Be’er, A., and Berman-Frank, I. (2018). Iron and phosphorus deprivation induce sociality in the marine bloom-forming cyanobacterium Trichodesmium. ISME J. 12, 1682–1693. doi: 10.1038/s41396-018-0073-5
Walworth, N., Pfreundt, U., Nelson, W. C., Mincer, T., Heidelberg, J. F., Fu, F., et al. (2015). Trichodesmium genome maintains abundant, widespread noncoding DNA in situ, despite oligotrophic lifestyle. Proc. Natl. Acad. Sci. U. S. A. 112, 4251–4256. doi: 10.1073/PNAS.1422332112
Wang, S., Koedooder, C., Zhang, F., Kessler, N., Eichner, M., Shi, D., et al. (2021). Colonies of the marine cyanobacterium Trichodesmium optimize dust utilization by selective collection and retention of nutrient-rich particles. iScience 25:103587. doi: 10.1016/J.ISCI.2021.103587
Keywords: Trichodesmium, hetR, cyanobacteria, morphotype, Red Sea, taxonomy, metagenome, biodiversity
Citation: Koedooder C, Landou E, Zhang F, Wang S, Basu S, Berman-Frank I, Shaked Y and Rubin-Blum M (2022) Metagenomes of Red Sea Subpopulations Challenge the Use of Marker Genes and Morphology to Assess Trichodesmium Diversity. Front. Microbiol. 13:879970. doi: 10.3389/fmicb.2022.879970
Edited by:
Francisco Rodriguez-Valera, Miguel Hernández University of Elche, SpainReviewed by:
Luis Miguel Rodríguez, University of Innsbruck, AustriaEric Daniel Becraft, University of North Alabama, United States
Copyright © 2022 Koedooder, Landou, Zhang, Wang, Basu, Berman-Frank, Shaked and Rubin-Blum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Coco Koedooder, Y29jby5rb2Vkb29kZXJAbWFpbC5odWppLmFjLmls