 Haiwei Wang
Haiwei Wang Xingyang Cui
Xingyang Cui Xuehui Cai
Xuehui Cai Tongqing An
Tongqing An- State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Harbin, China
RNA recombination is a major driver of genetic shifts tightly linked to the evolution of RNA viruses. Genomic recombination contributes substantially to the emergence of new viral lineages, expansion in host tropism, adaptations to new environments, and virulence and pathogenesis. Here, we review some of the recent progress that has advanced our understanding of recombination in positive-strand RNA viruses, including recombination triggers and the mechanisms behind them. The study of RNA recombination aids in predicting the probability and outcome of viral recombination events, and in the design of viruses with reduced recombination frequency as candidates for the development of live attenuated vaccines. Surveillance of viral recombination should remain a priority in the detection of emergent viral strains, a goal that can only be accomplished by expanding our understanding of how these events are triggered and regulated.
Introduction
Background
RNA viruses cause significant morbidity and mortality to both humans and livestock on a global scale. This extensive group of viruses includes severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) (Wang et al., 2020; Zhou et al., 2020), Ebola virus, poliovirus, foot-and-mouth disease virus (FMDV), and porcine reproductive and respiratory syndrome virus (PRRSV), among many others. Due to the low fidelity and lack of proofreading capacity of the virus-encoded RNA-dependent RNA polymerase (RdRp) (Steinhauer et al., 1992), the genetic heterogeneity of RNA virus populations enables efficient selection of progeny virus variants with newly acquired properties (Duffy et al., 2008). Within populations, recombination is a pervasive phenomenon of RNA viruses and an important strategy for accelerating the evolution of RNA virus populations.
Recombination is critical in the biology and evolution of RNA viruses. Accumulated evidence has shown that RNA virus recombination is one of the main approaches to virus rapid adaptation and evolution. However, there is a lack of in-depth understanding of the effects and the methods of studying virus recombination. This review provides a detailed summary of RNA virus recombination.
Genetic Recombination in RNA Viruses
Recombination in RNA viruses can be defined as an exchange of genetic material between at least two separate viral genomes. Recombination occurs when two or more viruses co-infect a single cell and exchange segments, often giving rise to viable hybrid progeny. To date, recombinant RNA viruses have been found in both positive-strand RNA viruses and negative-strand viruses (Han and Worobey, 2011; Bentley and Evans, 2018). Recombination is more common in positive single-stranded RNA viruses than in negative single-stranded RNA ones (Patiño-Galindo et al., 2021). Occasionally, RNA viruses exploit recombination to acquire host cellular sequences, largely impacting their functionality (Becher and Tautz, 2011; Zou et al., 2019).
Various animal and plant viruses exhibit different forms of recombination, some of which are unique to RNA viruses and different from DNA viruses. Based on the structure of viral RNA molecules and the sites where recombination occurs, RNA virus recombination can be classified into three types: homologous recombination, aberrant homologous recombination, and non-homologous recombination (Lai, 1992a). Homologous recombination occurs between parental RNA molecules that are similar in sequence, with the recombination region being located on the matching, homologous sequences. The resulting recombinant RNA contains the sequence and structure of the parental RNA molecules (Ji et al., 2020; Alatorre-García et al., 2021; Crook et al., 2021). Similarly, aberrant homologous recombination also requires two parental RNA molecules with similar sequences. However, in this case, the recombination event takes place in an unrelated region near the homologous sequence. Aberrant homologous recombination can result in RNA molecules with insertions, deletions, or duplications and, because of this, it often yields defective viral genomes (Mura et al., 2017; Genoyer et al., 2020; Felt et al., 2021). One specific mechanism of aberrant homologous recombination is copy-back RNA synthesis (Sun et al., 2019; Vignuzzi and López, 2019; Janissen et al., 2021), in which the 3′-end of nascent RNA base pairs to an internal position of the same RNA and continues synthesis, deleting the intervening sequences in the process. In contrast with the previous two cases, non-homologous recombination does not depend on sequence homology between the recombined parental RNA molecules, and the selection of recombined sites remains unclear.
Recombination can occur within the same or between different viral subtypes. For example, the hepatitis C virus (HCV) is a positive-sense, single-stranded RNA genome of the Flaviviridae family. HCV recombinants could occur at all levels, including between genotypes, between subtypes of the same genotype, and even between strains of the same subtype (González-Candelas et al., 2011), and a similar pattern of recombination has occurred in poliovirus (Xiao et al., 2022). The recombination rates vary markedly for different viral species. For example, positive single-stranded RNA viruses, such as picornaviruses and coronaviruses, show a high frequency of recombination. In contrast, the Zika virus displays a low recombination frequency (Han et al., 2016). Notably, the estimation of RNA virus recombination frequencies should be taken with caution because current RNA recombination assays require virus viability as an endpoint, thus imparting a strong selection and technical bias.
Consequences of Virus Recombination
Viral RNA recombination facilitates the ontogeny of viral variants with increased virulence and pathogenesis. The emergence of new variants could be the result of selective pressure or a positive selection. The new variant may respond to host immune pressure and develops advantageous mutations or recombination to make it more transmissible in susceptible populations or better able to escape our natural immune responses. Meanwhile, specific variants may be favored by having higher fitness in that context. The variant with higher replicative capacity can be selected and could be less virulent to the host, and the result does not necessarily have to be detrimental to the host. The impact of the new variant on the host may be double-sided. Understanding the propensity for RNA recombination between similar viruses infecting different host species, for example, establishes an important framework in the control of disease spread between endemic regions and could be used to model and predict the risks of future RNA virus infections.
Expansion of Host Species or Cell Tropism
Accumulating evidence supports the hypothesis that recombination could result in an expansion of cellular tropism and even host tropism. For example, studies have suggested that Western equine encephalitis virus (WEEV), a zoonotic virus that causes encephalitis in humans and severe diseases in horses, seems to be the result of the recombination between an Eastern equine encephalitis virus (EEEV)-like virus and a Sindbis-like virus (Figure 1A), exhibiting the encephalitogenic properties of EEEV and the antigenicity of Sindbis virus (Hahn et al., 1988; Forrester et al., 2012).
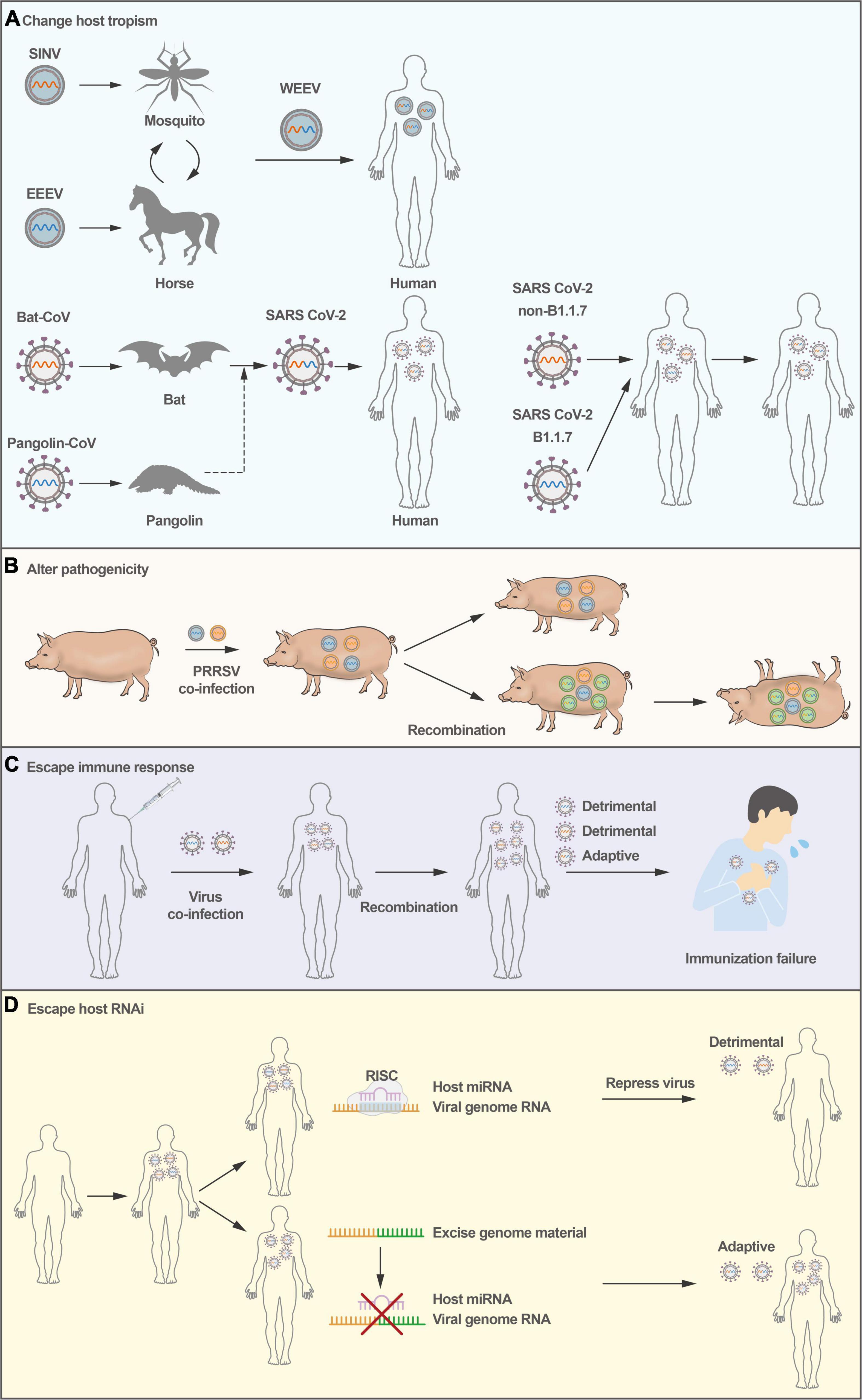
Figure 1. Consequences of virus recombination. (A) Virus recombination causes host species expansion; WEEV and SARS-CoV-2 were transferred from animals to humans via recombination. (B) PRRSV recombination causes pathogenicity alterations in pigs. (C) Virus recombination makes the virus escape immunity, and the replication of the virus in the host eventually results in immunological failure. (D) RNA viruses could excise genomic material through recombination as an intrinsic defense against antiviral RNA interference.
More recently, the SARS-CoV-2 pandemic has resulted in high morbidity and mortality in the human population. Despite extensive research, the origin of SARS-CoV-2 remains unclear. The receptor-binding domain (RBD) in its spike protein is almost identical in sequence to that of Pangolin-CoV (Zhang et al., 2020), suggesting that SARS-CoV-2 could have originated from recombination between coronaviruses with different host tropism (Lam et al., 2020; Figure 1A). Notably, genomic recombination analyses have shown that the circulating strains of SARS-CoV-2 from late 2020 and early 2021 in the United Kingdom are inter-lineage recombinants (Jackson et al., 2021). Subsequently, the SARS-CoV-2 epidemic has been under several distinct waves. The first was associated with a mix of SARS-CoV-2 lineages, while the second and third waves were driven by the Beta (B.1.351) and Delta (B.1.617.2) variants (Tegally et al., 2021a,b,c). In November 2021, a new SARS-CoV-2 variant associated with a rapid resurgence of infections was detected, and it was designated a variant of concern (Omicron, B.1.1.529) (Viana et al., 2022). The Omicron has three major lineages BA.1, BA.2, and BA.3. Given a large number of mutations between BA.1, BA.2, and BA.3 and other known SARS-CoV-2 lineages, it is thought that this may be associated with recombination. However, weak statistical and phylogenetic evidence could be used to determine the recombination of BA.1, BA.2, or BA.3 (Viana et al., 2022). Moreover, recombinant viruses are products that could be under a series of multiple partially overlapping recombination events. The existing evidence can not fully prove the complex recombination patterns.
Recombination in beta coronaviruses, including natural populations of MERS-CoV (Dudas and Rambaut, 2016; Kim et al., 2016; Schroeder et al., 2021), SARS, and SARS-like coronaviruses (Hon et al., 2008; Boni et al., 2020), occurs with increased frequency, and the recombinant viruses tend to be highly adaptable. RNA recombination allows viruses to drive the generation of new viral variants to be able to adapt to hosts in nature. Such recombination events may result in the emergence of a viable and highly virulent virus with altered tropism restrictions. In addition, recombination events could lead to exchangeable neutralization epitopes, facilitating the immune escape of highly contagious recombinant viruses in previously protected populations (Walsh et al., 2009).
Emerging Virulent Variants
Recombinant viruses can preserve or increase the pathogenicity of the parental strains (Shi et al., 2013; Mandary and Poh, 2018). For instance, etiological investigations for an outbreak of paralytic poliomyelitis that occurred in the Dominican Republic and Haiti between 2000 and 2001 revealed that the responsible viruses were recombinants between the poliovirus Sabin 1 strain and species C enteroviruses (Kew et al., 2002). In addition, a poliomyelitis outbreak was caused by type 2 vaccine-derived poliovirus recombinants in the Central African Republic in 2019 (Joffret et al., 2021). Finally, hand, foot, and mouth disease (HFMD) is usually a mild childhood disease caused mainly by coxsackievirus A16 and enterovirus A71 (EV-A71). In 2018, intra- and inter-typic recombination events amongst co-circulating EV-A71 strains contributed to the outbreak and epidemic of HFMD in the Western Pacific countries (Ang et al., 2021).
Virulent variants of animal viruses can be generated by recombination. Recombination occurs between wild-type PRRSV strains and between the modified live vaccine (MLV) strain and wild-type PRRSV (Zhou et al., 2018b; Wang et al., 2019). In 2009 and 2010, there was a marked increase in PRRSV infections in China, and the viruses for this outbreak were found to originate from a single recombination event (Shi et al., 2013). In 2013 and 2014, a recombinant NADC30-like PRRSV strain emerged, the product of recombination between a North America PRRSV NADC30 strain and a local strain in China, which caused 76.7% deaths in a pig farm and showed higher pathogenicity than its parental NADC30 PRRSV strain (Zhao et al., 2015; Figure 1B). More importantly, recombination between two MLV PRRSV strains produced pathogenic PRRSV, leading to disease outbreaks in many pig farms in France and Denmark (Eclercy et al., 2019; Kvisgaard et al., 2020). In addition, virulent recombinants have also been observed in cases of FMDV (Abd El Rahman et al., 2020), enterovirus EV-B83 (Xiao et al., 2020), and rodent coronavirus (Li et al., 2021). In addition, RNA virus recombination can significantly accelerate adaptation and virus spread, dramatically enhancing virulence (Xiao et al., 2016). Novel emerging virulent recombinants make these new strains more complex than their parental strains, posing many challenges in the prevention of epidemic diseases.
Host Selection and Virus Fitness
Recombination can create considerable changes in the viral genome, allowing for antigenic shifts, host jumps, and fitness modifications (Simon-Loriere and Holmes, 2011). For viruses with large genomes, the fitness of novel recombinant viruses may be particularly susceptible to the effects of recombination (Pérez-Losada et al., 2015). For example, the spike glycoprotein of coronaviruses has previously been identified as a recombination hotspot (Graham and Baric, 2010). Recombinants with new spikes may broaden or alter receptor usage, enabling host switches or expanding their host range (Goldstein et al., 2021). Chikungunya virus (CHIKV), an important human pathogen that has caused a long history of epidemics in Africa and Asia, has rapidly spread in Europe and the Americas (Lanciotti and Valadere, 2014). RNA copy-choice recombination of CHIKV is responsible for genome diversification. Moreover, RNA recombination can generate new viral variants that are positively selected in mosquito vectors, working as an efficient strategy to allow CHIKV to overcome “tight” genetic bottlenecks or even providing an advantage for viral replication (Filomatori et al., 2019). For poliovirus, a virus with a significantly smaller genome than coronavirus, recombination accelerates adaptation in the short time frame of acute poliovirus infection and enriches beneficial mutations while purging deleterious mutations (Xiao et al., 2016). Two high-frequency inter-lineage recombination hotspots have been located in the RdRp and GP2–GP3 regions of PRRSV-2 (Zhou et al., 2018a). The RdRp is involved in PRRSV replication, while GP2–GP4 are the major determinants of virus entry into host cells (Das et al., 2010; Liu et al., 2016; Zhao et al., 2018). The recombination hotspots of PRRSV-2 might be associated with an increase in replication capacity and cellular tropism, which presumably contributes to virus survival and interhost spread.
Failure of Vaccines Protection
Recombination can generally generate virus variants to be able to escape host immune responses. For example, poliovirus recombinants have been isolated from vaccine-associated poliomyelitis patients. Certain recombinants, particularly between type 3 and type 2 poliovirus vaccine strains, are frequently isolated (Minor et al., 1986). Recombination contributes to fitness and virulence in circulating vaccine-derived polioviruses (cVDPVs), just as it does for wild-type viruses. However, some recombinant cVDPVs do not escape host immune responses more than wild-type poliovirus. In addition, highly pathogenic porcine epidemic diarrhea virus (PEDV) strains emerged in China in 2012, circumventing existing vaccines, and RNA recombination events between wild-type and live-attenuated PEDV have seeded new outbreaks (Sun et al., 2012). The recombinant virus can escape from host adaptive immune responses, leading to failure in vaccine protection (Figure 1C). Therefore, RNA virus recombination could pose a potentially serious problem in the administration of MLV vaccines. For poliovirus, adaptive alleles can rapidly accumulate with effective recombination to alleviate the deleterious mutational load. Changing the adaptability may alter virus infection, which presents several possibilities for therapeutic strategies and rational vaccine design (Xiao et al., 2017).
Generation of Recombination Deficient Viruses
Accumulating evidence has shown that RNA recombination occurs at specific RNA secondary structures or homologous sequences between the two parental viruses (Rowe et al., 1997; Chuang and Chen, 2009; Runckel et al., 2013). Therefore, recombination rates could be reduced by altering stem-loop sequences or polymerase fidelity, generating recombination-deficient viruses with the potential for vaccine candidates. Some non-recombinogenic vaccines are developing, such as poliovirus type 2 vaccines. Yeh et al. (2020) introduced modifications within the 5′ untranslated region (UTR) of the Sabin 2 genome to stabilize attenuation determinants, 2C coding region to prevent recombination, and 3D polymerase to limit viral adaptability. Nonetheless, recombination deficient viruses may exhibit lower fitness. Thus, RNA recombination could produce selective disadvantages in progeny viruses, and care must be taken when interpreting such results.
Evasion of Host RNA Interference Defense Mechanism
In addition to the host interferon system combating viruses early upon infection, vertebrate organisms employ RNA interference (RNAi) as an antiviral defense mechanism. In this regard, positive-stranded RNA viruses can undergo strand-switching to rapidly excise genomic material through recombination as an intrinsic defense against antiviral RNAi (Aguado et al., 2018), rendering the virus capable of evading host RNAi-mediated defenses (Figure 1D). The rapid recombination capacity of positive-stranded RNA viruses provides them a unique feature that would significantly enable diversity in the context of evading RNAi, in addition to other innate cellular responses. There is some literature suggesting that RNAi is an antiviral mechanism for invertebrate hosts (Lecellier et al., 2005; Triboulet et al., 2007; Castanotto and Rossi, 2009), and plays a crucial role in the antiviral defense of invertebrates. However, the contribution of RNAi to mammalian antiviral innate immune defense has been underestimated and disputed, and more research may be needed in the future.
The Mechanisms of RNA Virus Recombination
Replicative and Non-replicative Mechanisms of RNA Virus Recombination
Various molecular mechanisms have been proposed to describe the recombination events in RNA viruses. To date, both the replicative copy-choice model and the non-replicative breakage-rejoining model have gained widespread acceptance. Most RNA viruses recombine through a copy-choice mechanism, which involves the formation of recombinant RNA molecules when the RdRp switches from one template to another during genome replication (Figure 2A; Lai, 1992b). However, whether the RdRp switches from one template to another is still a debatable issue. A new RdRp may engage the nascent RNA products after they align on a new template. Non-replicative recombination, a different process in which RNA molecules are cut at specific sites and then joined to form recombinant molecules (Figure 2B), has also been demonstrated in multiple viruses (Becher and Tautz, 2011; Holmblat et al., 2014; Imai et al., 2019), such as recombination between defective poliovirus and coxsackievirus genomes. Non-replicative recombination events may be a transient, intermediate step in the generation and selection of the fittest recombinants.
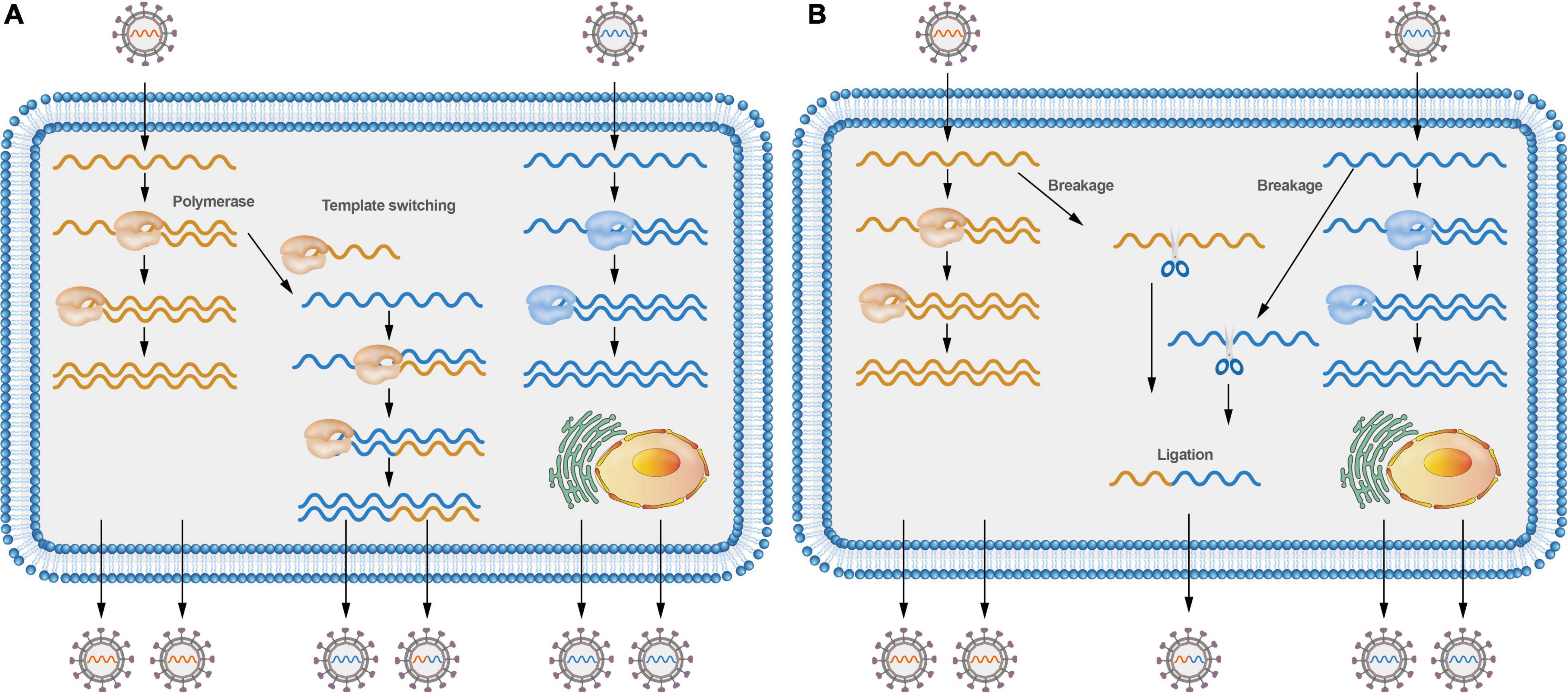
Figure 2. Models of positive-sense RNA viruses’ recombination. (A) Different viruses co-infecting a cell can switch viral templates, and RdRP can switch from donor template to receptor template to generate recombinant molecules. (B) Different viruses co-infecting a cell can lead to virus RNA breakage; RNA molecules can be cut at specific sites and then joined to form recombinant molecules.
Polymerase Fidelity Influences Viral Recombination
The viral RdRp plays a central role in mediating template-switching recombination. The fidelity of viral RdRp has shown to be a fundamental determinant of recombination frequency in both cell-based and biochemical assays (Fitzsimmons et al., 2018). Viral RdRp with low fidelity displays higher recombination frequencies, while viral RdRp with higher fidelity shows lower recombination frequencies (Poirier et al., 2015; Figure 3A). Interestingly, it has been shown that a ribavirin-resistant mutant (with a high-fidelity polymerase) rendered a virus resistant to ribavirin-induced error catastrophe only when recombination occurred at wild-type levels; and, accordingly, this variant became susceptible to ribavirin-induced error catastrophe when recombination was impaired (Kempf et al., 2019). These results support that recombination counteracts the negative consequences of replicative misincorporation, likely by purging genomes harboring deleterious mutations through recombination events. This process is consistent with a previously proposed model for recombination in enteroviruses (Lowry et al., 2014), a biphasic replicative process involving the generation of greater-than genome length “imprecise” intermediates followed by deletion of additional RNA sequences to maximize viral fitness. Whether RdRp errors trigger recombination is disputable.
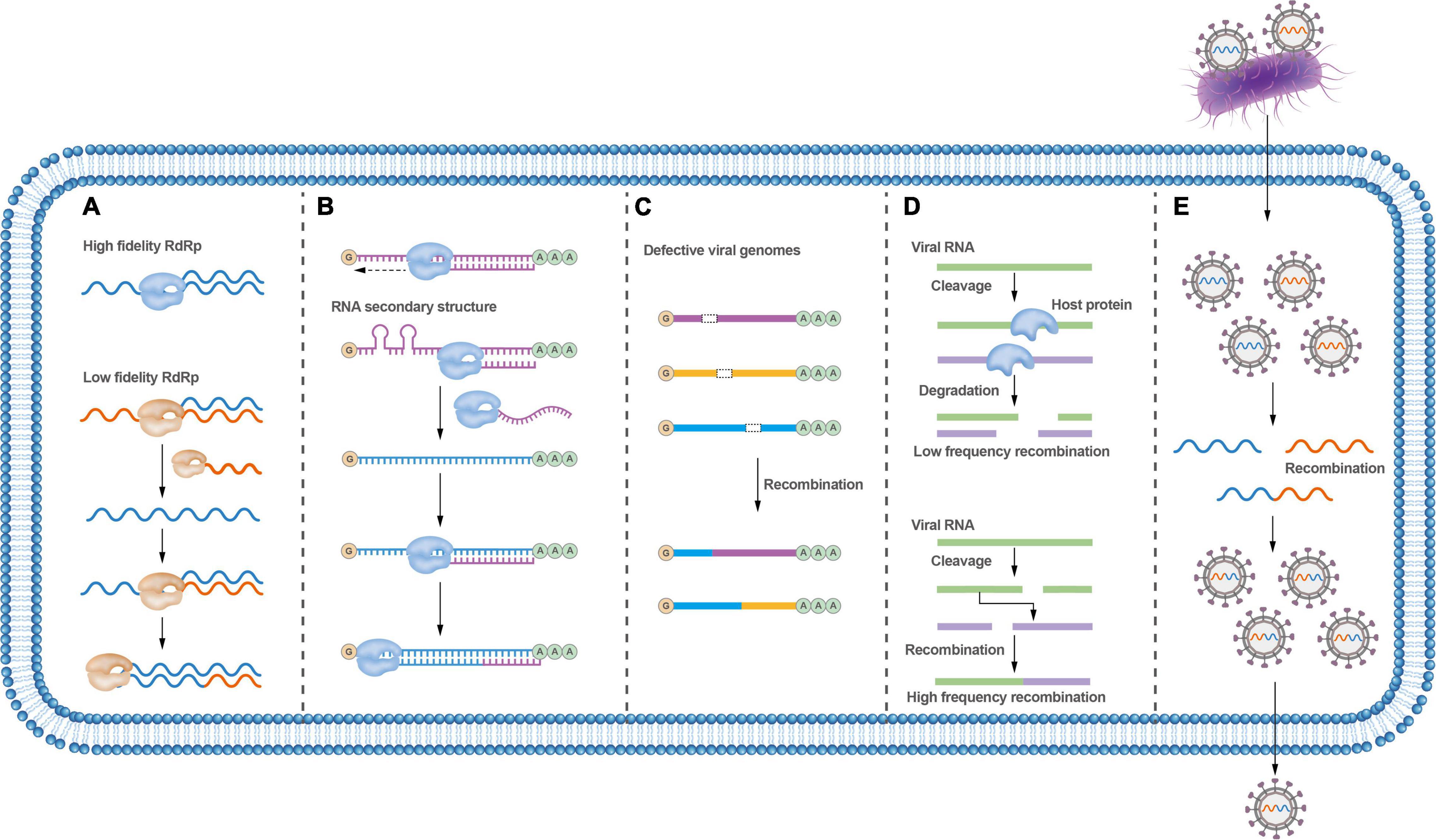
Figure 3. Factors that affect RNA virus recombination. (A) RdRp fidelity affects the recombination rate; low-fidelity RdRp variants are more prone to recombination. (B) Secondary RNA structures between distinct viruses may block strand-switching and lead to virus recombination. (C) Defective viruses can undergo recombination to exchange gene fragments to require the whole viral genome; viruses can recombine with unrelated viral genes or cellular genes. (D) Host proteins may influence virus recombination. (E) Bacteria can carry viruses during co-infection, thus facilitating virus recombination.
Nucleotide Sequences Affect Viral Recombination
Nucleotide homology, nucleotide composition, and secondary structures of viral genomic sequences could all affect the frequency of recombination (Marquez-Jurado et al., 2015). Recombination analysis of PRRSV revealed that most recombinations take place in regions with high genetic identity, and intra-lineage recombination is more frequent than that within inter-lineage (Das et al., 2010). For brome mosaic virus (BMV), the recombinant breakpoints cluster within or close to AU-rich regions (Nagy and Bujarski, 1997), and it is thought that the RdRp likely stops at an AU-rich location in the template chain and then jumps to another template chain during viral replication (Simon-Loriere and Holmes, 2011). Nucleotide composition has also been found to play an important role in determining the sites of homologous recombination of poliovirus (Runckel et al., 2013).
RNA secondary structures were confirmed to convey critical functional information during the recombination process (Figure 3B). It has been demonstrated that the recombination hot spots are located around hairpin structures in the genome of the turnip crinkle virus (TCV). Such RNA secondary structures are thought to inhibit the movement of RdRp and promote its switch between templates (Carpenter et al., 1995; Nagy et al., 1999). Similarly, gene synthesis and deep sequencing analysis of the breakpoints throughout the poliovirus genome have revealed that most breakpoints are associated with RNA secondary structures, including hairpins and stem-loop structures (Runckel et al., 2013). RNA secondary structures also seem to promote recombination in HIV and increase the viability and replication ability of recombinant HIV progeny (Simon-Loriere et al., 2010). Nonetheless, a recent study argues that recombination is not influenced by the RNA donor or acceptor sequence or its structure (Alnaji et al., 2020), which seems to contradict previous studies that recombinant virus genomes are related to RNA structure and sequence identity. Importantly, more recent research indicates that viruses undergo frequent and continuous recombination events over a prolonged period until the fittest and wild-type length of the recombinant genome is dominant (Bentley et al., 2021). These conflicting results may reflect variation in the recombination processes involved between different viruses. In the meantime, it also supports that recombination is a promiscuous event that is not significantly influenced by any single factor.
The Effects of Host or Bacteria Factors on Viral Recombination
Virus recombination can be affected by hosts. Several plant viruses, including TCV, tomato bushy stunt virus, cucumber necrosis virus, and potato virus X, have been shown to undergo non-homologous RNA recombination between a defective viral RNA and a helper wild-type virus RNA (Sztuba-Solińska et al., 2011; Figure 3C). As such, the host and other extrinsic factors can play important roles in this type of recombination. Host factors, combined with environmental factors that affect the Met22/Xrn1 pathway, seem to also play a role in viral RNA recombination (Jaag and Nagy, 2010). Host cellular RNA helicase RH20 and proteasomal Rpn11 protein can affect RNA recombination and viral replication (Prasanth et al., 2015; Wu and Nagy, 2020; Molho et al., 2021; Figure 3D).
Nidoviruses, including SARS-CoV-2, exhibit discontinuous transcription and produce a set of sub-genomic RNAs that contribute to high recombination rates (Yang et al., 2021). These viruses can incorporate either unrelated viral genes or cellular genes into the viral genome, possibly by a non-homologous recombination mechanism. Occasionally, cellular genes have been incorporated into viral genomes, such as the ubiquitin gene, which is regularly incorporated into the genome of bovine viral diarrhea virus, contributing to the cytopathogenic function displayed by the virus (Meyers et al., 1991).
In addition, bacterial infection may also affect viral recombination. The ability of enteric RNA viruses to undergo homologous recombination is variable and depends, at least in part, on bacterial infections that facilitate co-infection of different viral genotypes within the same cell (Erickson et al., 2018; Figure 3E).
Development of Assays to Quantify Virus Recombination Rates
Plaque-Purification and DNA Sequencing
Researchers often used a method of plaque purification to isolate recombinant viruses. For example, the investigation of intra- and inter-typic recombinant crosses of poliovirus was possible by this approach, as they were able to produce plaques in vitro (Kirkegaard and Baltimore, 1986). Such recombinants with a growth or plaque formation advantage can often be easily identified and differentiated from the two parental viruses.
For feline calicivirus, recombination rates have been detected in co-infected cells using quantitative reverse transcription-polymerase chain reaction amplification (RT-qPCR) (Symes et al., 2015). This method utilizes virus-specific primers upstream and downstream of the recombination hotspot that hybridized with only one of the strains during the co-infection, and the site of recombination is then confirmed by DNA sequencing. A similar approach has been used to detect recombination in poliovirus (Jarvis and Kirkegaard, 1992). The direct and quantitative detection of recombinant RNA molecules by PCR allows the study of recombination in RNA viruses without the requirement of a viable progeny, an endpoint that can bias the interpretation of predictive conclusions.
The above approach is a more classic recombination detection method, and while the results are more accurate, the process is time-consuming with a huge workload.
Cell-Based Recombination Assay
A cell-based recombination assay (termed CRE-REP) established by the Evans laboratory has drastically advanced studies of enterovirus recombination (Lowry et al., 2014). This approach is based on the co-transfection of two in vitro transcribed RNAs that alone cannot produce viable viruses. In this system, the “donor” RNA is a replication-competent, sub-genomic RNA encoding a luciferase reporter gene in place of the viral capsid sequences. In turn, the “acceptor” RNA is a replication-incompetent, genomic RNA that has a defective cis-acting replication element, and only after successful recombination between these two parental genomes, viable progeny recombinant viruses can be produced and quantified.
Since its emergence, this technique is currently extensively utilized for virus recombination studies in the poliovirus (Kempf et al., 2016; Kim et al., 2019; Woodman et al., 2019). Researchers have further adapted the CRE-REP assay, making it suitable for other viruses, such as the Seneca Valley virus (SVV) (Li et al., 2019). For instance, a lethal catalytic mutation can be introduced into the RdRp active site (Kempf et al., 2016; Li et al., 2019), the active site of the RdRp can be removed from the “acceptor” R NA (Woodman et al., 2019), or a lethal capsid deletion of the “donor” RNA (Kempf et al., 2016; Li et al., 2019) can be used to make both parental genomes non-viable. In addition, the use of fluorescent proteins as reporter genes (Kim et al., 2019) has enabled researchers to visualize and quantify viruses that are unable to form plaque efficiently by monitoring recombination events through imaging or cell sorting. CRE-REP assay is a particularly sophisticated method for detecting virus recombination at the cellular level. However, only some enteroviruses belonging to the family Picornaviridae have used this kind of assay to identify the recombination rate. Whether this method can be applied to other virus families has not been investigated.
In vitro Biochemically Defined Recombination Assay
A recent study of poliovirus recombination utilizes two different biochemical assays to distinguish copy-choice recombination from forced copy-choice recombination (Kim et al., 2019). In both assays, the purified RdRp engages a primed template, elongates that primer, disengages from the first template, engages a second (acceptor) template, and continues RNA synthesis. One assay uses oligo (dT) or oligo (rU) to prime poly (rU) RNA synthesis on oligo (rA) or poly (rA) RNA templates to mimic copy-choice recombination, which was first used to show template-switching of the RdRp during elongation (Arnold and Cameron, 1999). The RdRp-nascent RNA complex then moves to a new template from internal positions or, as a consequence of reaching the end of the template, creates greater-than-template-length products. The second assay uses a heteropolymeric, symmetrical, primed-template substrate (sym/sub) and is based on template-switching from the end of the template, thus mimicking forced copy-choice recombination (Woodman et al., 2016). This method can analyze recombination qualitatively and quantitatively in a precise and intuitive manner. However, performing these biochemical tests can be technically challenging.
Third-Generation Sequencing Approach Combined Sequence Analysis
Recent advances in genome sequencing by third-generation sequencing technologies (TGS), including PacBio single-molecule real-time sequencing and nanopore sequencing technology, can generate reads of >10 kb in length (Lu et al., 2016; Magi et al., 2017). The long reads enable to distinguish the recombinant viruses from the mixed parental viruses. A direct RNA sequencing approach based on nanopore sequencing technology has been used to characterize viral RNAs produced in cells infected with the HCoV-229E coronavirus, and several unexpected recombination sites were observed in the ORF1a 5′- and 3′-terminal regions (Viehweger et al., 2019). Although TGS showed a high error rate compared to next-generation sequencing (NGS) or Sanger methods, most of the errors in the assay were nucleotide-substitutions that did not affect recombination analysis. In addition, Tiled-ClickSeq has been developed to sequence the complete genome coverage, including the 5′UTR, at high depth and specificity to the virus on both Illumina and Nanopore platforms (Jaworski et al., 2021). Besides, a direct RNA sequencing approach was performed with samples from total RNA obtained from virus-infected cells where there is no bias from reverse transcription and amplification, allowing the quantification and analysis of the numbers of recombinant or parental molecules (Viehweger et al., 2019).
With the advancement and maturation of sequencing technology, an increasing number of experiments are opting for third-generation sequencing for the research. Although third-generation sequencing is certainly speedy and convenient for recombinant virus analysis, its high cost and greater error rate have led to some laboratories using Sanger sequencing instead.
Bioinformatics Assay
Bioinformatics analysis of genomic sequence data contributes to an in-depth understanding of the evolutionary process, including the impact of genomic structure (Reich et al., 2001), phenotypic variation (Zhang et al., 2002), and its relationship with genetic disease research (Daly et al., 2001). RNA virus recombination is one of the most important aspects of evolutionary biology, and bioinformatic methods can aid in adequately detecting its occurrence and understanding how it influences phylogenetic relationships (Posada et al., 2002). Phylogenetic tree analysis is generally a bioinformatics method to evaluate virus recombination (Posada et al., 2002). In addition, a phylogenetic compatibility matrix (PCM) for different regions of the enterovirus genome and correlations with sequence assignments to analyze recombination among complete genome sequences can be used (Simmonds and Welch, 2006). Determination of the variation in viral recombination and whether they are related to other biological characteristics is essential for future research. With the development of sequencing technology, the application of high-throughput sequencing technology combined with biological analysis is also a common method to analyze recombination. In the recombination analysis of SARS-CoV-2, Illumina and Nanopore reads and Python scripts were used to analyze the data and map RNA recombination events (Jaworski et al., 2021). Besides, sequences obtained from NGS and bioinformatics tools were used to analyze the recombination events within enterovirus C serotypes (Amona et al., 2020).
Summary and Outlook
Most RNA viruses can recombine either by homologous or non-homologous recombination, both of which play an important role in the biology and evolution of RNA viruses. Recombination is a major driving force for viral diversity and has significant biological consequences on viral biology. Recombination generates novel viral strains, which expands virus genetic diversity, increases virulence and pathogenesis, and promotes the emergence of new circulating viruses. In addition, recombination can facilitate the evolution of antiviral resistance, host immunity escape, and associate with alterations or expansions of viral host tropism. Thus, it is important to emphasize that viral recombination detection and surveillance need to be prioritized for viral disease control and prevention. Understanding the mechanisms of virus recombination and the factors that affect recombination can ultimately help us design live attenuated vaccines unable to recombine with circulating strains.
Simultaneously, recombination can be used as a beneficial tool for research. Reporter viruses are useful tools for screening susceptibility and resistance to antiviral compounds. However, the stability of reporter viruses has been challenged by viral RNA recombination, leading to the deletion of the reporter gene during viral replication. Lethal mutations upon recombination have been engineered to create a stable Zika virus harboring a NanoLuc-reporter gene (Baker et al., 2020). The developed recombination-dependent lethal approach could produce stable reporter viruses used for rapid diagnosis, vaccine evaluation, and antiviral discovery.
Author Contributions
TA and XHC contributed to conception of the study. HW and XYC wrote the draft. TA revised the draft. All authors read and approved the final manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (32072851) and the State Key Laboratory of Veterinary Biotechnology Research Fund (SKLVBF201902 and SKLVBF202110).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Craig E. Cameron and Dr. Hyejeong Kim (University of North Carolina School of Medicine) for preparing the manuscript and giving critical comments.
References
Abd El Rahman, S., Hoffmann, B., Karam, R., El-Beskawy, M., Hamed, M. F., Forth, L. F., et al. (2020). Sequence analysis of egyptian foot-and-mouth disease virus field and vaccine strains: intertypic recombination and evidence for accidental release of virulent virus. Viruses 12:990. doi: 10.3390/v12090990
Aguado, L. C., Jordan, T. X., Hsieh, E., Blanco-Melo, D., Heard, J., Panis, M., et al. (2018). Homologous recombination is an intrinsic defense against antiviral RNA interference. Proc. Natl. Acad. Sci. U.S.A. 115, E9211–E9219. doi: 10.1073/pnas.1810229115
Alatorre-García, T. A., Fonseca-Coronado, S., and González-Candelas, F. (2021). Homologous recombination as a mechanism of genetic changes in bovine parainfluenza-3 virus. Vet. Microbiol. 261:109185. doi: 10.1016/j.vetmic.2021.109185
Alnaji, F. G., Bentley, K., Pearson, A., Woodman, A., Moore, J. D., Fox, H., et al. (2020). Recombination in enteroviruses is a ubiquitous event independent of sequence homology and RNA structure. bioRxiv [Preprint] doi: 10.1101/2020.09.29.319285
Amona, I., Medkour, H., Akiana, J., Davoust, B., Tall, M. L., Grimaldier, C., et al. (2020). Enteroviruses from humans and great apes in the republic of congo: recombination within Enterovirus C serotypes. Microorganisms 8:1779. doi: 10.3390/microorganisms8111779
Ang, P. Y., Chong, C. W. H., and Alonso, S. (2021). Viral determinants that drive Enterovirus-A71 fitness and virulence. Emerg. Microbes Infect. 10, 713–724. doi: 10.1080/22221751.2021.1906754
Arnold, J. J., and Cameron, C. E. (1999). Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem. 274, 2706–2716. doi: 10.1074/jbc.274.5.2706
Baker, C., Xie, X., Zou, J., Muruato, A., Fink, K., and Shi, P. Y. (2020). Using recombination-dependent lethal mutations to stabilize reporter flaviviruses for rapid serodiagnosis and drug discovery. EBioMedicine 57:102838. doi: 10.1016/j.ebiom.2020.102838
Becher, P., and Tautz, N. (2011). RNA recombination in pestiviruses: cellular RNA sequences in viral genomes highlight the role of host factors for viral persistence and lethal disease. RNA Biol. 8, 216–224. doi: 10.4161/rna.8.2.14514
Bentley, K., Alnaji, F. G., Woodford, L., Jones, S., Woodman, A., and Evans, D. J. (2021). Imprecise recombinant viruses evolve via a fitness-driven, iterative process of polymerase template-switching events. PLoS Pathog. 17:e1009676. doi: 10.1371/journal.ppat.1009676
Bentley, K., and Evans, D. J. (2018). Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 99, 1345–1356. doi: 10.1099/jgv.0.001142
Boni, M. F., Lemey, P., Jiang, X., Lam, T. T., Perry, B. W., Castoe, T. A., et al. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417. doi: 10.1038/s41564-020-0771-4
Carpenter, C. D., Oh, J. W., Zhang, C., and Simon, A. E. (1995). Involvement of a stem-loop structure in the location of junction sites in viral RNA recombination. J. Mol. Biol. 245, 608–622. doi: 10.1006/jmbi.1994.0050
Castanotto, D., and Rossi, J. J. (2009). The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433. doi: 10.1038/nature07758
Chuang, C. K., and Chen, W. J. (2009). Experimental evidence that RNA recombination occurs in the Japanese encephalitis virus. Virology 394, 286–297. doi: 10.1016/j.virol.2009.08.030
Crook, J. M., Murphy, I., Carter, D. P., Pullan, S. T., Carroll, M., Vipond, R., et al. (2021). Metagenomic identification of a new sarbecovirus from horseshoe bats in Europe. Sci. Rep. 11:14723. doi: 10.1038/s41598-021-94011-z
Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J., and Lander, E. S. (2001). High-resolution haplotype structure in the human genome. Nat. Genet. 29, 229–232. doi: 10.1038/ng1001-229
Das, P. B., Dinh, P. X., Ansari, I. H., de Lima, M., Osorio, F. A., and Pattnaik, A. K. (2010). The minor envelope glycoproteins GP2a and GP4 of porcine reproductive and respiratory syndrome virus interact with the receptor CD163. J. Virol. 84, 1731–1740. doi: 10.1128/jvi.01774-09
Dudas, G., and Rambaut, A. (2016). MERS-CoV recombination: implications about the reservoir and potential for adaptation. Virus Evol. 2:vev023. doi: 10.1093/ve/vev023
Duffy, S., Shackelton, L. A., and Holmes, E. C. (2008). Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9, 267–276. doi: 10.1038/nrg2323
Eclercy, J., Renson, P., Lebret, A., Hirchaud, E., Normand, V., Andraud, M., et al. (2019). A field recombinant strain derived from two type 1 porcine reproductive and respiratory syndrome virus (PRRSV-1) modified live vaccines shows increased viremia and transmission in SPF pigs. Viruses 11:296. doi: 10.3390/v11030296
Erickson, A. K., Jesudhasan, P. R., Mayer, M. J., Narbad, A., Winter, S. E., and Pfeiffer, J. K. (2018). Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe 23, 77–88.e5. doi: 10.1016/j.chom.2017.11.007
Felt, S. A., Sun, Y., Jozwik, A., Paras, A., Habibi, M. S., Nickle, D., et al. (2021). Detection of respiratory syncytial virus defective genomes in nasal secretions is associated with distinct clinical outcomes. Nat. Microbiol. 6, 672–681. doi: 10.1038/s41564-021-00882-3
Filomatori, C. V., Bardossy, E. S., Merwaiss, F., Suzuki, Y., Henrion, A., Saleh, M. C., et al. (2019). RNA recombination at Chikungunya virus 3’UTR as an evolutionary mechanism that provides adaptability. PLoS Pathog. 15:e1007706. doi: 10.1371/journal.ppat.1007706
Fitzsimmons, W. J., Woods, R. J., McCrone, J. T., Woodman, A., Arnold, J. J., Yennawar, M., et al. (2018). A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 16:e2006459. doi: 10.1371/journal.pbio.2006459
Forrester, N. L., Palacios, G., Tesh, R. B., Savji, N., Guzman, H., Sherman, M., et al. (2012). Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J. Virol. 86, 2729–2738. doi: 10.1128/jvi.05591-11
Genoyer, E., Kulej, K., Hung, C. T., Thibault, P. A., Azarm, K., Takimoto, T., et al. (2020). The viral polymerase complex mediates the interaction of viral ribonucleoprotein complexes with recycling endosomes during Sendai virus assembly. mBio 11:e02028-20. doi: 10.1128/mBio.02028-20
Goldstein, S. A., Brown, J., Pedersen, B. S., Quinlan, A. R., and Elde, N. C. (2021). Extensive recombination-driven coronavirus diversification expands the pool of potential pandemic pathogens. bioRxiv [Preprint] doi: 10.1101/2021.02.03.429646
González-Candelas, F., López-Labrador, F. X., and Bracho, M. A. (2011). Recombination in hepatitis C virus. Viruses 3, 2006–2024. doi: 10.3390/v3102006
Graham, R. L., and Baric, R. S. (2010). Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 84, 3134–3146. doi: 10.1128/jvi.01394-09
Hahn, C. S., Lustig, S., Strauss, E. G., and Strauss, J. H. (1988). Western equine encephalitis virus is a recombinant virus. Proc. Natl. Acad. Sci. U.S.A. 85, 5997–6001. doi: 10.1073/pnas.85.16.5997
Han, G. Z., and Worobey, M. (2011). Homologous recombination in negative sense RNA viruses. Viruses 3, 1358–1373. doi: 10.3390/v3081358
Han, J. F., Jiang, T., Ye, Q., Li, X. F., Liu, Z. Y., and Qin, C. F. (2016). Homologous recombination of Zika viruses in the Americas. J. Infect. 73, 87–88. doi: 10.1016/j.jinf.2016.04.011
Holmblat, B., Jegouic, S., Muslin, C., Blondel, B., Joffret, M. L., and Delpeyroux, F. (2014). Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. mBio 5:e01119-14. doi: 10.1128/mBio.01119-14
Hon, C. C., Lam, T. Y., Shi, Z. L., Drummond, A. J., Yip, C. W., Zeng, F., et al. (2008). Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826. doi: 10.1128/jvi.01926-07
Imai, R., Nagai, M., Oba, M., Sakaguchi, S., Ujike, M., Kimura, R., et al. (2019). A novel defective recombinant porcine enterovirus G virus carrying a porcine torovirus papain-like cysteine protease gene and a putative anti-apoptosis gene in place of viral structural protein genes. Infect. Genet. Evol. 75:103975. doi: 10.1016/j.meegid.2019.103975
Jaag, H. M., and Nagy, P. D. (2010). The combined effect of environmental and host factors on the emergence of viral RNA recombinants. PLoS Pathog. 6:e1001156. doi: 10.1371/journal.ppat.1001156
Jackson, B., Boni, M. F., Bull, M. J., Colleran, A., Colquhoun, R. M., Darby, A. C., et al. (2021). Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 184, 5179–5188.e8. doi: 10.1016/j.cell.2021.08.014
Janissen, R., Woodman, A., Shengjuler, D., Vallet, T., Lee, K. M., Kuijpers, L., et al. (2021). Induced intra- and intermolecular template switching as a therapeutic mechanism against RNA viruses. Mol. Cell 81, 4467–4480.e7. doi: 10.1016/j.molcel.2021.10.003
Jarvis, T. C., and Kirkegaard, K. (1992). Poliovirus RNA recombination: mechanistic studies in the absence of selection. EMBO J. 11, 3135–3145. doi: 10.1002/j.1460-2075.1992.tb05386.x
Jaworski, E., Langsjoen, R. M., Mitchell, B., Judy, B., Newman, P., Plante, J. A., et al. (2021). Tiled-ClickSeq for targeted sequencing of complete coronavirus genomes with simultaneous capture of RNA recombination and minority variants. eLife 10:e68479. doi: 10.7554/eLife.68479
Ji, W., Wang, W., Zhao, X., Zai, J., and Li, X. (2020). Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 92, 433–440. doi: 10.1002/jmv.25682
Joffret, M. L., Doté, J. W., Gumede, N., Vignuzzi, M., Bessaud, M., and Gouandjika-Vasilache, I. (2021). Vaccine-derived polioviruses, central African republic, 2019. Emerg. Infect. Dis. 27, 620–623. doi: 10.3201/eid2702.203173
Kempf, B. J., Peersen, O. B., and Barton, D. J. (2016). Poliovirus polymerase Leu420 facilitates RNA recombination and ribavirin resistance. J. Virol. 90, 8410–8421. doi: 10.1128/jvi.00078-16
Kempf, B. J., Watkins, C. L., Peersen, O. B., and Barton, D. J. (2019). Picornavirus RNA recombination counteracts error catastrophe. J. Virol. 93:e00652-19. doi: 10.1128/jvi.00652-19
Kew, O., Morris-Glasgow, V., Landaverde, M., Burns, C., Shaw, J., Garib, Z., et al. (2002). Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 296, 356–359. doi: 10.1126/science.1068284
Kim, H., Ellis, V. D. III, Woodman, A., Zhao, Y., Arnold, J. J., and Cameron, C. E. (2019). RNA-dependent RNA polymerase speed and fidelity are not the only determinants of the mechanism or efficiency of recombination. Genes 10:968. doi: 10.3390/genes10120968
Kim, J. I., Kim, Y. J., Lemey, P., Lee, I., Park, S., Bae, J. Y., et al. (2016). The recent ancestry of Middle East respiratory syndrome coronavirus in Korea has been shaped by recombination. Sci. Rep. 6:18825. doi: 10.1038/srep18825
Kirkegaard, K., and Baltimore, D. (1986). The mechanism of RNA recombination in poliovirus. Cell 47, 433–443. doi: 10.1016/0092-8674(86)90600-8
Kvisgaard, L. K., Kristensen, C. S., Ryt-Hansen, P., Pedersen, K., Stadejek, T., Trebbien, R., et al. (2020). A recombination between two Type 1 porcine reproductive and respiratory syndrome virus (PRRSV-1) vaccine strains has caused severe outbreaks in Danish pigs. Transbound. Emerg. Dis. 67, 1786–1796. doi: 10.1111/tbed.13555
Lai, M. M. (1992a). RNA recombination in animal and plant viruses. Microbiol. Rev. 56, 61–79. doi: 10.1128/mr.56.1.61-79.1992
Lai, M. M. (1992b). Genetic recombination in RNA viruses. Curr. Top. Microbiol. Immunol. 176, 21–32. doi: 10.1007/978-3-642-77011-1_2
Lam, T. T., Jia, N., Zhang, Y. W., Shum, M. H., Jiang, J. F., Zhu, H. C., et al. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285. doi: 10.1038/s41586-020-2169-0
Lanciotti, R. S., and Valadere, A. M. (2014). Transcontinental movement of Asian genotype Chikungunya virus. Emerg. Infect. Dis. 20, 1400–1402. doi: 10.3201/eid2008.140268
Lecellier, C. H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., et al. (2005). A cellular microRNA mediates antiviral defense in human cells. Science 308, 557–560. doi: 10.1126/science.1108784
Li, C., Wang, H., Shi, J., Yang, D., Zhou, G., Chang, J., et al. (2019). Senecavirus-specific recombination assays reveal the intimate link between polymerase fidelity and RNA recombination. J. Virol. 93:e00576-19. doi: 10.1128/jvi.00576-19
Li, X., Wang, L., Liu, P., Li, H., Huo, S., Zong, K., et al. (2021). A novel potentially recombinant rodent coronavirus with a polybasic cleavage site in the spike protein. J. Virol. 95:e0117321. doi: 10.1128/JVI.01173-21
Liu, L., Tian, J., Nan, H., Tian, M., Li, Y., Xu, X., et al. (2016). Porcine reproductive and respiratory syndrome virus nucleocapsid protein interacts with Nsp9 and cellular DHX9 to regulate viral RNA synthesis. J. Virol. 90, 5384–5398. doi: 10.1128/JVI.03216-15
Lowry, K., Woodman, A., Cook, J., and Evans, D. J. (2014). Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS Pathog. 10:e1004191. doi: 10.1371/journal.ppat.1004191
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinformatics 14, 265–279. doi: 10.1016/j.gpb.2016.05.004
Magi, A., Giusti, B., and Tattini, L. (2017). Characterization of MinION nanopore data for resequencing analyses. Brief. Bioinform. 18, 940–953. doi: 10.1093/bib/bbw077
Mandary, M. B., and Poh, C. L. (2018). Changes in the EV-A71 genome through recombination and spontaneous mutations: impact on virulence. Viruses 10:320. doi: 10.3390/v10060320
Marquez-Jurado, S., Nogales, A., Zuniga, S., Enjuanes, L., and Almazan, F. (2015). Identification of a gamma interferon-activated inhibitor of translation-like RNA motif at the 3’ end of the transmissible gastroenteritis coronavirus genome modulating innate immune response. mBio 6:e00105. doi: 10.1128/mBio.00105-15
Meyers, G., Tautz, N., Dubovi, E. J., and Thiel, H. J. (1991). Viral cytopathogenicity correlated with integration of ubiquitin-coding sequences. Virology 180, 602–616. doi: 10.1016/0042-6822(91)90074-l
Minor, P. D., John, A., Ferguson, M., and Icenogle, J. P. (1986). Antigenic and molecular evolution of the vaccine strain of type 3 poliovirus during the period of excretion by a primary vaccinee. J. Gen. Virol. 67(Pt 4), 693–706. doi: 10.1099/0022-1317-67-4-693
Molho, M., Lin, W., and Nagy, P. D. (2021). A novel viral strategy for host factor recruitment: the co-opted proteasomal Rpn11 protein interaction hub in cooperation with subverted actin filaments are targeted to deliver cytosolic host factors for viral replication. PLoS Pathog. 17:e1009680. doi: 10.1371/journal.ppat.1009680
Mura, M., Combredet, C., Najburg, V., Sanchez David, R. Y., Tangy, F., and Komarova, A. V. (2017). Nonencapsidated 5’ copy-back defective interfering genomes produced by recombinant measles viruses are recognized by RIG-I and LGP2 but not MDA5. J. Virol. 91:e00643-17. doi: 10.1128/jvi.00643-17
Nagy, P. D., and Bujarski, J. J. (1997). Engineering of homologous recombination hotspots with AU-rich sequences in brome mosaic virus. J. Virol. 71, 3799–3810. doi: 10.1128/jvi.71.5.3799-3810.1997
Nagy, P. D., Pogany, J., and Simon, A. E. (1999). RNA elements required for RNA recombination function as replication enhancers in vitro and in vivo in a plus-strand RNA virus. EMBO J. 18, 5653–5665. doi: 10.1093/emboj/18.20.5653
Patiño-Galindo, J., Filip, I., and Rabadan, R. (2021). Global patterns of recombination across human viruses. Mol. Biol. Evol. 38, 2520–2531. doi: 10.1093/molbev/msab046
Pérez-Losada, M., Arenas, M., Galán, J. C., Palero, F., and González-Candelas, F. (2015). Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30, 296–307. doi: 10.1016/j.meegid.2014.12.022
Poirier, E. Z., Mounce, B. C., Rozen-Gagnon, K., Hooikaas, P. J., Stapleford, K. A., Moratorio, G., et al. (2015). Low-fidelity polymerases of alphaviruses recombine at higher rates to overproduce defective interfering particles. J. Virol. 90, 2446–2454. doi: 10.1128/jvi.02921-15
Posada, D., Crandall, K. A., and Holmes, E. C. (2002). Recombination in evolutionary genomics. Annu. Rev. Genet. 36, 75–97. doi: 10.1146/annurev.genet.36.040202.111115
Prasanth, K. R., Barajas, D., and Nagy, P. D. (2015). The proteasomal Rpn11 metalloprotease suppresses tombusvirus RNA recombination and promotes viral replication via facilitating assembly of the viral replicase complex. J. Virol. 89, 2750–2763. doi: 10.1128/JVI.02620-14
Reich, D. E., Cargill, M., Bolk, S., Ireland, J., Sabeti, P. C., Richter, D. J., et al. (2001). Linkage disequilibrium in the human genome. Nature 411, 199–204. doi: 10.1038/35075590
Rowe, C. L., Fleming, J. O., Nathan, M. J., Sgro, J. Y., Palmenberg, A. C., and Baker, S. C. (1997). Generation of coronavirus spike deletion variants by high-frequency recombination at regions of predicted RNA secondary structure. J. Virol. 71, 6183–6190. doi: 10.1128/jvi.71.8.6183-6190.1997
Runckel, C., Westesson, O., Andino, R., and DeRisi, J. L. (2013). Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog. 9:e1003164. doi: 10.1371/journal.ppat.1003164
Schroeder, S., Mache, C., Kleine-Weber, H., Corman, V. M., Muth, D., Richter, A., et al. (2021). Functional comparison of MERS-coronavirus lineages reveals increased replicative fitness of the recombinant lineage 5. Nat. Commun. 12:5324. doi: 10.1038/s41467-021-25519-1
Shi, M., Holmes, E. C., Brar, M. S., and Leung, F. C. (2013). Recombination is associated with an outbreak of novel highly pathogenic porcine reproductive and respiratory syndrome viruses in China. J. Virol. 87, 10904–10907. doi: 10.1128/jvi.01270-13
Simmonds, P., and Welch, J. (2006). Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 80, 483–493. doi: 10.1128/jvi.80.1.483-493.2006
Simon-Loriere, E., and Holmes, E. C. (2011). Why do RNA viruses recombine? Nat. Rev. Microbiol. 9, 617–626. doi: 10.1038/nrmicro2614
Simon-Loriere, E., Martin, D. P., Weeks, K. M., and Negroni, M. (2010). RNA structures facilitate recombination-mediated gene swapping in HIV-1. J. Virol. 84, 12675–12682. doi: 10.1128/jvi.01302-10
Steinhauer, D. A., Domingo, E., and Holland, J. J. (1992). Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122, 281–288. doi: 10.1016/0378-1119(92)90216-c
Sun, R. Q., Cai, R. J., Chen, Y. Q., Liang, P. S., Chen, D. K., and Song, C. X. (2012). Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 18, 161–163. doi: 10.3201/eid1801.111259
Sun, Y., Kim, E. J., Felt, S. A., Taylor, L. J., Agarwal, D., Grant, G. R., et al. (2019). A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog. 15:e1007707. doi: 10.1371/journal.ppat.1007707
Symes, S. J., Job, N., Ficorilli, N., Hartley, C. A., Browning, G. F., and Gilkerson, J. R. (2015). Novel assay to quantify recombination in a calicivirus. Vet. Microbiol. 177, 25–31. doi: 10.1016/j.vetmic.2015.02.017
Sztuba-Solińska, J., Urbanowicz, A., Figlerowicz, M., and Bujarski, J. J. (2011). RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 49, 415–443. doi: 10.1146/annurev-phyto-072910-095351
Tegally, H., Wilkinson, E., Althaus, C. L., Giovanetti, M., San, J. E., Giandhari, J., et al. (2021a). Rapid replacement of the Beta variant by the Delta variant in South Africa. medRxiv [Preprint] doi: 10.1101/2021.09.23.21264018
Tegally, H., Wilkinson, E., Lessells, R. J., Giandhari, J., Pillay, S., Msomi, N., et al. (2021b). Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 27, 440–446. doi: 10.1038/s41591-021-01255-3
Tegally, H., Wilkinson, E., Giovanetti, M., Iranzadeh, A., Fonseca, V., Giandhari, J., et al. (2021c). Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592, 438–443. doi: 10.1038/s41586-021-03402-9
Triboulet, R., Mari, B., Lin, Y. L., Chable-Bessia, C., Bennasser, Y., Lebrigand, K., et al. (2007). Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315, 1579–1582. doi: 10.1126/science.1136319
Viana, R., Moyo, S., Amoako, D. G., Tegally, H., Scheepers, C., Althaus, C. L., et al. (2022). Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603, 679–686. doi: 10.1038/s41586-022-04411-y
Viehweger, A., Krautwurst, S., Lamkiewicz, K., Madhugiri, R., Ziebuhr, J., Hölzer, M., et al. (2019). Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 29, 1545–1554. doi: 10.1101/gr.247064.118
Vignuzzi, M., and López, C. B. (2019). Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 4, 1075–1087. doi: 10.1038/s41564-019-0465-y
Walsh, M. P., Chintakuntlawar, A., Robinson, C. M., Madisch, I., Harrach, B., Hudson, N. R., et al. (2009). Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis. PLoS One 4:e5635. doi: 10.1371/journal.pone.0005635
Wang, A., Chen, Q., Wang, L., Madson, D., Harmon, K., Gauger, P., et al. (2019). Recombination between vaccine and field strains of porcine reproductive and respiratory syndrome virus. Emerg. Infect. Dis. 25, 2335–2337. doi: 10.3201/eid2512.191111
Wang, C., Horby, P. W., Hayden, F. G., and Gao, G. F. (2020). A novel coronavirus outbreak of global health concern. Lancet 395, 470–473. doi: 10.1016/s0140-6736(20)30185-9
Woodman, A., Arnold, J. J., Cameron, C. E., and Evans, D. J. (2016). Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 44, 6883–6895. doi: 10.1093/nar/gkw567
Woodman, A., Lee, K. M., Janissen, R., Gong, Y. N., Dekker, N. H., Shih, S. R., et al. (2019). Predicting intraserotypic recombination in enterovirus 71. J. Virol. 93:e02057-18. doi: 10.1128/jvi.02057-18
Wu, C. Y., and Nagy, P. D. (2020). Role reversal of functional identity in host factors: dissecting features affecting pro-viral versus antiviral functions of cellular DEAD-box helicases in tombusvirus replication. PLoS Pathog. 16:e1008990. doi: 10.1371/journal.ppat.1008990
Xiao, J., Zhang, Y., Hong, M., Han, Z., Zhang, M., Song, Y., et al. (2020). Phylogenetic characteristics and molecular epidemiological analysis of novel enterovirus EV-B83 isolated from Tibet, China. Sci. Rep. 10:6630. doi: 10.1038/s41598-020-63691-4
Xiao, T., Leng, H., Zhang, Q., Chen, Q., Guo, H., and Qi, Y. (2022). Isolation and characterization of a Sabin 3/Sabin 1 recombinant vaccine-derived poliovirus from a child with severe combined immunodeficiency. Virus Res. 308:198633. doi: 10.1016/j.virusres.2021.198633
Xiao, Y., Dolan, P. T., Goldstein, E. F., Li, M., Farkov, M., Brodsky, L., et al. (2017). Poliovirus intrahost evolution is required to overcome tissue-specific innate immune responses. Nat. Commun. 8:375. doi: 10.1038/s41467-017-00354-5
Xiao, Y., Rouzine, I. M., Bianco, S., Acevedo, A., Goldstein, E. F., Farkov, M., et al. (2016). RNA recombination enhances adaptability and is required for virus spread and virulence. Cell Host Microbe 19, 493–503. doi: 10.1016/j.chom.2016.03.009
Yang, Y., Yan, W., Hall, A. B., and Jiang, X. (2021). Characterizing transcriptional regulatory sequences in coronaviruses and their role in recombination. Mol. Biol. Evol. 38, 1241–1248. doi: 10.1093/molbev/msaa281
Yeh, M. T., Bujaki, E., Dolan, P. T., Smith, M., Wahid, R., Konz, J., et al. (2020). Engineering the live-attenuated polio vaccine to prevent reversion to virulence. Cell Host Microbe 27, 736–751.e8. doi: 10.1016/j.chom.2020.04.003
Zhang, T., Wu, Q., and Zhang, Z. (2020). Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 30, 1346–1351.e2. doi: 10.1016/j.cub.2020.03.022
Zhang, Y. X., Perry, K., Vinci, V. A., Powell, K., Stemmer, W. P., and del Cardayré, S. B. (2002). Genome shuffling leads to rapid phenotypic improvement in bacteria. Nature 415, 644–646. doi: 10.1038/415644a
Zhao, K., Gao, J. C., Xiong, J. Y., Guo, J. C., Yang, Y. B., Jiang, C. G., et al. (2018). Two residues in NSP9 contribute to the enhanced replication and pathogenicity of highly pathogenic porcine reproductive and respiratory syndrome virus. J. Virol. 92:e02209-17. doi: 10.1128/jvi.02209-17
Zhao, K., Ye, C., Chang, X. B., Jiang, C. G., Wang, S. J., Cai, X. H., et al. (2015). Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J. Virol. 89, 10712–10716. doi: 10.1128/jvi.01446-15
Zhou, L., Kang, R., Zhang, Y., Ding, M., Xie, B., Tian, Y., et al. (2018a). Whole genome analysis of two novel type 2 porcine reproductive and respiratory syndrome viruses with complex genome recombination between lineage 8, 3, and 1 strains identified in Southwestern China. Viruses 10:328. doi: 10.3390/v10060328
Zhou, L., Kang, R., Yu, J., Xie, B., Chen, C., Li, X., et al. (2018b). Genetic characterization and pathogenicity of a novel recombined porcine reproductive and respiratory syndrome virus 2 among Nadc30-like, Jxa1-like, and Mlv-like strains. Viruses 10:551. doi: 10.3390/v10100551
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. doi: 10.1038/s41586-020-2012-7
Keywords: RNA-dependent RNA polymerase, template switching, homologous recombination, copy-back RNA synthesis, defective viral genome
Citation: Wang H, Cui X, Cai X and An T (2022) Recombination in Positive-Strand RNA Viruses. Front. Microbiol. 13:870759. doi: 10.3389/fmicb.2022.870759
Received: 07 February 2022; Accepted: 21 April 2022;
Published: 18 May 2022.
Edited by:
Israel Pagan, Polytechnic University of Madrid, SpainReviewed by:
Ana Grande-Pérez, Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora” (IHSM-UMA-CSIC), SpainLauro Velazquez-Salinas, Plum Island Animal Disease Center, Agricultural Research Service (USDA), United States
Copyright © 2022 Wang, Cui, Cai and An. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongqing An, YW50b25ncWluZ0BjYWFzLmNu
†These authors have contributed equally to this work