 Karen Fong
Karen Fong Yu Tong Lu
Yu Tong Lu Thomas Brenner
Thomas Brenner Justin Falardeau
Justin Falardeau Siyun Wang
Siyun Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 22 July 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.853703
Prophages have long been regarded as an important contributor to the evolution of Salmonella and Verotoxin-producing E. coli (VTEC), members of the Enterobacteriaceae that cause millions of cases of foodborne illness in North America. In S. Typhimurium, prophages provide many of the genes required for invasion; similarly, in VTEC, the Verotoxin-encoding genes are located in cryptic prophages. The ability of prophages to quickly acquire and lose genes have driven their rapid evolution, leading to highly diversified populations of phages that can infect distantly-related bacterial hosts. To defend against foreign genetic materials (i.e., phages), bacteria have evolved Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) immunity, consisting of variable spacer regions that match short nucleic acid sequences of invaders previously encountered. The number of spacer regions varies widely amongst Enterobacteriaceae, and there is currently no clear consensus if the accumulation of spacers is linked to genomic prophage abundance. Given the immense prophage diversity and contribution to bacterial host phenotypes, we analyzed the prophage sequences within 118 strains of Salmonella and VTEC, 117 of which are of agricultural origin. Overall, 130 unique prophage sequences were identified and they were found to be remarkably diverse with <50% nucleotide similarity, particularly with the Gifsy-1 group which was identified in several Salmonella serovars and interestingly, a strain of VTEC. Additionally, we identified a novel plasmid-like phage that carried antibiotic resistance and bacteriocin resistance genes. The strains analyzed carried at least six distinct spacers which did not possess homology to prophages identified in the same genome. In fact, only a fraction of all identified spacers (14%) possessed significant homology to known prophages. Regression models did not discern a correlation between spacer and prophage abundance in our strains, although the relatively high number of spacers in our strains (an average of 27 in Salmonella and 19 in VTEC) suggest that high rates of infection may occur in agricultural niches and be a contributing driver in bacterial evolution. Cumulatively, these results shed insight into prophage diversity of Salmonella and VTEC, which will have further implications when informing development of phage therapies against these foodborne pathogens.
Prophages, temperate phages that are integrated into a bacterial host genome, are recognized as one of the greatest drivers of bacterial diversity and evolution (Ramisetty and Sudhakari, 2019). Investigation of bacterial genomes have revealed that temperate phages are diverse, common, and abundant (Brueggemann et al., 2017; Crispim et al., 2018). For example, 53 complete prophages were identified in 47 analyzed genomes of Desulfovibrio (Crispim et al., 2018), and prophages were identified in all 482 strains of Pneumococci studied (Brueggemann et al., 2017). Under various experimental environments, prophages also afforded their bacterial host with a range of fitness benefits, such as integration of antibiotic resistance genes encoding resistance to kanamycin, chloramphenicol and ampicillin (Wendling et al., 2020). Excision and release of free prophages that then lysed competing bacteria was also advantageous from a population standpoint (Wendling et al., 2020). Interestingly, prophage carriage often impedes superinfection from other phages; a phenomenon which provides a great survival benefit in the event the invading phage is massively virulent (Ramisetty and Sudhakari, 2019). Indeed, prophages possess novel genetic materials which have contributed greatly to host phenotype diversity. For example, the Verotoxin-encoding genes of Verotoxin-producing Escherichia coli (VTEC) are located in lambdoid prophages and the production of these toxins occur following prophage excision from the bacterial genome (Łos et al., 2013). Strains of Salmonella enterica serovar Typhimurium also contain an array of prophages that are critical for host virulence and fitness, such as the Gifsy phages (Hiley et al., 2014).
Non-typhoidal Salmonella and VTEC are foodborne pathogens which cause significant burdens on both the Canadian health care system and economy, ranking third and fourth, respectively, for the number of deaths attributed to foodborne illness (Government of Canada, 2016). In recent years, antibiotic resistance has risen to dangerously high levels and deaths from common infections are increasingly attributed to antibiotic resistance (World Health Organization, 2018). The growing inadequacy of antibiotics has paved the way for novel therapies that rely on antibiotic alternatives. Many studies have focused on application of lytic phages for direct killing of their bacterial hosts, however, host resistance still occurs (Fong et al., 2017, 2020).
Prophages may also be employed as antimicrobials (Hu et al., 2021). When environmental triggers inflict stress upon the host cell (e.g., UV light, antibiotic treatment) and induce the bacterial SOS response, prophage induction occurs, where the prophage excises from the genome, ultimately leads to host cell lysis and death (Fu et al., 2019). Somewhat paradoxically, prophage induction plays a critical role in the virulence of VTEC as its pathogenicity requires prophage release from the bacterial cell, which simultaneously releases the prophage-encoded Verotoxin (Łos et al., 2013). Because the host cell is killed upon prophage induction, strategies to induce prophage release have been explored as a means of biocontrol. In a study evaluating the effect of stresses on the production of prophage particles, inducing agents including heat, hydrogen chloride, lactic acid, hydrogen peroxide and high hydrostatic pressure reduced VTEC O104:H4 by 1 to 2 log CFU/ml by release of prophage (Fang et al., 2017). More recently, a proof-of-concept study by Cadieux et al. (2018) found that mitomycin C was the strongest inducing agent of VTEC and Salmonella prophages at a minimum concentration of 0.5 ug/mL. This resulted in a 1.5 and 3 log CFU/g reduction of Salmonella and VTEC on tomatoes, respectively. Similar reductions of 1 and 2 log CFU/g for VTEC and Salmonella, respectively, were also observed on spinach.
Clustered regularly interspaced short palindromic repeats (CRISPR) are repeat sequences that provide adaptive immunity against invasive foreign elements such as phages (Fu et al., 2017). The CRISPR-Cas system comprises two separate components: Cas-proteins that directly cleave foreign nucleic acid; and the CRISPR array that is an invader recognition tool (Touchon et al., 2011). Because CRISPRs are widespread in ~40% of bacteria (Touchon and Rocha, 2010), we probed the number and type of CRISPR arrays in our subset of bacterial strains in an effort to discern an association between lysogeny and carriage of CRISPRs. The CRISPR array affords the bacterial cell with “memory” for defense against foreign nucleic acid (e.g., plasmids and phages) by incorporating unique protospacers of invaders previously encountered (Touchon et al., 2011). Of particular interest are the spacer regions, as it is known these regions are usually identical to sequences possessed by mobile genetic elements (Louwen et al., 2014).
Given the survival advantages of prophage carriage and the advent of such novel approaches to phage-based biocontrol, it is important to gain a deeper understanding of the prophages of both VTEC and Salmonella. Previous work has highlighted the orthologous prophages of Escherichia and Salmonella (Bobay et al., 2014), and these genera include pathogens with highly plastic genomes (Touchon and Rocha, 2010). The enhanced availability of sequenced bacterial and phage genomes will undoubtedly offer additional insights into phage immunity. Here, we probe the prophage-related genetic elements of 118 strains (117 of agricultural origin) of VTEC and Salmonella in an effort to understand the abundance and diversity of the prophage repertoire in these bacterial pathogens. We also identify host CRISPR-Cas elements to discern an association between CRISPR arrays and the incidence of lysogeny.
Bacterial strains used in this study are listed in Table 1. Salmonella isolates (n=50) were previously isolated and analyzed from poultry and environmental samples by Brenner et al. (2020). Strains were paired-end sequenced on an Illumina NextSeq with a coverage of 15X. Reads were assembled with SPAdes v.3.10 and draft sequences assemblies annotated with the NCBI Prokaryotic Genome Annotation Pipeline (Bankevich et al., 2012). Salmonella sequences were deposited into Genbank under BioProject PRJNA224116.
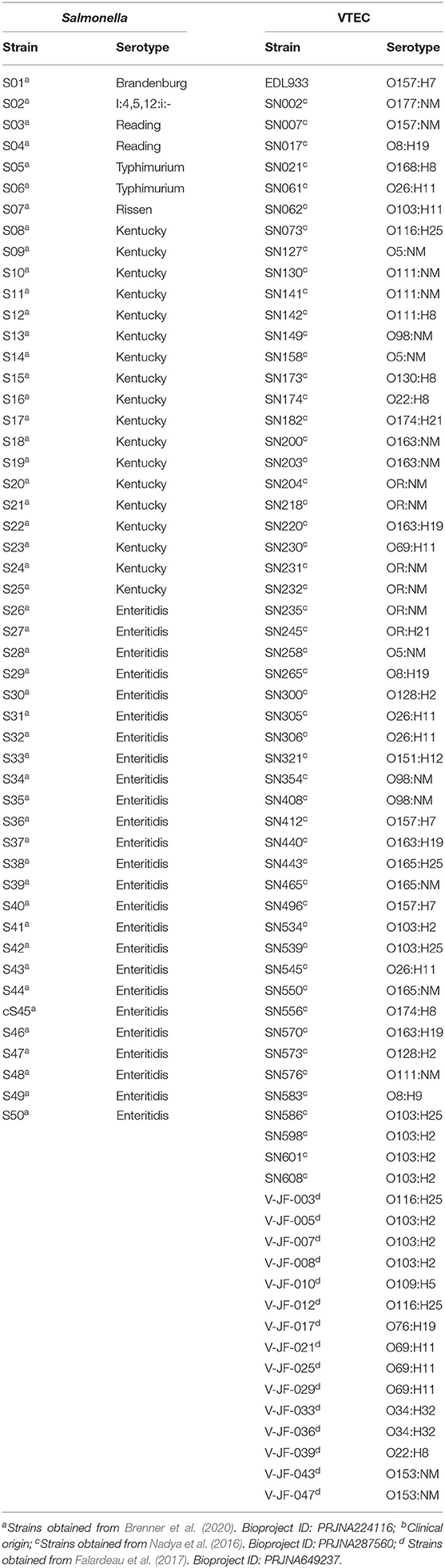
Table 1. Salmonella and VTEC strains used in this study.
The VTEC strains were isolated and analyzed previously in agricultural environments (irrigation water, sediment) by Nadya et al. (2016) and Falardeau et al. (2017) (Table 1). One clinical isolate from a previous foodborne outbreak, E. coli O157:H7 EDL933, was used as a reference strain because it was the first VTEC strain to be sequenced and has since been studied across many laboratories worldwide (Perna et al., 2001). VTEC strains beginning with “V-JF” were paired-end sequenced on an Illumina HiSeq with a coverage of 200X. Reads were assembled with SPAdes v.3.10 and draft sequence assemblies annotated with the NCBI Prokaryotic Genome Annotation Pipeline (PGAP). VTEC strains beginning with “SN” were subjected to paired-end sequencing on an Illumina MiSeq and assembled de novo with Spades v.3.1 (Bankevich et al., 2012). Draft sequence assemblies were annotated with Prokka v.1.10 (Seemann, 2014). VTEC sequences were deposited into Genbank under BioProject numbers PRJNA649237 and PRJNA287560.
PHASTER (PHAge Search Tool Enhanced Release) was used to identify intact prophages in the assembled draft genomes (Arndt et al., 2016). We used stringent criteria for prophage identification as described by Colavecchio et al. (2017b). Only prophages designated “intact” were used for further analysis. Hits to known phages were identified through PHASTER and subsequently confirmed with NCBI BLAST (Altschul et al., 1990).
All identified prophages were grouped if they were identified by BLAST best hit. If phages were identified by BLAST best hit and possessed 95–99.9% similarity over 50% query, they were designated variants and assigned a Roman numeral: I, II, III and so on. Nucleotide similarity was determined using the Clustal Omega alignment tool (Madeira et al., 2022), freely available at https://www.ebi.ac.uk/Tools/msa/clustalo/. Genes were annotated automatically with RAST (https://rast.nmpdr.org/) (Aziz et al., 2008).
The phylogenetic tree was constructed in R using the Ape package (Paradis and Schliep, 2019). Sequences were aligned using the ClustalW algorithm and the phylogenetic tree constructed using the Maximum-Likelihood method, employing 1,000 bootstrap replicates. The tree was imported into FigTree for additional rendering (Rambaut, 2007). Clusters were identified with ClusterPicker (Ragonnet-Cronin et al., 2013) using an inter-cluster threshold of 50% nucleotide identity (Fong et al., 2019). Comparative gene maps were constructed in R with the GeneplotR package (Guy et al., 2010) and Geneious Prime v.2022.0.2. Circular genomic visualizations and dotplot alignments were constructed with Geneious Prime v.2022.0.2 (www.geneious.com).
Bacterial CRISPR-Cas arrays were identified with CRISPRFinder (Grissa et al., 2007) using default parameters. Spacer sequences were assessed for homology to known phages using NCBI BLAST. The number of spacer sequences of VTEC and Salmonella were statistically compared with the Student's t-test in JMP version 11.1.1 (SAS Institute, Inc., Cary, NC, United States). A P-value of ≤0.05 was considered statistically significant.
Cumulatively, a total of 289 prophages were identified with bioinformatics analysis in our collection of Salmonella (n = 50) and VTEC (n = 68) strains (Supplementary Tables S1, S2). These were grouped into 130 unique (95% nucleotide identity over 50% query cover or lower) prophage sequences. Similar prophages within the same variant group were identified in diverse hosts (Supplementary Table S1) which may signify multiple phage/host interaction implications such as similar host ranges, common phage ancestry and recent or stable horizontal exchange and host acquisition (Hendrix et al., 1999). It is important to emphasize that phages were not induced experimentally, nevertheless, the intact prophages identified in this study would theoretically be capable of excision. As with all in silico prophage prediction tools, potential underrepresentation and misinterpretation of prophage diversity and abundance exists (i.e., split between two contigs in draft assemblies) and has been discussed extensively by Hurwitz et al. (2018). The prophages uncovered in this work provide a preliminary understanding of relationships in their respective hosts.
Prophages were clustered into 26 clades on the basis of 50% nucleotide similarity with 13 genomic singletons, highlighting the diversity in our subset of strains (Figure 1). Interestingly, prophages clustered together regardless of the bacterial genus, indicating that Salmonella and VTEC share genetically similar prophages with broad host ranges capable of inter-genus infectivity. Furthermore, these data suggest active dissemination of prophages amongst genetically distinct bacterial populations that may cohabitate similar niches, such as agricultural sites. We found prophages in Salmonella and VTEC that were most closely related to those of other hosts, such as Klebsiella, Haemophilus and Vibrio. The identification of genetically similar prophages amongst distinct bacteria may broaden the host range when designing future prophage therapies (e.g., engineering broad-range prophage induction agents) against foodborne pathogens (Hu et al., 2021). Furthermore, the discovery of distant hosts with closely-associated clinical outcomes may be attributed to the impact of the host environment in phage-host interactions.
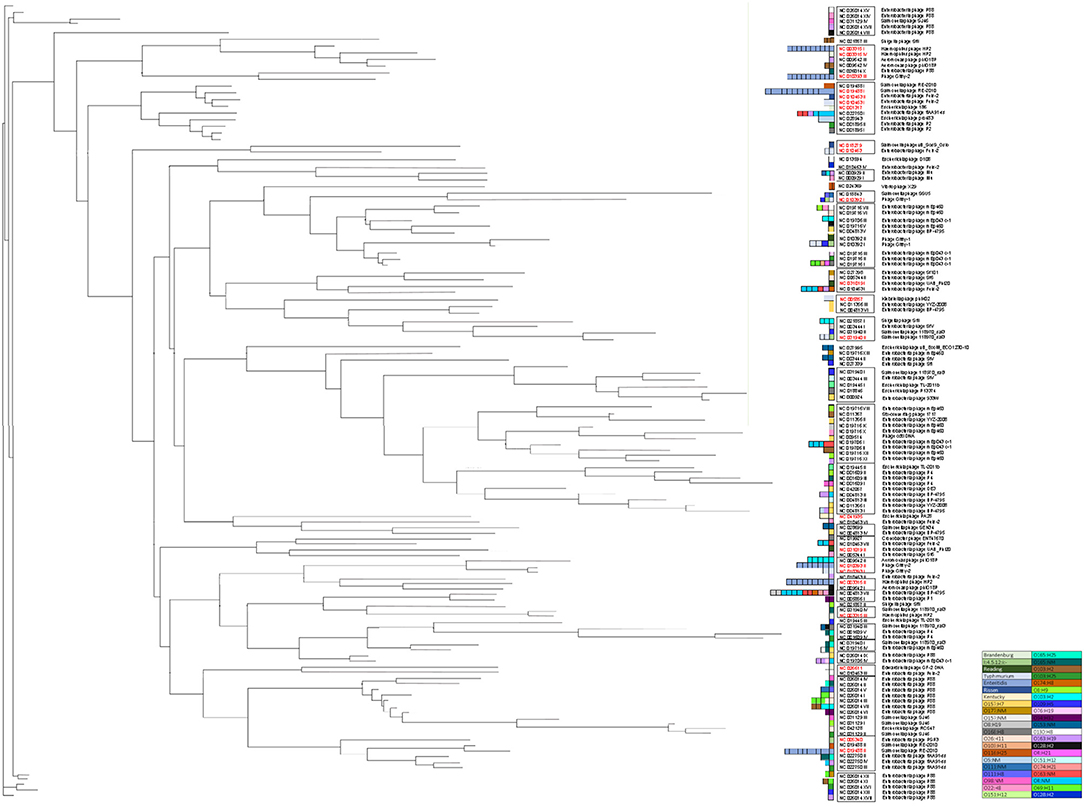
Figure 1. Maximum-likelihood tree of 130 prophages identified in this study. For clarity, bootstrap values (all >70%) are not shown. Scale bar represents the average number of nucleotide substitutions per site. Red font indicates prophages identified in our Salmonella strains. Colored boxes next to taxa names indicate serotype abundance and in which the prophages were identified.
We detected 13 unique prophages in 50 genomes of Salmonella (Supplementary Table S1). The abundance and type of prophage correlated with Salmonella serotype (i.e., prophage typing) (Figure 1, Thapa and Mohammed, 2019). Some prophages (NC 003315 I, NC 003315 II, NC 010393 II, NC 010393 III, NC 019488 I, NC 019488 II and NC 009542 II) were exclusively detected in strains of the same serotype (Figure 1). Polylysogeny (i.e., the carriage of more than one prophage) was also common; on average, 1.94 prophages were detected per Salmonella genome, which is lower than what has been reported (Bobay et al., 2013; Mottawea et al., 2018). Extremely divergent phages were also identified within the same genome; strain S01 contained five intact phages with an average nucleotide similarity of 51.5%. Similarly, S05 also contained five phages with an average similarity of 37.5%. These data indicate that phages with novel and diverse genetic materials are present in the genomes of our Salmonella collection. Compared to VTEC, we detected far fewer unique prophages in Salmonella. This may be due to several factors. For instance, E. coli may contain more preferable genomic integration sites and/or compatible genomic content (e.g., tRNAs, similar GC content) which has been demonstrated to favor phage infection and integration (Cardinale and Duffy, 2011). As part of the host immune response, superinfection exclusion systems also restrict superinfection by similar phages (Seed, 2015).
VTEC strain EDL933 was used in our prophage analyses as it is a well-characterized strain possessing genetically diverse prophages (Saile et al., 2016). Overall, 66 of 68 VTEC strains possessed at least one prophage. We did not identify any prophages in SN496 nor SN573, indicating these prophages may not be intact and/or defective. In a screening of 40 VTEC isolates by Zhang et al. (2020), 15.3 and 9.3% of prophages were incomplete and questionable, respectively. It currently remains unclear whether these prophages are inducible, however, Asadulghani et al. (2009) induced several defective prophages of VTEC O157 and found these phages maintained their virulence after the induction.
Clinical type strain EDL933 possessed 11 prophages, the most prophages of all strains tested. As the majority of our VTEC strains were isolated predominantly from the agricultural environment, these results suggest that these prophages may confer advantages to the host such that it allows for enhanced persistence in such environments.
On average, the incidence of prophage carriage in Salmonella was 1.94 per genome and 3.03 per VTEC genome (Figure 2). Prophage profiles clustered closely with serotype (i.e., number and type of phage correlated with bacterial serotype) (Figures 1, 2 and Supplementary Table S1), however, some prophages and their variants (e.g., NC_010392; Gifsy-1 and NC_010393; Gifsy-2) were not serotype-dependent and were identified in several strains representing different serotypes. Gifsy-1 and−2 -like phages are best known for contributing to the virulence of S. Typhimurium (Ho and Slauch, 2001), however in the present study they were identified in the genomes of S. Reading, S. Typhimurium, S. Enteritidis and VTEC. The variants differed greatly, with NC_010393 (I) possessing a genome size of ~16 kb, almost 50% smaller than variants (II) and (III). Further analysis revealed that these genes in variant (I) were mostly structural, while variants (II) and (III) also carried a slew of accessory genes for host invasion (e.g., sopE). Given that the annotation assigned mostly structural gene predictions, the carriage of variant (I) does not indicate the presence of any obvious host survival advantages but may provide an indirect mutualistic benefit if lysogeny is stably maintained. Diversity in Gifsy-1 was also greatly apparent (Figure 3) with variant (I) ~30% shorter than variant (II). An intact lysogeny cassette was not identified in this variant, although major structural and host recognition genes were identified (e.g., minor and major capsid proteins, portal proteins, tail fibers). Gifsy-1 was not only found in our subset of Salmonella strains but also in closely related to Gifsy-1 prophage in VTEC strain V-JF-010, which, to the best of our knowledge, has not been documented in E. coli before. Previous work has revealed that E. coli requires Salmonella OmpC protein for Gifsy prophage to bind (Ho and Slauch, 2001), however, upon homology analysis with NCBI BLAST no homologs were identified in strain V-JF-010. Further work is needed to elucidate the receptor of interest, and also to identify putative receptor sites in other bacterial genera, particularly in type phages such as Gifsy-like viruses that may confer virulence. The identification of such sites in taxonomically distinct species of bacteria is important for the design of prophage-engineered biocontrol strategies, an area of study still currently in its infancy (Cadieux et al., 2018).

Figure 2. Frequency histogram of strains carrying prophages.
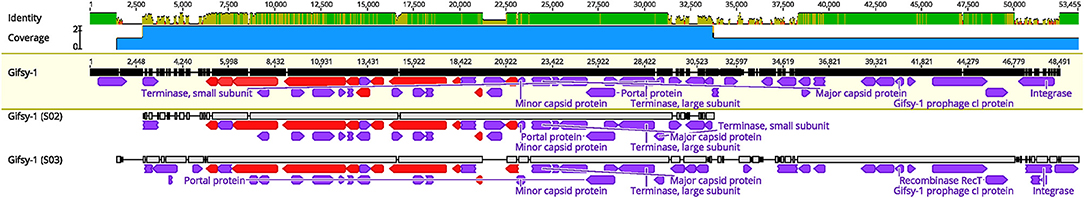
Figure 3. Comparisons between Gifsy-1 and related variants in S02 and S03. ORFs in red represent tail protein regions in the respective phages. Structural proteins and lysogeny-related genes are indicated.
Various genetic elements encoding an array of functions (i.e., host virulence, antimicrobial resistance) were identified in prophages of both Salmonella and VTEC (Table 2). Carriage of these virulence factors is problematic as these genes may be readily transduced throughout a bacterial population, potentially impacting host fitness such that it allows for greater survival in a variety of conditions. Phages may carry an array of genes that confer a host advantage (Colavecchio et al., 2017a; Gómez-Gómez et al., 2019; Wendling et al., 2020). For instance, prophages highly related to Gifsy-1, Gifsy-2 and Gifsy-3 are conserved within S. Typhimurium and contribute significantly to host virulence (Hiley et al., 2014). A previous study by Ross and Topp (2015) also observed the abundance of antibiotic-resistance elements in phages sourced from soil, suggesting that the agricultural environment may be a potent source of such genes.
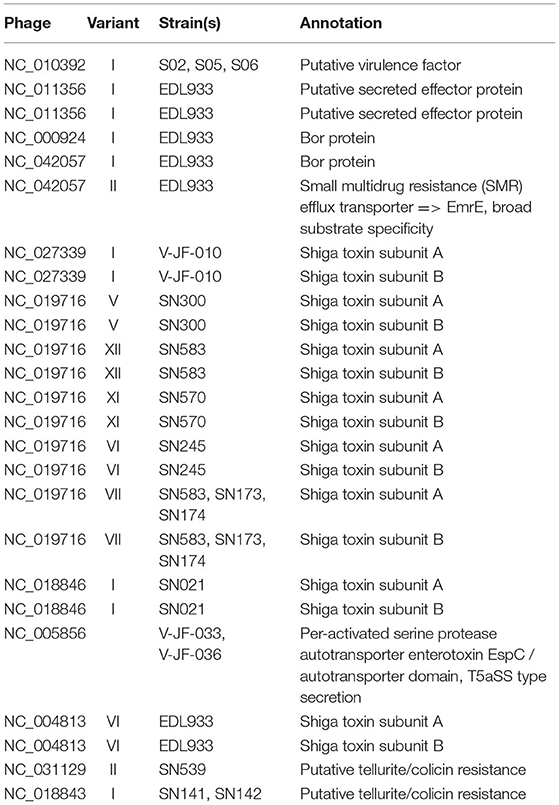
Table 2. Putative virulence genes carried by the prophages identified in this study.
A gene encoding putative tellurite and colicin resistance was functionally annotated in prophage SSU5 of SN141 and SN142 (Table 2). Interestingly, we also identified partitioning genes parA and parB. A high degree of homology to plasmid p4 was observed with demonstrated genomic rearrangements relative to each other (Figure 4). Plasmid p4 was originally isolated from an extended spectrum β-lactamase-producing strain of E. coli (Brolund et al., 2019; Accession: CP023851.1). Previous studies have observed the high degree of genomic similarity between SSU5 and plasmid pHCM2 (Kidgell et al., 2002; Kim et al., 2012), however, this variant of SSU5 appears to be most closely related to plasmid p4. SSU5 is 107,570 bp in length, with 130 open reading frames (Figure 5). Its putative roles as both a plasmid and phage may represent more avenues for transmission of genes that confer increased host fitness and warrants further investigation.
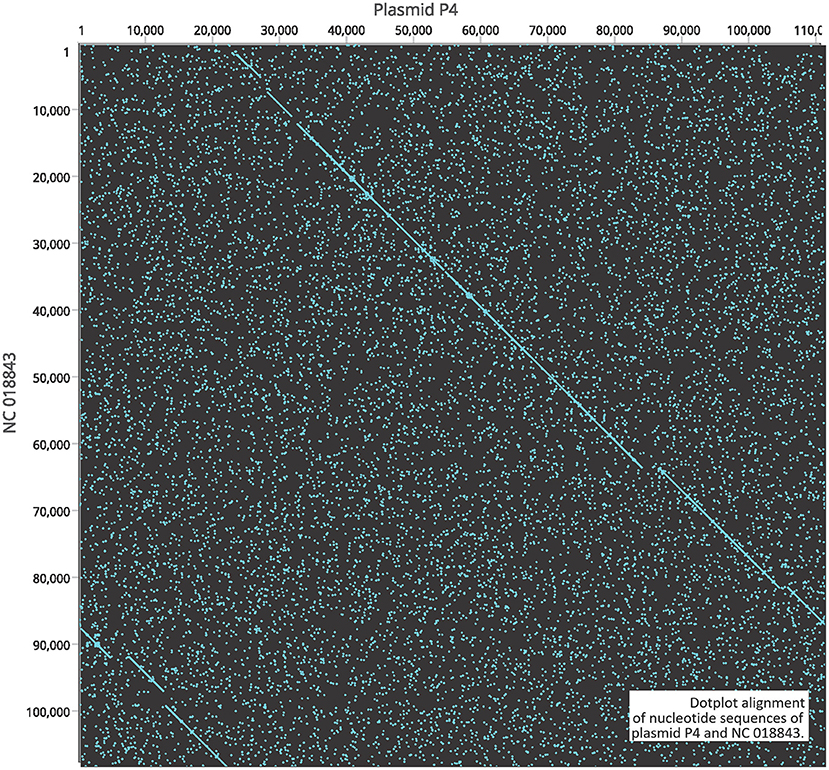
Figure 4. Dotplot alignment of nucleotide sequences of plasmid P4 and NC 018843.
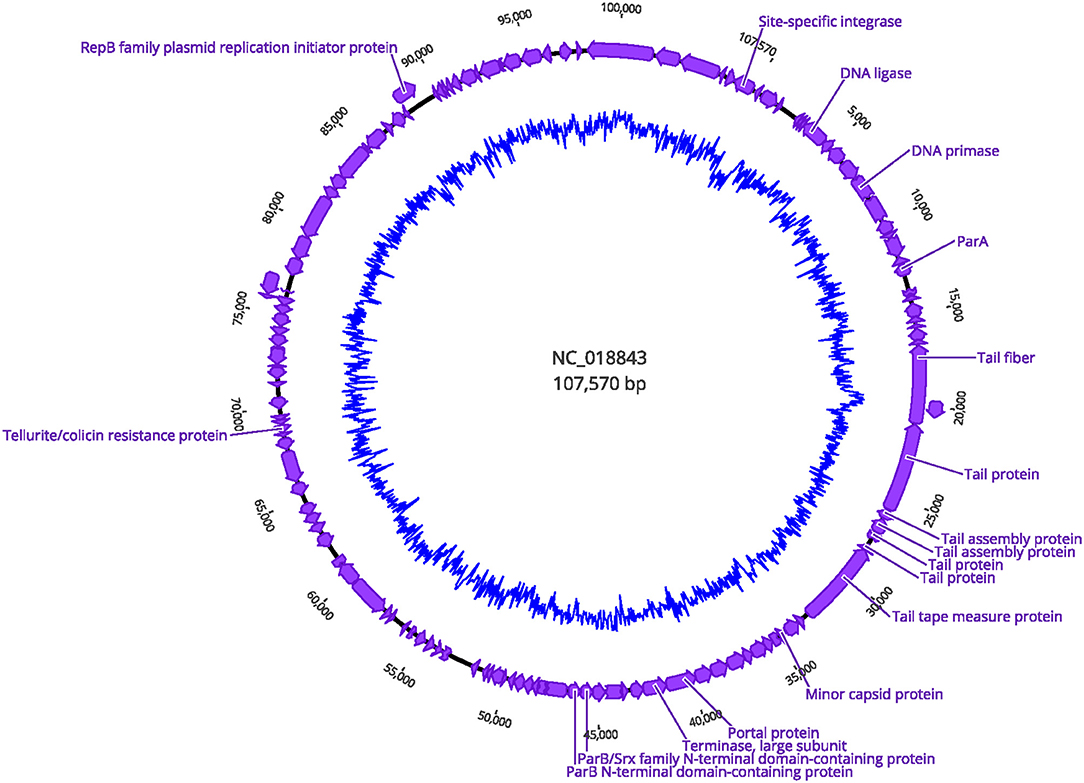
Figure 5. Circular genome visualization of prophage SSU5 identified in SN141 and SN142. Structural genes and genes encoding host genome integration are indicated. Inner circle represents GC content.
Tellurite resistance compounds have been found in several genera of bacteria, including E. coli, Salmonella, Shigella and Vibrio. Though uncommon, these compounds have been found in the chromosome or on plasmids (Chasteen et al., 2009). Experimentally, tellurite is used as a selective agent for the isolation of many pathogens, including E. coli O157:H7 (Turkovicova et al., 2016). Compared to the wild type strain, E. coli BL21 possessing the ter operon (terZABCDEF; responsible for conferring tellurite resistance) was able to grow in the presence of extremely high concentrations of tellurite (minimum inhibitory concentration of ~4 mM) (Turkovicova et al., 2016). In the environment, tellurium is relatively rare, however, its soluble salts such as potassium tellurite were used clinically as an antimicrobial agent in the past (Valkova et al., 2007). Additionally, it was shown previously that ter genes enhanced the ability of E. coli to survive in macrophages (Valkova et al., 2007).
It is interesting to note that the ter operon is functionally diverse and may encode for a variety of other bacterial phenotypes. The ter genes play a coordinated role in stress resistance and may offer resistance against a broad spectrum of agents, including colicins (small antimicrobial peptides (i.e., bacteriocins) produced by E. coli to kill non-host E. coli cells) (Jin et al., 2018) and phages by mounting a restriction or suicidal action upon phage infection (Anantharaman et al., 2012). Given the increasing demand for antibiotic alternatives in sectors such as food processing and clinical medicine, the dissemination of phage resistance genes in bacterial hosts can be problematic as it may limit the efficacy of phage-based treatments for host elimination (e.g., direct phage application). Because SSU5 may be able to act intracellularly as a plasmid and a phage, multiple avenues for dissemination throughout a bacterial population exist and may therefore amplify the spread of critically important genes such as those encoding antimicrobial resistance. Such phages may help to drive evolution of bacterial populations in certain environments.
The CRISPR loci is exploited for various analyses, such as microbial typing and tracking (Dion et al., 2021). Since spacer regions are hypervariable and provides historical information on phage resistance, we screened the spacer regions of identified CRISPR arrays with the aim to elucidate and characterize possible associations between spacer elements, prophage lysogeny and strain-specific differences in our dataset. All strains contained at least six unique spacers (Figure 6 and Supplementary Table S3). Salmonella contained significantly more spacers (an average of 27 regions) than VTEC (an average of 19 regions) (p <0.05). Previous work has revealed that in over >600 strains of Salmonella representing four serotypes, an average of 16 Class 1 and 20 Class 2 systems were identified (Shariat et al., 2015). More recent work also elucidated the diversity of spacers among various serotypes of Salmonella and identified a high number of spacers (440 and 330 unique spacers within 2221 and 2211 of CRISPR 1 and CRISPR2 arrays, respectively) in all Salmonella strains analyzed (Kushwaha et al., 2020). Many of the core CRISPR-associated (Cas) proteins that comprise the effector complex have evolved in line with the arms race against evolving phages (Koonin and Makarova, 2019). Thus, on the basis of Cas proteins and array organization, CRISPR defense systems have been categorized into two groups, Class 1 and Class 2 that are respectively subdivided into types I, III, IV and types II, V, VI (Makarova and Koonin, 2015). In a separate report surveying 100 strains of E. coli, 745 unique spacers were identified (Díez-Villaseñor et al., 2010). This is in stark contrast to a previous study by Touchon and Rocha (2010), where they observed no more than three CRISPRs in 51 genomes of Salmonella and Escherichia. These conflicting data may be because of several factors, not the least being differences in serotype, pathogenicity, and source of isolation of the strains tested. Zeng et al. (2017) observed that compared to clinical isolates (n = 17 spacers), food isolates of Cronobacter sakazakii possessed a significantly greater number of spacers in their CRISPR loci (n = 30). Correspondingly, foodborne isolates possessed less prophages (n = 2.81) compared to clinical isolates (n = 4.15). Enhanced evolution in CRISPR loci is important to consider when developing strategies such as phage cocktails for pathogen control because a key criterion for successful biocontrol is that resistance to phages is absent or delayed (Fong et al., 2019). It should be noted, however, that our bacterial strains of agricultural origin are draft genomes and may potentially underrepresent the true number of spacer regions. Nevertheless, our findings provide a preliminary basis for further probing of such genomes.
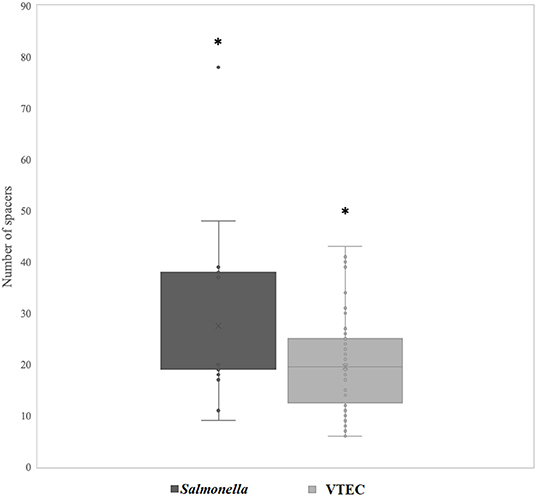
Figure 6. Number of spacer regions in Salmonella and VTEC. Asterisks indicate significance below α = 0.05 (Student's t-test).
Spacers with significant homology to the identified prophages in this study were not observed, and we did not identify any self-targeting spacers. Surprisingly, only a small subset of strains (14%) encoded spacers with homology to known prophages, and these were only identified in VTEC (Supplementary Table S3). It should be noted that the scarcity of sequenced prophages in public databases may have supported this observation (Zeng et al., 2017). We also did not observe a significant correlation between spacer and prophage abundance in our strains (Figure 7). In silico work by Wang et al. (2020) also did not discern an association between these two elements in Bifidobacterium pseudocatenulatum. Despite their high degree of conservation, the role of CRISPR-Cas systems in prophage immunity has been questioned previously (Shariat et al., 2015), which has led to the hypothesis that these systems in Salmonella and VTEC may have alternative functions yet to be clarified (Touchon and Rocha, 2010; Shariat et al., 2015).
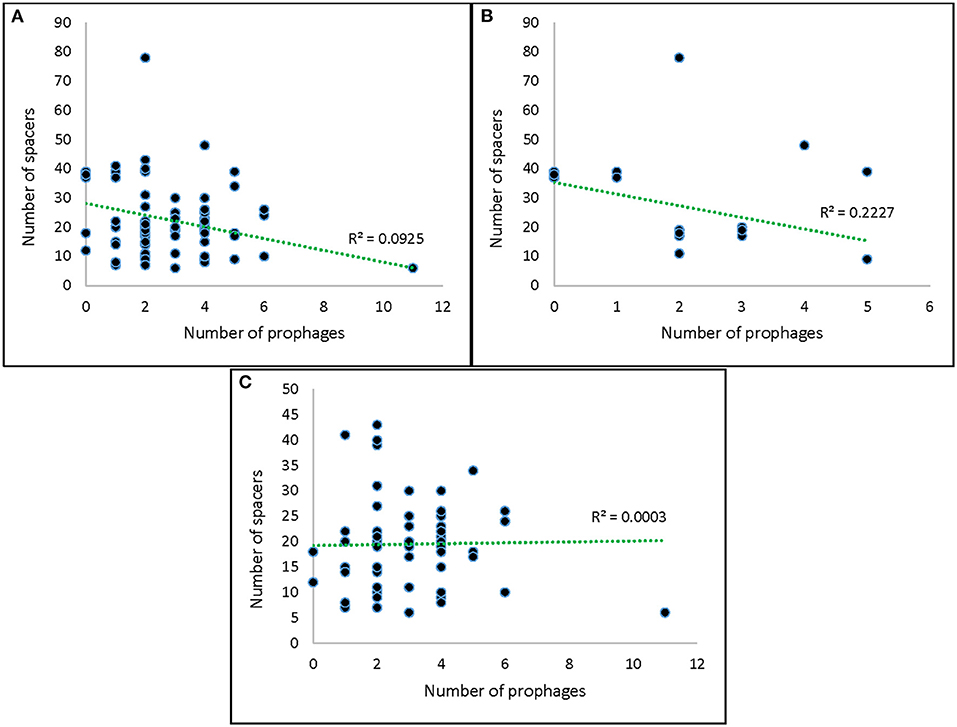
Figure 7. Correlation between number of spacers and number of prophages. (A) All strains; (B) Salmonella strains; (C) VTEC strains.
We have described here the identification and analysis of 130 unique prophages found in the genomes of 118 strains of Salmonella and VTEC, 117 of which are of agricultural origin. These prophages are diverse, with many prophages sharing a common host between Salmonella, VTEC and other bacteria, including Vibrio, Shigella and Klebsiella. The prophages identified contain novel attributes and genes that may have far-reaching impacts on host phenotype. Further probing of the host CRISPR elements revealed an abundance of spacers in our strains, which provides preliminary insights into prophage-host interactions in agricultural environments. Overall, our results shed insight into the prophage repertoire of agriculturally-sourced strains of Salmonella and VTEC, which will provide important considerations when developing prophage-based control strategies.
The data presented in the study are deposited in the GenBank repository. The names of the repository and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, BioProject IDs PRJNA6649825, PRJNA287560, and PRJNA649237.
KF and SW were responsible for study conception. SW provided funding for this research. KF and YL analyzed the data. TB and JF provided the bacterial strains used in this study. KF wrote the first draft of the manuscript and all authors provided feedback. All authors read and approved the final manuscript.
This work was supported by the National Sciences and Engineering Research Council of Canada, NSERC Discovery Grant RGPIN-2015-04871. Funding for this project has also been provided by the Governments of Canada and British Columbia through the Canadian Agricultural Partnership, a federal-provincial-territorial initiative. The program is delivered by the Investment Agriculture Foundation of BC (INV 133).
Opinions expressed in this document are those of the author and not necessarily those of the Governments of Canada and British Columbia or the Investment Agriculture Foundation of BC. The Governments of Canada and British Columbia, and the Investment Agriculture Foundation of BC, and their directors, agents, employees, or contractors will not be liable for any claims, damages, or losses of any kind whatsoever arising out of the use of, or reliance upon, this information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.853703/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, W. E., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 5. doi: 10.1016/S0022-2836(05)80360-2
Anantharaman, V., Lakshminarayan, M. I., and Aravind, L. (2012). Ter-dependent stress response systems: Novel pathways related to sensing, production of a nucleoside-like metabolite, and DNA-processing. Mol. Biosyst. 8, 3142–3165. doi: 10.1039/c2mb25239b
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Asadulghani, M. D., Ogura, Y., Ooka, T., Itoh, T., Sawaguchi, A., Iguchi, A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5, e10000408. doi: 10.1371/journal.ppat.1000408
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genom. 9, 75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bobay, L. M., Rocha, E. P. C., and Touchon, M. (2013). The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 30, 737–751. doi: 10.1093/molbev/mss279
Bobay, L. M., Touchon, M., and Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA. 111, 12127–12132. doi: 10.1073/pnas.140533611
Brenner, T., Fong, K., Lee, S., and Wang, S. (2020). A dynamic method for broad-spectrum bacteriophage cocktail formulation against poultry-associated Salmonella enterica. PHAGE. 1, 109–117. doi: 10.1089/phage.2020.0002
Brolund, A., Rajer, F., Giske, C. G., Melefors, O., Titelman, E., and Sandegren, L. (2019). Dynamics of resistance plasmids in extended-spectrum-beta-lactamase-producing Enterobacteriaceae during postinfection colonization. Antimicrob. Agents Chemother. 63, e02001–18. doi: 10.1128/AAC.02201-18
Brueggemann, A. B., Harrold, C. L., Rezaei Javan, R., van Tonder, A. J., McDonnell, A. J., and Edwards, B. A. (2017). Pneumococcal prophages are diverse, but not without structure or history. Sci. Rep. 7, 42976. doi: 10.1038/srep42976
Cadieux, B., Colavecchio, A., Jeukens, J., Freschi, L., Emond-Rheault, J. G., Kukavica-Ibrulj, I., et al. (2018). Prophage induction reduces Shiga toxin producing Escherichia coli (STEC) and Salmonella enterica on tomatoes and spinach: a model study. Food Control. 289, 250–259. doi: 10.1016/j.foodcont.2018.02.001
Cardinale, D. J., and Duffy, S. (2011). Single-stranded genomic architecture constrains optimal codon usage. Bacteriophage. 1, 219–224. doi: 10.4161/bact.1.4.18496
Chasteen, T. G., Fuentes, D. E., Tantaleán, J. C., and Vásquez, C. C. (2009). Tellurite: history, oxidative stress, and molecular mechanisms of resistance. FEMS Microbiol. Rev. 33, 820–832. doi: 10.1111/j.1574-6976.2009.00177.x
Colavecchio, A., Cadieux, B., Lo, A., and Goodridge, L. D. (2017a). Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family–a review. Front. Microbiol. 8:1108. doi: 10.3389/fmicb.2017.01108
Colavecchio, A., D'Souza, Y., Tompkins, E., Jeukens, J., Freschi, L., Emond-Rheault, J. G., et al. (2017b). Prophage integrase typing is a useful indicator of genomic diversity in Salmonella enterica. Front. Microbiol. 8, 1283. doi: 10.3389/fmicb.2017.01283
Crispim, J. S., Dias, R. S., Vidigal, P. M. P., de Sousa, M. P., da Silva, C. C., Santana, M. F., et al. (2018). Screening and characterization of prophages in Desulfovibrio genomes. Sci. Rep. 8, 9273. doi: 10.1038/s41598-018-27423-z
Díez-Villaseñor, C., Almendros, C., García-Martínez, J., and Mojica, F. J. (2010). Diversity of CRISPR loci in Escherichia coli. Microbiology. 156, 1351–1361. doi: 10.1099/mic.0.036046-0
Dion, M. B., Plante, P. L., Zufferey, E., Shah, S. A., Corbeil, J., and Moineau, S. (2021). Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter. Nucl. Acids Res. 49, 3127–3138. doi: 10.1093/nar/gkab133
Falardeau, J., Johnson, R. P., Pagotto, F., and Wang, S. (2017). Occurrence, characterization, and potential predictors of verotoxigenic Escherichia coli, Listeria monocytogenes, and Salmonella in surface water used for produce irrigation in the Lower Mainland of British Columbia, Canada. PLoS ONE. 12, e0185437. doi: 10.1371/journal.pone.0185437
Fang, Y., Mercer, R. G., McMullen, L. M., and Gänzle, M. (2017). Induction of Shiga toxin-encoding prophage by abiotic environmental stress in food. Appl. Environ. Microbiol. 83, e01378–e01317. doi: 10.1128/AEM.01378-17
Fong, K., LaBossiere, B., Switt, A. I. M., Delaquis, P., Goodridge, L., Levesque, R. C., et al. (2017). Characterization of four novel bacteriophages isolated from British Columbia for control of non-typhoidal Salmonella in vitro and on sprouting alfalfa seeds. Front. Microbiol. 8, 2193. doi: 10.3389/fmicb.2017.02193
Fong, K., Mu, K., Rheault, J. G., Levesque, R. C., Kitts, D., Delaquis, P., et al. (2020). Bacteriophage-insensitive mutants of antimicrobial-resistant Salmonella enterica are altered in their tetracycline resistance and virulence in Caco-2 intestinal cells. Int. J. Mol. Sci. 21, 1883. doi: 10.3390/ijms21051883
Fong, K., Tremblay, D. M., Delaquis, P., Goodridge, L., Levesque, R. C., Moineau, S., et al. (2019). Diversity and host specificity revealed by biological characterization and whole genome sequencing of bacteriophages infecting Salmonella enterica. Viruses. 11, 854. doi: 10.3390/v11090854
Fu, B. X. H., Wainberg, M., Kundaje, A., and Fire, A. Z. (2017). High-throughput characterization of cascade type I-E CRISPR guide efficacy reveals unexpected PAM diversity and target sequence preferences. Genetics. 206, 1727–1738. doi: 10.1534/genetics.117.202580
Fu, Y., Wu, Y., Yuan, Y., and Gao, M. (2019). Prevalence and diversity analysis of candidate prophages to provide an understanding on their roles in Bacillus thuringiensis. Viruses. 11, 388. doi: 10.3390/v11040388
Gómez-Gómez, C., Blanco-Picazo, P., Brown-Jaque, M., Quirós, P., Rodríguez-Rubio, L., Cerdà-Cuellar, M., et al. (2019). Infectious phage particles packaging antibiotic resistance genes found in meat products and chicken feces. Sci. Rep. 9, 1–11. doi: 10.1038/s41598-019-49898-0
Government of Canada. (2016). Yearly Food-borne Illness Estimates for Canada. Available online at: https://www.canada.ca/en/public-health/services/food-borne-illness-canada/yearly-food-borne-illness-estimates-canada.html (accessed April 10, 2022).
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35,W52–W57. doi: 10.1093/nar/gkm360
Guy, L., Roat Kultima, J., and Andersson, S. G. E. (2010). GenoPlotR: comparative gene and genome visualization in R. Bioinformatics. 26, 2334–2335. doi: 10.1093/bioinformatics/btq413
Hendrix, R. W., Smith, M. C. M., Burns, R. N., Ford, M. E., and Hatfull, G. F. (1999). Evolutionary relationships among diverse bacteriophages and prophages: All the world's a phage. Proc. Natl. Acad. Sci. U.S.A. 96, 2192–2197. doi: 10.1073/pnas.96.5.2192
Hiley, L., Fang, N. X., Micalizzi, G. R., and Bates, J. (2014). Distribution of Gifsy-3 and of variants of ST64B and Gifsy-1 prophages amongst Salmonella enterica serovar Typhimurium isolates: evidence that combinations of prophages promote clonality. PLOS ONE. 9, e86203. doi: 10.1371/journal.pone.0086203
Ho, T. D., and Slauch, J. M. (2001). OmpC is the receptor for Gifsy-1 and Gifsy-2 bacteriophages of Salmonella. J. Bacteriol. 183, 1495–1498. doi: 10.1128/JB.183.4.1495-1498.2001
Hu, J., Ye, H., Wang, S., Wang, J., and Han, D. (2021). Prophage activation in the intestine: Insights into functions and possible applications. Front. Microbiol. 12, 785634. doi: 10.3389/fmicb.2021.785634
Hurwitz, B. L., Ponsero, A., Thornton, Jr. J., and U'Ren, J. M. (2018). Phage hunters: Computational strategies for finding phages in large-scale 'omics datasets. Virus Res. 244, 110–115. doi: 10.1016/j.virusres.2017.10.019
Jin, X., Kightlinger, W., Kwon, Y. C., and Jong, S. H. (2018). Rapid production and characterization of antimicrobial colicins using Escherichia coli-based cell free protein synthesis. Synth Biol. 3, ysy004. doi: 10.1093/synbio/ysy004
Kidgell, C., Pickard, D., Wain, J., James, K., Diem Nga, L. T., Song, D.iep, To., et al. (2002). Characterisation and distribution of a cryptic Salmonella typhi plasmid pHCM2. Plasmid. 47, 159–171. doi: 10.1016/S0147-619X(02)00013-6
Kim, M., Kim, S., and Ryu, S. (2012). Complete genome sequence of bacteriophage SSU5 specific for Salmonella enterica serovar Typhimurium rough strains. J. Virol. 86:10894. doi: 10.1128/JVI.01796-12
Koonin, E. V., and Makarova, K. S. (2019). Origins and evolution of CRISPR-Cas systems. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 274, 20180087. doi: 10.1098/rstb.2018.0087
Kushwaha, S. K., Bhavesh, N. L. S., Abdella, B., Lahiri, C., and Marathe, S. A. (2020). The phylogenomics of CRISPR-Cas system and revelation of its features in Salmonella. Sci. Rep. 10, 21156. doi: 10.1038/s41598-020-77890-6
Łos, J. M., Łos, M., Wegrzyn, A., and Wegrzyn, G. (2013). Altriusm of Shiga toxin-producing Escherichia coli: recent hypothesis versus experimental results. Front. Cell Infect. Microbiol. 2, 166. doi: 10.3389/fcimb.2012.00166
Louwen, R., Staals, R. H. J., Enditz, H. P., van Baarlen, P., and van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88. doi: 10.1128/MMBR.00039-13
Madeira, F., Pearce, M., Tivey, A. R. N., Basutkar, P., Lee, J., Edbali, O., et al. (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucl. Acids Res. 50, W276–279. doi: 10.1093/nar/gkac240
Makarova, K. S., and Koonin, E. V. (2015). Annotation and classification of CRISPR-Cas systems. Methods Mol. Biol. 1311, 47–75. doi: 10.1007/978-1-4939-2687-9_4
Mottawea, W., Duceppe, M. O., Dupras, A. A., Usongo, V., Jeukens, J., Freschi, L., et al. (2018). Salmonella enterica prophage sequence profiles reflect genome diversity and can be used for high discrimination subtyping. Front. Microbiol. 9, 836. doi: 10.3389/fmicb.2018.00836
Nadya, S., Delaquis, P., Chen, J., Allen, K., Johnson, R. P., Ziebell, K., et al. (2016). Phenotypic and genotypic characteristics of Shiga toxin-producing Escherichia coli isolated from surface waters and sediments in a Canadian urban-agricultural landscape. Front. Cell. Infect. Microbiol. 6, 36. doi: 10.3389/fcimb.2016.00036
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 35, 526–528. doi: 10.1093/bioinformatics/bty633
Perna, N. T., Plunkett, G. III., Burland, V., Mau, B., Glasner, J. D., Rose, D. J., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 409, 529–533. doi: 10.1038/35054089
Ragonnet-Cronin, M., Lycett, S. J., Hodcroft, E., Hue, S., Fearnhill, E., Dunn, D., et al. (2013). Automated analysis of phylogenetic clusters. BMC Bioinformatics. 14, 317. doi: 10.1186/1471-2105-14-317
Rambaut, A. (2007). FigTree, a Graphical Viewer of Phylogenetic Trees. Available online at: http://tree.bio.ed.ac.uk/software/figtree (accessed May 25, 2010).
Ramisetty, B. C. M., and Sudhakari, P. A. (2019). Bacterial ‘grounded' prophages: Hotspots for genetic renovation and innovation. Front. Genet. 10, 65. doi: 10.3389/fgene.2019.00065
Ross, J., and Topp, E. (2015). Abundance of antibiotic resistance genes in bacteriophage following soil fertilization with dairy manure or municipal biosolids, and evidence for potential transduction. Appl. Environ. Microbiol. 81, 7095–7913. doi: 10.1128/AEM.02363-15
Saile, N., Voigt, A., Kessler, S., Stressler, T., Klumpp, J., Fischer, L., et al. (2016). Escherichia coli O157:H7 strain EDL933 harbors multiple functional prophage-associated genes necessary for the utilization of 5-N-Acetyl-9-0-Acetyl Neuraminic acid as a growth substrate. Appl. Environ. Microbiol. 82, 5940–5950. doi: 10.1128/AEM.01671-16
Seed, K. D. (2015). Battling phages: How bacteria defend against viral attack. PLOS Pathog. 11, e1004847. doi: 10.1371/journal.ppat.1004847
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shariat, N., Timme, R. E., Pettengill, J. B., Barrangou, R., and Dudley, E. G. (2015). Characterization and evolution of Salmonella CRISPR-Cas systems. Microbiology. 161, 374–386. doi: 10.1099/mic.0.000005
Thapa, S., and Mohammed, M. (2019). Characterization of prophages in Salmonella Typhimurium definitive type 8. Acc. Microbiol. 1, 181. doi: 10.1099/acmi.ac2019.po0060
Touchon, M., Charpentier, S., Clermont, O., Rocha, E. P. C., Denamur, E., and Branger, C. (2011). CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection. J. Bacteriol. 193, 2460–2467. doi: 10.1128/JB.01307-10
Touchon, M., and Rocha, E. P. (2010). The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS ONE. 5, e11126. doi: 10.1371/journal.pone.0011126
Turkovicova, L. S., Smidak, R., Jung, G., Turna, J., Lubec, G., and Aradska, J. (2016). Proteomic analysis of the TerC interactome: Novel links to tellurite resistance and pathogenicity. J. Proteomics. 136, 167–173. doi: 10.1016/j.jprot.2016.01.003
Valkova, D., Valkovicova, L., Vavrova, S., Kovacova, E., Mravec, J., and Turna, J. (2007). The contribution of tellurite resistance genes to the fitness of Escherichia coli uropathogenic strains. Cent. Eur. J. Biol. 2, 182–191. doi: 10.2478/s11535-007-0019-9
Wang, S., Liu, Q., Pei, Z., Wang, L., Tian, P., Liu, Z., et al. (2020). The diversity of the CRISPR-Cas system and prophages present in the genome reveals the co-evolution of Bifidobacterium pseudocatenulatum and phages. Front. Microbiol. 11, 1088. doi: 10.3389/fmicb.2020.01088
Wendling, C. C., Refardt, D., and Hall, A. R. (2020). Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic-resistance genes peak in different environments. Evolution. 75, 515–528. doi: 10.1111/evo.14153
World Health Organization. (2018). Antibiotic Resistance. Available online at: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed April 10, 2020).
Zeng, H., Zhang, J., Li, C., Xie, T., Ling, N., Wu, Q., et al. (2017). The driving force of prophages and CRISPR-Cas system in the evolution of Cronobacter sakazakii. Sci. Rep. 7, 40206. doi: 10.1038/srep40206
Keywords: prophage, phage, Salmonella, VTEC, CRISPR
Citation: Fong K, Lu YT, Brenner T, Falardeau J and Wang S (2022) Prophage Diversity Across Salmonella and Verotoxin-Producing Escherichia coli in Agricultural Niches of British Columbia, Canada. Front. Microbiol. 13:853703. doi: 10.3389/fmicb.2022.853703
Received: 12 January 2022; Accepted: 22 June 2022;
Published: 22 July 2022.
Edited by:
Muhammad Kamruzzaman, Westmead Institute for Medical Research, AustraliaReviewed by:
Alexander P. Hynes, McMaster University, CanadaCopyright © 2022 Fong, Lu, Brenner, Falardeau and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siyun Wang, c2l5dW4ud2FuZ0B1YmMuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.