 Rachel L. Spietz
Rachel L. Spietz Devon Payne
Devon Payne Gargi Kulkarni2
Gargi Kulkarni2 William W. Metcalf
William W. Metcalf Eric E. Roden
Eric E. Roden Eric S. Boyd
Eric S. Boyd
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 09 May 2022
Sec. Microbiological Chemistry and Geomicrobiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.878387
This article is part of the Research Topic The Electromicrobiology of Anaerobic Ecosystems View all 4 articles
Pyrite (FeS2) has a very low solubility and therefore has historically been considered a sink for iron (Fe) and sulfur (S) and unavailable to biology in the absence of oxygen and oxidative weathering. Anaerobic methanogens were recently shown to reduce FeS2 and assimilate Fe and S reduction products to meet nutrient demands. However, the mechanism of FeS2 mineral reduction and the forms of Fe and S assimilated by methanogens remained unclear. Thermodynamic calculations described herein indicate that H2 at aqueous concentrations as low as 10–10 M favors the reduction of FeS2, with sulfide (HS–) and pyrrhotite (Fe1–xS) as products; abiotic laboratory experiments confirmed the reduction of FeS2 with dissolved H2 concentrations greater than 1.98 × 10–4 M H2. Growth studies of Methanosarcina barkeri provided with FeS2 as the sole source of Fe and S resulted in H2 production but at concentrations too low to drive abiotic FeS2 reduction, based on abiotic laboratory experimental data. A strain of M. barkeri with deletions in all [NiFe]-hydrogenases maintained the ability to reduce FeS2 during growth, providing further evidence that extracellular electron transport (EET) to FeS2 does not involve H2 or [NiFe]-hydrogenases. Physical contact between cells and FeS2 was required for mineral reduction but was not required to obtain Fe and S from dissolution products. The addition of a synthetic electron shuttle, anthraquinone-2,6-disulfonate, allowed for biological reduction of FeS2 when physical contact between cells and FeS2 was prohibited, indicating that exogenous electron shuttles can mediate FeS2 reduction. Transcriptomics experiments revealed upregulation of several cytoplasmic oxidoreductases during growth of M. barkeri on FeS2, which may indicate involvement in provisioning low potential electrons for EET to FeS2. Collectively, the data presented herein indicate that reduction of insoluble FeS2 by M. barkeri occurred via electron transfer from the cell surface to the mineral surface resulting in the generation of soluble HS– and mineral-associated Fe1–xS. Solubilized Fe(II), but not HS–, from mineral-associated Fe1–xS reacts with aqueous HS– yielding aqueous iron sulfur clusters (FeSaq) that likely serve as the Fe and S source for methanogen growth and activity. FeSaq nucleation and subsequent precipitation on the surface of cells may result in accelerated EET to FeS2, resulting in positive feedback between cell activity and FeS2 reduction.
Iron disulfide, or pyrite (FeS2), is the most abundant sulfide mineral in Earth’s crust and its formation and fate modulate the biogeochemical cycles of iron (Fe), sulfur (S), oxygen, and carbon (Berner, 1984; Benning et al., 2000; Schoonen, 2004). Under oxic conditions, aerobic microorganisms can accelerate the oxidative dissolution of FeS2 (Percak-Dennett et al., 2017), a process that represents the primary input of sulfur to both the marine (Konhauser et al., 2011) and terrestrial biospheres since at least 2.8 Gya (Stüeken et al., 2012) or even earlier (Crowe et al., 2013). However, far less is known of the fate of FeS2 in anoxic environments. Anaerobic oxidation of FeS2 coupled to manganese oxide reduction (Schippers and Jørgensen, 2001) or nitrate reduction (Jorgensen et al., 2009; Jakus et al., 2021) has been demonstrated by microorganisms inhabiting marine sediments and subsurface aquifers, respectively. However, the generation of substantial manganese oxide or nitrate to sustain such reactions requires oxygen (Tipping, 1984; Garvin et al., 2009) or, in the absence of oxygen, abiotic or biotic photochemical processes (Johnson et al., 2013; Daye et al., 2019; Liu et al., 2020). These factors, therefore, likely limit the distribution of anaerobic FeS2 oxidation processes, both biotic and abiotic, to near surface environments that are either suboxic to anoxic or where light is available.
In addition to oxidative pathways, studies have shown that FeS2 can be abiotically reduced at high temperature (>90°C) and in the presence of high hydrogen (H2) partial pressures (>8 bar, equivalent to 7 mM aqueous; Hall, 1986; Truche et al., 2010). During abiotic FeS2 reduction at high temperature, sulfide (HS–) is released into solution and the iron sulfide mineral pyrrhotite (Fe1–xS) precipitates on the surface of FeS2 (Truche et al., 2010). At lower temperatures, reduced chromium ions can also promote abiotic reduction of FeS2, a feature that is exploited to determine total S in FeS2-containing environmental samples (Canfield et al., 1986). However, high temperature (>90°C) environments with high concentrations of H2 (>7 mM aqueous) or environments that have high concentrations of reduced chromium ions are rare and, in the case of the former, are likely limited to deep subsurface systems where microbial life is highly restricted or not possible (Colwell and D’Hondt, 2013; Orcutt et al., 2013; Colman et al., 2017; Bradley et al., 2020).
Recently, pure cultures of methanogenic archaea (Methanosarcina barkeri strain MS and Methanococcus voltae strain A3) were shown to catalyze the reductive dissolution of FeS2 when grown with methanol and acetate or with formate, respectively, when incubated at 38°C, with FeS2 as the sole source of Fe and S for cell growth (Payne et al., 2021b). This same study showed that M. voltae required direct contact with FeS2 to catalyze its reduction and/or to acquire Fe and S dissolution products to meet nutritional demands. The FeS2 dissolution product Fe1–xS has a relatively high solubility of ∼2 μM (Davison, 1991) at the ionic strength and the circumneutral pH of the base salts medium used to cultivate M. voltae in the aforementioned study (Payne et al., 2021b). In the presence of a stoichiometric excess of HS– (>2 μM) and at circumneutral pH, the predominant form of Fe(II) in solution from Fe1–xS dissolution is iron monosulfide aqueous (FeSaq) clusters (Luther and Rickard, 2005; Rickard and Luther, 2007). Given that the measured concentration of HS– significantly exceeded 2 μM (reaching concentrations as high as 35 μM) in the methanogen cultures that were actively shown to be reducing FeS2, it was proposed that the cells assimilated soluble FeSaq clusters to meet biosynthetic demands (Payne et al., 2021b). These findings are significant since they point to the existence of a biological mechanism that can drive FeS2 reductive dissolution and mobilization of Fe and S under anoxic and lower temperature (38°C) conditions that, until recently (Payne et al., 2021b), were thought to stabilize FeS2. Further, this newly discovered process may provide an explanation for how methanogens meet their unusually high demand for Fe (Ronnow and Gunnarsson, 1981; Major et al., 2004; Liu et al., 2010, 2012; Johnson et al., 2021) in anoxic and sulfidic habitats, conditions that favor formation of FeSaq, Fe1–xS, and FeS2 phases (Luther and Rickard, 2005; Rickard and Luther, 2007). However, the mechanism(s) involved in the reductive dissolution of FeS2 by methanogens remains unknown.
Several methanogens have been shown to generate metabolic H2 (Valentine et al., 2000; Lupa et al., 2008; Kulkarni et al., 2018), suggesting the possibility that FeS2 reduction is indirectly mediated by biogenic H2, similar to what has been shown abiotically at high temperature (Hall, 1986; Truche et al., 2010). For example, M. barkeri contributes to its proton motive force during metabolism of methanol by producing H2 intracellularly and oxidizing it extracellularly (Kulkarni et al., 2018). Likewise, Methanococcus maripaludis produces H2 during growth with formate through the combined activities of formate dehydrogenase and F420-reducing [NiFe]-hydrogenase (Lupa et al., 2008). While the amounts of H2 produced by methanogens under such growth conditions are low (∼0.6 mbar, equivalent to ∼0.8 μM aqueous; Kulkarni et al., 2018) and are far lower than the concentrations tested in high temperature abiotic FeS2 reduction experiments (>7 mM aqueous; Truche et al., 2010), the possibility exists that H2 produced during methanogenic metabolism could mediate FeS2 reduction. Possible support for this mechanism comes from the recent discovery of extracellular [NiFe]-hydrogenases mediating extracellular electron transfer (EET) reactions in M. maripaludis (Deutzmann et al., 2015).
In the present study, the potential role for H2 in the reductive dissolution of FeS2 was investigated using a combination of thermodynamic modeling and abiotic experiments. Next, the involvement of biogenic H2 in the reduction of FeS2 was evaluated in experiments using multiple strains of M. barkeri (Fusaro and MS). M. barkeri strains were evaluated for H2 production during growth with methanol and acetate on FeS2 as the sole source of Fe and S. Further, a mutant strain of M. barkeri Fusaro with deletions in four operons encoding five [NiFe]-hydrogenases in its genome, rendering it incapable of H2 production or consumption (Mand et al., 2018), was used to determine whether biogenic H2 and/or [NiFe]-hydrogenases are required for biological FeS2 reduction. The necessity for cells to directly contact FeS2 to reduce the mineral and/or to assimilate Fe or S from Fe1–xS that likely precipitates on the surface of FeS2 during reduction was investigated in wild-type M. barkeri Fusaro cultures with defined minerals (FeS2 or Fe1–xS) sequestered in dialysis membranes. Additionally, a model quinone compound, anthraquinone-2,6-disulfonate (AQDS), previously shown to facilitate electron transfer between M. barkeri and iron hydroxide minerals (Bond and Lovley, 2002; Liu et al., 2011), was included in cultures provided with FeS2 sequestered in dialysis tubing. Lastly, transcriptomic analyses of M. barkeri strain MS, which can utilize cysteine or sulfide as the sole S source, were conducted to identify potential proteins or processes involved in FeS2 reduction. Collectively, the results from this study are presented in a multistep biogeochemical model to explain how methanogen cells obtain Fe and S from FeS2 as FeSaq to meet nutritional demands.
Recent studies have shown that methanogens can reductively dissolve FeS2 (Payne et al., 2021a,b). Given that (1) methanogens can produce H2 during methanogenesis (Lupa et al., 2008; Kulkarni et al., 2018) combined with (2) prior studies that have demonstrated abiotic FeS2 reduction by H2, albeit at high temperature (>90°C) and high H2 partial pressure (>8 bar, equivalent to >7 mM aqueous H2; Truche et al., 2010), it was necessary to first evaluate the potential for H2 to abiotically reduce FeS2 at lower temperature. High temperature (>90°C) abiotic reduction of FeS2 occurs according to Eq. 1:
where the value of x can range from 0 to 0.17 (Hall, 1986; Rickard and Luther, 2007; Truche et al., 2010). The thermodynamics of Eq. 1 were evaluated and abiotic reduction experiments with H2 were conducted using laboratory-synthesized nanoparticulate FeS2 to determine if abiotic FeS2 reduction can occur at temperatures lower than 90°C, to identify the potential products of FeS2 dissolution, to quantify the threshold concentration of H2 required to generate detectable products of this reaction, and to determine the sensitivity of the reaction to product (i.e., HS–) accumulation.
Thermodynamic calculations were performed at pH 7.0 and an ionic strength of 0.05 M, a temperature of 38°C, and a free energy of formation for FeS2 of –160.2 kJ mol–1 (Chareev et al., 2014). These conditions were chosen because they are similar to those used or measured in methanogen cultures demonstrated to reduce FeS2 (Payne et al., 2021b). At 38°C, reduction of FeS2 by H2 was shown to be favorable across a range of aqueous H2 concentrations as low as 10–10 M and HS– concentrations (10–7 to 10–2 M), if one assumes that Fe1–xS [Fe0.86S; free energy of formation of –136 kJ mol–1 (Chareev et al., 2014)] is the end product of the reaction (Truche et al., 2010) and using the Davies equation (Stumm and Morgan, 1996) to account for the influence of ionic strength on soluble ion activities (Figure 1). Interestingly, the reaction was not thermodynamically favorable if the end product, Fe1–xS, was replaced with the iron monosulfide phase mackinawite [FeS; free energy of formation of –89.2 kJ mol–1 (Berner, 1967)] as a product (data not shown). This observation is consistent with the detection of Fe1–xS as the primary product of abiotic FeS2 reduction, albeit at high temperature [>90°C; (Truche et al., 2010)]. These calculations confirm that abiotic reduction of FeS2 to HS– and Fe1–xS, but not FeS, by H2 is thermodynamically feasible under the conditions of prior experiments (Payne et al., 2021a,b).
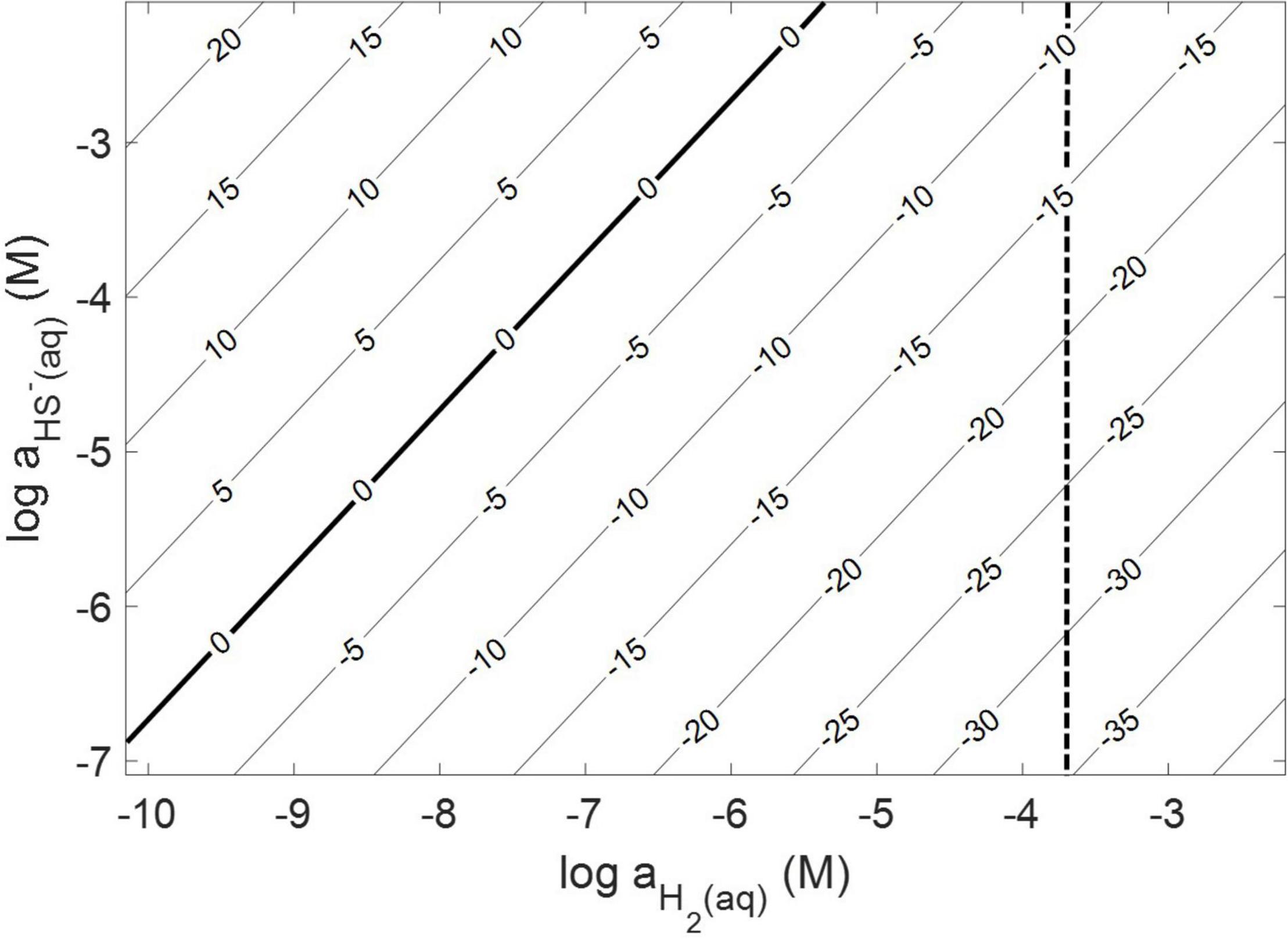
Figure 1. Gibb’s free energy calculated for abiotic pyrite (FeS2) reduction to pyrrhotite (Fe1–xS) and HS– by H2 at 38°C, an ionic strength of 0.05 M, and at a pH of 7.0. The vertical dashed line depicts the minimum H2 concentration that abiotic FeS2 reduction was detected experimentally (see Figure 2).
Abiotic experiments were conducted using synthetic FeS2 (2 mM in FeS2 formula unit) nanoparticles in reactors incubated at 38°C in low salinity, carbonate-buffered medium (2 g L–1 NaHCO3) at pH 7.0, similar to the conditions under which biological FeS2 reduction has been demonstrated (Payne et al., 2021a,b) and for which the thermodynamic parameters above were calculated. In reactors with a 100% H2 headspace (1.98 × 10–3 M aqueous H2), rapid abiotic reduction of FeS2 was observed, as determined by the production of total sulfide (sum of aqueous HS– and gaseous H2S) according to Eq. 1 (Figure 2). Significantly less, albeit still quantifiable, total sulfide was generated in abiotic reactors with a headspace comprising 10% H2 (1.98 × 10–4 M aqueous). Reactors with headspace H2 of 1% (1.98 × 10–5 M aqueous) or lower did not yield detectable total sulfide (minimum detection limit = 1.5 μM aqueous concentration) during the 4-day incubation. Nonetheless, this indicates that synthetic FeS2 can be reduced at temperatures as low as 38°C, but the products of this reaction are only detectable by colorimetric methods above a threshold H2 concentration that exists between 1.98 × 10–5 and 1.98 × 10–4 M. To confirm that abiotic reduction by H2 is not limited to synthetic nanoparticulate FeS2, 1.5 g of ground (63–125 μm size fraction) specimen-grade, high-purity FeS2 was incubated in 75 mL of base salts medium under 100% H2 (1.98 × 10–3 M aqueous concentration) or 100% N2. After 5 days of incubation at 38°C, significant total sulfide (7.19 ± 0.01 μmol) was generated through abiotic reduction by H2 and no sulfide was detected in N2 control reactors (data not shown). The concentration of dissolved H2 required to reduce FeS2 in abiotic experiments was far higher than the minimum (10–10 M) estimated by thermodynamic calculations. This suggests that factors other than the free energy for the reduction alone [e.g., particle interfacial energy (Stumm, 1992)] likely play a role in controlling the favorability of the reduction reaction.
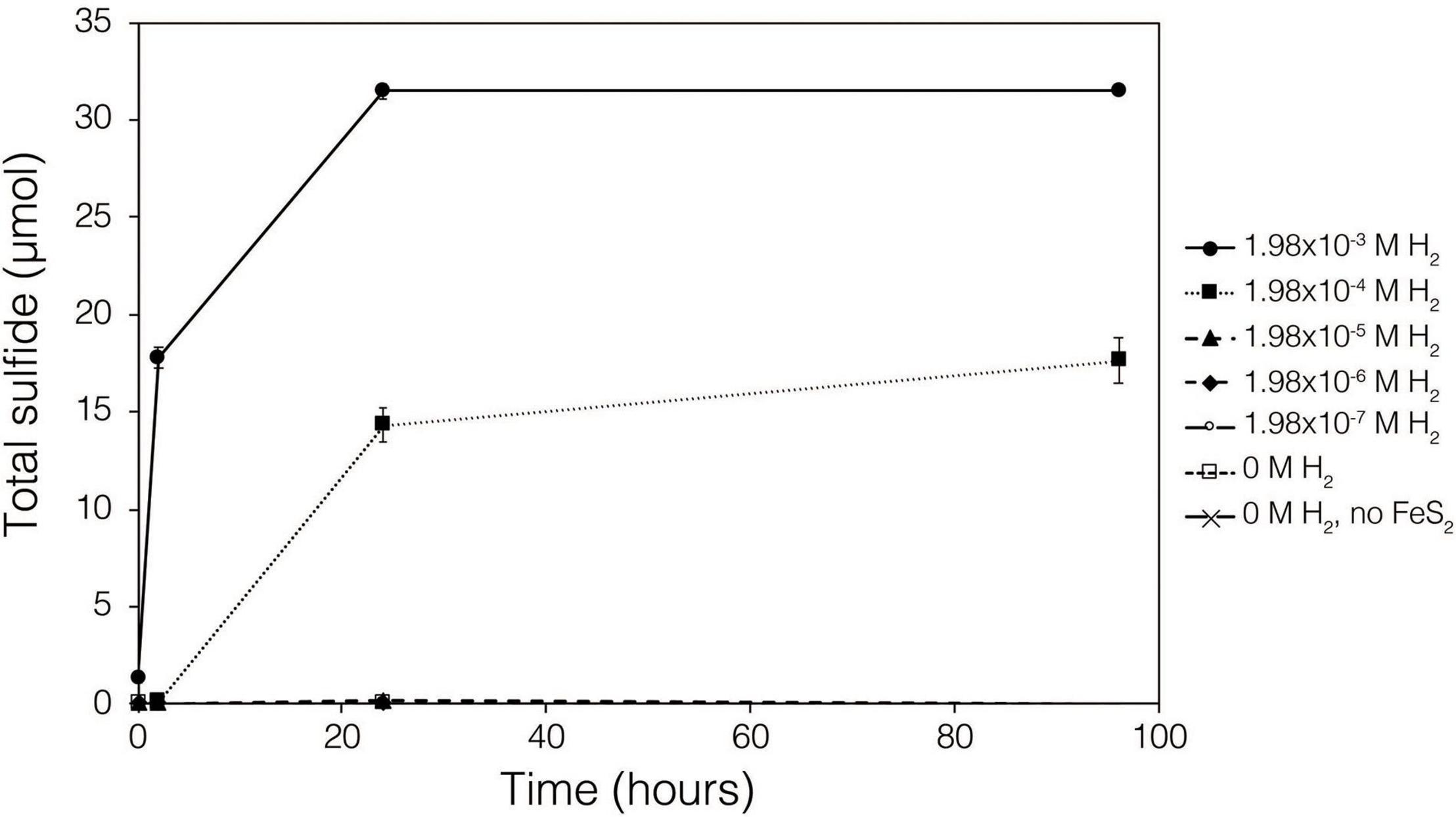
Figure 2. Production of total sulfide (aqueous plus gas phase) in reactors containing 2 mM synthetic FeS2 nanoparticles when incubated at 38°C in the presence of H2 ranging from 0 to 1.98 × 10– 3 M aqueous H2 (equivalent to 0 to 2.5 bar). All abiotic reactors contained 35 mL of base salts medium and 35 mL of headspace (balance of headspace as N2 gas).
In line with previous high-temperature studies of FeS2 reduction (Truche et al., 2010), thermodynamic calculations predicted that the favored product of FeS2 reduction is Fe1–xS. To examine the potential solid phase(s) of Fe(II) and S formed during low temperature FeS2 reduction in the presence of H2, X-ray diffraction (XRD) analyses of FeS2 reacted abiotically under 100% H2 headspace for 24 h at 38°C were conducted. The XRD spectra for the H2-reacted FeS2 was nearly identical to that for the unreacted FeS2 (Supplementary Figures 1A,B). Reference peaks for Fe1–xS (PDF #97-015-1765) and FeSmack (PDF #97-063-3302) were manually searched against the XRD spectra, for both reacted and unreacted FeS2, without matches. It is likely that the abundance of Fe1–xS, if produced during abiotic H2-driven FeS2 reduction, remains below the detection limit of XRD (∼10% relative abundance by weight). Additional characterization of FeS2 surfaces following reduction by H2 using higher resolution spectroscopic methods, such as X-ray absorption spectroscopy, are needed to identify the low abundance secondary mineral(s) that may form on the FeS2 surface.
While XRD analyses did not identify Fe1–xS produced during abiotic reduction of FeS2 reduction by H2, it was still the favored secondary mineral to form on the surface of FeS2 based on thermodynamic calculations and observations from studies conducted at higher temperature (Truche et al., 2010). Thus, the solubility of Fe1–xS was examined to identify its plausibility as a source of soluble Fe and S. Specimen Fe1–xS obtained from Aymar quarry, Gualba mines, Gualba, Montseny, Barcelona Spain, which has previously been shown to be of high purity (de Aldecoa et al., 2013), was used in experiments. XRD of the mineral indicated that the only FeS phase present was Fe1–xS (∼80%) with the balance as quartz (Supplementary Figure 1D). Dissolved (<0.2 μm filtered) Fe(II) was detected in abiotic incubations of 0.1 g of ground (63–125 μm) Fe1–xS following 7 days incubation (Figure 3). While this amount of Fe1–xS is greater than what one would expect to form in cultures of M. barkeri actively reducing FeS2, it does demonstrate that Fe(II) can be solubilized from Fe1–xS and is a possible source of Fe used by cells reducing FeS2. Interestingly, HS– was not detected (detection limit of 1.5 μM) in solution following this incubation period. This observation is consistent with a previous study that indicated that Fe [as Fe(II)], but not S (as HS–), in Fe1–xS is mobile and can solubilized from the mineral surface leaving behind a metal-deplete surface layer (Mikhlin, 2000).
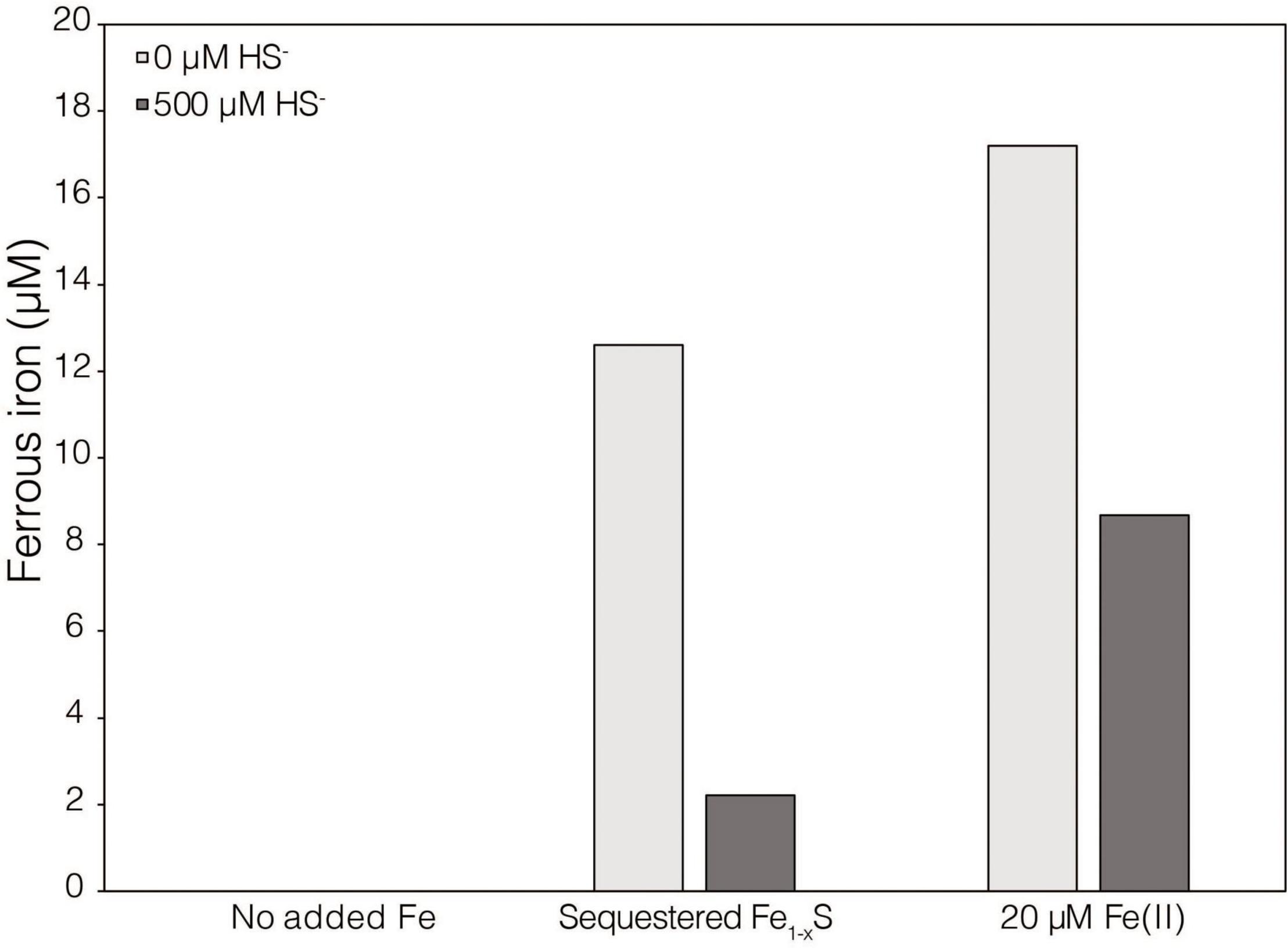
Figure 3. Dissolved ferrous iron [Fe(II)] concentration in abiotic dialysis experiment reactors following 7 days of incubation at 38°C. One hundred sixty-five milliliters serum bottles containing base salts medium were provided with no Fe (No added Fe), 0.1 g of specimen pyrrhotite sequestered in 50 kDa dialysis tubing (Sequestered Fe1–xS), or 20 μM FeCl2. Reactors were either provided with no sulfur source (0 μM HS–) or 500 μM Na2S (500 μM HS–). Following incubation, subsamples were collected under anoxic conditions and were leached with 1N HCl at 4°C for 16 h before quantifying Fe(II).
Importantly, during FeS2 reduction, the total number of moles of HS– formed should be equivalent to the total moles of Fe1–xS formed per Eq. 1. Since the dissolution of Fe from Fe1–xS is incomplete, the yield of Fe(II) in solution from Fe1–xS dissolution is far lower than the yield of total sulfide (>1 μM) during FeS2 reduction. Based on thermodynamic data (Luther and Rickard, 2005; Rickard and Luther, 2007), aqueous solutions with HS– in excess of Fe(II) favors hydrated FeSaq clusters as the predominant form of Fe(II) in solution. Experiments have shown that the methanogen M. voltae, when grown with FeS2 as the sole source of Fe and S, simultaneously exhibited evidence indicative of Fe limitation [i.e., up-expression of the Fe(II) transporter FeoB and the metal regulator DtxR] but at the same time hyperaccumulated Fe as a thioferrate-like mineral (Payne et al., 2021a). To explain this paradox, it was suggested that cells incorrectly sensed Fe(II) limitation during growth due to assimilation of Fe(II) complexed with sulfide (i.e., FeSaq). In this model, excess Fe(II) that was assimilated as hydrated FeSaq clusters (cells require more S than Fe) was then sequestered as thioferrate-like nanoparticles to limit toxicity (Payne et al., 2021a). FeSaq is predicted to be uncharged at the circumneutral pH of the culture medium used to cultivate M. voltae and M. barkeri (Jordan et al., 2019a,b). Thus, it is plausible that slightly hydrophobic and uncharged FeSaq can either passively diffuse or be actively transported across cellular membranes to provide Fe and S to cells. Additional growth experiments with Fe1–xS are described below.
The demonstration of abiotic reduction of synthetic and specimen FeS2 by H2, even at low temperature and low H2 concentrations, led to an investigation into the potential role of biogenic H2 in FeS2 reduction by M. barkeri strains Fusaro and MS. M. barkeri Fusaro was capable of reducing and growing on FeS2 as the sole Fe and S source, as indicated by the production of CH4, DNA, and total sulfide (Figures 4A,C,D). Cultures of FeS2-grown M. barkeri Fusaro generated H2 at concentrations that were similar to cultures grown with Fe(II) and HS– (Figure 4B). M. barkeri strain MS produced slightly more H2 during growth on FeS2 compared to growth on Fe(II) and cysteine, consistent with increased growth as indicated by higher DNA and CH4 production (Supplementary Figure 3B). Regardless, the maximum amount of H2 generated by M. barkeri strains Fusaro and MS grown with FeS2 (2.65 and 2.79 μM aqueous, respectively) was nearly two orders of magnitude below the lowest concentration of H2 (198 μM) that was experimentally found to drive abiotic FeS2 reduction (Figure 2). While this points to biogenic H2 not acting as the mediator of FeS2 reduction, it cannot be ruled out that the interface between FeS2 minerals and actively growing M. barkeri cells could sustain a locally elevated concentration of H2 that facilitates indirect, abiotic reduction of the mineral. This is particularly true considering that thermodynamic calculations indicate that far lower concentrations of H2 (10–10 M aqueous) may be able to drive abiotic FeS2 reduction with Fe1–xS as the end-product of the reaction (Eq. 1).
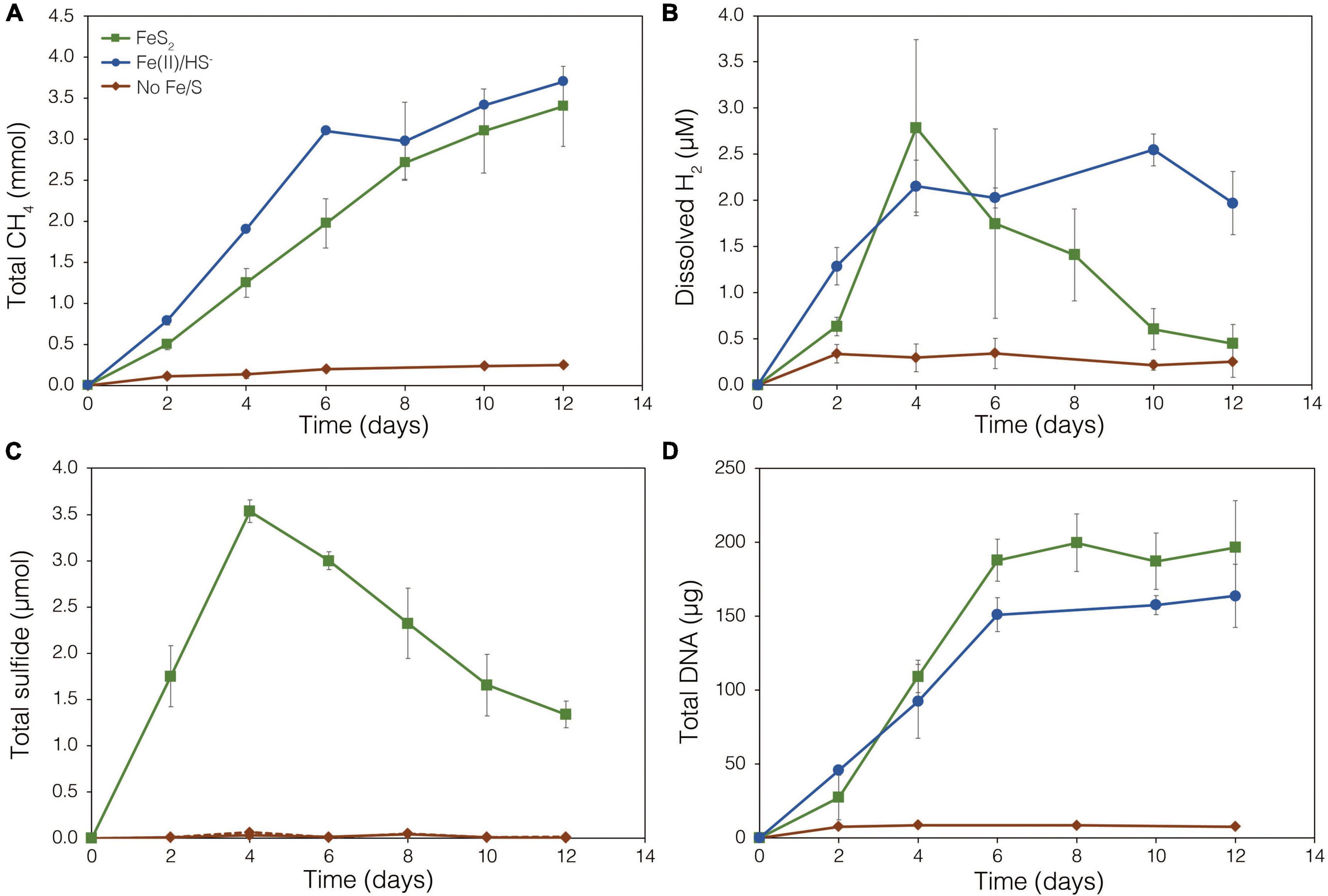
Figure 4. Production of (A) total CH4, (B) dissolved H2, (C) total sulfide (aqueous plus gas phase), and (D) total biomass (DNA) by Methanosarcina barkeri strain Fusaro during growth with 2 mM synthetic pyrite (FeS2) nanoparticles, 20 μM ferrous iron [Fe(II)] and 2 mM HS–, or no added Fe or S source (No Fe/S). The H2 concentration is reported for the dissolved phase. Averages and standard deviations for triplicates are shown. Protein data as an additional proxy for growth is presented in Supplementary Figure 3. Data depicting growth kinetics and activities of M. barkeri strain MS with FeS2 are presented in Supplementary Figure 4.
To further test whether FeS2 reduction is mediated by biogenic H2, a strain of M. barkeri Fusaro with mutations in all [NiFe]-hydrogenase operons encoded in its genome, rendering the strain unable to consume or produce H2 (Mand et al., 2018), was evaluated for its ability to reduce FeS2. The strain has mutations in one membrane-associated energy converting [NiFe]-hydrogenase (Ech), two F420-reducing [NiFe]-hydrogenases (Frh and Fre), and two membrane-associated methanophenazine-reducing [NiFe]-hydrogenases (Vht and Vhx; Kulkarni et al., 2018). The M. barkeri Fusaro [NiFe]-hydrogenase mutant maintained the ability to reduce FeS2 as indicated by significant accumulation of HS– in the medium during growth with methanol as the methanogenesis substrate and with FeS2 as the sole Fe and S source (Figure 5B). Growth was not detected when a source of Fe or S was not provided (Figures 5A,C). Further, growth of the M. barkeri Fusaro [NiFe]-hydrogenase mutant was slightly, albeit significantly (p < 0.05), enhanced on FeS2 relative to non-mineral sources of Fe and S [Fe(II) and HS–] (Figure 5C).
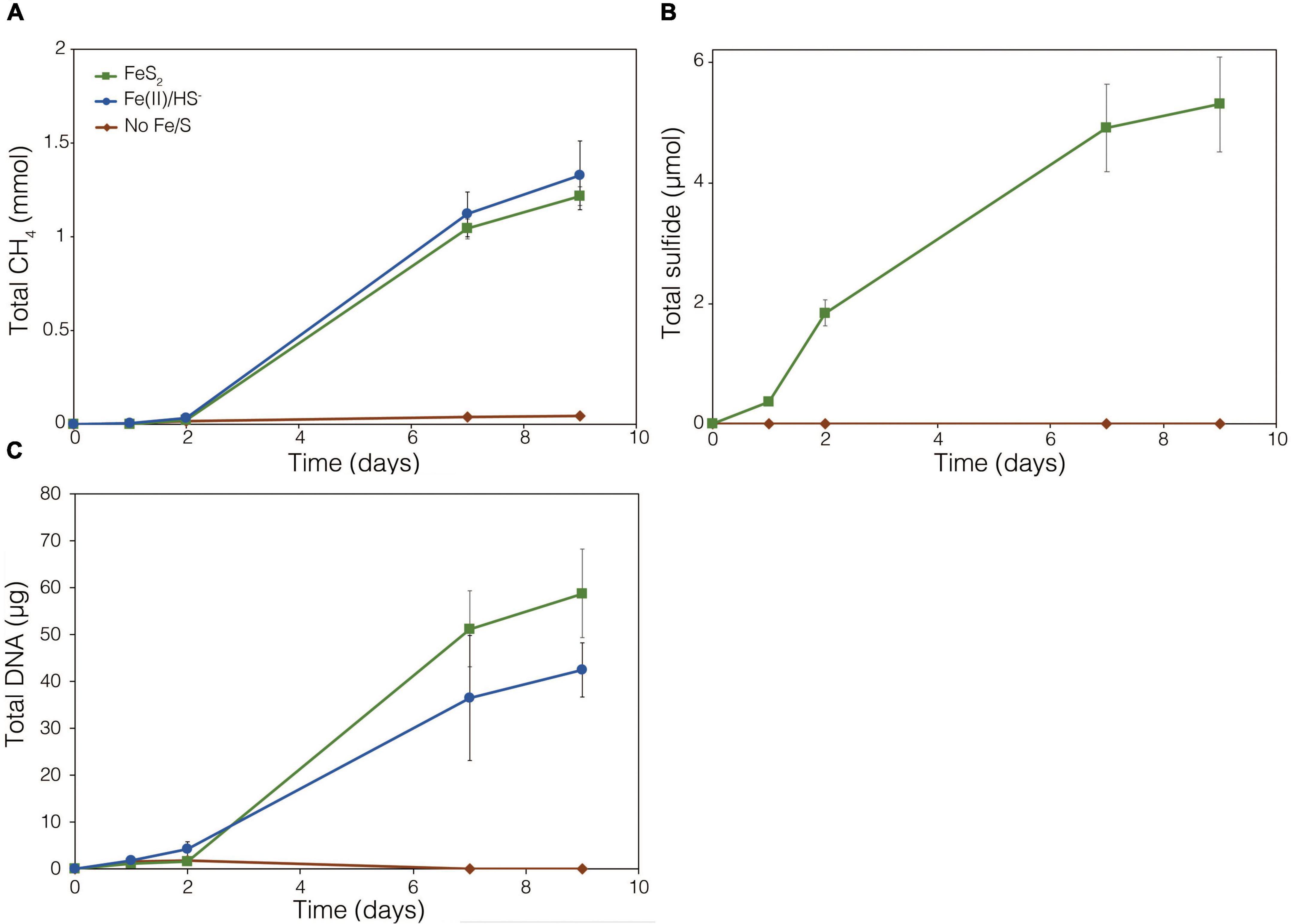
Figure 5. Production of (A) methane (CH4), (B) total sulfide (aqueous plus gas phase), and (C) total biomass (DNA) in cultures of Methanosarcina barkeri Fusaro with deletions in each of the four operons encoding all five [NiFe]-hydrogenases in its genome. The M. barkeri Fusaro hydrogenase mutant was grown either with no provided iron (Fe) or sulfur (S) source (No Fe/S); with 20 μM ferrous iron [Fe(II)] and 0.4 mM sulfide (HS–) [Fe(II)/HS–]; or with 2 mM synthetic pyrite (FeS2) nanoparticles as the sole sources of Fe and S. Hydrogen (H2) was not detected (detection limit 0.1 μM) in the headspace of any of the culture conditions tested (data not shown). Averages and standard deviations for triplicates are shown.
Throughout the growth experiment with the M. barkeri Fusaro [NiFe]-hydrogenase mutant, H2 remained below the limit of detection (0.1 μM aqueous). This indicates the [NiFe]-hydrogenase mutant strain of M. barkeri Fusaro did not produce H2, yet it maintained the ability to reduce FeS2 and assimilate dissolution products to meet Fe and S biosynthetic demands. It follows that indirect reduction of FeS2 through biogenic H2 is not the mechanism of biological FeS2 reduction. Further, these results indicate that [NiFe]-hydrogenases (Ech, Frh, Fre, Vht, and/or Vhx) themselves are not involved in FeS2 reduction, ruling out H2 or [NiFe]-hydrogenase mediated EET as a mechanism to drive FeS2 reduction. This is potentially consistent with previous results suggesting that H2 and [NiFe]-hydrogenases do not play a role in EET from the cells to external electron acceptors in M. barkeri strain Fusaro (Rowe et al., 2019). However, the directionality of EET from M. barkeri cells to FeS2 differs from that of the previous study which focused on electron acquisition from cathodes to M. barkeri Fusaro cells via EET (Rowe et al., 2019). Further, while electron acquisition from cathodes via EET was apparently respiratory in nature (Rowe et al., 2019), the reduction of FeS2 as described herein fulfills the purpose of generating bioavailable forms of Fe and S to meet nutritional demands. It is thus possible that the mechanisms of EET for M. barkeri in the growth conditions described herein, versus those described previously, differ.
Prior work showed that M. voltae A3 could not grow or reduce synthetic FeS2 when physical access to the mineral was restricted using dialysis tubing (Payne et al., 2021b). However, it remained unclear whether cells required direct access to the mineral to (1) carryout reduction and/or to (2) acquire Fe and S from the Fe1–xS that is predicted to precipitate on the surface of FeS2 following reduction. Field emission microscopy (FEM) was used to visualize physical associations between M. barkeri Fusaro cells and minerals during growth with FeS2 as the sole source of Fe and S. FEM shows that M. barkeri cells directly associated with FeS2 surfaces during growth (Supplementary Figure 5). M. barkeri is known to associate with other solid phases, such as cathodes, via extracellular polymeric substances (EPS; Rowe et al., 2019), thus a similar mechanism may be employed to attach to FeS2.
To examine the requirement for M. barkeri Fusaro cells to directly contact FeS2 in order to grow, synthetic FeS2 was sequestered in dialysis tubing with 100 kDa diameter pore size to prohibit physical contact of the cells with the mineral surface. This also excluded extracellular organic or inorganic molecular complexes with diameters larger than 100 kDa from interacting with the mineral surface. Under a subset of conditions, the synthetic quinone, AQDS, previously shown to act as an electron shuttle between M. barkeri and Fe-oxide minerals (Bond and Lovley, 2002), was provided to cells grown with FeS2 in solution and sequestered in dialysis tubing. This allowed for the determination of whether (1) direct electron transfer from the methanogen cell surface was required to reduce the mineral, (2) AQDS could enable FeS2 reduction by acting as an electron shuttle between the cell and FeS2, and/or (3) the products of FeS2 reduction could pass through the dialysis membrane to support growth. Cells that were provided with direct access to FeS2 rapidly reduced the mineral as indicated by total sulfide accumulation up to 3.53 μmol (30.5 μM aqueous sulfide) before the concentration slowly started to decrease, presumably due to its ultimate utilization by the cells (Figure 6B). Concurrently, cells generated CH4 and biomass, as determined by DNA production (Figures 6A,C) and protein production (Supplementary Figure 3). When FeS2 was sequestered in dialysis membranes and no AQDS was provided, M. barkeri Fusaro could not reduce FeS2 as indicated by the lack of sulfide accumulation in the growth medium (Figure 6B). Furthermore, production of CH4 and biomass (DNA and protein) were 1–2 orders of magnitude less when access to FeS2 was restricted by sequestering FeS2 in dialysis tubing when compared to when FeS2 was not sequestered and were not significantly different from controls where no Fe or S were provided (Figure 6A).
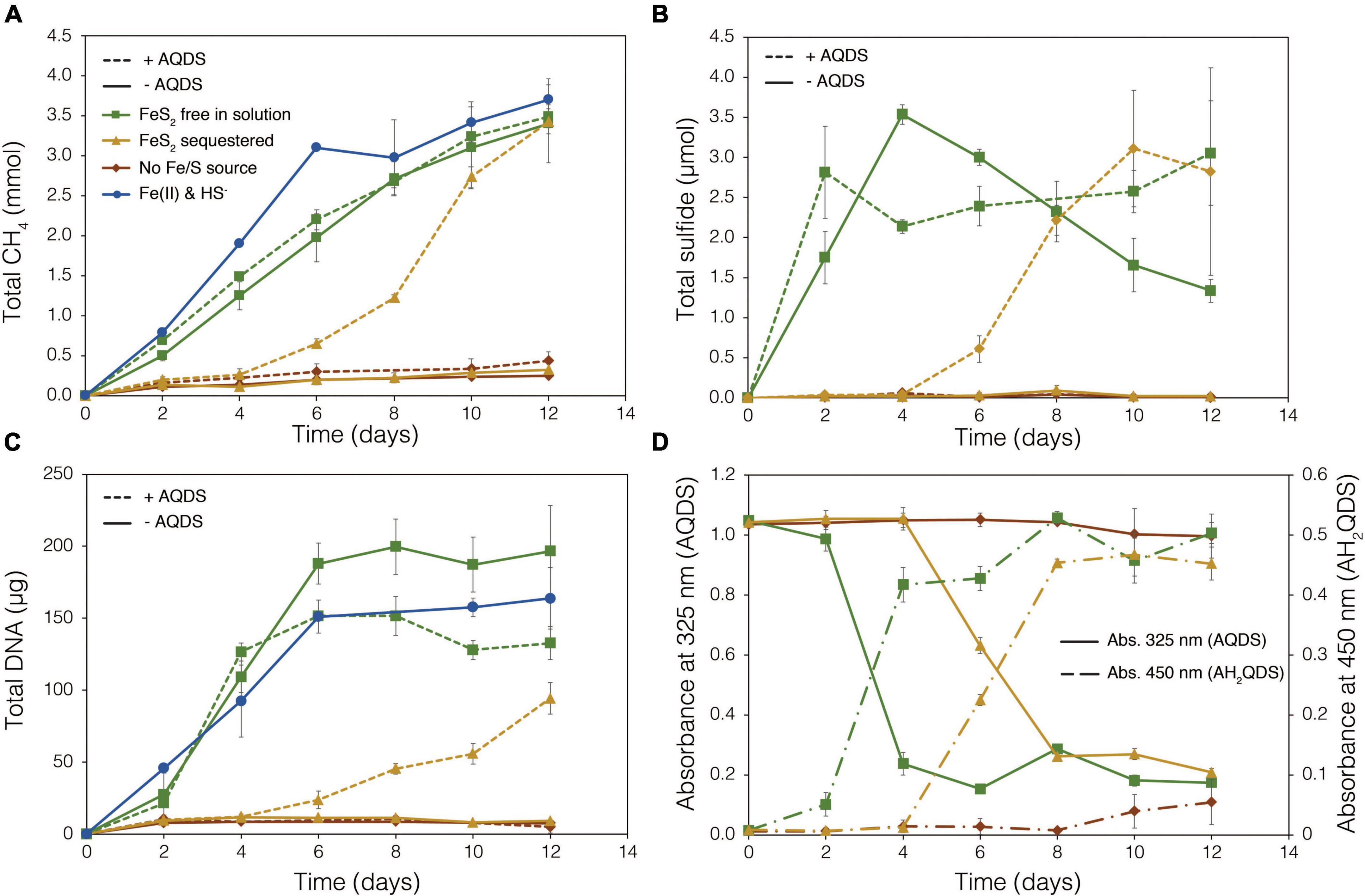
Figure 6. Production of (A) total methane (CH4), (B) total sulfide (aqueous plus gas phase), (C) total biomass (DNA), and (D) reduced anthrahydroquinone-2,6-disulfonate (AH2QDS) from anthraquinone-2,6-disulfonate (AQDS) by Methanosarcina barkeri strain Fusaro when grown with pyrite (FeS2) free in solution (green) or sequestered in 100 kDa dialysis tubing (orange) to prevent physical association. Negative control cultures contained no added Fe or S source (red), while positive control cultures were provided with 20 μM Fe(II) and 2 mM HS– (red; same data as presented in Figure 2). AQDS was provided at a final concentration of 20 mM. In (A–C), culture conditions with added AQDS are represented by dashed lines and culture conditions that do not contain AQDS are shown by solid lines. Only conditions provided AQDS are shown panel d, and absorbance at 325 nm (AQDS) is depicted by solid lines while absorbance at 450 nm (AH2QDS) depicted by long-dashed lines. Averages and standard deviations for triplicates are shown. Protein data as an additional proxy for growth is presented in Supplementary Figure 3.
Methanogens can transfer electrons to AQDS, generating the reduced form anthrahydroquinone-2,6-disulfonate (AH2QDS). The conversion of AQDS to AH2QDS was monitored spectrophotometrically by observing a shift in absorbance from 325 to 450 nm (Bond and Lovley, 2002) in cultures provided with direct access to FeS2 or when physical contact between cells and FeS2 was prevented using dialysis tubing. M. barkeri rapidly reduced AQDS to AH2QDS in cultures with FeS2 free in solution (Figure 6D), and this corresponded to accelerated HS– production from FeS2 reduction and enhanced cell growth and CH4 production activity (Figures 6A–C). Furthermore, the concentration of HS– remained steady during growth and even increased slightly toward the end of the methanogen growth phase, suggesting that cells continued to transfer electrons to AQDS, which in turn continued to reduce FeS2 even after cell growth had ceased. When FeS2 was sequestered in dialysis tubing, the conversion of AQDS to AH2QDS by M. barkeri lagged by approximately 2–4 days. However, following this lag phase, AQDS was reduced, and production of sulfide and cells occurred (Figures 6B,C). Despite the lag in activity and growth, the addition of AQDS as a soluble electron shuttle enabled comparable CH4 and biomass (DNA and protein) production in cultures where physical contact between cells and FeS2 was limited to those provided with FeS2 free in solution. This indicated that M. barkeri requires physical contact with FeS2 to directly transfer electrons to the mineral surface invoking EET as the primary mechanism for FeS2 reduction. However, soluble electron shuttles such as AQDS (a humic acid analog) can allow for mineral reduction when direct contact between cells and minerals is restricted. Further, this indicates that M. barkeri does not produce endogenous soluble electron shuttles capable of FeS2 reduction but may take advantage of exogenous shuttles (e.g., humic acids, quinones) in natural systems if they are available.
Thermodynamics experiments described above predicted that the byproducts of FeS2 reduction are HS– and a secondary mineral, Fe1–xS, which is consistent with previous observations of abiotic FeS2 reduction products at high temperature (Truche et al., 2010). To evaluate if M. barkeri Fusaro can use Fe1–xS as a source of Fe and/or S for growth and to determine if direct contact with this mineral is required for its acquisition and assimilation, dialysis experiments were conducted using ground (63–125 μm) specimen grade Fe1–xS particles as the sole Fe source. Ground Fe1–xS particles were sequestered in 100 kDa dialysis tubing and reactors were then amended with either 0 or 500 μM HS– as a S source, since abiotic dissolution of Fe1–xS was shown to release Fe(II) but not HS–, as discussed above. When 0 μM HS– was provided, no CH4 production or growth was detected in any condition (Figure 7). However, when cultures were provided with 500 μM HS–, activity and growth with sequestered Fe1–xS was comparable to growth with 20 μM Fe(II) and 500 μM HS– (Figure 7). Cultures provided with no Fe and 500 μM HS– demonstrated activity and growth, albeit CH4 production was 53–59% lower and biomass 40–41% lower than in cultures grown with 20 μM Fe(II) or provided with Fe1–xS sequestered in dialysis tubing, respectively. This apparent growth under Fe-limiting conditions is comparable to what has been reported previously for methanogens and is attributed to trace Fe contamination in reagents, in particular Na2S, despite its American Chemical Society (ACS) grade (Payne et al., 2021b). Collectively, these data indicate that cells require direct access to the surface of FeS2 to reduce the mineral but not to acquire dissolution products, as the solubility of Fe1–xS with respect to Fe(II) can support growth of M. barkeri. Further, these observations indicate that the source of S for growth is HS– from the initial FeS2 reduction step, not Fe1–xS. As stated above, the predominant form of Fe(II) solubilized from Fe1–xS in sulfidic solutions would be as FeSaq, which is readily produced in aqueous solutions containing excess HS– relative to Fe(II), making this the likely form of Fe and S that is assimilated.
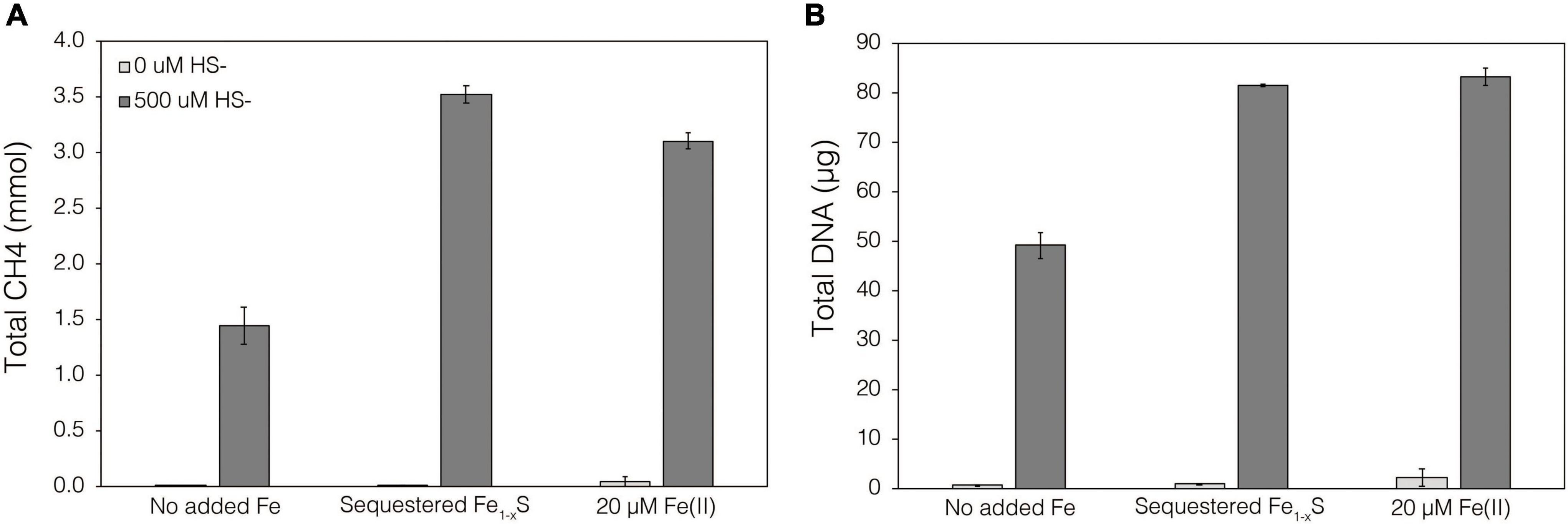
Figure 7. Production of (A) total CH4 and (B) total biomass (DNA) of M. barkeri Fusaro wild-type grown with no added Fe (No added Fe), with 0.1 g specimen pyrrhotite (Fe1–xS; 63–125 μm size fraction) sequestered in 100 kDa dialysis tubing (Sequestered Fe1–xS), or with 20 μM FeCl2. For each of these conditions, no sulfide (0 μM HS–) or 500 μM sulfide (500 μM HS–) was added as Na2S. The data represent the difference for each analyte between the initial measurement (day 0) and the final measurement (day 10). Averages and standard deviations for triplicates are shown. Sampling was infrequent to minimize potential damage to dialysis membranes from sharp, freshly fractured, Fe1–xS grains and their edges.
Because FeS2 is insoluble in water (equilibrium solubility at 25°C, 0.05 M ionic strength, and pH 7 = ca. 0.1 μM), as summarized by Rickard and Luther (2007), the reduction of persulfides in FeS2 by methanogens must occur extracellularly through EET mediated by large (>100 kDa) redox-active molecules or direct electron transfer (e.g., involving electronic conduits within pili, membrane-bound oxidoreductases, secreted molecules, etc.). Genes encoding proteins involved in reducing the persulfide in FeS2 through EET might be expected to be upregulated during growth on FeS2 as the sole source of Fe and S relative to growth soluble Fe and S sources. Shotgun transcriptomics data collected from M. barkeri strain MS were examined to identify protein-encoding genes whose expression was differentially regulated during growth on FeS2 (Supplementary Figures 4A–D). Strain MS was selected as it can use cysteine as its sole sulfur source in addition to HS–. This provides a suitable control for examining possible deleterious effects of minimal HS– generated from FeS2 reduction on growth relative to growth in the presence of 2 mM Na2S [Fe(II)/HS–], an important consideration given reports of HS– toxicity to cells (Edgcomb et al., 2004). M. barkeri strain MS encodes the same complement of [NiFe]-hydrogenases as the wild-type M. barkeri strain Fusaro (Mand et al., 2018; Mand and Metcalf, 2019). Consistent with the results described above indicating that H2 and [NiFe]-hydrogenases are not involved in FeS2 reduction, none of the five [NiFe]-hydrogenases were differentially expressed during growth on FeS2 compared to growth on Fe(II)/Cys or Fe(II)/HS– (Supplementary Figure 4E).
Of the 3,413 genes covered in the differential transcriptomics experiment, transcripts of only 30 were significantly upregulated [p value < 0.05, log2 fold change (LFC) > 0.5] and met abundance thresholds (>0.005% of total normalized read count) when cells were grown on FeS2 relative to those grown with either Fe(II)/cysteine or Fe(II)/HS– (Figure 8A and Supplementary Table 1). This collection of genes included those that code for 12 hypothetical proteins and seven proteins annotated as being involved in translation, ribosomal processing, and other growth-related functions. Nine of the identified FeS2-upregulated genes coded for proteins that are predicted to be membrane-associated. This included genes encoding ABC transporters of organic compounds (MSBRM_0980, MSBRM_0837, and MSBRM_2090) and ferrous iron in its hexaquo Fe(II) form (FeoB; MSBRM_0201; Lau et al., 2016). A gene coding for the transcriptional regulator for FeoB, feoA (MSBRM_0200; Lau et al., 2016), was also identified in this subset of genes upregulated during growth on FeS2. A model to rationalize the upregulation of transcripts of FeoAB when FeS2 is provided as the sole Fe source versus Fe(II) was outlined in a recently published paper that focused on M. voltae A3 (Payne et al., 2021a). Briefly, and as stated above, FeS2-grown M. voltae cells (and possibly M. barkeri cells) were proposed to transport Fe(II) that is complexed with sulfide (FeSaq; Truche et al., 2010). It was hypothesized that assimilation of Fe(II) as FeSaq led cells to sense limited availability of free/dissociable Fe(II) inside the cell via Fe(II)-binding transcription factors (i.e., DtxR, FeoA). As such, cells upregulated the hexaquo Fe(II) transporter (FeoB), and to a lesser extent, its transcriptional regulator (FeoA) and global metal regulator (DtxR), to overcome perceived Fe(II) limitation. Evidence indicated that excess transported Fe (cells require more S than Fe) was sequestered intracellularly as thioferrate-like molecules (Payne et al., 2021a). To this end, FeoAB are not hypothesized to be directly involved in FeS2 reduction, FeSaq assimilation, or in supplying reducing equivalents for EET reactions involving FeS2.
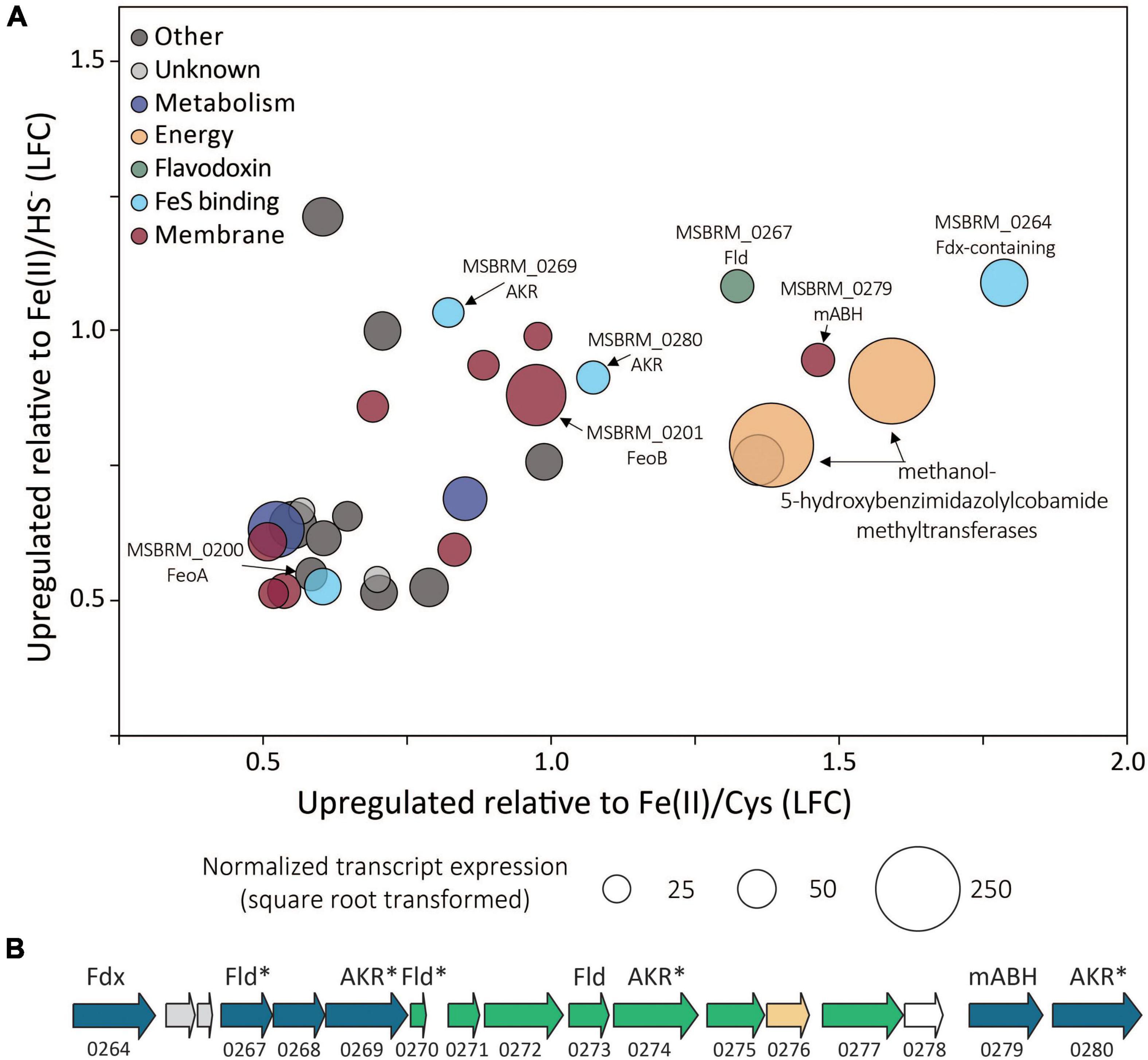
Figure 8. (A) Methanosarcina barkeri MS gene transcripts that were significantly upregulated in cells provided with synthetic pyrite (FeS2) nanoparticles compared to those provided with ferrous iron [Fe(II)]/cysteine (Cys) and those provided with Fe(II)/sulfide (HS–) as the sole iron and sulfur source. The log fold change (LFC) of transcript abundance in FeS2- versus Fe(II)/HS–-grown cells (y-axis) is plotted as a function of the LFC of transcript abundance in FeS2- versus Fe(II)/Cys-grown cells (x-axis). Each point represents a single gene and the size of the point represents the relative mean normalized transcript abundance detected across FeS2 replicate cultures. (B) A cassette of genes whose expression (transcripts) were upregulated when Methanosarcina barkeri MS cells were provided with synthetic FeS2 nanoparticles when compared to cells provided with 20 μM ferrous iron [Fe(II)] and 2 mM Cys or 20 μM ferrous iron [Fe(II)] and 2 mM HS– as sole sources of Fe and S. Truncated locus tags and annotations of gene functions as assessed by UniProt are provided (“MSBRM_” has been removed due to space limitations). Gene arrows colored blue represent significant differential expression in M. barkeri MS cells grown on FeS2 compared to cells grown with Fe(II)/Cys or Fe(II)/HS– (p < 0.05, LFC > 0.5). The green gene arrows were significantly upregulated (p < 0.05) in the FeS2 growth condition relative to both the Fe(II)/Cys or Fe(II)/HS– growth conditions but the LFC was below 0.5. The gene depicted in yellow was upregulated on the FeS2 growth condition relative to both the Fe(II)/Cys or Fe(II)/HS– growth conditions but not significantly (p > 0.05). The gene depicted in white was not differentially regulated among growth conditions. Importantly, an analysis of proteomes of Methanococcus voltae A3 cells provided with 2 mM FeS2 or 26 μM Fe(II) and 2 mM HS– independently identified homologs of many of the same proteins as being up-expressed under the FeS2 condition (Payne et al., 2021a), and these are indicated with an asterisk (*). Gene abbreviations: Fld, flavodoxin; AKR, aldo/keto reductase; Fdx, ferrodoxin; mABH, membrane-associated alpha/beta hydrolase; FeoAB, ferrous iron transporter subunits (A,B).
A cluster of differentially expressed genes that are co-localized on the M. barkeri MS genome that included several oxidoreductases was identified (Figure 8B). Among the 15 genes that comprise this cluster, six were significantly upregulated (p value < 0.05, LFC > 0.5) during growth on FeS2. An additional eight genes were moderately upregulated with a LFC below 0.5, and one additional gene was upregulated though its expression was not significantly different from Fe(II)/HS–- or Fe(II)/cysteine-grown cells. This cluster of genes codes for several enzymes predicted to be cytoplasmic, including one ferredoxin-domain containing oxidoreductase (Fdx; MSBRM_0264), three paralogous flavodoxin domain-containing oxidoreductases (Fld; MSBRM_0267, MSBRM_0270, MSBRM_0273), and three paralogous aldo-keto reductases (AKR; MSBRM_0269, MSBRM_0274 and MSBRM_0280). Two proteins were predicted to be membrane-associated including a putative alpha-beta hydrolase (mABH; MSBRM_0279) and an additional membrane-associated protein of unknown function (MSBRM_0268). Furthermore, a recently published shotgun proteomics study of M. voltae A3 cells grown with 2 mM FeS2 as the sole Fe and S source compared to cells grown with 26 μM Fe(II) and 2 mM HS– revealed significant up-expression (p < 0.05, LFC > 1) of two homologs of Fld (Mvol_0029, Mvol_0089) and three homologs of AKR (Mvol_0066, Mvol_0126, and Mvol_0416) in FeS2-grown cells (Payne et al., 2021a), all of which were predicted to be cytoplasmic. Homologs of mABH and the protein of unknown function (MSBRM_0268) were not upregulated in FeS2-grown M. voltae cells. The Fld and AKR homologs are not co-localized in the genome of M. voltae A3 although the majority are co-localized in the genome of M. barkeri Fusaro. Yet, the similarities in their regulation and expression in M. barkeri MS and M. voltae A3 and their cytoplasmic cellular location point to the potential role of these putatively redox-active enzymes in FeS2-dependent growth, as discussed in detail below.
To date, five strains representing four methanogen species, M. voltae A3 (Payne et al., 2021b), M. barkeri MS (Payne et al., 2021b), M. barkeri Fusaro (this work), an unnamed Methanothermobacter strain isolated from a New Zealand hot spring (provided by Matthew Stott, unpublished data), and M. maripaludis S2 (unpublished data) have been shown to reduce FeS2 to meet biosynthetic demands for Fe and S. These four species represent evolutionarily and ecologically distinct lineages of methanogens, including more deeply diverging members (M. voltae A3, M. maripaludis S2, uncharacterized Methanothermobacter) and more recently diverging members (M. barkeri MS, M. barkeri Fusaro; Bapteste et al., 2005; Brochier-Armanet et al., 2011; Petitjean et al., 2015). This suggests that the ability of methanogens to reduce FeS2 may be widespread among this group of microorganisms. As such, homologs of the 30 genes upregulated on FeS2 relative to Fe(II)/cysteine and Fe(II)/HS– were surveyed in the genomes of four of the five known FeS2 reducers (a genome is not available for the uncharacterized Methanothermobacter strain).
Homologs for 19 of the 30 upregulated FeS2 genes were detected in all four FeS2-reducing methanogens with available genomes (Supplementary Table 1). Five of these 19 genes encoded proteins that were related to ribosomal function, five were related to membrane transport, and two were related to energy or metabolism (Gene IDs provided in Supplementary Table 1). Five of the remaining 19 conserved genes were predicted to encode oxidoreductases (Fdx, Fld, and two AKR) and one of the genes is predicted to encode the hydrolase, mABH (Figure 8). Collectively, these data suggest that one or more proteins encoded in this gene cluster (i.e., Fdx, Fld, AKR, and/or mABH) may be involved in a function associated with FeS2 reduction as they are conserved across methanogens capable of this functionality.
Several dedicated mechanisms for EET have been proposed to explain the ability of microorganisms to transfer electrons to minerals, cathodes, or other microorganisms (Shi et al., 2016; Jorgensen, 2021; Rotaru et al., 2021). Notably, all the examples of EET to date have been described for their role in cellular respiration. However, EET to FeS2 by methanogens for mineral reduction is likely related to nutrient acquisition as FeS2 can serve as the sole source of Fe and S.
Methanogens, including those from the Methanosarcina genus, are capable of Fe(III) oxide mineral reduction as a means for energy conservation (Bond and Lovley, 2002; Sivan et al., 2016; Shang et al., 2020). It has been suggested that the mechanism of Fe(III)-oxide reduction mimics that for bacterial species, whereby Methanosarcina acetivorans cells use transmembrane, multiheme c-type cytochromes (MHC) encoded by mmcA to transfer electrons extracellularly to Fe(III) oxides (Holmes et al., 2019), thereby diminishing the rate of methanogenesis, possibly due to diversion of electrons away from methanogenic pathways. However, neither of the genomes of the M. barkeri strains used in this study (Fusaro or MS) or the genome of M. voltae A3 (Payne et al., 2021b) encode homologs of MHC (data not shown). Further, it has recently been shown that while MHC have been implicated in direct interspecies electron transfer between syntrophic Geobacter and Methanosarcinales, these molecular complexes are not actually required (Yee and Rotaru, 2020). The possibility exists for indirect EET via electron shuttling molecules such as flavins or enzymes such as [NiFe]-hydrogenase and formate dehydrogenase that are excreted from methanogen cells that then drive FeS2 reduction. Indeed, M. maripaludis has been shown to excrete extracellular enzymes, including both [NiFe]-hydrogenases and formate dehydrogenases, that are capable of driving corrosion of metallic iron (Deutzmann et al., 2015). However, in the present study it was shown that when M. barkeri was grown in medium with FeS2 sequestered in dialysis tubing with a pore size of 100 kDa, cells were unable to reduce FeS2, as evinced by the lack of HS– production and growth (Figure 5). This was also observed for M. voltae grown with formate as methanogenesis substrate (Payne et al., 2021b). Thus, a secreted electron shuttling molecule or enzyme would need to have a hydrodynamic diameter large enough to be blocked by the 100 kDa pore, which excludes involvement of low-molecular-weight endogenously produced electron shuttling molecules like flavins. Growth studies with the [NiFe]-hydrogenase M. barkeri Fusaro mutant rule out involvement of hydrogenase enzymes in FeS2 reduction. Nevertheless, at this time, enzymes and enzyme complexes with large molecular weights (>100 kDa) cannot necessarily be ruled out as being involved in FeS2 reduction.
A recent study showed that M. barkeri is capable of assimilating and/or precipitating nanoparticulate magnetite intracellularly, and these conductive nanoparticles enhanced methanogenesis activity presumably by acting as solid electron shuttles across the cell membrane (Fu et al., 2019). Given the production of FeS phases (e.g., Fe1–xS and/or FeS) during FeS2 reduction (Eq. 1), it seems plausible that they may participate in EET in methanogens in a manner similar to that of Shewanella (Kondo et al., 2015). Follow-up studies using high-resolution imaging and spectroscopic approaches combined with electrochemical approaches will advance understanding of the likely role of FeS nanoparticles in EET and FeS2 reduction.
Without a clear mechanism to describe biological FeS2 reduction emerging from experimental data, a model for spontaneous electron transfer from the cell surface to FeS2 is considered. In this model, reduction of FeS2 may occur by diversion of low potential electrons from the membrane, extracellular proteins, or other components of the extracellular milieu. While it is not yet clear what membrane and extracellular components are involved in FeS2 reduction, experiments that showed both AH2QDS and H2 can abiotically reduce FeS2 allow for an estimate of the reduction potential of electrons involved in mineral reduction.
The calculated non-standard state electrochemical potential (E) of H2 [E°= –414 mV (Thauer et al., 1977)] at a headspace partial pressure of 0.25 bar (198 μM dissolved H2) in pH 7.0 medium at 38°C is –318 mV, indicating that electrons under such conditions would need to have a midpoint potential equal to or lower than this to reduce FeS2. However, thermodynamic calculations conducted herein indicate that FeS2 reduction is favorable at H2 concentrations as low as 1 μbar (10–10 M), which equates to electrons with midpoint potentials of –123 mV. Furthermore, the standard state reduction potential of AH2QDS has been estimated to be –184 mV (Clark, 1960), which is lower than that for H2 at a concentration of 10–10 M, consistent with presented evidence indicating it can facilitate EET to FeS2 (Figure 6). Numerous membrane-associated oxidoreductases in methanogens are involved in redox reactions involving electrons that are far more reduced than –184 mV (Thauer et al., 1977), suggesting that their diversion via an undefined or even undedicated mechanism toward FeS2 is feasible. Alternatively, small redox-active molecules produced by methanogens, such as methanophenazine [E = –165 mV (Beifuss and Tietze, 2005)] may be involved in shuttling electrons across the cell membrane to the FeS2 surface. This would imply, however, that the mechanism involved in biological FeS2 reduction is not universal across FeS2-reducing methanogens since M. voltae does not produce methanophenazine (Deppenmeier et al., 1999).
Abiotic reduction of specimen and synthetic, nanoparticulate FeS2 by H2 at concentrations as low as 1.98 × 10–4 M was demonstrated herein. While the initial reduction reaction of FeS2 is necessary to generate HS–, the solubility of the likely FeS2 reduction product, Fe1–xS, controls the availability of Fe(II). Production of excess sulfide (>1 μM) via FeS2 reduction favors formation of FeSaq as the dominant form of Fe(II) in solution, which is hypothesized to be the form of Fe and S assimilated by FeS2-reducing methanogen cells. Dialysis experiments indicate that M. barkeri requires direct contact to reduce FeS2 through EET but not to obtain dissolution products to meet nutrient demands. Growth experiments with wild-type and mutant strains of M. barkeri indicate that H2 and [NiFe]-hydrogenase are not involved in EET and that the cells do not naturally produce electron shuttles capable of reducing EET. Synthetic electron shuttles such as AQDS can facilitate EET to FeS2, suggesting exogenous compounds with similar reduction potentials (quinones, humic acids) could serve this role in natural systems. Transcriptomic experiments did not identify differential expression of genes putatively involved in EET, although a cassette of genes that encodes several oxidoreductases, including Fdx, Fld, and AKRs, and mABH, was found to be significantly upregulated during growth on FeS2 relative to Fe(II)/cysteine and Fe(II)/HS–. It is possible that these proteins are involved in either supplying reducing equivalents for EET to FeS2 or involved in processing, trafficking, storing, or transforming FeSaq that is likely assimilated as the sole source of Fe and S during growth with FeS2 reduction. As such, the mechanism(s) of EET from methanogen cells to FeS2 requires direct contact between cells and the mineral and may involve electrically conductive components of EPS or electrically conductive FeS nanoparticles that self-assemble into conductive conduits within EPS that interfaces cells and the mineral surface.
A multi-step model is proposed to describe the reduction of FeS2 and the assimilation of Fe and S during growth with FeS2 and to guide future experiments (Figure 9). Cells first attach to the FeS2 surface, allowing for microbial reduction of FeS2 via a cell surface mediated mechanism of EET (Step 1). During FeS2 reduction, soluble HS– is generated and Fe1–xS precipitates on the surface of FeS2 (Step 2). Dissolution of Fe(II; but not S) from Fe1–xS occurs, and because dissolution is incomplete, the concentration of HS– in solution exceeds that of Fe(II; Step 3). This favors the formation of FeSaq clusters as the primary form of Fe(II) in solution (Step 4). FeSaq clusters may passively diffuse or be actively transported across the membrane to meet Fe and S nutritional demands. FeSaq clusters, which themselves are likely to be conductive, may also associate with the surface of the cell, EPS, or mineral, thereby enhancing EET to FeS2 (Step 4). This model clarifies the surface-requirement of methanogens during growth on FeS2 to that of EET, rather than for Fe or S acquisition from Fe1–xS. Further, it highlights the complexities methanogens face in obtaining Fe and S to meet nutritional demands in anoxic environments where FeS, Fe1–xS, and FeS2 are likely to be prevalent forms of these elements (Rickard and Luther, 2007).
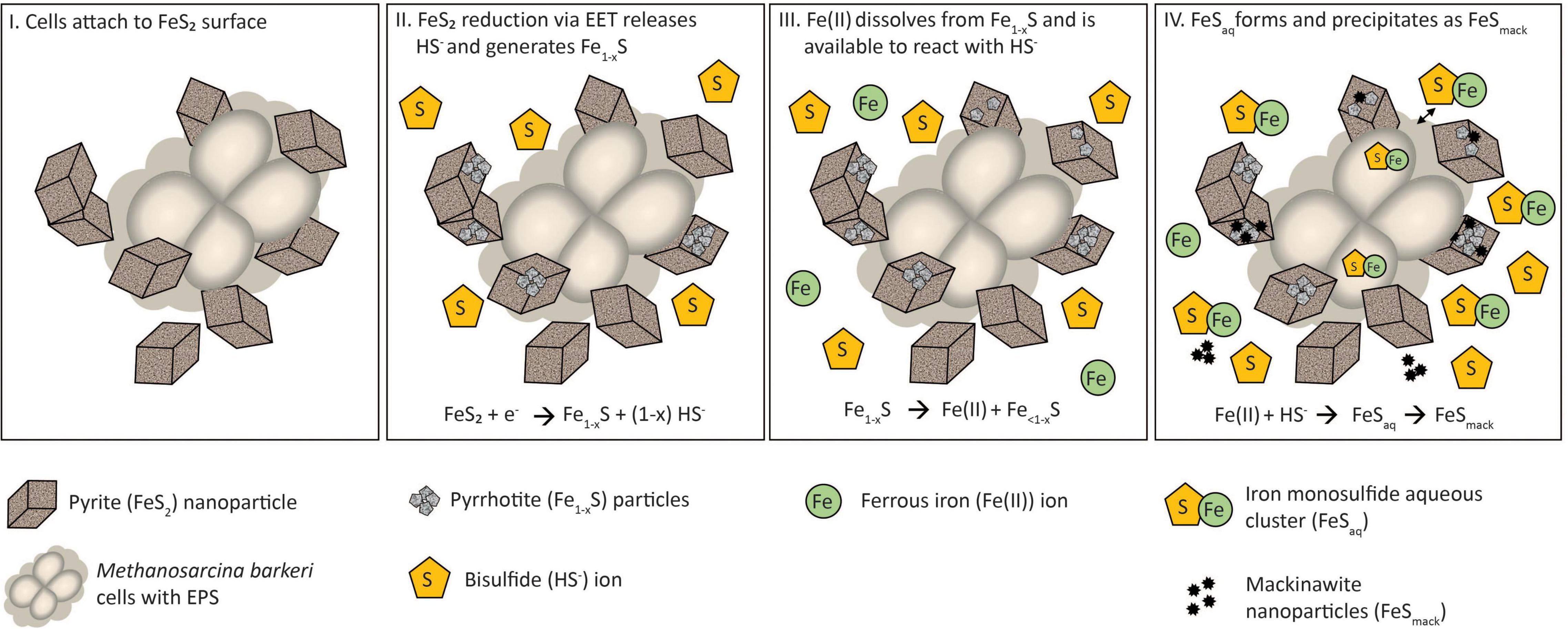
Figure 9. A model for Methanosarcina barkeri growth on pyrite (FeS2). See main text for description.
Synthetic nanoparticulate pyrite (FeS2) was prepared as previously described (Payne et al., 2021b) and XRD analysis is provided in Supplementary Figure 1. ACS grade chemicals were used in mineral syntheses (and preparation of cultivation medium, described below) and glassware was washed with 10% trace-metal grade nitric acid (HNO3) to remove potential trace metal impurities. Briefly, the initial FeS2 synthesis reaction was conducted in an anaerobic chamber (2.5% H2/balance N2) and was then bubbled with sterile N2 gas passed over a heated (200°C) and H2-reduced column containing native copper shavings for 1 h L–1. Following bubbling, FeS2 was incubated anoxically (N2 headspace) in a sealed, serum bottle for 4 days at 65°C followed by an additional 4 days at 85°C to complete the synthesis. Finally, to remove unreacted HS–, Fe(II), FeS, and S0, the synthesized nanoparticulate FeS2 was washed (via centrifugation and decanting) four times with 1N HCl, once with boiling 6N HCl, twice with MilliQ (MQ) H2O, three times with >99.5% acetone, and finally three times with sterile, anoxic MQ H2O inside of an anaerobic chamber. Following the final wash step, the FeS2 was resuspended in sterile, anoxic MQ H2O and transferred to a sterile serum bottle that was then capped and sealed prior to removal from the anaerobic chamber. Upon removal from the anaerobic chamber, the headspace of the serum bottle was flushed with 0.22 μm filtered N2 gas. To determine the concentration of the synthesized FeS2 slurry, triplicate 1 mL aliquots were transferred to pre-weighed serum bottles, dried anoxically under N2 gas, and weighed to calculate the weight percent and molar concentration. The concentration of the FeS2 stock was adjusted to 0.2 M by diluting into sterile, anoxic MQ H2O.
Specimen grade FeS2 was obtained from Zacatecas, Mexico (Ward’s Science, Rochester, NY, United States) and specimen grade Fe1–xS was obtained from Aymar quarry, Gualba mines, Gualba, Montseny, Barcelona Spain. Within a laminar flow hood, specimen minerals were crushed with a MQ-cleaned and ethanol-sterilized jaw crusher (Gilson, Lewis Center, OH, United States) then applied to a cleaned and sterile sieve stack (United States Standard #10/2,000 μm, United States Standard #35/500 μm, United States Standard #60/250 μm, United States Standard #120/125 μm, United States Standard #230/63 μm, and catch pan; all 8” diameter). The 63–125 μm fractions were collected and washed. Specimen FeS2 was washed as described above for synthetic FeS2 while specimen Fe1–xS was washed three times with >99.5% acetone and then three times with sterile, anoxic MQ within an anaerobic chamber. The washed minerals were transferred to sterilized, N2 purged, sealed serum bottles then dried under a stream of 0.22 μm filtered N2 gas. The dried minerals were characterized using a SCINTAG X-1 system X-ray powder diffraction (XRD) spectrometer (XRD Eigenmann GmbH, Mannheim, Germany).
Synthetic, nanoparticulate FeS2 (2 mM) was added to triplicate reactors that contained 35 mL of low salinity basal medium (in g L–1: NaCl, 1.00; MgCl2 ⋅ 6H2O, 0.40; NH4Cl, 0.50; KCl, 0.50; CaCl2 ⋅ 2H2O, 0.10; KH2PO4, 0.15; NaHCO3, 2.0) in 70 mL glass serum bottles capped with blue butyl rubber stoppers (Bellco, Vineland, NJ, United States). This medium is similar to growth medium for M. barkeri MS described below, but without methanol, acetate, vitamins, or trace metal solutions. The reactors were then purged for 20 min with 0.2 μm filtered N2 that had been passed over a heated (200°C) and H2-reduced column containing native copper shavings before pressurizing the reactors by adding H2 to the specified concentration, replacing the appropriate gas volume, and then pressurizing to 2.5 bar with N2. In the case of the 100% H2 condition, the headspace was purged with H2 for 20 min and was then pressurized to 2.5 bar with H2. The reactors were incubated at 38°C statically in the dark.
Reactors were sampled for HS– via the methylene blue assay (Fogo and Popowsky, 1949) and Fe(II) via the ferrozine assay (Stookey, 1970). Samples for HS– determination were analyzed immediately whereas samples for Fe(II)/(III) determination were first incubated for 16 h in 1N HCl at room temperature (∼20°C) before centrifugation (5,000 × g, 5 min, 4°C) to pellet the mineral. The supernatant was split in half, with one half used to measure the concentration of Fe(II). The other half of the sample was used to quantify Fe(III) by first reducing the sample with 0.2 M hydroxylamine hydrochloride for 30 min and then determining total Fe, from which Fe(II) was subtracted to arrive at Fe(III) concentration. Fe(III) was not detected in any incubation experiment.
To further examine the mineral product that forms during reductive dissolution of FeS2, 65 μmol of laboratory synthesized nanoparticulate FeS2 was incubated in 5 mL (6.5 mM final concentration) in MQ H2O undisturbed at 38°C for 24 h under 2.5 bar H2 (1.98 × 10–3 M aqueous). The pelleted mineral was dried anoxically under a stream of N2 gas (∼10 psi delivered through a 22-gauge needle) for ∼2 h while sitting in a warm water bath on low heat to promote drying. The dried mineral was characterized using XRD, as described above, within 2 h of preparation.
Methanosarcina barkeri strain MS and M. barkeri strain Fusaro were obtained from the American Type Culture Collection (ATCC-43569 and ATCC-BAA-2329, respectively). A mutant strain of M. barkeri strain Fusaro with deletions in four operons encoding for its five [NiFe]-hydrogenases was constructed as part of a prior study and was used herein (Mand et al., 2018). For all strains, growth medium was prepared without added Fe and S in MQ water and using acid (10% HNO3) washed glassware. Specifically, M. barkeri was grown in low-salinity medium, as described above for abiotic experiments. The base salts solution was boiled for 10 min then purged with N2 gas passed over the heated copper column containing reduced copper shavings for 1 h L–1. After sparging, base salts medium was moved to an anaerobic chamber and allowed to cool to room temperature. Once cooled, NaHCO3 (2.00 g L–1) was added, and the pH was adjusted to 7.0 with anoxic 2N HCl. Next, 75 mL of the base salts medium were dispensed into 165 mL serum bottles, sealed with blue butyl stoppers, and capped with aluminum crimp caps. Sealed serum bottles were removed from the anaerobic chamber, the headspace was exchanged for 15 min with N2:CO2 (80%:20%) gas passed over a heated copper column, and then they were autoclaved. After autoclaving, a sterile and anoxic 100X phosphate solution containing 0.35 g L–1 K2HPO4 and 0.23 g L–1 KH2PO4 was added to each bottle of base salts medium to a final dilution of 1X. Prior to inoculation, the base salts medium used to cultivate both methanogen strains was amended with 1% (v/v) Wolfe’s vitamins and 1% SL-10 trace metals (no added Fe containing components). All M. barkeri cultures were provided with 0.5% (v/v) methanol and 40 mM acetate as a methanogenesis substrates and grown anaerobically with a N2:CO2 (80%:20%) headspace pressurized to 2.5 bar.
Maintenance cultures were transferred every 4–7 days when they reached late log-phase. Cultures were maintained by providing aqueous sources of Fe and S, which consisted of 20 μM FeCl2 and 2 mM HS–. The M. barkeri Fusaro mutant strain was also provided with 2 mM cysteine to maintain cultures. To inoculate experimental cultures, mid-log phase grown cells were washed by centrifugation (4,696 × g for 20 min at 4°C) under anoxic conditions. Spent medium was decanted and washed cells were resuspended in sterile, anoxic base salts medium in a sealed serum bottle. Cultures were handled within an anaerobic chamber throughout the washing procedure. A 10% (v/v) transfer of washed cells was used to inoculate freshly prepared medium. All cultures were grown statically on their sides at 38°C in the dark.
Growth of M. barkeri was monitored by quantification of DNA and protein. M. barkeri grows in aggregates and strongly associates with FeS2 (Supplementary Figure 5), which prohibits accurate cell enumeration. Further, the presence of FeS2 minerals in the growth medium interferes with OD measurements, commonly used to quantify growth of M. barkeri (Sowers et al., 1993). Therefore, DNA and protein were used as proxies for cell production. In the case of DNA quantification, it is important to note that other Methanosarcina species contain fewer genome copies per cell during slow growth compared to fast growth (Hildenbrand et al., 2011) and thus DNA could underestimate biomass production during early and late log phase. It is also not known if cells produce excess EPS or protein during attachment to mineral phases, which could influence protein-based estimates of biomass production. Nonetheless, to quantify DNA, 2 mL of M. barkeri MS or Fusaro cultures were centrifuged at 20,000 × g for 20 min at 4°C, the supernatant was removed, and the cell pellet was resuspended in a lysis buffer solution containing 489 μL sodium phosphate buffer (MP Biomedicals, Irvine, CA, United States) and 61 μL MT buffer (MP Biomedicals). Once the lysis buffer was added to the cell pellet, the solution was mixed by gentle agitation and subjected to three rounds of freezing at –80°C and heating/thawing at 70°C in a heat block. The mixture was then transferred to a Lysis E tube (MP Biomedicals) and homogenized on a bead beater (Biospec Products, Bartlesville, OK, United States) for 40 s. Finally, the tube and its contents were centrifuged for 15 min at 14,000 × g to separate cell debris from DNA. The concentration of DNA in the supernatant was quantified fluorometrically with a Qubit HS Double Stranded DNA kit and Qubit fluorimeter (Invitrogen, Carlsbad, CA, United States). For protein quantification, a 1 mL aliquot of M. barkeri cultures was centrifuged at 20,000 × g for 20 min at 4°C, the supernatant was removed, and the cell pellet was resuspended in 0.5 M NaOH. The solution was incubated at 99°C in a heat block for 10 min, allowed to cool to room temperature, and then centrifuged for 15 min at 14,000 × g to separate debris from protein. The concentration of protein was quantified fluorometrically with a Qubit Protein Assay kit and Qubit fluorometer (Invitrogen, Carlsbad, CA, United States).
Headspace gas from cultures was sampled with a N2-flushed syringe to monitor CH4 and H2 and was diluted with ultra-high purity N2 into gas-tight CaliBond bags (Calibrated Instruments Inc., Manhasset, NY, United States). CH4 and H2 were determined by gas chromatography via injection of a 5 mL of sample into an injector valve set at 55°C on an SRI 8610C gas chromatograph (SRI instruments, Torrance, CA, United States). The gas chromatroph was equipped with a 4.5 m × 0.125″ OD Hayesep DB 100/120 packed column with the oven set to 44°C (Valco Instrument Company Inc., Houston, TX, United States). CH4 was detected by a flame-ionization detection at 156°C with ultra-high purity He as carrier gas and H2 was measured by a pulse-discharge He-ionizer detector at 100°C. Methane and H2 peak area values were converted to ppm using pre-mixed gas standards (EGAS Depot, Nampa, ID, United States). Dissolved HS– and headspace CH4 measurements were converted to total sulfide and total CH4 (dissolved and gas phase) using Henry’s Law.
For dialysis experiments, 100 kDa dialysis tubing was prepared for FeS2 and Fe1–xS experiments, as previously described (Payne et al., 2021b) using a series of ethanol and MQ H2O rinses to wash and sterilize the membranes. Once sterilized, tied dialysis membranes were transferred to an anaerobic chamber where synthetic FeS2 (final concentration of 2 mM Fe) or specimen Fe1–xS (0.1 g) was added to each dialysis bag then the ends were secured using monofilament lines. The same concentrations of FeS2 and Fe1–xS were added to reactors where minerals were not sequestered in dialysis membranes. The dialysis bags were rinsed with sterile, anoxic MQ H2O before transferring to a prepared bottle of medium amended with methanogenesis substrates, trace metals, and vitamins, which was then capped and sealed before removing from the anaerobic chamber. Unamended controls contained dialysis tubing prepared the same as above but without any mineral or Fe added. Upon removing the sealed medium bottles from the anaerobic chamber, the headspace was purged with an 80:20 mixture of 0.2 μm filtered N2:CO2 for 45 min to remove gas originating from the anaerobic chamber and were then pressurized with N2:CO2 (final pressure of 1.72 atm) before inoculating with mid-log M. barkeri Fusaro wild-type cells grown with sulfide and FeCl2 (10% v/v inoculum) that were washed by pelleting cells via centrifugation and gently resuspending in anoxic base salts medium within an anaerobic chamber. Anoxic AQDS was added to a final concentration of 2 mM and monitored spectrophotometrically at 325 nm for AQDS and 450 nm for AH2QDS.
Cultures of M. barkeri MS were grown in quadruplicate on three different sources of Fe and S for RNA-Seq analysis: 2 mM FeS2, 20 μM FeCl2 and 2 mM L-cysteine, or 20 μM FeCl2 and 2 mM Na2S. Quadruplicate cultures for each condition were kept separate throughout sample collection, RNA sequencing, and analysis. Cells were harvested at mid-log growth phase, as determined by CH4 production, and final biomass was estimated by DNA quantification. To harvest biomass, M. barkeri MS cells were subjected to vacuum filtration at low pressure (5 psi) onto 47 mm 0.2-μm Supor 200 PES filters (Pall, Port Washington, NY, United States) within an anaerobic chamber. Using flame-sterilized scissors, each filter was cut in half and each half transferred to a 1.5 mL cryotube that was then sealed before being removed from the anaerobic chamber. The tube and its contents were immediately flash frozen in liquid nitrogen and frozen cells were stored at –80°C until processing.
Total RNA from M. barkeri strain MS was extracted using TRIzol reagent (Invitrogen) following the manufacturer’s protocol with slight modification. Individual frozen half filters containing M. barkeri cells were transferred to a Lysis E tube (MP Biomedicals) on ice and 1 mL of TRIzol was immediately added to the frozen filter. TRIzol-treated M. barkeri cells were subjected to three cycles of 40 s of bead beating followed by resting for 5 min at room temperature (∼20°C). Two hundred microliter of molecular grade chloroform was added to each tube and each tube was inverted to mix, incubated at room temperature for 3 min, and centrifuged for 15 min at 12,000 × g at 4°C. The upper aqueous phase containing RNA was carefully transferred to a clean 2 mL tube. To precipitate RNA, 0.5 mL of pre-chilled (4°C) 100% molecular grade isopropanol was added to each tube and tubes and their contents were incubated on ice for 10 min followed by centrifugation for 10 min at 12,000 × g at 4°C. The supernatant was discarded, and 1 mL of 75% molecular grade ethanol was added to wash the RNA. After centrifugation for 5 min at 7,500 × g at 4°C, the supernatant was removed, and the RNA pellet was air dried for 5–10 min. Total RNA was dissolved in 50 μL RNA-grade water (Thermo Fisher Scientific, Waltham, MA, United States) by incubating in a 55°C heat block for 10 min. RNA was treated with Turbo DNase (Invitrogen) to remove residual DNA per manufacturer’s instructions. After DNase treatment, the remaining total RNA was subjected to a second round of isopropanol precipitation, ethanol wash, and resuspension as described above. The concentration of total RNA was quantified fluorometrically using the Qubit BR RNA kit and a Qubit fluorometer (Invitrogen) and the quality checked using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). Absence of DNA was verified by subjecting the RNA extract to 40 cycles of PCR using archaeal-specific 16S rRNA primers (344F/915R) as previously described (Boyd et al., 2013) and checking for amplification products via gel electrophoresis. Total RNA was sent to the University of Wisconsin’s Genome Expression Center for quality control, rRNA depletion using custom M. barkeri strain MS-specific oligos designed using the sequences for M. barkeri MS’s large and small ribosomal subunits, and sequencing on an Illumina NovaSeq 2 × 150 bp.
Paired-end reads were processed using default settings in TrimGalore!,1 a wrapper that implements CutAdapt (Martin, 2011) and FastQC2 to remove adapter sequences and filter reads, respectively. Reads were aligned to the reference M. barkeri MS genome (ASM97002v1) using Bowtie2 (Langmead and Salzberg, 2012). Reads were counted for each locus using HTSeq (Anders et al., 2015) followed by normalization and analysis in DESeq2 (Love et al., 2014) implemented in R v3.6.0.
The RNA-sequencing data reported in this article have been deposited in the NCBI GEO database (GSE168895).
RS and EB conceived and designed the study. RS, DP, ER, and EB collected, interpreted, and analyzed experimental data. GK and WM designed and provided mutant methanogen strains for experiments. RS and EB wrote the manuscript. All authors reviewed the manuscript before submission.
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the United States Department of Energy through Grant DE-SC0020246 to EB for geochemical, physiological, and molecular work and DE-FG02-02ER15296 to WM for construction of the M. barkeri Fusaro mutant used in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.878387/full#supplementary-material
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Bapteste, E., Brocheir, C., and Boucher, Y. (2005). Higher-level classification of the archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363. doi: 10.1155/2005/859728
Beifuss, U., and Tietze, M. (2005). “Methanophenazine and other natural biologically active phenazines,” in Natural Products Synthesis II. Topics in Current Chemistry, ed. J. Mulzer (Berlin: Springer), 77–113. doi: 10.1007/b96889
Benning, L. G., Wilkin, R. T., and Barnes, H. L. (2000). Reaction pathways in the Fe-S system below 100°C. Chem. Geol. 167, 25–51. doi: 10.1016/s0009-2541(99)00198-9
Berner, R. A. (1967). Thermodynamic stability of sedimentary iron sulfides. Am. J. Sci. 265, 773–785. doi: 10.2475/ajs.265.9.773
Berner, R. A. (1984). Sedimentary pyrite formation: an update. Geochim. Cosmochim. Acta 48, 605–615. doi: 10.1016/0016-7037(84)90089-9
Bond, D. R., and Lovley, D. R. (2002). Reduction of Fe(III) oxide by methanogens in the presence and absence of extracellular quinones. Environ. Microbiol. 4, 115–124. doi: 10.1046/j.1462-2920.2002.00279.x
Boyd, E. S., Hamilton, T. L., Wang, J., He, L., and Zhang, C. (2013). The role of tetraether lipid composition in the adaptation of thermophilic archaea to acidity. Front. Microbiol. 4:62. doi: 10.3389/fmicb.2013.00062
Bradley, J. A., Arndt, S., Amend, J. P., Burwicz, E., Dale, A. W., Egger, M., et al. (2020). Widespread energy limitation to life in global seafloor sediments. Sci. Adv. 6, 1–9. doi: 10.1126/sciadv.aba0697
Brochier-Armanet, C., Forterre, P., and Gribaldo, S. (2011). Phylogeny and evolution of the archaea: one hundred genomes later. Curr. Opin. Microbiol. 14, 274–281. doi: 10.1016/j.mib.2011.04.015
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M., and Berner, R. A. (1986). The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155. doi: 10.1016/0009-2541(86)90078-1
Chareev, D. A., Voronin, M. V., and Osadchii, E. G. (2014). Thermodynamic study of monoclinic pyrrhotite in equilibrium with pyrite in the Ag-Fe-S system by solid-state electrochemical cell technique. Am. Mineral. 99, 2031–2034. doi: 10.2138/am-2014-4753
Clark, W. M. (1960). Oxidation-Reduction Potentials of Organic Systems. Baltimore: Baltimore, Williams & Wilkins.
Colman, D. R., Poudel, S., Stamps, B. W., Boyd, E. S., and Spear, J. R. (2017). The deep, hot biosphere: twenty-five years of retrospection. Proc. Natl. Acad. Sci. U.S.A. 114, 6895–6903. doi: 10.1073/pnas.1701266114
Colwell, F. S., and D’Hondt, S. (2013). Nature and extent of the deep biosphere. Rev. Mineral. Geochem. 75, 547–574. doi: 10.1515/9781501508318-019
Crowe, S. A., Dossing, L. N., Beukes, N. J., Bau, M., Kruger, S. J., Frei, R., et al. (2013). Atmospheric oxygenation three billion years ago. Nature 501, 535–538. doi: 10.1038/nature12426
Davison, W. (1991). The solubility of iron sulphides in synthetic and natural waters at ambient temperature. Aquat. Sci. 53, 309–329. doi: 10.1007/bf00877139
Daye, M., Klepac-Ceraj, V., Pajusalu, M., Rowland, S., Farrell-Sherman, A., Beukes, N., et al. (2019). Light-driven anaerobic microbial oxidation of manganese. Nature 576, 311–314. doi: 10.1038/s41586-019-1804-0
de Aldecoa, A. L., Roldan, F. V., and Menor-Salvan, C. (2013). Natural pyrrhotite as a catalyst in prebiotic chemical evolution. Life 3, 502–517. doi: 10.3390/life3030502
Deppenmeier, U., Lienard, T., and Gottschalk, G. (1999). Novel reactions involved in energy-conservation by methanogenic archaea. FEBS Lett. 457, 291–297. doi: 10.1016/s0014-5793(99)01026-1
Deutzmann, J. S., Sahin, M., and Spormann, A. M. (2015). Extracellular enzymes facilitate electron uptake in biocorrosion and bioelectrosynthesis. mBio 6, 1–8. doi: 10.1128/mBio.00496-15
Edgcomb, V. P., Molyneaux, S. J., Saito, M. A., Lloyd, K., Boer, S., Wirsen, C. O., et al. (2004). Sulfide ameliorates metal toxicity for deep-sea hydrothermal vent archaea. Appl. Environ. Microbiol. 70, 2551–2555. doi: 10.1128/AEM.70.4.2551-2555.2004
Fogo, J. K., and Popowsky, M. (1949). Spectrophotometric determination of hydrogen sulfide. Anal. Chem. 21, 732–734. doi: 10.1021/ac60030a028
Fu, L., Zhou, T., Wang, J., You, L., Lu, Y., Yu, L., et al. (2019). NanoFe3O4 as Solid electron shuttles to accelerate acetotrophic methanogenesis by Methanosarcina barkeri. Front. Microbiol. 10:388. doi: 10.3389/fmicb.2019.00388
Garvin, J., Buick, R., Anbar, A. D., Arnold, G. L., and Kaufman, A. J. (2009). Isotopic evidence for an aerobic nitrogen cycle in the latest archean. Science 323, 1045–1048. doi: 10.1126/science.1165675
Hall, A. J. (1986). Pyrite-pyrrhotine redox reactions in nature. Mineral. Mag. 60, 223–229. doi: 10.1180/minmag.1986.050.356.05
Hildenbrand, C., Stock, T., Lange, C., Rother, M., and Soppa, J. (2011). Genome copy numbers and gene conversion in methanogenic archaea. J. Bacteriol. 193, 734–743. doi: 10.1128/JB.01016-10
Holmes, D. E., Ueki, T., Tang, H.-Y., Zhou, J., Smith, J. A., Chaput, G., et al. (2019). A membrane-bound cytochrome enables Methanosarcina acetivorans to conserve energy from extracellular electron transfer. mBio 10, 1–12. doi: 10.1128/mBio.00789-19
Jakus, N., Mellage, A., Hoschen, C., Maisch, M., Byrne, J. M., Mueller, C. W., et al. (2021). Anaerobic neutrophilic pyrite oxidation by a chemolithoautotrophic nitrate-reducing iron(II)-oxidizing culture enriched from a fractured aquifer. Environ. Sci. Technol. 55, 9876–9884. doi: 10.1021/acs.est.1c02049
Johnson, C., England, A., Munro-Ehrlich, M., Colman, D. R., Dubois, J. L., and Boyd, E. S. (2021). Pathways of iron and sulfur acquisition, cofactor assembly, destination, and storage in diverse archaeal methanogens and alkanotrophs. J. Bacteriol. 203:e0011721. doi: 10.1128/JB.00117-21
Johnson, J. E., Webb, S. M., Thomas, K., Ono, S., Kirschvink, J. L., and Fischer, W. W. (2013). Manganese-oxidizing photosynthesis before the rise of cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 11238–11243. doi: 10.1073/pnas.1305530110
Jordan, S. F., Nee, E., and Lane, N. (2019a). Isoprenoids enhance the stability of fatty acid membranes at the emergence of life potentially leading to an early lipid divide. Interface Focus 9:20190067. doi: 10.1098/rsfs.2019.0067
Jordan, S. F., Rammu, H., Zheludev, I. N., Hartley, A. M., Marechal, A., and Lane, N. (2019b). Promotion of protocell self-assembly from mixed amphiphiles at the origin of life. Nat. Ecol. Evol. 3, 1705–1714. doi: 10.1038/s41559-019-1015-y
Jorgensen, B. B. (2021). Do methanogenic archaea cause reductive pyrite dissolution in subsurface sediments? ISME J. 16, 1–2. doi: 10.1038/s41396-021-01055-0
Jorgensen, C. J., Jacobsen, O. S., Elberling, B., and Aamand, J. (2009). Microbial oxidation of pyrite coupled to nitrate reduction in anoxic groundwater sediment. Environ. Sci. Technol. 43, 4851–4857. doi: 10.1021/es803417s
Kondo, K., Okamoto, A., Hashimoto, K., and Nakamura, R. (2015). Sulfur-mediated electron shuttling sustains microbial long-distance extracellular electron transfer with the aid of metallic iron sulfides. Langmuir 31, 7427–7434. doi: 10.1021/acs.langmuir.5b01033
Konhauser, K. O., Lalonde, S. V., Planavsky, N. J., Pecoits, E., Lyons, T. W., Mojzsis, S. J., et al. (2011). Aerobic bacterial pyrite oxidation and acid rock drainage during the great oxidation event. Nature 478, 369–373. doi: 10.1038/nature10511
Kulkarni, G., Mand, T. D., and Metcalf, W. W. (2018). Energy conservation via hydrogen cycling in the methanogenic archaeon Methanosarcina barkeri. mBio 9, e1256–e1218. doi: 10.1128/mBio.01256-18
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lau, C. K., Krewulak, K. D., and Vogel, H. J. (2016). Bacterial ferrous iron transport: the Feo system. FEMS Microbiol. Rev. 40, 273–298. doi: 10.1093/femsre/fuv049
Liu, D., Wang, H., Dong, H., Qiu, X., Dong, X., and Cravotta, C. A. (2011). Mineral transformations associated with goethite reduction by Methanosarcina barkeri. Chem. Geol. 288, 53–60. doi: 10.1016/j.chemgeo.2011.06.013
Liu, W., Hao, J., Elzinga, E. J., Piotrowiak, P., Nanda, V., Yee, N., et al. (2020). Anoxic photogeochemical oxidation of manganese carbonate yields manganese oxide. Proc. Natl. Acad. Sci. U.S.A. 117, 22698–22704. doi: 10.1073/pnas.2002175117
Liu, Y., Beer, L. L., and Whitman, W. B. (2012). Methanogens: a window into ancient sulfur metabolism. Trends Microbiol 20, 251–258. doi: 10.1016/j.tim.2012.02.002
Liu, Y., Sieprawska-Lupa, M., Whitman, W. B., and White, R. H. (2010). Cysteine is not the sulfur source for iron-sulfur cluster and methionine biosynthesis in the methanogenic archaeon Methanococcus maripaludis. J. Biol. Chem. 285, 31923–31929. doi: 10.1074/jbc.m110.152447
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lupa, B., Hendrickson, E. L., Leigh, J. A., and Whitman, W. B. (2008). Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. Appl. Environ. Microbiol. 74, 6584–6590. doi: 10.1128/AEM.01455-08
Luther, G. W., and Rickard, D. T. (2005). Metal sulfide cluster complexes and their biogeochemical importance in the environment. J. Nanopart. Res. 7, 389–407. doi: 10.1007/s11051-005-4272-4
Major, T. A., Burd, H., and Whitman, W. B. (2004). Abundance of 4Fe-4S motifs in the genomes of methanogens and other prokaryotes. FEMS Microbiol. Lett. 239, 117–123. doi: 10.1016/j.femsle.2004.08.027
Mand, T. D., Kulkarni, G., and Metcalf, W. W. (2018). Genetic, biochemical, and molecular characterization of Methanosarcina barkeri mutants lacking three distinct classes of hydrogenase. J. Bacteriol. 200, 1–16. doi: 10.1128/JB.00342-18
Mand, T. D., and Metcalf, W. W. (2019). Energy conservation and hydrogenase function in methanogenic archaea, in particular the genus Methanosarcina. Microbiol. Mol. Biol. Rev. 83, 1–22. doi: 10.1128/MMBR.00020-19
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 3. doi: 10.1089/cmb.2017.0096
Mikhlin, Y. (2000). Reactivity of pyrrhotite surfaces: an electrochemical study. Phys. Chem. Chem. Phys. 2, 5672–5677. doi: 10.1039/b005373m
Orcutt, B. N., Larowe, D. E., Biddle, J. F., Colwell, F. S., Glazer, B. T., Reese, B. K., et al. (2013). Microbial activity in the marine deep biosphere: progress and prospects. Front. Microbiol. 4:189. doi: 10.3389/fmicb.2013.00189
Payne, D., Spietz, R. L., and Boyd, E. S. (2021b). Reductive dissolution of pyrite by methanogenic archaea. ISME J. 15, 3498–3507. doi: 10.1038/s41396-021-01028-3
Payne, D., Shepard, E. M., Spietz, R. L., Steward, K., Brumfield, S., Young, M., et al. (2021a). Examining pathways of iron and sulfur acquisition, trafficking, deployment, and storage in mineral-grown methanogen cells. J. Bacteriol. 203, 1–18. doi: 10.1128/JB.00146-21
Percak-Dennett, E., He, S., Converse, B., Konishi, H., Xu, H., Corcoran, A., et al. (2017). Microbial acceleration of aerobic pyrite oxidation at circumneutral pH. Geobiology 15, 690–703. doi: 10.1111/gbi.12241
Petitjean, C., Deschamps, P., Lopez-Garcia, P., Moreira, D., and Brochier-Armanet, C. (2015). Extending the conserved phylogenetic core of archaea disentangles the evolution of the third domain of life. Mol. Biol. Evol. 32, 1242–1254. doi: 10.1093/molbev/msv015
Rickard, D., and Luther, G. W. I. (2007). Chemistry of iron sulfides. Chem. Rev. 107, 514–562. doi: 10.1021/cr0503658
Ronnow, P. H., and Gunnarsson, L. A. H. (1981). Sulfide-dependent methane production and growth of a thermophilic methanogenic bacterium. Appl. Environ. Microbiol. 42, 580–584. doi: 10.1128/aem.42.4.580-584.1981
Rotaru, A. E., Yee, M. O., and Musat, F. (2021). Microbes trading electricity in consortia of environmental and biotechnological significance. Curr. Opin. Biotechnol. 67, 119–129. doi: 10.1016/j.copbio.2021.01.014
Rowe, A. R., Xu, S., Gardel, D., Bose, A., Girguis, P. R., Amend, J. P., et al. (2019). Methane-linked mechanisms of electron uptake from cathodes by Methanosarcina barkeri. mBio 10, 1–12. doi: 10.1128/mBio.02448-18
Schippers, A., and Jørgensen, B. B. (2001). Oxidation of pyrite and iron sulfide by manganese dioxide in marine sediments. Geochim. Cosmochim. Acta 65, 915–922. doi: 10.1016/s0016-7037(00)00589-5
Schoonen, M. A. A. (2004). “Mechanisms of sedimentary pyrite formation,” in Sulfur Biogeochemistry—Past and Present, eds J. P. Amend, K. J. Edwards, and T. W. Lyons (Boulder, CO: Geological Society of America), 117–134.
Shang, H., Daye, M., Sivan, O., Borlina, C. S., Tamura, N., Weiss, B. P., et al. (2020). Formation of zerovalent iron in iron-reducing cultures of Methanosarcina barkeri. Environ Sci Technol 54, 7354–7365. doi: 10.1021/acs.est.0c01595
Shi, L., Dong, H., Reguera, G., Beyenal, H., Lu, A., Liu, J., et al. (2016). Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 14, 651–662. doi: 10.1038/nrmicro.2016.93
Sivan, O., Shusta, S. S., and Valentine, D. L. (2016). Methanogens rapidly transition from methane production to iron reduction. Geobiology 14, 190–203. doi: 10.1111/gbi.12172
Sowers, K. R., Boone, J. E., and Gunsalus, R. P. (1993). Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl. Environ. Microbiol. 59, 3832–3839. doi: 10.1128/aem.59.11.3832-3839.1993
Stookey, L. L. (1970). Ferrozine—a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781. doi: 10.1021/ac60289a016
Stüeken, E. E., Catling, D. C., and Buick, R. (2012). Contributions to late archaean sulphur cycling by life on land. Nat. Geosci. 5, 722–725. doi: 10.1038/ngeo1585
Stumm, W. (1992). Chemistry of the Solid-Water Interface: Processes at the Mineral-Water And Particle-Water Interface In Natural Systems. New York, NY: John Wiley & Son Inc.
Thauer, R. K., Jungermann, K., and Decker, K. (1977). Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41, 100–180. doi: 10.1128/br.41.1.100-180.1977
Tipping, E. (1984). Temperature dependence of Mn(II) oxidation in lakewater: a test of biological involvement. Geochim. Cosmochim. Acta 48, 1353–1356. doi: 10.1016/0016-7037(84)90069-3
Truche, L., Berger, G., Destrigneville, C., Guillaume, D., and Giffaut, E. (2010). Kinetics of pyrite to pyrrhotite reduction by hydrogen in calcite buffered solutions between 90 and 180°C: implications for nuclear waste disposal. Geochim. Cosmochim. Acta 74, 2894–2914. doi: 10.1016/j.gca.2010.02.027
Valentine, D. L., Blanton, D. C., and Reeburgh, W. S. (2000). Hydrogen production by methanogens under low-hydrogen conditions. Arch. Microbiol. 174, 415–421. doi: 10.1007/s002030000224
Keywords: methanogens, pyrrhotite (Fe1–xS), dissolution, hydrogen, extracellular electron transfer, pyrite (FeS2)
Citation: Spietz RL, Payne D, Kulkarni G, Metcalf WW, Roden EE and Boyd ES (2022) Investigating Abiotic and Biotic Mechanisms of Pyrite Reduction. Front. Microbiol. 13:878387. doi: 10.3389/fmicb.2022.878387
Received: 18 February 2022; Accepted: 31 March 2022;
Published: 09 May 2022.
Edited by:
David Jeffrey Fraser Walker, University of Texas at Austin, United StatesReviewed by:
Gary Sawers, Martin Luther University of Halle-Wittenberg, GermanyCopyright © 2022 Spietz, Payne, Kulkarni, Metcalf, Roden and Boyd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric S. Boyd, ZXJpYy5ib3lkQG1vbnRhbmEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.