
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 May 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.855317
This article is part of the Research Topic Community Series in Plants and Microbial Communities: Diversity, Pathogens and Biological Control, volume II View all 25 articles
 Chunbo Dong1
Chunbo Dong1 Qiuyu Shao1Yulian Ren1Wei Ge1Ting Yao2Haiyan Hu3Jianzhong Huang4Zongqi Liang1Yanfeng Han1,5*
Qiuyu Shao1Yulian Ren1Wei Ge1Ting Yao2Haiyan Hu3Jianzhong Huang4Zongqi Liang1Yanfeng Han1,5*
Medicinal plants are inhabited by diverse microbes in every compartment, and which play an essential role in host growth and development, nutrient absorption, synthesis of secondary metabolites, and resistance to biological and abiotic stress. However, the ecological processes that manage microbiota assembly and the phenotypic and metabolic characteristics of the core microbiota of Eucommia ulmoides remain poorly explored. Here, we systematically evaluated the effects of genotypes, compartment niches, and environmental conditions (climate, soil nutrition, and secondary metabolites) on the assembly of rhizosphere soil and bark associated bacterial communities. In addition, phenotypic and metabolic characteristics of E. ulmoides core microbiota, and their relationship with dominant taxa, rare taxa, and pharmacologically active compounds were deciphered. Results suggested that microbiota assembly along the two compartments were predominantly shaped by the environment (especially pH, relative humidity, and geniposide acid) and not by host genotype or compartment niche. There were 690 shared genera in the rhizosphere soil and bark, and the bark microbiota was mainly derived from rhizosphere soil. Core microbiota of E. ulmoides was a highly interactive “hub” microbes connecting dominant and rare taxa, and its phenotypic characteristics had a selective effect on compartment niches. Metabolic functions of the core microbiota included ammonia oxidation, nitrogen fixation, and polyhydroxybutyrate storage, which are closely related to plant growth or metabolism. Moreover, some core taxa were also significantly correlated with three active compounds. These findings provide an important scientific basis for sustainable agricultural management based on the precise regulation of the rhizosphere soil and bark microbiota of E. ulmoides.
Under natural conditions, plant growth and behavior are strongly dependent on related microbial communities called microbiome (Agler et al., 2016; Trivedi et al., 2020). These microorganisms inhabit various compartments of plant, including root, stems, leave, flower, and fruit, and influence host physiology and fitness by providing plants with nutrients and improving their resistance to biological and abiotic stresses (Mendes et al., 2013; Agler et al., 2016; Li et al., 2019). The phyllosphere and rhizosphere are vital interfaces for the interaction between host and environment and perform important biological functions in plants (Beckers et al., 2017). The rhizosphere microbiota is regarded as the second genome of plants, controlling the activation, uptake and utilization of nutrients in plants (Shen et al., 2021). Plant rhizosphere microbiota have functions similar to animal gut microbiota, which can not only regulate host performance local space, but also affect the host remotely and across spatial scales (Hou et al., 2021). Bark, an important organ of the plant, is always exposed to the oligotrophic environment with various temperatures and UV radiation (Dong et al., 2021c). It plays many roles in transporting nutrients and preventing the pests and diseases (Pellitier et al., 2019), however, the functions and construction processes of its associated microbiota are poorly understood.
Eucommia ulmoides Oliv., a medicinal and edible plant that produces an abundance of active compounds, is the source of E. ulmoides rubber used in industry and has high economic value. The bark contains pinoresinol diglucoside, geniposide acid, aucubin, chlorogenic acid, and other compounds with important clinical effects (He et al., 2014). Moreover, the gutta percha from E. ulmoides rubber is an excellent polymer material that contributes to the supply of rubber resources and the sustainable development of the rubber industry (Wei et al., 2021). Previous studies revealed that E. ulmoides bark harbored members of the microbial genera Cladosporium, Sphingomonas, Alternaria, Devosia, Marmoricola, Rhodococcus, and Teratosphaeria, which were affected by climatic conditions, altitude, and secondary metabolites (Dong et al., 2020, 2021a,c). However, most of the related studies mainly considered one or a few aspects. We are still working to develop a systematic understanding of how compartment niches, host genotypes, soil factors, and microbial metabolites affect the construction of the E. ulmoides microbiota.
Studies suggested that plant-associated microbiota was predominantly determined by niche and host species/genotypes (Wagner et al., 2016; Cregger et al., 2018; Qian et al., 2019). Host genetic characteristics are an important driving force for the assembly of the microbiota. Composition and structure of the microbiota was affected by vertical transmission of endophytic or seed microbes, especially in the early stages of plant growth (Tkacz et al., 2015; Wagner et al., 2016; Xiong et al., 2021), but conversely, plants can also exert strong selective effects on their microbes through their own immune system, genetic network, and secondary metabolites (Cordovez et al., 2019). In addition, different compartments of plant were confirmed to have strong selectivity for the microbiota (Bai et al., 2015). For example, the phyllosphere community mainly comprises bacteria belonging to Proteobacteria, Bacteroidetes, and Firmicutes, where members of Proteobacteria constitute ∼50% of the community composition (Trivedi et al., 2020). In addition to this biological pressure, assembly of the plant-associated microbiota is also controlled by climate, soil properties, and human disturbance (Carlström et al., 2019).
Not all members of the microbiota are beneficial to plants, and a significant proportion of microbiota members are neutral and even a small number are harmful (Berg, 2009; Mendes et al., 2013). Particular microbes, termed “hub microbes or core microbes” due to their central position in a microbial network, are disproportionally important in shaping microbial communities on plant hosts (Agler et al., 2016). The core microbiota of medicinal plants may be instrumental in determining the quality of medicinal materials (Huang et al., 2018; Dong et al., 2021a; Xu et al., 2021). The core microbiota not only directly play a beneficial role (Chen et al., 2018) but can also influence the wider microbial community through the community cascade effect, thereby driving evolution and function of the microbial community (Agler et al., 2016). The bark of E. ulmoides was reported to be rich in fungal diversity, especially the rare fungal taxa that cannot be ignored (Dong et al., 2021c). Increasing numbers of studies have suggested that the relationship between rare taxa and ecosystem functions may be closer than previously expected (Pascoal et al., 2021; Zhao et al., 2022). Nevertheless, the function of the core microbiota and its relationship with dominant microbiota and rare microbiota has yet to be elucidated.
This study selected 66 E. ulmoides samples (33 rhizosphere soil and 33 bark samples) from 11 regions with obvious geographical distribution. We aimed to assess how host (compartment niches and genotypes) and environmental conditions (longitude and latitude, altitude, climate, soil nutrition, and metabolites) interactively shape assembly of microbiota with different compartments of E. ulmoides; and to explore the composition and function of the core microbiota of E. ulmoides.
A total of 66 soil rhizosphere and bark samples from 11 representative regions (Zunyi, Panzhou, Wangcang, Heishui, Cili, Liuyang, Shuanglong, Shiyan, Linbao, Qimen, and Longshi) of E. ulmoides in China were used in the study (Supplementary Figure 1). Three healthy E. ulmoides, approximately 10 m from one another, were selected in each region, and bark samples from 1.5 m above the ground of each tree were collected (Dong et al., 2020). Sub-samples for chemical analysis were transported to the laboratory, dried, and crushed, followed by the measurement of pinoresinol diglucoside, aucubin, and geniposidic acid by tandem liquid chromatography quadrupole time of flight mass spectrometry (LC-QTOF MS/MS). Sub-samples for DNA extraction were frozen in dry ice during transportation and stored at −80°C until further processing. Subsequent chemical analysis and DNA extraction were performed as described previously Dong et al. (2020).
Similarly, three healthy E. ulmoides trees were chosen from each region, and rhizosphere soil sample was collected from each tree. For each tree, rhizosphere soil samples from three vertical directions (approximately 1 m away from the trunk) at a depth of 20 cm were collected, and were uniformly mixed into a sample (Xu et al., 2018). Soil samples were used to extract the total genomic DNA and measure physiochemical factors.
Total genomic DNA of E. ulmoides rhizosphere soil samples was extracted according to FastDNA Spin Kit for Soil (MP, United States). In this study, DNA concentration between 40 and 60 ng/μL was considered qualified. According to universal primer pair 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), the 16S rRNA gene was amplified. The conditions and mixtures for PCR amplification referred to our previous method (Dong et al., 2021b). PCR products were sequenced by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China) based on an Illumina MiSeq PE300 platform (Illumina, San Diego, United States). Most of these analysis were performed on BMK Cloud.1 The raw data was uploaded to the NCBI database (BioProject ID: PRJNA771480; BioSample: SAMN22311478).
The bark sampling method was according to our previous study (Dong et al., 2021b), and the current work is a follow-up study to explore the construction mechanism of the rhizosphere soil and bark microbiota and the function of the core microbiota. The bark sample was corresponding to the rhizosphere soil sample. The diversity of bark bacterial communities was characterized based on universal primer pair 338F and 806R. The sequencing data of E. ulmoides bark (BioProject ID: PRJNA771480; BioSample: SAMN22311479) was collated and compiled based on our previous study (Dong et al., 2021b).
The approach of genotype characteristics, active compounds, and soil physicochemical properties were described in our previous study (Dong et al., 2021b). The genetic characteristics of E. ulmoides were analyzed based on the chloroplast gene trnH-psbA (forward: 5′-CGCGCATGGTGGATTCACAATCC-3′; Reverse: 5′-GTTATGCATGAACGTAATGCTC-3′) and trnL-F (forward: 5′-ATTTGAACTGGTGACACGAG-3′; Reverse: 5′-CGAAATCGGTAGACGCTACG-3′), and these analyses were performed by the software on BioEdit v7.0.9.0, MAFFT v 7.037, MEGA6, SequenceMatrix1.7.8, and DnaSP version 6. The content of active compounds was determined based on LC-QTOF MS/MS. The determination of soil physicochemical properties mainly refers to “Physical and Chemical Analysis of Soil Properties.” In this study, the data of host genotypes, active compounds, and soil physicochemical properties were placed in Supplementary Tables 1, 2.
Core microbiota was defined with reference to the method of Dong et al. (2021a). The operational taxonomic units (OTUs) with all samples were screened out to construct a co-occurrence network. Highly significant (r > 0.6 or r < −0.6; p < 0.05) spearman correlations between bacterial co-occurrence network were visualized by Gephi, version 0.9.2. Similarly, network topology parameters (e.g., degree, betweenness centrality, closeness centrality, hub, clustering, and modularity) were calculated in the software Gephi, version 0.9.2. In the study, the threshold for core microbiota identification was degree ≥ 20, and closeness centrality ≥ 0.29. In addition, OTUs with relative abundance above 0.1% were placed within the dominant taxa (Xiong et al., 2021), while rare taxa refer to OTUs with relative abundance below 0.01% (Pascoal et al., 2021).
The habitat, energy source, and metabolic functions of the core microbiota were annotated based on METAGENassist2 (Arndt et al., 2012). The phenotypic information of 11,000 microorganisms and the full-sequence of approximately 1,800 microorganisms were included in METAGENassist. Moreover, the phenotypic information contains approximately 20 categories for each microorganism, such as metabolism, habitat, energy source(s), oxygen requirements, preferred temperature range.
The weighted UniFrac distance matrix was calculated to evaluate beta-diversity (β-diversity), and then non-metric multidimensional scaling (NMDS) was used for ranking analysis (Xiong et al., 2021). The significance of different factors on community dissimilarity was tested with permutational multivariate analysis of variance (PERMANOVA) or nested PERMANOVA using the “adonis” function of the VEGAN package in R 3.6.3 (Oksanen et al., 2015). Null model analysis was performed “picante” package (Zhao et al., 2021), Structural equation model was analyzed the “lavaan” package in R 3.6.3 (Rosseel, 2012). Traceability analysis refers to the fast expectation-maximization for microbial source tracking (FEAST), which is a ready-to-use scalable framework that can simultaneously estimate the contribution of thousands of potential source environments in a timely manner, thereby helping unravel the origins of complex microbial communities3 (Shenhav et al., 2019).
Non-metric multidimensional scaling and PERMANOVA showed that variation of bacterial communities in the rhizosphere soil (R2 = 78.84%, p = 0.001) and bark (R2 = 67.14%, p = 0.001) of E. ulmoides was predominantly determined by environmental conditions (Figures 1A,B and Table 1). The compartment niche did not have a significant effect on E. ulmoides-associated bacterial communities (R2 = 1.73%, p = 0.28) (Table 1), but the rhizosphere soil and bark samples clustered clearly and independently (Figure 1C and Supplementary Figure 2). Similarly, different genotypes had no significant effects on the rhizosphere soil and bark bacteria of E. ulmoides (Figures 1D–F and Table 1).
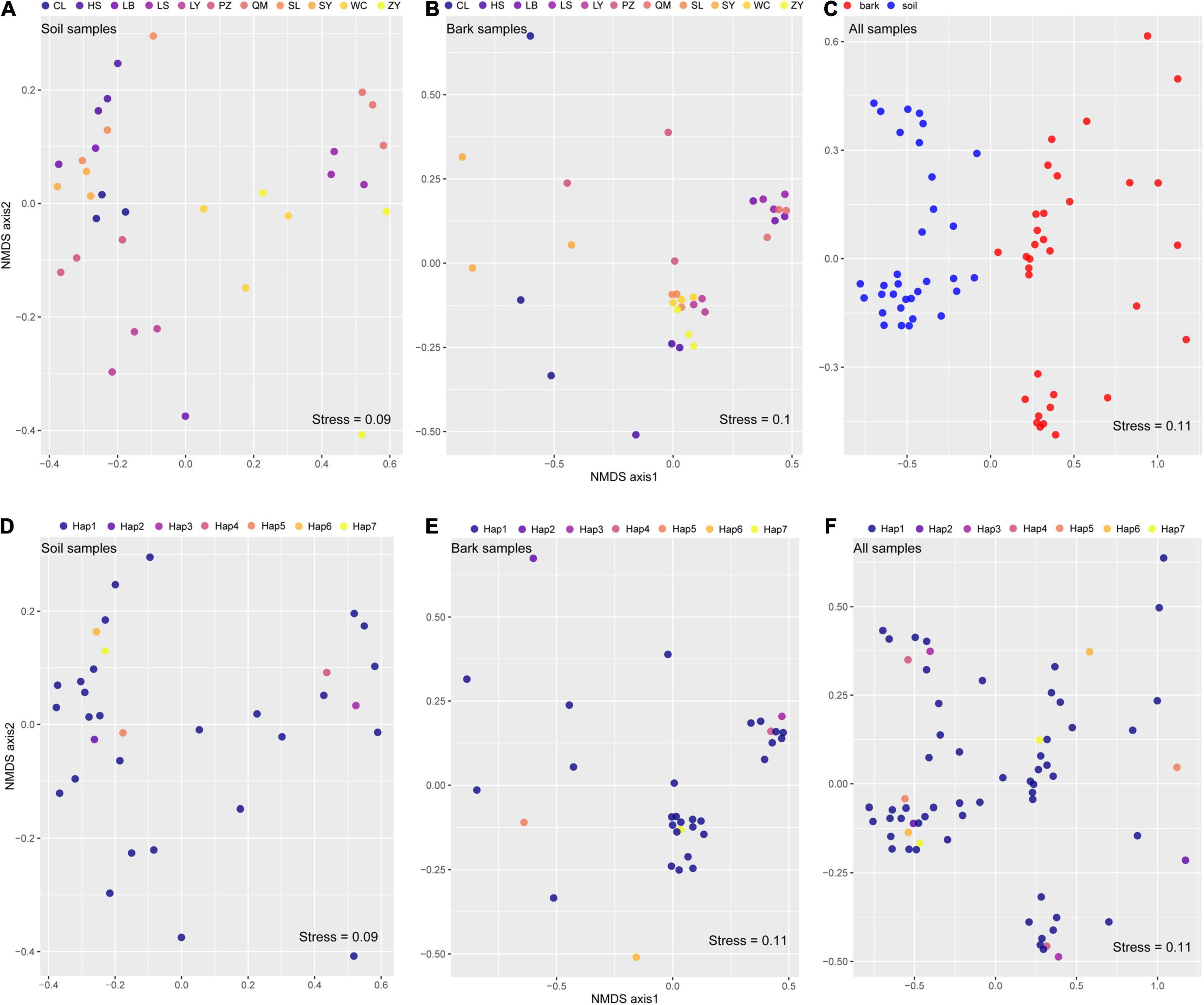
Figure 1. Microbiome assembly was shaped more strongly by environmental conditions than by host niches or genotypes of E. ulmoides. Nonmetric multidimensional scaling (NMDS) ordinations based on weighted UniFrac distances matrices of bacterial communities for: (A) rhizosphere soil (n = 33); (B) bark (n = 33); (C) all samples of different niches (n = 66); (D) soil samples of different genotypes (n = 33); (E) bark samples of different genotypes (n = 33); (F) all samples of different genotypes (n = 33).
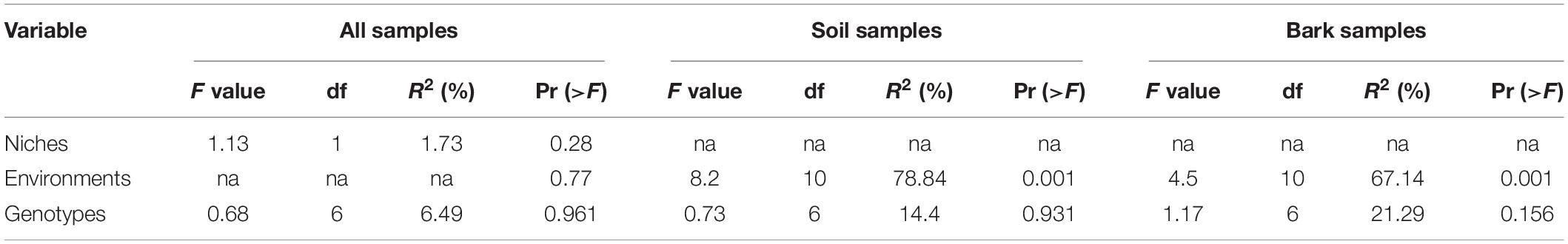
Table 1. The influences of host niche, genotype, and environmental factors on the bacterial community of E. ulmoides based on PERMANOVA.
According to the unweighted β-NTI statistics, the β-NTI values of the rhizosphere soil and bark bacterial communities of E. ulmoides mainly ranged from −1 to 9 (Figures 2A,B), suggesting that the assembly of bacterial community was affected by both stochastic and deterministic factors (β-NTI value > 2, then the deterministic process was dominant; β-NTI value < 2, then the stochastic process was dominant). SEM further suggested that the bacterial α-diversity in the rhizosphere soil and bark of E. ulmoides was affected to varying degrees by host active components, soil physicochemical properties, and climatic factors (Figures 2C,D). For example, pH (with an estimated value of 0.56, p < 0.001), aucubin (0.31, p < 0.001), and relative humidity (0.30, p < 0.001) had significant positive effects on the Shannon diversity index, while geniposide acid had a significant negative effect on the Shannon diversity index (−0.47, p < 0.001). Altitude (0.61, p < 0.001) and relative humidity (0.46, p < 0.001) had significant positive effects on the Chao index, while rainfall had a significant negative effect on the Chao index (Figure 2C).
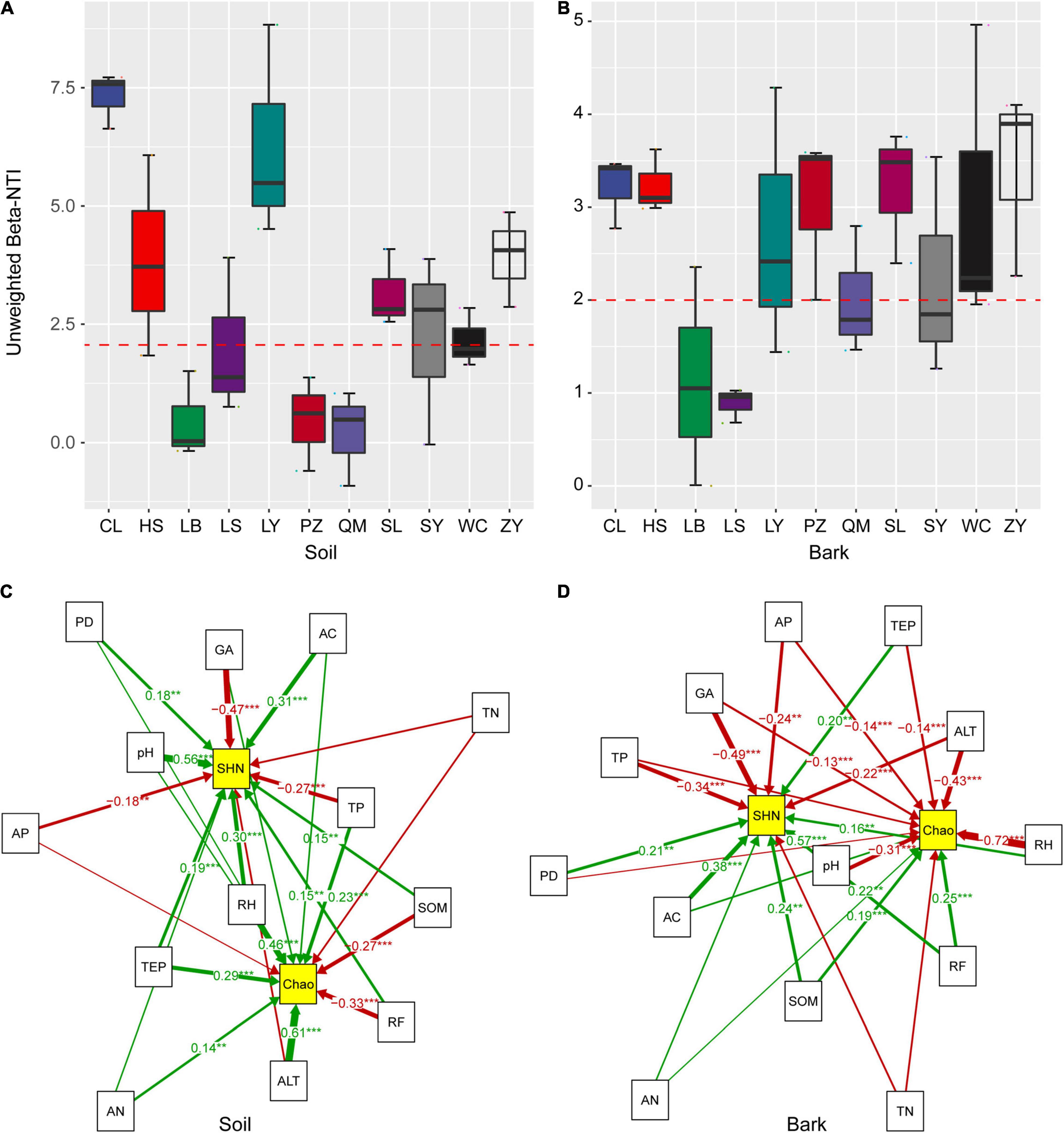
Figure 2. Null model and structural equation model analysis reveals the bacterial community of assembly process of E. ulmoides. β-NTI values of bark (A) and rhizosphere soil (B) bacterial communities were distributed in 11 producing areas (C), Fitting relationships between active compounds, soil physicochemical factors, and climatic factors, and diversity index of soil bacterial community. (D) Fitting relationships between active compounds, soil physicochemical factors, and climatic factors, and diversity index of bark bacterial community. Active compounds included geniposidic acid (GA), aucubin (AC), and pinoresinol diglucosid (PD). Soil physicochemical factors included available phosphorus (AP), soil organic matter (SOM), pH, total phosphorus (TP), available nitrogen (AN), and total nitrogen (TN). Climatic factors included Rainfall (RF), relative humidity (RH), altitude (ALT), and temperature (TEP). Diversity index includes Shannon diversity index (SHN), Chao diversity index (Chao). Solid lines and arrows show significant and non-significant relationships between different variables (variables with normalized path coefficients > 0.1). Significance levels were **p < 0.01, ***p < 0.001.
The bark and rhizosphere soil microbiota respond similarly to environmental factors. pH (0.57, p < 0.001) and aucubin (0.38, p < 0.001) had significant positive effects on the Shannon diversity index; geniposide acid (−0.49, p < 0.001) and total phosphorus (−0.34, p < 0.001) had significant negative effects on the Shannon diversity index; and pH (−0.72, p < 0.001), altitude (−0.43, p < 0.001), and relative humidity (−0.31, p < 0.001) had significant negative effects on the Chao index (Figure 2D). Overall, the composition and structure of the E. ulmoides microbiota were driven by multiple environmental factors.
Although compartment niches were not the main factor affecting the microbiota of E. ulmoides, there were still similarities and differences in the composition of the microbial communities of rhizosphere soil and bark. At the genus level, 690 taxa were shared between rhizosphere soil and bark microbial communities, among which the top five in relative abundance were Enterobacter, an unidentified genus of Acidobacteria, an unidentified genus of Muribaculaceae, Sphingomonas, and Lactobacillus. Bark contained five unique taxa, which were all unidentified genera of Verrucomicrobiae, Actinophytocola, Planctomycetacia, Myxococcales, and Armatimonadeles. In contrast, rhizosphere soil contained 43 unique taxa, among which the top five relative abundances comprised unidentified genera of Bacteroidia, Clostridiales, and Saccharibacillus, Spirochaeta, and Erysipelotrichaceae (Figure 3A).

Figure 3. Genus-level differences and potential source tracking of rhizosphere soil and bark bacterial communities. (A) Venn diagrams showing shared and unique bacterial genera in different niches. (B) FEAST showing that the rhizosphere soil bacterial community was a potential source of the bark bacterial community.
Fast expectation-maximization microbial source tracking (FEAST) suggested that 54.92% of the members of the bark bacterial community were derived from rhizosphere soil, and 45.08% of the members were derived from other unknown environments (Figure 3B).
Here, we identified dominant taxa, core taxa and rare taxa (Figures 4A,B and Supplementary Figures 3, 4). A total of 201 OTUs (0.1% of total OTU number) were defined as the dominant microbiota, with Enterobacter, Actinobacteria, and Sphingomonas containing the most OTUs. The dominant microbiota accounted for 78.89% of the soil samples, and 70.61% of the bark samples (Supplementary Figure 4a). A total of 185 OTUs (0.01% of total OTU number) were defined as rare microbiota, of which the Proteobacteria Deltaproteobacteria, Burkholderiaceae, and Rhizobiales contained the most OTUs (Supplementary Figure 4b). Based on the network parameters, 18 OTUs were defined as core microbiota (Supplementary Figure 3), and these belonged to the genera Escherichia, Enterococcus, Methylobacterium, and Ramlibacter. The proportion of core microbiota was higher in bark samples and lower in soil samples compared to the dominant microbiota (Figures 4A,B).
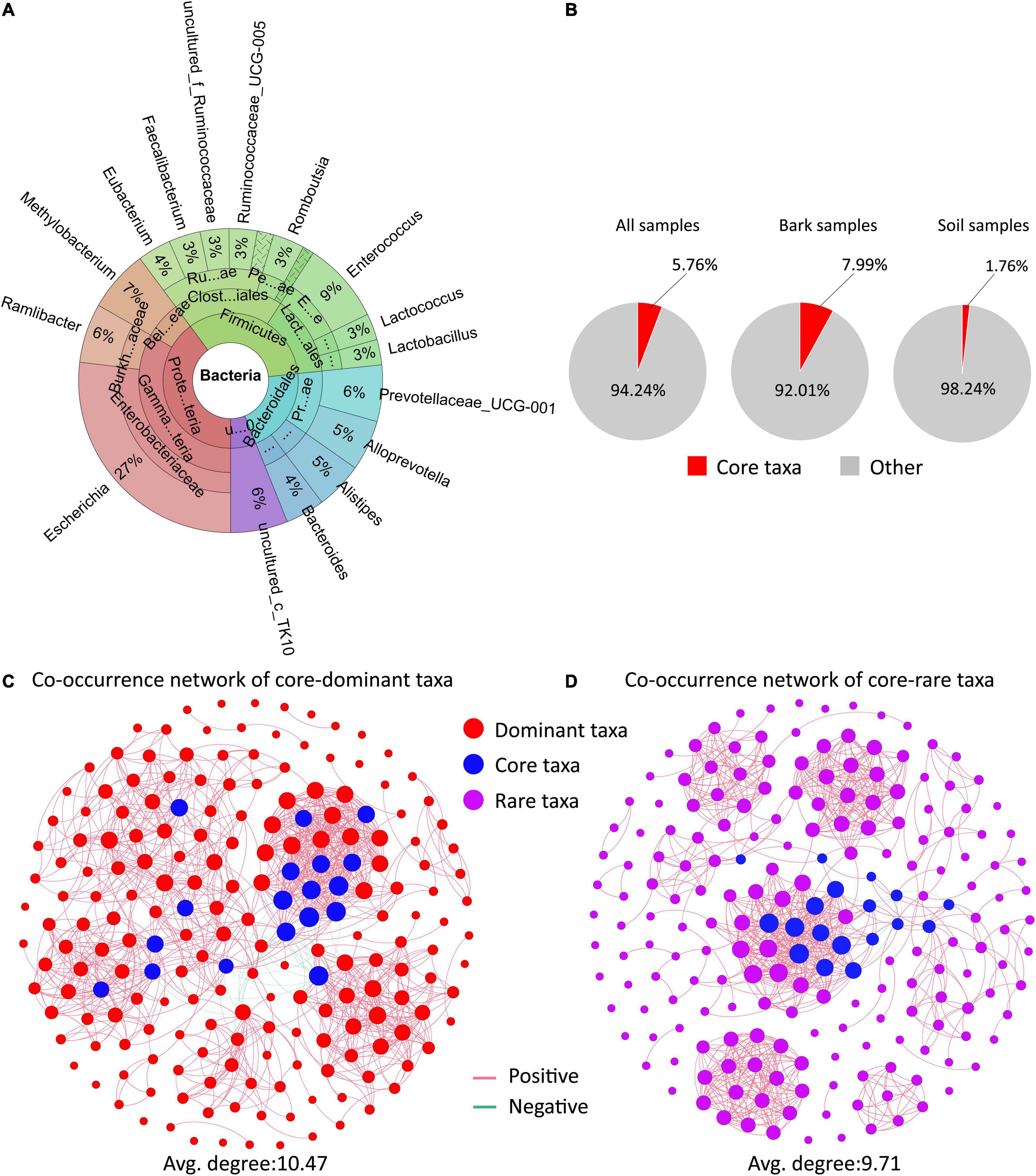
Figure 4. The interactions of the core microbiota with the rare microbiota. (A) Composition and classification information of the core microbiota. (B) Distribution of core microbiota in different samples. (C,D) Bacterial co–occurrence networks along the core taxa–dominant microbiota (C) and core taxa–rare microbiota (D).
To further characterize the impact of the core microbiota on other members of the host microbiota, potential interrelationships between the core microbiota and the dominant and rare microbiota were evaluated (Figures 4C,D). There was no significant difference in the complexity of the two microbial networks (average degree representing network complexity). Within co-occurrence networks, we found that correlations were usually positive (core-dominant taxa, 97.35%, n = 1057; core-rare, 99.1%, n = 783) (Figures 4C,D). Core microbiota was a highly interactive “hub” microbes connecting dominant and rare taxa, play an important role in determining microbiome structures.
Phenotypic and functional characteristics of the core microbiota of E. ulmoides were deciphered based on the METAGENassist platform and statistical analysis (Figure 5). The core microbiota with multiple habitat characteristics mostly exists in the soil rhizosphere, while the core microbiota related to the host predominantly exist in the bark of E. ulmoides. Based on energy source utilization, the core microbiota present in the rhizosphere soil were mainly methylotrophs, while the core microbiota existing in the bark were mostly autotrophs (Figure 5).
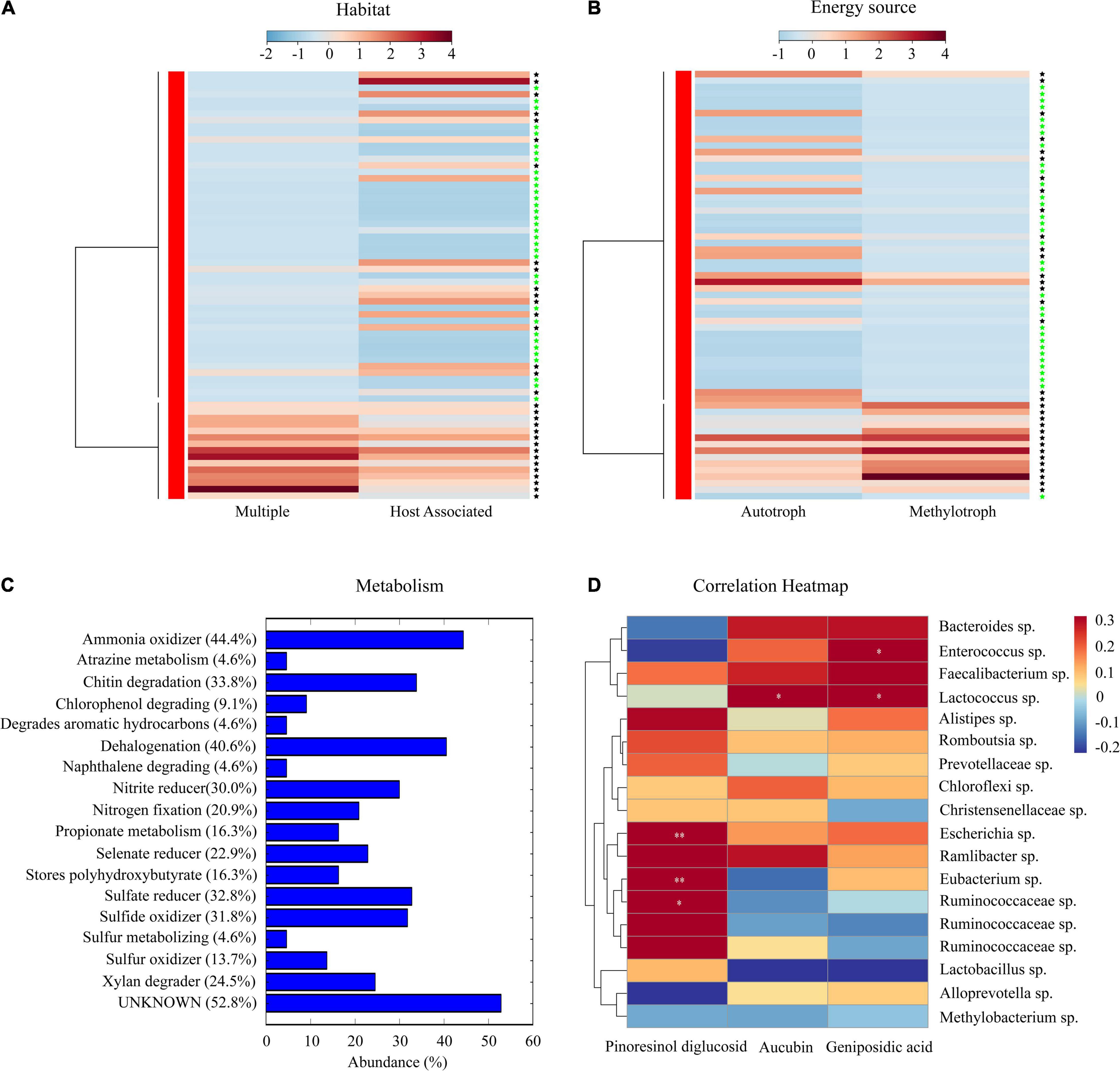
Figure 5. Phenotypic characteristics and potential metabolic functions of the core microbiota of E. ulmoides. (A) Habitat information of the core microbiota. (B) Energy source of the core microbiota. (C) Potential metabolism of the core microbiota. (D) Heatmap of the correlation between core microbiota and active compounds. The “black asterisk” represents rhizosphere soil samples, “green asterisk” represents bark samples. Significance levels were *p < 0.05, **p < 0.01.
Metabolic functions of the core microbiota of E. ulmoides predominantly comprised ammonia oxidizer, dehalogenation, chitin degradation, sulfate reducer, and sulfide oxidizer. In addition, some metabolic functions closely related to the biological properties of E. ulmoides were worthy of attention, such as xylan degrader, nitrogen fixation, and stores polyhydroxybutyrate (Figure 5C). The correlation heatmap showed that Escherichia, Eubacterium, and an unidentified genus of the family Ruminococcaceae were significantly correlated with pinoresinol diglucoside, Lactococcus exhibited a significant correlation with aucubin, and Enterococcus and Lactococcus were significantly correlated with geniposidic acid (Figure 5).
The microbiota of medicinal plants is an important factor affecting the health and productivity of the host (Maggini et al., 2020), and can be directly or indirectly involved in the production of biologically active compounds by the host (Koberl et al., 2013). The medicinal plant-associated microbiota has been gradually characterized with the omics technology (Chen et al., 2018; Ulloa-Muñoz et al., 2020; Dong et al., 2021c; Sauer et al., 2021). However, the mechanism of medicinal plant-associated microbiota construction and the phenotype and function of the core microbiota have yet to be elucidated. Findings from the current study demonstrated that assembly of the rhizosphere soil and bark microbiota was shaped predominantly by environmental factors, rather than by host genotype or compartment niche. Moreover, the core microbiota were key taxonomic units for the construction of microbial networks and were closely related to the growth and the accumulation of secondary metabolites of E. ulmoides.
Assembly of plant-associated microbiota is regulated by the host’s metabolism, genotype, soil physicochemical properties, climate, and niche (Wagner et al., 2016; Cregger et al., 2018; Sun et al., 2020; Xiong et al., 2021). In the current study, assembly of the rhizosphere soil and bark microbiota was predominantly shaped by environmental factors and not by genotype or compartment niche (Table 1). Environmental factors are generally accepted to be closely related to the composition of plant-associated microbiota (Breidenbach et al., 2015; Trivedi et al., 2020; Zhang et al., 2020; Bona et al., 2021). Relative humidity was significantly correlated with the diversity index of the soil and bark bacterial communities, which was consistent with previous studies (Dong et al., 2021c). As reported by Cruz-Paredes et al. (2021), environmental humidity within a certain range significantly impacts the respiration and growth of microorganisms. Moreover, pH could also significantly affect assembly of microbial communities in soil and bark (Figure 2). This may be because pH combines multiple soil chemical variables and is the result of many chemical reactions between organic (biological and abiotic) and inorganic soil components (Lopes et al., 2021). The bark microbiota was not directly in contact with soil pH but was still significantly affected by pH in the current study (Figure 2B). It was previously reported that the indirect impact of soil pH on the microbiota in other compartments of the plant is because microbes in the other compartments were derived from rhizosphere soil (Fitzpatrick et al., 2018; Lopes et al., 2021). Similarly, FEAST analysis suggested that 54.92% of the members of the bark bacterial community were derived from rhizosphere soil (Figure 3B).
Venn diagram analysis showed that 690 genera, such as Enterobacter, Sphingomonas, and Lactobacillus (Figure 3A), were shared between soil and bark communities. These genera were also found in previous studies on E. ulmoides (Dong et al., 2020, 2021a) and are beneficial microorganisms with potential ecological functions for medicinal plants (Quijada et al., 2018; Asaf et al., 2020; Webster et al., 2020). Highly abundant unique bacteria of the rhizosphere soil samples included Bacteroidia, Clostridiales, Saccharibacillus, Spirochaeta, and Erysipelotrichaceae, which have specific selective effects on the soil, predominantly exist in the soil, and play an important role in geo-biological-chemical circulation (Wilms et al., 2006; Namirimu et al., 2019). Most of the bacteria unique to the bark are those with relatively older evolutionary status, such as the Planctomycetes, which are important for the nitrogen cycle (Cavalier-Smith, 2002). Myxococcus, which is only detected in the bark of rhododendrons, has broad application potential in agriculture, biomedicine, and environmental protection due to its ability to produce a variety of natural products (Wang et al., 2021).
Core microbiota, an important subset of the host-associated microbiota, can not only promote the growth of host plants but also recruit functional species and block pathogens/pests (Banerjee et al., 2018; Hassani et al., 2018; Toju et al., 2018). In recent years, the core microbiota of medicinal plants has been accepted as a processing plant or reservoir for secondary metabolites (Koberl et al., 2013; Chen et al., 2018; Xu et al., 2021). Using microbial network analysis, 18 core bacteria of E. ulmoides belonging to the genera Escherichia, Enterococcus, Methylobacterium, Eubacterium, and Lactococcus were identified (Figure 4A). Among them, species of Escherichia, Eubacterium, and Ruminococcaceae were significantly related to pinoresinol diglucoside; a species of Lactococcus had a significant correlation with aucubin; and species of Enterococcus and Lactococcus were significantly correlated with geniposide acid (Figure 4D). Previous studies have shown that Escherichia sp. (Leonard et al., 2008), Enterococcus sp. (Müller et al., 2001), Eubacterium sp. (Newman and Cragg, 2020), and Lactococcus sp. (Golomb and Marco, 2015), Ruminococcaceae sp. (Yin et al., 2020) could promote plant growth or accumulate secondary metabolites. The phenotypic information indicated that the core microbiota mainly contained methylotrophic bacteria in the rhizosphere soil and mostly autotrophic bacteria in the bark (Figure 5B). This indicated that the core microbiota from the rhizosphere soil may play the role of consumers, while the core microbiota from the bark may predominantly play producer roles (Thakur and Geisen, 2019; El-Sayed et al., 2021). The metabolome of the 18 core bacteria was further analyzed, and in addition to some common metabolic functions of the plant microbiota (Xu et al., 2018; Trivedi et al., 2019), the functions of xylan degradation, nitrogen fixation, and polyhydroxybutyrate storage were worthy of additional attention. Xylan-degrading and nitrogen-fixing bacteria were reported to potentially exhibit direct or indirect effects on plant growth and metabolism (Gopalakrishnan et al., 2017; Jung et al., 2018). Polyhydroxybutyrate was a type of polyhydroxyalkanoate (PHA), a polyester polymer (Sirohi et al., 2021) that has similar structure and function to E. ulmoides rubber, such as strong plasticity (He et al., 2014; Sirohi et al., 2021). Therefore, the core taxa with polyhydroxybutyrate metabolism functions may be involved in the synthesis of E. ulmoides rubber. In general, these findings reveal that the core microbiota may participate in soil organic matter decomposition and metabolite synthesis in E. ulmoides.
Network analysis can be used to identify the core or hub microbiota at the center of the microbial network (Banerjee et al., 2018; Dong et al., 2021a). Core microbiota is composed by important taxonomic units that dominate host microbial community construction, influencing community structure through strong interactions with the host or other microbes rather than simply being present in high abundance (Banerjee et al., 2018; Hamonts et al., 2018; Trivedi et al., 2020; Dong et al., 2021c). A comprehensive investigation of the Arabidopsis phyllosphere microbiota found that the core microorganisms Albugo sp. and Dioszegia sp. regulate the entire microbial community by inhibiting the growth and diversity of other microbes (Agler et al., 2016). The current study found that the core microbiota was highly connected with the dominant microbiota, forming closely related microbial clusters (Figure 4C). Dominant microbiota and core microbiota have vital ecological roles in microbiota assembly and ecosystem functions (Banerjee et al., 2018). Core microbiota can selectively adjust and assist these dominant microbiota (Banerjee et al., 2018), such as by forming a highly specific microbiota with the dominant microbiota for biological nitrogen fixation (Fierer, 2017). In contrast with the dominant microbiota, rare microbiota was often overlooked. Recent studies confirmed that rare microbiota was instrumental in the construction, function, and stability of microbiota (Jousset et al., 2017; Dong et al., 2021c; Pascoal et al., 2021). Similarly, significant interactions between core and rare microbiota of E. ulmoides were detected in the current study (Figure 4D). This is congruent with previous findings that rare microbiota often interacts with other species although they occupy narrow ecological niches (Fierer et al., 2007). Together these data suggest the core microbiota not only has important functions but may also be a keystone species that determines the network structure of the E. ulmoides microbial community.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YH, TY, and JH: conceptualization and funding acquisition. CD, QS, YR, and WG: data acquisition. CD and HH: formal analysis. QS: writing the first draft. CD, YH, and ZL: writing, review, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
This work was supported by “Hundred” Talent Projects of Guizhou Province [Qian Ke He (2020)6005]; the Key Areas of Research and Development Program of Guangdong Province (No. 2018B020205003); the Natural Science Foundation of China (No. 32060011); and Construction Program of Biology First-class Discipline in Guizhou [GNYL(2017)009].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.855317/full#supplementary-material
Supplementary Figure 1 | Geographic distribution of 11 regions of E. ulmoides in China.
Supplementary Figure 2 | Hierarchical clustering of rhizosphere soil and bark samples of E. ulmoides based on the unweighted paired average method (UPGMA).
Supplementary Figure 3 | The core microbiota of E. ulmoides was defined based on the network connection method. (a) The topology of the correlation co-occurrence network. (b) Core microbiota were defined based on closeness centrality and degree.
Supplementary Figure 4 | The composition and distribution of dominant (a) and rare taxa (b).
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14:e1002352. doi: 10.1371/journal.pbio.1002352
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., et al. (2012). METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 40, W88–W95. doi: 10.1093/nar/gks497
Asaf, S., Numan, M., Khan, A. L., and Al-Harrasi, A. (2020). Sphingomonas: from diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 40, 138–152. doi: 10.1080/07388551.2019.1709793
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., and Rott, M. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Banerjee, S., Schlaeppi, K., and van der Heijden, M. G. A. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Beckers, B., Op De Beeck, M., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5:25. doi: 10.1186/s40168-017-0241-2
Berg, G. (2009). Plant–microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biot. 84, 11–18. doi: 10.1007/s00253-009-2092-7
Bona, E., Massa, N., Toumatia, O., Novello, G., Cesaro, P., Todeschini, V., et al. (2021). Climatic zone and soil properties determine the biodiversity of the soil bacterial communities associated to native plants from desert areas of north-central algeria. Microorganisms 9:1359. doi: 10.3390/microorganisms9071359
Breidenbach, B., Pump, J., and Dumont, M. G. (2015). Microbial community structure in the rhizosphere of rice plants. Front. Microbiol. 6:1537. doi: 10.3389/fmicb.2015.01537
Carlström, C. I., Field, C. M., Bortfeld-Miller, M., Müller, B., Sunagawa, S., and Vorholt, J. A. (2019). Synthetic microbiota reveal priority effects and keystone strains in the Arabidopsis phyllosphere. Nat. Ecol. Evol. 3, 1445–1454. doi: 10.1038/s41559-019-0994-z
Cavalier-Smith, T. (2002). The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int. J. Syst. Evol. Microbiol. 52, 7–76. doi: 10.1099/00207713-52-1-7
Chen, H., Wu, H., Yan, B., Zhao, H., Liu, F., Zhang, H., et al. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: a rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci 19:672. doi: 10.3390/ijms19030672
Cordovez, V., Dini-Andreote, F., Carrión, V. J., and Raaijmakers, J. M. (2019). Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 73, 69–88. doi: 10.1146/annurev-micro-090817-062524
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., Vilgalys, R., Tuskan, G. A., et al. (2018). The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8
Cruz-Paredes, C., Tájmel, D., and Rousk, J. (2021). Can moisture affect temperature dependences of microbial growth and respiration? Soil Biol. Biochem. 156:108223. doi: 10.1016/j.soilbio.2021.108223
Dong, C., Zhang, Z., Shao, Q., Yao, T., Liang, Z., and Han, Y. (2021c). Mycobiota of Eucommia ulmoides bark: diversity, rare biosphere and core taxa. Fungal Ecol. 53:101090. doi: 10.1016/j.funeco.2021.101090
Dong, C., Shao, Q., Zhang, Q., Yao, T., Huang, J., Liang, Z., et al. (2021a). Preferences for core microbiome composition and function by different definition methods: evidence for the core microbiome of Eucommia ulmoides bark. Sci. Total Environ. 790:148091. doi: 10.1016/j.scitotenv.2021.148091
Dong, C., Zhang, Z., Shao, Q., Yao, T., Hu, H., Huang, J., et al. (2021b). Deciphering the effects of genetic characteristics and environmental factors on pharmacological active components of Eucommia ulmoides. Ind. Crop. Prod. 175:114293. doi: 10.1016/j.indcrop.2021.114293
Dong, C., Yao, T., Zhang, Z., Chen, W., Liang, J., Han, Y., et al. (2020). Structure and function of bacterial microbiota in Eucommia ulmoides bark. Curr. Microbiol. 77, 3623–3632. doi: 10.1007/s00284-020-02157-2
El-Sayed, A. S. A., Shindia, A. A., Ali, G. S., Yassin, M. A., Hussein, H., Awad, S. A., et al. (2021). Production and bioprocess optimization of antitumor Epothilone B analogue from Aspergillus fumigatus, endophyte of Catharanthus roseus, with response surface methodology. Enzyme Microb. Tech. 143:109718. doi: 10.1016/j.enzmictec.2020.109718
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fitzpatrick, C. R., Copeland, J., Wang, P. W., Guttman, D. S., Kotanen, P. M., and Johnson, M. T. J. (2018). Assembly and ecological function of the root microbiome across angiosperm plant species. P. Natl. Acad. Sci. USA. 115:E1157. doi: 10.1073/pnas.1717617115
Golomb, B. L., and Marco, M. L. (2015). Lactococcus lactis metabolism and gene expression during growth on plant tissues. J. Bacteriol. 197, 371–381. doi: 10.1128/jb.02193-14
Gopalakrishnan, S., Srinivas, V., and Samineni, S. (2017). Nitrogen fixation, plant growth and yield enhancements by diazotrophic growth-promoting bacteria in two cultivars of chickpea (Cicer arietinum L.). Biocatal. Agric. Biotechnol. 11, 116–123. doi: 10.1016/j.bcab.2017.06.012
Hamonts, K., Trivedi, P., Garg, A., Janitz, C., Grinyer, J., Holford, P., et al. (2018). Field study reveals core plant microbiota and relative importance of their drivers. Environ. Microbiol. 20, 124–140. doi: 10.1111/1462-2920.14031
Hassani, M. A., Duran, P., and Hacquard, S. (2018). Microbial interactions within the plant holobiont. Microbiome 6:58. doi: 10.1186/s40168-018-0445-0
He, X., Wang, J., Li, M., Hao, D., Yang, Y., Zhang, C., et al. (2014). Eucommia ulmoides Oliv.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 151, 78–92. doi: 10.1016/j.jep.2013.11.023
Hou, S., Thiergart, T., Vannier, N., Mesny, F., Ziegler, J., and Pickel, et al. (2021). A microbiota–root–shoot circuit favours Arabidopsis growth over defence under suboptimal light. Nat. Plants 7, 1078–1092. doi: 10.1038/s41477-021-00956-4
Huang, W., Long, C., and Lam, E. (2018). Roles of plant-associated microbiota in traditional herbal medicine. Trends Plant Sci. 23, 559–562. doi: 10.1016/j.tplants.2018.05.003
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Jung, B. K., Hong, S.-J., Jo, H. W., Jung, Y., Park, Y.-J., Park, C. E., et al. (2018). Genome sequencing to develop Paenibacillus donghaensis strain JH8T (KCTC 13049T=LMG 23780T) as a microbial fertilizer and correlation to its plant growth-promoting phenotype. Mar. Genom. 37, 39–42. doi: 10.1016/j.margen.2017.11.006
Koberl, M., Schmidt, R., Ramadan, E. M., Bauer, R., and Berg, G. (2013). The microbiome of medicinal plants: diversity and importance for plant growth, quality and health. Front. Microbiol. 4:400. doi: 10.3389/fmicb.2013.00400
Leonard, E., Yan, Y., Fowler, Z. L., Li, Z., Lim, C.-G., Lim, K.-H., et al. (2008). Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Mol. Pharmaceut. 5, 257–265. doi: 10.1021/mp7001472
Li, J., Zhou, L., and Lin, W. (2019). Calla lily intercropping in rubber tree plantations changes the nutrient content, microbial abundance, and enzyme activity of both rhizosphere and non-rhizosphere soil and calla lily growth. Ind. Crop. Prod. 132, 344–351. doi: 10.1016/j.indcrop.2019.02.045
Lopes, L. D., Hao, J., and Schachtman, D. P. (2021). Alkaline soil pH affects bulk soil, rhizosphere and root endosphere microbiomes of plants growing in a Sandhills ecosystem. FEMS Microbiol. Ecol 97:fiab028. doi: 10.1093/femsec/fiab028
Maggini, V., Mengoni, A., Bogani, P., Firenzuoli, F., and Fani, R. (2020). Promoting model systems of microbiota–medicinal plant interactions. Trends Plant Sci. 25, 223–225. doi: 10.1016/j.tplants.2019.12.013
Mendes, R., Garbeva, P., and Raaijmakers, J. M. (2013). The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663. doi: 10.1111/1574-6976.12028
Müller, T., Ulrich, A., Ott, E. M., and Müller, M. (2001). Identification of plant-associated enterococci. J. Appl. Microbiol. 91, 268–278. doi: 10.1046/j.1365-2672.2001.01373.x
Namirimu, T., Kim, J., and Zo, Y.-G. (2019). Isolation and identification of alkali-tolerant bacteria from near-shore soils in Dokdo island. Microbiol. Biotech. Lett. 47, 105–115. doi: 10.4014/mbl.1807.07018
Newman, D. J., and Cragg, G. M. (2020). Plant endophytes and epiphytes: burgeoning sources of known and “unknown” cytotoxic and antibiotic agents? Planta Med. 86, 891–905. doi: 10.1055/a-1095-1111
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2015). Vegan: Community Ecology Package. R Package Vegan, Vers. 2.2-1. Worl. Agro. Cent. Available online at: https://CRAN.R-project.org/package=vegan
Pascoal, F., Costa, R., and Magalhaes, C. (2021). The microbial rare biosphere: current concepts, methods and ecological principles. FEMS Microbiol. Ecol 97:fiaa227. doi: 10.1093/femsec/fiaa227
Pellitier, P. T., Zak, D. R., and Salley, S. O. (2019). Environmental filtering structures fungal endophyte communities in tree bark. Mol. Ecol. 28, 5188–5198. doi: 10.1111/mec.15237
Qian, X., Li, H., Wang, Y., Wu, B., Wu, M., Chen, L., et al. (2019). Leaf and root endospheres harbor lower fungal diversity and less complex fungal co-occurrence patterns than rhizosphere. Front. Microbiol. 10:1015. doi: 10.3389/fmicb.2019.01015
Quijada, N. M., De Filippis, F., Sanz, J. J., García-Fernández, M.d.C, Rodríguez-Lázaro, D., Ercolini, D., et al. (2018). Different Lactobacillus populations dominate in “Chorizo de León” manufacturing performed in different production plants. Food Microbiol. 70, 94–102. doi: 10.1016/j.fm.2017.09.009
Rosseel, Y. (2012). Lavaan: an R package for structural equation modeling and more. Version 0.5–12 (BETA). J. stat. softw. 48, 1–36. doi: 10.1002/9781119579038.ch1
Sauer, S., Dlugosch, L., Kammerer, D. R., Stintzing, F. C., and Simon, M. (2021). The microbiome of the medicinal plants Achillea millefolium L. and Hamamelis virginiana L. Front. Microbiol. 12:696398–696398. doi: 10.3389/fmicb.2021.696398
Shen, J. B., Bai, Y., Wei, Z., Chu, C. C., Yuan, L. X., and Zhang, et al. (2021). Rhizobiont: an interdisciplinary innovation and perspective for harmonizing resources, environment, and food security. Acta Pedologica Sinica 58, 805–813. doi: 10.11766/trxb202012310722
Shenhav, L., Thompson, M., Joseph, T. A., Briscoe, L., Furman, O., Bogumil, D., et al. (2019). FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632. doi: 10.1038/s41592-019-0431-x
Sirohi, R., Lee, J. S., Yu, B. S., Roh, H., and Sim, S. J. (2021). Sustainable production of polyhydroxybutyrate from autotrophs using CO2 as feedstock: challenges and opportunities. Bioresource Technol. 341:125751. doi: 10.1016/j.biortech.2021.125751
Sun, X., Pei, J., Lin, Y. L., Li, B. L., Zhang, L., Ahmad, B., et al. (2020). Revealing the impact of the environment on Cistanche salsa: from global ecological regionalization to soil microbial community characteristics. J. Agric. Food. Chem. 68, 8720–8731. doi: 10.1021/acs.jafc.0c01568
Thakur, M. P., and Geisen, S. (2019). Trophic regulations of the soil microbiome. Trends Microbiol. 27, 771–780. doi: 10.1016/j.tim.2019.04.008
Tkacz, A., Cheema, J., Chandra, G., Grant, A., and Poole, P. S. (2015). Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J. 9, 2349–2359. doi: 10.1038/ismej.2015.41
Toju, H., Peay, K. G., Yamamichi, M., Narisawa, K., Hiruma, K., Naito, K., et al. (2018). Core microbiomes for sustainable agroecosystems. Nat. Plant 4, 247–257. doi: 10.1038/s41477-018-0139-4
Trivedi, C., Reich, P. B., Maestre, F. T., Hu, H.-W., Singh, B. K., and Delgado-Baquerizo, M. (2019). Plant-driven niche differentiation of ammonia-oxidizing bacteria and archaea in global drylands. ISME J. 13, 2727–2736. doi: 10.1038/s41396-019-0465-1
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., and Singh, B. K. (2020). Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Ulloa-Muñoz, R., Olivera-Gonzales, P., Castañeda-Barreto, A., Villena, G. K., and Tamariz-Angeles, C. (2020). Diversity of endophytic plant-growth microorganisms from Gentianella weberbaueri and Valeriana pycnantha, highland Peruvian medicinal plants. Microbiol. Res. 233:126413. doi: 10.1016/j.micres.2020.126413
Wagner, M. R., Lundberg, D. S., Del Rio, T. G., Tringe, S. G., Dangl, J. L., and Mitchell-Olds, T. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7, 12151. doi: 10.1038/ncomms12151
Wang, J., Wang, J., Wu, S., Zhang, Z., and Li, Y. (2021). Global geographic diversity and distribution of the Myxobacteria. Microbiol. Spectr. 9, e00012–e21. doi: 10.1128/Spectrum.00012-21
Webster, G., Mullins, A. J., Cunningham-Oakes, E., Renganathan, A., Aswathanarayan, J. B., Mahenthiralingam, E., et al. (2020). Culturable diversity of bacterial endophytes associated with medicinal plants of the Western Ghats, India. FEMS Microbiol. Ecol. 96:fiaa147. doi: 10.1093/femsec/fiaa147
Wei, X., Peng, P., Peng, F., and Dong, J. (2021). Natural polymer Eucommia ulmoides Rubber: a novel material. J. Agric. Food Chem. 69, 3797–3821. doi: 10.1021/acs.jafc.0c07560
Wilms, R., Köpke, B., Sass, H., Chang, T. S., Cypionka, H., and Engelen, B. (2006). Deep biosphere-related bacteria within the subsurface of tidal flat sediments. Environ. Microbiol. 8, 709–719. doi: 10.1111/j.1462-2920.2005.00949.x
Xiong, C., Zhu, Y. G., Wang, J. T., Singh, B., Han, L. L., Shen, J. P., et al. (2021). Host selection shapes crop microbiome assembly and network complexity. N. Phytol. 229, 1091–1104. doi: 10.1111/nph.16890
Xu, J., Zhang, Y., Zhang, P., Trivedi, P., Riera, N., Wang, Y., et al. (2018). The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9:4894. doi: 10.1038/s41467-018-07343-2
Xu, M., Bai, H. Y., Fu, W. Q., Sun, K., Wang, H. W., Xu, D. L., et al. (2021). Endophytic bacteria promote the quality of Lyophyllum decastes by improving non-volatile taste components of mycelia. Food. Chem. 336:127672. doi: 10.1016/j.foodchem.2020.127672
Yin, J., Ren, W., Wei, B., Huang, H., Li, M., Wu, X., et al. (2020). Characterization of chemical composition and prebiotic effect of a dietary medicinal plant Penthorum chinense Pursh. Food. Chem. 319:126568. doi: 10.1016/j.foodchem.2020.126568
Zhang, Y., Zheng, L., Zheng, Y., Xue, S., Zhang, J., Huang, P., et al. (2020). Insight into the assembly of root-associated microbiome in the medicinal plant Polygonum cuspidatum. Ind. Crop. Prod 145:112163. doi: 10.1016/j.indcrop.2020.112163
Zhao, W., Wang, X. B., Shi, L. Y., Zhu, W. Y., Ma, L., Wang, J. J., et al. (2021). Calculation method for stochastic and deterministic assembly processes of prokaryotic communities. Bio- 101:e2003400. doi: 10.21769/BioProtoc.2003400
Keywords: Eucommia ulmoides Oliv, plant-microbe interactions, community assembly, core microbiota, microbial functions
Citation: Dong C, Shao Q, Ren Y, Ge W, Yao T, Hu H, Huang J, Liang Z and Han Y (2022) Assembly, Core Microbiota, and Function of the Rhizosphere Soil and Bark Microbiota in Eucommia ulmoides. Front. Microbiol. 13:855317. doi: 10.3389/fmicb.2022.855317
Received: 15 January 2022; Accepted: 13 April 2022;
Published: 03 May 2022.
Edited by:
Jesús Navas-Castillo, La Mayora Experimental Station (CSIC), SpainReviewed by:
Ziting Wang, Guangxi University, ChinaCopyright © 2022 Dong, Shao, Ren, Ge, Yao, Hu, Huang, Liang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanfeng Han, eWZoYW5AZ3p1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.