 Diana Diaz-Cánova
Diana Diaz-Cánova Ugo L. Moens
Ugo L. Moens Annika Brinkmann
Annika Brinkmann Andreas Nitsche2
Andreas Nitsche2 Malachy Ifeanyi Okeke
Malachy Ifeanyi Okeke
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 May 2022
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.868887
This article is part of the Research Topic Emerging and Re-emerging Viral Zoonoses View all 9 articles
Orthopoxviruses (OPXVs) not only infect their natural hosts, but some OPXVs can also cause disease in humans. Previously, we partially characterized an OPXV isolated from an 18-year-old male living in Northern Norway. Restriction enzyme analysis and partial genome sequencing characterized this virus as an atypical cowpox virus (CPXV), which we named CPXV-No-H2. In this study, we determined the complete genome sequence of CPXV-No-H2 using Illumina and Nanopore sequencing. Our results showed that the whole CPXV-No-H2 genome is 220,276 base pairs (bp) in length, with inverted terminal repeat regions of approximately 7 kbp, containing 217 predicted genes. Seventeen predicted CPXV-No-H2 proteins were most similar to OPXV proteins from the Old World, including Ectromelia virus (ECTV) and Vaccinia virus, and North America, Alaskapox virus (AKPV). CPXV-No-H2 has a mosaic genome with genes most similar to other OPXV genes, and seven potential recombination events were identified. The phylogenetic analysis showed that CPXV-No-H2 formed a separate clade with the German CPXV isolates CPXV_GerMygEK938_17 and CPXV_Ger2010_MKY, sharing 96.4 and 96.3% nucleotide identity, respectively, and this clade clustered closely with the ECTV-OPXV Abatino clade. CPXV-No-H2 is a mosaic virus that may have arisen out of several recombination events between OPXVs, and its phylogenetic clustering suggests that ECTV-Abatino-like cowpox viruses form a distinct, new clade of cowpox viruses.
Poxvirus is a family of double-stranded DNA viruses that can infect a broad range of hosts, including mammals, birds, reptiles, and insects (International Committee on Taxonomy of Viruses, ICTV1). Based on the host, Poxviridae is divided into two subfamilies: Chordopoxvirinae (poxviruses that infect vertebrates) and Entomopoxvirinae (poxviruses that infect insects) (MacLachlan and Dubovi, 2017). Within the subfamily Chordopoxvirinae, there is the genus Orthopoxvirus (OPXV). They are viruses with large, linear, double-stranded DNA genomes ranging in size from 170 to 250 kbp (Hendrickson et al., 2010).
One of the best-known species among OPXV is Variola virus (VARV), the causative agent of smallpox. It was one of the deadliest viruses in human history and was declared to be successfully eradicated in 1980 after a worldwide smallpox vaccination campaign (Strassburg, 1982). Other members of the OPXV genus also cause human diseases, such as Cowpox virus (CPXV), Monkeypox virus (MPXV), and vaccinia-like virus (Vora et al., 2015; Reynolds et al., 2018; Diaz, 2021; Silva et al., 2021), but those are zoonotic OPXVs. Variola virus is the only OPXV that exclusively infected humans in nature. Among the most studied members of OPXVs, Vaccinia virus (VACV) is the prototype species. Several VACV strains were used as smallpox vaccines during the world vaccination campaign (Jacobs et al., 2009).
OPXVs can be further divided into New World and Old World OPXVs according to their endemism. The Old World or African-Eurasian OPXV group contains seven species: VARV, VACV, MPXV, CPXV, Camelpox virus (CMLV), Ectromelia virus (ECTV), and Taterapox virus (TATV). The New World OPXV group comprises three species that are endemic to North America: Raccoonpox virus (RCNV), Volepox virus (VPXV), and Skunkpox virus (SKPV) (Smithson et al., 2017b).
In recent times, the increased number of reported OPXV infections as well as the emergence of new OPXVs or re-emergence of existing OPXVs has been reported in several countries across the world (Abrahão et al., 2015; Kalthan et al., 2018). Three novel OPXV species have recently been discovered: Abatino macacapox virus (OPXV Abatino) in Italy (Cardeti et al., 2017), Ahkmeta virus (AKMV) in Georgia (Gao et al., 2018), and Alaskapox virus (AKPV) in the United States (Gigante et al., 2019).
The increasing number of OPXV infections in humans could be due to low population immunity against smallpox after the cessation of smallpox vaccination. The vaccinia-like virus infections were reported in different places and host species (Dumbell and Richardson, 1993; Abrahão et al., 2015; Miranda et al., 2017), including humans (Damaso et al., 2007; Megid et al., 2012). In different countries in Africa, human cases of MPXV infections have been reported (Nakoune et al., 2017; Durski et al., 2018; Yinka-Ogunleye et al., 2019; Alakunle et al., 2020); imported MPXV cases were as well reported in Israel, the United Kingdom and Singapore (Vaughan et al., 2018; Erez et al., 2019; Ng et al., 2019). In Europe, cases of cowpox were reported (Tryland et al., 1998; Kalthoff et al., 2014; Ferrier et al., 2021). The distribution of CPXV is in Eurasia (Chantrey et al., 1999; Wolfs et al., 2002; Laakkonen et al., 2006; Vorou et al., 2008; Popova et al., 2017; Diaz, 2021; Ferrier et al., 2021). The natural reservoirs of CPXV are wild rodents (Chantrey et al., 1999; Kinnunen et al., 2011). CPXV has a wide host spectrum, including humans, monkeys, cats, dogs, horses, and farmed llamas (Tryland et al., 1998; Smith et al., 1999; Girling et al., 2011; Prkno et al., 2017; Diaz, 2021). CPXV’s broad range is associated with its large genome, which is the largest genome among OPXVs (Gubser et al., 2004; Carroll et al., 2011). CPXV is polyphyletic (Carroll et al., 2011; Okeke et al., 2014; Franke et al., 2017; Mauldin et al., 2017), and their strains cluster in at least five clades (Mauldin et al., 2017; Jeske et al., 2019). Among them, some clades are more genetically similar to VACV (VACV-like virus) and VARV (VARV-like virus), whereas other CPXV strains appear as single branches and have a mosaic genome that contains genomic parts from different clades (Franke et al., 2017). The genetic heterogeneity inside CPXV could partially be due to recombination processes with other OPXV species or between CPXV clades (Okeke et al., 2012, 2014; Franke et al., 2017).
A poxvirus was isolated from an 18-year-old man living in the county Nordland, Norway (Hansen et al., 2009). Based on the detection of A-type inclusion (ATI) bodies, the sequence and phylogenetic analysis of hemagglutinin (HA) gene, cytokine response modifier B (crmB) gene, and Chinese hamster ovary host range (CHOhr) genes as well as Hind III restriction map, this virus was classified as a CPXV and was tentatively named CPXV-No-H2 (Hansen et al., 2009; Okeke et al., 2012). This isolate produces an atypical ATI phenotype, V+/, in which the virions are encrusted only in the periphery of ATI (Okeke et al., 2012). The sequencing of two of the three genes (atip, p4c, and A27L) involved in the production of ATI with virions embedded into ATI (V+) (Patel and Pickup, 1987; McKelvey et al., 2002; Howard et al., 2010) showed that it has intact atip and p4c genes. Furthermore, interestingly, the atip gene of CPXV-No-H2 closely related to that of ECTV with a bootstrap support of 100%, whereas the p4c gene was more diverse compared to the orthologs in other OPXVs (Okeke et al., 2012, 2014).
In this study, we report the whole sequence and genomic characterization of a Norwegian human CPXV isolate, CPXV-No-H2. We annotated the open reading frames, performed recombination analysis, and determined phylogenetic relationships with other OPXV genomes.
The Fennoscandian CPXV No-H2 strain was isolated in 2001 from a human patient from Northern Norway (Hansen et al., 2009; Okeke et al., 2012). CPXV-No-H2 was cultured on a monolayer of Vero cells (ATCC No. CCL-81) in 175-cm2 flasks (NUNC Sweden) as previously described (Okeke et al., 2012). Viral DNA was extracted from semi-purified virions using QIAGEN Genomic-tip 100/G and QIAGEN Genomic DNA Buffer Set, following the manufacturer’s instructions (Qiagen, Hilden, Germany). DNA concentration was measured using NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific™, Waltham, MA, United States).
The genome of CPXV-No-H2 was sequenced using Illumina and Oxford Nanopore Technologies (ONT; Oxford, United Kingdom), respectively. The preparation of sequencing libraries and next-generation sequencing with Illumina was performed at the Norwegian Sequencing Centre, Oslo. ThruPLEX DNA-Seq kit with an input DNA of 50 ng was used for the library preparation. Whole-genome sequencing was performed on an Illumina MiSeq instrument (Illumina Inc., San Diego, CA, United States) using MiSeq Reagent v3 (600 cycles), producing 2×300-bp paired-end reads. For nanopore sequencing, sequencing libraries were prepared using the Ligation Sequencing Kit SQK-LSK109 (ONT, Oxford, United Kingdom) and native barcoding expansion kit EXP-NBD104 and EXP-NBD114 (ONT). Up to 14 samples were multiplexed on R9.4 flow cells (FLO-MIN106). The run was performed on GridION X5 (Oxford, United Kingdom) using MinKNOW v20.10.6. Library preparation and nanopore sequencing were performed at the Genomics Support Centre Tromsø at UiT-The Arctic University of Norway.
Raw sequencing data from Illumina MiSeq were evaluated for their quality using FastQC software v0.11.8 (Andrews, 2010). Adapter removal and quality filtering were conducted using Trimmomatic v0.39 (Parameters: ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36) (Bolger et al., 2014). In order to remove reads corresponding to host cells, filtered reads were mapped against Chlorocebus sabaeus (GCF_000409795.2) using FastQ Screen v0.14.1 (Wingett and Andrews, 2018) with BWA v.0.7.17 (Li and Durbin, 2009). The remaining reads were used in the genome assembly. Raw nanopore data (fast5 files) were base called using Guppy 4.2.3 in MinKNOW 20.10.6, with a qscore of 7 as filter, to produce Fastq formatted sequence files. Fastq sequences were demultiplexed using Guppy 4.2.3—likewise with barcode removal. Host sequences were filtered out using FastQ Screen v0.14.1 (Wingett and Andrews, 2018) with BWA v.0.7.17 (Li and Durbin, 2009) as described above. SPAdes v3.15.3 (Bankevich et al., 2012) was used to combine the ONT long reads and the Illumina reads to produce a hybrid assembly (with nanopore option and default parameters). Contigs were screened using BLAST2 to remove host contamination. In order to assemble the complete genome, the Illumina reads were mapped to the contigs using Geneious mapper implemented in Geneious Prime 2020.2.4 (Biomatters, Inc., Newark, NJ, United States). Then, the extended contigs were merged into one by Geneious assembler in Geneious Prime 2020.2.4.
The assembled genome was annotated using Genome Annotation Transfer Utility (GATU) software from the Viral Bioinformatics Resource Centre (Tcherepanov et al., 2006). ECTV Moscow strain (ECTV_Mos), CPXV Brighton Red strain (CPXV_Br), and VACV Copenhagen strain (VACV_Cop) were used as reference genomes. These reference sequences were retrieved from the Viral Orthologous Clusters (VOCs) database (Ehlers et al., 2002). The GATU parameters included open reading frames (ORFs) longer than 30 amino acids, with a maximum overlap of 25%. Gene annotations from the reference genomes were transferred to the CPXV-No-H2 genome when the level of similarity was ≥80%. The putative coding sequences (CDS) with low similarity to the reference genes were subjected to a BLASTp analysis against the proteins belonging to the Poxviridae family from the NCBI database. Putative CDS with high similarity to other poxviruses were annotated. Similarly, the unassigned ORFs were investigated using BLASTp searches to find orthologous genes. In cases where more than one CDS were found in the same genomic region, the CDS with the highest similarity was selected. Geneious Prime 2020.2.4 was used to visualize, edit, and correct the annotations, if needed.
For phylogenetic analysis, 75 OPXV genomes were retrieved from the VOCs database (Ehlers et al., 2002), except for CPXV_GerMygEK938_17, which was retrieved from GenBank. The OPXV genomes used in this study are listed in Supplementary Table 1. The alignments of (1) the genomes, excluding the inverted terminal repeats (ITRs; called core genome), (2) the genomic region from the first gene until the last gene (referred to as the whole genome), and (3) the orthologous genes of the 76 OPXVs (including CPXV-No-H2) were performed using MAFFT v1.4.0 (with default parameters; Katoh and Standley, 2013) implemented in Geneious Prime 2020.2.4. The poorly aligned positions were removed from the alignments (1 and 2) with Gblocks 0.91b using default parameters (Talavera and Castresana, 2007). The orthologous genes were identified using OrthoFinder v2.5.2 (Emms and Kelly, 2015). The orthologs (present in ≥95% of the genomes) were aligned as described above and concatenated in Geneious Prime 2020.2.4.
The phylogenetic relationship among these OPXVs was inferred by the maximum likelihood (ML) and Bayesian inference (BI) methods. ML trees were constructed in RAxML v.8.2.12 (Stamatakis, 2014) using the best-fitting nucleotide substitution model and 1,000 bootstrap replicates. The best-fit nucleotide substitution model for the alignment data was selected using the modelTest-NG v.0.1.6 (Darriba et al., 2020). BI analyses were performed using MrBayes v.3.2.7 (Ronquist et al., 2012) under the best-fitting substitution model with the following parameters: 2 million generations, nchains = 4, samplefreq = 500, and burninfrac = 0.25. The phylogenetic trees were visualized using FigTree v1.4.4 (Rambaut, 2018).
Predicted CDS from isolate CPXV-No-H2 were extracted, translated into amino acid sequences, and compared to the CPXV_Br, ECTV_Mos, or VACV_Cop proteins using BLASTp (ncbi-blast+ v2.11.0) (Camacho et al., 2009). To find the closest annotated proteins for all predicted CPXV-No-H2 CDS, every translated CPXV-No-H2 CDS was analyzed by BLASTp search against proteins of the Poxviridae family. A BLASTn identity analysis was performed on predicted CPXV-No-H2 CDS that encode proteins with a higher identity to other OPXV proteins than CPXV proteins. When the first hit in BLASTp or BLAStn was CPXV-No-H2 protein or genome, the second hit was used.
The genome sequence of CPXV-No-H2 was examined for potential recombination events using recombination detection program 4 (RPD4) (Martin et al., 2015) and SimPlot v3.5.1 (Lole et al., 1999). A putative recombinant event was taken into account if it was identified by RDP4 and/or Simplot analysis and the sequence was most similar to the possible minor parental. The whole genome of CPXV-No-H2 was aligned to other OPXV genomes used as putative parentals (AKPV, CPXV_Gri, CPXV_GerMygEK938_17, ECTV_Mos, MPXV_Zaire, and VACV_LC16m8), with MAFFT v1.4.0 (Katoh and Standley, 2013) implemented in Geneious Prime 2020.2.4. Gaps were not removed from the multiple alignments. Similarity plots were performed on the multiple alignments using the SimPlot program (Lole et al., 1999) with default settings. Putative recombination breakpoints were determined by maximization of χ2 analysis (Lole et al., 1999; Lim et al., 2011). For recombination analysis with RPD4, seven methods [RDP (Martin and Rybicki, 2000), GENECONV (Padidam et al., 1999), Bootscan (Martin et al., 2005), MaxChi (Smith, 1992), Chimaera (Posada and Crandall, 2001), SiScan (Gibbs et al., 2000), and 3Seq (Boni et al., 2007)] were used to detect potential recombination events. RDP4 was used with the default parameters, except for the option “require topological evidence.” The recombination events that were identified by 6 of 7 methods with significant p-values (p ≤ 0.01) were considered potential recombinant events. The beginning and end of the breakpoints of these events suggested by RPD4 were used to identify the potential recombinant sequence. When the breakpoints were not identified by RDP4, the range of positions of the breakpoints obtained by Simplot analysis was used. Those potential recombinant sequences were utilized to build an ML tree using RAxML v.8.2.12 (Stamatakis, 2014). Phylogenetic tree incongruence was further used to map potential recombination sequences. Furthermore, a BLASTn identity analysis was performed on those potential recombinant sequences.
Two large contigs (>1000 bp) were obtained with the hybrid assembly and after removing the host contamination. The average coverage of the major and minor contig was 1502X and 735X, respectively. The mean genomic coverage of CPXV-No-H2 was 1370X. The assembled whole-genome length of CPXV-No-H2 was 220,276 bp. The ITR regions were approximately 7 kbp, and the central region was 206,204 bp. The A+T content of the CPXV-No-H2 genome was 66.6%. Genome annotation predicted 217 potential genes in the CPXV-No-H2 genome (Figure 1 and Supplementary Table 2). The overlapping genes were excluded from the annotation process. However, there were 20 predicted overlapping genes (Supplementary Table 3). Some of them were homologs of CPXV_Br genes (CPXV004, CXPV47, CPXV51A, CPXV058, CPXV078A, CPXV096, CPXV116, CPXV119A, CPXV130, CPXV152A, CPXV160, CPXV170, and CPXV214). The whole genome sequence is deposited in GenBank with accession number OM460002.
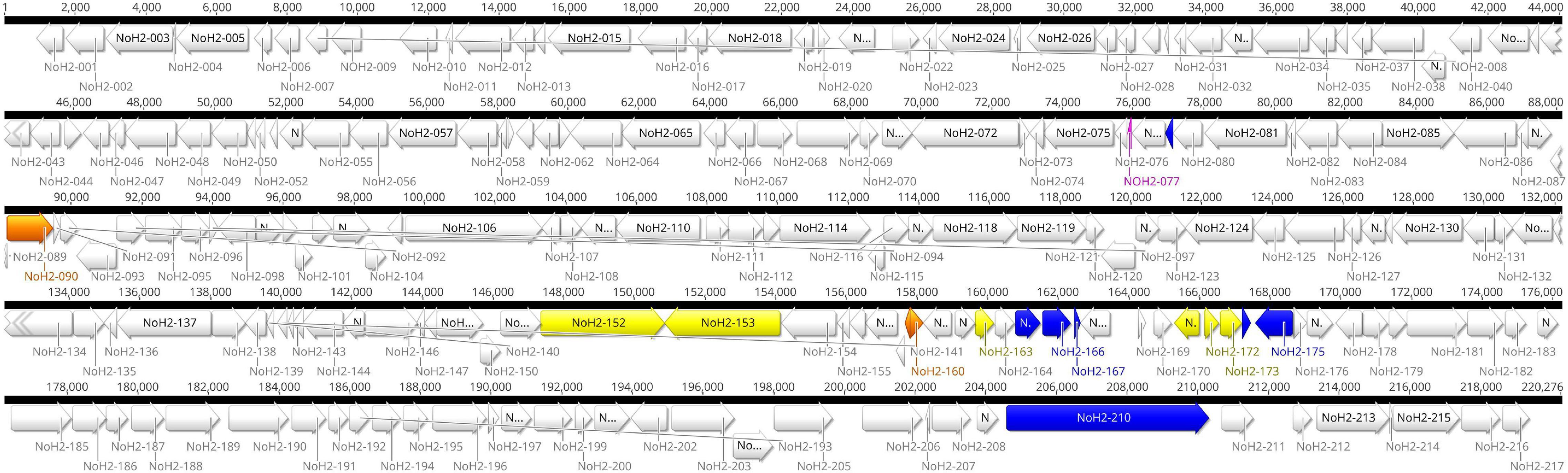
Figure 1. Genome map of CPXV-No-H2. Localization of 217 predicted coding sequences (CDS) and nine putative recombination events in the CPXV-No-H2 genome. Green blocks represent the putative recombination events. Other colors were used to visualize the amino acid sequence similarity between translated CDS to other OPXV proteins: blue blocks represent CDS with a higher similarity to Alaskapox virus proteins, yellow blocks represent CDS with a higher similarity to Ectromelia virus (ECTV) proteins, orange blocks represent CDS with a higher similarity to Vaccinia virus (VACV) proteins, and fuchsia block represents the CDS with a higher similarity to ECTV, VACV, and Horsepox virus proteins.
The phylogenetic analysis showed that the ML tree topologies were similar to the phylogenetic trees generated by the BI method, regardless of the alignments used. The BI phylogenetic trees had strong posterior probabilities in most nodes (≥0.95) (Figures 2–4). Unlike the BI trees, the ML trees had low clade support (<70%) in some of the nodes (Supplementary Figures 2–4). The BI phylogenetic trees of 76 OPXV whole genomes, 76 OPXV core genomes, and 134 OPXV orthologous genes are shown in Figures 2–4, respectively. The Old World and New World OPXV were separated into two groups in the phylogenetic trees generated from 76 OPXV whole genomes (Figure 2), 76 OPXV core genomes (Figure 3), and 134 OPXV orthologous genes (Figure 4). Within the Old World OPXV, the strains from the same OPXV species were grouped into clusters, except for CPXV strains that formed more than one cluster. CPXV was divided into clusters: CPXV-like 1, CPXV-like 2, VARV-like, VACV-like, and new clade (Franke et al., 2017). Although the strains of VACV-like did not form a proper cluster, they were closely related VACV (Figures 2–4).
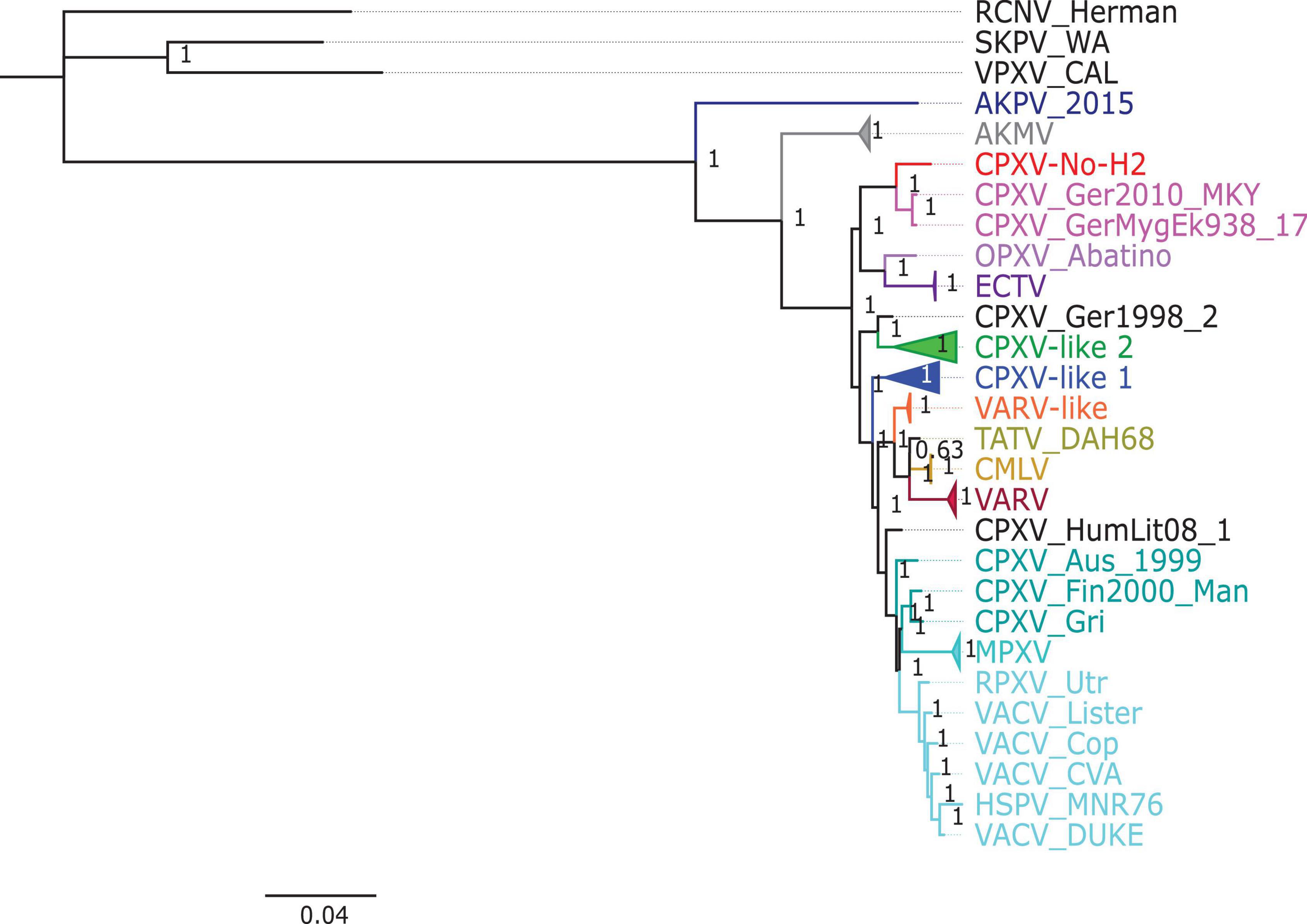
Figure 2. Bayesian inference phylogenetic tree based on 76 orthopoxvirus whole genomes. Posterior probabilities are shown on the right side of each node, and only posterior probabilities above 0.6 are shown. The cowpox virus strains were grouped into different clades: CPXV-like 1, CPXV-like 2, and VARV-like (Franke et al., 2017). The scale bar represents the expected substitutions per site.
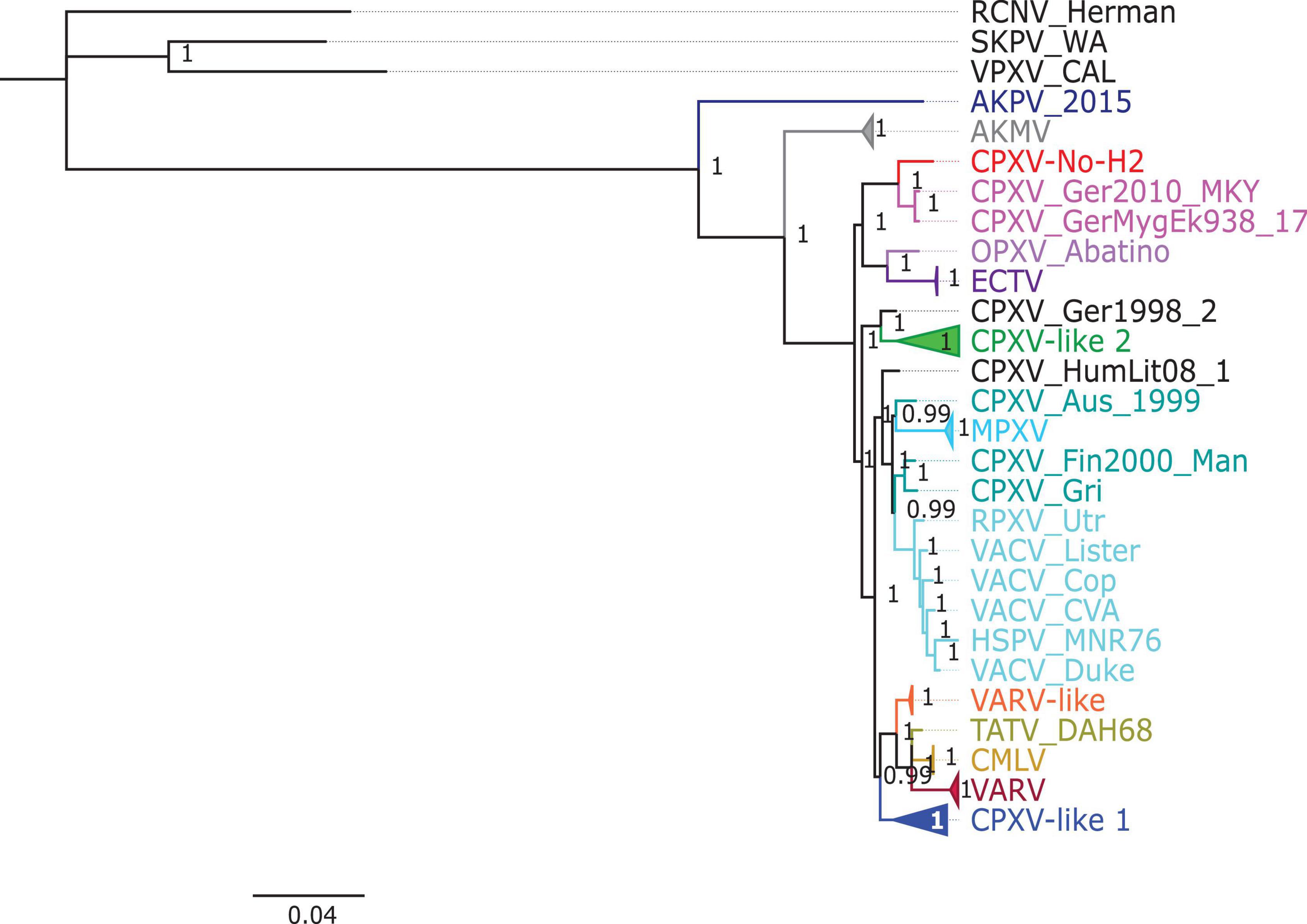
Figure 3. Bayesian inference phylogenetic tree based on 76 orthopoxvirus core genomes. Posterior probabilities are shown on the right side of each node, and only posterior probabilities above 0.6 are shown. The cowpox virus (CPXV) strains were grouped into different clades: CPXV-like 1, CPXV-like 2, and VARV-like (Franke et al., 2017). The scale bar represents the expected substitutions per site.
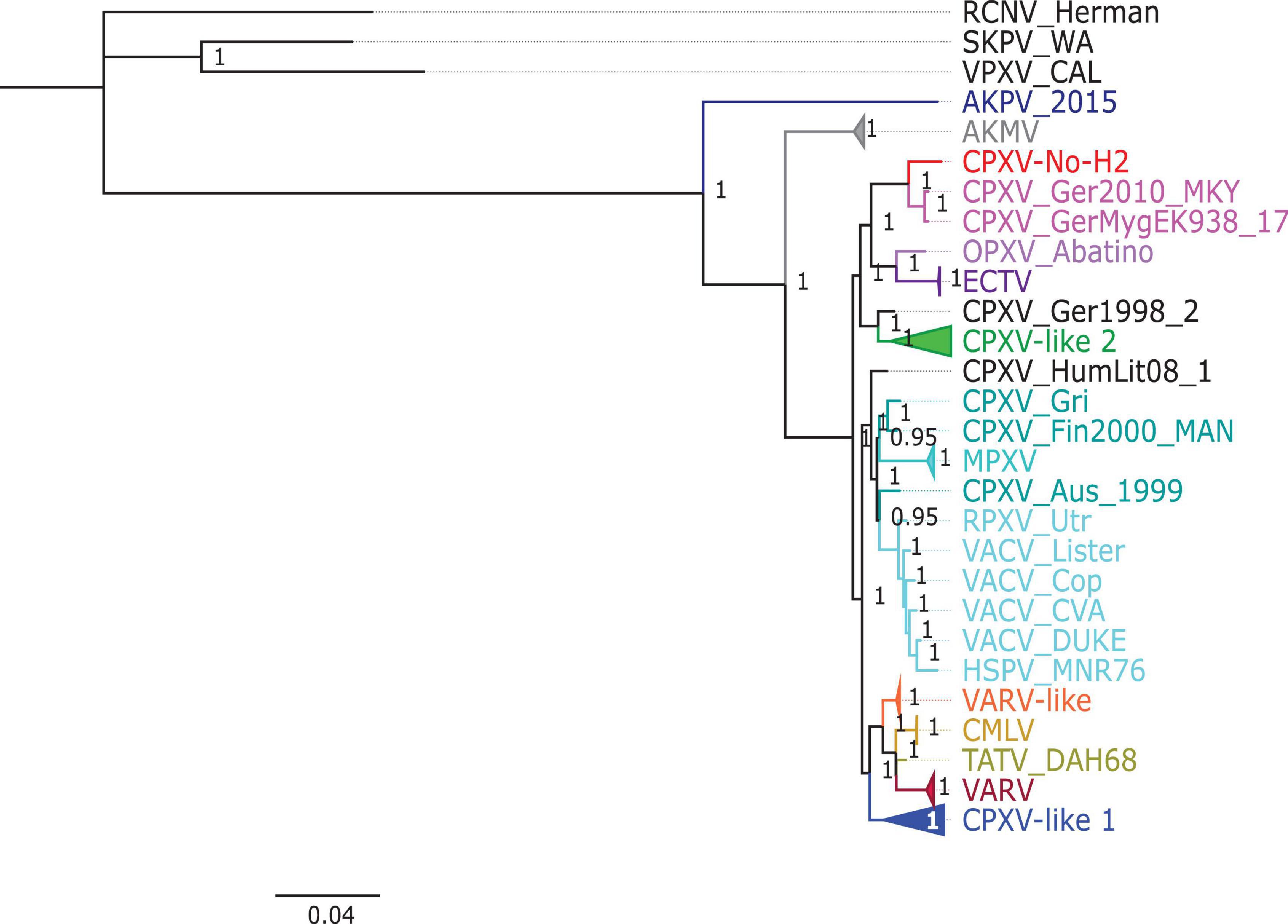
Figure 4. Bayesian inference phylogenetic tree based on 134 orthopoxvirus orthologous genes. Posterior probabilities are shown on the right side of each node, and only posterior probabilities above 0.6 are shown. The cowpox virus (CPXV) strains were grouped into different clades: CPXV-like 1, CPXV-like 2, and VARV-like (Franke et al., 2017). The scale bar represents the expected substitutions per site.
The new clade comprised CPXV-No-H2 and two German CPXV isolates: CPXV_GerMygEK938_17 and CPXV_Ger2010_MKY (posterior probabilities of 1.0 and bootstrap values of 100%) (Figures 2–4 and Supplementary Figures 2–4). The CPXV-No-H2 genome was most similar to the CPXV_GerMygEK_938_17 genome (96.38% identical), and the second most similar virus was CPXV_Ger2010_MKY (96.26% identical), based on the alignment of 76 OPXV whole genomes. The new clade was closely related to the ECTV/Abatino clade. Both clades formed a major clade together (posterior probabilities of 1.0 and bootstrap values > 89%) (Figures 2–4 and Supplementary Figures 2–4). In this study, the new clade (CPXV-No-H2/CPXV_GerMygEK938_17/CPXV_Ger2010_MKY) was tentatively named “ECTV-Abatino-like.”
In phylogenetic trees derived from the 134 OPXV orthologous genes, the ECTV-Abatino-like/ECTV/OPXV Abatino clade clustered with CPXV_Ger1998/CPXV-like 2 clade with a strong posterior probability (1.0), but with a low bootstrap support value (46%) (Figure 4 and Supplementary Figure 4), whereas the phylogeny of the 76 OPXV whole and core genomes showed that the ECTV-Abatino-like/ECTV/OPXV Abatino clade was separated from the other Old World OPXV, which formed a major polyphyletic clade (posterior probability of 1.0 and bootstrap values > 81%) (Figures 2, 3 and Supplementary Figures 2, 3). This major polyphyletic clade was further resolved in two groups: the CPXV_Ger1998/CPXV-like 2 clade (posterior probability of 1.0 and bootstrap values of 100%) and a larger group containing CPXV-like 1, VARV-like, VARV-TATV-CMLV, CPXV_HumLit08, VACV-like, MPXV, RPXV, and VACV clades (posterior probabilities of 1.0 and bootstrap values of 100%) (Figures 2, 3 and Supplementary Figures 2, 3). The clustering within this monophyletic group apparently differs between the tree based on 76 OPXV whole genomes and the trees built from 76 OPXV core genomes and 134 OPXV orthologous genes (Figures 2–4 and Supplementary Figures 2–4). In the former, CPXV-like 1 branches separated from other members of the large polyphyletic group (Figure 2 and Supplementary Figure 2). These members formed a cluster and were further split into two clusters: the VARV-like/TATV/CMLV/VARV cluster and the CPXV_HumLit08/VACV-like/MPXV/RPXV/VACV cluster. Both clusters were supported by strong posterior probabilities (1.0) and bootstrap values (100%) (Figure 2 and Supplementary Figure 2), while 76 OPXV core genomes and 134 OPXV orthologous gene phylogenies grouped CPXV-like 1 into the same cluster with VARV-like/TATV/CMLV/VARV, with posterior probabilities of 0.99 and 1.0 and bootstrap values of 51 and 99%, respectively (Figures 3, 4 and Supplementary Figures 3, 4). Additionally, CPXV_HumLit08, VACV-like, MPXV, RPXV, and VACV were grouped into the same cluster, with posterior probabilities of 1.0 and bootstrap values of 100%.
However irrespective of the aforementioned differences between the whole genome tree on one hand and the core genome and the concatenated 134 orthologous genes on the other, the following topologies were consistent in all the trees generated from the three distinct datasets: (i) ECTV-Abatino-like CPXV clustered closely with ECTV-OPXV Abatino clade, (ii) VACV-like CPXV grouped together with VACV, (iii) VARV-like CPXV clustered closely with VARV-TATV-CMLV clade, (iv) CPXV-like 1 clade is sister to VACV-like clade, and (vi) AKPV/AKMV are intermediate between Old World and New World OPXV.
The gene content and organization of the CPXV-No-H2 genome were similar to that of the CPXV_Br and ECTV_Mos genomes. All CPXV_Br genes (excluding ITR genes) were found in the CPXV-No-H2 genome, except for CPXV221 (encodes CrmD protein) and CPXV192 (encodes CPXV192 protein). The last gene is truncated in CPXV-No-H2 and overlapped to a major predicted gene. Similarly, comparing CPXV-No-H2 and ECTV_Mos, it was shown that EVM003/170 (homolog to CPXV221) was missing in the CPXV-No-H2 genome. Additionally, the EVM006 gene (encodes C-type lectin) was absent in the CPXV-No-H2 genome.
The predicted gene NoH2-154 encodes an intact p4c protein compared to CPXV_Br, whose p4c gene is disrupted in two fragments (CPXV159 and CPXV161). Similarly, this gene is fragmented in ECTV_Mos (Chen et al., 2003). The BLASTp analysis for NoH2-154 revealed that the best hit was an inclusion protein III from Buffalopox virus, with 87.7% identity. This protein (501 aa) was smaller than the p4c protein from CPXV-No-H2 (512 aa). The next best BLASTp hits were longer proteins of 527 aa from CPXV_Ger2010_MKY and 523 aa from CPXV_GerMygEK938_17, which shared 88.6 and 89.12% identity with p4c protein from CPXV-No-H2, respectively. BLASTn showed that the CPXV-No-H2 p4c gene was most similar to the p4c gene from CPXV_GerMygEK938_17.
Within ITRs of CPXV-No-H2, five of eight duplicate CPXV_Br genes were found (CPXV003/227, CPXV005/226, CPXV006/225, CPXV007/224, and CPXV008/223). The terminal CPXV004 gene was also found in both ITRs of CPXV-No-H2 (NoH2-A and NoH2-T) (Supplementary Table 3), but they overlapped two major predicted genes (NoH2-002 and NoH2-216). Interestingly, NoH2-006, the ortholog of CPXV009/222, was found as a single copy downstream of the left ITR of the CPXV-No-H2 genome (Figure 1).
All predicted genes in CPXV-No-H2 were found to have homologs in either CPXV_Br, ECTV_Mos, or VACV_Cop, except for NoH2-008 and NoH2-212. The translated NoH2-008 CDS shared 100% amino acid identity with the hypothetical protein CPXV0285 of CPXV_FM2292 (CRL86746.1) and CPXV_Ger2007_Vole (SBN49117.1). The predicted gene NoH2-212 was a homolog of CPXV-GRI-K3R (encodes CrmE protein). The BLASTp analysis of this translated CDS showed that it shared the highest amino acid identity (95.2%) with a CPXV_GerMygEK938_17 protein (hypothetical protein pCPXV003 CAB5514210.1). The NoH2-212 gene was located upstream of the right ITR of CPXV-No-H2 (Figure 1).
Of the 217 predicted genes of CPXV-No-H2, 17 coded for proteins that were most similar to other OPXV proteins than CPXV proteins. Seven of them shared high similarity to North American OPXV proteins, AKPV, and 10 genes were most similar to Old World OPXV proteins, including ECTV and VACV (Supplementary Table 4). The seven predicted CPXV-No-H2 proteins were most similar (i.e., >92% amino acid identity) to AKPV proteins, including NoH2-079, NoH2-165, NoH2-166, NoH2-167, NoH2-174, NoH2-175, and NoH2-210. The BLASTn analysis of their seven predicted CPXV-No-H2 genes revealed that NoH2-079, NoH2-165, NoH2-166, NoH2-167, NoH2-174, and NoH2-210 shared the highest similarity (i.e., > 97% nucleotide identity) with AKPV-076, AKPV-162, AKPV-163, AKPV-164, AKPV-171, and AKPV-203, respectively, whereas NoH2-175 shared the highest nucleotide similarity with CPXV_GerMygEK938_17/CPXV_Ger2010_MKY. However, the BLASTn analysis of NoH2-175 with the intergenic region between NoH2-174 and NoH2-175 revealed the highest similarity with AKPV (93.98% nucleotide identity).
The six predicted proteins most identical (i.e., > 94% amino acid identity) to ECTV proteins were NoH2-152, NoH2-153, NoH2-163, NoH2-171, NoH2-172, and NoH2-173. At the nucleotide level, NoH2-152, NoH2-153, NoH2-171, NoH2-172, and NoH2-173 had > 95% identity with the corresponding ECTV EVM127, EVM128, EVM140, EVM141, and EVM142 genes. The NoH2-163 gene, however, was most similar to CPXV169 from CPXV_GerMygEK938_17 and CPXV_Ger2010_MKY (98.6% identity), whereas the next best BLASTn hit was an ECTV gene with 98.4% identity. The difference between their percent identities was due to one identical nucleotide (Supplementary Figure 1).
The three CPXV-No-H2 predicted proteins most similar to VACV proteins included NoH2-090, NoH2-159, and NoH2-160. The BLASTn search of these predicted genes revealed that NoH2-159 shared 100% nucleotide identity with VACV, BPXV, and CPXV genomes, and NoH2-160 was 98.4% identical to VACV LC16m8 (m8197R) and VACV LC16mO genes (mO197R). The predicted protein of the gene NoH2-077 was 100% identical to ECTV, HSPV, and VACV proteins. However, the BLASTn of this predicted gene showed that it was 100% identical to CPXV_GerMygEK938_17 genome, but this region was not annotated.
Overlapping genes were excluded from the annotation process. There were 20 overlapping predicted genes (Supplementary Table 3). Fourteen of them were homologs of CPXV_Br (CPXV004, CXPV47, CPXV51A, CPXV058, CPXV078A, CPXV096, CPXV116, CPXV119A, CPXV130, CPXV152A, CPXV160, CPXV170, and CPXV214). Another six overlapping genes did not correspond to any annotated CPXV gene. The BLASTp analysis of the protein encoded by the six overlapping genes revealed that five shared the highest similarity (> 83% amino acid identity) to CMLV_0408151v (NoH2-B), OPXV Abatino (NoH2-H), VACV_CEyV1 (NoH2-G and NoH2-N), or VACV_Lister (NoH2-R) proteins (Supplementary Table 3).
Previous studies by our group had identified a putative crossover event downstream of the atip gene (Okeke et al., 2012). Consequently, the complete CPXV-No-H2 genome was examined for recombination because it contained genomic regions with predicted genes similar to AKPV, ECTV, or VACV genomes. Nine putative recombination events were predicted by RDP4 and Simplot analysis for the CPXV-No-H2 genome (Table 1 and Figure 1). Six potential recombination regions were a result of recombination events between the parentals of AKPV and CPXV (putative recombination events 1–6), two originated from recombination events between the parental ECTV and CPXV (putative recombination events 7 and 8), and one was a product of a recombination event between the parental VACV and CPXV (putative recombination event 9) (Table 1). Within the nine putative recombinant regions in CPXV-No-H2, only one recombinant region (putative recombination event 6) was close to terminal regions, whereas the other eight recombinant regions were located in the central region of the genome.
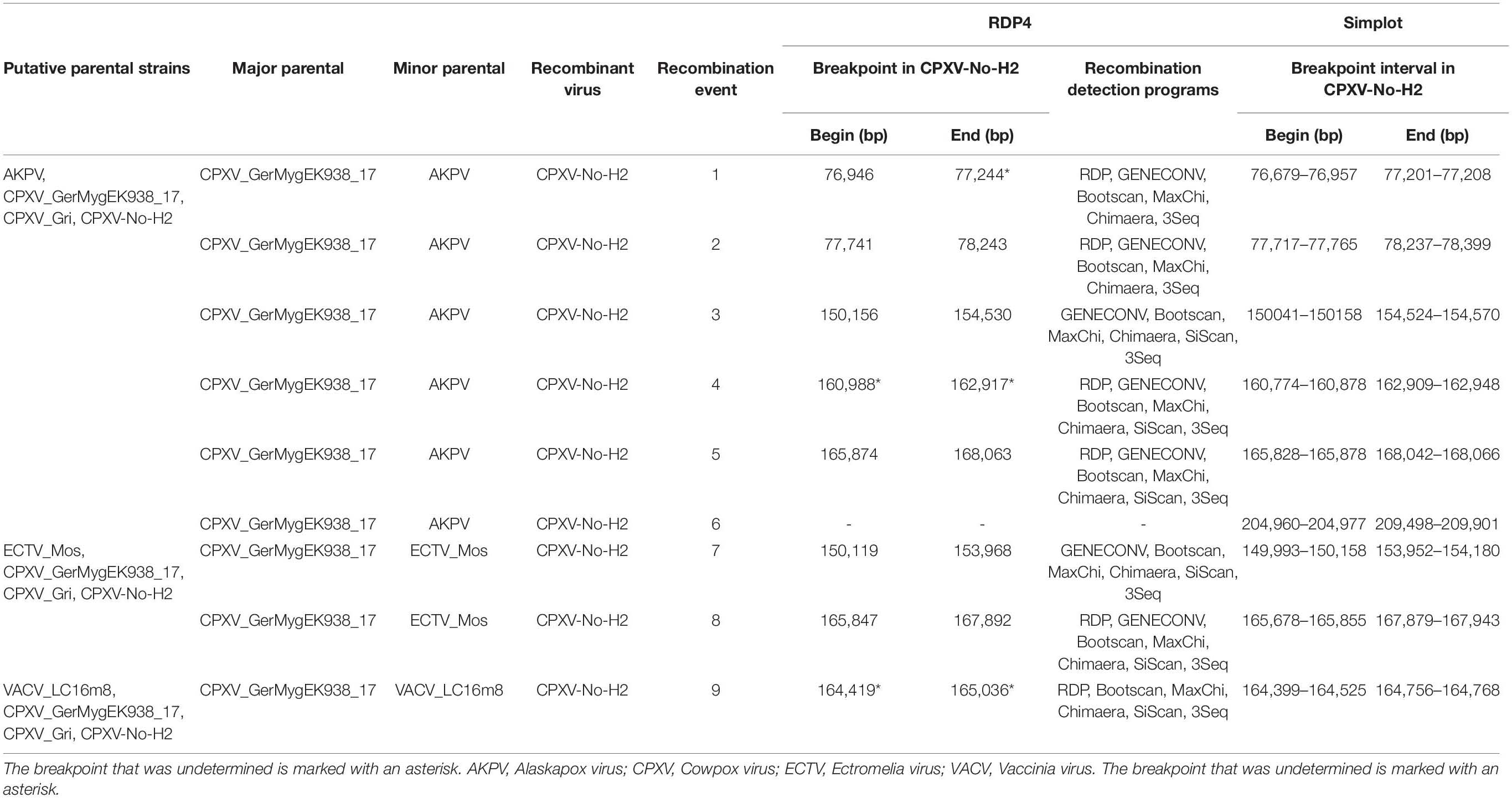
Table 1. Predicted recombination events in the CPXV-No-H2 genome using recombination detection program 4 (RPD4) and Simplot analysis.
The first potential recombinant region in the CPXV-No-H2 genome (putative recombination event 1) comprised the NoH2-079 gene and started from position 76,946 bp in the CPXV-No-H2. The ending breaking could be between positions 77,201 and 77,208 bp in the CPXV-No-H2 genome based on Simplot analysis. The next potential recombinant region (putative recombination event 2) was almost 500 bp downstream of the first one. It was located between 77,741 and 78,243 bp in the CPXV-No-H2 genome and contained parts of NoH2-080 and NoH2-081. These two putative recombinant regions shared the highest similarity to the AKPV genome (>98% identical) (Supplementary Table 5).
The third potential recombinant region (putative recombination event 3) spanned approximately 4,500 bp, from position 150,156 to 154,530 bp in the CPXV-No-H2 genome. However, it overlapped with the predicted recombinant region between the parental ECTV and CPXV (putative recombination event 7), located between 150,119 and 153,968 bp in the CPXV-No-H2 genome. The latter encompassed only two genes, NoH2-152 and No-153, compared to the former that also contained part of the NoH2-154 gene. The BLASTn analysis of the third potential recombinant region revealed the highest similarity with the AKPV genome (96.89% identical), whereas the putative recombinant region between the parental ECTV and CPXV was most similar to the ECTV genomes, with 97.93% nucleotide identity (Supplementary Table 5).
The fourth potential recombinant region in the CPXV-No-H2 genome (putative recombination event 4) included the genes NoH2-165, NoH2-166, and NoH2-167 and part of NoH2-168. The Simplot analysis revealed that the beginning and ending breakpoints were located between 160,774 and 160,878 bp and between 162,909 and 162,948 bp in the CPXV-No-H2 genome, respectively. This genomic region was most similar to the AKPV genome, sharing 97.66% nucleotide identity (Supplementary Table 5). The fifth potential recombinant region (putative recombination event 5) started from 165,874 to 168,063 bp in the CPXV-No-H2 genome. It overlapped with another putative recombinant region between the parental ECTV and CPXV (putative recombination event 8), which was located between 165,847 and 167,892 bp in the CPXV-No-H2 genome. Both regions contained part of NoH2-171, NoH2-172, NoH2-173, and NoH2-174 and part of NoH2-175. The BLASTn analysis of these two putative recombination regions revealed that the first hit was the AKPV genome, with > 97% nucleotide identity (Supplementary Table 5).
A sixth potential recombinant event between the parental AKPV and CPXV (putative recombination event 6) was detected only by Simplot analysis. The cross-over points lay between 204,960 and 204,977 bp and between 209,488 and 209,901 bp in the CPXV-No-H2 genome. It contained a major part of the NoH2-210 gene, which was most similar to AKPV-203 (97.15% identical) and also shared similarity with the Murmansk-007 gene (91.44% identical). Furthermore, this putative recombinant sequence showed its highest identity with the AKPV genome (98.4% identical), followed by the Murmansk microtuspox virus genome (91.44% identical).
One putative recombination event between the parental VACV and CPXV (putative recombination event 9) was detected. The breakpoints were undetermined by RDP4, but the Simplot analysis revealed that the putative recombinant sequence started from position 164,400–164,525 bp and ended at position 164,756–164,768 bp in the CPXV-No-H2 genome. This region contained a small part of NoH2-169 and a major part of NoH2-170. The latter gene shared 94.44% identity with the genes of CPXV and RPXV and the VACV strains, such as Lister, Cantagalo, CVA, and NYCBH.
The phylogenetic analysis of the six putative recombinant regions between the parental AKPV and CPXV showed that CPXV-No-H2 clustered with AKPV with a bootstrap support of > 91%, except for the phylogenetic tree based on the fifth putative recombinant region (165,874–168,063 bp in the CPXV-No-H2 genome), where CPXV-No-H2 clustered with ECTV with a low bootstrap support (55%), and they were grouped with AKPV (bootstrap value of 100%) (Supplementary Figures 5–10). CPXV-No-H2 likewise clustered with ECTV in the phylogenetic tree generated from the potential recombinant region between the parental ECTV and CPXV (165,847–167,892 bp in the CPXV-No-H2 genome) that overlapped the fifth putative recombinant region (Supplementary Figure 12). Unlike the previous phylogenetic tree, the bootstrap support for this clade was higher (93%) though.
Based on the phylogenetic analysis of the putative recombinant sequence between the parental ECTV and CPXV (150,119–153,968 bp in the CPXV-No-H2 genome), CPXV-No-H2 formed a cluster with ECTV (Supplementary Figure 11). This cluster was most closely related to AKPV and formed a major clade, with AKPV and AKMV, separating them from other Old World OPXV. However, the phylogenetic tree of the recombinant region between the parental AKPV and CPXV (150,156–154,530 bp in the CPXV-No-H2 genome), which overlapped that recombinant region, clustered CPXV-No-H2 with AKPV, and both isolates were closely related to ECTV (Supplementary Figure 7). The phylogenetic tree based on the putative recombinant sequence between the parental VACV and CPXV placed CPXV-No-H2 inside the VACV cluster (Supplementary Figure 13).
CPXV-No-H2 is an isolate from a human in Northern Norway that was classified as an atypical CPXV based on ATI phenotype, sequence of the atip and p4c genes, and Hind III restriction map (Hansen et al., 2009; Okeke et al., 2012). Our phylogeny analysis indicated that CPXV-No-H2 is most closely related to the German CPXV isolates CPXV_GerMygEK938_17 and CPXV_Ger2010_MKY (Figures 2–4). Similarly, phylogenetic analysis based on the HA gene also resolved CPXV_Ger2010_MKY and CPXV-No-H2 in the same cluster (Kalthoff et al., 2014). The three CPXV isolates (CPXV_GerMygEK938_17, CPXV_Ger2010_MKY, and CPXV-No-H2) may be part of a novel CPXV lineage separated from the other CPXV strains. It was previously suggested that CPXV_Ger2010_MKY and CPXV_GerMygEK938_17 were part of a new cluster provisionally called CPXV-like 3 (Franke et al., 2017; Jeske et al., 2019). However, this cluster was supported by a low bootstrap value (Jeske et al., 2019). The phylogenetic analysis reported in our study indicated that the new clade (CPXV-No-H2/CPXV_GerMygEK938_17/CPXV_Ger2010_MKY) was more closely related to ECTV and OPXV Abatino than other OPXVs, with strong posterior probabilities and bootstrap values (Figures 2–4 and Supplementary Figures 2–4). Thus, we tentatively named this clade as “ECTV-Abatino-like.”
The ECTV-Abatino-like/ECTV/OPXV Abatino clade was separated from the Old World OPXV in 76 OPXV whole- and core-genome phylogenetic trees, while a phylogenetic tree based on 134 OPXV orthologous genes showed that this clade clustered closely with CPXV_Ger1998/CPXV-like 2 but with poor bootstrap support (46%). We suggest that the separation of ECTV-Abatino-like/ECTV/OPXV Abatino from the other Old World OPXV may be due to the presence of some genes (or genomic regions) located in the core genome, which are not included within the 134 OPXV orthologous genes. A previous study showed that CPXV_GerMygEK938_17/CPXV_Ger2010_MKY/ECTV/OPXV Abatino clustered with CPXV-like 2 but with low bootstrap support (< 70%) (Jeske et al., 2019), although AKPV was not included in their phylogenetic analysis compared to our study that included AKPV and more OPXV strains. When AKPV was excluded from the construction of our phylogenetic trees, the ECTV-Abatino-like/ECTV/OPXV Abatino clade clustered with CPXV_Ger1998/CPXV-like 2 in 75 OPXV whole- and core-genome phylogenetic trees but with low bootstrap support (Supplementary Figures 14–17). In contrast, the bootstrap value in the node that clustered these clades increased from 46 to 82% in the phylogenetic tree based on 134 OPXV orthologous genes (Supplementary Figures 18, 19). We suspect that the genes or genomic regions that separated those clades have homologs in AKPV—for instance, homologs of NoH2-166, NoH2-167, NoH2-174, and NoH2-210 genes, which were most similar to the AKPV genes, were not included in the construction of the phylogenetic tree based on 134 OPXV orthologous genes.
In fact, CPXV-No-H2 has a mosaic genome with genes most similar to the OPXV genes from the Old World, including ECTV and VACV, and the North America, AKPV. Previously, we have shown that the atip gene from CPXV-No-H2 displayed the highest similarity to the corresponding ECTV gene, and the insertion of the ECTV atip gene may be a result of the recombination between CPXV and ECTV or an ECTV-like virus (Okeke et al., 2012). Our present study suggested similar findings and indicated that CPXV-No-H2 has also undergone recombination events between AKPV and VACV. A recombination between OPXVs has been reported by others (Gubser et al., 2004; Coulson and Upton, 2011; Qin et al., 2011, 2015; Okeke et al., 2012; Smithson et al., 2014, 2017a; Franke et al., 2017; Gao et al., 2018; Gigante et al., 2019).
CPXV-No-H2 displays recombination events with OPXVs that were isolated from different places and species. CPXV-No-H2 is a strain from Northern Norway (Okeke et al., 2012). Its closest relatives CPXV_GerMygEK938_17 and CPXV_Ger2010_MKY were isolated in Germany, but they were isolated from different species: bank vole and cotton-top tamarin, respectively (Kalthoff et al., 2014; Jeske et al., 2019). It was suggested that the infection of cotton-top tamarin was mediated by bank vole infected with CPXV (Jeske et al., 2019; Weber et al., 2020). In contrast, AKPV was isolated from a human patient in North America (Alaska, the United States). The patient‘s infection source is unknown, but it is presumable that she was infected by a small mammal (Springer et al., 2017; Gigante et al., 2019). VACV and ECTV have been reported around the world (Dumbell and Richardson, 1993; Miranda et al., 2017; Mavian et al., 2021). ECTV infects laboratory mice worldwide (Trentin and Briody, 1953; Mavian et al., 2021; Wang et al., 2021). The first discovered ECTV strain, ECTV_Hampstead, was isolated in the United Kingdom and was the progenitor of the European outbreaks. Only one ECTV strain (ECTV_MouKre) was isolated from a wild mouse in Germany (Mavian et al., 2021). The worldwide presence of ECTV in animals suggests their presence also in Norwegian fauna and hence the possibility to recombine with CPXV.
Among the nine potential recombination events in the CPXV-No-H2 genome, two potential recombination events with the parental AKPV (putative recombination events 3 and 5) overlap with two potential recombination events with the parental ECTV (putative recombination events 6 and 7). Interestingly, in the same position of these recombinant regions, AKPV has undergone a potential recombination with ECTV, and it was suggested that ECTV contains an AKPV-like sequence (Gigante et al., 2019).
These recombinant regions (putative recombination events 5 and 8) contain the atip gene, which is one of the three genes (atip, p4c, and A27L) required for the formation of the V+ ATI phenotype (Patel and Pickup, 1987; McKelvey et al., 2002; Howard et al., 2010). CPXV-No-H2 contains an intact ECTV-like atip, p4c, and A27L genes. Those latter genes were most similar to CPXV_GerMygEK938_17 genes. CPXV-No-H2 produces mainly virions encrusted on the surface of ATI (V+/) similar to ECTV_Hampstead, which produces both V+ and V+/ ATI phenotype (Ichihashi and Matsumoto, 1966; Okeke et al., 2012; Mavian et al., 2021). ECTV_Hampstead encodes a full-length p4c protein compared to other ECTV isolates with V– ATI phenotype. Besides this, it contains the atip and A27L genes (Mavian et al., 2021). AKPV and CPXV_Ger2010_MKY also comprise these three genes and produce the V+ ATI phenotype (Franke et al., 2017; Springer et al., 2017; Gigante et al., 2019). There is no report of the production of ATI bodies in CPXV_GerMygEK938_17; however, its atip, p4c, and A27L genes are most similar to those of CPXV_Ger2010_MKY.
The potential recombination event between the parental AKPV and CPXV (putative recombinant event 6) located close to the terminal region contains part of the NoH2-210 gene that shared similarity with AKPV-203 and the Murmansk gene. AKPV-203 is one of the three AKPV genes that may be introduced from/to Murmansk poxvirus by recombination (Gigante et al., 2019). Murmansk is a non-OPXV that belongs to the genus Centapoxvirus that was isolated in Murmansk, Russia (Smithson et al., 2017a). In three of the six recombination events with the parental AKPV (putative recombination events 1, 4, and 6), it seems that CPXV-No-H2 contains AKPV-like sequences rather than AKPV containing CPXV-No-H2-like sequences because the phylogenetic trees showed that CPXV-No-H2 is not part of the ECTV-Abatino-like clade and was placed next to AKPV (Supplementary Figures 5, 8, 10). In contrast, the overlapping recombinant regions seem to be CPXV-No-H2-like sequences that were introduced to AKPV based on the phylogenetic tree and the sequence similarity (Supplementary Figures 7, 9, 11, 12).
Reconstructing the evolutionary history of CPXV-No-H2 is difficult since it displays several potential recombination events with different OPXVs, especially when it is suspected that recombination events occurred between these OPXVs (such as AKPV and ECTV) (Gigante et al., 2019). Additionally, these OPXVs were isolated from different continents (Springer et al., 2017; Mavian et al., 2021). One plausible hypothesis about the mosaic genome of CPXV-No-H2 is that the CPXV_GerMygEK938_17-like virus was probably circulating in a population of rodents in Europe, and it underwent recombination with the AKPV-like virus. The resultant virus, CPXV-No-H2-like virus, could have suffered genomic changes and adapted to mice, which could be the possible ancestor of ECTV. The origin of ECTV from the CPXV-like ancestor was previously proposed (Jeske et al., 2019) since ECTV has a shorter genome (ranging from 204 to 208 kbp) and reduced number of genes compared to CPXV that has the largest genome among OPXVs, about 220 kbp (Chen et al., 2003; Hendrickson et al., 2010; Carroll et al., 2011; Dabrowski et al., 2013; Mavian et al., 2014, 2021). Our results suggest that CPXV-No-H2 could be derived from a CPXV_GerMygEK938_17-like virus because (1) CPXV_GerMygEK938_17 shares the highest similarity with CPXV-No-H2, (2) it did not show any significant recombination event (Kalthoff et al., 2014; Jeske et al., 2019), (3) none of the seven recombination regions in CPXV-No-H2 was highly similar to either CPXV_GerMygEK938_17 or CPXV_Ger2010_MKY, (4) there is high similarity between their p4c and A27L genes, (5) its place of isolation was also in Europe, and (6) we speculated that it has V+ ATI phenotype similar to CPXV_Ger2010_MKY due to the similarity between their atip, p4c, and A27L genes.
The recombination may have occurred between CPXV_GerMygEK938_17-like virus and AKPV-like virus rather than ECTV-like virus because, aside from two recombination events with the parental AKPV that overlapped a recombination event with the parental ECTV, there are other four recombinant events with the parental AKPV which cannot be viewed as a simple coincidence. In addition, the two suspected recombination regions in the ECTV genome (Gigante et al., 2019) were more similar to CPXV-No-H2 than AKPV (data not shown). Furthermore, hypothetically, CPXV_GerMygEK938_17 may produce V+ ATI similar to AKPV, while CPXV-No-H2 and ECTV_Hampstead produce both V+ and V+/ ATI phenotypes (Ichihashi and Matsumoto, 1966; Okeke et al., 2012; Mavian et al., 2021). It seems that the putative progeny virus, CPXV-No-H2-like virus, may have reduced its ability to embed virions into ATI bodies. This was also observed in the derivates of ECTV_Hampstead that produces the V– ATI phenotype (Mavian et al., 2021). We speculated that the recombination between CPXV_GerMygEK938_17-like virus and AKPV-like virus could take place in a rodent in Europe because AKPV contains genes from a Russian poxvirus, Murmansk, which was isolated from a root vole (Smithson et al., 2017a; Gigante et al., 2019), and CPXV_GerMygEK938_17 was isolated from bank vole in Europe (Jeske et al., 2019). Furthermore, CPXV-No-H2 was isolated in Europe, likewise with CPXV_Ger2010_MKY and ECTV_Hampstead (the source of the European outbreaks) (Hansen et al., 2009; Okeke et al., 2012; Kalthoff et al., 2014; Mavian et al., 2021).
However, it is pertinent to note that recombination detection programs predict hypothetical recombination events across genomes, and the outputs are sensitive to input parameter settings, particularly the sliding window size. To increase the likelihood of putative recombination events being real, we recommend the following: (i) use of these programs at default settings, (ii) identification of the exact recombination event by at least two different programs and algorithms, (iii) discountenance of recombination events without very high statistical support, (iv) confirmation of recombination breakpoints by manual inspection of similarity plots, and (v) incongruence of phylogenetic trees.
Another explanation for the presence of the OPXV-like genomic regions in CPXV-No-H2 could be symplesiomorphy because most genomic regions were similar to more than one taxon—for instance, the two CPXV-No-H2 genomic regions that were similar to ECTV and AKPV may be inherited from a common ancestral virus, likewise with the AKPV-like genomic region that contains part of the NoH2-210 similar to AKPV and Murmansk. However, symplesiomorphy does not explain the presence of the AKPV-like genomic region of 2,150 bp in CPXV-No-H2, which did not share high similarity with other taxa. The only plausible explanation is that CPXV-No-H2 may have obtained this sequence from an AKPV-like virus by recombination.
Overall, the genetic analysis of the atypical CPXV-No-H2 suggested that it contains sequences similar to other OPXVs, and one of the plausible explanations for their presence was recombination events with other OPXVs. In addition, CPXV-No-H2 is part of a new CPXV clade that was more phylogenetically related to ECTV and OPXV Abatino than other CPXV strains. Our findings provide some insight into the evolutionary history of CPXV and strongly support the genetic heterogeneity of the species CPXV. The discovery of new CPXV isolates and their phylogenetic relationship with OPXVs as well their genomic characterization will contribute to the further elucidation of the complex evolutionary history of CPXV.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/genbank/, OM460002.
DD-C conducted the experiments, analyzed the data, and wrote the manuscript. MO and UM conceptualized the study, supervised the design and execution of the project, and wrote the manuscript. AB and AN contributed to data interpretation and revision of the manuscript for improved intellectual content. All authors contributed to the article and approved the submitted version.
This study was supported by the University of Tromsø, the Arctic University of Norway (project A212100108) and the National Graduate School in Infection Biology and Antimicrobials (grant no. 249062). Article processing charge was paid UiT - The Arctic University of Norway.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Jessin Janice and Juan Daniel Montenegro Cabrera for their assistance during the bioinformatics analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.868887/full#supplementary-material
Abrahão, J. S., Campos, R. K., De Souza Trindade, G., Da Fonseca, F. G., Ferreira, P. C. P., and Kroon, E. G. (2015). Outbreak of Severe Zoonotic Vaccinia Virus Infection, Southeastern Brazil. Emerg. Infect. Dis. 21:695. doi: 10.3201/EID2104.140351
Alakunle, E., Moens, U., Nchinda, G., and Okeke, M. I. (2020). Monkeypox Virus in Nigeria: infection Biology, Epidemiology, and Evolution. Viruses 12:1211157. doi: 10.3390/V12111257
Andrews, S. (2010). A Quality Control tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed October 16, 2020).
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19:477. doi: 10.1089/CMB.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2020. doi: 10.1093/BIOINFORMATICS/BTU170
Boni, M. F., Posada, D., and Feldman, M. W. (2007). An Exact Nonparametric Method for Inferring Mosaic Structure in Sequence Triplets. Genetics 176, 1035–1047. doi: 10.1534/GENETICS.106.068874
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Cardeti, G., Gruber, C. E. M., Eleni, C., Carletti, F., Castilletti, C., Manna, G., et al. (2017). Fatal Outbreak in Tonkean Macaques Caused by Possibly Novel Orthopoxvirus, Italy, January 2015 - Volume 23, Number 12—December 2017 - Emerging Infectious Diseases journal - CDC. Emerg. Infect. Dis. 23, 1941–1949. doi: 10.3201/EID2312.162098
Carroll, D. S., Emerson, G. L., Li, Y., Sammons, S., Olson, V., Frace, M., et al. (2011). Chasing Jenner’s vaccine: Revisiting Cowpox virus classification. PLoS One 6, 4–9. doi: 10.1371/journal.pone.0023086
Chantrey, J., Meyer, H., Baxby, D., Begon, M., Bown, K. J., Hazel, S. M., et al. (1999). Cowpox: reservoir hosts and geographic range. Epidemiol. Infect. 122:455. doi: 10.1017/S0950268899002423
Chen, N., Danila, M. I., Feng, Z., Buller, R. M. L., Wang, C., Han, X., et al. (2003). The genomic sequence of ectromelia virus, the causative agent of mousepox. Virology 317, 165–186. doi: 10.1016/S0042-6822(03)00520-8
Coulson, D., and Upton, C. (2011). Characterization of indels in poxvirus genomes. Virus Genes 42, 171–177. doi: 10.1007/S11262-010-0560-X/FIGURES/4
Dabrowski, P. W., Radoniæ, A., Kurth, A., and Nitsche, A. (2013). Genome-wide comparison of cowpox viruses reveals a new clade related to variola virus. PLoS One 8, 1–9. doi: 10.1371/journal.pone.0079953
Damaso, C. R. A., Reis, S. A., Jesus, D. M., Lima, P. S. F., and Moussatché, N. (2007). A PCR-based assay for detection of emerging vaccinia-like viruses isolated in Brazil. Diagn. Microbiol. Infect. Dis. 57, 39–46. doi: 10.1016/J.DIAGMICROBIO.2006.07.012
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 37:294. doi: 10.1093/MOLBEV/MSZ189
Diaz, J. H. (2021). The Disease Ecology, Epidemiology, Clinical Manifestations, Management, Prevention, and Control of Increasing Human Infections with Animal Orthopoxviruses. Wilderness Environ. Med. 32, 528–536. doi: 10.1016/J.WEM.2021.08.003
Dumbell, K., and Richardson, M. (1993). Virological investigations of specimens from buffaloes affected by buffalopox in Maharashtra State, India between 1985 and 1987. Arch. Virol. 128, 257–267. doi: 10.1007/BF01309438
Durski, K. N., McCollum, A. M., Nakazawa, Y., Petersen, B. W., Reynolds, M. G., Briand, S., et al. (2018). Emergence of Monkeypox — West and Central Africa, 1970–2017. Morb. Mortal. Wkly. Rep. 67:310. doi: 10.15585/MMWR.MM6710A5
Ehlers, A., Osborne, J., Slack, S., Roper, R. L., and Upton, C. (2002). Poxvirus Orthologous Clusters (POCs). Bioinformatics 18, 1544–1545. doi: 10.1093/BIOINFORMATICS/18.11.1544
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:2. doi: 10.1186/S13059-015-0721-2
Erez, N., Achdout, H., Milrot, E., Schwartz, Y., Wiener-Well, Y., Paran, N., et al. (2019). Diagnosis of Imported Monkeypox, Israel, 2018. Emerg. Infect. Dis. 25:980. doi: 10.3201/EID2505.190076
Ferrier, A., Frenois-Veyrat, G., Schvoerer, E., Henard, S., Jarjaval, F., Drouet, I., et al. (2021). Fatal cowpox virus infection in human fetus, france, 2017. Emerg. Infect. Dis. 27, 2570–2577. doi: 10.3201/eid2710.204818
Franke, A., Pfaff, F., Jenckel, M., Hoffmann, B., Höper, D., Antwerpen, M., et al. (2017). Classification of cowpox viruses into several distinct clades and identification of a novel lineage. Viruses 9, 1–14. doi: 10.3390/v9060142
Gao, J., Gigante, C., Khmaladze, E., Liu, P., Tang, S., Wilkins, K., et al. (2018). Genome sequences of Akhmeta virus, an early divergent old world orthopoxvirus. Viruses 10:50252. doi: 10.3390/v10050252
Gibbs, M. J., Armstrong, J. S., and Gibbs, A. J. (2000). Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16, 573–582. doi: 10.1093/BIOINFORMATICS/16.7.573
Gigante, C. M., Gao, J., Tang, S., McCollum, A. M., Wilkins, K., Reynolds, M. G., et al. (2019). Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska. Viruses 11:1080708. doi: 10.3390/V11080708
Girling, S. J., Pizzi, R., Cox, A., and Beard, P. M. (2011). Fatal cowpox virus infection in two squirrel monkeys (Saimiri sciureus). Vet. Rec. 169, 156–156. doi: 10.1136/VR.D4005
Gubser, C., Hué, S., Kellam, P., and Smith, G. L. (2004). Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 85, 105–117. doi: 10.1099/VIR.0.19565-0/CITE/REFWORKS
Hansen, H., Okeke, M. I., Nilssen, O., and Traavik, T. (2009). Comparison and phylogenetic analysis of cowpox viruses isolated from cats and humans in Fennoscandia. Arch. Virol. 154, 1293–1302. doi: 10.1007/S00705-009-0442-5/FIGURES/3
Hendrickson, R. C., Wang, C., Hatcher, E. L., and Lefkowitz, E. J. (2010). Orthopoxvirus Genome Evolution: the Role of Gene Loss. Viruses 2, 1933–1967. doi: 10.3390/V2091933
Howard, A. R., Weisberg, A. S., and Moss, B. (2010). Congregation of Orthopoxvirus Virions in Cytoplasmic A-Type Inclusions Is Mediated by Interactions of a Bridging Protein (A26p) with a Matrix Protein (ATIp) and a Virion Membrane-Associated Protein (A27p). J. Virol. 84:7592. doi: 10.1128/JVI.00704-10
Ichihashi, Y., and Matsumoto, S. (1966). Studies on the nature of marchal bodies (A-type inclusion) during ectromelia virus infection. Virology 29, 264–275. doi: 10.1016/0042-6822(66)90033-X
Jacobs, B. L., Langland, J. O., Kibler, K. V., Denzler, K. L., White, S. D., Holechek, S. A., et al. (2009). Vaccinia virus vaccines: past, present and future. Antiviral Res. 84, 1–13. doi: 10.1016/J.ANTIVIRAL.2009.06.006
Jeske, K., Weber, S., Pfaff, F., Imholt, C., Jacob, J., Beer, M., et al. (2019). Molecular Detection and Characterization of the First Cowpox Virus Isolate Derived from a Bank Vole. Viruses 11:1111075. doi: 10.3390/V11111075
Kalthan, E., Tenguere, J., Ndjapou, S. G., Koyazengbe, T. A., Mbomba, J., Marada, R. M., et al. (2018). Investigation of an outbreak of monkeypox in an area occupied by armed groups, Central African Republic. Méd. Mal. Infect. 48, 263–268. doi: 10.1016/J.MEDMAL.2018.02.010
Kalthoff, D., Bock, W. I., Hühn, F., Beer, M., and Hoffmann, B. (2014). Fatal cowpox virus infection in cotton-top tamarins (Saguinus oedipus) in Germany. Vector-Borne Zoonotic Dis. 14, 303–305. doi: 10.1089/VBZ.2013.1442
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/MOLBEV/MST010
Kinnunen, P. M., Henttonen, H., Hoffmann, B., Kallio, E. R., Korthase, C., Laakkonen, J., et al. (2011). Orthopox Virus Infections in Eurasian Wild Rodents. Vector Borne Zoonotic Dis. 11, 1133–1140. doi: 10.1089/VBZ.2010.0170
Laakkonen, J., Kallio-Kokko, H., Öktem, M. A., Blasdell, K., Plyusnina, A., Niemimaa, J., et al. (2006). Serological Survey for Viral Pathogens in Turkish Rodents. J. Wildl. Dis. 42, 672–676. doi: 10.7589/0090-3558-42.3.672
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/BIOINFORMATICS/BTP324
Lim, T. H., Lee, H. J., Lee, D. H., Lee, Y. N., Park, J. K., Youn, H. N., et al. (2011). An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect. Genet. Evol. 11, 678–685. doi: 10.1016/J.MEEGID.2011.01.007
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 73:160. doi: 10.1128/jvi.73.1.152-160.1999
MacLachlan, N. J., and Dubovi, E. J. (eds) (2017). “Poxviridae,” in Fenner’s Veterinary Virology, (Boston: Academic Press), 157–174. doi: 10.1016/B978-0-12-800946-8.00007-6
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:3. doi: 10.1093/VE/VEV003
Martin, D. P., Posada, D., Crandall, K. A., and Williamson, C. (2005). A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retrovir. 21, 98–102. doi: 10.1089/AID.2005.21.98
Martin, D., and Rybicki, E. (2000). RDP: detection of recombination amongst aligned sequences. Bioinformatics 16, 562–563. doi: 10.1093/BIOINFORMATICS/16.6.562
Mauldin, M. R., Antwerpen, M., Emerson, G. L., Li, Y., Zoeller, G., Carroll, D. S., et al. (2017). Cowpox virus: What’s in a Name? Viruses 2017:101. doi: 10.3390/V9050101
Mavian, C., López-Bueno, A., Bryant, N. A., Seeger, K., Quail, M. A., Harris, D., et al. (2014). The genome sequence of ectromelia virus Naval and Cornell isolates from outbreaks in North America. Virology 462–463, 218–226. doi: 10.1016/j.virol.2014.06.010
Mavian, C., López-Bueno, A., Martín, R., Nitsche, A., and Alcamí, A. (2021). Comparative pathogenesis, genomics and phylogeography of mousepox. Viruses 13:13061146. doi: 10.3390/v13061146
McKelvey, T. A., Andrews, S. C., Miller, S. E., Ray, C. A., and Pickup, D. J. (2002). Identification of the Orthopoxvirus p4c Gene, Which Encodes a Structural Protein That Directs Intracellular Mature Virus Particles into A-Type Inclusions. J. Virol. 76:11216. doi: 10.1128/JVI.76.22.11216-11225.2002
Megid, J., Borges, I. A., Abrahão, J. S., Trindade, G. S., Appolinário, C. M., Ribeiro, M. G., et al. (2012). Vaccinia Virus Zoonotic Infection, São Paulo State, Brazil. Emerg. Infect. Dis. 18:189. doi: 10.3201/EID1801.110692
Miranda, J. B., Borges, I. A., Campos, S. P. S., Vieira, F. N., De Ázara, T. M. F., Marques, F. A., et al. (2017). Serologic and Molecular Evidence of Vaccinia Virus Circulation among Small Mammals from Different Biomes, Brazil. Emerg. Infect. Dis. 23:931. doi: 10.3201/EID2306.161643
Nakoune, E., Lampaert, E., Ndjapou, S. G., Janssens, C., Zuniga, I., Van Herp, M., et al. (2017). A Nosocomial Outbreak of Human Monkeypox in the Central African Republic. Open Forum Infect. Dis. 4:168. doi: 10.1093/OFID/OFX168
Ng, O. T., Lee, V., Marimuthu, K., Vasoo, S., Chan, G., Lin, R. T. P., et al. (2019). A case of imported Monkeypox in Singapore. Lancet. Infect. Dis. 19:1166. doi: 10.1016/S1473-3099(19)30537-7
Okeke, M. I., Hansen, H., and Traavik, T. (2012). A naturally occurring cowpox virus with an ectromelia virus A-type inclusion protein gene displays atypical A-type inclusions. Infect. Genet. Evol. 12, 160–168. doi: 10.1016/J.MEEGID.2011.09.017
Okeke, M. I, Okoli, A. S., Nilssen, O., Moens, U., Tryland, M., Bøhn, T., et al. (2014). Molecular characterization and phylogenetics of Fennoscandian cowpox virus isolates based on the p4c and atip genes. Virol. J. 11, 1–16. doi: 10.1186/1743-422X-11-119/TABLES/5
Padidam, M., Sawyer, S., and Fauquet, C. M. (1999). Possible Emergence of New Geminiviruses by Frequent Recombination. Virology 265, 218–225. doi: 10.1006/VIRO.1999.0056
Patel, D. D., and Pickup, D. J. (1987). Messenger RNAs of a strongly-expressed late gene of cowpox virus contain 5’-terminal poly(A) sequences. EMBO J. 6:3787. doi: 10.1002/J.1460-2075.1987.TB02714.X
Popova, A. Y., Maksyutov, R. A., Taranov, O. S., Tregubchak, T. V., Zaikovskaya, A. V., Sergeev, A. A., et al. (2017). Cowpox in a human, Russia, 2015. Epidemiol. Infect. 145, 755–759. doi: 10.1017/S0950268816002922
Posada, D., and Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. 98, 13757–13762. doi: 10.1073/PNAS.241370698
Prkno, A., Hoffmann, D., Goerigk, D., Kaiser, M., van Maanen, A. C. F., Jeske, K., et al. (2017). Epidemiological investigations of four cowpox virus outbreaks in alpaca herds, Germany. Viruses 9, 1–15. doi: 10.3390/v9110344
Qin, L., Favis, N., Famulski, J., and Evans, D. H. (2015). Evolution of and Evolutionary Relationships between Extant Vaccinia Virus Strains. J. Virol. 89:1809. doi: 10.1128/JVI.02797-14
Qin, L., Upton, C., Hazes, B., and Evans, D. H. (2011). Genomic Analysis of the Vaccinia Virus Strain Variants Found in Dryvax Vaccine. J. Virol. 85:13049. doi: 10.1128/JVI.05779-11
Rambaut, A. (2018). FigTree. Available online at: http://tree.bio.ed.ac.uk/software/figtree/ [accessed February 19, 2021]
Reynolds, M. G., Guagliardo, S. A. J., Nakazawa, Y. J., Doty, J. B., and Mauldin, M. R. (2018). Understanding orthopoxvirus host range and evolution: from the enigmatic to the usual suspects. Curr. Opin. Virol. 28, 108–115. doi: 10.1016/J.COVIRO.2017.11.012
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61, 539–542. doi: 10.1093/SYSBIO/SYS029
Silva, N. I. O., de Oliveira, J. S., Kroon, E. G., Trindade, G., de, S., and Drumond, B. P. (2021). Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2021:13. doi: 10.3390/V13010043
Smith, J. M. (1992). Analyzing the mosaic structure of genes. J. Mol. Evol. 342, 126–129. doi: 10.1007/BF00182389
Smith, K. C., Bennett, M., and Garrett, D. C. (1999). Skin lesions caused by orthopoxvirus infection in a dog. J. Small Anim. Pract. 40, 495–497. doi: 10.1111/J.1748-5827.1999.TB03003.X
Smithson, C., Tang, N., Sammons, S., Frace, M., Batra, D., Li, Y., et al. (2017b). The genomes of three North American orthopoxviruses. Virus Genes 53, 21–34. doi: 10.1007/S11262-016-1388-9/TABLES/2
Smithson, C., Meyer, H., Gigante, C. M., Gao, J., Zhao, H., Batra, D., et al. (2017a). Two novel poxviruses with unusual genome rearrangements: NY_014 and Murmansk. Virus Genes 53, 883–897. doi: 10.1007/S11262-017-1501-8/FIGURES/5
Smithson, C., Purdy, A., Verster, A. J., and Upton, C. (2014). Prediction of Steps in the Evolution of Variola Virus Host Range. PLoS One 9:e91520. doi: 10.1371/JOURNAL.PONE.0091520
Springer, Y. P., Hsu, C. H., Werle, Z. R., Olson, L. E., Cooper, M. P., Castrodale, L. J., et al. (2017). Novel Orthopoxvirus Infection in an Alaska Resident. Clin. Infect. Dis. An Off. Publ. Infect. Dis. Soc. Am. 64:1737. doi: 10.1093/CID/CIX219
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1313. doi: 10.1093/BIOINFORMATICS/BTU033
Strassburg, M. A. (1982). The global eradication of smallpox. Am. J. Infect. Control 10, 53–59. doi: 10.1016/0196-6553(82)90003-7
Talavera, G., and Castresana, J. (2007). Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tcherepanov, V., Ehlers, A., and Upton, C. (2006). Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Gen. 7:150. doi: 10.1186/1471-2164-7-150
Trentin, J. J., and Briody, B. A. (1953). An outbreak of mouse-pox (infectious ectromelia) in the United States: II. Definit. Diag. 1953, 227. doi: 10.1126/science.117.3035.227
Tryland, M., Myrmel, H., Holtet, L., Haukenes, G., and Traavik, T. (1998). Clinical cowpox cases in Norway. Scand. J. Infect. Dis. 30, 301–303. doi: 10.1080/00365549850160972
Vaughan, A., Aarons, E., Astbury, J., Balasegaram, S., Beadsworth, M., Beck, C. R., et al. (2018). Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 23:1800509. doi: 10.2807/1560-7917.ES.2018.23.38.1800509
Vora, N. M., Li, Y., Geleishvili, M., Emerson, G. L., Khmaladze, E., Maghlakelidze, G., et al. (2015). Human Infection with a Zoonotic Orthopoxvirus in the Country of Georgia. N. Engl. J. Med. 372:1223. doi: 10.1056/NEJMOA1407647
Vorou, R. M., Papavassiliou, V. G., and Pierroutsakos, I. N. (2008). Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 21, 153–156. doi: 10.1097/QCO.0B013E3282F44C74
Wang, J., Liu, X., Zhu, Q., Wu, Q., Tang, S., Zhang, L., et al. (2021). Identification, Isolation, and Characterization of an Ectromelia Virus New Strain from an Experimental Mouse. Virol. Sin. 36, 155–158. doi: 10.1007/s12250-020-00263-w
Weber, S., Jeske, K., Ulrich, R. G., Imholt, C., Jacob, J., Beer, M., et al. (2020). In vivo characterization of a bank vole-derived cowpox virus isolate in natural hosts and the rat model. Viruses 12:12020237. doi: 10.3390/v12020237
Wingett, S. W., and Andrews, S. (2018). FastQ Screen: a tool for multi-genome mapping and quality control. F1000Research 7:1338. doi: 10.12688/F1000RESEARCH.15931.2
Wolfs, T. F. W., Wagenaar, J. A., Niesters, H. G. M., and Osterhaus, A. D. M. E. (2002). Rat-to-Human Transmission of Cowpox Infection. Emerg. Infect. Dis. 8:1495. doi: 10.3201/EID0812.020089
Keywords: poxvirus, phylogenetics, Fennoscandian, Norway, recombination
Citation: Diaz-Cánova D, Moens UL, Brinkmann A, Nitsche A and Okeke MI (2022) Genomic Sequencing and Analysis of a Novel Human Cowpox Virus With Mosaic Sequences From North America and Old World Orthopoxvirus. Front. Microbiol. 13:868887. doi: 10.3389/fmicb.2022.868887
Received: 03 February 2022; Accepted: 24 February 2022;
Published: 03 May 2022.
Edited by:
Vladimir N. Uversky, University of South Florida, United StatesReviewed by:
David Hugh Evans, University of Alberta, CanadaCopyright © 2022 Diaz-Cánova, Moens, Brinkmann, Nitsche and Okeke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ugo L. Moens, dWdvLm1vZW5zQHVpdC5ubw==; Malachy Ifeanyi Okeke, bWFsYWNoeS5va2VrZUBhdW4uZWR1Lm5n
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.