 Yanan Zhang
Yanan Zhang Honglong Chu
Honglong Chu Liqiong Yu1
Liqiong Yu1 Fei He
Fei He Yong Gao
Yong Gao Lizhou Tang
Lizhou Tang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 13 July 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.868709
This article is part of the Research TopicCelebrating 20 Years of Microbial Plant Pathogen GenomesView all 5 articles
Bacterial soft rot is a devastating disease for a wide range of crops, vegetables, and ornamental plants including konjac (Amorphophallus konjac). However, the pangenome and genomic plasticity of the konjac soft rot pathogens is little explored. In this study, we reported the complete genome sequences of 11 bacterial isolates that can cause typical soft rot symptoms in konjac by in vitro and in vivo pathogenicity tests. Based on in silico DNA–DNA hybridization, average nucleotide identity and phylogenomic analysis, all 11 isolates were determined to be Pectobacterium aroidearum. In addition, synteny analysis of these genomes revealed considerable chromosomal inversions, one of which is triggered by homologous recombination of ribose operon. Pangenome analysis and COG enrichment analysis showed that the pangenome of P. aroidearum is open and that accessory genes are enriched in replication, recombination, and repair. Variations in type IV secretion system and type VI secretion system were found, while plant cell wall degrading enzymes were conserved. Furthermore, sequence analyses also provided evidence for the presence of a type V secretion system in Pectobacterium. These findings advance our understanding of the pathogenicity determinants, genomic plasticity, and evolution of P. aroidearum.
Bacterial soft rot is a disease of agricultural ecosystems, caused by multiple genera of Gram-negative and Gram-positive bacteria including Pseudomonas, Bacillus, Burkholderia, Pantoea, Enterobacter, Klebsiella, Leuconostoc and Clostridium (Charkowski, 2018). Soft rot Pectobacteriaceae (SRP), belonging to the genera Pectobacterium and Dickeya, are the most widely studied soft rot bacterial pathogens. They infect a broad range of important crop and ornamental plants, leading to economic and yield losses in the field and in storage (Toth et al., 2021a). Konjac (A. konjac), a perennial plant belonging to the family Araceae, is widely grown as a cash crop in tropical and subtropical Asian countries such as China, India, and Japan. The konjac glucomannan (KGM), extracted from the corm, is a water-soluble polysaccharide (dietary fiber) with diverse applications in food science and nutrition, biotechnology, pharmacology and fine chemicals (Behera and Ray, 2016; Zhu, 2018). However, bacterial sot rot is becoming a major threat to konjac production in China (Wu et al., 2015).
Based on 16S rRNA gene sequence analysis and biochemical tests, the causal agent of konjac soft rot was identified as Pectobacterium aroidearum in the Yunnan province of southwestern China (Wei et al., 2020). In recent years, P. aroidearum was also reported to cause soft rot in Chinese cabbage (Xie et al., 2018), Cucurbita pepo (Moraes et al., 2017), Syngonium podophyllum (Xu et al., 2020) and Olecranon Honey Peach (Liang et al., 2022). It should be noted that P. aroidearum was not proposed as a novel species until 2013 (Nabhan et al., 2013). Earlier studies identified P. carotovorum subsp. carotovorum and P. chrysanthemi as the soft rot pathogen of Konjac (Wu et al., 2011). In general, the taxonomy of SRP has been in a state of flux especially over the last two decades due to the development of genome-based taxonomic tools (Toth et al., 2021b). According to the List of Prokaryotic names with Standing in Nomenclature (LPSN), the genus Pectobacterium has 19 child taxa with a validly published and correct name (Parte et al., 2020).
Advances in DNA sequencing technology have made large volumes of genomic data available (Land et al., 2015). The use of genome data promises increasing precision and accuracy for the taxonomy of prokaryotes especially at species and subspecies levels (Maiden et al., 2013; Chun et al., 2018). Based on average nucleotide identity (ANI), in silico DNA–DNA hybridization (isDDH) and phylogenomic analysis, new studies have illustrated inconsistencies between established taxonomies and evidence from completely sequenced isolates such as subspecies of P. carotovorum (Pritchard et al., 2016; Zhang et al., 2016). Given the importance of correct taxonomy and the increasing availability of whole-genome sequences, using genomic data will likely become routine for microbial taxonomy in the near future (Meier-Kolthoff et al., 2021).
Comparative genomics, including pangenome analysis, has also been used to reveal the basis of pathogenicity, genomic diversity, pathogenic evolution and host adaptation (Toth et al., 2006; Sheppard et al., 2018; Amir et al., 2020). For Pectobacterium spp., genome-wide analyses have indicated considerable variation in the pathogenicity determinants including phytotoxins, polysaccharides, iron uptake systems, asecretion systems (type IV-VI), antimicrobial compounds, and CRISPR-Cas systems, whereas the plant cell wall degrading enzymes (PCWDEs) are highly conserved (Li et al., 2018, 2019; Arizala and Arif, 2019). Of particular interest to soft rot, pangenome analyses have also been performed for some species of Pectobacterium such as P. actinidiae (Lu et al., 2021) and P. parmentieri (Zoledowska et al., 2018).
The present study aims to further investigate the causal agent of konjac soft rot and determine its taxonomy based on genomic data. We achieved this by isolating the soft rot pathogens of konjac and assembling 11 complete genomes using Nanopore and Illumina sequencing. From this, we performed comparative genomics and pangenome-oriented analyses for Pectobacterium spp. and P. aroidearum strains. Overall, the obtained results provide new insights into the pathogenicity determinants, genomic structure and evolution of P. aroidearum.
Konjac corms with symptoms of soft rot were collected from Qujing City, Yunnan Province and Ankang City, Shaanxi Province, China in 2019 and 2020 (Supplementary Table 1). Infected tissues were cut into small pieces and were sterilized in 75% (v/v) ethanol for 30 s, followed by three times rinses with sterile distilled water. The tissues were then mashed and diluted in sterile distilled water. A volume of 200 μl bacterial suspension from each dilution was spread on nutrient agar (NA) medium and incubated at 28°C for 48 h. A single colony was then picked and subcultured in nutrient broth (NB) medium. Pure cultures were obtained through successive streaking on NA.
For konjac slice assay, bacterial strains were grown overnight, washed and resuspended into sterile water with an OD600 = 0.2. The tubers were sliced 0.5 cm thick and placed in a plastic food container containing wet paper tissues. Tuber slices were then inoculated with 20 μl of bacterial solution and incubated at 28°C for 48 h. Sterile water was used as a negative control. For in vivo assay, bacterial suspension was prepared with a concentration as mentioned above. Stems of 6-months-old konjac seedlings were then inoculated using a pin-prick inoculation method under greenhouse conditions. Similarly, sterile water was used as a negative control. The assay was repeated twice independently.
Pectinolytic activity assay for bacterial isolates was performed on crystal violet pectate (CVP) medium (Hélias et al., 2012). Briefly, pure bacterial suspensions were diluted into 102–103 CFU/ml. A volume of 100 μl dilution was then plated on a CVP medium and incubated at 28°C for 48 h.
Eleven pathogenic strains (per isolate per sample) were selected and then sequenced. Genomic DNA extraction, sequence library construction and sequencing were conducted at the Beijing Novogene Bioinformatics Technology Co., Ltd. Briefly, total DNA from each isolate at the exponential stage was extracted and assessed using agarose gel electrophoresis and Qubit 2.0 Fluorometer (Thermo Scientific). After library construction, sequencing was performed using a Nanopore PromethION platform and an Illumina NovaSeq PE150 platform. For most bacterial isolates, a hybrid assembly pipeline was conducted by Unicycler v0.4.8 using both Illumina reads and long reads (Wick et al., 2017). The genome of strain QJ003 was assembled by Raven v1.5.1 (Vaser and Šikić, 2021) and further polished by Pilon v1.24 (Walker et al., 2014). The quality of all genome assemblies was assessed by BUSCO v5.2.2 based on the dataset bacteria_odb10 (Manni et al., 2021). Prokka v1.14.5 was used for genome annotation (Seemann, 2014).
Pairwise average nucleotide identity (ANI) values were calculated by a Python module pyani v0.2.11 using ANIm method.1 isDDH was estimated via the Genome-to-Genome Distance Calculator 3.0 web server using the recommended formula 22 (Meier-Kolthoff et al., 2021).
In addition to our 11 new assemblies, 53 complete genome sequences of Pectobacterium were retrieved from GenBank in May 2021.3 The accession numbers and other basic information for these downloaded genomes are provided (Supplementary Table 2). After confirming that all 11 P. aroidearum assembly sequences began with the gene dnaA, bacterial synteny was evaluated and visualized by multiple whole-genome alignment using progressiveMauve (Darling et al., 2010). Dot plot between two genomes was generated by D-Genies web server4 (Cabanettes and Klopp, 2018). A comparative genomic ring plot was generated using BLAST Ring Image Generator (BRIG; Alikhan et al., 2011).
Parsimony trees were inferred with kSNP v3.1.2 based on pan-genome SNPs (Gardner et al., 2015). The optimum k-mer size used for the Pectobacterium species and P. aroidearum isolates was 21 and 19, respectively, which was determined by subcommand Kchooser.
A maximum-likelihood (ML) tree for the P. aroidearum strains was inferred by FastTree 2.1.11 using generalized time-reversible (GTR) models based on core-gene alignment (Price et al., 2010). The Newick tree files were visualized using the online program iTOL v5 (Letunic and Bork, 2021) and MEGA11 (Tamura et al., 2021).
The Roary pipeline was used to infer the pangenome and a gene presence/absence matrix of Pectobacterium spp. and P. aroidearum, respectively (Page et al., 2015). The parameters for minimum blastp percentage identity (−i) and core gene (−cd) were adjusted to 90% and 100%, respectively.
For enrichment analysis, a COG functional category was assigned to the protein-coding genes of P. aroidearum QJ036 using eggNOG-mapper v2.1.4 (Cantalapiedra et al., 2021). Functional enrichment analysis was performed using Fisher’s exact test within the R environment (v4.0.0). p values for multiple comparisons were adjusted with the Benjamini and Hochberg method (Benjamini and Hochberg, 1995).
Protein secretion systems (types I, II, III, IV, V, and VI) were detected by uploading the protein sequences to TXSScan/MacSyFinder (Abby et al., 2014, 2016). SecRet65 was used to predict the T6SS-associated proteins (Li et al., 2015). Domain identification and protein family classification was conducted by InterProScan (Blum et al., 2021). Multiple sequence alignment was then performed by T-Coffee (Di Tommaso et al., 2011) and visualized by ENDscript (Robert and Gouet, 2014).
PCWDE proteins of the genera Pectobacterium and Dickeya were downloaded from the UniProt database and used for building a reference database. BLAST+ (v2.6.0) was applied to find putative PCWDEs in 14 P. aroidearum genomes using an E-value threshold (E-value < 1e − 5). Only hits with at least 35% identity and 50% query coverage were kept. If a protein annotation from BLAST+ results was not consistent with that derived from eggNOG-mapper (v2.1.4), this protein annotation was examined manually.
Genomic islands (GIs) were predicted using the IslandViewer 4 webserver.6 IslandViewer 4 integrated three GI prediction methods (IslandPath-DIMOB, SIGI-HMM and IslandPick) as well as annotations of virulence factors, pathogen-associated genes, and antimicrobial resistance (AMR) genes.
To further determine the causal agent of konjac soft rot in China, A. konjac tubers with foul-smelling rot symptoms were collected from Qujing, Yunnan Province and Ankang, Shaanxi Province, China. After bacterial isolation and purification, a total of 11 isolates were shown to cause typical soft rot symptoms on konjac tubers in vitro (Figure 1A; Supplementary Figure 1). As expected, the pectolytic ability of these candidate pathogens was further confirmed based on the formation of deep cavities on crystal violet pectate (CVP) media (Figure 1B; Supplementary Figure 2). 16S rRNA gene sequencing and BLAST analysis showed that all 11 16S rRNA genes share at least 99% identity with that of P. aroidearum (NR_159926), which is consistent with previous findings (Xie et al., 2020). Since all candidate pathogens belonged to the same species based on 16S rRNA gene sequences and morphological similarity, only one strain OJ036 was selected for pathogenicity test in vivo. Stem rot and wilting symptoms were visible only within 2 days for seedlings inoculated with strain QJ036 while no symptoms were observed for the control group (Figure 1C). This represents strong evidence that the causal agent of konjac soft rot is P. aroidearum.

Figure 1. Pathogenicity tests and host range determination of Pectobacterium aroidearum QJ036. (A) Bacterial cultures of QJ036 strain (108 CFU/ml) were inoculated on konjac slice tubers for 24 h. Sterile water was also used as negative control. (B) The formation of pits caused by QJ036 strain on CVP medium incubated at 28°C for 48 h. (C) Bacterial cultures of QJ036 strain (108 CFU/ml) and sterile water were inoculated into stems of 6-months-old konjac seedlings, respectively. Pictures were taken at the indicated time points. (D) Bacterial cultures of QJ036 strain (108 CFU/ml) were inoculated on slice tubers of indicated species for 24 h. All these experiments were repeated at least twice independently with similar results.
In addition, we did pathogenicity tests in vitro to explore the host range of P. aroidearum. Interestingly, P. aroidearum was found to cause typical rot symptoms on sweet potato (Ipomoea batatas (L.) Lam.), jícama (Pachyrhizus erosus (L.) Urb.), yacón (Smallanthus sonchifolius (Poepp.) H.Rob.) and taro (Colocasia esculenta (L.) Schott), which has not been reported (Figure 1D). Although in vivo pathogenicity tests are needed, these results suggest a broader host range of P. aroidearum.
To further confirm the taxonomic status and investigate the genomic diversity of konjac soft rot pathogens, whole-genome sequencing was performed for the 11 strains using a Nanopore PromethION platform and an Illumina NovaSeq platform with more than 100x coverage depth (Supplementary Table 3). After hybrid genome assembly using filtered data, one circular chromosome without gaps was obtained for each isolate. High scores (>99%) were achieved from a BUSCO assessment of genomic completeness, indicating the high quality of our assemblies (Supplementary Figure 3). The parsimony tree based on genome-wide single nucleotide polymorphisms (SNPs) clustered these Pectobacterium spp. into seven well-resolved clades, of which clade VII can be further clustered into three subclades (Figure 2). Notably, all konjac soft rot pathogens form a monophyletic group (clade IV) with P. aroidearum L6, P. carotovorum subsp. carotovorum PC1 and P. carotovorum subsp. carotovorum PCCS1, indicating that these strains belong to the same species (Figure 2). This result is further supported by average nucleotide identity (ANI) and digital DNA–DNA hybridization (dDDH) analyses, two widely used methods for the taxonomy of prokaryotes (Chun et al., 2018). Sequence comparisons between the trains in clade IV show that ANI is greater than 95% and dDDH greater than 70%, which are above the cut-off values for species delineation (Supplementary Table 4). Taken together, these results clearly indicate that all these strains in clade IV should be classified as P. aroidearum and that three strains (P. carotovorum subsp. carotovorum PC1, P. carotovorum subsp. carotovorum PCCS1 and P. carotovorum subsp. carotovorum PCC21) are incorrectly named.
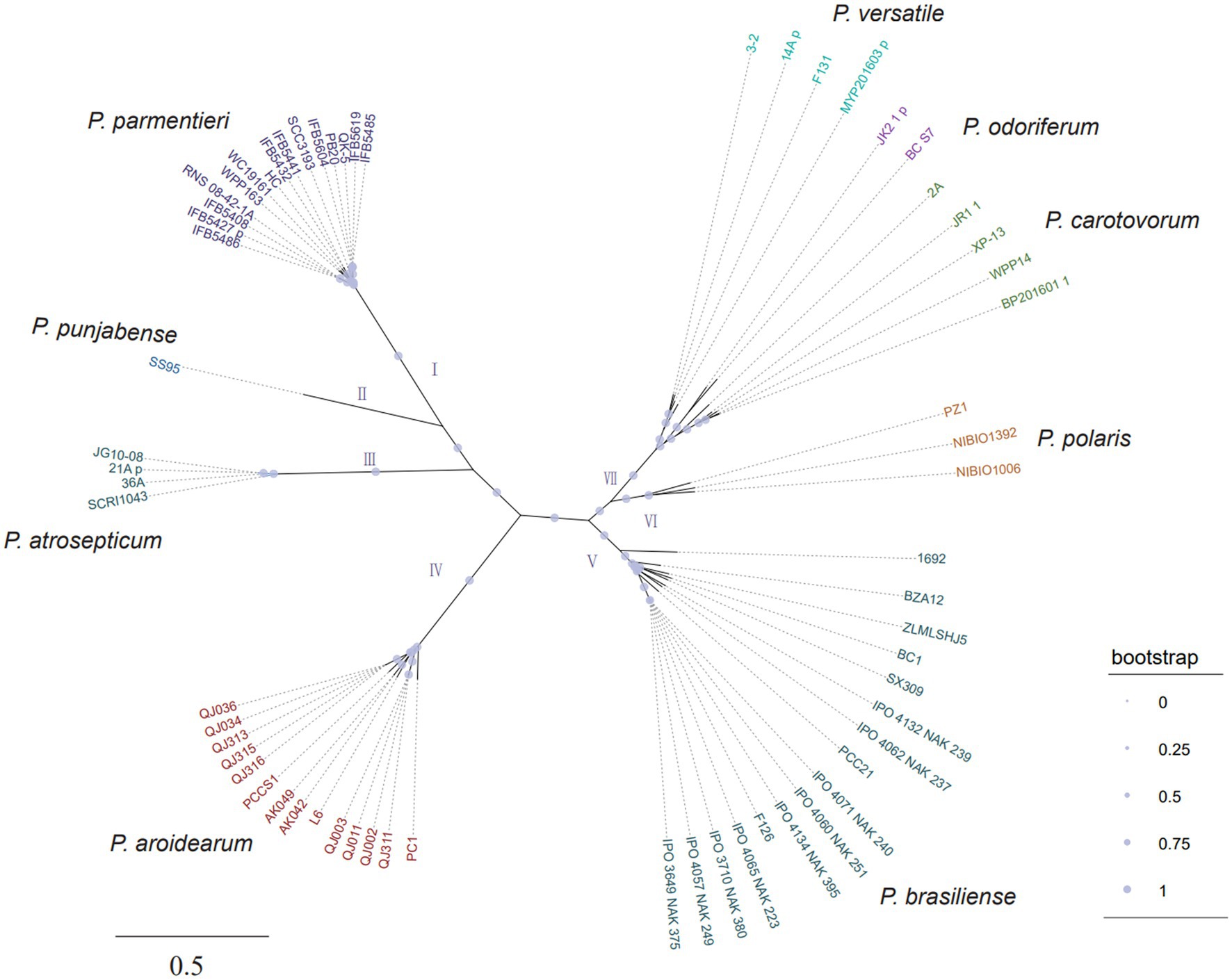
Figure 2. The unrooted parsimony tree of 64 Pectobacterium strains based on all SNPs. The consensus parsimony tree was constructed by kSNP3 and visualized by iTOL. The support values were calculated by FastTreeMP. Branch lengths are expressed in terms of changes per number of SNPs.
Overall genomic features of P. aroidearum strains, including genome size, GC (guanine-cytosine) content, number of protein-coding sequences (CDS), and number of RNA genes, were quantified (Table 1). The length of P. aroidearum genomes ranges from 4,865,541 bp (QJ315) to 5,057,072 bp (QJ003). GC content varies between 51.6% and 51.9%. The highest number of putative CDS was observed in AK042 (4,469), whereas the lowest number of CDS was found in QJ311 (4,277). In addition, the number of tRNA, rRNA and CRISPRs (Clustered Regularly Interspaced Short Palindromic Repeats) is the same among our 11 genome assemblies. We found no evidence of plasmids in P. aroidearum.
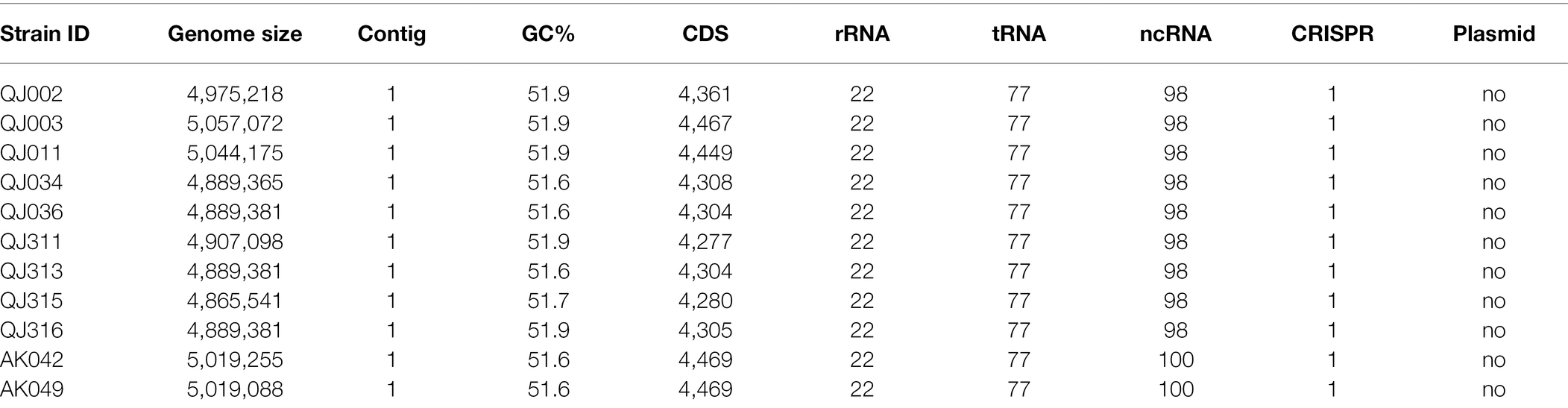
Table 1. General genome characteristics of sequenced Pectobacterium aroidearum strains.
For synteny analysis, the program Mauve was used to perform whole-genome multiple alignment to detect genomic rearrangement. Interestingly, a couple of large-scale chromosomal inversions were observed among these 11 P. aroidearum isolates (Figure 3A). These strains could be clustered into three groups based on genomic rearrangement, which is further supported by the phylogenetic analysis using genome-wide SNPs (Figure 3A) and BLAST comparisons of the whole genome (Supplementary Figure 4). Except for strain QJ311, the grouping of the other strains is consistent with their geographical distribution (Supplementary Table 1). To further confirm the above results, a total of three strains (QJ002, QJ036 and AK042) selected from each group were used for pairwise genome alignment. As expected, dot plots also revealed the genomic inversions between QJ036 and either QJ002 or AK042 (Figure 3B).
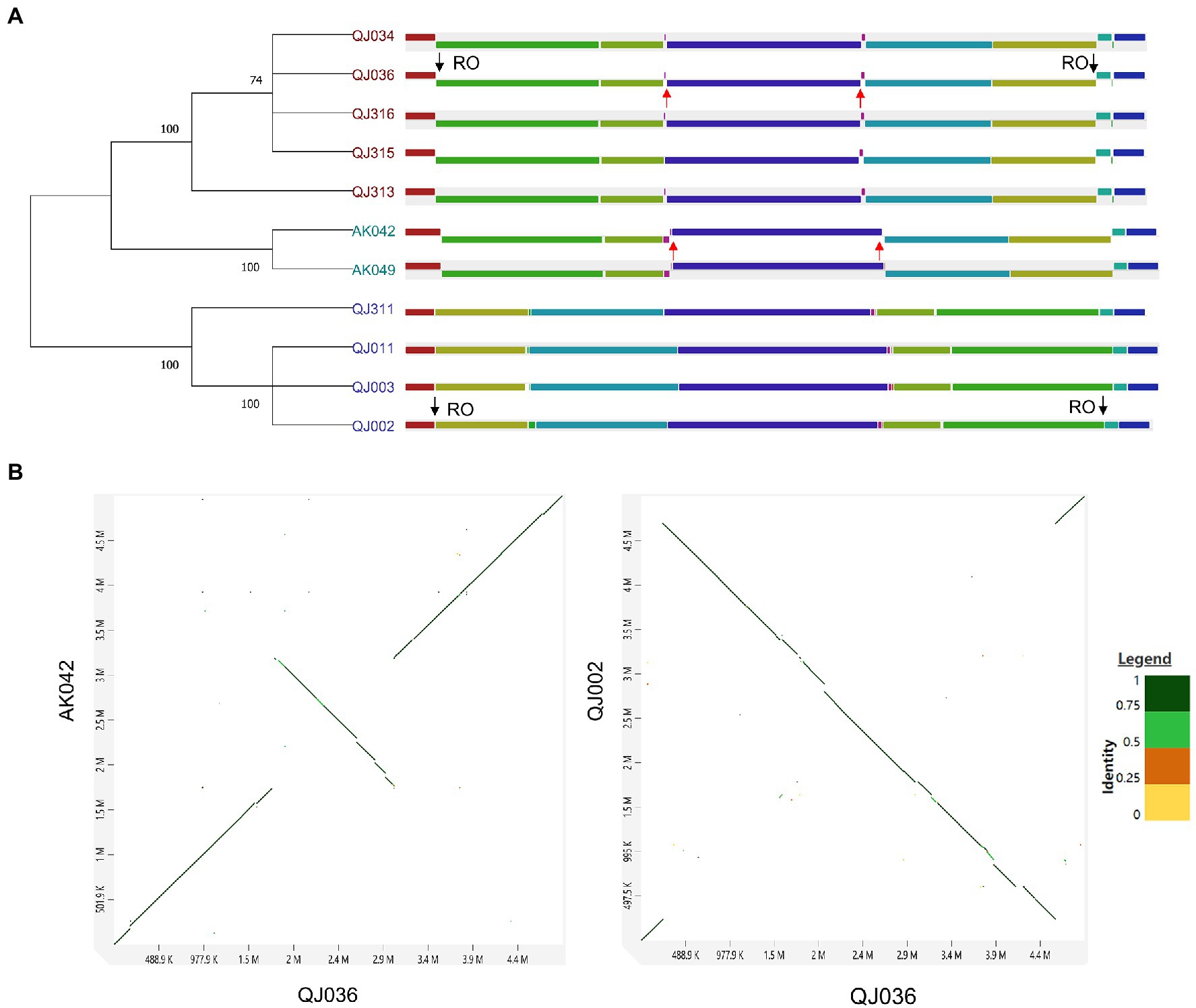
Figure 3. Synteny analysis of Pectobacterium aroidearum strains. (A) Genome alignment using the progressiveMauve algorithm. Each locally collinear block (LCB) is assigned a unique color. The black arrows indicate the inversion region between QJ036 and QJ002 while the red arrows indicate the inversion region between QJ036 and AK042. RO stands for ribosome operon. SNPs-based parsimony tree of 11 Pectobacterium strains was constructed by kSNP3 and the condensed tree with a root at midpoint was computed by MEGA11 with the default cutoff value (≥50). (B) Pairwise genome alignment for the selected strains is shown in the dot plots generated by an online tool D-GENIES.
The major mechanisms underlying chromosomal rearrangements are recombinational exchanges between homologous sequences such as ribosomal operons and mobile genetic elements (MEGs) including transposons, insertion sequence (IS) elements, and prophages (Raeside et al., 2014). The origins of the chromosomal inversion were investigated by examining the sequences bordering each inversion. The inversion that occurred between QJ036 and QJ002 resulted from homologous recombination of ribosome operons which are located exactly at the border regions of this inversion (Figure 3A). However, the reason for the inversion between QJ036 and AK042 is unknown.
The pangenomes of plant pathogens are often associated with species- or strain-specific virulence, host specificity or adaptive potential, or evolutionary history (Amir et al., 2020). To investigate the genomic plasticity of genus Pectobacterium, the 64 complete genomes from 9 Pectobacterium spp. including our 11 assemblies were used to identify the core- and pan-genomes. The number of core genes was 2,228, which only accounts for 11.31% of the pangenome, which contained a total of 19,698 genes (Supplementary Figure 5A). As more genomes were added, the pangenome trend showed a gradual expansion, implying an open pangenome of Pectobacterium strains (Supplementary Figure 5B).
Similarly, a pangenome of 6,630 genes was identified from the 14 P. aroidearum genomes, with a core genome of 3,575 genes (53.92% of the pangenome; Figure 4A). The number of accessory genes of each strain varied from 701 (PC1) to 898 (QJ003; Figure 4B). The pangenome fitted cumulative curve showed that with the addition of more genomes, the number of core genes remained relatively stable while the number of total genes continued to increase, indicating that the pangenome is still open (Figure 4C). In addition, a maximum likelihood (ML) tree based on core gene alignment is congruent with the matrix representing the presence and absence of core and accessory genes (Figure 4D).
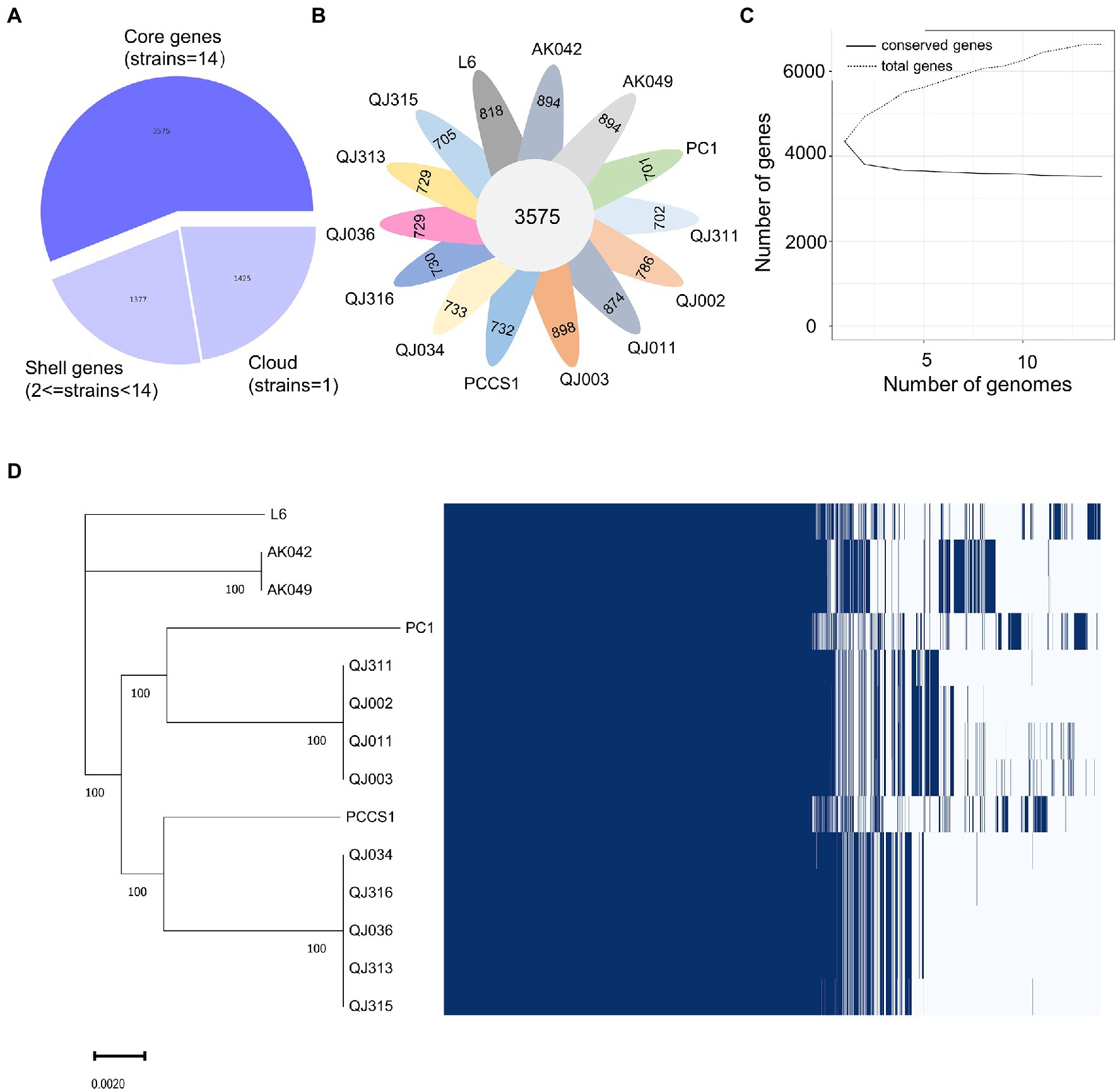
Figure 4. Pangenome analysis of 14 Pectobacterium aroidearum strains conducted with the Roary pipeline. (A) A pie chart displays the proportion of genes in the core, shell, and cloud of the pangenome. (B) A follower plot shows the number of core genes and accessory genes in each P. aroidearum strain. (C) The size of the core genome and pangenome with the increasing numbers of P. aroidearum genomes. (D) Gene presence–absence matrix shows the distribution of genes in each genome. The maximum likelihood (ML) tree is based on all the core gene alignment of the 14 P. aroidearum genomes. Each column represents an orthologous gene family. Dark blue blocks and light gray indicate the presence or absence of a gene, respectively.
Functional enrichment analysis was performed to uncover the biological roles of core and accessory genes using the QJ036 genome as an example. We classified all genes in the QJ036 genome into three groups including genus-core genes (2228), species-core genes (1347) and accessory genes (729). Enrichment analysis based on the COG (Clusters of Orthologous Groups of proteins) database showed that genus-core genes are overrepresented in categories associated with essential life activity such as energy production and conversion, the transport and metabolism of amino acid, nucleotide and lipid, and translation, ribosomal structure and biogenesis (Table 2). The species-core genes are enriched for virulence-associated categories, including the transport and metabolism of carbohydrate and inorganic ion, cell motility, and intracellular trafficking and secretion (Table 2). Interestingly, the category of replication, recombination and repair is the only enriched COG for the accessory genes (Table 2). Further investigation listed all genes belonging to this category and found that a majority of these genes were annotated as recombinase/integrase and mobile genetic elements such as transposons, insertion sequence (IS) elements (Supplementary Table 5).
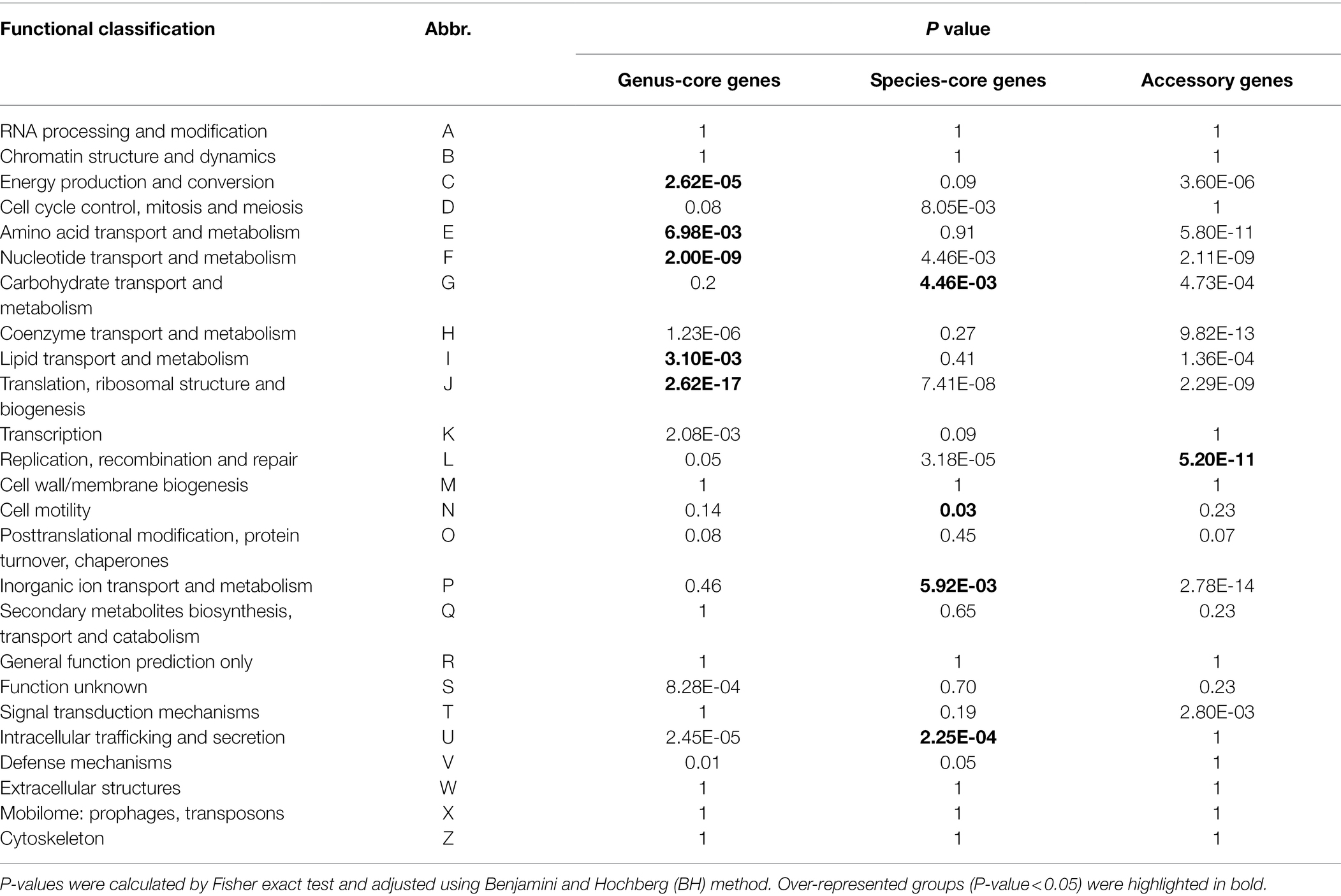
Table 2. Functional enrichment analyses of genes in Pectobacterium aroidearum QJ036.
Bacterial secretion systems and plant cell wall degrading enzymes (PCWDEs) play a key role in the interaction between soft rot bacteria and host plants. We mined the 14 P. aroidearum genomes to identify and compare these determinants of pathogenicity. All six secretion systems (types I–VI) were detected using the program TXSScan. Type I secretion system (T1SS), type II secretion system (T2SS), type III secretion system (T3SS) and type VI secretion system (T6SS) were conserved in all tested P. aroidearum strains (Figure 5A). However, type IV secretion system (T4SS), a versatile secretion system that is involved in protein translation, bacterial conjugation and DNA uptake/release, is not detected in several P. aroidearum genomes (Figure 5A). Notably, P. aroidearum L6 has two copies of each T4SS subtypes (typeG and typeT) that are classified according to different mating pair formation complexes (MPF; Guglielmini et al., 2013). For T6SS, although each P. aroidearum strain has a conserved T6SS cluster, the number of predicted effectors and immunity proteins varies considerably (Supplementary Table 6).
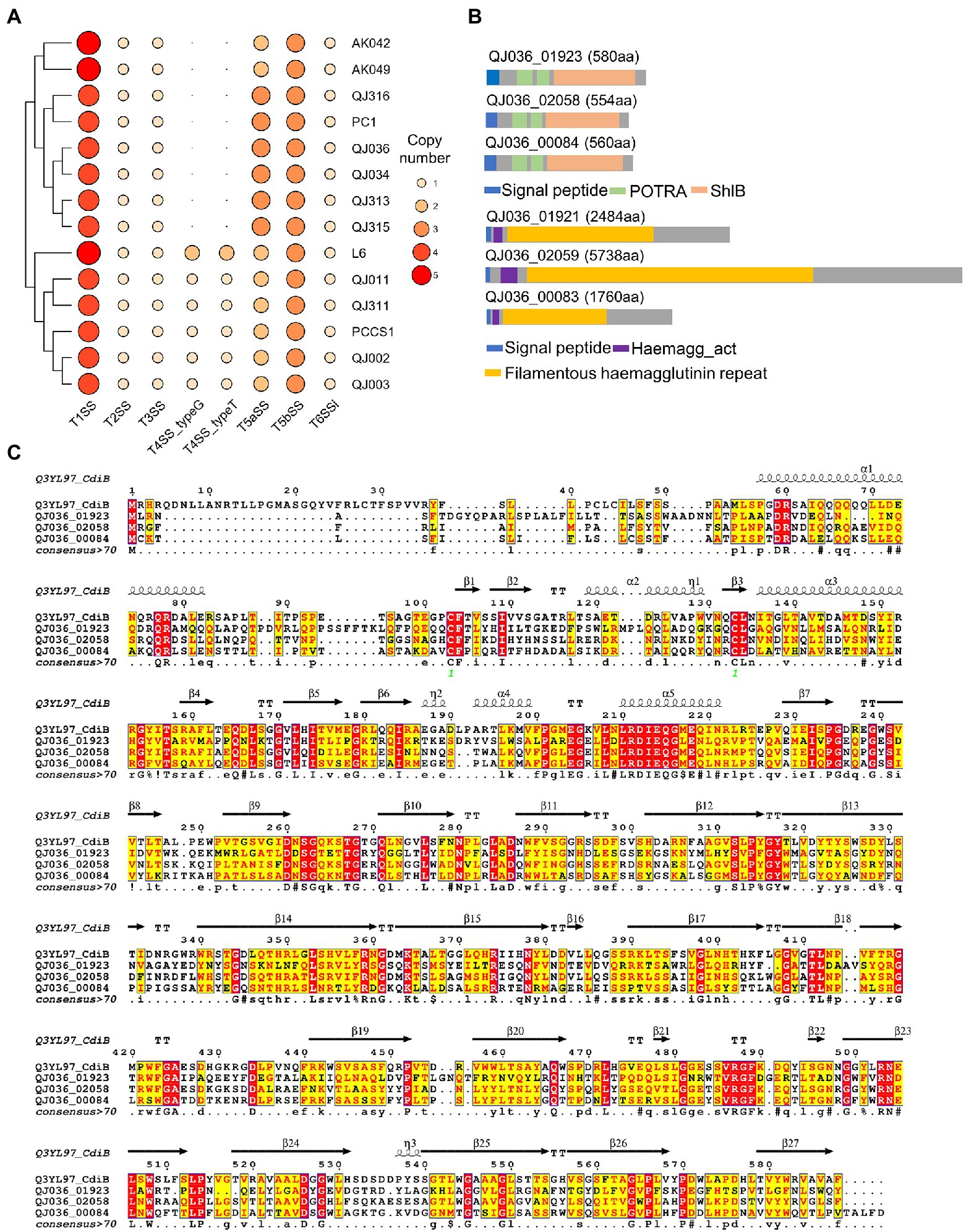
Figure 5. Prediction of bacterial secretion systems in Pectobacterium aroidearum strains. (A) The heatmap shows the distribution of bacterial secretion systems in P. aroidearum strains. (B) Predicted domains in three putative T5bSS proteins of P. aroidearum QJ036 using the online software InterProScan. (C) Sequence similarities and secondary structure elements from aligned sequences of putative T5bSS proteins and CdiB were rendered by the webserver ESPript 3. The secondary structure depiction is based on CdiB (PDB: 6WIM).
Although the presence of type V secretion system (T5SS) in Pectobacterium spp. is still inconclusive (Li et al., 2018; Van Gijsegem et al., 2021), two subtypes of T5SS were identified by TXSScan in our assemblies. To further confirm the prediction, sequence analysis was performed using putative T5SS proteins from the QJ036 strain. In two-partner secretion (TPS or type 5b), the passenger domain (TpsA protein) and the β-domain (TpsB protein) are encoded by two separate genes which are frequently, but not always, encoded in an operon (Wells and Henderson, 2013). Domain analysis based on the InterPro database showed that each putative T5bSS protein (QJ036_01923, QJ036_02058 and QJ036_00084) identified by TXSScan contains a signal sequence, two polypeptide-transport-associated (POTRA) domains and a shlB domain (β-domain; Figure 5B). In addition, these proteins displayed high similarity in primary and secondary structure with the known TpsB protein CdiB of Escherichia coli (Figure 5C). These results strongly indicate that these three proteins are members of TpsB family. To find the TpsA proteins in P. aroidearum, TpsA protein fhaB (Uniprot: P12255) of Bordetella pertussis was used as a query to search against protein database of QJ036 using BLASTP. Domain analysis of the three BLASTP hits (QJ036_01921, QJ036_02059, and QJ036_00083) shows that each contains a signal sequence, a haemagg_act domain and a filamentous hemagglutinin repeat region (Figure 5B). As expected for a type 5b, these three TpsB proteins are very close to their corresponding TpsA proteins according to the locus tags. These results therefore provide evidence for the existence of three copies of T5bSS in P. aroidearum QJ036. However, the presence of T5aSS is not well supported by our sequence analysis results.
PCWDEs seem to be very conserved in the P. aroidearum strains examined in this study. In total, 21 PCWDEs genes were found in each strain including two cellulase genes (celV, celS), one oligogalacturonate lyase gene (ogl), eight pectate lyase genes (pel1, pel2, pel3, pelX, pelY, pelW, pelE, pelL), two pectin acetylesterase genes (paeX, paeY), one pectin lyase gene (pnl), two pectinesterase genes (pemA, pemB), three polygalacturonase genes (pehX, pehK, pehN, pehA), two rhamnogalacturonan lyase genes (rhiE, rhiN) and one protease gene (prtC; Supplementary Table 7).
Genomic islands (GIs) are clusters of consecutive genes likely acquired via horizontal gene transfer (HGT), which may facilitate microbial adaptation by disproportionately encoding factors involved in virulence or antimicrobial resistance (Bertelli et al., 2019). Here, GIs of P. aroidearum isolates were identified using the online webserver IslandViewer 4 (Bertelli et al., 2017). The location of GIs in each genome was visualized, which revealed that P. aroidearum isolates with similar GIs distribution patterns tended to be from the same sampling site (Supplementary Figure 6). In addition, the number of GI genes in each genome ranged from 454 (PC1) to 866 (AK042), accounting for about 10%–20% of the total genes of a genome (Figure 6A). However, none of these genes were annotated as virulence/resistance factors, and the majority of these genes were annotated as hypothetical proteins. To further explore the gene content of GIs, we collected all GI-derived genes with a gene ID and calculated the occurrence number of each gene (Supplementary Table 8). The proteins encoded by the top 11 genes include prophage integrase (IntA, IntS), tyrosine recombinase XerC, transporter (YflS, CitN), major exported protein HcpA, DNA-binding transcriptional repressor YiaJ, Acetyl-CoA:oxalate CoA-transferase YfdE, ribose import permease RbsC, Tyrocidine synthase 3 TycC and DNA topoisomerase 3 TopB (Figure 6B).
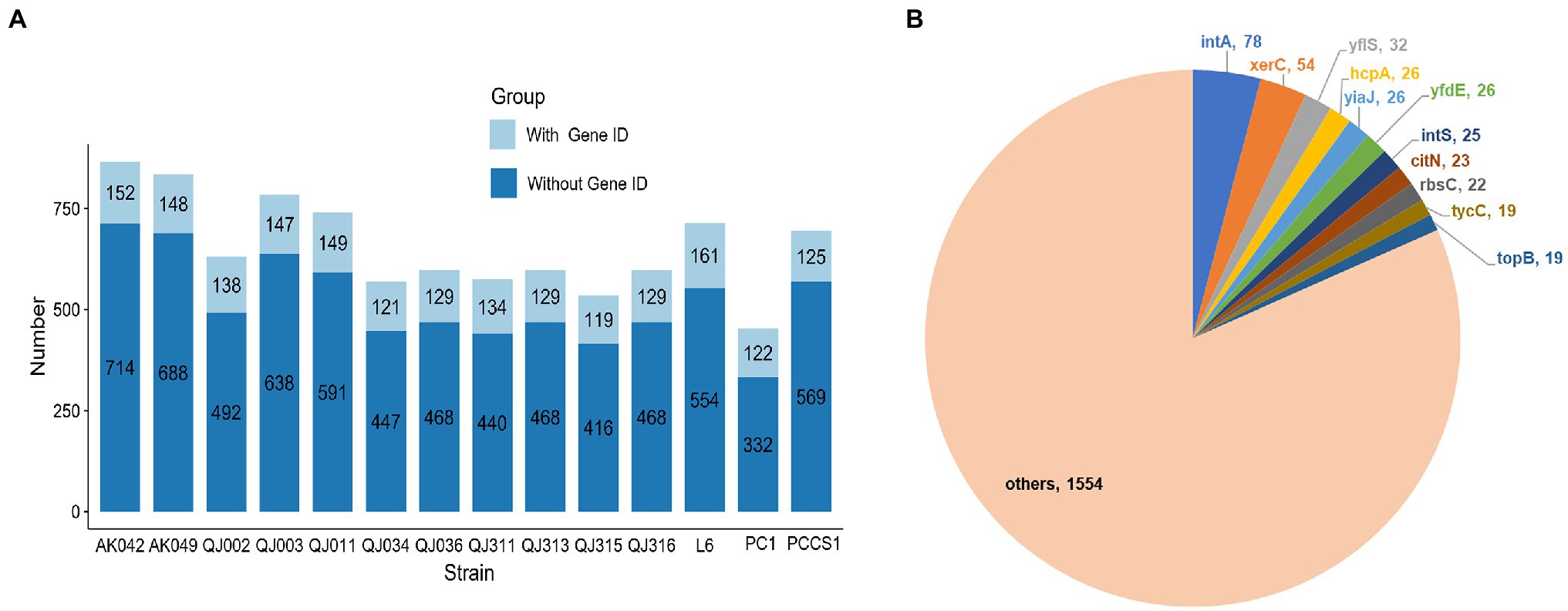
Figure 6. Prediction of genomic islands (GIs) in 14 Pectobacterium aroidearum strains. (A) The bar plot shows the number of genes from GIs in each P. aroidearum strain. (B) The pie plot shows the frequency of top 11 genes. The values represent the total occurrence number of each gene in 14 P. aroidearum strains.
The identification and classification of the causal agent of konjac soft rot is a prerequisite for effective management of this disease. A nationwide survey in China indicated that konjac soft rot is caused by P. carotovora subsp. carotovora and P. chrysanthemi, with P. chrysanthemi acting as the major pathogen (Wu et al., 2015). However, our study isolated 11 pathogenic strains from three sampling sites of two provinces (Yunnan and Shaanxi) in China, and proved that all these soft rot pathogens belong to P. aroidearum, although it remains possible that konjac soft rot is caused by more than one bacterial pathogen. In addition, considerable variation exists in regard to host specificity for Soft Rot Pectobacteriaceae (SRP; Khadka et al., 2020). Some SRP have wide host ranges, while others have only one or a few plant species (Toth et al., 2021a). P. aroidearum can infect multiple monocotyledonous and dicotyledonous plant species, especially from the families Araceae and Solanaceae such as Zantedeschia aethiopica, S. podophyllum, A. konjac, C. pepo and Solanum tuberosum (Wei et al., 2020; Xu et al., 2020; Toth et al., 2021a). Our in vitro pathogenicity tests were the first to show that P. aroidearum also caused typical rot symptoms on sweet potato, jícama, yacón and taro, suggesting a broader host range of P. aroidearum. Further research comparing P. aroidearum to other Pectobacterium species with few host plants will help reveal how P. aroidearum can infect so many plants.
The development of next-generation sequencing technologies (NGS) has made the pangenome a new tool for analyzing pathogenic bacteria (Rouli et al., 2015). To date, there are only a couple of pangenome studies for individual Pectobacterium species (Zoledowska et al., 2018; Lu et al., 2021). The core genome size (2,228 genes) of Pectobacterium spp. is much smaller than that of P. aroidearum, and the reduced genes are enriched in virulence-associated COG categories (e.g., the transport and metabolism of carbohydrate and inorganic ion, cell motility, and intracellular trafficking and secretion), which indicates some variation in virulence-related genes among Pectobacterium species. As more genomes of Pectobacterium species become available, it will be important to directly compare the core genome of each species. Interestingly, our comparative genomic analyses strongly suggest that strains including P. carotovorum subsp. carotovorum PC1, P. carotovorum subsp. carotovorum PCCS1 and P. carotovorum subsp. carotovorum PCC21 are misnamed. Instead, P. carotovorum subsp. carotovorum PC1 and P. carotovorum subsp. carotovorum PCCS1 should be classified as P. aroidearum.
Bacterial genomes are considerably stable in the short term but are plastic from an evolutionary perspective, which creates a delicate balance between genome integrity and instability that is essential for survival and adaptation (Darmon and Leach, 2014). In fact, genomic rearrangements are not only detected across species, but also present in members of the same species for some organisms (Darling et al., 2008). In a long-term evolution experiment using E. coli, a total of 110 rearrangement events including 19 inversions were detected, and about 70% of rearrangements were associated with recombination between insertion sequence (IS) elements (Raeside et al., 2014). The importance of IS elements is also highlighted by another study which showed that large inversions were only detected in Bordetella species with genomes harboring multicopy IS elements (Weigand et al., 2019). Interestingly, the pangenome analyses and functional enrichment revealed that many accessory genes of P. aroidearum QJ036 encoded mobile genetic elements, which are likely associated with the dynamics of genome rearrangement. It is known that mobile genetic elements can provide novel genotypes for evolution by facilitating genomic rearrangements and the capture of new genes for bacterial pathogens (Frost et al., 2005; Jackson et al., 2011). Although the effect of these observed chromosomal inversions on fitness and virulence of P. aroidearum appears to be minor, genome architecture should be taken into consideration when comparing phylogenetically close bacterial pathogens with virulence variation.
The primary virulence determinant of SRP is a large arsenal of plant cell wall-degrading enzymes (Davidsson et al., 2013). Although the number of PCWDE-encoding genes varies slightly across studies, including ours, for the most part these genes seem to be highly conserved (Li et al., 2018, 2019; Arizala and Arif, 2019). Unlike other secreted substrates, T5SS substrates secrete themselves by forming a channel in outer membrane, through which either the remainder of the protein or a separate protein is transported (Green and Mecsas, 2016). For two-partner secretion (T5bSS), the TpsA serves as the secreted protein, which plays an important role in bacterial virulence in Pseudomonas fluorescens (Sun et al., 2016) and B. pertussis (Melvin et al., 2014).
Here, our sequence analyses provide strong evidence of the existence of three copies of T5bSS in P. aroidearum. Further research is needed to explore whether these different TpsA proteins, exhibiting substantial size variation, contribute to bacterial pathogenicity. Although it is known that the primary roles of T6SS are associated with both host manipulation and interbacterial competition, how exactly T6SS contributes to virulence is still elusive in Pectobacterium (Bernal et al., 2018). In addition, our study suggests that the number of T6SS effectors and immunity proteins varies among P. aroidearum strains.
Bacterial evolution is dominated by the relative rates of two processes: mutations from DNA replication errors and horizontal gene transfer (HGT; Sheppard et al., 2018). The development of genome sequencing has promoted the realization that HGT is a major evolutionary force reshaping bacterial genomes and therefore influencing bacterial adaptation (Daubin and Szöllősi, 2016). For example, horizontally acquired quorum-sensing regulators expand the host adaptation repertoire in the phytopathogen P. brasiliense (Bellieny-Rabelo et al., 2020). Here, IslandViewer 4 was used to find genomic islands including pathogenicity islands (PAIs) in P. aroidearum. Although 10%–20% of genes appeared in genomic islands, no gene was annotated as a virulence factor, indicating that key pathogenicity determinants of P. aroidearum are not acquired via HGT. For the genes with high frequency such as intA, xerC, yflS, citN, further studies are needed to address why these genes are often acquired by HGT as well as their function in P. aroidearum.
The raw reads and complete genome assemblies were deposited in the NCBI SRA database and NCBI WGS database, respectively, which is under the bio-project accession number PRJNA794971.
YZ and HC designed the experiments and wrote the manuscript. YZ, HC, LY, and FH performed sample collection, experiments, and bioinformatic analysis. YG and LT revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities (202001BA070001-231) and the National Natural Science Foundation of China (31860057 and 31901468).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.868709/full#supplementary-material
Supplementary Figure 1 | Pathogenicity tests of other bacterial isolates on konjac slice tuber.
Supplementary Figure 2 | The formation of pits caused by other bacterial isolates on CVP medium.
Supplementary Figure 3 | BUSCO assessment results of the 11 P. aroidearum assemblies.
Supplementary Figure 4 | Genome comparison of the 11 bacterial isolates using P. aroidearum QJ002 as the reference by the BLAST Ring Image Generator (BRIG).
Supplementary Figure 5 | Pangenome analysis of 64 Pectobacterium strains using the Roary pipeline. (A) A pie chart displays the proportion of genes in the core, shell, and cloud of the pangenome. (B) The size of the core genome and pangenome with the increasing numbers of Pectobacterium genomes.
Supplementary Figure 6 | Linear visualization of predicted GIs in 14 P. aroidearum genomes with blocks colored according to the prediction method: IslandPick (green), IslandPath-DIMOB (blue) and SIGI-HMM (orange). Geographic location of these strains are shown on the right side of the figure.
Supplementary Table 1 | Basic information of collected isolates P. aroidearum.
Supplementary Table 2 | Basic information for 53 downloaded genomes of Pectobacterium spp.
Supplementary Table 3 | Basic statistics of short and long reads.
Supplementary Table 4 | Pairwise comparisons of isDDH and ANI values of Pectobacterium strains in clade IV.
Supplementary Table 5 | List of accessory genes that belongs to the category of replication, recombination and repair.
Supplementary Table 6 | Type VI secretion system (T6SS) prediction for P. aroidearum genomes using web server SecReT6 v3.
Supplementary Table 7 | Predicted plant cell wall-degrading enzymes (PCWDEs) in 14 P. aroidearum strains.
Supplementary Table 8 | The total numbers of each gene derived from GIs.
1. ^https://github.com/widdowquinn/pyani
2. ^http://ggdc.dsmz.de/ggdc.php#
3. ^https://www.ncbi.nlm.nih.gov/genome/browse/?report=5#!/overview/Pectobacterium
4. ^http://dgenies.toulouse.inra.fr/
Abby, S. S., Cury, J., Guglielmini, J., Néron, B., Touchon, M., and Rocha, E. P. C. (2016). Identification of protein secretion systems in bacterial genomes. Sci. Rep. 6:23080. doi: 10.1038/srep23080
Abby, S. S., Néron, B., Ménager, H., Touchon, M., and Rocha, E. P. C. (2014). MacSyFinder: A program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9. doi: 10.1371/journal.pone.0110726
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Amir, R., Sani, Q.-A., Maqsood, W., Munir, F., Fatima, N., Siddiqa, A., et al. (2020). “Pan-genomics of plant pathogens and its applications,” in Pan-Genomics: Applications, Challenges, and Future Prospects. eds. D. Barh, S. Soares, S. Tiwari, and V. Azevedo (San Diego, CA, USA: Elsevier), 121–145.
Arizala, D., and Arif, M. (2019). Genome-wide analyses revealed remarkable heterogeneity in pathogenicity determinants, antimicrobial compounds, and CRISPR-cas systems of complex phytopathogenic genus pectobacterium. Pathogens 8:247. doi: 10.3390/pathogens8040247
Behera, S. S., and Ray, R. C. (2016). Konjac glucomannan, a promising polysaccharide of Amorphophallus konjac K. Koch in health care. Int. J. Biol. Macromol. 92, 942–956. doi: 10.1016/j.ijbiomac.2016.07.098
Bellieny-Rabelo, D., Nkomo, N. P., Shyntum, D. Y., and Moleleki, L. N. (2020). Horizontally acquired quorum-sensing regulators recruited by the phop regulatory network expand the host adaptation repertoire in the phytopathogen Pectobacterium brasiliense. mSystems 5:e00650-19. doi: 10.1128/msystems.00650-19
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bernal, P., Llamas, M. A., and Filloux, A. (2018). Type VI secretion systems in plant-associated bacteria. Environ. Microbiol. 20, 1–15. doi: 10.1111/1462-2920.13956
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bertelli, C., Tilley, K. E., and Brinkman, F. S. L. (2019). Microbial genomic island discovery, visualization and analysis. Brief. Bioinform. 20, 1685–1698. doi: 10.1093/bib/bby042
Blum, M., Chang, H. Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A., et al. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. doi: 10.1093/nar/gkaa977
Cabanettes, F., and Klopp, C. (2018). D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6:e4958. doi: 10.7717/peerj.4958
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Charkowski, A. O. (2018). The changing face of bacterial soft-rot diseases. Annu. Rev. Phytopathol. 56, 269–288. doi: 10.1146/annurev-phyto-080417-045906
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Darling, A. E., Miklós, I., and Ragan, M. A. (2008). Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 4:e1000128. doi: 10.1371/journal.pgen.1000128
Darmon, E., and Leach, D. R. F. (2014). Bacterial genome instability. Microbiol. Mol. Biol. Rev. 78, 1–39. doi: 10.1128/mmbr.00035-13
Daubin, V., and Szöllősi, G. J. (2016). Horizontal gene transfer and the history of life. Cold Spring Harb. Perspect. Biol. 8:a018036. doi: 10.1101/cshperspect.a018036
Davidsson, P. R., Kariola, T., Niemi, O., and Tapio Palva, E. (2013). Pathogenicity of and plant immunity to soft rot pectobacteria. Front. Plant Sci. 4:191. doi: 10.3389/fpls.2013.00191
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J. M., et al. (2011). T-coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17. doi: 10.1093/nar/gkr245
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: An overview. Microbiol. Spectr. 4, 1–19. doi: 10.1128/microbiolspec.vmbf-0012-2015
Guglielmini, J., De La Cruz, F., and Rocha, E. P. C. (2013). Evolution of conjugation and type IV secretion systems. Mol. Biol. Evol. 30, 315–331. doi: 10.1093/molbev/mss221
Hélias, V., Hamon, P., Huchet, E., Wolf, J. V. D. D., and Andrivon, D. (2012). Two new effective semiselective crystal violet pectate media for isolation of Pectobacterium and Dickeya. Plant Pathol. 61, 339–345. doi: 10.1111/j.1365-3059.2011.02508.x
Jackson, R. W., Vinatzer, B., Arnold, D. L., Dorus, S., and Murillo, J. (2011). The influence of the accessory genome on bacterial pathogen evolution. Mob. Genet. Elements 1, 55–65. doi: 10.4161/mge.1.1.16432
Khadka, N., Joshi, J. R., Reznik, N., Chriker, N., Nudel, A., Zelinger, E., et al. (2020). Host specificity and differential pathogenicity of pectobacterium strains from dicot and monocot hosts. Microorganisms 8, 1–22. doi: 10.3390/microorganisms8101479
Land, M., Hauser, L., Jun, S. R., Nookaew, I., Leuze, M. R., Ahn, T. H., et al. (2015). Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 15, 141–161. doi: 10.1007/s10142-015-0433-4
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, J., Yao, Y., Xu, H. H., Hao, L., Deng, Z., Rajakumar, K., et al. (2015). SecReT6: a web-based resource for type VI secretion systems found in bacteria. Environ. Microbiol. 17, 2196–2202. doi: 10.1111/1462-2920.12794
Li, L., Yuan, L., Shi, Y., Xie, X., Chai, A., Wang, Q., et al. (2019). Comparative genomic analysis of pectobacterium carotovorum subsp. Brasiliense SX309 provides novel insights into its genetic and phenotypic features. BMC Genomics 20, 1–17. doi: 10.1186/s12864-019-5831-x
Li, X., Ma, Y., Liang, S., Tian, Y., Yin, S., Xie, S., et al. (2018). Comparative genomics of 84 Pectobacterium genomes reveals the variations related to a pathogenic lifestyle. BMC Genomics 19:889. doi: 10.1186/s12864-018-5269-6
Liang, Z., Liu, H., Xu, Z., and Zhang, L.-H. (2022). First report of Pectobacterium aroidearum causing soft rot in olecranon honey peach (Prunus persica) in China. Plant Dis. doi: 10.1094/PDIS-10-21-2238-PDN [Epub ahead of print].
Lu, Q., Yan, F., Liu, Y., Li, Q., Yang, M., and Liu, P. (2021). Comparative genomic analyses reveal functional insights Into key determinants of the pathogenesis of Pectobacterium actinidiae in kiwifruit. Phytopathology 111, 789–798. doi: 10.1094/phyto-07-20-0287-r
Maiden, M. C. J., Van Rensburg, M. J. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2021). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Melvin, J. A., Scheller, E. V., Miller, J. F., and Cotter, P. A. (2014). Bordetella pertussis pathogenesis: current and future challenges. Nat. Rev. Microbiol. 12, 274–288. doi: 10.1038/nrmicro3235
Moraes, A. J. G., Souza, E. B., Mariano, R. L. R., Silva, A. M. F., Lima, N. B., Peixoto, A. R., et al. (2017). First report of Pectobacterium aroidearum and Pectobacterium carotovorum subsp. brasiliensis causing soft rot of Cucurbita pepo in Brazil. Plant Dis. 101:379. doi: 10.1094/PDIS-08-16-1168-PDN
Nabhan, S., de Boer, S. H., Maiss, E., and Wydra, K. (2013). Pectobacterium aroidearum sp. nov., a soft rot pathogen with preference for monocotyledonous plants. Int. J. Syst. Evol. Microbiol. 63, 2520–2525. doi: 10.1099/ijs.0.046011-0
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parte, A. C., Carbasse, J. S., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/c5ay02550h
Raeside, C., Gaffé, J., Deatherage, D. E., Tenaillon, O., Briska, A. M., Ptashkin, R. N., et al. (2014). Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. mBio 5, e01377–e01314. doi: 10.1128/mBio.01377-14
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microb. New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sheppard, S. K., Guttman, D. S., and Fitzgerald, J. R. (2018). Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. doi: 10.1038/s41576-018-0032-z
Sun, Y. Y., Chi, H., and Sun, L. (2016). Pseudomonas fluorescens filamentous hemagglutinin, an iron-regulated protein, is an important virulence factor that modulates bacterial pathogenicity. Front. Microbiol. 7:1320. doi: 10.3389/fmicb.2016.01320
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Toth, I. K., Barny, M., Brurberg, M. B., Condemine, G., Czajkowski, R., Elphinstone, J. G., et al. (2021a). “Pectobacterium and Dickeya: environment to disease development,” in Plant Diseases Caused by Dickeya and Pectobacterium Species. eds. F. Van Gijsegem, J. M. van der Wolf, and I. K. Toth (Cham, Switzerland: Springer), 39–84.
Toth, I. K., Barny, M., Czajkowski, R., Elphinstone, J. G., Li, X. S., Pédron, J., et al. (2021b). “Pectobacterium and Dickeya: taxonomy and evolution,” in Plant Diseases Caused by Dickeya and Pectobacterium Species. eds. F. Van Gijsegem, J. M. van der Wolf, and I. K. Toth (Cham, Switzerland: Springer), 13–37.
Toth, I. K., Pritchard, L., and Birch, P. R. J. (2006). Comparative genomics reveals what makes an enterobacterial plant pathogen. Annu. Rev. Phytopathol. 44, 305–336. doi: 10.1146/annurev.phyto.44.070505.143444
Van Gijsegem, F., Hugouvieux-Cotte-Pattat, N., Kraepiel, Y., Lojkowska, E., Moleleki, L. N., Gorshkov, V., et al. (2021). “Molecular interactions of Pectobacterium and Dickeya with plants,” in Plant Diseases Caused by Dickeya and Pectobacterium Species. eds. F. VanGijsegem, J. M. van der Wolf, and I. K. Toth (Cham: Springer International Publishing), 85–147.
Vaser, R., and Šikić, M. (2021). Time- and memory-efficient genome assembly with raven. Nat. Comput. Sci. 1, 332–336. doi: 10.1038/s43588-021-00073-4
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wei, H., Yang, M., Pei, W., Wei, W., Huang, F., Liu, J., et al. (2020). First report of Pectobacterium aroidearum causing soft rot of Amorphophallus konjac in China. Plant Dis. 104:969. doi: 10.1094/PDIS-09-19-1960-PDN
Weigand, M. R., Peng, Y., Batra, D., Burroughs, M., Davis, J. K., Knipe, K., et al. (2019). Conserved patterns of symmetric inversion in the genome evolution of bordetella respiratory pathogens. mSystems 4:e00702-19. doi: 10.1128/msystems.00702-19
Wells, T. J., and Henderson, I. R. (2013). “Type 1 and 5 secretion systems and associated toxins,” in Escherichia coli. ed. M S. Donnenberg (San Diego, CA, USA: Academic Press), 499–532.
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genomics 3, e000132–e000137. doi: 10.1099/mgen.0.000132
Wu, J., Diao, Y., Gu, Y., and Hu, Z. (2011). Molecular detection of Pectobacterium species causing soft rot of Amorphophallus konjac. World J. Microbiol. Biotechnol. 27, 613–618. doi: 10.1007/s11274-010-0496-2
Wu, J., Yang, C., Jiao, Z., Hu, Z., Ding, Z., and Qiu, Z. (2015). Genetic relationships of soft rot Bacteria isolated from Konjac in China by amplified fragment length polymorphism (AFLP) and 16S rDNA gene sequences. Agric. Sci. 6, 717–723. doi: 10.4236/as.2015.67069
Xie, H., Li, X. Y., Ma, Y. L., and Tian, Y. (2020). First report of Pectobacterium aroidearum causing soft rot of Chinese cabbage in China. Plant Dis. 102:674. doi: 10.1094/PDIS-07-17-1059-PDN
Xie, H., Li, X. Y., Ma, Y. L., and Tian, Y. (2018). First report of Pectobacterium aroidearum causing soft rot of Chinese cabbage in China. Plant Dis. 102:674. doi: 10.1094/PDIS-07-17-1059-PDN
Xu, P. D., Wei, D. D., Li, Z. P., Qin, C., Li, X., Lin, C. H., et al. (2020). First report of bacterial soft rot on Syngonium podophyllum caused by Pectobacterium aroidearum in China. Plant Dis. 104:2720. doi: 10.1094/PDIS-03-20-0552-PDN
Zhang, Y., Fan, Q., and Loria, R. (2016). A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole-genome sequencing data. Syst. Appl. Microbiol. 39, 252–259. doi: 10.1016/j.syapm.2016.04.001
Zhu, F. (2018). Modifications of konjac glucomannan for diverse applications. Food Chem. 256, 419–426. doi: 10.1016/j.foodchem.2018.02.151
Keywords: bacterial soft rot, Pectobacterium aroidearum, comparative genomics, genomic rearrangement, Amorphophallus konjac
Citation: Zhang Y, Chu H, Yu L, He F, Gao Y and Tang L (2022) Analysis of the Taxonomy, Synteny, and Virulence Factors for Soft Rot Pathogen Pectobacterium aroidearum in Amorphophallus konjac Using Comparative Genomics. Front. Microbiol. 13:868709. doi: 10.3389/fmicb.2022.868709
Edited by:
Jesús Navas-Castillo, La Mayora Experimental Station (CSIC), SpainReviewed by:
Margarita Gomila, University of the Balearic Islands, SpainCopyright © 2022 Zhang, Chu, Yu, He, Gao and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizhou Tang, YmlvbG9neXRhbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.