 Tianjiao Li1,2Yiming Li1,2Xiuguo Ma1Xue Dan1Xianjiao Huang1Qinying Li3Shimin Lei1
Tianjiao Li1,2Yiming Li1,2Xiuguo Ma1Xue Dan1Xianjiao Huang1Qinying Li3Shimin Lei1 Zhengchun Zhang4
Zhengchun Zhang4 Sheng Huang2Wei Jiang2
Sheng Huang2Wei Jiang2 Yanhua Yu2
Yanhua Yu2 Yong-Qiang He1,2*
Yong-Qiang He1,2*- 1College of Agriculture, Guangxi University, Nanning, China
- 2State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresource and College of Life Science and Technology, Nanning, China
- 3Napo Agricultural and Rural Bureau, Napo County, Baise, China
- 4New Rural Development Institute of Guangxi University, Nanning, China
Xanthomonas oryzae pv. textitoryzae (Xoo) is a causal agent of rice bacterial leaf blight (BLB), the major rice disease, which is seriously constraining rice production in Asia. The interaction between Xoo and rice is in a dynamic process, essentially the co-evolution. Tracking the occurrence of plant diseases and identifying the epidemic pathogens in time are critical to assessing the epidemic disease status and understanding the pathogen evolution. In 2020, the occurrences of rice BLB were spotted in many places of Guangxi, the major rice growing region in China. Two of the 2020-epidemic Xoo strains, namely, GXO20-01 and GXO20-06, were isolated from low land and high mountain paddies in Guangxi, respectively, and were demonstrated to be race R8 of Chinese Xoo strains, but with significantly different virulence on certain susceptible varieties of rice. The HiFi PacBio sequencing revealed that GXO20-01 and GXO20-06 share the highly syntenic genome structures and the major genome contents, but only differ in <10 genes, including one gene encoding for transcription activator-like effector (TALE). A phylogenomic analysis grouped GXO20-01 and GXO20-06 into the PX-A lineage, stood close to PXO563 and PXO71 strains, but stood away from the other Chinese Xoo strains; for example, the JL25 and YC11. A comparative genomic analysis revealed that the major pathogenicity/virulence genes are conserved in two, newly isolated Xoo strains and the other Xoo strains in PX-A lineage, including the majority genes for the TALomes. The genomic differences between the Xoo strains were pinpointed to a few tal genes, which were variable in both their numbers and sequences, even between GXO20-01 and GXO20-06, the two 2020-epidemic Xoo strains. The study further revealed the instability and variability of tal genes in Xoo and highlighted the utility of HiFi long-read sequencing in TALE analysis and pathogen tracking.
Introduction
The gram-negative bacterium Xanthomonas oryzae pv. oryzae (Xoo) causes the rice bacterial leaf blight (BLB), one of the most destructive rice diseases in the major rice-growing regions worldwide, seriously affecting the quantity and quality of the staple crop rice (Niño-Liu et al., 2006). The interactions between Xoo and rice plants follow the gene for gene hypothesis to some extent, enabling Xoo-rice pathosystem as an excellent model platform in plant sciences (Zhang and Wang, 2013). The Xoo infects host plants depending on a series of virulence factors, including adhesins, degradative enzymes, polysaccharides, and especially the proteinous effectors, translocated via the type III secretion system, which is also known as type III secretion effectors (T3SEs) (An et al., 2020). Among the T3SEs, the transcription activator-like effectors (TALEs), which are presumed as one of the highest developed and most complex pathogenic factors in phytopathogenic bacteria, play crucial roles in pathogenicity and racial discriminations in Xoo-rice interaction (Erkes et al., 2017). A typical TALE consists of three parts: The N-terminal domain containing the type III secretion signal, the C-terminal domain that is acting a role in nuclear localization and transcriptional activation, and the central highly conserved repeat units. Each repeat unit contains 33–35 amino acids, in which the 12th and 13th amino acids are variable, called the repeat variable di-residue (RVD). Different RVDs can bind different bases of target DNA, thus ensuring that TALEs contain multiple RVDs that can recognize specific DNA sequences (Mak et al., 2012). The main function of a TALE is binding to the effector binding element (EBE) in the host genome to induce the expression of downstream susceptibility genes (Kay and Bonas, 2009; Moscou and Bogdanove, 2009). In most cases, the target genes are sugar–transporter genes, that is, the SWEET genes (Streubel et al., 2013). Alternatively, some TALEs, especially the iTALE, may trigger plant defense by activating an executor resistance gene or neutralizing R-gene-mediated plant disease resistance (Ji et al., 2016; Read et al., 2016).
The TALE-encoding genes (tal genes) in Xoo are presumed to be highly dynamic and variations in TALE repeats, such as recombination, duplication, InDels, and SNPs in RVDs, have greatly contributed to the production of new toxicity and the escape of plant immunity by the pathogens (Erkes et al., 2017). Tracking and identifying the variation of tal gene in the emerging or reemerging Xoo strains are helpful for accurate disease diagnosis and disease resistance breeding. However, the highly repetitive structure and the extreme similarity among the different members of tal genes in an individual Xoo strain greatly constrict correctly sequencing and assembling of such genes in the genome by using previous sequencing methods, e.g., the Sanger and the second-generation sequencing methods. Recently, long-read sequencing technologies are overcoming the early limitations in accuracy and throughput, thereby broadening their application domains in complex genomics. In GenBank, there are about 113 Xoo strains whose genomes were claimed to have been completely sequenced, but their sequencing qualities are of significant differences. The tal gene sequences and their distributions are of significant diversity amongst Xoo strains. An individual Xoo strain usually contains 9–21 tal genes, 9 per genome for African strains, and 13–21 per genome for Asian strains (Oliva et al., 2019). To date, about 100 tal genes composing the pan-TALome of Xoo, divided into 30 types, have been identified in Xoo strains. In rice plants, there are more than 20 OsSWEET genes, but in nature, only three of them, namely, OsSWEET11, OsSWEET13, and OsSWEET14 are demonstrated to be induced by Xoo (Oliva et al., 2019). In view of this, there are still a lot of unknown parts to be excavated in the updating Xoo-rice pathosystem.
In the time duration between the 1970s and 1990s, the rice BLB disease was rampant in the vast rice growing area, the south of the Yangtze River, which caused great losses to rice production in China. However, in the early 21st century, the rice BLB suddenly disappeared in most of the rice growing areas in China for some period of time, supposedly due to the breeding and promotion of disease-resistant rice varieties and the intensive applications of chemical germicides and antibiotics. However, in the last decade, the cases of BLB disease have been observed or/and reported increasingly in major rice growing areas in China, even serious occurrences in some certain areas were witnessed (Chen et al., 2012, 2016, 2017, 2019, 2021). The roller coaster pattern of the BLB incidences just reflects the dynamic zig-zag model in the interaction of pathogen and host plants while their long-term co-evolution in the virulence factors and the resistance genes (Jones and Dangl, 2006). Under certain environmental conditions, the occurrence of plant diseases indicates that the balance between plants and pathogens has been broken. The reason is either the loss of plant disease resistance genes or the evolution of virulence factors of pathogens. Therefore, the investigation and documentation of the occurrences or re-occurrences of Xoo strains were conducive to the effective control of BLB and targeted the disease-resistant breeding in rice production.
Guangxi is located in southern China at low latitude, which shares borders with Vietnam, and the Tropic of Cancer passes across its center. With a subtropical monsoon humid climate in Guangxi, the abundant rainfall and fairly high temperature ensure most of Guangxi is a highly suitable double cropping rice region in China. However, Guangxi has many mountainous areas where the lower accumulated temperature can only sustain the single-cropping rice production. The complex cropping rice production systems (such as mixed single- and double-cropping) pose potential challenges in plant protection. Previously, it was noticed that the differences in occurrences and incidences of rice bacterial blight in between high mountain and low land paddies in Guangxi, but little is known about the differences in their pathogens. In 2020, the occurrence of rice BLB was spotted in many places in Guangxi. We carried out a systematic investigation on rice BLB of Guangxi in 2020 and isolated the representative isolates from the typical disease area (Li et al., 2021). In this study, we focused on the comparative studies and genomic analysis on the representative Xoo strains of 2020 BLB occurrences isolated from low land and high mountain paddies in Guangxi, China.
Materials and Methods
Bacterial Strains and Rice Varieties Used in This Study
In 2020, we carried out a systematic survey about rice BLB in Guangxi (Li et al., 2021) and had isolated and identified the Xoo from the BLB frequent onset areas. The representative Xoo strains were named as GXO20-serial number; e.g., GXO20-01 in which GXO indicates the Guangxi Xanthomonas oryzae strain and 20-01 indicates chronologically the No. 1 strain isolated in 2020 (Table 1). To comparatively study Xoo strains isolated from the low land and the high mountain paddies in Guangxi, China, we have selected the representative Xoo strain GXO20-01 from Heng County, the typical low land area in Guangxi, and GXO20-06 from Napo County, the typical high mountain area in Guangxi. The distance between the two places is about 500 km, along the Tropic of Cancer. The bacterial strains used in this study are listed in Table 1. The Xoo international type strain Xoo PXO99A was provided by the Institute of Plant Protection of Nanjing Agricultural University. The race R5 of Chinese strain YC11 was provided by the College of Life Science and Technology, Huazhong Agricultural University (Zheng et al., 2020).
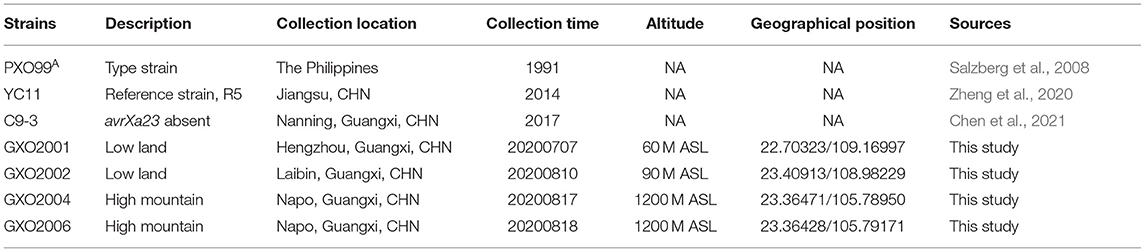
Table 1. Xanthomonas oryzae pv. oryzae strains used in this study.
The differential rice varieties used in this study were a simplified combination of near-isogenic lines (Fang et al., 1990; Liu et al., 2007) (Supplementary Table S1), IRBB2, IRBB3, IRBB5, IRBB13, IRBB14, and IR24, provided by the International Rice Research Institute (IRRI), Philippines (Ogawa et al., 1991), via the Institute of Plant Protection of Nanjing Agricultural University, China.
Plant Assays and Pathotypic Analysis
The Xoo strains stored at −80°C were activated by streak culture on PSG medium (10 g l-1 peptone, 10 g l-1 sucrose, 1 g l-1 glutamic acid, pH 7.0) agar plates. The batch cultures of Xoo strains were grown in PSG medium for 18 h, about to the mid-log phase and then collected, and resuspended to the optical density at 600 nm (OD600) of 0.5.in sterile water.
The rice seeds were sown in plastic pots in a greenhouse under a 12-h light/dark cycle at 25–28°C, and the 45-day-old seedlings were clip-inoculated with a bacterial suspension of 0.5 OD600 and maintained under conditions of 80% relative humidity. Twenty leaves of each differential were inoculated with each isolate or sterilized water (as control). The disease severity was scored based on the visual assessment of the lesion length 14 days post-inoculation. The leaves with a lesion length of <1/4 and ≥1/4 were classified as resistant (R), and susceptible (S), respectively (Chen et al., 2019). All experiments were repeated three times. The least significant difference method (LSD method) was used for data analysis and the box-plot was generated using Origin 2018. A pathotypic analysis of Xoo strains was performed according to their reactions on NIL rice (Supplementary Table S1).
Genome Sequencing and Assemblage
For the whole genome sequencing, Xoo strains were grown in PSG medium for 36 h, about to the early stationary phase, and then harvested. The genomic DNA of each strain was isolated and purified by using QIAGEN genomic-tip 20G following the manufacturer's protocol. The DNA samples were fragmented using g-TUBE. HiFi sequencing libraries were prepared using SMRTbell™ Express Template Prep Kit 2.0. The libraries were further size selected using BluePippin Systems. The average fragment length of the sequencing library is about 15 kb for circular consensus sequencing (CCS) to achieve long and high-fidelity (HiFi) reads (Tang, 2019). The DNA sequencing was performed using PacBio Sequel II System (Pacific Biosciences, USA) at Biomarker Technologies CO., LTD (Beijing, China). The HiFi reads have been de novo assembled using the Hifiasm software (Cheng et al., 2021). The Pilon v1.22 software was used to correct the assembly with Illumina data, and Circulator v1.5.5 software was used to cyclize and adjust the starting site. The genome completeness and contamination were evaluated by CheckM v1.1.3 (Parks et al., 2015).
Genome Annotation and the Categorical Analysis
The preliminary genome annotation was carried out by NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016) and GeneMarkS-2 software (Lomsadze et al., 2018). The gene function and pathway analysis (Supplementary Table S2) were performed by BLASTP searches against functional databases, including Cluster of Orthologous Groups of proteins (COG) database (https://www.ncbi.nlm.nih.gov/COG/), Gene Ontology (GO) (http://geneontology.org/), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, http://www.genome.jp/kegg/).
To decipher the Xoo genome from the phytopathological perspective, the confirmed and putative pathogenicity/virulence genes in Xoo were grouped into certain categories (Supplementary Table S3) in accordance with the genome categorical system for Xanthomonas first used by da Silva et al. (2002) and further improved by Thieme et al. (2005). The TAL effectors of each genome were identified and classified by AnnoTALE (Grau et al., 2016).
Comparative Genomics and Phylogenomic Analysis
For structural comparison, complete genomes were aligned using progressive Mauve (Darling et al., 2010) with default settings. For phylogenomic analysis, complete genomes were aligned using Mauve. The tree was annotated and visualized by using the CVTree3 web server (http://cvtree.online/v3/cvtree/). The phylogenetic trees were constructed by using the neighbor-joining (NJ) method. The K-tuple length is 6 (Zuo and Hao, 2015). Orthologous genes of GXO20-01 and GXO20-06 were extracted from Mauve in pairwise comparison (Darling et al., 2010).
Results
Pathotyping of Xoo Strains From Low Land and High Mountain Paddies in Guangxi, China
To comparatively study the 2020-epidemic Xoo strains isolated from low land and high mountain paddies, we had chosen two Xoo strains, GXO20-01 and GXO20-02, from Heng County (the typical low land area) and two strains, GXO20-04 and GXO20-06, from Napo County (the typical high mountain area), respectively (Table 1). On the basis of molecular identification and back inoculation, the four Xoo strains were demonstrated to be quite similar in phenotypic characteristics, including EPS production, extracellular enzyme activity, motility, stress tolerance, and biofilm formation (data not shown).
The pathotypic analysis of Xoo strains was commonly performed by using a simplified combination of IRBB NILs, IRBB2, IRBB3, IRBB5, IRBB13, IRBB14, and IR24, in China (Liu et al., 2007; Chen et al., 2012). In this study, the positive control Xoo strain YC11 clearly showed the SSRRSS interaction results on the differential rice varieties (Zheng et al., 2020). The pathotyping results demonstrated that all of four representative Xoo strains from low land and high mountain paddies in Guangxi belong to race R8, indicated by the SSRSSS interaction results (Figure 1). Additionally, statistical analysis showed that there were significant differences between those Xoo strains on virulence in some certain rice varieties (Figure 1).
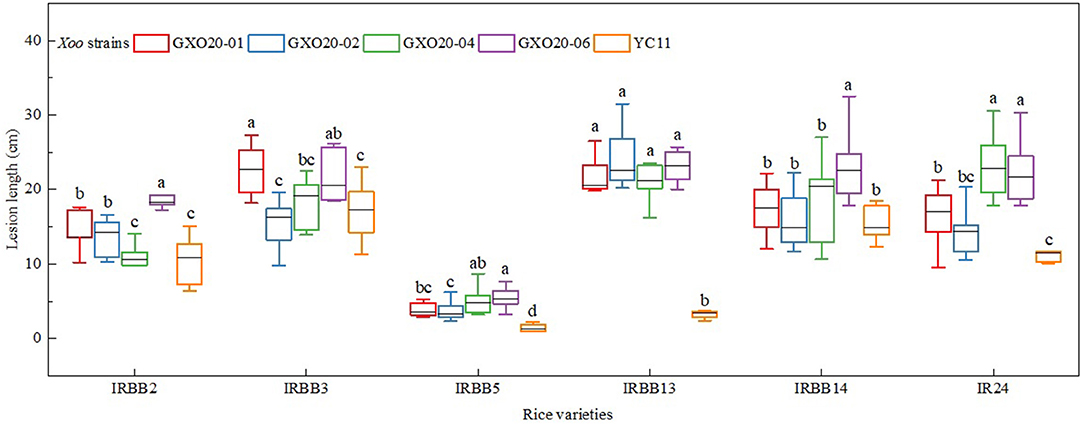
Figure 1. Responses of disease of newly isolated Xoo strains test on IRBB NILs. The least significant difference (LSD) test was used for data analysis at P < 0.05 level, showing as lowercase letters.
Complete Genome Characteristics of GXO20-01 and GXO20-06 Genomes
To quickly and effectively compare the genomic information of Xoo strains isolated from low land and high mountain paddies, GXO20-01 and GX20-06 were selected as the representative Xoo strains for low land and high mountain paddies, respectively (Table 1). Complete genome sequencing of two Xoo strains was performed using the Pacific Biosciences (PacBio) platform with high-fidelity (HiFi) long reads (see Materials and Methods). The CCS data for GXO20-01 and GX20-06 are as follows: SeqNum: 6357 and 5238 reads, SumBase: 6,106,727 and 53,252,594 bp, N50 length: 10,505 and 10,286 bp. The HiFi reads have been de novo assembled using the Hifiasm software (Cheng et al., 2021). The Pilon v1.22 software was used to correct the assembly with Illumina data, and Circulator v1.5.5 software was used to cyclize and adjust the starting site. One contig was assembled for each genome, with sizes 4,963,479 and 4,962,861 bp, respectively (Table 2). Two Xoo strains contain no plasmid. The DNA completeness for the two genomes was found to be 99.62 and 99.44%, respectively, and contamination for the two genomes was 0.37%. The basic genome features of GXO20-01 and GX20-06 and other sequenced Xoo strains were listed in Table 2. Also, the basic genome features of GXO20-01 and GX20-06 were annotated to encode 724 and 727 pseudogenes, similar to those found in PXO99A, 759 (Salzberg et al., 2008) and in MAFF 311018, 747 (Ochiai et al., 2005), respectively (Table 2). These results also indicated that the HiFi sequencing quality is relatively high.
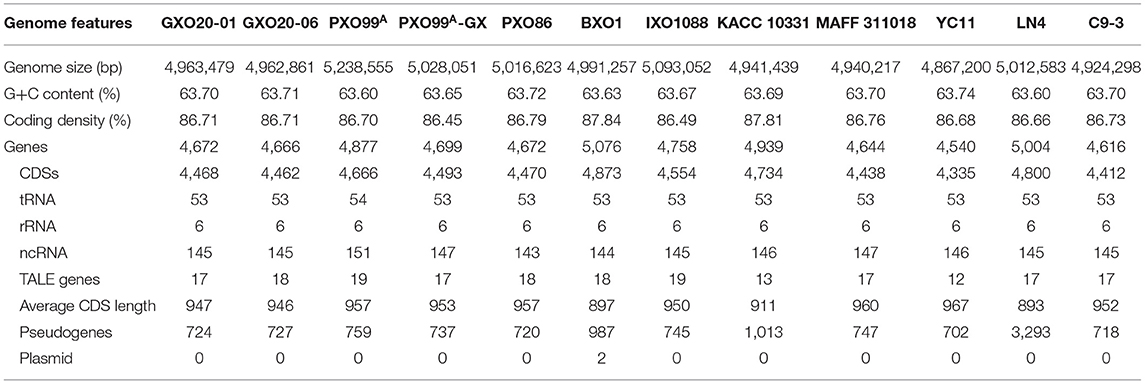
Table 2. The genomic features of Xoo GXO20-01 and GXO20-06 and other Xoo strains.
Genome Annotation and the Categorical Analysis of Pathogenicity/Virulence Genes
The genomes were annotated 4,468 and 4,462 protein encoding sequences (CDSs) in GXO20-01 and GXO20-06, respectively (Table 2; Supplementary Table S2), among which 628 and 629 in GXO20-01 and GXO20-06, respectively (Supplementary Table S3), were annotated as pathogenicity/virulence genes in accordance with the genome categorical system for Xanthomonas firstly used by da Silva et al. (2002) and further improved by Thieme et al. (2005). The Xoo genomes encode typical pathogenicity/virulence factors found in Xanthomonas spp., including extracellular hydrolases, extracellular polysaccharides, adhesins, type II secretion system, type III secretion system, and T3SEs (White et al., 2009; An et al., 2020).
In this study, the pathogenicity-related genes in Xoo were divided into 10 groups: (1) Type III secretion system (hrp/hrc genes) and its effectors, (2) other secretion systems, (3) extracellular enzymes, (4) surface polysaccharides, lipopolysaccharides, and antigens, (5) toxin and detoxification, (6) adhesion and biofilm, (7) quorum sensing and regulation of virulence factors, (8) adaptation to atypical conditions, (9) bacterial motility, and (10) other factors in bacterium–plant interaction (Supplementary Table S3). The detailed genome annotation might be helpful to elucidate the pathogenesis of this typical plant pathogenic bacterium and to pinpoint the certain genes in different infection stages (Supplementary Table S3).
Genome Structures and Genome Contents for GXO20-01 and GXO20-06 Are Strikingly Similar
To clarify the relationship between the two strains, we have conducted pairwise comparative analysis both in their genome structures and genome contents. The results showed that the chromosomes of GXO20-01 and GXO20-06 are entirely syntenous (Figure 2A). The positions of the 17 allelic tal genes show no duplications or rearrangements in one genome relative to the other (Figure 2B). The results of genome two-way alignments indicated that the two genomes are conserved both in gene contents and their allelic sites (Supplementary Table S2). Among the 4,468 and 4,462 annotated genes in GXO20-01 and GXO20-06, 4,442 genes are orthologous. Totally, there are 123 genomic variational sites between genomes, including 97 SNPs, 19 indels, and 7 complex variations. Among the SNPs, 76 were found in coding sequence genes, 33 SNPs were missense variants that resulted in 20 gene differences between two strains, as more than one SNPs in one single gene. The indels and complex variations also caused nine gene differences between the two strains (Table 3; Supplementary Tables S2, S3). Here, GXO20-01 has 6 strain-specific genes encoding a 5-amino-6-(5-phosphoribosylamino) uracil reductase (RibD), a GNAT family N-acetyltransferase, two adenylyl–sulfate kinase, outer membrane protein, and a hypothetical protein, and GXO20-06 has only one strain-specific gene, the tal5a encoding a TalDR-like TALE. Subsequently, it was found that the two TALE repertoires are nearly identical but only one TALE missing in GXO20-01. Consequently, the pairwise genomic comparison and phylogenetic analysis suggested that GXO20-01 and GXO20-06 are probably sibling Xoo strains.
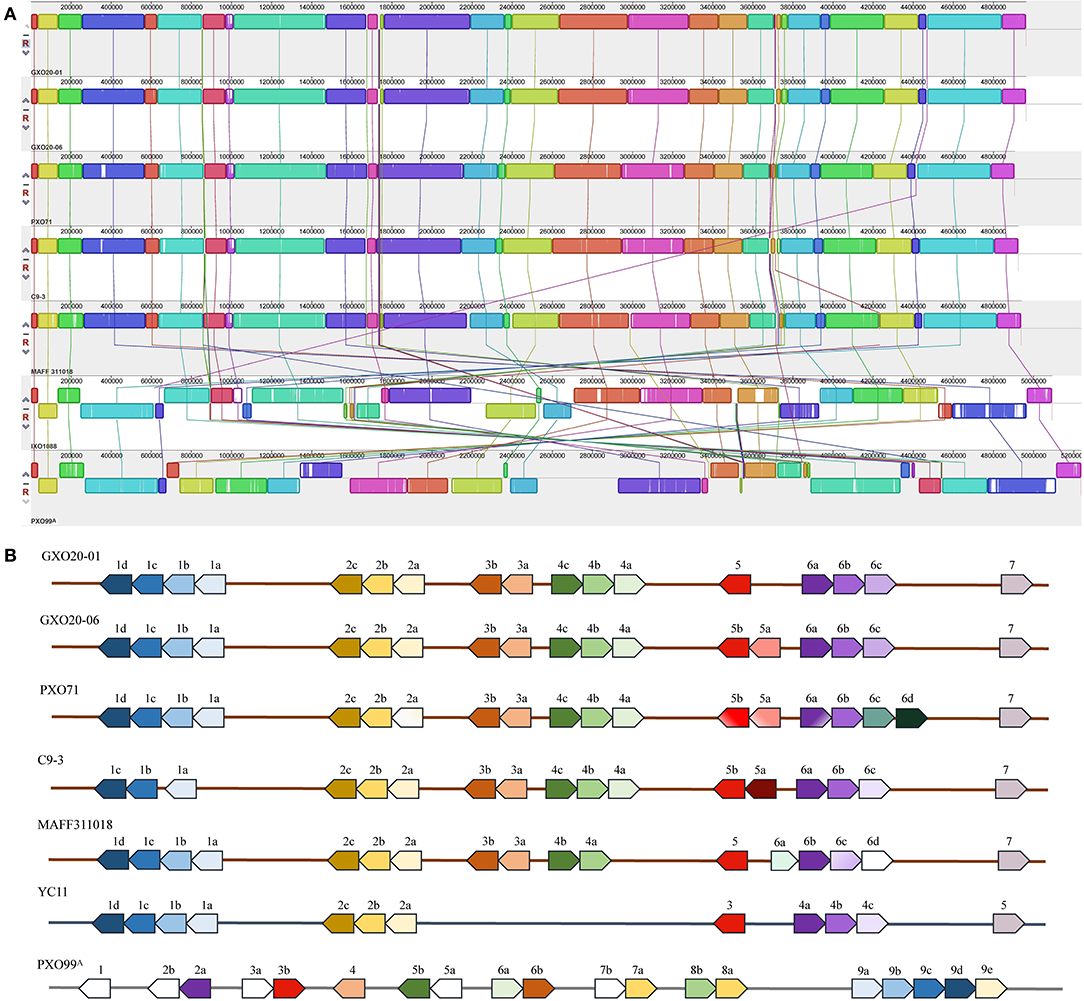
Figure 2. Comparison of whole genomes and tal genes of Xoo strains. (A) Progressive Mauve alignments chromosomes of Xoo strains. Colinear blocks are shown in green, with gray indicating regions of sequence dissimilarity. The ruler indicates distance from the annotated origin in base pairs. (B) The tal genes of Xoo strains. The genes are represented as arrows at their relative positions in the linearized chromosome. Arabic numerals indicate the serial number of tal gene clusters in an Xoo strain, and each lowercase letter indicates each tal gene in a certain gene cluster.

Table 3. The genomic variations and differential genes between Xoo strains GXO20-01 and GXO20-06.
The Relationships Analysis of the Two Guangxi Strains With Other Xoo Strains
Multiple alignments of the selected Xoo genomes showed that the genome structures of GXO20-01 and GXO20-06 are highly collinear with Xoo strains in lineage PX-A (Quibod et al., 2016) (Figure 2A) and the loci of the major tal genes are highly conserved (Figure 2B).
To further determine the relationships of the newly sequenced Xoo strains with other Xoo strains, we carried out a phylogenomic analysis in which representative Xoo strains were selected representing typical Xoo strains from around the world (Figure 3). The phylogenomic analysis clearly grouped GXO20-01 and GXO20-06 in the branch of PXO563 and XF89b in lineage PX-A (Quibod et al., 2016) or Xoo-A (Chien et al., 2019), but away from the Chinese mainland strains (Zheng et al., 2020).
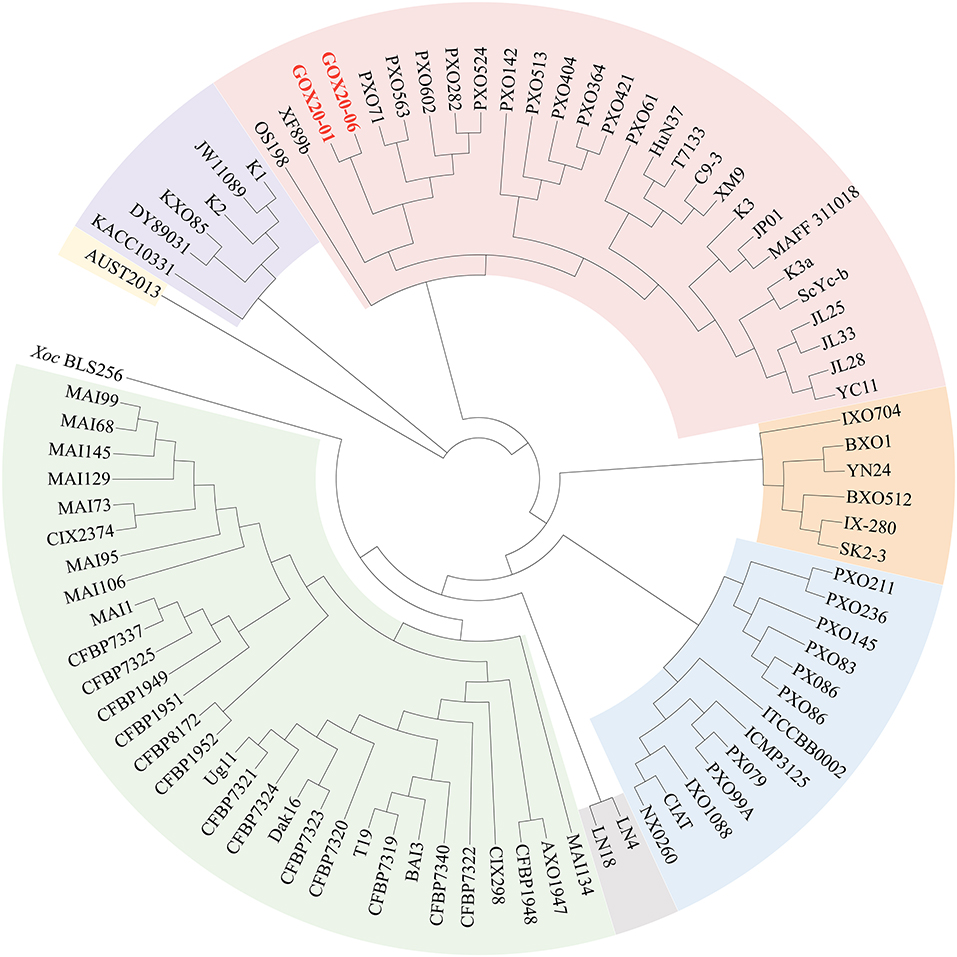
Figure 3. Phylogenomic analysis of Xoo strains based on genome sequences of representative Xoo strains. The genome sequences were obtained from NCBI. The tree was annotated and visualized by using CVTree3 web server. The phylogenetic trees by using the NJ method. The K-tuple length is 6.
The GXO20-06 Tal5a Gene Situated in the Variable tal Locus Encodes a TalDR-Like TALE
The TALEs are the most important virulence factor used by Xoo to interact with rice, enforcing virulence, proliferation, and dissemination against rice (Erkes et al., 2017). AnnoTALE analysis indicated that GXO20-01 and GXO20-06 contain 17 and 18 tal genes, respectively. Among the 17 shared TALEs, 15 typical TALEs are assigned to classes TalAF, TalAN, TalAD, TalAB, TalAL, TalAD, TalAO, TalAE, TalAM, TalAH, TalAA, TalAR, TalAG, TalAS, TalAQ, and the iTALE/truncTALEs are assigned to class TalAI. Compared with the closely related Xoo strains, GXO20-01 and GXO20-06 contain no TalFX that is found in C9-3, no TalBK that is found in LN4, and no TalDW that is found in XF89b (Table 4).

Table 4. RVD sequences in TALEs of Xoo strains GXO20-01 and GXO20-06.
The diversity in TALE repertoires of Xoo actually underpins the complexity of the virulence of Xoo races. A general survey of Xoo genomes in GenBank indicated that there are not two Xoo strains sharing the identical TALome. In our case, the closely related sibling Xoo strains GXO20-01 and GXO20-06 still have one TALE gene difference.
It is worth mentioning that GXO20-06 contains a tal5a gene situated in the variable tal locus that encodes a TalDR-like TALE, which is missing in GXO20-01. TalDR is a particular TALE that is found only in the PXO563 and PXO71. The tal5 locus (here referred to as GXO20-06 genome) contains a number of insertion sequences and multiple repeat sequences (Figure 4), which is considered to be the most unstable section in Xoo genomes. The genomic comparison indicated that in most Xoo genomes, tal5 locus or its homologous sections encode one or two iTALE/truncTALEs, but their CDS sequences differ largely (Table 4). In most cases, talDR/talDR-like gene totally disappeared in certain Xoo genomes. Such TALE diversities might provide a novel marker or reference for the genotyping or tracking of pathogens. Also, GXO20-06 is isolated from high mountain paddies, which still carry the talDR/talDR-like gene in the chromosome, suggesting the slow variation probably due to mild selection pressure from a single cropping rice system. As epidemic Xoo strains, it is expected GXO20-01 and GXO20-06 strains with only one TALE distinct to be the ideal reference strains in the research on the pathogenesis, virulence evolution, and pathogen tracking of rice BLB in China and even in many regions worldwide.
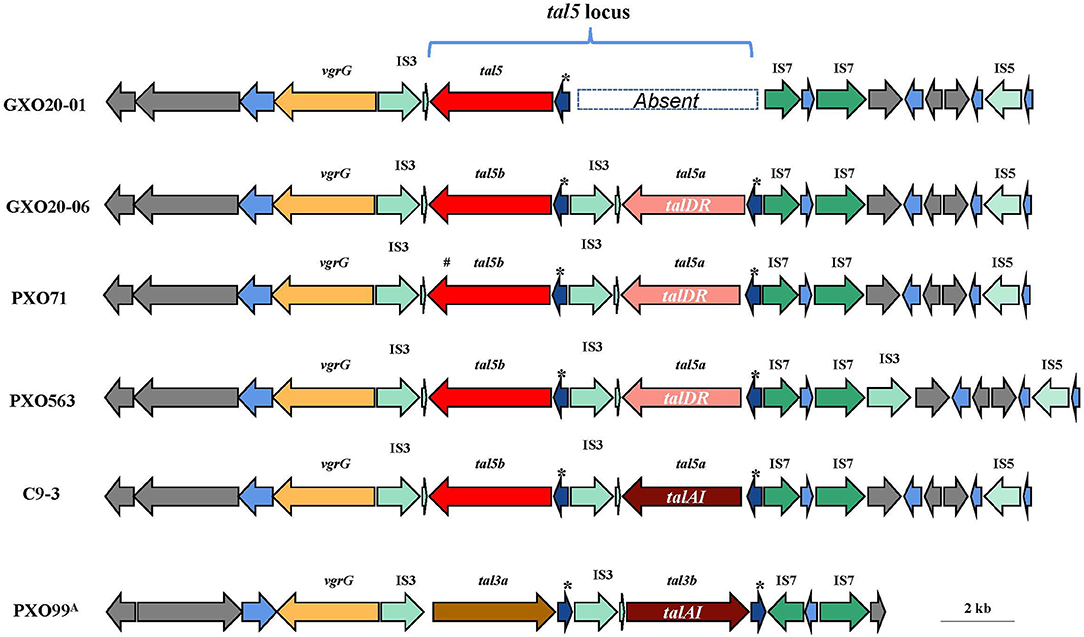
Figure 4. Comparison of the genomic location that contains the tal5 gene cluster. Genes are depicted by arrows indicating the transcription direction. The “*” indicates the direct repeat sequences. tal5a of C9-3 is a truncated tal gene encoding a TalAI effector.
Discussion
Guangxi is the major rice producing region in China and is presumed to be one of the central areas of ancient rice domestication (Huang et al., 2012). Guangxi is also known as one of the hardest hit areas of rice diseases in China (Lin et al., 2021). In 2020, we have carried out a systematic survey on rice BLB in Guangxi. A batch of epidemic Xoo strains was isolated and characterized. In this study, we have sequenced two Xoo strains GXO20-01 and GXO20-06, isolated from low land and high mountain paddies in Guangxi, respectively. Phylogenomic analysis and genome comparison indicated that GXO20-01 and GXO20-06 are the two Xoo sibling strains, with only differing few genes, including the talDR/talDR-like gene that was only found in PXO563 and PXO71, the Philippines strain (Quibod et al., 2016). As the talDR/talDR-like gene is absent from other Chinese Xoo strains, the relationships between the GXO20-06 and these two Philippines strains are interesting. As rice BLB is a frequent disease in many countries, it is helpful for effective prevention and control of the disease to establish multiple ways for diagnosing and tracking the virulence variations, e.g., the TALE typing.
The two strains GXO20-01 and GXO20-06 were chronologically isolated from low land and high mountain paddies in Guangxi, respectively. GXO20-01 is earlier than GXO20-06 to be collected from the paddies, however, GXO20-06 is assumed to be closer to their common ancestor according to their TALE repertoires. In view of the genome structure in the tal5 locus, GXO20-01 might easily lose the talDR/talDR-like gene, other than GXO20-06 acquired the gene. Taking together the climates and cropping systems, it is tempting to conclude that the genomic variation pace of Xoo GXO20-06 in high mountain paddy is slower than that of GXO20-01 in low land paddy.
Pathotyping analysis showed that both GXO20-01 and GXO20-06 belong to the race R8 group which was demonstrated to be the predominant strain in the paddies of South China (Chen et al., 2017, 2019). It was shown that GXO20-01 and GXO20-06 cannot overcome the resistance of xa5, indicating that rice materials or varieties carrying the recessive xa5 gene are of favorable superiority and wide application prospect in controlling rice BLB in Guangxi.
The TALEs are found only in a small number of bacterial species are believed to be one of the most complex virulence factors both in their molecular structures and their functional mechanisms (Erkes et al., 2017). With nearly identical, tandem-repeat-filled sequences in a single tal gene and with the multiple tal gene copies scattered in Xanthomonas genome, sequencing the genome of a Xanthomonas oryzea can be challenging (Booher et al., 2015). Elucidating Xo genomes require sequencing data that is both accurate and complete. Single Molecule, Real-Time (SMRT) Sequencing technology has evolved to a different type of long read, known as highly accurate long reads, or HiFi reads (Tang, 2019). Although the CCS read numbers, total sequencing bases and sequencing depth are significantly reduced, the PacBio® HiFi sequencing method yields highly accurate long-read sequencing datasets with read lengths averaging 10–25 kb and accuracies >99.5% (Tang, 2019; Cheng et al., 2021). In this study, the HiFi sequencing allowed quickly, effectively, and accurately assembling the complete gap-free genomes of Xoo strains.
In conclusion, by the utility of HiFi long-read sequencing in Xoo genome sequencing, this study further revealed the instability and variability of tal genes in the epidemic Xoo strains from different rice paddies. The approach and genome data provide information for discovering the new virulence factors and elucidating the functional mechanisms of TALEs in the Xoo-rice pathosystem.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
Y-QH and TL designed the research and drafted the manuscript. TL, XH, QL, and ZZ isolated the bacterial strains. TL, XD, and Y-QH isolated the genome DNA and performed the genomic data analysis. TL, XM, and SL conducted the plant assays and pathotyping. YL, SH, and WJ conducted bioinformatic analysis. WJ and YY contributed to revising the manuscript. All authors made their contributions to the manuscript, contributed to the interpretation of data, and approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (2018YFD0200302), the Natural Science Foundation of Guangxi (2019GXNSFAA245052), and the Graduate Education Innovation Fund Project in Guangxi (YCBZ2020016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.867633/full#supplementary-material
References
An, S. Q., Potnis, N., Dow, M., Vorhölter, F. J., He, Y. Q., Becker, A., et al. (2020). Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 44, 1–32. doi: 10.1093/femsre/fuz024
Booher, N. J., Carpenter, S. C. D., Sebra, R. P., Wang, L., Salzberg, S. L., Leach, J. E., et al. (2015). Single molecule real-time sequencing of Xanthomonas oryzae genomes reveals a dynamic structure and complex TAL (transcription activator like) effector gene relationships. Microb. Genomics. 1, e000032. doi: 10.1099/mgen.0.000032
Chen, S., Wang, C. Y., Su, J., Feng, A. Q., Zhu, X. Y., and Zeng, L. X. (2017). Differential detection and analysis of pathotypes and differentiation against Xanthomonas oryzae pv. oryzae in southern China. J. Plant Prot. 44, 217–222. doi: 10.13802/j.cnki.zwbhxb.2017.2015165
Chen, X. L, Yu, L., Gao, L. L., Jiang, T., Li, Q. Y., et al. (2012). Elevational variation in diversity of Xanthomonas oryzae pv. oryzae in South-West China. J. Phytopathol. 160, 261–268. doi: 10.1111/j.1439-0434.2012.01892.x
Chen, X., Li, Q., Wang, J., Zhang, Y., Tang, L., Huang, S., et al. (2021). Genome Resource of a hypervirulent strain C9-3 of Xanthomonas oryzae pv.oryzae causing bacterial blight of rice. Plant Dis. 106, 741–744. doi: 10.1094/PDIS-07-21-1565-A
Chen, X., Wei, S., Yan, Q., Huang, F., Ma, Z., and Li, R. (2019). Virulence and DNA fingerprinting analysis of Xanthomonas oryzae pv. oryzae identify a new pathotype in Guangxi, South China. J. Basic Microbiol. 59, 1082–1091. doi: 10.1002/jobm.201900354
Chen, X., Yan, Q., Li, R., Li, K., and Gao, L. (2016). First report of a new and highly virulent race of Xanthomonas oryzae pv. oryzae, the causal agent of bacterial leaf blight of rice in Guangxi Province, China. Plant Dis. 100, 1492. doi: 10.1094/PDIS-12-15-1400-PDN
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods.18, 170–175. doi: 10.1038/s41592-020-01056-5
Chien, C. C., Chou, M. Y., Chen, C. Y., and M. C Shih (2019). Analysis of genetic diversity of Xanthomonas oryzae pv. oryzae populations in Taiwan. Sci. Rep. 9, 316. doi: 10.1038/s41598-018-36575-x
da Silva, A. C., Ferro, J. A., Reinach, F. C., Farah, C. S., Furlan, L. R., Quaggio, R. B., et al. (2002). Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 417, 459–463. doi: 10.1038/417459a
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 5, e11147. doi: 10.1371/journal.pone.0011147
Erkes, A., Reschke, M., Boch, J., and Grau, J. (2017). Evolution of transcription activator-like effectors in Xanthomonas oryzae. Genome Biol. Evol. 9, 1599–1615. doi: 10.1093/gbe/evx108
Fang, C. T., Xu, Z. G., Guo, C. J., Yin, S. Z., Xu, X. M., and Zhang, Q. (1990). Study on the pathotypes of Xanthomonas oryzae pv. oryzae in China. Acta Phytopath. Sin. 20, 80–81.
Grau, J., Reschke, M., Erkes, A., Streubel, J., Morgan, R. D., Wilson, G. G., et al. (2016). AnnoTALE: bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Sci. Rep. 6:21077. doi: 10.1038/srep21077
Huang, X., Kurata, N., Wei, X., Wang, Z. X., Wang, A., Zhao, Q., et al. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nature. 490, 497–501. doi: 10.1038/nature11532
Ji, Z., Ji, C., Liu, B., Zou, L., Chen, G., and Yang, B. (2016). Interfering TAL effectors of Xanthomonas oryzae neutralize R-gene-mediated plant disease resistance. Nat. Commun. 7, 13435. doi: 10.1038/ncomms13435
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature. 444, 323–329. doi: 10.1038/nature05286
Kay, S., and Bonas, U. (2009). How Xanthomonas type III effectors manipulate the host plant. Curr. Opin. Microbiol. 12, 37–43. doi: 10.1016/j.mib.2008.12.006
Li, T., Huang, X., Zhang, Z., Ling, Y., Li, Y., Ma, X., et al. (2021). The occurrence of rice white leaf blight in the alpine mountain rice area of Napo County in 2020 and the prevention and control recommendations. Guangxi Plant Prot. 34, 29–32. (In Chinese).
Lin, Z., Long, M., Tang, J., Xin, D., and Huang, C. (2021). Occurrence of the main pests on crops in 2020 in Guangxi. Guangxi Plant Prot. 34, 25–32. (In Chinese).
Liu, H., Yang, W., Hu, B., and Liu, F. (2007). Virulence analysis and race classification of Xanthomonas oryzae pv. oryzae in China. J. Phytopathol. 155, 129–135. doi: 10.1111/j.1439-0434.2007.01197.x
Lomsadze, A., Gemayel, K., Tang, S., and Borodovsky, M. (2018). Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 29, 1079–1089. doi: 10.1101/gr.230615.117
Mak, A. N., Bradley, P., Cernadas, R. A., Bogdanove, A. J., and Stoddard, B. L. (2012). The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 335, 716–719. doi: 10.1126/science.1216211
Moscou, M. J., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science. 326, 1501. doi: 10.1126/science.1178817
Niño-Liu, D. O., Ronald, P. C., and Bogdanove, A. J. (2006). Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol. Plant Pathol. 7, 303–324. doi: 10.1111/j.1364-3703.2006.00344.x
Ochiai, H., Inoue, Y., Takeya, M., Sasaki, A., and Kaku, H. (2005). Genome sequence of Xanthomonas oryzae pv. oryzae suggests contribution of large numbers of effector genes and insertion sequences to its race diversity. Jpn. Agric. Res. Quart. 39, 275–287. doi: 10.6090/jarq.39.275
Ogawa, T., Yamamoto, T., Khush, G. S., and Mew, T. W. (1991). Breeding of near-isogenic lines of rice with single genes for resistance to bacterial blight pathogen (Xanthomonas campestris pv. oryzae). Jpn J. Breed. 41, 523–529.
Oliva, R., Ji, C., Atienza-Grande, G., Huguet-Tapia, J. C., Perez-Quintero, A., Li, T., et al. (2019). Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. 37, 1344–1350. doi: 10.1038/s41587-019-0267-z
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Quibod, I. L., Perez-Quintero, A., Booher, N. J., Dossa, G. S., Grande, G., Szurek, B., et al. (2016). Effector diversification contributes to Xanthomonas oryzae pv. oryzae phenotypic adaptation in a semi-isolated environment. Sci Rep. 6, 34137. doi: 10.1038/srep34137
Read, A. C., Rinaldi, F. C., Hutin, M., He, Y.-Q., Triplett, L. R., and Bogdanove, A. J. (2016). Suppression of Xo1-mediated disease resistance in rice by a truncated, non-DNA-binding TAL effector of Xanthomonas oryzae. Front. Plant Sci. 7, 1516. doi: 10.3389/fpls.2016.01516
Salzberg, S. L., Sommer, D. D., Schatz, M. C., Phillippy, A. M., Rabinowicz, P. D., Tsuge, S., et al. (2008). Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genom. 9, 204. doi: 10.1186/1471-2164-9-534
Streubel, J., Pesce, C., Hutin, M., Koebnik, R., Boch, J., and Szurek, B. (2013). Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 200, 808–819. doi: 10.1111/nph.12411
Tang, L.. (2019). Circular consensus sequencing with long reads. Nat. Methods. 16, 958. doi: 10.1038/s41592-019-0605-6
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thieme, F., Koebnik, R., Bekel, T., Berger, C., Boch, J., Buttner, D., et al. (2005). Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 187, 7254–7266. doi: 10.1128/JB.187.21.7254-7266.2005
White, F. F., Potnis, N., Jones, J. B., and Koebnik, R. (2009). The type III effectors of Xanthomonas. Mol. Plant Pathol. 10, 749–766. doi: 10.1111/j.1364-3703.2009.00590.x
Zhang, H., and Wang, S. (2013). Rice versus Xanthomonas oryzae pv. oryzae: a unique pathosystem. Curr. Opin. Plant Biol. 16, 188–195. doi: 10.1016/j.pbi.2013.02.008
Zheng, J., Song, Z., Zheng, D., Hu, H., Liu, H., Zhao, Y., et al. (2020). Population genomics and pathotypic evaluation of the bacterial leaf blight pathogen of rice reveals rapid evolutionary dynamics of a plant pathogen. bioRxiv. 704221. doi: 10.1101/704221
Keywords: Xanthomonas oryzae pv. oryzae, HiFi PacBio sequencing, genome, tal, high mountain paddy
Citation: Li T, Li Y, Ma X, Dan X, Huang X, Li Q, Lei S, Zhang Z, Huang S, Jiang W, Yu Y and He Y-Q (2022) Comparative Genomic Analysis of Two Xanthomonas oryzae pv. oryzae Strains Isolated From Low Land and High Mountain Paddies in Guangxi, China. Front. Microbiol. 13:867633. doi: 10.3389/fmicb.2022.867633
Received: 01 February 2022; Accepted: 15 March 2022;
Published: 28 April 2022.
Edited by:
Teresa Ann Coutinho, University of Pretoria, South AfricaReviewed by:
Ravi Patel, Connecticut Agricultural Experiment Station, United StatesPrabhu B. Patil, Institute of Microbial Technology (CSIR), India
Copyright © 2022 Li, Li, Ma, Dan, Huang, Li, Lei, Zhang, Huang, Jiang, Yu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Qiang He, eXFoZUBneHUuZWR1LmNu