 Pynhunlang Kharnaior
Pynhunlang Kharnaior Jyoti Prakash Tamang
Jyoti Prakash Tamang- Department of Microbiology, School of Life Sciences, Sikkim University, Gangtok, India
Kinema is a popular sticky fermented soybean food of the Eastern Himalayan regions of North East India, east Nepal, and south Bhutan. We hypothesized that some dominant bacteria in kinema may contribute to the formation of targeted and non-targeted metabolites for health benefits; hence, we studied the microbiome–metabolite mining of kinema. A total of 1,394,094,912 bp with an average of 464,698,304 ± 120,720,392 bp was generated from kinema metagenome, which resulted in the identification of 47 phyla, 331 families, 709 genera, and 1,560 species. Bacteria (97.78%) were the most abundant domain with the remaining domains of viruses, eukaryote, and archaea. Firmicutes (93.36%) was the most abundant phylum with 280 species of Bacillus, among which Bacillus subtilis was the most dominant species in kinema followed by B. glycinifermentans, B. cereus, B. licheniformis, B. thermoamylovorans, B. coagulans, B. circulans, B. paralicheniformis, and Brevibacillus borstelensis. Predictive metabolic pathways revealed the abundance of genes associated with metabolism (60.66%), resulting in 216 sub-pathways. A total of 361 metabolites were identified by metabolomic analysis (liquid chromatography-mass spectrophotometry, LC-MS). The presence of metabolites, such as chrysin, swainsonine, and 3-hydroxy-L-kynurenine (anticancer activity) and benzimidazole (antimicrobial, anticancer, and anti-HIV activities), and compounds with immunomodulatory effects in kinema supports its therapeutic potential. The correlation between the abundant species of Bacillus and primary and secondary metabolites was constructed with a bivariate result. This study proves that Bacillus spp. contribute to the formation of many targeted and untargeted metabolites in kinema for health-promoting benefits.
Introduction
Fermented soybean products are one of the oldest food items of the Oriental Asians (Shurtleff and Aoyagi, 2017), which are either fungal-fermented products used as condiments and taste makers, such as shoyu of Japan, doenjang of Korea, and douchi of China, and also eaten as foods such as miso of Japan, tempe of Indonesia, and sufu/furu of China (Kadar et al., 2020; Takeshita, 2020), or bacteria-fermented foods such as natto, kinema, cheonggukjang, thua-nao, etc. (Tamang et al., 2016, 2020; Kwon et al., 2019). Historically and anthropologically, spontaneously fermented sticky soybean foods with umami flavor (Hartley et al., 2019) are confined to few Asian countries, namely, Japan (natto), Korea (cheonggukjang), Laos (sieng), northern Thailand (thua nao), Myanmar (pe poke), eastern Nepal and southern Bhutan (kinema), and North East India (kinema, hawaijar, bekang, tungrymbai, peha, peron-namsing, peruyaan, and aakhonii) (Kwon et al., 2019; Tamang et al., 2021a). Sticky fermented soybean foods are still produced traditionally by natural fermentation (Tamang, 2015), except Japanese natto, which is now produced by the commercial starter of Bacillus subtilis variety natto strain (Ju et al., 2019). Colossal microbial community structures with inter–intra species diversity have been observed in some fermented soybean foods of Asia (Kharnaior and Tamang, 2021; Ren et al., 2021), which are also the major sources of biological active compounds (Cao et al., 2019).
Shotgun metagenome sequence analysis is one of the most advance sequence-based taxonomic tools, which profiles the microbial domains up to species level (Durazzi et al., 2021), including culturable and unculturable bacteria, yeasts, fungi, viruses, and archaea in food samples (Leech et al., 2020). Metabolomics approach, based on liquid chromatography-mass spectrophotometry (LC-MS)-based chromatographic analysis, has been extensively applied in fermented soybean foods (Liu et al., 2021; Song et al., 2021) to profile targeted and untargeted metabolites (Eudy et al., 2020) with multiple applications in nutrition (González-Peña and Brennan, 2019). Among the untargeted metabolites, immunomodulators (Cambeiro-Pérez et al., 2018) are considered as the vital biomolecules for health-promoting benefits (Shahbazi et al., 2021). Integrated metagenomics–metabolomics approaches have been reported in few fermented foods such as Chinese pu-erh tea (Zhao et al., 2019), kombucha (Villarreal-Soto et al., 2020), kefir (Blasche et al., 2021), and doenjang and ganjang of Korea (Song et al., 2021).
Kinema (Figure 1A) is a sticky and umami-flavored solid fermented soybean food, which is conventionally prepared by natural fermentation, and is consumed as curry (Figure 1B) by the Nepali/Gorkha community of the Eastern Himalayan regions of North East India, east Nepal, and south Bhutan (Nikkuni et al., 1995; Tamang, 2021) for more than 2,000 years (Tamang and Samuel, 2010). During kinema preparation, local varieties of soybean seeds are soaked overnight, boiled, and the excess water is discarded; cooked beans are wrapped in fern fronds or paddy straw inside bamboo-made baskets and placed above earthen kitchen for natural fermentation for 1–3 days (Tamang, 2015). During the natural fermentation, soybean grits are covered with whitish viscous sticky materials, presumably poly-glutamic acid (Chettri et al., 2016), with umami taste and is commonly eaten as a side dish with steamed rice (Tamang, 2015). Microbiological investigation of kinema samples by culture method showed Bacillus subtilis as the dominant bacterium during natural fermentation (Sarkar et al., 1994; Tamang and Nikkuni, 1996; Tamang, 2003). Targeted high-throughput sequencing was applied to study the metataxonomic of kinema samples collected from India, Nepal, and Bhutan, which revealed the abundance of B. subtilis, few other non-Bacillus bacteria, yeasts, and molds detected in low abundances (Kharnaior and Tamang, 2021). However, metagenome–metabolite mining of fermented foods may help to understand the roles of beneficial microorganisms for production of primary and secondary metabolites. We hypothesized that some dominant bacteria present in kinema may contribute to the formation of targeted and non-targeted metabolites. Hence, this paper is aimed to decipher the integrative metagenomic and metabolomic approach to profile the abundant domains in kinema samples from three different countries, namely, India, Nepal, and Bhutan, and to determine both targeted and non-targeted metabolites by LC-MS. Functional profiles of metagenomes were also predicted using the SqueezeMeta pipeline and Kyoto Encyclopedia of Genes and Genomes (KEGG) database and were validated with the metabolomics profiles of the samples to find the relationship between predominant species and metabolite formation in kinema.

Figure 1. (A) Freshly prepared kinema; (B) Kinema curry.
Materials and Methods
Sample Collection
Eighteen sun-dried samples of kinema were aseptically collected from different places of India, Nepal, and Bhutan (Supplementary Table 1) in a pre-sterile container, sealed, labeled, and brought to laboratory for further analysis.
Genomic DNA Extraction
Ten grams of each sample were homogenized with 90 ml sterile 0.1 M phosphate buffer (pH 6.4) and kept undisturbed for 5 min to settle down the sample debris. Homogenate was collected, and genomic DNA was extracted using Nucleospin Food kit (Macherey-Nagel GmbH & Co., KG, Düren, Germany) following the manufacturer’s instructions. Quantification of DNA concentration was checked using a spectrophotometer (Eppendorf, United States), and quality of DNA was observed in 0.8% agarose gel electrophoresis followed by visualization under Gel Doc EZ imager (Bio-Rad, United States). Following the method described by Ray et al. (2019), the DNA from 18 samples of three countries were pooled in equal quantity (pooled from six initial samples of each country i.e., India, Nepal, and Bhutan), and the mixture was considered as one sample from each country and were used for metagenomic analysis.
MinION Library Preparation and Metagenomics Sequencing
The MinION gDNA sequencing Ligation Kit SQK-LSK109 (Oxford Nanopore Technologies, Oxford, United Kingdom) was used to prepare the metagenome libraries (Sevim et al., 2019) according to the manufacturer’s instruction. Genomic DNA (10 μg) was sheared using the Covaris g-tubes to produce the fragments of the size > 10 kb (Covaris Inc., MA, United States). The sheared genomic DNA with appropriate size was then selected using the BluePippin instrument (Sage Science, Beverly, MA, United States). End-repairing was performed on the sheared DNA using the NEBNext FFPE DNA Repair kit following the manufacturer’s protocol (New England BioLabs, Ipswich, MA, United States). Qubit HS DNA kit was used to quantify the gDNA. Ligation Sequencing Kit SQK-LSK109 was used for the clean-up and adapter ligation. The ligated sample was purified and eluted using the AMPure XP beads (Beckman Coulter Inc., United States) along with wash buffer and kit. Libraries were then sequenced (1D sequencing) on MinION MK1 device using R9 flow cell chemistry. The device was controlled through the MinKNOW software version 1.0.5 (Oxford Nanopore Technologies, Oxford, United Kingdom); Metrichor platform of ONT (Oxford Nanopore Technologies) was used to perform the 1D base calling.
Bioinformatics Analysis
Metataxonomic
The generated fast5 data from MinION ONT was processed by converting into fastq using poretools software version 0.6.0 for kinema metagenome (Loman and Quinlan, 2014). Furthermore, the sequence quality was subjected and examined using NanoPlot version 1.30.1 (De Coster et al., 2018) and was assembled using canu-assembler (Koren et al., 2017). Taxonomy classification of the assembled quality sequences were assigned using the Kaiju taxonomic pipeline (Menzel et al., 2016), mapped against the GenBank-derived database (NCBI nr + euk) containing millions of protein sequences from bacteria, viruses, eukaryotes, and archaea (Chen et al., 2017). Default “greedy” algorithm was preferred to map the sequences against the database (Zhang et al., 2000). A minimum required match length, minimum match score, and E-value of m = 11, s = 80, and E = 0.05, respectively, was filtered to avoid mismatches, chimera, low-complexity regions, and false positive taxon assigned (Menzel et al., 2016). Additionally, amino acid substitution was performed in amino acid sequence, and multiple matches were ranked, and taxon was classified from the database. After translation of open reading frames (ORFs) into a set of amino acid fragments, the same fragments were ranked by BLOSUM62 (BLOcks SUbstitution Matrix) score and the database search with the highest scoring was started (Lazar et al., 2019). Burrows–Wheeler transform (BWT) algorithm was used to search the fragments backward against the database (Pokrzywa and Polanski, 2010). The fragments with higher score were used for classifying and outputting the taxon identifier (Menzel et al., 2016).
Predictive Functional Features
Predictive metabolic pathways of kinema metagenome were derived using SqueezeMeta v1.3.0 (Tamames and Puente-Sánchez, 2019). Imported metagenomic data with contigs of < 500 bp were removed using prinseq (Schmieder and Edwards, 2011). Prediction of gene was performed using Prodigal v2.6.2 (Hyatt et al., 2012), and the homologies of predicted genes were searched against the functional database using DIAMOND, a computational tool for the alignment of sequencing reads against a protein reference database, which aligned the sequence against a protein database (Buchfink et al., 2015). After running the DIAMOND, the method assigned as functions to each ORF was carried out using the fun3 method (fun3 method produced functional assignments to compare gene sequences against the functional database) for clusters of orthologous groups/non-supervised orthologous groups (COGs/NOGs) using evolutionary genealogy of genes: non-supervised orthologous groups (eggNOG) database (Huerta-Cepas et al., 2016) and KEGG database (Kanehisa and Goto, 2000). The highest-scoring ORFs exceeding 30% (default) were considered for annotation (Tamames and Puente-Sánchez, 2019). Annotations were processed for predictive pathways and enzyme classification (Ye and Doak, 2009). Additionally, high-level function (level 1), lower-level function (level 2), and the sub-pathways (level 3) were categorized (Scala et al., 2019), and relative abundance of > 1% was visualized by bar plot using STAMP v2.1.3 (Parks et al., 2014). The inter distribution of predictive functional profiles among samples was performed using Mann–Whitney test (Fong and Huang, 2019) considering the sub-pathways (level 3).
Metabolite Profiling by Liquid Chromatography-Mass Spectrophotometry Analysis
LC-MS coupled to a Q-Exactive Orbitrap (Dionex Ultimate 3000, Thermo Fisher Scientific, United States) was used for the metabolite profiling of kinema samples (three samples each from India, Nepal, and Bhutan) following the method of Ramakrishnan et al. (2016). A 200-mg amount of each powdered sample was mixed with 1 ml of 80% methanol and sonicated in ice, the mixture was centrifuged at 14,000 rpm for 5 min at 4°C, and the supernatant was filtered using a 0.2-μm PTFE filter. Then, 10 μl of each sample was injected into the LC-MS system coupled to a Q-Exactive Orbitrap, and chromatography was performed on a hydrophilic interaction liquid chromatography (HILIC) (5 μ, 150 mm × 4.6 mm, Phenomenex Luna) with a flow rate of 0.4 ml/min at 40°C. The mobile phase A contained 10 mM ammonium acetate with acetonitrile in the ratio of 1:1 (0.1% FA), and phase B contained acetonitrile (0.1% FA) for positive mode. Whereas, for the negative mode, the mobile phase A contained 5 mM ammonium acetate in water, and the phase B contained 5 mM ammonium acetate in water with acetonitrile in the ratio of 1:9. The following gradient gave optimal resolution and was used in both ionization modes (0–2 min: 100% B, 2–15 min: 100–90% B, 15–25 min: 90–80% B, 25–30 min: 80–75% B, 30–35 min: 75–20% B, 35–40 min: 20–0% B, 40–45 min: 0% B, 45–45.1: 0–100% B, and 45.1–55 min: 100% B) at a 400-μl/min flow rate. The Q-Exactive Orbitrap (Thermo Fisher Scientific, United States) was set up for data acquisition in the full scan/data-dependent scan (FS/DDS) mode in a mass range of 70–1,050 m/z, alternating between MS and MS/MS scans. The MS operating conditions for all samples were as follows: spray voltage, 4,000 V (2,500 V for negative); vaporizer temperature, 320 °C; sheath gas flow rate of 30 arbitrary units (40 for negative); and auxiliary gas flow rate of 10 arbitrary units. Injector settings were as follows: 0–2 min: waste, 2–45 min: load, and 45–55 min: waste. The spiked reserpine and taurocholate were used as internal standards and later used for intensity normalization of data. The raw data files were imported into SIEVE 2.2 for the generation of peak list and component extraction (Hao et al., 2018), and the Human Metabolome DataBase (HMDB), KEGG (Zhang B. et al., 2018), and PlantCyc (Zhang et al., 2010) were used for possible identification of the compounds. The coefficient of variation (CV) was calculated from the pooled quality control sample data, and the data with CV_QC > 20% were removed.
Statistical Analysis
Pooled Sequences
Nucleotide diversity (pi) analysis and different indices of neutrality test, based on sequence polymorphism in DnaSP software version 6 (Rozas et al., 2017), were performed to justify the pooling of DNA from six samples from each country (India, Nepal, and Bhutan) for pooled sequence in terms of intra-sample (within the sample) diversity. For each sample, the intra-species diversity was calculated by one-sample t-test using IBM SPSS v20.0 (Statistical Package for the Social Sciences) to check the significant differences within the sample. Statistical relations among the samples were performed using Mann–Whitney test (Fong and Huang, 2019).
Diversity Indices
Non-parametric Shannon index and Simpson’s index of diversity (1-D) were calculated for beta diversity indices using PAST software version 4.0 (Martino et al., 2019). Bray–Curtis index of beta diversity was also calculated using PAST version 4.0 and visualized via principal coordinates analysis (PCoA) plot (Martino et al., 2019). The clustering pattern of microbial population was calculated using the unweighted pair group method with arithmetic mean (UPGMA) (Parks and Beiko, 2012). Fisher’s exact test (non-parametric) was performed through the Statistical Analysis of Metagenomic Profiles (STAMP) software version 2.1.3 (Parks et al., 2014) to check the significance in the distribution of different microbial taxa among the samples.
Metabolite Profiles
The significant differences of metabolites were calculated by Student t-test (two-tailed) (Kwon et al., 2019). Metabolites were log transformed (log xi + 1) and analyzed by PCoA using Bray–Curtis dissimilarity (Djeni et al., 2020). The hierarchical clustering of metabolites was calculated using UPGMA (Parks and Beiko, 2012). The correlation between the significant differentiated metabolites of more than fourfold and predominant species was measured by a non-parametric Spearman’s rank correlation using IBM SPSS v20.0. The network-based correlation was visualized using MetScape v3.1.3 in Cytoscape v3.8.2. Metabolites identified by LC-MS analysis were imported in MetaboAnalyst (v5.0) and further carried out to describe the association of metabolites with KEGG pathways referring to HMDB (Pang et al., 2021). Additionally, metabolites associated with predictive pathways were analyzed by network-functional analysis modules and visualized using KEGG global metabolic network (Pang et al., 2021). The variation of metabolites among the samples was analyzed based on the fold change (FC) ratio. The outcome was transformed into log value (log2 FC) (Hyeon et al., 2020), and significant differences were tested using analysis of variance (ANOVA) (Song et al., 2021).
Results
Pooled Sequence Statistics
Nucleotide diversity (Pi) per site of pooled samples was 0.72668 (India), 0.7243 (Nepal), and 0.72687 (Bhutan), respectively. The neutrality indices were performed using two tests: Tajima’ D with values of 1.363 (India), 1.347 (Nepal), and 1.364 (Bhutan), respectively, and Fu’s Fs statistic representing the values of -46.669, -31.869, and -43.373 of kinema from India, Nepal, and Bhutan, respectively. Pooled DNA samples were not significantly (p > 0.1) different in terms of sequence variation within the sample.
Metagenomic Sequencing and Microbial Community
A total of 1,394,094,912 bp with an average of 464,698,304 ± 120,720,392 bp was generated from kinema metagenome. In comparison, among the samples, kinema from Nepal has the longest contig of 533,203 bp, whereas kinema from Bhutan has the shortest contig of 1,001 bp (Table 1). Raw reads from the taxonomic classification analysis were then normalized as relative percentages for visualization, and taxonomic abundances with > 1% and those with < 1% abundances were grouped as “others.” The overall taxonomy classification resulted in the identification of 47 phyla, 331 families, 709 genera, and 1,560 species. In the organismal diversity, the most abundant domain was bacteria (97.78%) in kinema metagenome, and the remaining domains were viruses, eukaryote, and archaea (Figure 2A). Firmicutes (93.36%) was the most abundant bacterial phylum (Figure 2B and Supplementary Table 2). Further classification led to the identification of abundant families represented by Bacillaceae (81.03%), followed by Paenibacillaceae, Planococcaceae, and Enterococcaceae (Figure 2C and Supplementary Table 3). Bacillus was the most abundant genus followed by Brevibacillus, Kurthia, Nit1virus (virus), Proteus, Acinetobacter, and other genera (Figure 2D and Supplementary Table 4). At the species level, B. subtilis was detected as the most abundant species in kinema, followed by B. glycinifermentans, B. cereus, Brevibacillus borstelensis, B. licheniformis, B. thermoamylovorans, Kurthia sp. 11kri321, B. coagulans, B. circulans, B. paralicheniformis, and others (Figure 2E and Supplementary Table 5). No eukaryote and archaea species were found with > 1% relative abundance in kinema metagenomes.
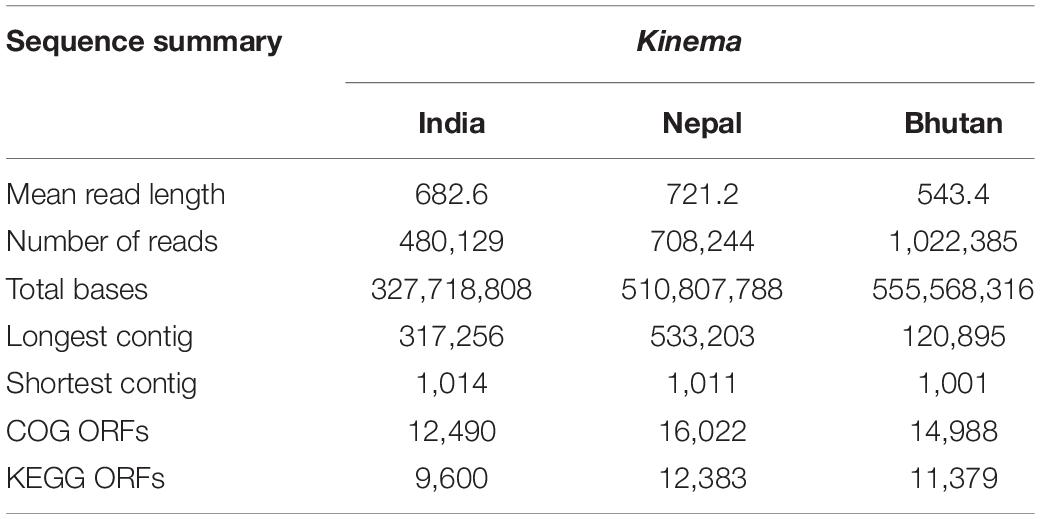
Table 1. General summary and details of metagenomic sequences and assembly statistics.
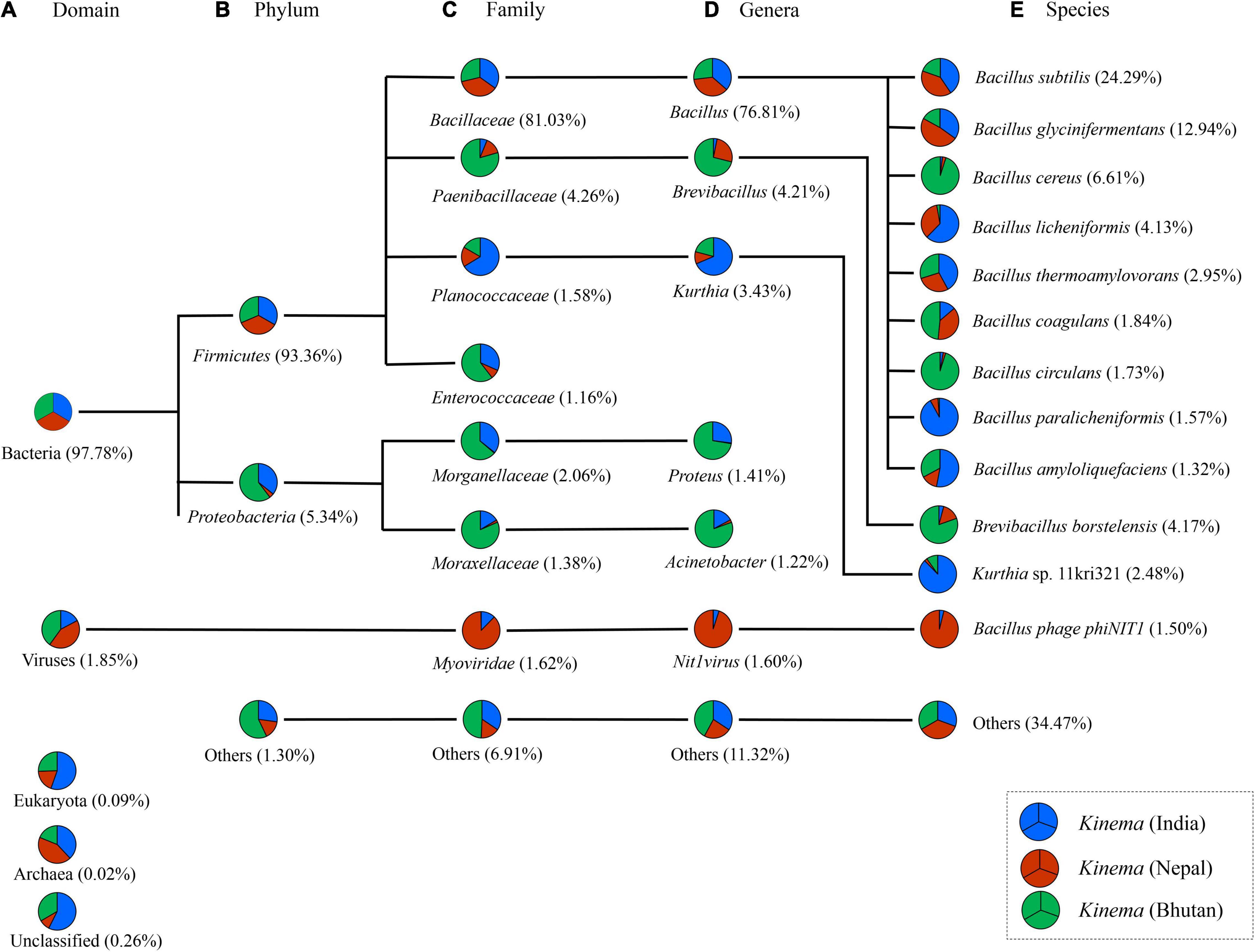
Figure 2. Differences in relative abundance (%) of predominant species in kinema metagenome at different taxonomic levels (A) domain, (B) phylum, (C) family, (D) genus, and (E) species levels. The minor species with a relative percentage of less than 1% were grouped and represented as others.
The result showed that B. subtilis was predominant in kinema samples of India and Nepal (Figure 3A). Contrastingly, B. cereus and B. borstelensis were significantly (p < 0.05) abundant in kinema samples of Bhutan (Figure 3A). In the overall distribution of species of all domains, bacterial species were the dominant domain in which 280 species of Bacillus (Supplementary Table 6) and 121 species of lactic acid bacteria (LAB) (Supplementary Table 7) were detected with < 1% abundances in kinema metagenomes. Apart from bacteria, the second abundant domain was viruses with a total of 42 phage(s), out of which 26 were Bacillus phage and 16 were others (Supplementary Table 8). Myoviridae was the predominant viral family, among which Bacillus phage phiNIT1 was the abundant phage observed in kinema. Among eukaryotes, a total number of 56 species were identified; 17 species each were classified as molds and yeasts, respectively, and 22 species were grouped as other microbial eukaryotes (Supplementary Table 9). Additionally, 11 species were identified under the domain archaea (Supplementary Table 10).
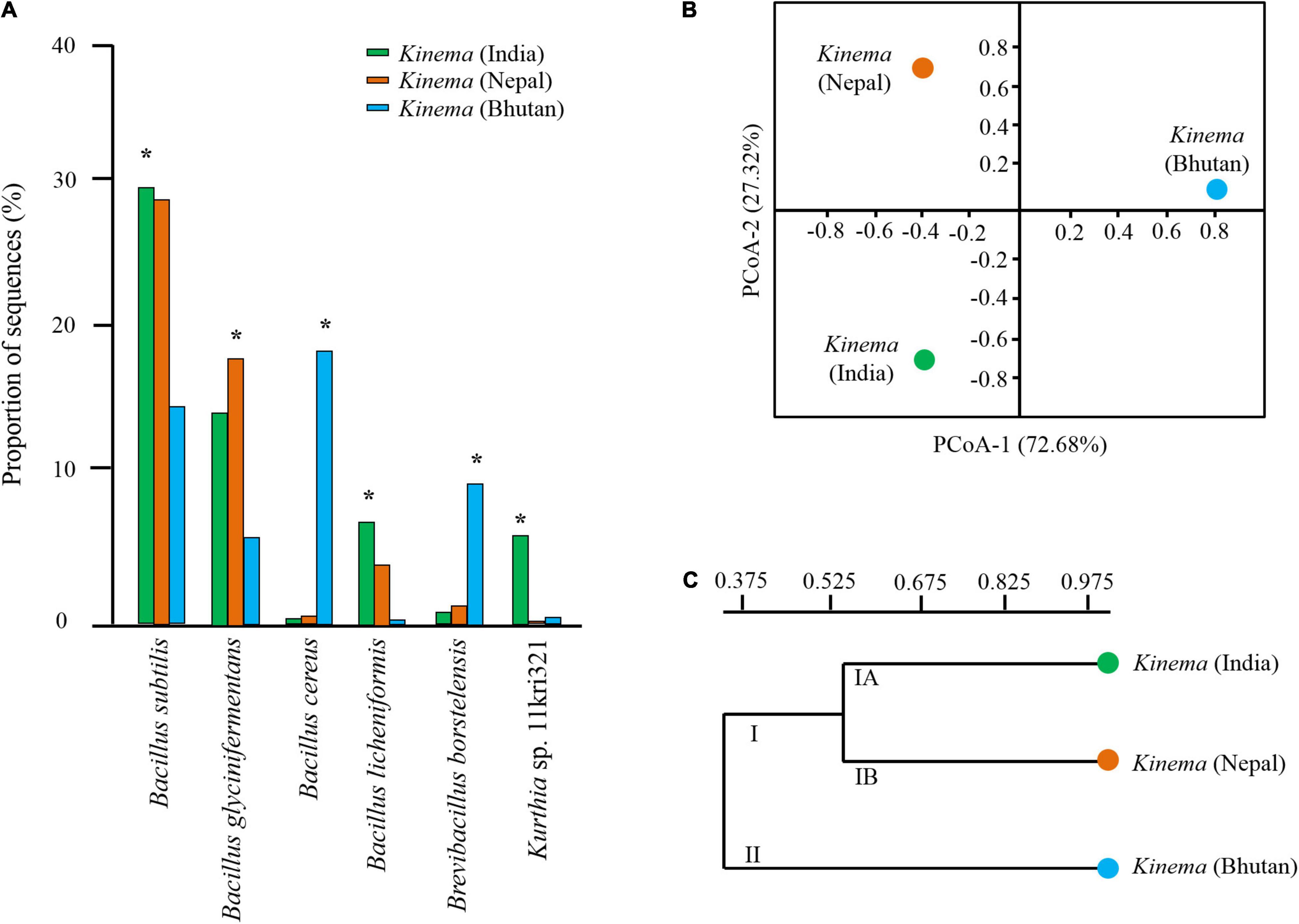
Figure 3. (A) Significant differences* of species abundance with a relative abundance of more than 1% were calculated among samples by a non-parametric Fisher’s exact statistical test. (B) Principal coordinates analysis (PCoA) plot using Bray–Curtis dissimilarity. (C) Unweighted pair group method with arithmetic mean (UPGMA) hierarchical cluster constructed based on species level to observe the differences in the overall microbial structure and diversity in kinema samples collected from different geographical locations.
Diversity Indices and Shared and Unique Species
Good’s coverage ranging from 0.98 to 0.99 was observed in all samples. The alpha diversity indices revealed the diversity in the following order: Bhutan (0.92) > India (0.71) > Nepal (0.57) (data not shown). Similarly, a non-parametric Shannon index also followed the same pattern, Bhutan (5.45) > India (3.92) > Nepal (2.89). Intra-diversity showed no significant differences in pooled samples of kinema from India (p = 0.54), Nepal (p = 0.55), and Bhutan (p = 0.48). Based on the species distribution and its relative abundance of all domains, the inter-diversity, calculated by PCoA, was distinctly different among samples (Figure 3B). The hierarchical clustering UPGMA revealed two clusters: cluster I (kinema from India and Nepal) and cluster II (kinema from Bhutan). Cluster I was observed with two subclusters, cluster IA and cluster IB of kinema from India and Nepal, respectively (Figure 3C). Statistical variation in the distribution of species among samples showed that kinema from India was significantly different from Nepal (p = 0.01945) and Bhutan (p = 0.000127), respectively. Similarly, kinema from Nepal was also significantly (p = 0.00007398) different from Bhutan.
It was observed that 485 overall species including bacteria, viruses, eukaryotes, and archaea were common in all samples. We observed that 479 bacterial species were shared in all samples, whereas 56 species were unique to India, 95 species to Nepal, and 229 species to Bhutan, respectively (Figure 4 and Supplementary Table 11). Among viruses, four species were found common such as Bacillus phage Shbh1, Bacillus phage SP-10, Bacillus phage SPG24, and Bacillus phage SPP1, whereas 13, 5, and 10 viral species were unique to kinema from India, Nepal, and Bhutan, respectively (Figure 4 and Supplementary Table 12). Among eukaryotes, two species (Pichia kudriavzevii and Mucor ambiguousi) were the common species, whereas 27, 8, and 10 species were unique species to India, Nepal, and Bhutan, respectively (Figure 4 and Supplementary Table 13). No common and shared archaea species were detected in kinema samples, whereas two, six, and three species of archaea were found unique to India, Nepal, and Bhutan, respectively (Figure 4 and Supplementary Table 14).
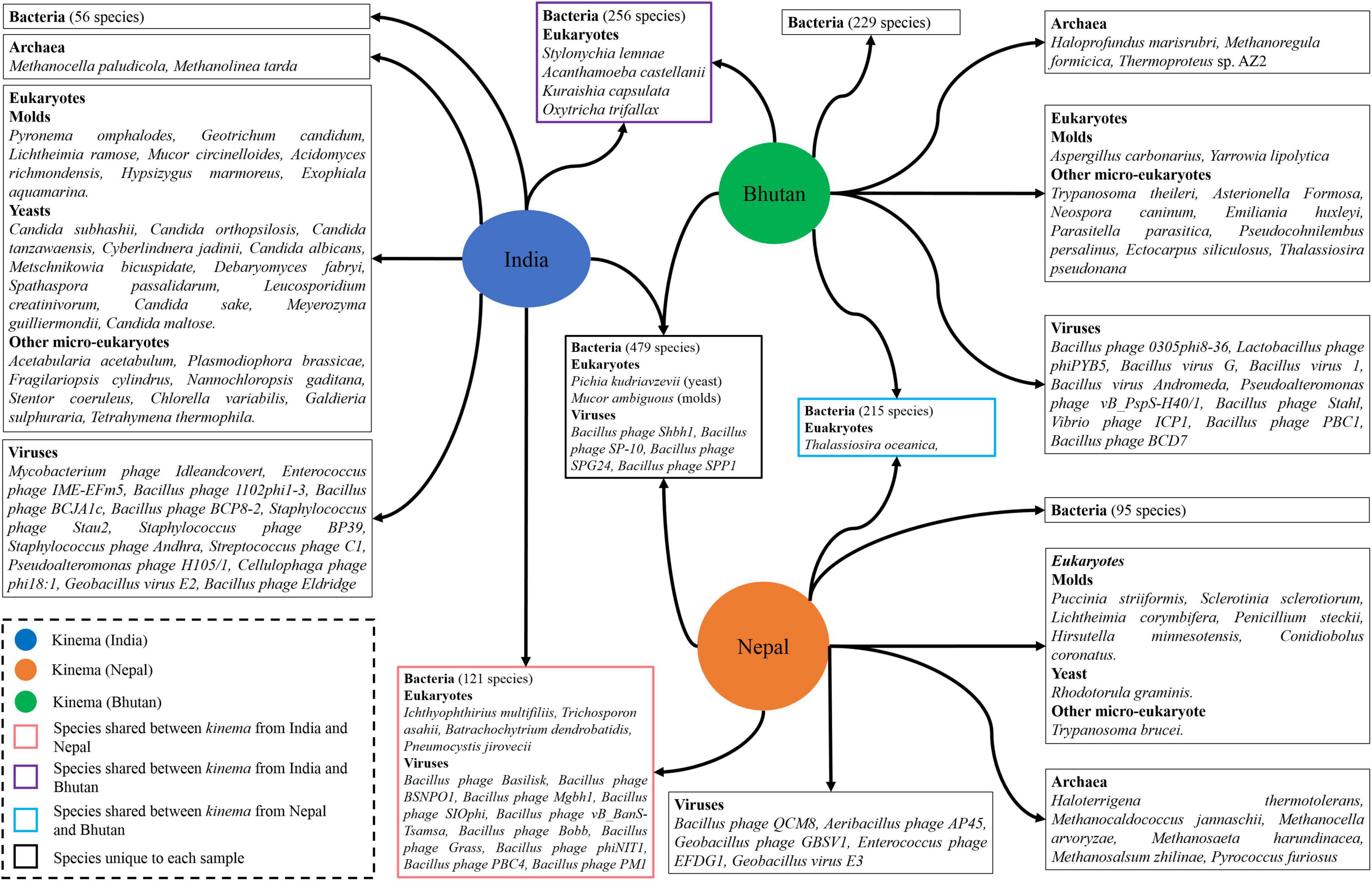
Figure 4. Representation of shared and unique species identified from different domains in kinema samples.
Predictive Functional Profiles
Different enhanced functional pathways were observed after mapping metagenomic ORFs against eggNOG and KEGG databases. Around 56.41–56.84% of the total mapped ORFs were assigned to COG genes, and 43.16–43.59% were assigned as KEGG pathway genes. Metagenome ORFs were aligned with genes associated to 216 KEGG metabolic pathways and categorized into three different levels. In level 1, metabolism was the most abundant category followed by environmental information processing, genetic information processing, cellular processes, human diseases, and organismal systems (Figure 5A). At level 2, there were 17 super-pathways with > 1% abundance and minor super-pathways with a relative abundance < 1% (Figure 5B), and at level 3, there were 36 super-pathways with > 1% abundance (Figure 5C) with 180 minor super-pathways with a relative abundance of < 1% (Supplementary Table 15). The distribution of the functional features in kinema metagenome was compared, and significant differences among the functional features of kinema of India, Nepal (p = 0.04182), and Bhutan (p = 0.02698) were found. Similarly, significant differences between kinema from Nepal and Bhutan (0.00007833) were observed. Exploring the enzyme classification by predictive analysis revealed the presence of protease, glucosidase, galactosidase, amylase, lipase, and γ-PGA proteins and enzymes involved in its biosynthesis (Supplementary Table 16). Additionally, further analysis was carried out on enzymes involved in aminoacyl-tRNA biosynthesis (Figure 6 and Supplementary Table 17) and phosphotransferase system (PTS) (Figure 7 and Supplementary Table 18), which revealed the enzymes responsible in the biosynthesis of amino acids and sugar components present in kinema.
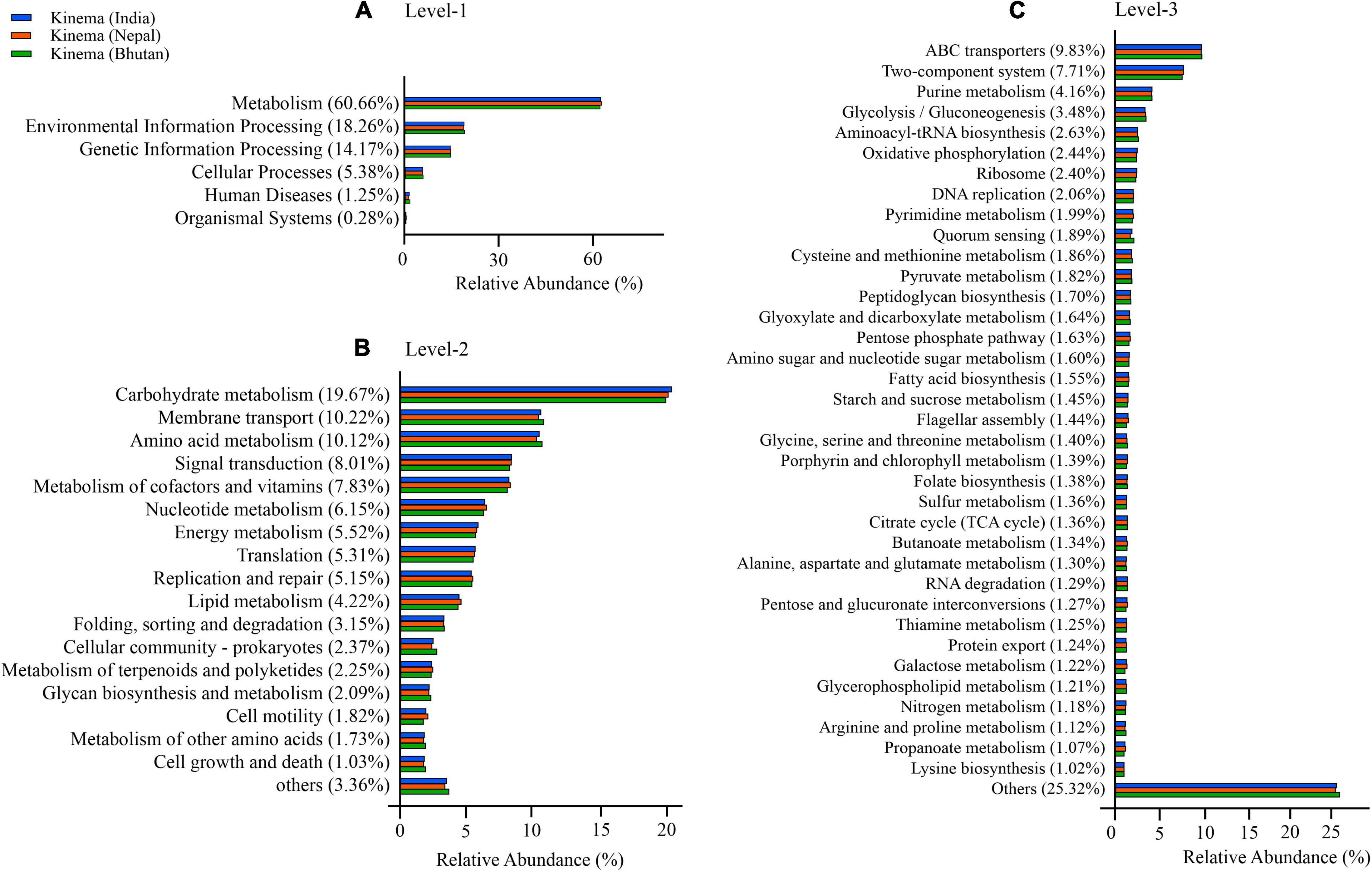
Figure 5. Predictive functional features mapped against Kyoto Encyclopedia of Genes and Genomes (KEGG) database were represented by relative abundance (%) at three different categories: (A) Level 1, (B) level 2, and (C) level 3. The functional features with less than 1% were grouped as others.
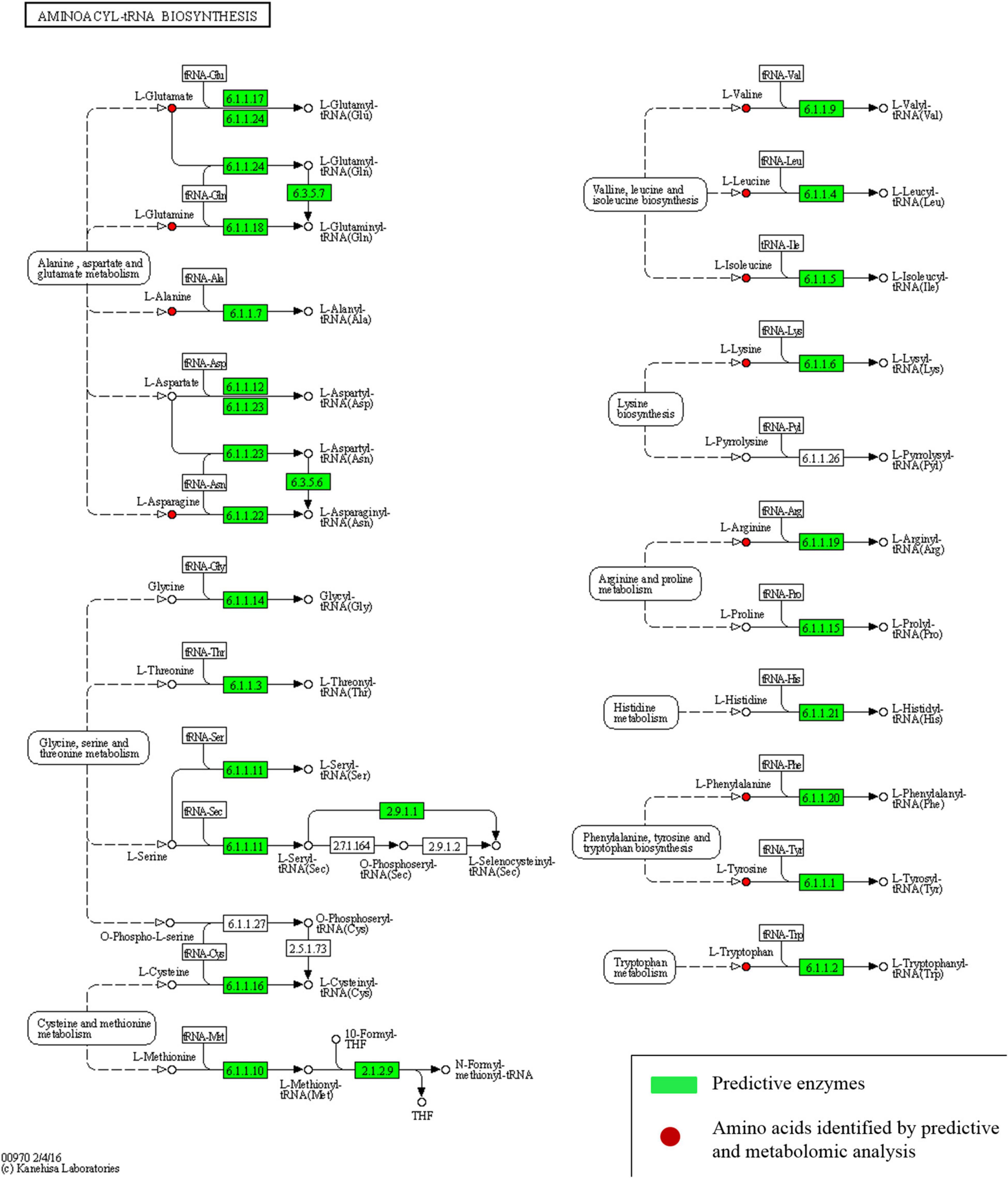
Figure 6. Predictive enzymes involved in aminoacyl-tRNA biosynthesis of different amino acids.

Figure 7. Predictive enzymes involved in phosphotransferase system (PTS).
Metabolites Associated With Predictive Kyoto Encyclopedia of Genes and Genomes Pathways
The analysis of metabolites associated with predictive functional pathways using MetaboAnalyst v5.0 network revealed that many compounds were found associated with different metabolic pathways (Figure 8 and Supplementary Table 19). Amino acids such as glutamine, asparagine, valine, alanine/sarcosine, arginine, lysine, phenylalanine, tyrosine, glutamate, leucine/isoleucine, tryptophan, ornithine, and 4-aminobutanoate (γ-aminobutyric acid, GABA) were associated with predictive pathways related to amino acid metabolism. Fatty acids were associated with fatty acid biosynthesis, biosynthesis of unsaturated fatty acids, and alpha-linolenic acid metabolism (Figure 8). Sugars were mostly found to be associated with carbohydrate metabolism. Whereas, in citrate cycle (TCA cycle) pathways of the carbohydrate metabolism, organic acids such as citrate and isocitrate were involved, which were also found to be associated with glyoxylate and dicarboxylate metabolism. Among metabolites, daidzein, genistein, isovitexin, genistin, maackiain, biochanin-A, (+)-pisatin, apigenin, chrysin, and chrysophanol were found to be associated with flavonoid and isoflavonoid biosynthesis. Biotin, nicotinamide, pyridoxamine, pyridoxine, pantothenate, and riboflavin were found to be associated with metabolism of vitamins and cofactors. (R)-lipoate was associated with lipoic acid metabolism. Additionally, sarpagine and catharanthine were associated with indole alkaloid biosynthesis; swainsonine with tropane, piperidine, and pyridine alkaloid biosynthesis; and solavetivone with sesquiterpenoid and triterpenoid biosynthesis (Supplementary Table 19).
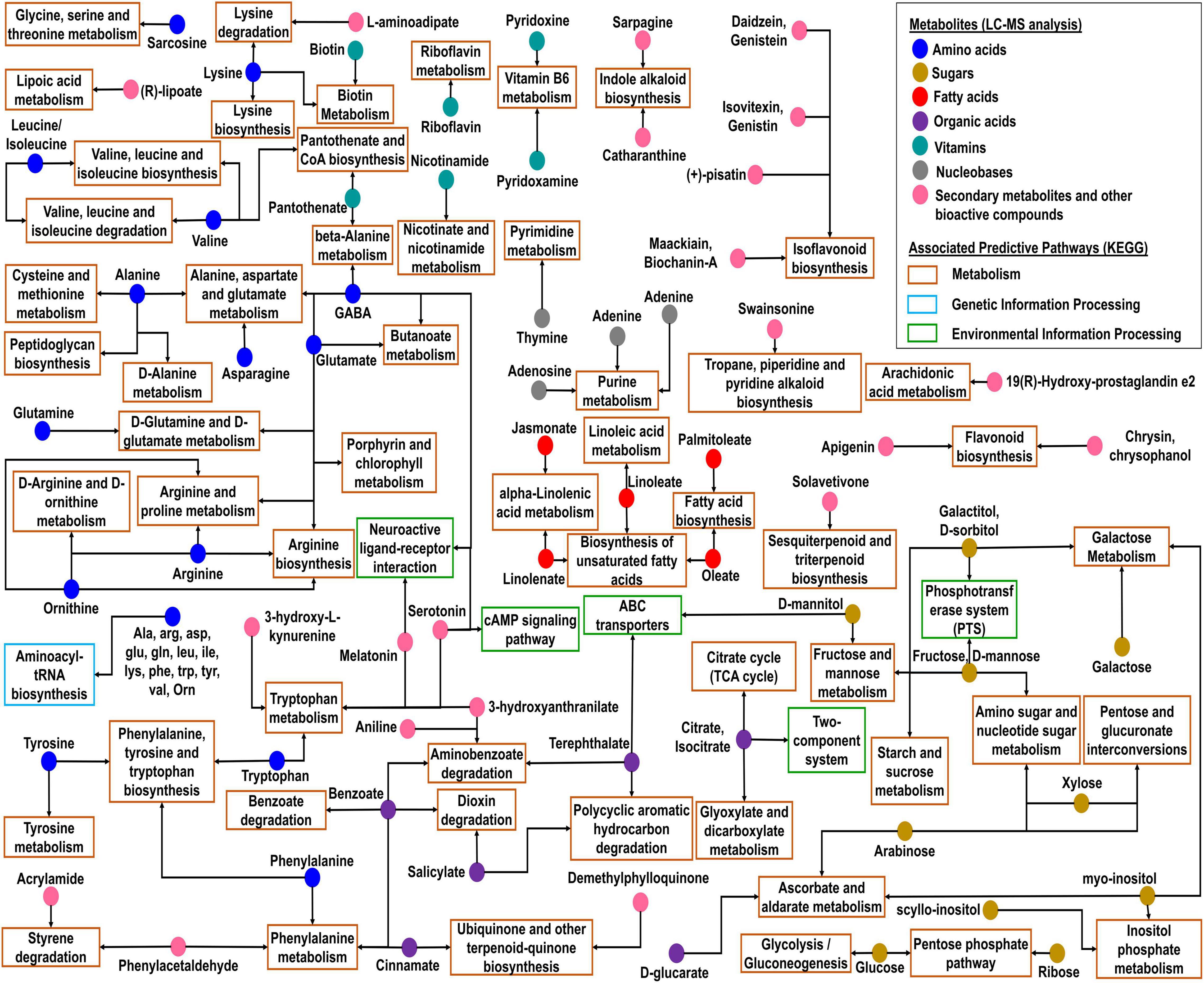
Figure 8. Network and functional analysis modules of metabolites associated with predictive KEGG pathways represented via KEGG global metabolic network using MetaboAnalyst.
Targeted and Untargeted Metabolites
A total of 361 metabolites were identified using the PlantCyc database. PCoA scores were plotted with a total cumulative variance of 77.1% (PCoA-1, 54.7%; PCoA-2, 22.5%) with distinct formation of metabolites (Figure 9A). The hierarchical clustering analysis of significant metabolites revealed the formation of two clusters (Figure 9B), which followed the same pattern as shown in the microbial distribution. Cluster I constituted kinema from India and Nepal and was further sub-divided into two sub-clusters, clusters IA and IB, respectively. Cluster II constituted kinema from Bhutan and was observed to be distinctly different from the other samples. In metabolomic analysis, 36 primary metabolites (Figure 10A and Supplementary Table 20A) and 37 secondary metabolites were identified (Figure 10B and Supplementary Table 20B). The presence of isoflavones and various bioactive compounds associated with immunomodulatory effects, immuno-stimulatory effects, phytoalexins, and anti-viral properties was detected (Table 2). The abundance of secondary metabolites and other bioactive compounds was compared among kinema samples based on log-transformed fold change (Figure 11). Among isoflavones identified in kinema, daidzein, chrysin, and chrysophanol were significantly (p = 0.014651) higher in Nepal than in India and Bhutan. Other than isoflavones, compounds such as p-coumaroyltyramine, melatonin, 3-hydroxy-L-kynurenine, and catharanthine were higher in India compared to Bhutan and Nepal (p < 0.05). Serotonin, solavetivone, swainsonine, N-acetylaspartic acid, and sarpagine were higher (p < 0.05) in Bhutan than in India and Nepal, and soyasaponin III was found to be higher in Nepal than Bhutan and India (p = 0.0000396). The metabolites with at least fourfold differences among samples from different locations were detected (Figure 11 and Supplementary Table 21).
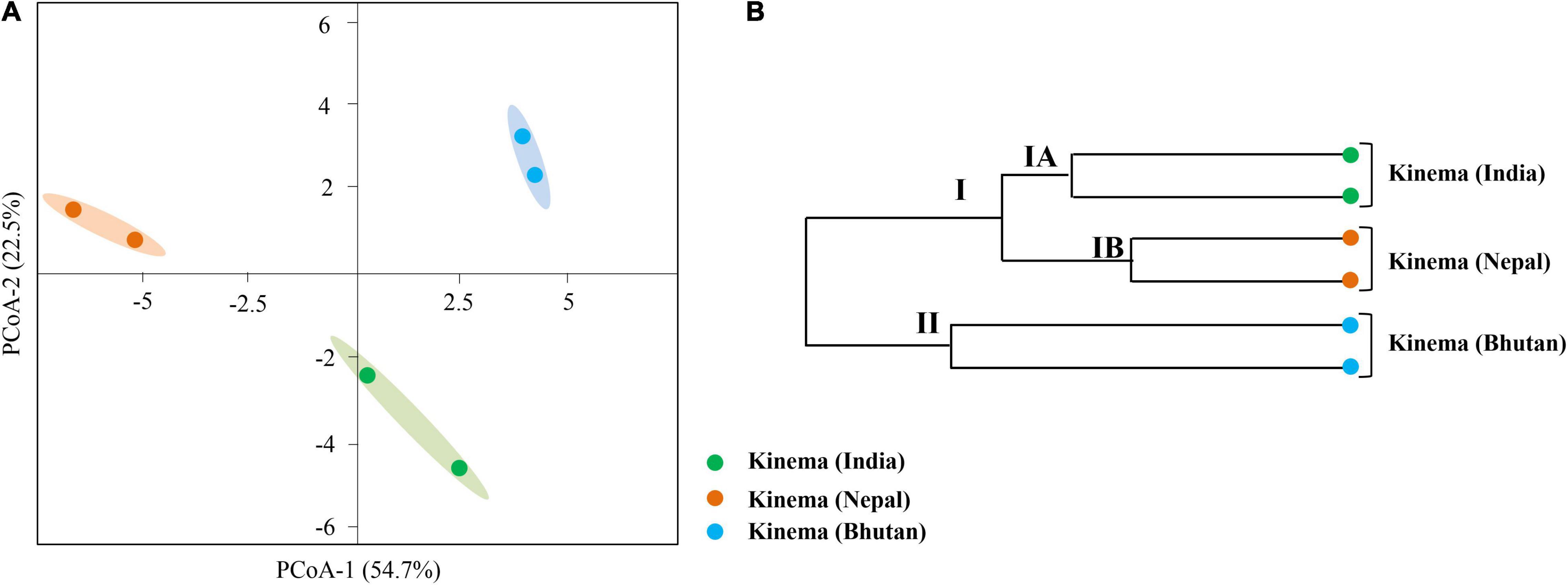
Figure 9. Comparison of metabolite formation in kinema samples from India, Nepal, and Bhutan. (A) PCoA using Bray–Curtis dissimilarity. (B) UPGMA hierarchical clustering was plotted for the distribution of metabolites analyzed by liquid chromatography-mass spectrophotometry (LC-MS).
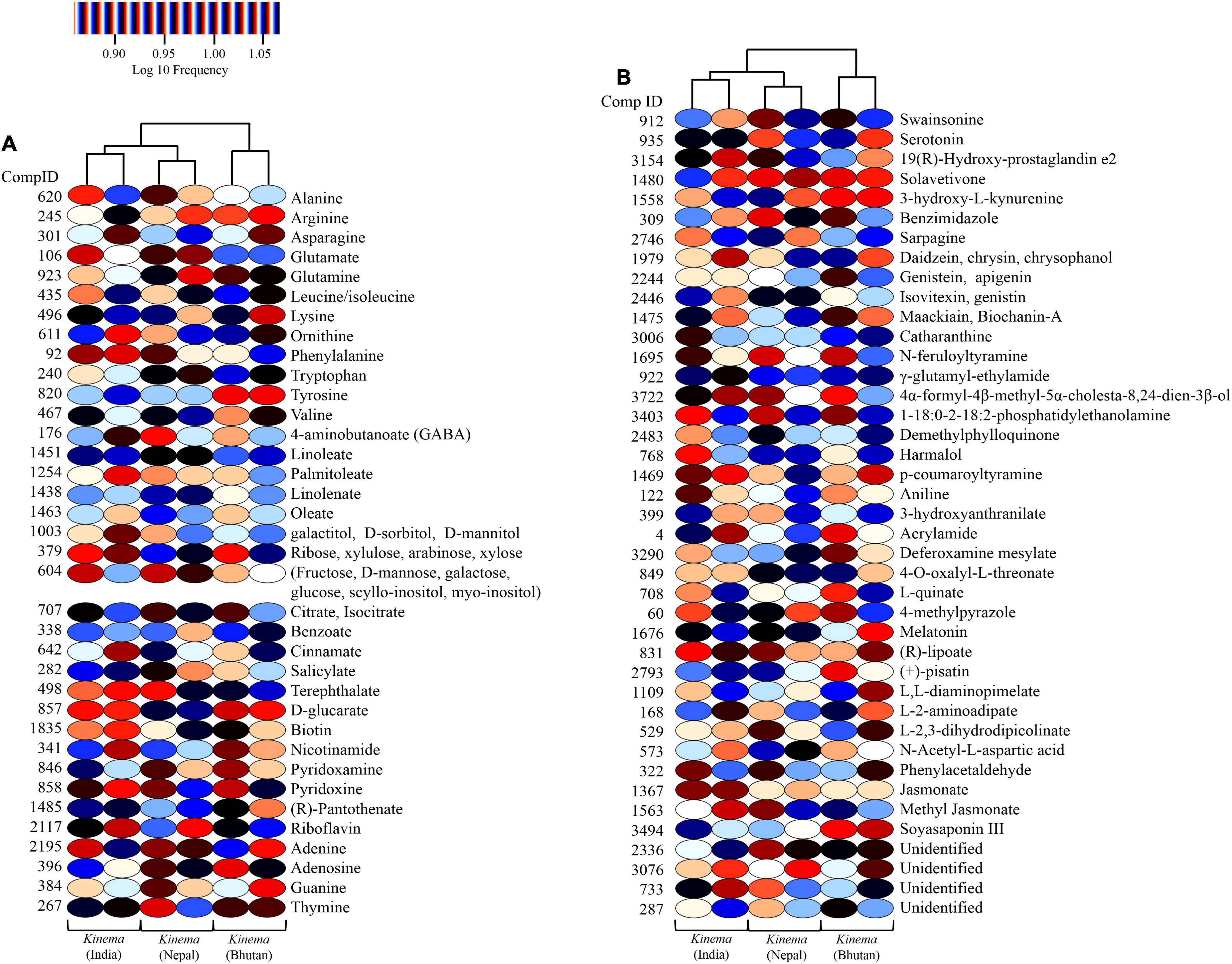
Figure 10. Heatmap representation of significant metabolites (p < 0.05) in kinema samples. (A) Significant primary metabolites include amino acids, fatty acids, sugars, organic acids, vitamins, and nucleobases. (B) Significant secondary metabolites such as isoflavones, soyasaponin, flavoring agents, and other bioactive compounds with potential therapeutic properties.
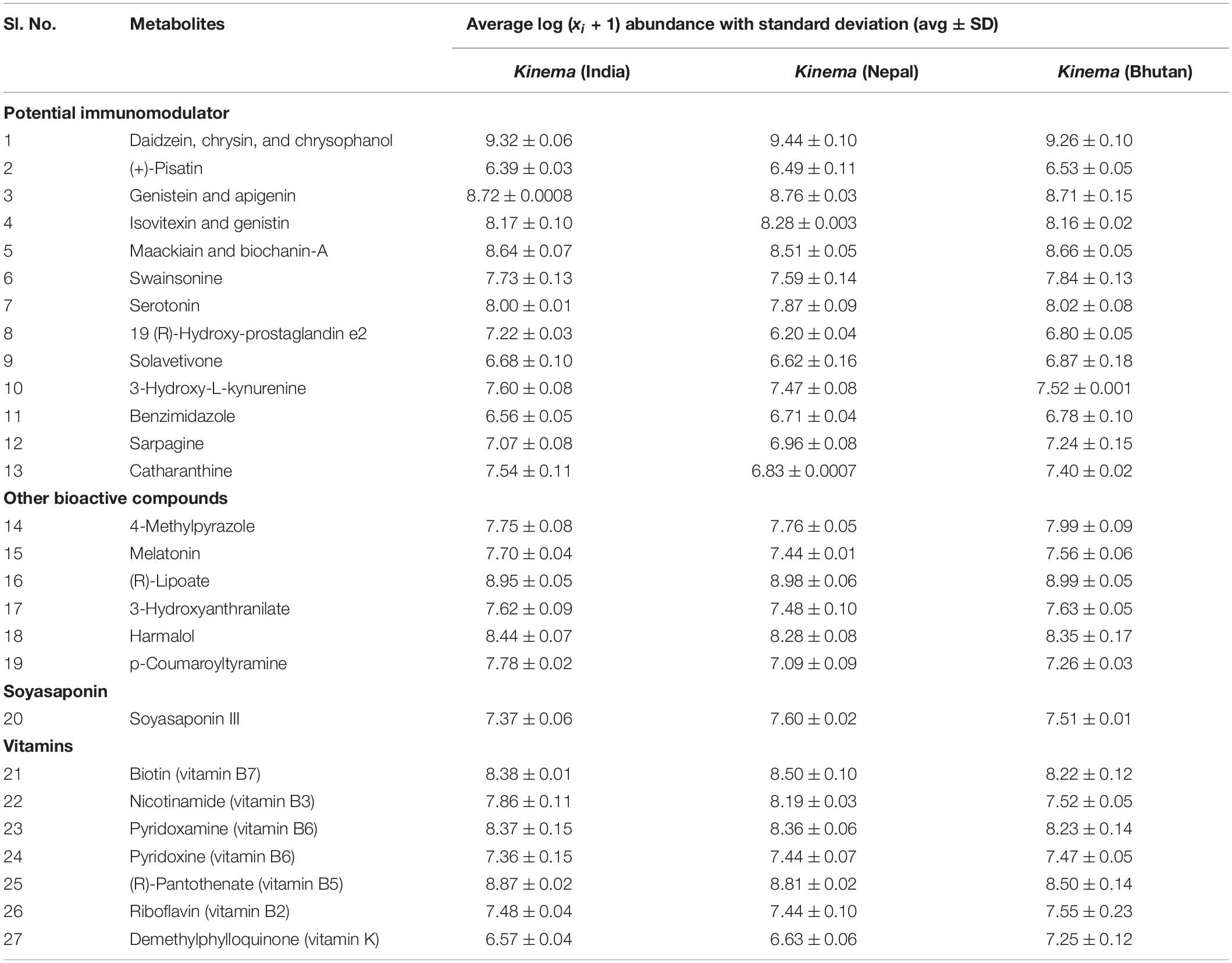
Table 2. Metabolites with potential immunomodulatory effects, other bioactive compounds, soyasaponin, and vitamins (p < 0.05).
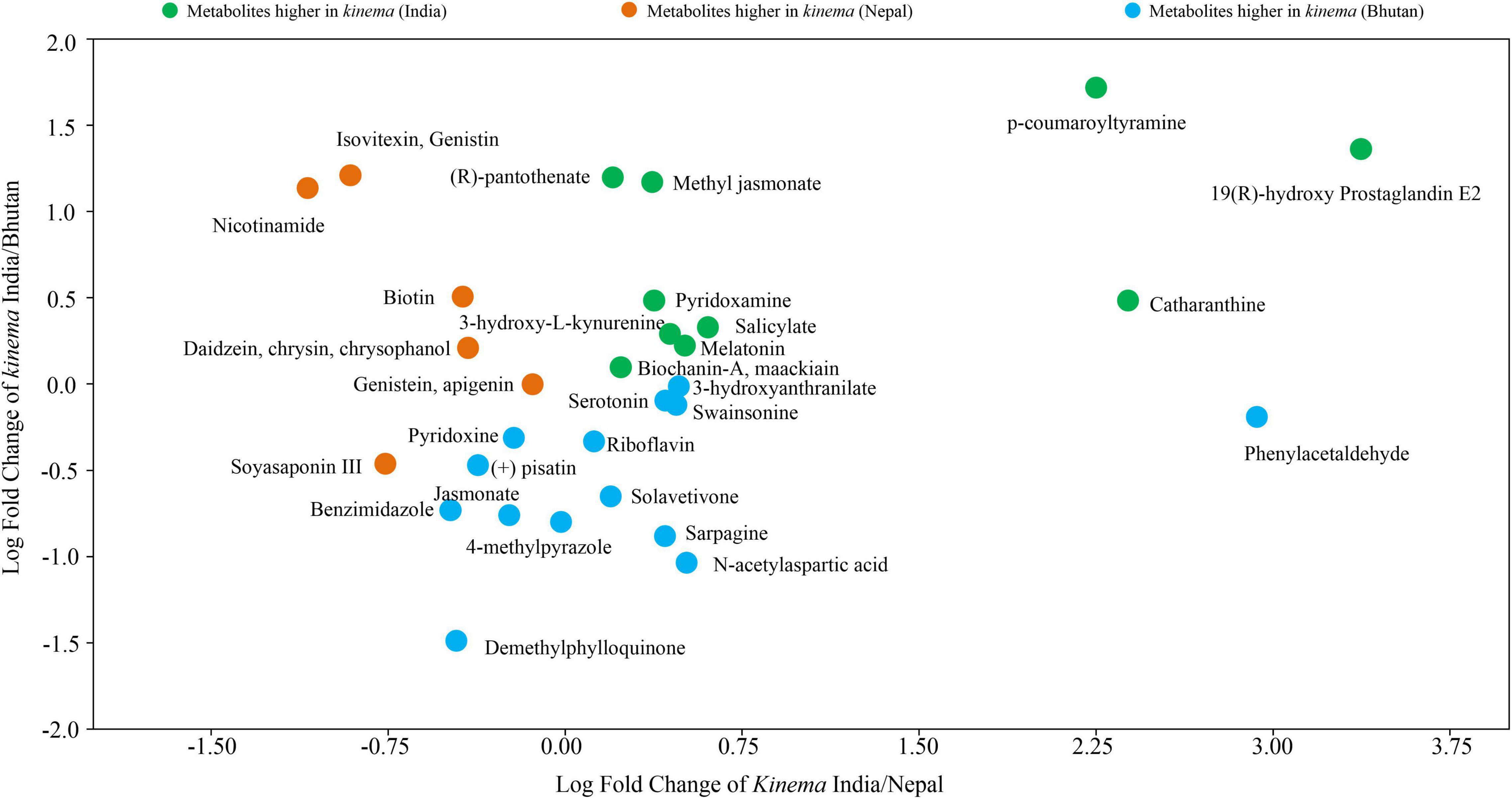
Figure 11. The abundance of secondary metabolites, vitamins, flavoring agents, and other bioactive compounds of kinema based on the log-transformed fold change, and the metabolites were found significantly different (p < 0.05) among samples.
Correlation of Metabolites With Abundant Metagenomes
The correlation between the abundant species and primary metabolites (amino acids, fatty acids, sugars, organic acids, vitamins, and nucleobases) was constructed with a bivariate result obtained from Spearman’s correlation (Figure 12). B. subtilis, B. paralicheniformis, and B. licheniformis were positively correlated (p < 0.01) with arginine, phenylalanine, glutamate, leucine, isoleucine, tryptophan, and 4-aminobutanoate (GABA) among amino acids; linoleate and linolenate among fatty acids; galactitol, sorbitol, mannitol, ribose, xylulose, arabinose, xylose, fructose, mannose, galactose, glucose, L-gulose, scyllo-inositol, and myo-inositol among sugars; and citrate, isocitrate, and salicylate among organic acids. B. borstelensis, B. cereus, and B. coagulans were positively correlated (p < 0.01) with alanine, valine, lysine, palmitoleate, benzoate, and pyridoxine. Furthermore, the correlation analysis observed the formation of galactitol, sorbitol, mannitol, ribose, xylulose, arabinose, xylose, leucine/isoleucine, and salicylate with B. amyloliquefaciens, B. thermoamylovorans, and Kurthia sp. 11kri321 (p < 0.01). B. circulans was found to be significant and positively correlated (p < 0.01) with ornithine, glutamine, cinnamate, terephthalate, riboflavin, thymine, GABA, and glutamate. B. glycinifermentans was the only species found to be positively correlated (p < 0.01) with the compounds glutamate, phenylalanine, arginine, tryptophan, linoleate, linolinate, fructose, mannose, galactose, glucose, scyllo-inositol, myo-inositol, citrate/isocitrate, biotin, nicotinamide, adenosine, and adenine.
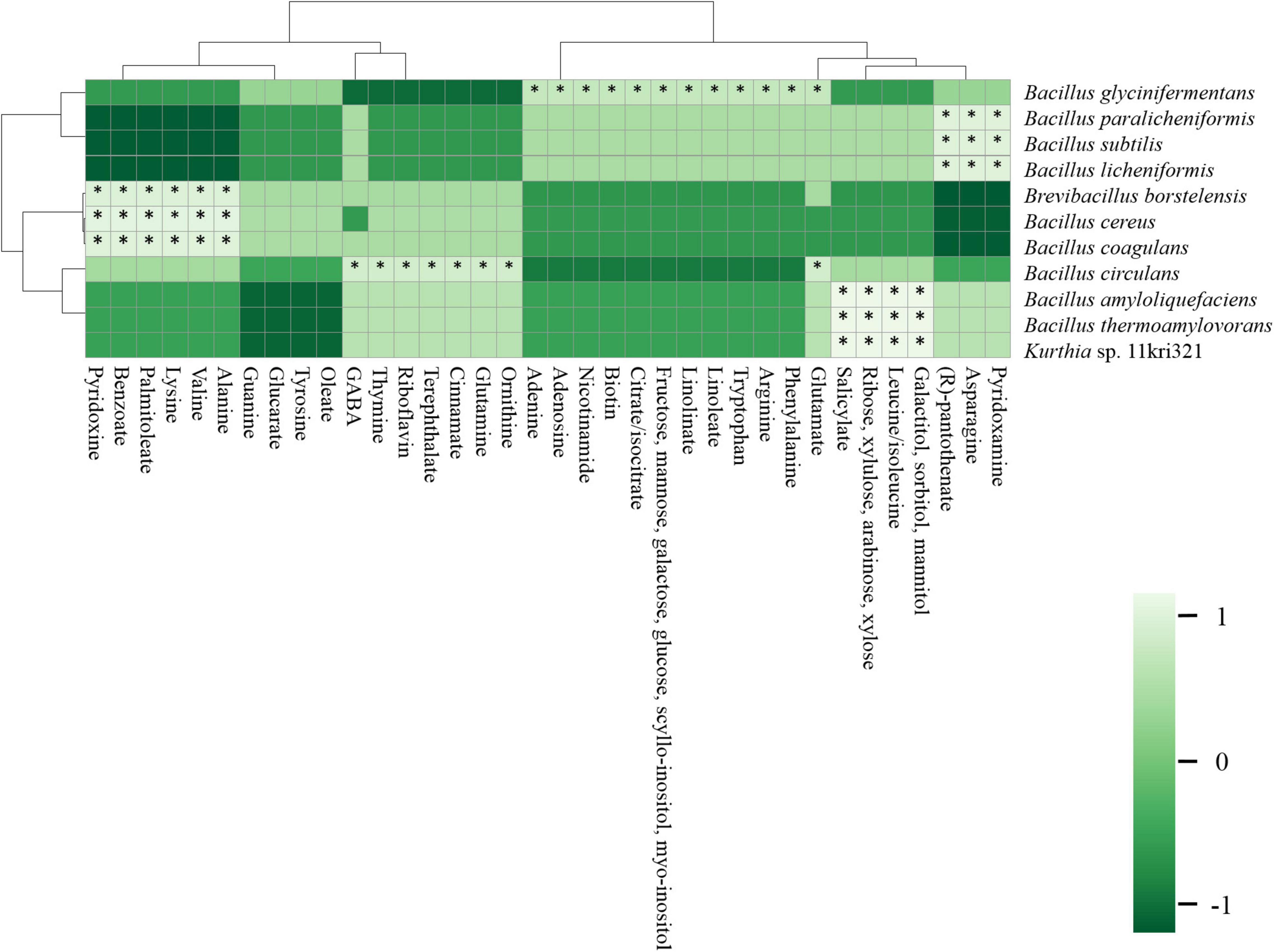
Figure 12. A correlation analysis between abundant species (> 1%) and primary metabolites (amino acids, fatty acids, organic acids, sugars, vitamins, and nucleobases) using Spearman’s correlation constructed by Clustvis.
Similarly, the relationship of abundant species identified in kinema with secondary metabolites and other bioactive compounds was explored using a bivariate analysis results obtained from Spearman’s correlation (Figure 13). The correlation of B. glycinifermentans with the formation of daidzein, chrysin, chrysophanol, genistein, apigenin, isovitexin, and genistin was positively significant (p < 0.01). B. borstelensis, B. cereus, and B. coagulans were positively correlated (p < 0.01) with the formation of jasmonate, benzimidazole, demethylphylloquinone, 4-methylpyrazole, and (+)-pisatin. Phenylacetaldehyde, swainsonine, serotonin, solavetivone, sarpagine, maackiain, biochanin-A, 3-hydroxyanthranilate, and N-acetyl-L-aspartic acid showed a positive correlation (p < 0.01) with B. circulans, whereas 19 (R)-hydroxy-prostaglandin e2, 3-hydroxy-L-kynurenine, catharanthine, harmalol, p-coumaroyltyramine, melatonin, and salicylate showed a positive correlation (p < 0.01) with B. thermoamylovorans, Kurthia sp. 11kri321, and B. amyloliquefaciens. Among other metabolites, methyl jasmonate was the only metabolite showing a significant correlation (p < 0.01) with B. subtilis, B. licheniformis, and B. paralicheniformis.
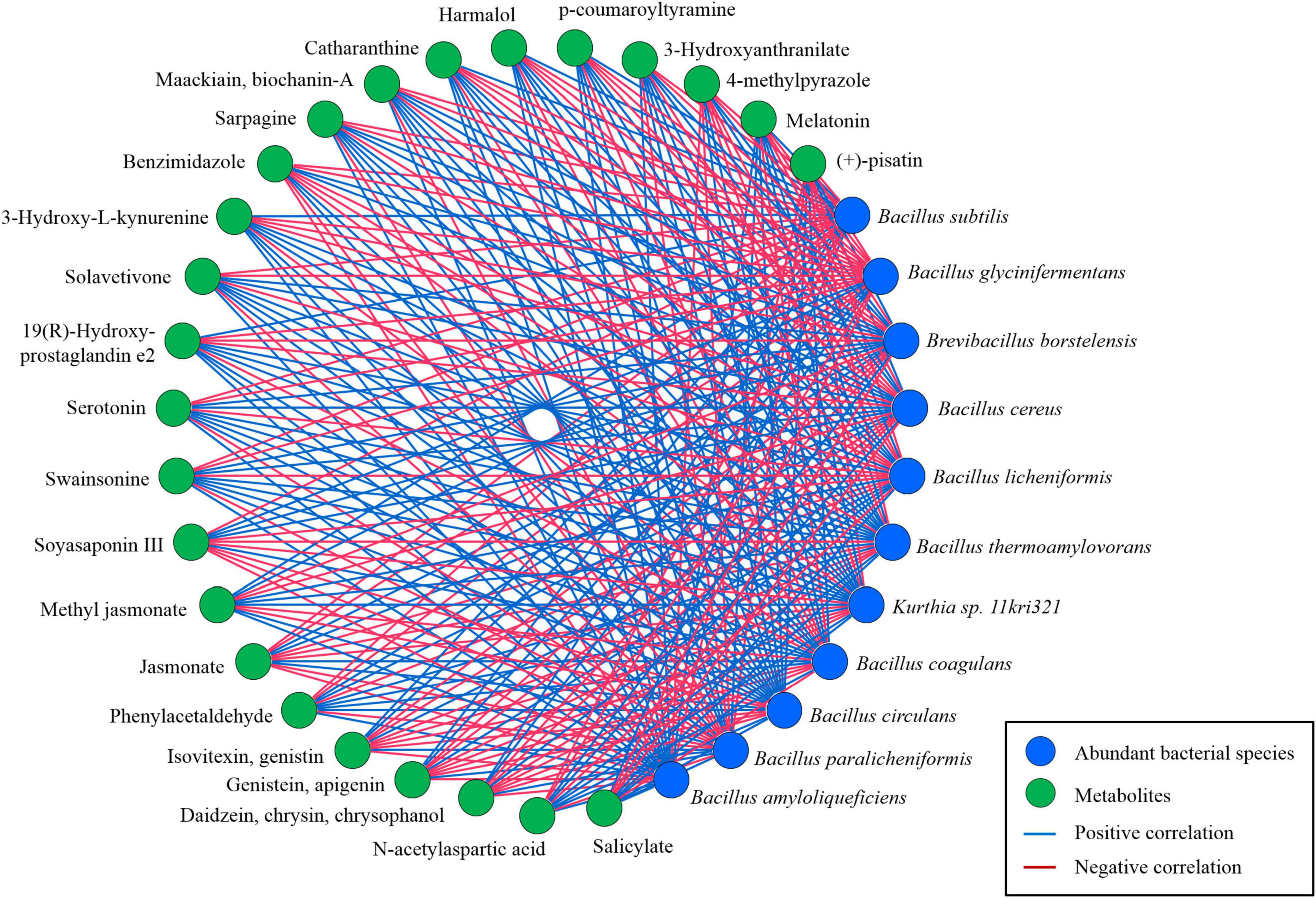
Figure 13. The correlation-based network between predominant microbial species (> 1%) and significant secondary metabolites based on a non-parametric Spearman’s correlation data constructed using MetScape v3.1.3 (Cytoscape v3.8).
Discussion
Microbial Structure
Complex microbial community of major or minor domains exists or co-exists during uncontrolled natural fermentation of plant or animal substrates into fermented foods (Capozzi et al., 2017; Tamang et al., 2020). Kinema is also a fermented product of uncontrolled natural fermentation of soybean with existence of diverse microbial communities. The amplicon sequence of OTUs of kinema samples were earlier analyzed targeting the 16S rRNA gene for bacteria and ITS gene for fungi and yeasts (Kharnaior and Tamang, 2021). However, the profile of the microbial domains that appeared during kinema fermentation is important to know the entire microbial community structure of naturally fermented kinema. Hence, the shotgun metagenomic analysis was performed to explore the microbial community structure of kinema samples collected from India, Nepal, and Bhutan. DNA from the samples of the three countries were pooled in equal quantity and were used for metagenomic analysis. Pooled DNA samples were not significantly (p > 0.1) different in terms of sequence variation within the sample, since the pooled sequence could represent an actual sequence from each initial DNA sample (Anand et al., 2016).
In the organismal diversity, classified bacteria were the most abundant domain in kinema metagenomes, which was also observed in other naturally fermented soybean foods such as pe poke of Myanmar (Tamang et al., 2021b) and cheonggukjang of Korea (Tamang et al., 2022). In terms of relative abundance and occurrence, a colossal diversity of species of Bacillus was detected in kinema samples with 280 species including those detected at < 1% abundances. Previously, by amplicon sequence analysis, only 27 species of Bacillus were reported in kinema samples (Kharnaior and Tamang, 2021). Depth learning of genome mining of kinema has depicted huge microbial ecology, which is possible only by metagenomics using random shotgun sequence of DNA to highlight the higher resolution on taxonomic analyses at low abundance genera or species level (Tovo et al., 2020). Intra/inter-species diversity of Bacillus, both culturable and unculturable, in kinema exists and co-exists for the survival and competition among different species. The dominance of Bacillus in fermented soybean foods is probably due to the alkalinity of the product during fermentation (Gopikrishna et al., 2021) and also its proteolytic activity (Nguyen and Nguyen, 2020). Predominant Bacillus with different species has been reported in other Asian fermented soybean foods such as cheonggukjang (Nam et al., 2012; Tamang et al., 2022), natto (Kubo et al., 2011), pe poke (Tamang et al., 2021b), douchi (Hu et al., 2019), and thua nao (Pakwan et al., 2020). B. subtilis does not cause any health risk to consumers; hence, it is a safe and beneficial bacterium (Su et al., 2020). Besides B. subtilis, the next abundant species of Bacillus detected in kinema metagenome was B. glycinifermentans, which is known to produce many bioactive compounds (Stadermann et al., 2017). Some other major beneficial species of Bacillus, namely, B. thermoamylovorans, a thermostable enzyme producer (Yamada et al., 2017), B. coagulans, a probiotic bacterium (Cao et al., 2020), and B. licheniformis, functional bacterium (Muras et al., 2021), were also detected in kinema samples. However, the abundant species of Bacillus in samples of Bhutan was B. cereus, which was also detected by amplicon sequence analysis of kinema samples (Kharnaior and Tamang, 2021). B. cereus might have existed in fermenting soybeans during the uncontrolled spontaneous fermentation. Though B. cereus is a food-borne pathogen, its pathogenicity varies from harmless to lethal (Kavanaugh et al., 2022). B. borstelensis was also an abundant rod-shaped and Gram-positive bacterium in kinema samples, which is mostly thermoresistant, enzyme producing, and alkaline tolerant (Cotta et al., 2021). About 121 species of LAB (<1% relative abundance) were also detected from kinema samples, showing their co-existence with Bacillus spp. Although their abundance is low (<1%), their existence in naturally fermented soybean food may have fermentative or other functional roles, which may be investigated further.
Abundance of viruses, mostly bacteriophages, was only 1.85% in kinema metagenomes; their appearance might suppress the growth of some pathogenic bacterial species, which were detected in low abundance, probably combating their multiplication for food safety to extend the shelf life of the product (Endersen and Coffey, 2020). Few archaeal species were also detected in kinema; though their abundance was very low, their contributions to health and disease remain unknown (Adam et al., 2017).
Microbial communities in kinema samples of Bhutan showed the highest diversity index compared to those of India and Nepal, probably due to environmental factors (Lee S. et al., 2017), seasonal changes (Kumar et al., 2019), geographical locations (Lee et al., 2019), and inconsistency in traditional preparation practices adopted by the local people. We observed that a set of species was common and unique to each country, and the reason could be the unusual associations of microbial communities and environmental conditions including temperature and humidity. However, the predominance of beneficial microbes with various antimicrobial properties could be the reason for the reduction of the abundance of unwanted microorganisms.
Predictive Functional Profiles
The predictive functional analysis of kinema metagenomes by KEGG annotation revealed the abundance of several metabolic pathways, where carbohydrate metabolism was predominant. The abundance of genes related to galactose metabolism could involve in the degradation of complex sugars in soybeans during fermentation (Chun et al., 2021). In amino acid metabolism, the abundance of genes associated to sub-pathways alanine, aspartate, and glutamate metabolism in kinema metagenome could enhance the aroma and flavor of the product (Yi and Hong, 2021). Glutamate is known as the precursor for the biosynthesis of GABA (Dhakal et al., 2012), which has several bio-functional properties (Diez-Gutiérrez et al., 2020). The presence of genes related to branched-chain amino acids was identified, which could be involved in the metabolism of carbon or nitrogen by B. subtilis (Belitsky, 2015). β-glucosidase detected in kinema could be involved in the biosynthesis of isoflavones and hydrolysis of oligosaccharides (Khosravi and Razavi, 2021).
Metabolite Profiles
Metabolite profiled by LC-MS analysis revealed the presence of several bioactive compounds indicating kinema has several bio-functional properties, which may impart health-promoting benefits to the consumers. Although raw soybeans have various functional compounds, the presence of microorganisms can enhance the levels of production of amino acids and secondary metabolites (Hyeon et al., 2020). The abundance of B. subtilis during the natural fermentation processes of kinema could enhance the nutrient content because of the ability to release hydrolase enzymes that breaks down macromolecules into small molecules and increases the content of metabolites, as also observed in cheonggukjang (Lee et al., 2017; Ali et al., 2018). Some metabolites such as amino acid, namely, alanine, leucine, isoleucine, phenylalanine, and valine, and phenylacetaldehyde were identified in metabolomic analysis of kinema, which are known to contribute to taste and flavor development in the product (Lioe et al., 2018). Different types of isoflavones such as daidzein and genistein were detected in kinema, which are mostly present in soybeans (Choi et al., 2020a,b) and are associated with many health-promoting or immune-stimulating properties (Kim, 2021). The content of isoflavones in kinema could be indirectly influenced by the presence of microbes in the rhizosphere of soybean plants that secretes various secondary metabolites, since soybean is known to produce specialized metabolites such as isoflavones and soyasaponins (Sugiyama, 2019). Group B soyasaponin (soyasaponin III) was also detected in kinema samples, which has an antitumor effect, anti-inflammatory activity, and antimicrobial and cardiovascular-protective activities (Omar et al., 2020). Metabolomics result revealed the presence of vitamin B complex such as biotin, riboflavin, nicotinamide, pyridoxamine, pyridoxine, and (R)-pantothenate in kinema, which are the sources of micronutrients required for bio-functional activities including host immunity (Yoshii et al., 2019). Vitamin K, also known as demethylphylloquinone, was also detected in kinema, which has an anti-inflammatory effect (Harshman and Shea, 2016). The presence of metabolites such as lipoate, 3-hydroxyanthranilate, and melatonin in kinema may be related to antioxidant activities (Galano et al., 2016), and detection of chrysophanol in kinema may have immuno-stimulating effects, anti-inflammatory effects, and anticancer activities (Wen et al., 2018; Prateeksha et al., 2019). Also, p-coumaroyltyramine and 4-methylpyrazole were detected in kinema, and these bio-active compounds are used as anti-diabetic and anti-acetylcholinesterase (Masi et al., 2021). Benzimidazole, as an anti-inflammatory agent (Hashem and Bakri, 2021), is also detected in kinema samples. Among the samples, kinema samples of Bhutan depicted a higher diversity and secondary metabolites comparable to those of India and Bhutan. Therefore, we believe that microorganisms play a crucial role in secreting and changing the chemical diversity of various metabolites under certain environmental conditions including temperature and pH. We also observed that the metabolite abundance differs among kinema from different locations, which suggested that the differences in cultivars or soybean seeds may vary the composition of metabolites (Wang et al., 2019), and also, the formation of metabolites could enhance or affect the accumulation of secondary metabolites by various environmental factors such as regional variation, climates, and methods of preparation (Iqbal et al., 2018).
Validation of Metabolomics With Metagenomics
Predictive functionality generated by KEGG annotation is a hypothetical claim, and prediction of genes is associated with functional profiles. Validation with real-time experiment may verify the predictive metabolic pathways to claim the presence or absence of some metabolites in the samples. Hence, we compared the KEGG database generated by metabolomics analysis with that of predictive metabolic pathways of the abundant metagenomes in kinema samples of India, Nepal, and Bhutan. Stickiness in kinema is due to γ-polyglutamic acid (γ-PGA) (Chettri et al., 2016), which determines the quality of the product preferred by the consumers. B. subtilis and B. licheniformis were both correlated with glutamate production in kinema, probably associated with γ-PGA production (Cai et al., 2017; Zhang C. et al., 2018). The positive correlation between B. amyloliquefaciens and many amino acids in kinema may enhance the antioxidant activity and formation of bioactive compounds and increase the levels of isoflavones (Shahzad et al., 2020). B. coagulans showed a positive correlation with tryptophan, tyrosine, and genistin in this study, which may improve the amino acid absorption (Stecker et al., 2020). The correlation between amino acids and B. borstelensis observed in kinema metagenomes might be the ability of thermostable D-amino acid amidase to produce amino acids at higher temperature (Baek, 2003). Unsaturated fatty acids (UFAs) such as oleate, linoleate, and linolenate were detected in kinema, the biosynthetic pathway of which were also predicted in kinema metagenomes. UFAs are important untargeted metabolites in fermented soybean foods (Park and Kim, 2021), which play an important role in biotic and abiotic stresses (He et al., 2020). The co-existence of B. subtilis with some fungi in kinema might be correlated with increase of unsaturated fatty acids in fermented soybeans (Kanghae et al., 2017). Pantothenate and pyridoxamine were positively correlated with B. subtilis strain, which may enhance the biosynthesis of vitamins and cofactors in kinema, imparting health-promoting benefits to consumers. Serotonin, a neurotransmitter (Sahu et al., 2018), was found to be correlated with the neuroactive ligand–receptor interaction via predictive function, and it could control the signaling pathways either intracellularly by serotonylation or extracellularly via membrane receptors (Bockaert et al., 2021). Swainsonine predicted in KEGG database was also detected in metabolomics analysis in kinema samples, which is involved in the biosynthesis of tropane, piperidine, and pyridine alkaloid (Li and Lu, 2019). A positive correlation between the predominant Bacillus species and the secondary metabolites such as aglycones and glycosides was observed, with the potential ability of Bacillus species in producing secondary metabolites that mediate antibiosis and exhibit as antimicrobial compounds (Caulier et al., 2019). A variation in microbial and metabolite formation in naturally fermented kinema samples of three different countries has been observed, and this may be due to different cultivars of soybean seeds used for fermentation, soil types, geographical and ago-climatic variation, and community- or region-specific conventional methods of preparation. However, the results obtained from the metagenome–metabolite interaction by correlation analysis may provide a better understanding in the selection of strains or targeting metabolites of interest for further studies.
Conclusion
The novel outcome of this study is the metabolomics-driven metagenome mining of kinema, a naturally fermented low-cost plant high protein food in local diet of India, Nepal, and Bhutan, with more than 1,560 species of microbial communities, and the detection of several targeted and untargeted metabolites. The presence of metabolites such as chrysin, swainsonine, and 3-hydroxy-L-kynurenine (anticancer activity); benzimidazole (antimicrobial and anticancer); and compounds with immunomodulatory effects in kinema supports its therapeutic potential. Therefore, this study proves that kinema, a unique Himalayan fermented soybean food, is a rich source of several bioactive compounds, immunomodulators, and vitamins for nutritional supplements as well as for therapeutic uses.
Data Availability Statement
The metagenomic sequences of kinema were submitted and available in the National Center for Biotechnology Information (NCBI) under the Bio-project ID PRJNA695113 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA695113) with the Sequence Read Archive (SRA) numbers SRR13573809 kinema (India) (https://www.ncbi.nlm.nih.gov/sra/SRR13573809), SRR13573808 kinema (Bhutan) (https://www.ncbi.nlm.nih.gov/sra/SRR13573808), and SRR13573807 kinema (Nepal) (https://www.ncbi.nlm.nih.gov/sra/SRR13573807).
Author Contributions
PK: methodology, software, investigation, data curation, and writing—original draft preparation. JT: conceptualization, visualization, supervision, validation, and writing—reviewing and editing. Both authors contributed to the article and approved the submitted version.
Funding
The authors are grateful to the Department of Biotechnology (DBT), Ministry of Science and Technology, New Delhi for financial support (Project No: BT/PR16706/NER/95/259/2015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.868383/full#supplementary-material
References
Adam, P., Borrel, G., Brochier-Armanet, C., and Gribaldo, S. (2017). The growing tree of Archaea: new perspectives on their diversity, evolution and ecology. ISME J. 11, 2407–2425. doi: 10.1038/ismej.2017.122
Ali, M. W., Sjahzad, R., Bilal, S., Adhikari, B., Kim, I. D., Lee, J. D., et al. (2018). Comparison of antioxidants potential, metabolites, and nutritional profiles of Korean fermented soybean (Cheonggukjang) with Bacillus subtilis KCTC 13241. J. Food Sci. Technol. 55, 2871–2880. doi: 10.1007/s13197-018-3202-2
Anand, S., Mangano, E., Barizzone, N., Bordoni, R., Sorosina, M., Clarelli, F., et al. (2016). Next generation sequencing of pooled samples: guideline for variants’ filtering. Sci. Rep. 6:33735. doi: 10.1038/srep33735
Baek, D. H. (2003). Characterization of a thermostable D-stereospecific alanine amidase from Brevibacillus borstelensis BCS-1. Appl. Environ. Microbiol. 69, 980–986. doi: 10.1128/AEM.69.2.980-986.2003
Belitsky, B. R. (2015). Role of branched-chain amino acid transport in Bacillus subtilis CodY activity. J. Bacteriol. 197, 1330–1338. doi: 10.1128/JB.02563-14
Blasche, S., Kim, Y., Mars, R. A. T., Machado, D., Kafkia, E., Milanese, A., et al. (2021). Metabolic cooperation and spatiotemporal niche partitioning in a kefir microbial community. Nat. Microbiol. 6, 196–208. doi: 10.1038/s41564-020-00816-5
Bockaert, J., Bécamel, C., Chaumont-Dubel, S., Claeysen, S., Vandermoere, F., and Marin, P. (2021). Novel and atypical pathways for serotonin signalling. Facul. Rev. 10:52. doi: 10.12703/r/10-52
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cai, D., He, P., Lu, X., Zhua, C., Zhu, J., Wang, Q., et al. (2017). A novel approach to improve poly-γ-glutamic acid production by NADPH regeneration in Bacillus licheniformis WX-02. Sci. Rep. 7:43404. doi: 10.1038/srep43404
Cambeiro-Pérez, N., Hidalgo-Cantabrana, C., Moro-García, M. A., Alonso-Arias, R., Simal-Gándara, J., Sánchez, B., et al. (2018). A metabolomics approach reveals immunomodulatory effects of proteinaceous molecules derived from gut bacteria over human peripheral blood mononuclear cells. Front. Microbiol. 9:2701. doi: 10.3389/fmicb.2018.02701
Cao, J., Yu, Z., Liu, W., Zhao, J., Zhang, H., Zhai, Q., et al. (2020). Probiotic characteristics of Bacillus coagulans and associated implications for human health and diseases. J. Funct. Foods 64:103643. doi: 10.1016/j.jff.2019.103643
Cao, Z. H., Green-Johnson, J. M., Buckley, N. D., and Lin, Q. Y. (2019). Bioactivity of soy-based fermented foods: a review. Biotechnol. Adv. 37, 223–238. doi: 10.1016/j.biotechadv.2018.12.001
Capozzi, V., Fragasso, M., Romaniello, R., Berbegal, C., Russo, P., and Spano, G. (2017). Spontaneous food fermentations and potential risks for human health. Ferment 3:49. doi: 10.3390/fermentation3040049
Caulier, S., Nannan, C., Gillis, A., Licciardi, F., Bragard, C., and Mahillon, J. (2019). Overview of the antimicrobial compounds produced by members of the Bacillus subtilis group. Front. Microbiol. 10:302. doi: 10.3389/fmicb.2019.00302
Chen, C., Huang, H., and Wu, C. H. (2017). Protein bioinformatics databases and resources. Methods Mol. Biol. (Clifton, N.J.) 1558, 3–39. doi: 10.1007/978-1-4939-6783-4_1
Chettri, R., Bhutia, M., and Tamang, J. P. (2016). Poly-γ-glutamic acid (PGA)-producing Bacillus species isolated from Kinema, Indian fermented soybean food. Front. Microbiol. 7:971. doi: 10.3389/fmicb.2016.00971
Choi, Y. M., Yoon, H., Lee, S., Ko, H. C., Shin, M. J., Lee, M. C., et al. (2020a). Isoflavones, anthocyanins, phenolic content, and antioxidant activities of black soybeans (Glycine max (L.) Merrill) as affected by seed weight. Sci. Rep. 10:19960. doi: 10.1038/s41598-020-76985-4
Choi, Y. R., Shim, J., and Kim, M. J. (2020b). Genistin: a novel potent anti-adipogenic and anti-lipogenic agent. Molecules 25:2042. doi: 10.3390/molecules25092042
Chun, B. H., Han, D. M., Kim, H. M., Park, D., Jeong, D. M., Kang, H. A., et al. (2021). Metabolic features of ganjang (a Korean Traditional Soy Sauce) fermentation revealed by genome-centered metatranscriptomics. mSystems 6:e0044121. doi: 10.1128/mSystems.00441-421
Cotta, S. P. M., Marins, M. S., Marriel, I. E., Lana, U. G. P., Gomes, E. A., Figueiredo, J. E. F., et al. (2021). Thermo-resistant enzyme-producing microorganisms isolated from composting. Brazilian J. Biol. 83:e244205. doi: 10.1590/1519-6984.244205
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dhakal, R., Bajpai, V. K., and Baek, K. H. (2012). Production of GABA (γ - aminobutyric acid) by microorganisms: a review. Brazilian J. Microbiol. 43, 1230–1241. doi: 10.1590/S1517-83822012000400001
Diez-Gutiérrez, L., Vicente, L. S., Barrón, L. J. R., del Carmen, Villarán, M., and Chávarri, M. (2020). Gamma-aminobutyric acid and probiotics: multiple health benefits and their future in the global functional food and nutraceuticals market. J. Funct. Foods 64:103669. doi: 10.1016/j.jff.2019.103669
Djeni, T. N., Kouame, K. H., Ake, F. D. M., Amoikon, L. S. T., Dje, M. K., and Jeyaram, K. (2020). Microbial diversity and metabolite profiles of palm wine produced from three different palm tree species in Côte d’Ivoire. Sci. Rep. 10:1715. doi: 10.1038/s41598-020-58587-58582
Durazzi, F., Sala, C., Castellani, G., Manfreda, G., Remondini, D., and De Cesare, A. (2021). Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 11:3030. doi: 10.1038/s41598-021-82726-y
Endersen, L., and Coffey, A. (2020). The use of bacteriophages for food safety. Cur. Opin. Food Sci. 36, 1–8. doi: 10.1016/j.cofs.2020.10.006
Eudy, B. J., McDermott, C. E., Liu, X., and da Silva, R. P. (2020). Targeted and untargeted metabolomics provide insight into the consequences of glycine-N-methyltransferase deficiency including the novel finding of defective immune function. Physiol. Rep. 8:e14576. doi: 10.14814/phy2.14576
Fong, Y., and Huang, Y. (2019). Modified Wilcoxon-Mann-Whitney test and power against strong null. Am. Stat. 73, 43–49. doi: 10.1080/00031305.2017.1328375
Galano, A., Castañeda-Arriaga, R., Pérez-González, A., Tan, D. X., and Reiter, R. J. (2016). Phenolic melatonin-related compounds: their role as chemical protectors against oxidative stress. Molecules 21:1442. doi: 10.3390/molecules21111442
González-Peña, D., and Brennan, L. (2019). Recent advances in the application of metabolomics for nutrition and health. Ann. Rev. Food Sci. Technol. 10, 479–519. doi: 10.1146/annurev-food-032818-121715
Gopikrishna, T., Kumar, H. K. S., Perumal, K., and Elangovan, E. (2021). Impact of Bacillus in fermented soybean foods on human health. Ann. Microbiol. 71, 1–16. doi: 10.1186/s13213-021-01641-9
Hao, L., Wang, J., Page, D., Asthana, S., Zetterberg, H., Carlsson, C., et al. (2018). Comparative evaluation of MS-based metabolomics software and its application to preclinical Alzheimer’s disease. Sci. Rep. 8:9291. doi: 10.1038/s41598-018-27031-x
Harshman, S. G., and Shea, M. K. (2016). The role of vitamin k in chronic aging diseases: inflammation, cardiovascular disease, and osteoarthritis. Cur. Nutri. Rep. 5, 90–98. doi: 10.1007/s13668-016-0162-x
Hashem, H. E., and Bakri, Y. E. (2021). An overview on novel synthetic approaches and medicinal applications of benzimidazole compounds. Arab. J. Chem. 14, 103418.
Hartley, I. E., Liem, D. G., and Keast, R. (2019). Umami as an ‘alimentary’ taste. a new perspective on taste classification. Nutrients 11:182. doi: 10.3390/nu11010182
He, M., Qin, C. X., Wang, X., and Ding, N. Z. (2020). Plant unsaturated fatty acids: biosynthesis and regulation. Front. Plant Sci. 11:390. doi: 10.3389/fpls.2020.00390
Hu, Y., Yu, D., Wang, Z., Hou, J., Tyagi, R., Liang, Y., et al. (2019). Purification and characterization of a novel, highly potent fibrinolytic enzyme from Bacillus subtilis DC27 screened from Douchi, a traditional Chinese fermented soybean food. Sci. Rep. 9:9235. doi: 10.1038/s41598-019-45686-y
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Hyatt, D., LoCascio, P. F., Hauser, L. J., and Uberbacher, E. C. (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230. doi: 10.1093/bioinformatics/bts429
Hyeon, H., Min, C. W., Moon, K., Cha, J., Gupta, R., Park, S. U., et al. (2020). Metabolic profiling-based evaluation of the fermentative behavior of Aspergillus oryzae and Bacillus subtilis for soybean residues treated at different temperatures. Foods 9:117. doi: 10.3390/foods9020117
Iqbal, N., Zhang, Q., Wu, H., Yang, C., Deng, J., Qin, W., et al. (2018). Soybean (Glycine max L.) germplasm screening and geographical determination based on targeted isoflavone metabolomics. Appl. Ecol. Environ. Res. 16, 3933–3953. doi: 10.15666/aeer/1604_39333953
Ju, S., Cao, Z., Wong, C., Liu, Y., Foda, M. F., Zhang, Z., et al. (2019). Isolation and optimal fermentation condition of the Bacillus subtilis subsp. natto strain WTC016 for nattokinase production. Ferment 5:92. doi: 10.3390/fermentation5040092
Kadar, A. D., Astawan, M., Putri, S. P., and Fukusaki, E. (2020). Metabolomics-based study of the effect of raw materials to the end product of tempe—an Indonesian fermented soybean. Metabolites 10:367. doi: 10.3390/metabo10090367
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30.
Kanghae, A., Eungwanichayapant, D. P., and Chukeatirote, E. (2017). Fatty acid profiles of fermented soybean prepared by Bacillus subtilis and Rhizopus oligosporus. Environ. Exp. Biol. 15, 173–176.
Kavanaugh, D. W., Glasset, B., Dervyn, R., Guérin, C., Plancade, S., Herbin, S., et al. (2022). New genetic biomarkers to differentiate non-pathogenic from clinically relevant Bacillus cereus strains. Clin. Microbiol. Infect. 28, 137.e1–137.e8. doi: 10.1016/j.cmi.2021.05.035
Kharnaior, P., and Tamang, J. P. (2021). Bacterial and fungal communities and their predictive functional profiles in kinema, a naturally fermented soybean food of India, Nepal and Bhutan. Food Res. Int. 140:110055. doi: 10.1016/j.foodres.2020.110055
Khosravi, A., and Razavi, S. H. (2021). Therapeutic effects of polyphenols in fermented soybean and black soybean products. J. Function. Foods 81:104467. doi: 10.1016/j.jep.2020.113236
Kim, I. S. (2021). Current perspectives on the beneficial effects of soybean isoflavones and their metabolites for humans. Antioxidants 10:1064. doi: 10.3390/antiox10071064
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kubo, Y., Rooney, A. P., Tsukakoshi, Y., Nakagawa, R., Hasegawa, H., and Kimura, K. (2011). Phylogenetic analysis of Bacillus subtilis strains applicable to natto (fermented soybean) production. Appl. Environ. Microbiol. 77, 6463–6469. doi: 10.1128/AEM.00448-11
Kumar, J., Sharma, N., Kaushal, G., Samurailatpam, S., Sahoo, D., Rai, A. K., et al. (2019). Metagenomic insights into the taxonomic and functional features of Kinema, a traditional fermented soybean product of Sikkim Himalaya. Front. Microbiol. 10:1744. doi: 10.3389/fmicb.2019.01744
Kwon, Y. S., Lee, S., Lee, S. H., Kim, H. J., and Lee, C. H. (2019). Comparative evaluation of six traditional fermented soybean products in East Asia: a metabolomics approach. Metabolites 9:183. doi: 10.3390/metabo9090183
Lazar, I. M., Karcini, A., Ahuja, S., and Estrada-Palma, C. (2019). Proteogenomic analysis of protein sequence alterations in breast cancer cells. Sci. Rep. 9:10381. doi: 10.1038/s41598-019-46897-z
Lee, D. H., Kim, M. J., Ahn, J., Lee, S. H., Lee, H., Kim, J. H., et al. (2017). Nutrikinetics of isoflavone metabolites after fermented soybean product (cheonggukjang) ingestion in ovariectomized mice. Mol. Nutr. Food Res. 61:1700322. doi: 10.1002/mnfr.201700322
Lee, E. M., Park, S. J., Lee, J. E., Lee, B. M., Shin, B. K., Kang, D. J., et al. (2019). Highly geographical specificity of metabolomic traits among Korean domestic soybeans (Glycine max). Food Res. Int. 120, 12–18. doi: 10.1016/j.foodres.2019.02.021
Lee, S., Lee, S., Singh, D., Oh, J. Y., Jeon, E. J., Ryu, H. S., et al. (2017). Comparative evaluation of microbial diversity and metabolite profiles in doenjang, a fermented soybean paste, during the two different industrial manufacturing processes. Food Chem. 221, 1578–1586. doi: 10.1016/j.foodchem.2016.10.135
Leech, J., Cabrera-Rubio, R., Walsh, A. M., Macori, G., Walsh, C. J., Barton, W., et al. (2020). Fermented-food metagenomics reveals substrate-associated differences in taxonomy and health-associated and antibiotic resistance determinants. mSystems 5:00522-20 doi: 10.1128/mSystems.00522-20
Li, X., and Lu, P. (2019). Transcriptome profiles of Alternaria oxytropis provides insights into swainsonine biosynthesis. Sci. Rep. 9:6021. doi: 10.1038/s41598-019-42173-2
Lioe, H. N., Kinjo, A., Yasuda, S., Kuba-Miyara, M., Tachibana, S., and Yasuda, M. (2018). Taste and chemical characteristics of low molecular weight fractions from tofuyo-Japanese fermented soybean curd. Food Chem. 252, 265–270. doi: 10.1016/j.foodchem.2018.01.117
Liu, J., Chen, J., Li, S., Tian, W., Wu, H., and Han, B. (2021). Comparison of volatile and non-volatile metabolites in sufu produced with Bacillus licheniformis by rapid fermentation. Int. J. Food Prop. 24, 553–563. doi: 10.1080/10942912.2021.1901733
Loman, N. J., and Quinlan, A. R. (2014). Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401. doi: 10.1093/bioinformatics/btu555
Martino, C., Morton, J. T., Marotz, C. A., Thompson, L. R., Tripathi, A., Knight, R., et al. (2019). A Novel sparse compositional technique reveals microbial perturbations. mSystems 4:e00016-19. doi: 10.1128/mSystems.00016-19
Masi, M., Koirala, M., Delicato, A., Di Lecce, R., Merindol, N., Ka, S., et al. (2021). Isolation and biological characterization of homoisoflavanoids and the alkylamide N-p-coumaroyltyramine from Crinum biflorum Rottb., an amaryllidaceae species collected in Senegal. Biomolecules 11:1298. doi: 10.3390/biom11091298
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257. doi: 10.1038/ncomms11257
Muras, A., Romero, M., Mayer, C., and Otero, A. (2021). Biotechnological applications of Bacillus licheniformis. Crit. Rev. Biotechnol. 41, 609–627. doi: 10.1080/07388551.2021.1873239
Nam, Y. D., Yi, S. H., and Lim, S. I. (2012). Bacterial diversity of cheonggukjang, a traditional Korean fermented food, analyzed by barcoded pyrosequencing. Food Control 28, 135–142. doi: 10.1016/j.foodcont.2012.04.028
Nguyen, T., and Nguyen, C. H. (2020). Determination of factors affecting the protease content generated in fermented soybean by Bacillus subtilis 1423. Energy Rep. 6, 831–836. doi: 10.1016/j.egyr.2019.11.011
Nikkuni, S., Karki, T. B., Vilku, K. S., Suzuki, T., Shindoh, K., Suzuki, C., et al. (1995). Mineral and amino acid contents of kinema, a fermented soybean food prepared in Nepal. Food Sci. Technol. Int. 1, 107–111. doi: 10.3136/fsti9596t9798.1.107
Omar, A., Kalra, R. S., Putri, J., Elwakeel, A., Kaul, S. C., Wadhwa, R., et al. (2020). Soyasapogenol-A targets CARF and results in suppression of tumor growth and metastasis in p53 compromised cancer cells. Sci. Rep. 10:6323. doi: 10.1038/s41598-020-62953-5
Pakwan, C., Chitov, T., Chantawannakul, P., Manasam, M., Bovonsombut, S., and Disayathanoowa, T. (2020). Bacterial compositions of indigenous Lanna (Northern Thai) fermented foods and their potential functional properties. PLoS One 15:e0242560. doi: 10.1371/journal.pone.0242560
Pang, Z., Chong, J., Zhou, G., de Lima Morais, D. A., Chang, L., Barrette, M., et al. (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 39, W388–W396. doi: 10.1093/nar/gkab382
Park, M. K., and Kim, Y. S. (2021). Mass spectrometry based metabolomics approach on the elucidation of volatile metabolites formation in fermented foods: a mini review. Food Sci. Biotechnol. 30, 881–890. doi: 10.1007/s10068-021-00917-9
Parks, D. H., and Beiko, R. G. (2012). Measuring community similarity with phylogenetic networks. Mol. Biol. Evol. 29, 3947–3958. doi: 10.1093/molbev/mss200
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Pokrzywa, R., and Polanski, A. (2010). BWtrs: a tool for searching for tandem repeats in DNA sequences based on the Burrows-Wheeler transform. Genomics 96, 316–321. doi: 10.1016/j.ygeno.2010.08.001
Prateeksha, M. A. Y., Singh, B. N., Sudheer, S., Kharwar, R. N., Siddiqui, S., et al. (2019). Chrysophanol: a natural anthraquinone with multifaceted biotherapeutic potential. Biomolecules 9:68. doi: 10.3390/biom9020068
Ramakrishnan, P., Nair, S., and Rangiah, K. (2016). A method for comparative metabolomics in urine using high resolution mass spectrometry. J. Chromatogr. A 1443, 83–92. doi: 10.1016/j.chroma.2016.02.080
Ray, K. J., Cotter, S. Y., Arzika, A. M., Kim, J., Boubacar, N., Zhou, Z., et al. (2019). High-throughput sequencing of pooled samples to determine community-level microbiome diversity. Ann. Epidemiol. 39, 63–68. doi: 10.1016/j.annepidem.2019.09.002
Ren, F., Yan, D. H., Liu, Y., Wang, C., and Guo, C. (2021). Bacterial and fungal communities of traditional fermented Chinese soybean paste (Doujiang) and their properties. Food Sci. Nutr. 9, 5457–5466. doi: 10.1002/fsn3.2505
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sahu, A., Gopalakrishnan, L., Gaur, N., Chatterjee, O., Mol, P., Modi, P. K., et al. (2018). The 5-Hydroxytryptamine signalling map: an overview of serotonin-serotonin receptor mediated signalling network. J. Cell Com. Signal 12, 731–735. doi: 10.1007/s12079-018-0482-2
Sarkar, P. K., Tamang, J. P., Cook, P. E., and Owens, J. D. (1994). Kinema—a traditional soybean fermented food: proximate composition and microflora. Food Microbiol. 11, 47–55. doi: 10.1006/fmic.1994.1007
Scala, G., Serra, A., Marwah, V. S., Saarimaki, L. A., and Greco, D. (2019). FunMappOne: a tool to hierarchically organize and visually navigate functional gene annotations in multiple experiments. BMC Bioinform. 20:79. doi: 10.1186/s12859-019-2639-2
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Sevim, V., Lee, J., Egan, R., Clum, A., Hundley, H., Lee, J., et al. (2019). Shotgun metagenome data of a defined mock community using Oxford nanopore, PacBio and Illumina technologies. Sci. Data 6:285. doi: 10.1038/s41597-019-0287-z
Shahbazi, R., Sharifzad, F., Bagheri, R., Alsadi, N., Yasavoli-Sharahi, H., and Matar, C. (2021). Anti-inflammatory and immunomodulatory properties of fermented plant foods. Nutrients 13:1516. doi: 10.3390/nu13051516
Shahzad, R., Shehzad, A., Bilal, S., and Lee, I. J. (2020). Bacillus amyloliquefaciens RWL-1 as a new potential strain for augmenting biochemical and nutritional composition of fermented soybean. Molecules 25:2346. doi: 10.3390/molecules25102346
Shurtleff, W., and Aoyagi, A. (2017). History of Fermented Soyfoods in the Mekong Basin (1637-2017): Extensively Annotated Bibliography and Sourcebook. Lafayette, CA: Soyinfo Center.
Song, D. H., Chun, B. H., Lee, S., Son, S. Y., Reddy, C. K., Mun, H. I., et al. (2021). Comprehensive metabolite profiling and microbial communities of Doenjang (fermented soy paste) and Ganjang (fermented soy sauce): a comparative study. Foods 10:641. doi: 10.3390/foods10030641
Stadermann, K. B., Blom, J., Borgmeier, C., Sciberras, N., Herbold, S., Kipker, M., et al. (2017). First complete genome sequence of Bacillus glycinifermentans B-27. J. Biotechnol. 257, 187–191. doi: 10.1016/j.jbiotec.2017.04.021
Stecker, R. A., Moon, J. M., Russo, T. J., Mumford, P. W., Jager, R., Purpura, M., et al. (2020). Bacillus coagulans GBI-30, 6086 improves amino acid absorption from milk protein. Nutr. Metab. (Lond.) 17:93. doi: 10.1186/s12986-020-00515-2
Su, Y., Liu, C., Fang, H., and Zhang, D. (2020). Bacillus subtilis: a universal cell factory for industry, agriculture, biomaterials and medicine. Microb. Cell Fact. 19:173. doi: 10.1186/s12934-020-01436-8
Sugiyama, A. (2019). The soybean rhizosphere: metabolites, microbes, and beyond—a review. J. Adv. Res. 19, 67–73. doi: 10.1016/j.jare.2019.03.005
Takeshita, N. (2020). Fungal research in Japan: tradition and future. Fungal Bio. Biotechnol. 7:14. doi: 10.1186/s40694-020-00104-1
Tamames, J., and Puente-Sánchez, F. (2019). SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 9:3349. doi: 10.3389/fmicb.2018.03349
Tamang, J. P. (2003). Native microorganisms in fermentation of kinema. Indian J. Microbiol. 43, 127–130.
Tamang, J. P. (2015). Naturally fermented ethnic soybean foods of India. J. Ethnic Foods 2, 8–17. doi: 10.1016/j.jef.2015.02.003
Tamang, J. P. (2021). “Ethno-microbiology” of ethnic Indian fermented foods and alcoholic beverages. J. Appl. Microbiol 1–17. doi: 10.1111/jam.15382
Tamang, J. P., and Nikkuni, S. (1996). Selection of starter cultures for the production of kinema, a fermented soybean food of the Himalaya. World J. Microbiol. Biotechnol. 12, 629–635. doi: 10.1007/BF00327727
Tamang, J. P., and Samuel, D. (2010). “Dietary culture and antiquity of fermented foods and beverages,” in Fermented Foods and Beverages of the World, eds J. P. Tamang and K. Kailasapathy (New York, NY: CRC Press, Taylor & Francis Group). doi: 10.3389/fmicb.2021.797295
Tamang, J. P., Cotter, P., Endo, A., Han, N. S., Kort, R., Liu, S. Q., et al. (2020). Fermented foods in a global age: east meets west. Compre. Rev. Food Sci. Food Safe. 19, 184–217. doi: 10.1111/1541-4337.12520
Tamang, J. P., Das, D., Kharnaior, P., Pariyar, P., Thapa, N., Jo, S. W., et al. (2022). Shotgun metagenomics of cheonggukjang, a fermented soybean food of Korea: community structure, predictive functionalities and amino acids profile. Food Res. Int. 151:110904. doi: 10.1016/j.foodres.2021.110904
Tamang, J. P., Jeyaram, K., Rai, A. K., and Mukherjee, P. K. (2021a). Diversity of beneficial microorganisms and their functionalities in community-specific ethnic fermented foods of the Eastern Himalayas. Food Res. Int. 148:110633. doi: 10.1016/j.foodres.2021.110633
Tamang, J. P., Kharnaior, P., Pariyar, P., Thapa, N., Lar, N., Win, K. S., et al. (2021b). Shotgun sequence- based metataxonomic and predictive functional profiles of Pe poke, a naturally fermented soybean food of Myanmar. PLoS One 16:e0260777. doi: 10.1371/journal.pone.0260777
Tamang, J. P., Shin, D. H., Jung, S. J., and Chae, S. W. (2016). Functional properties of microorganisms in fermented foods. Front. Microbiol. 7:578. doi: 10.3389/fmicb.2016.00578
Tovo, A., Menzel, P., Krogh, A., Lagomarsino, M. C., and Suweis, S. (2020). Taxonomic classification method for metagenomics based on core protein families with Core-Kaiju. Nuclei Acids Res. 48:e93. doi: 10.1093/nar/gkaa568
Villarreal-Soto, S. A., Bouajila, J., Pace, M., Leech, J., Cotter, P. D., Souchard, J. P., et al. (2020). Metabolome-microbiome signatures in the fermented beverage, Kombucha. Int. J. Food Microbiol. 333:108778. doi: 10.1016/j.ijfoodmicro.2020.108778
Wang, J., Zhou, P., Shi, X., Yang, N., Yan, L., Zhao, Q., et al. (2019). Primary metabolite contents are correlated with seed protein and oil traits in near-isogenic lines of soybean. Crop J. 7, 651–659. doi: 10.1016/j.cj.2019.04.002
Wen, Q., Mei, L., Ye, S., Liu, X., Xu, Q., Miao, J., et al. (2018). Chrysophanol demonstrates anti-inflammatory properties in LPS-primed RAW 264.7 macrophages through activating PPAR-γ. Int. Immunopharmacol. 56, 90–97. doi: 10.1016/j.intimp.2018.01.023
Yamada, C., Sawano, K., Iwase, N., Matsuoka, M., Arakawa, T., Nishida, S., et al. (2017). Isolation and characterization of a thermostable lipase from Bacillus thermoamylovorans NB501. J. Gen. Appl. Microbiol. 62, 313–319. doi: 10.2323/jgam.2016.06.002
Ye, Y., and Doak, T. G. (2009). A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 5:e1000465. doi: 10.1371/journal.pcbi.1000465
Yi, S. H., and Hong, S. P. (2021). Characteristics of bacterial strains with desirable flavor compounds from Korean traditional fermented soybean paste (Doenjang). Molecules 26:5067. doi: 10.3390/molecules26165067
Yoshii, K., Hosomi, K., Sawane, K., and Kunisawa, J. (2019). Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front. Nutr. 6:48. doi: 10.3389/fnut.2019.00048
Zhang, B., Hu, S., Baskin, E., Patt, A., Siddiqui, J. K., and Mathé, E. A. (2018). RaMP: a comprehensive relational database of metabolomics pathways for pathway enrichment analysis of genes and metabolites. Metabolites 8:16. doi: 10.3390/metabo8010016
Zhang, C., Wu, D., and Qiu, X. (2018). Stimulatory effects of amino acids on γ-polyglutamic acid production by Bacillus subtilis. Sci. Rep. 8: 17934.
Zhang, P., Dreher, K., Karthikeyan, A., Chi, A., Pujar, A., Caspi, R., et al. (2010). Creation of a genome-wide metabolic pathway database for Populus trichocarpa using a new approach for reconstruction and curation of metabolic pathways for plants. Plant Physiol. 153, 1479–1491. doi: 10.1104/pp.110.157396
Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214. doi: 10.1089/10665270050081478
Keywords: fermented soybean, metagenomics, metabolomics, immunodomulators, metataxonomic
Citation: Kharnaior P and Tamang JP (2022) Metagenomic-Metabolomic Mining of Kinema, a Naturally Fermented Soybean Food of the Eastern Himalayas. Front. Microbiol. 13:868383. doi: 10.3389/fmicb.2022.868383
Received: 02 February 2022; Accepted: 24 February 2022;
Published: 29 April 2022.
Edited by:
Fatih Ozogul, Çukurova University, TurkeyCopyright © 2022 Kharnaior and Tamang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jyoti Prakash Tamang, anlvdGlfdGFtYW5nQGhvdG1haWwuY29t