 Laura E. Tibbs-Cortes
Laura E. Tibbs-Cortes Bienvenido W. Tibbs-Cortes
Bienvenido W. Tibbs-Cortes Stephan Schmitz-Esser
Stephan Schmitz-Esser- 1Department of Agronomy, Iowa State University, Ames, IA, United States
- 2Interdepartmental Genetics and Genomics Graduate Program, Iowa State University, Ames, IA, United States
- 3Department of Animal Science, Iowa State University, Ames, IA, United States
- 4Interdepartmental Microbiology Graduate Program, Iowa State University, Ames, IA, United States
The microbiome of tardigrades, a phylum of microscopic animals best known for their ability to survive extreme conditions, is poorly studied worldwide and completely unknown in North America. An improved understanding of tardigrade-associated bacteria is particularly important because tardigrades have been shown to act as vectors of the plant pathogen Xanthomonas campestris in the laboratory. However, the potential role of tardigrades as reservoirs and vectors of phytopathogens has not been investigated further. This study analyzed the microbiota of tardigrades from six apple orchards in central Iowa, United States, and is the first analysis of the microbiota of North American tardigrades. It is also the first ever study of the tardigrade microbiome in an agricultural setting. We utilized 16S rRNA gene amplicon sequencing to characterize the tardigrade community microbiome across four contrasts: location, substrate type (moss or lichen), collection year, and tardigrades vs. their substrate. Alpha diversity of the tardigrade community microbiome differed significantly by location and year of collection but not by substrate type. Our work also corroborated earlier findings, demonstrating that tardigrades harbor a distinct microbiota from their environment. We also identified tardigrade-associated taxa that belong to genera known to contain phytopathogens (Pseudomonas, Ralstonia, and the Pantoea/Erwinia complex). Finally, we observed members of the genera Rickettsia and Wolbachia in the tardigrade microbiome; because these are obligate intracellular genera, we consider these taxa to be putative endosymbionts of tardigrades. These results suggest the presence of putative endosymbionts and phytopathogens in the microbiota of wild tardigrades in North America.
Introduction
Tardigrades are a poorly-studied but globally ubiquitous phylum of microscopic animals. They are members of the superphylum Ecdysozoa, a group that also includes arthropods and nematodes (Aguinaldo et al., 1997). All tardigrades are aquatic; however, while some live in bodies of fresh or salt water, they are most commonly collected from moss or lichen, where they live in interstitial films of water. When this water dries up, tardigrades survive by dehydrating and entering a state of dramatically reduced metabolism known as cryptobiosis (Kinchin, 1994). In this state, they are famously able to survive extreme conditions, ranging from temperatures near absolute zero (Becquerel, 1950) to the vacuum of space (Jönsson et al., 2008). Despite extensive study of tardigrades’ survival abilities, little is known about many aspects of their biology, including their microbiota (Vecchi et al., 2018). This is particularly important because tardigrades’ presence in moss and lichen, which often grow on tree bark, brings them into close contact with trees, including important orchard crops such as apple trees (Malus domestica L. Borkh). Therefore, any plant pathogens present in the tardigrade microbiota have the potential to affect these crops, underscoring the importance of understanding the tardigrade microbiota in an agricultural context.
The first study of tardigrade-associated bacteria, published in 1999, found that bacteria of the phytopathogenic genus Xanthomonas could be grown from the feces of the tardigrade Macrobiotus hufelandi C.A.S. Schultze, 1834 isolated from the wild. However, attempts to inoculate M. hufelandi with Serratia marcescens were unsuccessful, suggesting a non-random relationship between M. hufelandi and Xanthomonas (Krantz et al., 1999). The following year, a second paper showed that M. hufelandi exposed to infected leaves could spread Xanthomonas campestris pv. raphani (the causal pathogen of radish leaf spot disease) to healthy radish seedlings in the laboratory. This demonstrated that M. hufelandi can act as a vector of radish leaf spot disease (Benoit et al., 2000).
Animal vectors are known to spread many plant diseases, with major consequences for crop production worldwide (Mew, 1993; Duveiller et al., 1997; Ng and Falk, 2006). Current research focuses on insect vectors (Ng and Falk, 2006), but the work of Krantz et al. and Benoit et al. demonstrate that members of at least one tardigrade species (M. hufelandi) can spread bacterial disease in plants (Benoit et al., 2000) and can act as reservoirs of plant pathogens (Krantz et al., 1999). Because M. hufelandi and many other tardigrade species live in close contact with plants, bacteria deposited in their feces may infect these plants, especially as other Ecdysozoans are known to spread phytopathogens in this manner (Stavrinides et al., 2009; Dutta et al., 2014). Tardigrades also have the potential to spread phytopathogenic bacteria over large areas because many tardigrade species are cosmopolitan (Meyer, 2013) and may be dispersed by wind or migratory birds (Mogle et al., 2018).
The genus Xanthomonas found in association with M. hufelandi includes pathovars that infect staple food crops including rice (Oryza sativa L.; Mew, 1993), wheat (Triticum aestivum L.; Duveiller et al., 1997), and maize (Zea mays L.; Karamura et al., 2007) with potentially devastating effects. For example, bacterial blight (X. oryzae pv. oryzae) can cause yield losses of up to 50% in rice infected as seedlings, impacting both economies and food security (Mew, 1993). Yet, although tardigrades are known vectors of this important genus, there has been no additional literature published on phytopathogens associated with tardigrades in the past two decades.
Recent studies of the tardigrade microbiome, while not focusing on phytopathogens, have leveraged advances in sequencing technology by using 16S rRNA gene amplicon sequencing. Vecchi et al. surveyed the microbial communities associated with six tardigrade species: Acutuncus antarcticus (Richters, 1904) collected from freshwater sediment in Antarctica, after which a subsample was raised in laboratory culture; Ramazzottius oberhaeuseri (Doyère, 1840), collected from lichen on two different trees in Italy; Macrobiotus macrocalix Bertolani & Rebecchi, 1993 and Richtersius coronifer (Richters, 1903), both collected from the same moss on a rock in Sweden; and Echiniscus trisetosus Cuénot, 1932, and Paramacrobiotus areolatus (Murray, 1907), both collected from the same moss on a rock in Italy (Vecchi et al., 2018). The authors found that the tardigrade microbiome is dominated by Proteobacteria and Bacteroidetes, is distinct from and usually less diverse than that of their substrates, differs among tardigrade species, and is altered by laboratory culturing of the tardigrades. Vecchi et al. (2018) also identified potential endosymbionts of the obligate intracellular order Rickettsiales within the tardigrade microbiome. This is particularly intriguing because the genera Wolbachia and Rickettsia, both members of Rickettsiales, are known to have reproductive effects on their hosts, including inducing parthenogenesis (Werren et al., 2008; Giorgini et al., 2010). Notably, parthenogenesis is common in tardigrades (Bertolani, 2001; Guil et al., 2022). A subsequent analysis of these data identified four putative endosymbionts in the order Rickettsiales, three of which belonged to Anaplasmataceae and one to Ca. Tenuibacteraceae. These were differentially associated with different tardigrade species, and fluorescence in situ hybridization (FISH) detected bacteria within the ovaries of some tardigrades, suggesting that tardigrade endosymbionts are vertically transmitted (Guidetti et al., 2020).
A second study surveyed the microbiota of a newly-described tardigrade species, Paramacrobiotus experimentalis Kaczmarek, Mioduchowska, Poprawa & Roszkowska, 2020, collected from two samples of moss growing on soil in Madagascar and subsequently raised in laboratory culture for 2 years before DNA extraction (Kaczmarek et al., 2020). This study again identified differences between the tardigrades’ microbiome and that of their environment and detected evidence of putative endosymbionts of the intracellular groups Rickettsiales and Polynucleobacter. Proteobacteria and Firmicutes were the dominant phyla in P. experimentalis, and 31 operational taxonomic units (OTUs) shared across tardigrade samples were identified as potential core microbiome members for this tardigrade species (Kaczmarek et al., 2020).
A third paper conducted 16S rRNA amplicon sequencing on four tardigrade species: Hypsibius exemplaris Gąsiorek, Stec, Morek & Michalczyk, 2018, collected from rotting leaves in a pond in the United Kingdom; Macrobiotus polypiformis Roszkowska, Ostrowska, Stec, Janko & Kaczmarek, 2017, collected from moss on a wall in Ecuador; Paramacrobiotus fairbanksi Schill, Förster, Dandekar & Wolf, 2010, collected from moss in Antarctica; and Paramacrobiotus sp. Guidetti et al., 2009, collected from moss on a wall, soil, and railroad tracks at two locations in Poland. Of these, all but P. fairbanksi were subsequently cultured prior to DNA extraction. This study identified Proteobacteria, Firmicutes, and Actinobacteria as the most abundant phyla in the studied tardigrades but primarily focused on putative endosymbionts of tardigrades, specifically OTUs assigned to Rickettsiales and Wolbachia. Members of Wolbachia were detected in adult P. sp. and M. polypiformis, and Rickettsiales were detected in eggs of P. fairbanksi as well as adult M. polypiformis and P. sp. Neither Rickettsiales nor Wolbachia were detected in Hys. exemplaris or the adult P. fairbanksi (Mioduchowska et al., 2021).
Most recently, Zawierucha et al. sequenced 16S rRNA, ITS1, and 18S rRNA genes to identify bacteria, fungi, and microeukaryotes, respectively, associated with the glacial tardigrade Cryobiotus klebelsbergi (Mihelčič, 1959; Zawierucha et al., 2022). Cryobiotus klebelsbergi were collected from cryoconite on the surface of Forni Glacier in Italy; DNA was extracted from four samples immediately and from another three after starving for 3 weeks. The authors found that relative richness of bacteria, fungi, and microeukaryotes was highest in cryoconite, followed by fed tardigrades and finally starved tardigrades. Polaromonas sp. was the most abundant bacterium in both fed and starved C. klebelsbergi, while Pseudomonas sp. and Ferruginibacter sp. were the second most abundant bacteria in fed and starved tardigrades, respectively (Zawierucha et al., 2022).
16S rRNA gene amplicon sequencing has allowed major advances in the study of the tardigrade microbiota. However, contamination is an ongoing issue in microbiome studies, especially in low microbial biomass samples such as tardigrades. In these cases, contaminants can make up a relatively large proportion of all sequence reads and therefore have a disproportionately large impact on results. A minimum standard developed for such studies is the RIDE checklist, which advises researchers to report the methodology used to reduce and assess contamination, to include three types of negative controls (sampling blank, DNA extraction blank, and no-template amplification controls), to determine contamination level by comparing these negative controls to the samples, and to explore contaminants’ impact on results (Eisenhofer et al., 2019). However, while recommended laboratory practices can reduce contamination, they cannot eliminate it. Therefore, in silico approaches have been developed to better accomplish the last two steps of the RIDE checklist. For example, the program decontam identifies contaminants based on their presence in negative controls and higher frequencies in low-concentration samples. It then removes them from further analysis, dramatically improving the accuracy of results (Davis et al., 2018; Karstens et al., 2019). In this work, we followed the RIDE checklist and utilized decontam for in silico contaminant removal.
This study represents the first survey of tardigrade microbiota in North America, as well as the first such survey in an agricultural setting (apple orchards). Rather than focusing on the microbiome of individual tardigrade species, this work is the first to study the microbiome of a full community of tardigrades, hereafter referred to as the tardigrade community microbiome. It is also only the fifth survey of the tardigrade microbiome ever conducted and leverages contamination mitigation methods not used in the previous studies. In addition to identifying putative plant pathogens and endosymbionts associated with tardigrade communities in apple orchards, this study examines whether the tardigrade microbiome differs in four contrasts: (1) across locations, (2) between substrates (moss vs. lichen), (3) between tardigrades and their substrates, and (4) across years.
Materials and Methods
Moss and Lichen Sample Collection
In summer 2019, lichen samples were collected from apple trees growing in six orchards (Locations 1–6) in Hardin and Franklin counties in north-central Iowa, United States (Supplementary Figures S1, S2). One of these (Location 1) had previously been surveyed for tardigrades (Tibbs-Cortes et al., 2020). One sample of lichen was collected from each tree, and three to five trees were sampled at each location. Moss was also present on the sampled apple trees at Location 2, so moss samples were collected from three of these trees. In June 2020, additional lichen samples were taken from the trees at Location 1 to enable comparison across years. All moss and lichen samples were placed in individual brown paper bags, which were stored in a cool, dry room to allow the samples to dehydrate. From each of the 2020 lichen samples, five subsamples of 0.25 g were placed in sterile 1.5 ml tubes and frozen at −20°C for substrate DNA extraction. Table 1 shows a summary of collected samples.
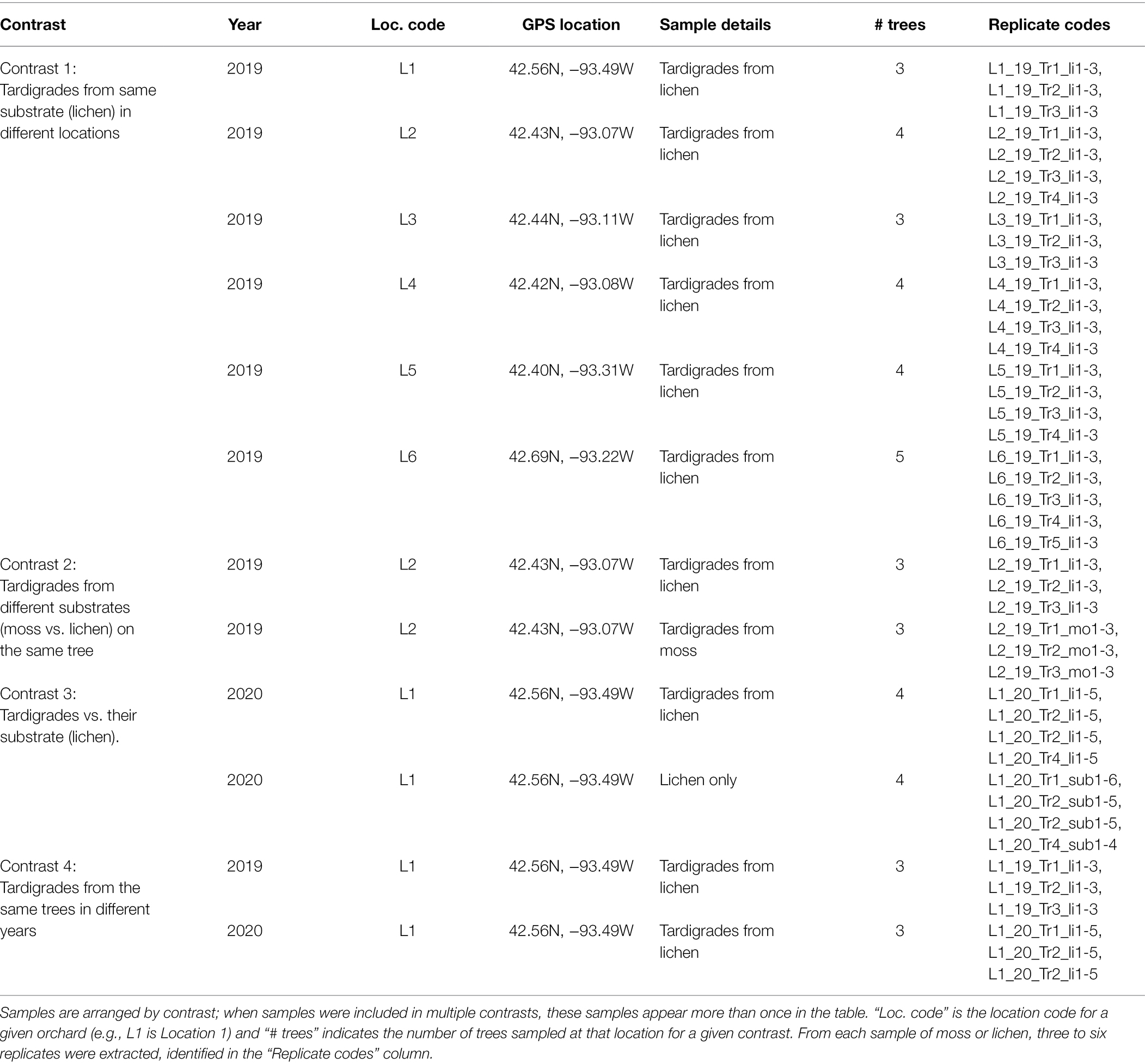
Table 1. Details of samples used in the experiment.
Aseptic Technique
16S rRNA gene amplicon sequencing studies focusing on low-biomass samples are prone to biases from external contamination during sample processing, DNA extraction, library preparation, and sequencing. Therefore, all subsequent tardigrade isolation and DNA extraction steps were carried out using barrier pipette tips (Axygen) and in a sterile work area dedicated to the project. A Bunsen burner was used to create a sterile field for all tardigrade isolations and DNA extractions.
Tardigrade Extraction
To extract tardigrades, each moss or lichen sample was soaked in glass-distilled water for a minimum of 4 h. Subsamples of this water were then examined under a dissecting microscope, and tardigrades were extracted with Irwin loops (Miller, 1997; Schram and Davison, 2012). The Irwin loop was disinfected by a flame between each collected tardigrade.
Next, isolated tardigrades were washed by immersion in droplets of PCR-grade water treated with diethyl pyrocarbonate (DEPC). Tardigrades were then transferred to a fresh drop of DEPC-treated water. This washing process was repeated for a total of three washes, and Irwin loops were sterilized between each wash. Three to six replicates of 30 tardigrades each were collected from each substrate sample (identified by replicate codes shown in Table 1) and were then stored in DEPC-treated water at −20°C.
DNA Extraction and Sequencing
The DNeasy PowerLyzer PowerSoil Kit (Qiagen) was utilized for DNA extraction. Substrate samples were first ground with a sterilized pestle before being transferred to the bead tubes, while tardigrades were directly transferred to bead tubes. Bead tubes were then transferred to a Bead Mill 24 homogenizer (Fisher Scientific). Tardigrades were homogenized using a single 30 s cycle at 5.00 speed, and substrate samples were homogenized using three 30 s cycles with 10 s between cycles at 5.50 speed. Homogenized bead tubes were then centrifuged at 10,000 × g (30 s for tardigrades and 3 min for substrate) before proceeding according to the manufacturer’s instructions; the optional 5 min incubation at 2–8°C was performed during steps 7 and 9. Following elution of DNA with 90 μl of elution buffer, DNA quality and concentration of a 1 μl sample was measured using a NanoDrop spectrometer. Extracted DNA was stored at −20°C.
DNA was loaded onto sterile 96 well plates for library preparation and sequencing. In addition to the tardigrade and substrate samples, three types of controls were included to account for contamination. Six tardigrade processing controls (TPC, equivalent to RIDE sampling blank controls) were created by applying the tardigrade extraction and subsequent DNA extraction protocols to blank samples. Ten DNA processing controls (DPC, equivalent to RIDE DNA extraction blank controls) were created by conducting DNA extraction on 100 μl of DEPC-treated water. Finally, ten wells were loaded with DEPC-treated water to form the library processing controls (LPC, equivalent to RIDE no-template amplification controls). Controls and samples were then submitted for library preparation and 16S rRNA gene amplicon sequencing targeting the V4 region at the Iowa State University DNA facility. Library preparation was conducted following the Earth Microbiome Project 16S Illumina amplicon protocol1 with the following modifications: (1) a single amplification was conducted for each sample rather than in triplicate, (2) PCR purification was conducted using the QIAquick PCR Purification Kit (Qiagen), and (3) all reactions and purification steps were conducted at half volume using a Mantis liquid handler (Formulamatrix) which was cleaned with isopropanol prior to library preparation. Libraries were loaded onto the MiSeq platform at a concentration of ~4pM, and paired-end sequencing was conducted at 500 cycles.
Data Analysis
Following sequencing, three paired end samples representing replicates L6_19_Tr2_li2, L6_19_Tr4_li1, and L5_19_Tr3_li3 (Table 1) were removed from the dataset due to poor quality. Raw reads were processed with mothur version 1.43.0. Sequences were screened to remove reads that contained any ambiguities, were shorter than 252 bases, and had homopolymeric sequences greater than 8 bases. In total, 1,157,089 reads were removed from the raw dataset of 7,805,248 reads. Screened reads were then aligned against the SILVA alignment version 138, and reads which aligned outside the region covered by 95% of the alignment were removed. The SILVA database was also used to remove 145,663 chimeric sequences and to classify remaining sequences. De novo OTU clustering was then conducted at a 99% similarity threshold.
R version 4.0.3 running packages decontam (Davis et al., 2018), phyloseq (McMurdie and Holmes, 2013), DivNet (Willis and Martin, 2020), and corncob (Martin et al., 2020), as well as a more efficient implementation of DivNet known as divnet-rs2 running in Rust, were used for subsequent analyses. Using decontam, contaminant OTUs were identified and removed based on their relative prevalence in control vs. true samples (prevalence method, threshold 0.25; Davis et al., 2018). Next, OTUs with fewer than 10 reads in experimental samples were removed. From these data, alpha diversity parameters (Shannon and Simpson) were calculated using DivNet and divnet-rs, which use a log-ratio model and share information across all samples to improve diversity estimates (Willis and Martin, 2020). Relative abundance, differential abundance, and differential variability of taxa were calculated in corncob, which uses a beta-binomial model (Martin et al., 2020). Differences were declared significant when False Discovery Rate (FDR)-corrected p values (Benjamini and Hochberg, 1995) were <0.05. Principal Coordinates Analysis (PCoA) was conducted in phyloseq using the default Bray-Curtis distance.
Identification of Unclassified Putative Plant Pathogens and Endosymbionts
In the cases where OTUs of interest were not classified by mothur to the genus level, BLAST and RDP Classifier (Wang et al., 2007; Camacho et al., 2009) results were used to provide additional information about taxonomic classification. First, the mothur command “get.oturep” was used to generate a FASTA file containing the representative sequence for each OTU; OTUs with fewer than 10 reads in experimental samples were removed. BLAST analysis was performed using BLAST+ v2.11.0. The NCBI 16S RefSeq collection (representing 22,061 taxa; O’Leary et al., 2016) was downloaded and converted into a BLAST database using the “makeblastdb” command. The “blastn” command was then run against this database using the representative sequence FASTA as the query. Results with the 15 lowest E-values were kept for each OTU. The representative sequence FASTA was also entered into the RDP Classifier web tool version 2.11 using 16S rRNA training set 18,3 and the assignment detail for all OTUs was downloaded.
Data and Code Availability
Raw sequencing files are deposited at the Sequence Read Archive under BioProject accession PRJNA801902 at: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA801902. Mothur output and code used for analysis is available at: https://github.com/LTibbs/tardigrade_microbiome.
Results
In total, 118 DNA samples and 26 controls were sequenced. The DNA samples consisted of 20 from lichen as well as 89 and 9 from tardigrades extracted from lichen and moss, respectively, collected from a total of 23 different apple trees in six Iowa orchards. Tardigrades collected included members of the genera Milnesium, Ramazzottius, and Paramacrobiotus. The controls consisted of 6 TPCs, 10 DPCs, and 10 LPCs. From these sequences, 248,493 OTUs were identified by mothur. The decontam package identified and removed 986 OTUs as contaminants. Of the remaining OTUs, 235,652 were removed because they were represented by fewer than 10 reads in the experimental samples, leaving 11,855 OTUs for further analysis.
Mothur classification and decontam scores for all OTUs with more than 10 reads in experimental samples are shown in Supplementary Table S1. BLAST results and RDP Classifier results for these OTUs can be found in Supplementary Tables S2 and S3, respectively. Relative abundance of OTUs by sample and by contrast are provided in Supplementary Tables S4–S8; significantly differentially abundant and variable phyla, genera, and OTUs across contrast levels are presented in Supplementary Table S9. Overall, the five most abundant phyla were Proteobacteria, Bacteroidota, Actinobacteriota, Firmicutes, and Acidobacteriota (Supplementary Figure S3), while the three most abundant genera were Pseudomonas, Bradyrhizobium, and an unclassified Enterobacteriaceae (Supplementary Figure S4). From the PCoA of all samples, the first principal coordinate clearly separates substrate samples from tardigrade samples, while the second coordinate tends to separate the 2020 from the 2019 samples. Samples from different locations and from moss and lichen are not clearly separated by the first two coordinates (Supplementary Figure S5).
Contrast 1: Location
The tardigrade community microbiome differed significantly across locations, as shown by the Simpson and Shannon indices, which differed significantly in most pairwise comparisons of locations (Table 2). Across locations, 13 phyla and 44 genera were both significantly differentially abundant and significantly differentially variable. Sixteen OTUs were significantly differentially abundant only, four OTUs were significantly differentially variable only, and three OTUs were both significantly differentially abundant and variable (Supplementary Table S9). These identified differential taxa included the aforementioned top five phyla (Proteobacteria, Bacteroidota, Actinobacteriota, Firmicutes, and Acidobacteriota) and top three genera (Pseudomonas, Bradyrhizobium, and unclassified Enterobacteriaceae) from the experiment as a whole. Despite these differences, the locations clustered together in the PCoA (Figure 1).
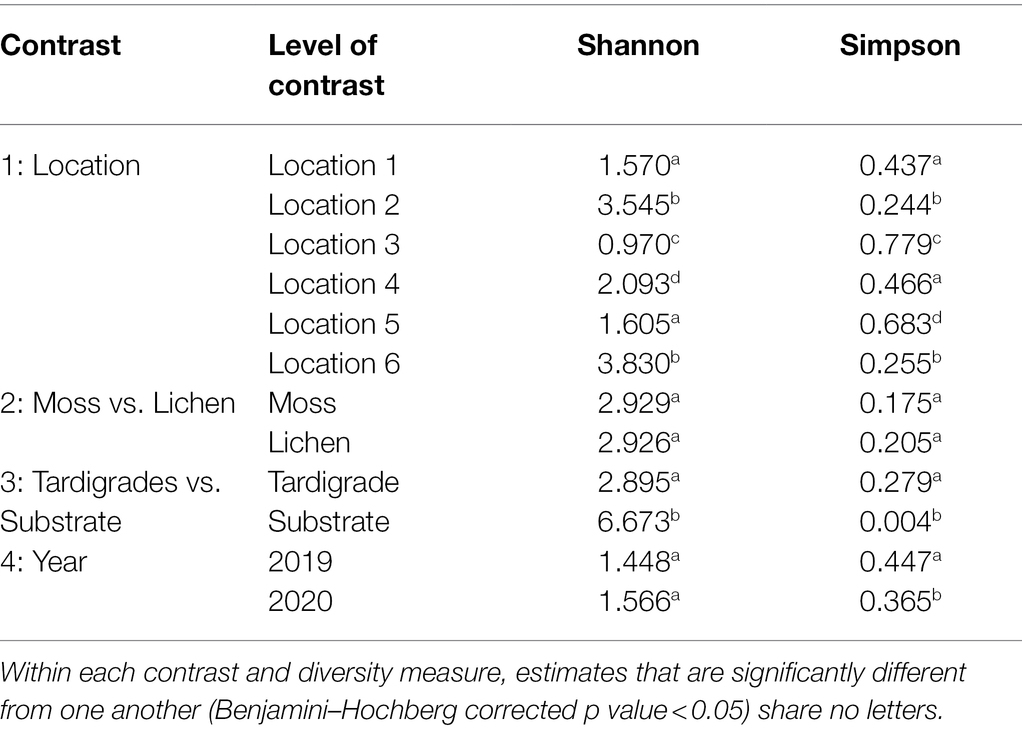
Table 2. Alpha diversity measures Shannon and Simpson estimated for each level of each contrast of interest.
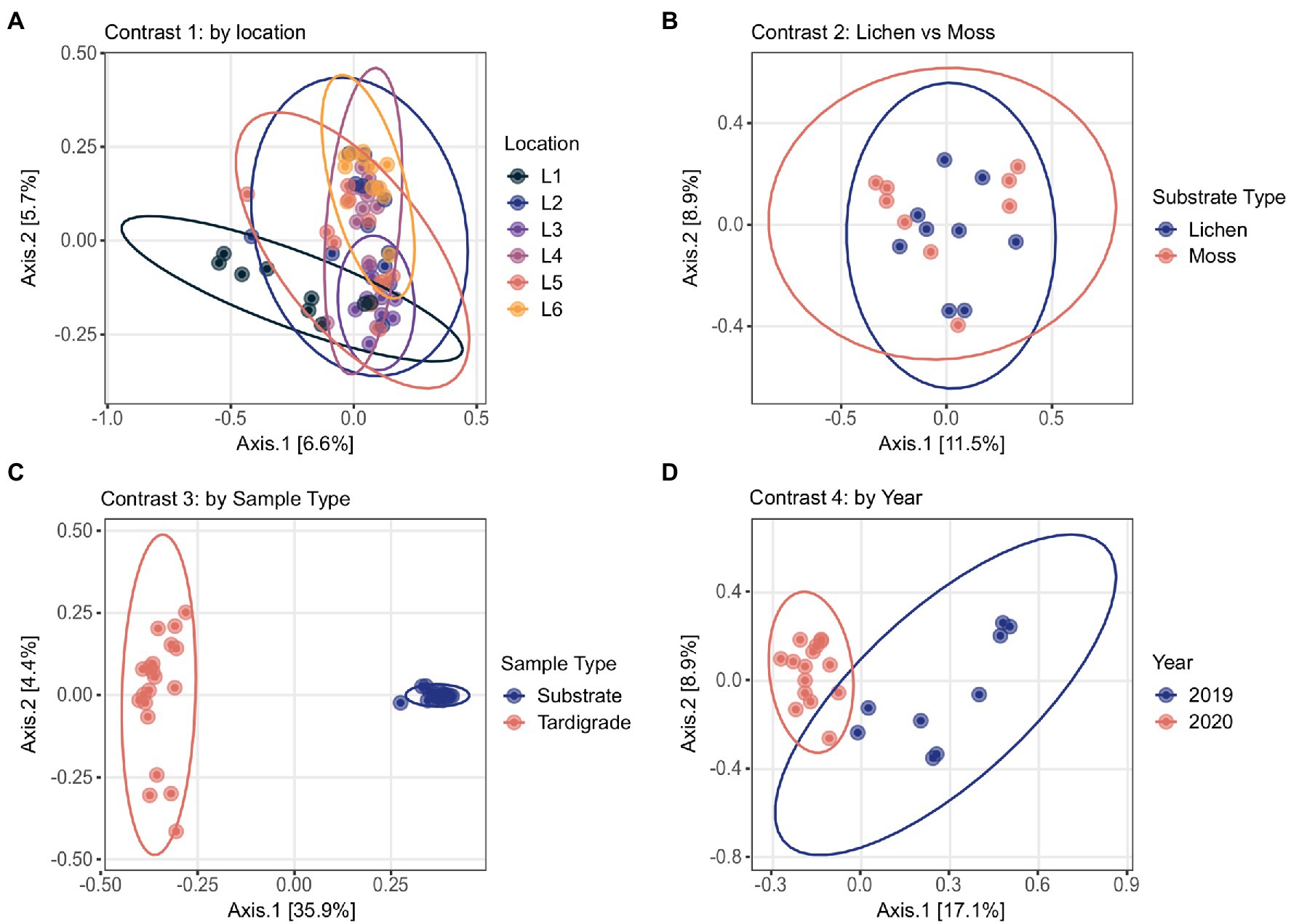
Figure 1. Principal Coordinates Analyses conducted by contrast based on Bray-Curtis distance. Location names (Location 1 through Location 6) are abbreviated as L1 through L6. (A) Tardigrade samples from different locations overlap one another, as do (B) tardigrades isolated from lichen and moss. However, (C) tardigrade and substrate samples are clearly separated, and (D) 2019 and 2020 tardigrade samples are mostly separated.
Contrast 2: Moss vs. Lichen
The community microbiome of tardigrades extracted from moss did not differ significantly in alpha diversity from that of tardigrades extracted from lichen as measured by the Shannon and Simpson indices (Table 2). PCoA further demonstrates that the overall microbial community did not differ by substrate type (Figure 1). However, between tardigrades collected from moss and those from lichen, five phyla and 11 genera were both significantly differentially abundant and significantly differentially variable, while three OTUs were significantly differentially abundant only (Supplementary Table S9). These included the common phyla Firmicutes and Bacteroidota; of these, Firmicutes were more abundant in moss-associated and Bacteroidota in lichen-associated tardigrades (Supplementary Table S10, Figure 2).
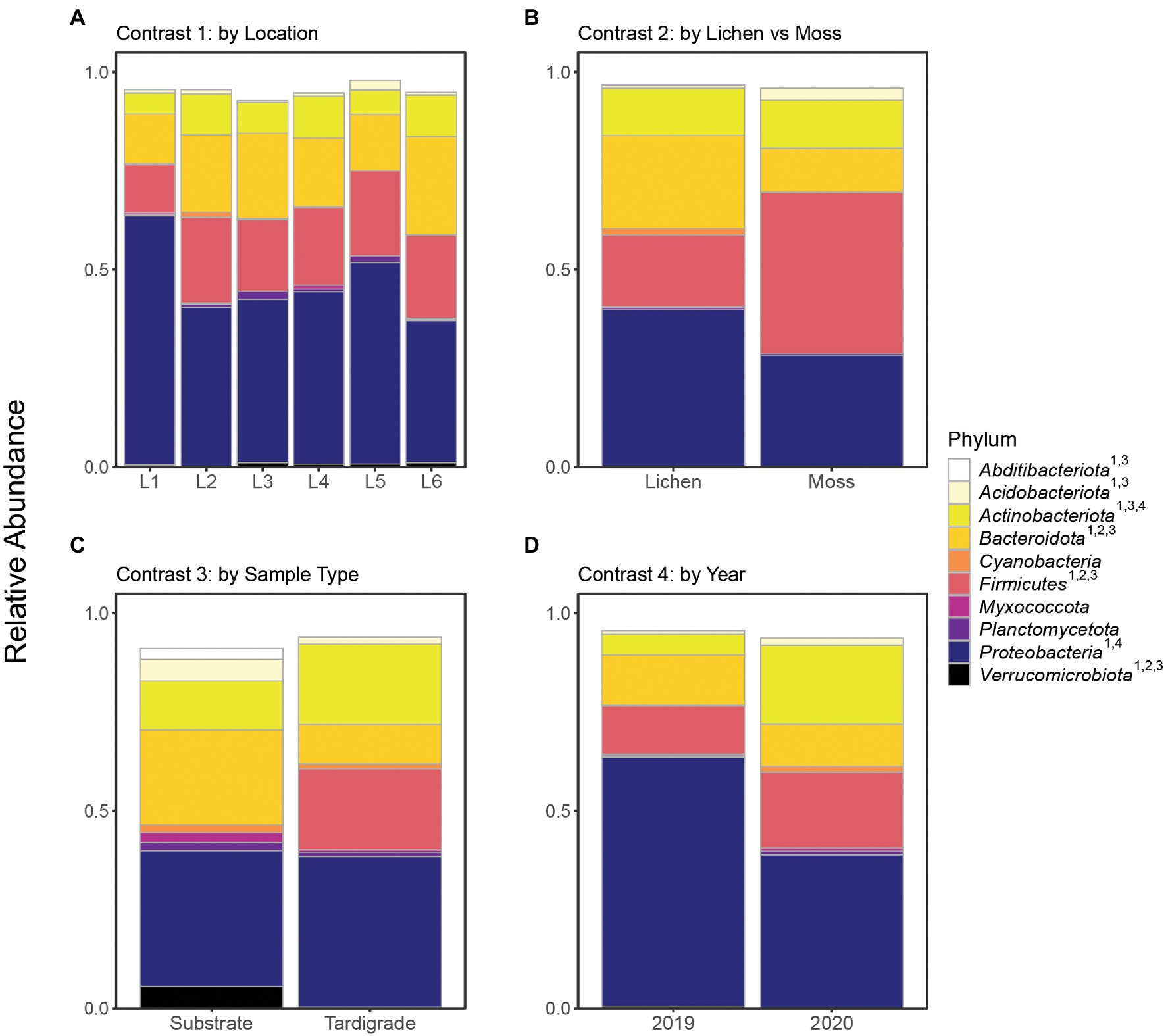
Figure 2. Relative abundance of top 10 identifiable phyla shown across all contrasts. Superscript numbers indicate the contrasts in which a given phylum was significantly differentially abundant. Location names (Location 1 through Location 6) are abbreviated as L1 through L6. (A) Contrast 1, (B) Contrast 2, (C) Contrast 3, and (D) Contrast 4.
Contrast 3: Tardigrades vs. Substrate
The microbiota of tardigrades was significantly less diverse than that of their lichen substrate, as measured by both Shannon and Simpson indices (Table 2); the tardigrade and substrate samples also formed distinct clusters as shown by PCoA (Figure 1). Between tardigrades and their substrate, 17 phyla, 181 genera, and 101 OTUs were significantly differentially abundant and variable, while 308 OTUs were significantly differentially abundant only and 124 OTUs were significantly differentially variable only (Supplementary Table S9). These differential taxa included four of the top five phyla (all except Proteobacteria) and all three of the three most abundant genera from the experiment as a whole. Remarkably, the relative abundance of Firmicutes was nearly a 1,000 times higher in the tardigrades (20.5%) than in their substrate (0.021%; Supplementary Table S10, Figure 2).
Contrast 4: Year
From 2019 to 2020, the tardigrade community microbiome increased in diversity as measured by the Simpson index, though no significant difference was found between the Shannon indices (Table 2). The two years also formed mostly distinct clusters in the PCoA (Figure 1). Between the two years, 44 genera and one OTU were significantly differentially variable and abundant, while two phyla and 26 OTUs were significantly differentially abundant only (Supplementary Table S9). These differential taxa included two of the five most common phyla (Proteobacteria and Actinobacteriota) and two of the three most common genera (Pseudomonas and unclassified Enterobacteriaceae). The unclassified Enterobacteriaceae had a particularly large change in relative abundance, decreasing more than 160-fold from 10.1% in 2019 to 0.062% in 2020 (Supplementary Table S11; Figure 3).
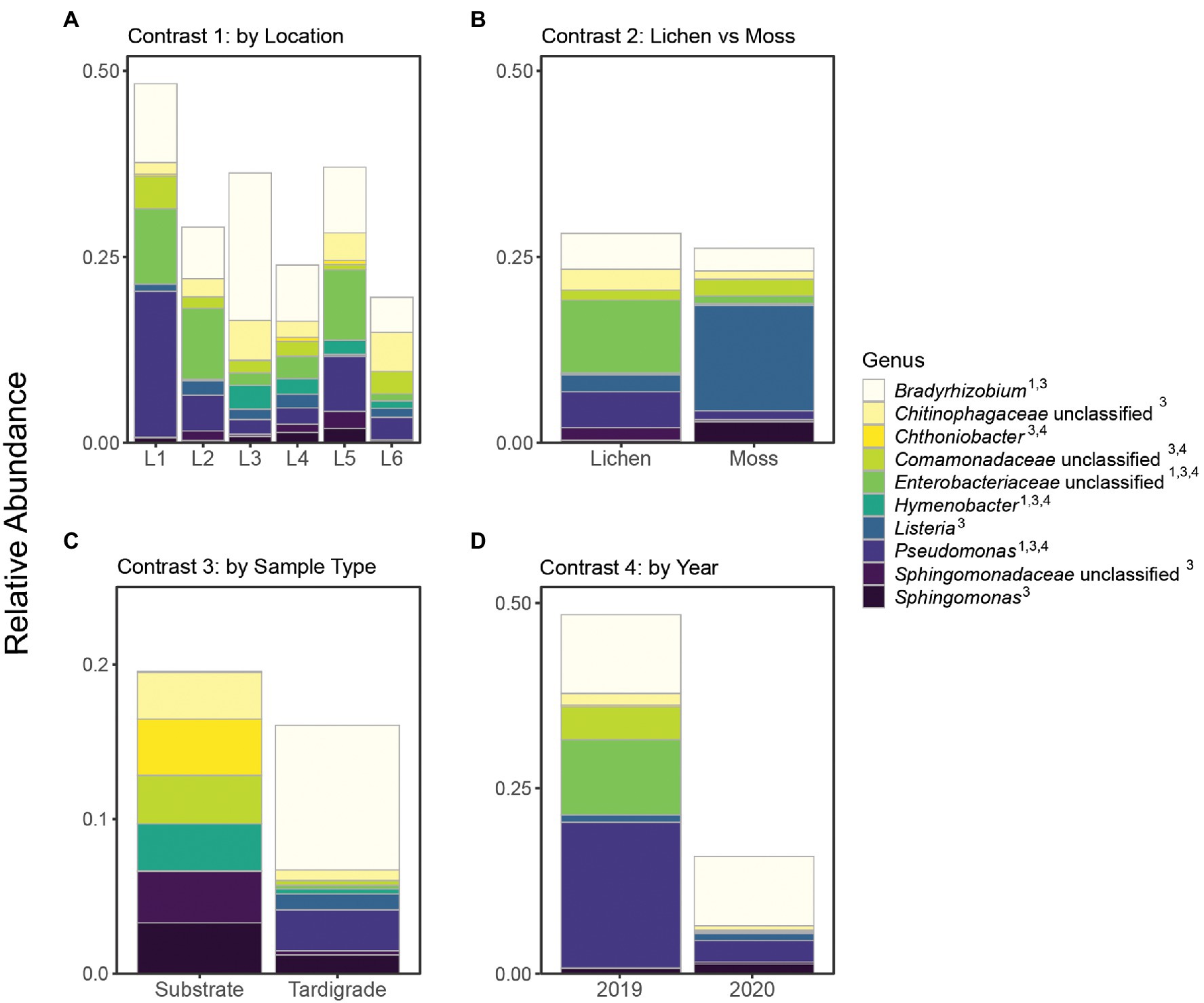
Figure 3. Relative abundance of top 10 genera (identifiable at least to family level) shown across all contrasts. Superscript numbers indicate the contrasts in which a given genus was significantly differentially abundant. Location names (Location 1 through Location 6) are abbreviated as L1 through L6. (A) Contrast 1, (B) Contrast 2, (C) Contrast 3, and (D) Contrast 4.
Discussion
Tardigrade Community
This study examined the microbiota of the full tardigrade community from a particular substrate sample, in contrast to previous surveys that studied isolated species, often from laboratory cultures (Vecchi et al., 2018; Kaczmarek et al., 2020; Mioduchowska et al., 2021; Zawierucha et al., 2022). While the focus of the current study was the bacteria associated with the full tardigrade community in a given sample, it is of course also desirable to identify species-specific tardigrade microbiota, as Vecchi et al. (2018) found that tardigrade-associated bacteria varied among tardigrade species, and we encourage further research on bacteria associated with individual tardigrade species. However, while it is occasionally possible in samples containing only a few tardigrade species to extract and immediately identify members of each for study (Vecchi et al., 2018), many environmental samples contain numerous species, including cryptic species that may be difficult or impossible to distinguish without molecular, life cycle, or other data (Cesari et al., 2013; Guidetti et al., 2016). For example, all three of the tardigrade genera observed in the current survey (Milnesium, Ramazzottius, and Paramacrobiotus) would require additional morphometric or egg observations to identify members to species level (Binda and Pilato, 1987; Kinchin, 1996; Guidetti et al., 2009; Michalczyk et al., 2012). While it would have been possible to culture collected tardigrades in the laboratory to collect eggs and generate enough individuals to both mount for morphometric species identification and to process for bacterial DNA, Vecchi et al. (2018) found that culturing significantly affects the tardigrade microbiome. Therefore, the current study has the unique advantage of better reflecting the tardigrade community microbiome in its natural state compared to studies that focus on cultured tardigrades.
While tardigrade species were not identified in this study, a December 2015 collection effort at Location 1 provides information on tardigrade diversity in the area. From lichen growing on some of the same apple trees used in the current study, the previous study identified Milnesium cf. barbadosense Meyer and Hinton 2012; Mil. burgessi Schlabach, Donaldson, Hobelman, Miller, and Lowman, 2018; Mil. swansoni Young, Chappell, Miller, and Lowman, 2016; and P. tonollii (Ramazzotti, 1956), as well as members of Milnesium Doyère, 1840; Ramazzottius Binda and Pilato, 1986; Paramacrobiotus Guidetti, Schill, Bertolani, Dandekar and Wolf, 2009; and Macrobiotidae Thulin, 1928 not identifiable to species (Tibbs-Cortes et al., 2020). While tardigrade communities are dynamic across both time (Schuster and Greven, 2007, 2013) and space (Meyer, 2006, 2008), the dominant species present in tardigrade communities in a given area can remain remarkably stable across years (Nelson and McGlothlin, 1996; Schuster and Greven, 2007). This suggests that species information from the 2015 survey may be relevant to the current study.
Reducing Effects of Contamination
All tardigrade microbiome surveys, including the current study, employed laboratory technique to reduce contamination by washing the tardigrades in sterile water before DNA extraction (Vecchi et al., 2018; Kaczmarek et al., 2020; Mioduchowska et al., 2021; Zawierucha et al., 2022). Working in a sterile environment further decreases contamination; therefore, Zawierucha et al. (2022) extracted tardigrades from substrate in a sterile environment (laminar flow chamber), and we worked in a sterile field created by a Bunsen burner throughout the experiment. Three previous studies included one type each of negative controls recommended by the RIDE standards for low biomass studies (Eisenhofer et al., 2019; DNA extraction blank in (Mioduchowska et al., 2021), sampling blank in (Kaczmarek et al., 2020), and no-template amplification control in (Zawierucha et al., 2022)). Kaczmarek et al. (2020) and Mioduchowska et al. (2021) did not sequence the negative controls; instead, they performed PCR amplification of these controls and determined that no contamination was present because no bands were visible. However, samples without visible bands from PCR can generate sequencing reads (Davis et al., 2018), and this method would not detect contaminants introduced during library preparation or sequencing steps. Zawierucha et al. (2022) removed all OTUs present in the no-template amplification control from analysis. However, low levels of true sequences, especially from high-abundance OTUs, are often present in negative controls due to cross-contamination of samples; these biologically important OTUs would therefore be removed from the analysis (Davis et al., 2018; Karstens et al., 2019). In our study, we included and sequenced all three RIDE-recommended types of negative controls.
No previous survey of the tardigrade microbiota has employed model-based in silico contaminant identification and removal, which we accomplished using the decontam package. Decontam removed 986 OTUs as contaminants, including five that would otherwise have been in the top 10 OTUs in the study by read count. Of course, further improvements are always possible. Contaminants are expected to differ in prevalence among negative control types depending on their point of introduction, but current in silico contamination removal methods treat all negative controls identically (Davis et al., 2018). Future development of a method that leverages the unique information provided by each type of negative control would therefore be desirable. However, by working under a flame, including and sequencing all recommended types of negative controls, and leveraging in silico contaminant identification and removal, we have produced what we expect to be the tardigrade microbiome survey least affected by contamination to date.
Tardigrade Community Microbiome by Contrast
We investigated the tardigrade community microbiome across four contrasts. First, we determined that the tardigrade community microbiome varied significantly in structure across locations (Table 2). Vecchi et al. (2018) found that an average of 15.4% of the microbial OTUs in a tardigrade collected from moss or lichen originate from its substrate. Therefore, known impacts of geographical location on microbial communities (Baldrian, 2017; Coller et al., 2019) could have resulted in different microbial communities present in each location to inoculate the tardigrades. Additionally, as the tardigrade microbiota is species-specific (Vecchi et al., 2018), the differences in microbial communities observed across locations may reflect spatial variation in tardigrade communities’ species composition (Meyer, 2008). Of course, these explanations are not mutually exclusive and could both play a role in shaping distinct tardigrade community microbiomes across locations. Vecchi et al. surveyed tardigrades of the same species collected from different locations, but they did not test for differences in the microbiome across locations. However, they did identify an OTU in the genus Luteolibacter that was significantly associated with Ram. oberhaeuseri collected from a location at 34 meters above sea level but not from another location 797 meters above sea level (Vecchi et al., 2018), suggesting that future work may detect differences in the microbiota of the same tardigrade species across locations.
In contrast two, the community microbiome of tardigrades collected from lichen was compared with that of tardigrades collected from moss on the same trees. While a few taxa were significantly differentially abundant and variable across substrates, the two substrates did not differ in alpha diversity and were not separated by PCoA (Figure 1). This similarity in the tardigrade community microbiome was initially surprising, as previous literature has demonstrated significant differences between the microbiota of moss and lichen even on the same tree (Aschenbrenner et al., 2017). However, this similarity across substrates could be due to the presence of similar tardigrade species, as previous studies have failed to demonstrate significant differences between tardigrade communities found in moss and lichen (Young and Clifton, 2015; Nelson et al., 2020). In future surveys, it would be interesting to compare the microbiota of tardigrades from additional substrate types (e.g., soil) and to determine if this similarity in tardigrade microbiome across substrates persists at the species as well as the community level.
Results of contrast three demonstrate that the tardigrade community microbiome is distinct from and significantly less diverse than that of its lichen substrate (Table 2, Figure 1). This result agrees with previous studies that found relatively higher diversity in substrates than in their resident Ecdysozoans, including wild tardigrades collected from moss and lichen (Vecchi et al., 2018), cultured tardigrades (Kaczmarek et al., 2020), and the nematodes Meloidogyne hapla (Adam et al., 2014) and Caenorhabditis elegans (Johnke et al., 2020). This study is the first to demonstrate this trend at the tardigrade community rather than species level. Vecchi et al. (2018) suggested that the lower microbial diversity in tardigrades with respect to their substrates may be due to the small size of tardigrades limiting the biomass and therefore the diversity of their microbiome (small host hypothesis) and/or to selectiveness of tardigrades inhibiting growth of some bacterial species and promoting growth of others (selective host hypothesis). Supporting the selective host hypothesis, earlier work found that tardigrades could be successfully inoculated with some bacteria (Xanthomonas) but not others (Serratia; Krantz et al., 1999). It is possible that some bacteria have co-evolved with tardigrades, becoming permanent residents of the gastrointestinal tract or cuticle, a hypothesis that has been suggested for the Ecdysozoan C. elegans (Zhang et al., 2017). The life cycle of tardigrades poses a unique selective pressure on any permanent residents of the microbiota, as these organisms would also have to survive within the tardigrade during cryptobiosis. This would be especially true for the obligate endosymbiotic taxa Rickettsiales and Polynucleobacter previously observed in tardigrades (Vecchi et al., 2018; Guidetti et al., 2020; Kaczmarek et al., 2020; Mioduchowska et al., 2021), as well as for the Rickettsia identified in the current study (see below).
Contrast four determined that the tardigrade community microbiome is temporally dynamic, changing significantly on the same trees from 2019 to 2020 (Table 2; Figure 1). Again, this may be due to changes in habitat microbiome, as microbiota of other substrates (e.g., soil and litter) are known to vary across years due to changing environmental factors such as nutrient availability (Martinović et al., 2021). This variation may also be due to temporal changes in the tardigrade community composition; although tardigrade species present may remain consistent in a location over years, their relative abundances shift in part due to changes in rainfall, humidity, and temperature (Schuster and Greven, 2007). This temporal variability raises important implications for future studies of the tardigrade community microbiome. For example, the relative abundance of putative phytopathogens differed significantly across years (Supplementary Table S9). Future work could identify temporal variables affecting the ability of tardigrades to act as potential reservoirs of phytopathogens and other bacteria. We also encourage further studies of the tardigrade microbiome to account for temporal changes and to investigate this variation with additional time points to increase resolution.
Tardigrade-Associated Taxa
In this study, the five most abundant phyla were Proteobacteria, Firmicutes, Bacteroidota, Actinobacteria, and Acidobacteria (Supplementary Table S10). All of these except Acidobacteria were previously reported as highly abundant in at least two of the three previous tardigrade microbiome surveys that presented results at a phylum level, with Proteobacteria identified as the most abundant phylum in all cases (Vecchi et al., 2018; Kaczmarek et al., 2020; Mioduchowska et al., 2021). Combined, the tardigrades in these studies represent a diverse set of species, including wild and laboratory-reared specimens isolated from multiple continents, suggesting that the predominance of these phyla is broadly characteristic of the microbiome of Tardigrada, regardless of species or location. These phyla, especially Proteobacteria, are also dominant in the microbiomes of other Ecdysozoans, including soil nematodes (Adam et al., 2014; Dirksen et al., 2016; Elhady et al., 2017), marine nematodes (Arcos et al., 2021), and insects (Colman et al., 2012; Engel and Moran, 2013). The tardigrade microbiota therefore appears similar to that of other Ecdysozoans at the phylum level.
A number of OTUs significantly more abundant in tardigrades than in their substrate in this study belong to taxa previously identified in the tardigrade microbiome. These include members of Enhydrobacter (Vecchi et al., 2018), Enterobacteriaceae (Kaczmarek et al., 2020; Mioduchowska et al., 2021), and Acinetobacter (Vecchi et al., 2018; Mioduchowska et al., 2021; Supplementary Table S9). In this study, OTU 22 was classified as Enhydrobacter, and its abundance in the tardigrade population varied over time, increasing significantly from 2019 (0.21%) to 2020 (2.1%; Supplementary Tables S8, S9). It is possible that Enhydrobacter is common to Ecdysozoan microbiomes, as it is also an abundant taxon in the gut contents of larval wood wasps (Li et al., 2021) and nematodes (Adam et al., 2014). Members of Enterobacteriaceae included OTUs 2 and 20. OTU 20 was further identified as a member of the Escherichia/Shigella complex, but OTU 2 could not be classified to the genus level (Supplementary Tables S2, S3). OTU 2 showed significant temporal variation, decreasing in relative abundance from 10.0 to 0.062% from 2019 to 2020 (Supplementary Tables S8, S9). Enterobacteriaceae is also highly represented in the gut microbiota of insects (Hernández-García et al., 2017; Moro et al., 2021) and nematodes (Zimmermann et al., 2020; Zhou et al., 2022). This suggests that Enterobacteriaceae may be residents of the tardigrade digestive tract. Finally, OTU 16 was a member of Acinetobacter that increased significantly in abundance from 2019 (0.000052%) to 2020 (2.3%) and was one of the three OTUs significantly differentially abundant across substrate type (Supplementary Tables S8, S9). Acinetobacter is associated with the cuticle of the nematodes M. hapla, M. incognita, and Pratylenchus penetrans (Adam et al., 2014; Elhady et al., 2017), suggesting that it may also be associated with the cuticle of tardigrades.
However, many of the tardigrade-associated taxa observed in this study have not been previously reported in the tardigrade microbiome. In fact, the most abundant OTU across all samples in this study (OTU 1) was a member of the genus Bradyrhizobium, which was not previously reported from the tardigrade microbiome. This OTU was also spatially dynamic, differing significantly in abundance across locations (Supplementary Table S9). Bradyrhizobium has been previously observed in the microbiota of plant pathogenic nematodes (Adam et al., 2014; Eberlein et al., 2016) and leaf hoppers (Horgan et al., 2019). This genus has also been found in the lichen microbiome (Bates et al., 2011; Erlacher et al., 2015; Graham et al., 2018), perhaps indicating that tardigrades acquire this bacterium from their habitat. Another tardigrade-associated genus, Micrococcus, was differentially abundant across both locations and years (Supplementary Table S9). This genus has been reported from the cuticles of soil nematodes (Adam et al., 2014) as well as fish parasitic nematodes (Arcos et al., 2021), suggesting that Micrococcus may be associated with the tardigrade cuticle. Another notable tardigrade-associated genus in this study was Nakamurella, represented primarily by OTU 33, which showed differential abundance across locations (Supplementary Table S9). Nakamurella intestinalis has been isolated from the feces of another Ecdysozoan, the katydid Pseudorynchus japonicus (Kim et al., 2017). Nakamurella endophytica and N. flava were identified as endophytes of mangroves and mint, respectively (Tuo et al., 2016; Yan et al., 2020), and N. albus and N. leprariae were originally discovered in lichens (Jiang et al., 2020; An et al., 2021). This suggests that tardigrades could obtain endophytic or lichen-dwelling Nakamurella from their habitat.
Putative Endosymbionts
Our survey corroborates previous observations of putative endosymbionts of the obligate intracellular order Rickettsiales associated with tardigrades. Three of the four previous surveys of the tardigrade microbiome have detected OTUs of this order (Vecchi et al., 2018; Guidetti et al., 2020; Kaczmarek et al., 2020; Mioduchowska et al., 2021); in addition to unclassified Rickettsiales, these OTUs included members of Wolbachia (Mioduchowska et al., 2021), Anaplasmataceae, and Ca. Tenuibacteraceae (Guidetti et al., 2020). Kaczmarek et al. also detected the obligate intracellular genus Polynucleobacter (2020). In the current survey, we identified two Rickettsiales OTUs. Of these, one was classified by mothur as Wolbachia (OTU 3606), and the other was further classified as Rickettsia (OTU 180) by BLAST and RDP analysis (Supplementary Tables S2, S3). The relative abundance of OTU 180 was significantly higher in tardigrades (0.88%) than in their lichen substrate (0.0012%; Supplementary Table S7) as well as significantly higher in 2020 (1.0%) than in 2019 (0.00026%; Supplementary Tables S8, S9). OTU 3606 was numerically more abundant in tardigrades (0.030%) than substrate (5.1 × 10−12%), though this difference was not statistically significant (Supplementary Table S7). Taken together, the intracellular nature of Rickettsiales and the higher abundance in tardigrades suggests that OTUs 180 and 3606 are endosymbionts of tardigrades.
The presence of endosymbionts may have implications for tardigrade reproduction and evolution, as members of Rickettsia and Wolbachia are known to manipulate host reproduction in other Ecdysozoans. Wolbachia is well-known for causing parthenogenesis in nematodes and arthropods, as well as feminization of males, cytoplasmic incompatibility, and male-killing (Werren et al., 2008; Kraaijeveld et al., 2011; Correa and Ballard, 2016; Kajtoch and Kotásková, 2018). Similarly, Rickettsia can induce parthenogenesis (Hagimori et al., 2006; Giorgini et al., 2010) and male-killing (Lawson et al., 2001) in arthropods. Parthenogenesis is common in tardigrades (Bertolani, 2001; Guil et al., 2022). Further investigation is necessary to determine if this is due to reproductive manipulators such as Rickettsia and Wolbachia. Future analysis could follow the example of Guidetti et al. (2020) by incorporating FISH to confirm the presence of these and other endosymbionts within tardigrade tissues.
Putative Phytopathogens
Our analysis also aimed to determine whether wild tardigrades living in apple orchards harbor phytopathogenic bacteria, and in fact, the second most abundant genus overall found in this survey was Pseudomonas, which contains more than 20 known plant pathogens (Höfte and De Vos, 2006). Pseudomonas was significantly associated with tardigrades (relative abundance in tardigrades and substrate of 2.7% and 0.051%, respectively; Supplementary Table S11) and was spatially and temporally dynamic in the tardigrade community microbiome, as relative abundance of Pseudomonas decreased significantly from 2019 (19.6%) to 2020 (3.0%) and differed significantly across locations (Supplementary Tables S9, S11). Pseudomonas was also detected in all four of the previous surveys of the tardigrade microbiome (Vecchi et al., 2018; Kaczmarek et al., 2020; Mioduchowska et al., 2021; Zawierucha et al., 2022), and Vecchi et al. (2018) identified it as part of the core tardigrade microbiome. Pseudomonas is also present in the microbiota of soil nematodes (Adam et al., 2014; Dirksen et al., 2016; Zimmermann et al., 2020) and insects (Hernández-García et al., 2017; Horgan et al., 2019; Xue et al., 2021). Notably, other Ecdysozoans (insects) act as vectors of P. syringae (Stavrinides et al., 2009; Donati et al., 2017), which is one of the most agriculturally damaging Pseudomonas species (Höfte and De Vos, 2006; Xin et al., 2018). However, Pseudomonas is very diverse, containing many non-pathogenic species (Silby et al., 2011; Passera et al., 2019). In fact, some Pseudomonas isolates from wild C. elegans confer resistance to fungal pathogens in their hosts (Dirksen et al., 2016), raising the possibility that Pseudomonas could be similarly beneficial to tardigrades.
Two additional putative phytopathogens were significantly more abundant in tardigrades than their substrate. The first, OTU 261, was identified by mothur as a member of Ralstonia, a genus containing the phytopathogenic R. solanacearum complex. In addition to being found at significantly higher abundance in tardigrades (0.018%) than their substrate (0.00017%; Supplementary Table S7), OTU 261 was temporally dynamic, decreasing significantly from 2019 (0.16%) to 2020 (0.00068%; Supplementary Tables S8, S9). Ralstonia has been previously observed in the tardigrade P. fairbanksi (Mioduchowska et al., 2021) and in nematodes (Eberlein et al., 2016; Elhady et al., 2017). The R. solanacearum complex causes major yield losses in food crops including tomatoes, bananas, and potatoes (Yuliar et al., 2015; Paudel et al., 2020). Two notable members of this complex are spread by insect vectors; the cercopoids Hindola fulva and H. strata act as vectors of R. syzygii, while the Blood Disease Bacterium is spread nonspecifically by pollinators (Eden-Green et al., 1992; Remenant et al., 2011).
The second, OTU 208, was classified by BLAST and RDP analysis to the Erwinia/Pantoea cluster (Supplementary Tables S2, S3), which includes a number of economically important phytopathogens (Kido et al., 2008; Zhang and Qiu, 2015; Dutkiewicz et al., 2016; Shapiro et al., 2016). Erwinia amylovora is of particular note as it causes fire blight in apple trees (Aćimović et al., 2015). This OTU had a significantly higher relative abundance of 0.046% in tardigrades compared to 0.0044% in their substrate (Supplementary Tables S7, S9). While neither Erwinia nor Pantoea have previously been identified in tardigrades, Erwinia has been found in arthropods (Xue et al., 2021) and nematodes (Eberlein et al., 2016). Additionally, multiple phytopathogens in Pantoea and Erwinia are transmitted by insect vectors (Basset et al., 2000; Sasu et al., 2010; Ordax et al., 2015; Walterson and Stavrinides, 2015; Dutkiewicz et al., 2016). However, it is also possible that OTU 208 represents a symbiont in tardigrades, as Erwinia also includes the olive fly obligate gut symbiont Candidatus Erwinia dacicola (Blow et al., 2020).
Additional putative plant pathogens were observed at lower abundances in the tardigrade community microbiome and were not significantly more abundant in tardigrades than their substrate. These include another Ralstonia (OTU 1556) and OTU 1620, which was classified as Pectobacterium by BLAST and RDP (Supplementary Tables S2, S3). Members of Pectobacterium cause soft rot diseases in economically important plants, and some strains are capable of infecting multiple plant species (Ma et al., 2007; Li et al., 2020). Additionally, prompted by previous observation of the tardigrade M. hufelandi acting as a vector of the plant pathogen X. campestris (Benoit et al., 2000), we searched the tardigrade community microbiome for members of Xanthomonas. OTUs 10,409 and 12,281 were classified as Xanthomonas (OTU 10,409 by BLAST and RDP analysis), but were both at extremely low abundance (Supplementary Table S4).
In summary, we observed the presence of multiple putative plant pathogens in the community microbiome of tardigrades isolated from apple orchards. Tardigrades could act as vectors or reservoirs of these putative pathogens, a possibility raised by the previous observation of M. hufelandi as a vector of X. campestris (Benoit et al., 2000). However, a major limitation of this study is the use of only 16S rRNA amplicon sequencing. Because multiple marker genes are required to distinguish among species within Pseudomonas, Ralstonia, Erwinia, and Pantoea (Gomila et al., 2015; Zhang and Qiu, 2015; Palmer et al., 2017; Paudel et al., 2020; Saati-Santamaría et al., 2021), we were unable to identify OTUs in our study to species level. Therefore, we are unable to determine whether the identified OTUs in plant pathogenic genera are themselves phytopathogens. We encourage future analyses of tardigrade-associated bacteria in these groups through techniques such as metagenome sequencing and multilocus sequence typing to clarify this point.
Conclusion
This study is the first microbiome analysis of wild tardigrade populations in an agricultural setting and is also the first microbiome study assessing North American tardigrades. Our methods reduced the effects of contamination compared to other tardigrade microbiome studies by including aseptic technique, all three recommended control types, and in silico contaminant removal. We found that the tardigrade community microbiome is distinct from the substrate microbiota and varies across location and time. In addition to identifying putative endosymbionts, we also observed multiple tardigrade-associated taxa that may represent phytopathogens. The results of this study both increase our knowledge of the tardigrade microbiome and prompt new avenues of research.
Data Availability Statement
The datasets presented in this study can be found online at the Sequence Read Archive repository, BioProject accession PRJNA801902: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA801902.
Author Contributions
LT-C: conceptualization, software, formal analysis, investigation, data curation, writing – original draft, review, and editing, and visualization. BT-C: conceptualization, investigation, data curation, writing – original draft, review, and editing, and visualization. SS-E: methodology, resources, and writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
LT-C and BT-C are supported by the National Science Foundation Graduate Research Fellowship Program (NSF GRFP grant no. 1744592). Funds were also provided by the Iowa State University Plant Sciences Institute, and the open access publication fees for this article were covered by the Iowa State University Library.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank the owners of the studied apple orchards, including Dennise Smith, Roland Newby, Koenigs’ Acres Farm, and others who wish to remain anonymous. We also thank Lucas Koester and Chiron Anderson for providing example phyloseq code and tutorials as well as useful input on the project. We thank the Iowa State University (ISU) DNA Facility for their input on sequencing, the ISU Department of Ecology, Evolution, and Organismal Biology for use of microscopes, and the Iowa Geological Survey for shapefiles used to produce the collection map.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articless/10.3389/fmicb.2022.866930/full#supplementary-material
Supplementary Figure S1 | Collection locations map. The map of Iowa, United States shows the sampled counties outlined in red. The inset shows collection sites within Hardin and Franklin counties identified by location number.
Supplementary Figure S2 | Representative images of sampled trees and substrates. (A) Tree 2, Location 1 (B) Lichen collected from Tree 2, Location 1 (C) Moss collected from Tree 1, Location 2.
Supplementary Figure S3 | Relative abundance of top 10 identifiable phyla shown across all samples. Location names (Location 1 through Location 6) are abbreviated as L1 through L6.
Supplementary Figure S4 | Relative abundance of top 10 genera (identifiable at least to family level) shown across all samples. Location names (Location 1 through Location 6) are abbreviated as L1 through L6.
Supplementary Figure S5 | Principal Coordinates Analysis of all samples based on Bray-Curtis distance. Location names (Location 1 through Location 6) are abbreviated as L1 through L6. Substrate samples are clearly separated from the tardigrade samples along Axis 1. On Axis 2, samples from 2020 are generally clustered away from 2019 samples. Samples from different locations and from lichen and moss overlap.
Supplementary Table S1 | Mothur classification and decontam score for all OTUs with more than 10 reads in experimental samples. OTUs with decontam score below the 0.25 threshold are marked as contaminants.
Supplementary Table S2 | BLAST results for OTUs with more than 10 total reads in experimental samples. BLAST+ v2.11.0 was used to query representative sequences for each OTU against a database generated from the NCBI 16S RefSeq collection. The 15 hits with the lowest E-values are given for each OTU.
Supplementary Table S3 | RDP Classifier results for OTUs with more than 10 total reads in experimental samples. Representative sequences for each OTU were uploaded to the RDP Classifier webtool (https://rdp.cme.msu.edu/classifier/classifier.jsp), version 2.11 using 16S rRNA training set 18.
Supplementary Table S4 | Relative abundance of all analyzed OTUs in each sample.
Supplementary Table S5 | Relative abundance of each OTU by level of Contrast 1 (Location).
Supplementary Table S6 | Relative abundance of each OTU in moss and in lichen (i.e., at each level of Contrast 2).
Supplementary Table S7 | Relative abundance of each OTU in tardigrades and in their lichen substrate (i.e., at each level of Contrast 3).
Supplementary Table S8 | Relative abundance of each OTU in tardigrades in 2019 and in 2020 (i.e., at each level of Contrast 4).
Supplementary Table S9 | Differentially abundant and variable taxa across contrasts. Corncob was used to identify significantly (Benjamini–Hochberg corrected p value <0.05) differentially abundant and variable taxa across four contrasts of interest. Genus names are presented including their phylum, class, order, and family names to prevent ambiguities.
Supplementary Table S10 | Relative abundance of top 10 identifiable phyla across levels of each contrast.
Supplementary Table S11 | Relative abundance of top 10 genera (identifiable at least to family level) across levels of each contrast.
Footnotes
1. ^https://earthmicrobiome.org/protocols-and-standards/16s/
References
Aćimović, S. G., Zeng, Q., McGhee, G. C., Sundin, G. W., and Wise, J. C. (2015). Control of ire blight (Erwinia amylovora) on apple trees with trunk-injected plant resistance inducers and antibiotics and assessment of induction of pathogenesis-related protein genes. Front. Plant Sci. 6:16. doi: 10.3389/fpls.2015.00016
Adam, M., Westphal, A., Hallmann, J., and Heuer, H. (2014). Specific microbial attachment to root knot nematodes in suppressive soil. Appl. Environ. Microbiol. 80, 2679–2686. doi: 10.1128/AEM.03905-13
Aguinaldo, A. M., Turbeville, J. M., Linford, L. S., Rivera, M. C., Garey, J. R., Raff, R. A., et al. (1997). Evidence for a clade of nematodes, arthropods and other Moulting animals. Nature 387, 489–493. doi: 10.1038/387489a0
An, D.-F., Yang, S.-J., Jiang, L.-Q., Wang, X.-Y., Huang, X.-Y., Lang, L., et al. (2021). Nakamurella leprariae Sp. Nov., isolated from a lichen sample. Arch. Microbiol. 204:19. doi: 10.1007/s00203-021-02626-7
Arcos, S. C., Lira, F., Robertson, L., González, M. R., Carballeda-Sangiao, N., Sánchez-Alonso, I., et al. (2021). Metagenomics analysis reveals an extraordinary inner bacterial diversity in Anisakids (Nematoda: Anisakidae) L3 larvae. Microorganisms 9:1088. doi: 10.3390/microorganisms9051088
Aschenbrenner, I. A., Cernava, T., Erlacher, A., Berg, G., and Grube, M. (2017). Differential sharing and distinct co-occurrence networks among spatially close bacterial microbiota of bark, mosses and lichens. Mol. Ecol. 26, 2826–2838. doi: 10.1111/mec.14070
Baldrian, P. (2017). Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol. Rev. 41, 109–130. doi: 10.1093/femsre/fuw040
Basset, A., Khush, R. S., Braun, A., Gardan, L., Boccard, F., Hoffmann, J. A., et al. (2000). The Phytopathogenic Bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc. Natl. Acad. Sci. U. S. A. 97:3376, –3381. doi: 10.1073/pnas.97.7.3376
Bates, S. T., Cropsey, G. W. G., Gregory Caporaso, J., Knight, R., and Fierer, N. (2011). Bacterial communities associated with the lichen Symbiosis. Appl. Environ. Microbiol. 77, 1309–1314. doi: 10.1128/AEM.02257-10
Becquerel, P. (1950). La Suspension de La Vie Au Dessous de 1/20 K Absolu Par Demagnetization Adiabatique de l’alun de Fer Dans Le Vide Les plus Eléve. Comptes Rendus Des Séances de l’Académie Des Sciences 231, 261–263.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Benoit, T. G., Locke, J., Marks, J. R., and Beasley, C. W. (2000). Laboratory transmission of Xanthomonas campestris pv. Raphani by a Tardigrade (Parachela = Macrobiotidae). Fla. Entomol. 83, 197–199. doi: 10.2307/3496157
Bertolani, R. (2001). Evolution of the reproductive mechanisms in Tardigrades — a review. Zool. Anz. J. Comp. Zool. 240, 247–252. doi: 10.1078/0044-5231-00032
Binda, M. G., and Pilato, G. (1987). Ramazzottius, Nuovo Genere Di Eutardigrado (Hypsibiidae). Animal 13, 159–166.
Blow, F., Gioti, A., Goodhead, I. B., Kalyva, M., Kampouraki, A., Vontas, J., et al. (2020). Functional genomics of a symbiotic community: shared traits in the olive fruit Fly gut microbiota. Genome Biol. Evol. 12, 3778–3791. doi: 10.1093/gbe/evz258
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10:421. doi: 10.1186/1471-2105-10-421
Cesari, M., Guidetti, R., Rebecchi, L., Giovannini, I., and Bertolani, R. (2013). A DNA barcoding approach in the study of Tardigrades. J. Limnol. 72, 182–198. doi: 10.4081/jlimnol.2013.s1.e23
Coller, E., Cestaro, A., Zanzotti, R., Bertoldi, D., Pindo, M., Larger, S., et al. (2019). Microbiome of vineyard soils is shaped by geography and management. Microbiome 7:140. doi: 10.1186/s40168-019-0758-7
Colman, D. R., Toolson, E. C., and Takacs-Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21, 5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x
Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing Against a master manipulator. Front. Ecol. Evol. 3:153. doi: 10.3389/fevo.2015.00153
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and Metagenomics data. Microbiome 6, 1–14. doi: 10.1186/s40168-018-0605-2
Dirksen, P., Marsh, S. A., Braker, I., Heitland, N., Wagner, S., Nakad, R., et al. (2016). The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol. 14:38. doi: 10.1186/s12915-016-0258-1
Donati, I., Mauri, S., Buriani, G., Cellini, A., and Spinelli, F. (2017). Role of Metcalfa pruinosa as a vector for Pseudomonas syringae pv. Actinidiae. Plant Pathol. J. 33, 554–560. doi: 10.5423/PPJ.OA.04.2017.0074
Dutkiewicz, J., Mackiewicz, B., Lemieszek, M. K., Golec, M., and Milanowski, J. (2016). Pantoea agglomerans: a mysterious bacterium of evil and good. Part III. Deleterious effects: infections of humans, animals and plants. Ann. Agric. Environ. Med. 23, 197–205. doi: 10.5604/12321966.1203878
Dutta, B., Barman, A. K., Srinivasan, R., Avci, U., Ullman, D. E., Langston, D. B., et al. (2014). Transmission of Pantoea ananatis and P. agglomerans, causal agents of center rot of onion (Allium cepa), by onion Thrips (Thrips tabaci) Through feces. Phytopathology 104, 812–819. doi: 10.1094/PHYTO-07-13-0199-R
Duveiller, E., Bragard, C., and Marite, H. (1997). “Bacterial leaf streak and black chaff caused by Xanthomonas translucens” in The Bacterial Disease of Wheat: Concepts and Methods of Disease Management. eds. A. McNab, E. Duveiller, L. Fucikovsky, and K. Rudolph (Mexico: CIMMYT), 25–47.
Eberlein, C., Heuer, H., Vidal, S., and Westphal, A. (2016). Microbial communities in Globodera pallida females raised in potato monoculture soil. Phytopathology 106, 581–590. doi: 10.1094/PHYTO-07-15-0180-R
Eden-Green, S. J., Balfas, R., Sutarjo, T., and Jamalius, (1992). Characteristics of the transmission of Sumatra disease of cloves by tube-building Cercopoids, Hindola spp. Plant Pathol. 41, 702–712. doi: 10.1111/j.1365-3059.1992.tb02553.x
Eisenhofer, R., Minich, J. J., Marotz, C., Cooper, A., Knight, R., and Weyrich, L. S. (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Elhady, A., Giné, A., Topalovic, O., Jacquiod, S., Sørensen, S. J., Sorribas, F. J., et al. (2017). Microbiomes associated with infective stages of root-knot and lesion nematodes in soil. PLoS One 12:e0177145. doi: 10.1371/journal.pone.0177145
Engel, P., and Moran, N. A. (2013). Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut Microbes 4, 60–65. doi: 10.4161/gmic.22517
Erlacher, A., Cernava, T., Cardinale, M., Soh, J., Sensen, C. W., Grube, M., et al. (2015). Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front. Microbiol. 6:53. doi: 10.3389/fmicb.2015.00053
Giorgini, M., Bernardo, U., Monti, M. M., Nappo, A. G., and Gebiola, M. (2010). Rickettsia symbionts cause parthenogenetic reproduction in the parasitoid wasp Pnigalio soemius (Hymenoptera: Eulophidae). Appl. Environ. Microbiol. 76, 2589–2599. doi: 10.1128/AEM.03154-09
Gomila, M., Peña, A., Mulet, M., Lalucat, J., and García-Valdés, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
Graham, L. E., Trest, M. T., Will-Wolf, S., Miicke, N. S., Atonio, L. M., Piotrowski, M. J., et al. (2018). Microscopic and metagenomic analyses of Peltigera ponojensis (Peltigerales, Ascomycota). Int. J. Plant Sci. 179, 241–255. doi: 10.1086/696534
Guidetti, R., Rebecchi, L., Bertolani, R., Ingemar, K. J., Kristensen, R. M., and Cesari, M. (2016). Morphological and molecular analyses on Richtersius (Eutardigrada) diversity reveal its new systematic position and Lead to the establishment of a new genus and a new family within Macrobiotoidea. Zool. J. Linn. Soc. 178, 834–845. doi: 10.1111/zoj.12428
Guidetti, R., Schill, R. O., Bertolani, R., Dandekar, T., and Wolf, M. (2009). New molecular data for tardigrade phylogeny, with the erection of Paramacrobiotus gen. Nov. J. Zool. Syst. Evol. Res. 47, 315–321. doi: 10.1111/J.1439-0469.2009.00526.X
Guidetti, R., Vecchi, M., Ferrari, A., Newton, I. L. G., Cesari, M., and Rebecchi, L. (2020). Further insights in the Tardigrada microbiome: phylogenetic position and prevalence of infection of four new Alphaproteobacteria putative Endosymbionts. Zool. J. Linnean Soc. 188, 925–937. doi: 10.1093/zoolinnean/zlz128
Guil, N., Guidetti, R., Cesari, M., Marchioro, T., Rebecchi, L., and Machordom, A. (2022). Molecular phylogenetics, speciation, and long distance dispersal in tardigrade evolution: a case study of the genus Milnesium. Mol. Phylogenet. Evol. 169:107401. doi: 10.1016/J.YMPEV.2022.107401
Hagimori, T., Abe, Y., Date, S., and Miura, K. (2006). The first finding of a Rickettsia bacterium associated with parthenogenesis induction Among insects. Curr. Microbiol. 52, 97–101. doi: 10.1007/s00284-005-0092-0
Hernández-García, J. A., Briones-Roblero, C. I., Rivera-Orduña, F. N., and Zúñiga, G. (2017). Revealing the gut Bacteriome of Dendroctonus bark beetles (Curculionidae: Scolytinae): diversity, Core members and co-evolutionary patterns. Sci. Rep. 7:13864. doi: 10.1038/s41598-017-14031-6
Höfte, M., and De Vos, P. (2006). “Plant pathogenic Pseudomonas species” in Plant-Associated Bacteria. ed. S. S. Gnanamanickam (Dordrecht: Springer), 507–533.
Horgan, F. G., Srinivasan, T. S., Crisol-Martínez, E., Maria Liberty, P., Almazan, A. F., Ramal, R. O., et al. (2019). Microbiome responses during virulence adaptation by a phloem-feeding insect to resistant near-isogenic Rice lines. Ecol. Evol. 9, 11911–11929. doi: 10.1002/ece3.5699
Jiang, L.-Q., An, D.-F., Zhang, K., Li, G.-D., Wang, X.-Y., Lang, L., et al. (2020). Nakamurella albus sp. nov.: a novel Actinobacterium isolated from a lichen sample. Curr. Microbiol. 77, 1896–1901. doi: 10.1007/s00284-020-01928-1
Johnke, J., Dirksen, P., and Schulenburg, H. (2020). Community assembly of the Native C. elegans microbiome is influenced by time, substrate and individual bacterial taxa. Environ. Microbiol. 22, 1265–1279. doi: 10.1111/1462-2920.14932
Jönsson, K. I., Rabbow, E., Schill, R. O., Harms-Ringdahl, M., and Rettberg, P. (2008). Tardigrades survive exposure to space in low earth orbit. Curr. Biol. 18, R729–R731. doi: 10.1016/j.cub.2008.06.048
Kaczmarek, Ł., Roszkowska, M., Poprawa, I., Janelt, K., Kmita, H., Gawlak, M., et al. (2020). Integrative Description of Bisexual Paramacrobiotus experimentalis sp. nov. (Macrobiotidae) from Republic of Madagascar (Africa) with Microbiome Analysis. Mol. Phylogenet. Evol. 145:106730. doi: 10.1016/j.ympev.2019.106730
Kajtoch, Ł., and Kotásková, N. (2018). Current state of knowledge on Wolbachia infection among Coleoptera: a systematic review. PeerJ 6:e4471. doi: 10.7717/peerj.4471
Karamura, G., Smith, J., Studholme, D., Kubiriba, J., and Karamura, E. (2007). Comparative pathogenicity studies of the Xanthomonas vasicola species on maize, sugarcane and Banana. African J. Plant Sci. 9, 385–400. doi: 10.5897/AJPS2015.1327
Karstens, L., Asquith, M., Davin, S., Damien Fair, W., Gregory, T., Wolfe, A. J., et al. (2019). Controlling for contaminants in low-biomass 16S RRNA gene sequencing experiments. MSystems 4, e00290–e00219. doi: 10.1128/mSystems.00290-19
Kido, K., Adachi, R., Hasegawa, M., Yano, K., Hikichi, Y., Takeuchi, S., et al. (2008). Internal fruit rot of netted melon caused by Pantoea ananatis (=Erwinia ananas) in Japan. J. Gen. Plant Pathol. 74, 302–312. doi: 10.1007/s10327-008-0107-3
Kim, S.-J., Cho, H., Joa, J.-H., Hamada, M., Ahn, J.-H., Weon, H.-Y., et al. (2017). Nakamurella intestinalis sp. nov., isolated from the Faeces of Pseudorhynchus japonicus. Int. J. Syst. Evol. Microbiol. 67, 2970–2974. doi: 10.1099/ijsem.0.002059
Kinchin, I. M. (1996). Morphometric analysis of Ramazzottius varieornatus (Hypsibiidae: Eutardigrada). Zool. J. Linnean Soc. 116, 51–60. doi: 10.1111/j.1096-3642.1996.tb02332.x
Kraaijeveld, K., Reumer, B. M., Mouton, L., Kremer, N., Vavre, F., and van Alphen, J. J. M. (2011). Does a parthenogenesis-inducing Wolbachia induce vestigial cytoplasmic incompatibility? Naturwissenschaften 98, 175–180. doi: 10.1007/s00114-010-0756-x
Krantz, S. L., Benoit, T. G., and Beasley, C. W. (1999). Phytopathogenic bacteria associated with Tardigrada. Zool. Anz. 238, 259–260.
Lawson, E. T., Mousseau, T. A., Klaper, R., Hunter, M. D., and Werren, J. H. (2001). Rickettsia associated with male-killing in a buprestid beetle. Heredity 86, 497–505. doi: 10.1046/j.1365-2540.2001.00848.x
Li, J., Li, C., Wang, M., Wang, L., Liu, X., Gao, C., et al. (2021). Gut structure and microbial communities in Sirex noctilio (Hymenoptera: Siricidae) and their predicted contribution to larval nutrition. Front. Microbiol. 12:641141. doi: 10.3389/fmicb.2021.641141
Li, X., Lu, F., Chen, C., Sun, W., Tian, Y., and Xie, H. (2020). Characteristics and rapid diagnosis of Pectobacterium carotovorum ssp. associated With bacterial soft rot of vegetables in China. Plant Dis. 104, 1158–1166. doi: 10.1094/PDIS-05-19-1033-RE
Ma, B., Hibbing, M. E., Kim, H.-S., Reedy, R. M., Yedidia, I., Breuer, J., et al. (2007). Host range and molecular phylogenies of the soft rot Enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
Martin, B. D., Witten, D., and Willis, A. D. (2020). Modeling microbial abundances and Dysbiosis with Beta-binomial regression. Ann. Appl. Stat. 14, 94–115. doi: 10.1214/19-aoas1283
Martinović, T., Odriozola, I., Mašínová, T., Bahnmann, B. D., Kohout, P., Sedlák, P., et al. (2021). Temporal turnover of the soil microbiome composition is guild-specific. Ecol. Lett. 24, 2726–2738. doi: 10.1111/ele.13896
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, H. A. (2006). Small-scale spatial distribution variability in terrestrial Tardigrade populations. Hydrobiologia 558, 133–139. doi: 10.1007/s10750-005-1412-x
Meyer, H. A. (2008). Distribution of tardigrades in Florida. Southeast. Nat. 7, 91–100. doi: 10.1656/S407.sl
Meyer, H. A. (2013). Terrestrial and freshwater Tardigrada of the Americas. Zootaxa 3747, 001–071. doi: 10.11646/zootaxa.3747.1.1
Michalczyk, Ł., Wełnicz, W., Frohme, M., Kaczmarek, Ł., Welnicz, W., Frohme, M., et al. (2012). Redescriptions of three Milnesium Doyère, 1840 taxa (Tardigrada: Eutardigrada: Milnesiidae), including the nominal species for the genus. Zootaxa 3154, 1–20. doi: 10.11646/zootaxa.3154.1.1
Mioduchowska, M., Nitkiewicz, B., Roszkowska, M., Kačarević, U., Madanecki, P., Pinceel, T., et al. (2021). Taxonomic classification of the bacterial Endosymbiont Wolbachia based on next-generation sequencing: is there molecular evidence for its presence in Tardigrades? Genome 64, 951–958. doi: 10.1139/gen-2020-0036
Mogle, M. J., Kimball, S. A., Miller, W. R., and McKown, R. D. (2018). Evidence of avian-mediated long distance dispersal in American Tardigrades. PeerJ 6:e5035. doi: 10.7717/peerj.5035
Moro, M. S., Xing, W., Wei, W., Mendes, L. W., Allen, K. C., Pinheiro, J. B., et al. (2021). Characterization and comparison of intestinal bacterial microbiomes of Euschistus heros and Piezodorus guildinii collected in Brazil and the United States. Front. Microbiol. 12:769965. doi: 10.3389/fmicb.2021.769965
Nelson, D. R., Bartels, P. J., and Fegley, S. R. (2020). Environmental correlates of Tardigrade community structure in mosses and lichens in the Great Smoky Mountains National Park (Tennessee and North Carolina, USA). Zool. J. Linnean Soc. 188, 913–924. doi: 10.1093/ZOOLINNEAN/ZLZ043
Nelson, D. R., and McGlothlin, K. L. (1996). A new species of Calohypsibius (phylum Tardigrada, Eutardigrada) from Roan Mountain, Tennessee-North Carolina, U.S.A. Zool. J. Linnean Soc. 116, 167–174. doi: 10.1111/j.1096-3642.1996.tb02341.x
Ng, J. C. K., and Falk, B. W. (2006). Virus-vector interactions mediating nonpersistent and Semipersistent transmission of plant viruses. Annu. Rev. Phytopathol. 44, 183–212. doi: 10.1146/annurev.phyto.44.070505.143325
O’Leary, N. A., Wright, M. W., Rodney Brister, J., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Ordax, M., Piquer-Salcedo, J. E., Santander, R. D., Sabater-Muñoz, B., Biosca, E. G., López, M. M., et al. (2015). Medfly Ceratitis capitata as potential vector for fire blight pathogen Erwinia amylovora: survival and transmission. PLoS One 10:e0127560. doi: 10.1371/journal.pone.0127560
Palmer, M., Steenkamp, E. T., Coetzee, M. P. A., Chan, W.-Y., van Zyl, E., De Maayer, P., et al. (2017). Phylogenomic resolution of the bacterial genus Pantoea and its relationship with Erwinia and Tatumella. Antonie Van Leeuwenhoek 110, 1287–1309. doi: 10.1007/s10482-017-0852-4
Passera, A., Compant, S., Casati, P., Maturo, M. G., Battelli, G., Quaglino, F., et al. (2019). Not just a pathogen? Description of a plant-beneficial Pseudomonas syringae strain. Front. Microbiol. 10:1409. doi: 10.3389/fmicb.2019.01409
Paudel, S., Dobhal, S., Alvarez, A. M., and Arif, M. (2020). Taxonomy and phylogenetic research on Ralstonia solanacearum species complex: a complex pathogen with extraordinary economic consequences. Pathogens (Basel, Switzerland) 9:886. doi: 10.3390/pathogens9110886
Remenant, B., de Cambiaire, J.-C., Cellier, G., Jacobs, J. M., Mangenot, S., Barbe, V., et al. (2011). Ralstonia syzygii, the blood disease bacterium and Some Asian R. solanacearum strains form a single genomic species despite divergent lifestyles. PLoS One 6:e24356. doi: 10.1371/journal.pone.0024356
Saati-Santamaría, Z., Peral-Aranega, E., Velázquez, E., Rivas, R., and García-Fraile, P. (2021). Phylogenomic analyses of the genus Pseudomonas Lead to the rearrangement of several species and the definition of new genera. Biology 10:782. doi: 10.3390/biology10080782
Sasu, M. A., Seidl-Adams, I., Wall, K., Winsor, J. A., and Stephenson, A. G. (2010). Floral transmission of Erwinia tracheiphila by cucumber beetles in a wild Cucurbita pepo. Environ. Entomol. 39, 140–148. doi: 10.1603/EN09190
Schram, M. D., and Davison, P. G. (2012). Irwin loops—a history and method of constructing homemade loops. Trans. Kans. Acad. Sci. 115, 35–40. doi: 10.1660/062.115.0106
Schuster, R., and Greven, H. (2007). A long-term study of population dynamics of Tardigrades in the Moss Rhytidiadelphus squarrosus (Hedw.) Warnst. J. Limnol. 66, 141–151. doi: 10.4081/jlimnol.2007.s1.141
Schuster, R., and Greven, H. (2013). Reproductive traits of Macrobiotus hufelandi during a long-term field study with notes on Paramacrobiotus richtersi and Diphascon pingue (Eutardigrada). J. Limnol. 72, 166–174. doi: 10.4081/jlimnol.2013.s1.e21
Shapiro, L. R., Scully, E. D., Straub, T. J., Park, J., Stephenson, A. G., Beattie, G. A., et al. (2016). Horizontal gene acquisitions, Mobile element proliferation, and genome decay in the host-restricted plant pathogen Erwinia tracheiphila. Genome Biol. Evol. 8, 649–664. doi: 10.1093/gbe/evw016
Silby, M. W., Winstanley, C., Godfrey, S. A. C., Levy, S. B., and Jackson, R. W. (2011). Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Stavrinides, J., McCloskey, J. K., and Ochman, H. (2009). Pea aphid as Both host and vector for the Phytopathogenic bacterium Pseudomonas syringae. Appl. Environ. Microbiol. 75, 2230–2235. doi: 10.1128/AEM.02860-08
Tibbs-Cortes, L. E., Tibbs-Cortes, B. W., and Miller, W. R. (2020). Tardigrades of Hardin County, Iowa: seven new records from Iowa, USA. Trans. Kans. Acad. Sci. 123, 203–212. doi: 10.1660/062.123.0117
Tuo, L., Li, F.-N., Pan, Z., Lou, I., Guo, M., Lee, S. M.-Y., et al. (2016). Nakamurella endophytica sp. nov., a novel Endophytic Actinobacterium isolated from the bark of Kandelia candel. Int. J. Syst. Evol. Microbiol. 66, 1577–1582. doi: 10.1099/ijsem.0.000923
Vecchi, M., Newton, I. L. G. G., Cesari, M., Rebecchi, L., and Guidetti, R. (2018). The microbial Community of Tardigrades: environmental influence and species specificity of microbiome structure and composition. Microb. Ecol. 76, 467–481. doi: 10.1007/s00248-017-1134-4
Walterson, A. M., and Stavrinides, J. (2015). Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 39, 968–984. doi: 10.1093/femsre/fuv027
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of RRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Willis, A. D., and Martin, B. D. (2020). Estimating diversity in networked ecological communities. Biostatistics 23, 207–222. doi: 10.1093/biostatistics/kxaa015
Xin, X.-F., Kvitko, B., and He, S. Y. (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat. Rev. Microbiol. 16, 316–328. doi: 10.1038/nrmicro.2018.17
Xue, H., Zhu, X., Wang, L., Zhang, K., Li, D., Ji, J., et al. (2021). Gut bacterial diversity in different life cycle stages of Adelphocoris suturalis (Hemiptera: Miridae). Front. Microbiol. 12:670383. doi: 10.3389/fmicb.2021.670383
Yan, X.-R., Chen, M.-S., Yang, C., An, M.-B., Li, H.-Y., Shi, H.-C., et al. (2020). Nakamurella flava sp. nov., a novel Endophytic Actinobacterium isolated from Mentha haplocalyx Briq. Int. J. Syst. Evol. Microbiol. 70, 835–840. doi: 10.1099/ijsem.0.003831
Young, A., and Clifton, K. (2015). Tardigrades inhabit lichen and moss in Smith Rock State Park, Oregon. Bull. Calif. Lichen Soc. 22, 48–53.
Yuliar, Y., Nion, A., and Toyota, K. (2015). Recent trends in control methods for bacterial wilt diseases caused by Ralstonia solanacearum. Microbes Environ. 30, 1–11. doi: 10.1264/jsme2.ME14144
Zawierucha, K., Trzebny, A., Buda, J., Bagshaw, E., Franzetti, A., Dabertid, M., et al. (2022). Trophic and symbiotic links between obligate-glacier water bears (tardigrada) and cryoconite microorganisms. PLoS One 17:e0262039. doi: 10.1371/JOURNAL.PONE.0262039
Zhang, F., Berg, M., Dierking, K., Félix, M.-A., Shapira, M., Samuel, B. S., et al. (2017). Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 8:485. doi: 10.3389/fmicb.2017.00485
Zhang, Y., and Qiu, S. (2015). Examining phylogenetic relationships of Erwinia and Pantoea species using whole genome sequence data. Antonie Van Leeuwenhoek 108, 1037–1046. doi: 10.1007/s10482-015-0556-6
Zhou, G.-W., Zheng, F., Fan, X.-T., Li, M.-J., Sun, Q.-Y., Zhu, Y.-G., et al. (2022). Host age increased conjugal plasmid transfer in gut microbiota of the soil invertebrate Caenorhabditis elegans. J. Hazard. Mater. 424:127525. doi: 10.1016/j.jhazmat.2021.127525
Keywords: tardigrade, microbiota, phytopathogen, endosymbiont, 16S rRNA amplicon sequencing
Citation: Tibbs-Cortes LE, Tibbs-Cortes BW and Schmitz-Esser S (2022) Tardigrade Community Microbiomes in North American Orchards Include Putative Endosymbionts and Plant Pathogens. Front. Microbiol. 13:866930. doi: 10.3389/fmicb.2022.866930
Edited by:
Amparo Latorre, University of Valencia, SpainReviewed by:
Laila Pamela Partida-Martinez, Instituto Politécnico Nacional de México (CINVESTAV), MexicoMichele Cesari, University of Modena and Reggio Emilia, Italy
Copyright © 2022 Tibbs-Cortes, Tibbs-Cortes and Schmitz-Esser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura E. Tibbs-Cortes, bHRpYmJzQGlhc3RhdGUuZWR1
†ORCID: Laura E. Tibbs-Cortes, orcid.org/0000-0003-3188-6820
Bienvenido W. Tibbs-Cortes, orcid.org/0000-0003-3435-4889
Stephan Schmitz-Esser, orcid.org/0000-0002-1907-0709