 Jessica A. Lee
Jessica A. Lee Sergey Stolyar1,2,3
Sergey Stolyar1,2,3 Christopher J. Marx
Christopher J. Marx- 1Department of Biological Sciences, University of Idaho, Moscow, ID, United States
- 2Institute for Modeling Collaboration and Innovation, University of Idaho, Moscow, ID, United States
- 3Institute for Interdisciplinary Data Sciences, University of Idaho, Moscow, ID, United States
- 4Space Biosciences Research Branch, NASA Ames Research Center, Moffett Field, CA, United States
Pink-pigmented facultative methylotrophs have long been studied for their ability to grow on reduced single-carbon (C1) compounds. The C1 groups that support methylotrophic growth may come from a variety of sources. Here, we describe a group of Methylobacterium strains that can engage in methoxydotrophy: they can metabolize the methoxy groups from several aromatic compounds that are commonly the product of lignin depolymerization. Furthermore, these organisms can utilize the full aromatic ring as a growth substrate, a phenotype that has rarely been described in Methylobacterium. We demonstrated growth on p-hydroxybenzoate, protocatechuate, vanillate, and ferulate in laboratory culture conditions. We also used comparative genomics to explore the evolutionary history of this trait, finding that the capacity for aromatic catabolism is likely ancestral to two clades of Methylobacterium, but has also been acquired horizontally by closely related organisms. In addition, we surveyed the published metagenome data to find that the most abundant group of aromatic-degrading Methylobacterium in the environment is likely the group related to Methylobacterium nodulans, and they are especially common in soil and root environments. The demethoxylation of lignin-derived aromatic monomers in aerobic environments releases formaldehyde, a metabolite that is a potent cellular toxin but that is also a growth substrate for methylotrophs. We found that, whereas some known lignin-degrading organisms excrete formaldehyde as a byproduct during growth on vanillate, Methylobacterium do not. This observation is especially relevant to our understanding of the ecology and the bioengineering of lignin degradation.
Introduction
Microbial processes for degrading lignin and lignin-derived aromatics are of intense interest in a diversity of fields, ranging from fundamental ecosystems science—which seeks to understand the processes by which the carbon fixed in plant biomass is mineralized (Lehmann and Kleber, 2015)—to bioenergy engineering—where the recalcitrance of lignin is a major hurdle in the processing of plant biomass (Ragauskas et al., 2014)—and the fossil fuel industry—which seeks to understand microbial transformations of coal (Welte, 2016). Lignin, which comprises approximately 20% of the carbon fixed by photosynthesis on land (Ruiz-Dueñas and Martínez, 2009), is an exceptionally complex and irregular, polycyclic aromatic polymer, in which many of the constituent aromatic rings are heavily substituted with methoxy (-OCH3) groups (Vanholme et al., 2010). While depolymerization of lignin in the environment is typically attributed to the activity of fungi, recent studies probing the biodiversity of the microbial lignin-degrading community in soils have inspired increased interest in the role of bacteria in both the depolymerization and the degradation of its monomeric aromatic constituents (Wilhelm et al., 2019; Díaz-García et al., 2021).
In the aerobic bacterial degradation of lignin-derived aromatic monomers such as vanillate, the degradation of the aromatic ring proceeds by the protocatechuate branch of the beta-ketoadipate pathway. The gene cluster encoding this pathway is widely distributed among soil microorganisms, and has a complex evolutionary history resulting in diverse patterns of gene organization and regulation (Harwood and Parales, 1996; Parke, 1997; Buchan et al., 2004). A commonly described configuration includes the genes pcaH and pcaG (encoding the enzymes that cleave the aromatic ring of protocatechuate to generate beta-carboxy-cis-cis-muconate), pcaB (to generate gamma-carboxymuconolactone), pcaC (to generate beta-ketoadipate enol-lactone), pcaD (to generate beta-ketoadipate), pcaI and pcaJ (to generate beta-ketoadipyl-CoA), and pcaF (to generate succinyl-CoA and acetyl-CoA). In the case of methoxylated aromatics, this pathway is preceded by the removal of the methoxy group; in most aerobic organisms, this occurs via vanillate monooxygenase, a Rieske [2Fe-2S] enzyme that demethylates vanillate to generate protocatechuate and formaldehyde (Brunel and Davison, 1988; Mitsui et al., 2003; Merkens et al., 2005; Sudtachat et al., 2009; Chen et al., 2012) (Figure 1). Pseudomonas putida growing on vanillate as a sole carbon source excretes formaldehyde into the medium in measurable amounts (Lee et al., 2021). Formaldehyde is a potent electrophile, and toxic to microorganisms due to its reactivity with DNA and proteins (Chen et al., 2016). Elimination of this toxin is therefore essential to lignin degradation. Multiple studies have demonstrated experimentally, through either engineered disruption or constitutive expression of formaldehyde oxidation capacity in Bradyrhizobium diazoefficiens (Sudtachat et al., 2009), P. putida (Hibi et al., 2005), Burkholderia cepacia (Mitsui et al., 2003), and Corynebacterium glutamicum (Lessmeier et al., 2013), that demethoxylation of vanillate is dependent on the activity of a functional formaldehyde detoxification system, and formaldehyde removal may be the rate-limiting step to the degradation of lignin-derived methoxylated aromatics.
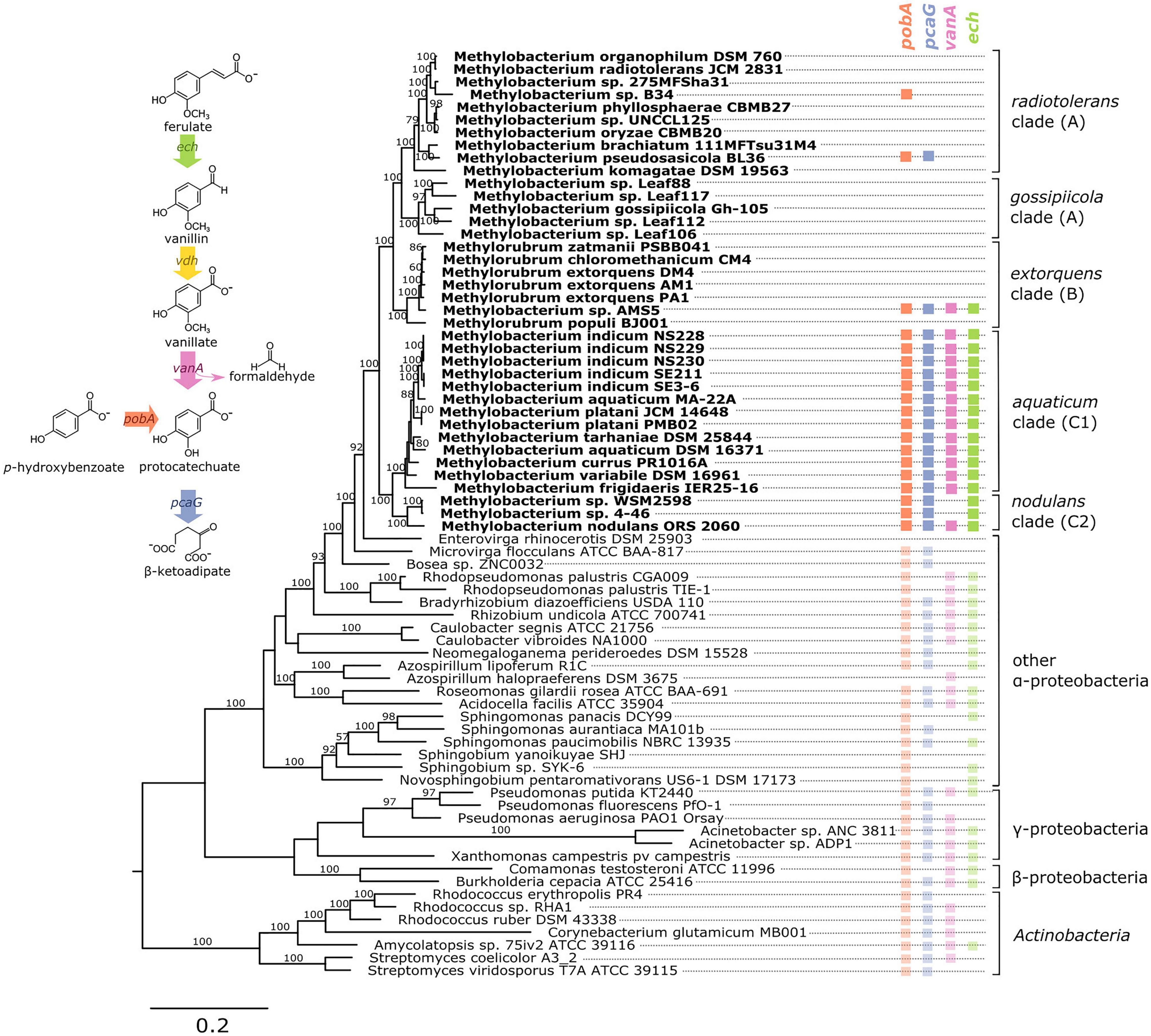
Figure 1. Genes associated with aromatic catabolism are present in two clusters of closely-related Methylobacterium species, with some exceptions. Genomes of Methylobacterium species and known aromatic-degrading bacteria were searched for four genes involved in different steps of the degradation of lignin-derived methoxylated aromatic compounds (upper left). Colored squares indicate the presence of each gene. Methylobacterium species are shown in bold text and colors, reference organisms (not Methylobacterium) in normal text and lighter colors. Note: among the reference organisms, all species that do not have pcaG do have the genes for protocatechuate 4,5-dioxygenase, indicating an alternative ring cleavage mechanism and different pathway for the catabolism of PCA. The phylogeny was composed using a concatenated alignment of four housekeeping genes (atpD, recA, rpob, and 16S rRNA), with Prochlorococcus marinus as an outgroup (not shown). Clade A/B/C designations are in accordance with (Green and Ardley, 2018; Alessa et al., 2021; Leducq et al., 2022). Branch labels indicate bootstrap percent; values < 50 are not shown. Sequence data are given in Supplementary Files 1–4. Due to space considerations, not all genomes searched are shown in this figure; the full set, with accession numbers, is given in Supplementary Table 1 (Methylobacterium) and 2 (reference species), and a phylogeny of all Methylobacterium species are shown in Supplementary Figure 2.
Formaldehyde detoxification pathways in lignin-degrading organisms are diverse, and in some organisms include more than one mechanism. In some cases formaldehyde is oxidized to CO2 via formate, such as in the thiol-dependent systems in C. glutamicum and Bradyrhizobium japonicum or the Zn-dependent aldehyde dehydrogenase in P. putida (Roca et al., 2009); in others it may be incorporated into biomass via the ribulose monophosphate (RuMP) pathway, as in B. cepacia (Kato et al., 2006). Furthermore, methanogens of the genus Methermicoccus are capable of using the methoxy groups of coal-derived aromatic compounds for the production of methane through a novel metabolic pathway (“methoxydotrophic methanogenesis”), a discovery that illuminated a previously-unknown link between C1 metabolism and the anaerobic degradation of recalcitrant aromatic compounds (Mayumi et al., 2016; Lloyd et al., 2021).
Methylotrophs, organisms that use reduced C1 compounds for growth, therefore make appealing candidates for efficient degradation of lignin-derived methyoxylated aromatics. Members of the pink-pigmented methylotrophic alpha-proteobacteria, and particularly the model organism Methylorubrum (formerly Methylobacterium) extorquens, have long been studied for their metabolism of simple C1 compounds such as methanol, formate, methylamine, and halogenated methanes (Chistoserdova, 2011). Formaldehyde is a central intermediate in the metabolism of many of these substrates, raising the possibility that, if these methylotrophs were capable of demethoxylating aromatic compounds, they could also use the resulting formaldehyde as a growth substrate (Lee et al., 2021). It has previously been documented that two members of the group are capable of growth on aromatic compounds (Ito and Iizuka, 1971; Jourand, 2004; Ventorino et al., 2014); however, the prevalence of this trait among Methylorubrum, Methylobacterium, and their close relatives in the Methylobacteriaceae, and the organization and evolutionary history of the genes involved, has not been described. And while formaldehyde metabolism has been extensively studied in the pink methylotrophs (Vorholt et al., 1998; Marx et al., 2003), aromatic compounds have traditionally not been discussed as a potential substrate for methylotrophic growth (Chistoserdova and Lidstrom, 2013; Kelly et al., 2014).
Here we report novel findings on the ecology and evolution of methoxydotrophic growth and catabolism of aromatic compounds by members of the Methylobacterium. We explored the genomic capacity of these species by searching published genomes, verified growth and formaldehyde production/consumption on aromatic compounds in the laboratory, and surveyed published metagenome data to assess the distribution and prevalence of aromatic-degrading Methylobacterium in the environment. For a thorough description of aerobic microbial degradation of lignin-derived aromatic compounds, we direct the reader to the excellent reviews published previously (Harwood and Parales, 1996; Masai et al., 2007); a simplified diagram of the pathway and the four genes of interest to this study (vanA, pcaG, pobA, and ech) is given in Figure 1. Because we were especially interested in the interaction between aromatic catabolism and methylotrophy, our metagenomic analysis focused especially on vanA (KO:K03862), the gene encoding the alpha subunit of vanillate monooxygenase.
Materials and Methods
Taxonomic Nomenclature
A recently proposed re-organization of the taxonomy of the pink-pigmented facultative methylotrophs suggested renaming the extorquens clade with the genus name “Methylorubrum” (Green and Ardley, 2018). However, others have pointed out that the remaining Methylobacterium clades are not monophyletic, leading to the suggestion that the entire group be returned to the name Methylobacterium, until more data is gathered to support splitting the genus (Hördt et al., 2020; Alessa et al., 2021). Therefore, in this manuscript, we will refer to the formally described Methylorubrum species using that name, but to describe the larger monophyletic group that is the focus of this study we will use the term Methylobacterium.
Phylogenetic Analysis
The genetic potential of Methylobacterium species (Kelly et al., 2014) (Supplementary Table 1) for the degradation of aromatic compounds was assessed through analysis of genomes available on the IMG/M database (Chen et al., 2019). We surveyed all the genomes that were classified in the genera Methylobacterium or Methylorubrum as of April 2020. Of the 134 genomes returned, 3 were omitted from analysis: Methylobacterium sp. CCH7-A2 is classified by the Genome Taxonomy Database (Parks et al., 2018) as a Bosea sp. and by IMG as forming a clique with Porphyrobacter donghaensis; Methylobacterium sp. ZNC0032 is also annotated as a Bosea sp. and does not cluster with the Methylobacterium (included in Figure 1 and Supplementary Table 1 but not in further analyses); and Methylobacterium platani JCM 14648 has 2 genome entries in IMG, of which we used the earlier, more complete assembly. We should note that we found that a genome currently named Streptomyces purpurogeneiscleroticus NRRL B-2952 is identified as Methylobacterium by GTDB and in IMG forms a clique with several Methylobacterium species, including Methylobacterium fujisawaense, Magnaporthe oryzae, and Methylobacterium phyllosphaerae; however, it does not carry the vanA gene and was not included in this analysis. Finally, it has been noted that the published genome of Methylobacterium organophilum used in this study may contain contaminants and a resequencing places it in closer relation to the extorquens (B) clade than shown here (Alessa et al., 2021), but it is not one of the vanA-carrying organisms of interest. In total we analyzed 131 genomes, described in Supplementary Table 1.
Genes of interest were identified by Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology ID: vanA (vanillate monooxygenase alpha subunit, KO:K03862); pcaG (protocatechuate 3,4-dioxygenase alpha subunit, KO:K00448); pobA (p-hydroxybenzoate 3-monooxygenase, KO:K00481); and ech (trans-feruloyl-CoA hydratase/vanillin synthase, KO:K18383). To identify ech genes, we also searched for IMG Term Object ID 8865, vanillin synthase, and we used IMG’s Conserved Neighborhood search tool to identify an ech homolog in that was syntenic across several Methylobacterium genomes but only sometimes annotated as Vanillin Synthase. Locus tags of all genes are given in Supplementary Table 1. Vanillin synthase, vdh (KO:K21802), was not studied because we were not easily able to identify homologs in all organisms. Known lignin-degrading organisms were added to the phylogenies as reference organisms, and Prochlorococcus marinus NATL2A and archaeal species were used as outgroups (Supplementary Table 2).
For the phylogeny in Figure 1, which shows all putative aromatic-degrading Methylobacterium strains in the context of selected other Methylobacterium and known lignin-degrading organisms, four gene alignments were concatenated: 16S rRNA (the RNA component of the small ribosomal subunit), rpoB (DNA-directed RNA polymerase subunit beta, KO:K03043), atpD (ATP Synthase F1 complex beta subunit, KO:K02112), and recA (recombination protein RecA, KO:K03553), all of which have been shown previously to be informative for phylogenetic analysis in Methylobacterium and closely related organisms (Gaunt et al., 2001; Menna et al., 2009; Zhang et al., 2012). Locus tags are given in Supplementary Table 1. The clade structure generally agrees with previous Methylobacterium phylogenies based on 16S rRNA, rpoB, or core genes, though some analyses find clade A not to be monophyletic (Green and Ardley, 2018; Alessa et al., 2021; Leducq et al., 2022).
Two additional phylogenies were constructed as Supplementary Information: one containing all the Methylobacterium strains surveyed for this study, using the rpoB gene (Leducq et al., 2022) with B. diazoefficiens USDA 110 as an outgroup; and one showing the relationships of the Rieske [2Fe-2S] oxygenase homologs. The rpoB tree includes five of the Methylobacterium strains carrying aromatic catabolism genes (sp. MIMD6, sp. 174MFSha1-1, sp. AM11, sp. YR596, and sp. CCH7-A2) for which the full-length 16S rRNA gene sequence was not available and therefore are not included in Figure 1. For 3 of the Methylobacterium strains in our study (Methylobacterium sp. CG08_land_8_20_14_0_20_71_15, M. phyllosphaerae JCM 16408, and M. thiocyanatum JCM 10893), no rpoB sequences were available and aromatic catabolism genes were also not detected; these strains do not appear in any of the phylogenies but are listed in Supplementary Table 1. The homolog tree was constructed using all the genes annotated as COG 4638 in IMG in the surveyed Methylobacterium strains, as well as vanA sequences from other species for reference.
All gene sequences were aligned using MUSCLE v. 3.7 (Edgar, 2004) (RRID:SCR_011812) on the CIPRES Science Gateway (Miller et al., 2010), using the following parameters at all iterations: ClustalW as the sequence weighting scheme, UPGMB clustering, SUEFF = 0.1, pseudo rooting, and sum-of-pairs scoring for refinement. Phylogenetic trees were generated using RAxML v. 8.2.12 (Stamatakis, 2014) (RRID:SCR_006086) on CIPRES using the RAxML-HPC2 on XSEDE tool, with the following parameters: 25 rate categories, rapid bootstrapping with GTRCAT model for 100 iterations, random seed = 12345 for bootstrapping and parsimony, and outgroups as given in Supplementary Table 2 (outgroups are not shown in figures). All alignments are provided in Supplementary Files 1–10.
Genomic Context of Aromatic Catabolism Genes
We used GeneHood v.0.15.0-0 (Ortega, 2020) to assess synteny in the gene neighborhoods surrounding the vanA and pcaH genes in Methylobacterium species. For each gene of interest, the database locus tag from the MIST 3.0 database (Gumerov et al., 2020) was provided and up to 15 genes upstream and downstream of the gene were plotted (in genomes that are not fully assembled, some scaffolds did not have 15 genes on either side of the target gene). Phylogenetic trees, gene annotations, and color-coding were added manually using Inkscape v. 0.91 (Inkscape Team, 2003) (RRID:SCR_014479).
GC content in the Methylobacterium sp. AMS5 genome was calculated in R v.4.0.2 (R Core Team, 2018) (RRID:SCR_001905) using the alphabetFrequency function from Biostrings v.2.46.0 (Pagès et al., 2019) (RRID:SCR_016949); for plotting, a sliding window of 5 kb was used for the full genome and 500 bp for the catabolic island region. Genome signature difference was calculated using a custom script in R, using the formula for the δ* difference (Karlin, 2001) with tetranucleotides rather than dinucleotides.
Growth Assays on Aromatic Substrates
All organisms were grown on MPIPES medium (Delaney et al., 2013) with the addition of 1× Wolfe’s Vitamins (Atlas, 2010), 25 μM LaCl3 [as has been found to facilitate growth in some Methylobacterium spp. (Skovran and Martinez-Gomez, 2015)], and carbon substrates as specified. All conditions contained 16 mM of carbon [16 mM methanol, 4 mM succinate, 2.67 mM glucose, 2.29 mM benzoate, 2.29 mM p-hydroxybenzoate (PHBA), 2.29 mM protocatechuic acid (PCA), 2 mM vanillate, 1.6 mM ferulate, or 0.4 g/L Kraft lignin]. While some soil microenvironments may experience higher substrate concentrations and some lower, 16 mM of carbon is in the range that has been commonly used for M. extorquens to ensure that final population size scales linearly with substrate concentration for both single- and multi-carbon substrates (Lee et al., 2009). Methylobacterium sp. 4–46 was also tested on methanol without LaCl3 to assess the effect of lanthanum on methanol growth. For each assay, one colony was inoculated from a culture plate into 5 mL MPIPES with 3.5 mM disodium succinate and grown 24 h to obtain a stationary-phase culture, then diluted 1:64 (v/v) in the same medium and grown again until stationary. Using that inoculum, the growth experiment was conducted as follows: each replicate was diluted 1:64 into MPIPES and aliquoted into several wells of a Costar 48-well tissue culture-treated plate (product #3548, Corning Inc., Corning, NY, United States) for substrates other than methanol, or into Balch-type glass culture tubes for methanol, along with 16 mM carbon substrate. Total volume was 640 μL per well in culture plates or 5 mL per tube in Balch tubes. Each strain was tested in biological triplicate (3 separate colonies). Balch tubes were sealed with butyl rubber stoppers to prevent escape of methanol.
Plates were incubated at 30°C in a LPX44 Plate Hotel (LiCONiC, Woburn, MA, United States) with shaking at 250 RPM. Optical density was assessed using a Wallac 1420 Victor2 Microplate Reader (PerkinElmer, Waltham, MA, United States), reading OD600 for 0.4 s at intervals of between 2 and 5 h. Culture tubes were incubated in a TC-7 tissue culture roller drum (New Brunswick Scientific, Edison, NJ, United States) at a speed setting of 7, and OD600 was measured in the culture tubes using a Spectronic 200 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). Outputs from the Wallac 1420 software were collated using Curve Fitter (Delaney et al., 2013); data cleaning and plotting were then conducted in R.
Formaldehyde Production During Growth on Vanillate
Growth of Methylobacterium strains on vanillate was initiated with stationary-phase MPIPES-succinate cultures as described above; these were diluted 1:64 (v/v) into 5 mL MPIPES with 2 mM vanillate and grown 2 days until stationary phase to allow cultures to acclimate to the substrate. This inoculum was diluted 1:32 into culture flasks containing 20 mL of MPIPES with 2 mM vanillate and incubated, shaking, at 30°C. Cultures were sampled every 4 h: OD600 was measured in a SmartSpec Plus Spectrophotometer (Bio-Rad Laboratories, Hercules, CA, United States), and the supernatant of 500 μL of centrifuged culture was used for the measurement of formaldehyde using the colorimetric assay described by Nash (1953). All strains were tested in biological triplicate. Although the active component in the Nash assay used has been found to react to several multi-carbon aldehydes, we have no reason to believe these compounds could be produced by vanillate metabolism, given what is known about the pathway; moreover, reactions with all compounds are slower than the reaction with formaldehyde (Compton and Purdy, 1980).
Formaldehyde production of non-methylotrophs during vanillate growth was assayed similarly. Stationary-phase vanillate-grown cultures of P. putida KT2440 and Rhodococcus jostii RHA1 [USDA Agricultural Research Service Culture Collection (NRRL)] were diluted 1:64 (v/v) into 5 mL MPIPES medium in culture tubes, with sampling every 2 h. Vanillate was provided at 4 mM; although this was greater than in the Methylobacterium experiment, we have found that vanillate concentration has little effect on the generation of formaldehyde in the medium (Lee et al., 2021).
Analysis of Methylobacterium vanA Genes in Published Metagenomes
To assess the abundance of Methylobacterium-encoded vanA genes in the environment, we searched all published metagenomes available through the IMG/M portal on December 20, 2017. The “Function Search” tool was used to identify all metagenomes containing genes annotated with KO:K03862 (vanA). To increase our chances of observing meaningful ecological patterns, we restricted our analysis to metagenomes containing >100 vanA genes. Genes on scaffolds with a phylogenetic lineage assignment of Methylobacterium were designated “Methylobacterium vanA.” and the Ecosystem Type field in the sample metadata was summarized into a “Sample Type” designation for analysis (e.g., Phyllosphere and Phylloplane were classified as “Leaf,” Rhizosphere and Rhizoplane as “Root,” etc.).
To provide a tree-based assessment of vanA diversity in addition to the USEARCH-based IMG/M phylogenetic lineage assignment (Huntemann et al., 2016), we downloaded the Methylobacterium vanA gene sequences and placed them in the reference vanA phylogeny (described above). pplacer v.1.1 (Matsen et al., 2010) (RRID:SCR_004737) was used with HMMer v.3.1b.2 (Eddy, 2007) (RRID:SCR_005305), RAxML v.8.2 (Stamatakis, 2014), and taxtastic v.0.8.8 for Python 3 (RRID:SCR_008394). The resulting tree was paired with metadata from sequence scaffolds in Phyloseq v.1.22.3 for R (McMurdie and Holmes, 2013) (RRID:SCR_013080) for the final phylogeny. Using pplacer, we found that only 182 out of 348 genes actually fell within the Methylobacterium clades, whereas 31 clustered more closely with Azospirillum halopraeferens, and the remaining 135 were distributed among more distantly related organisms (Supplementary Figure 1). This may be due in part to the overall complexity of the Rieske-type monooxygenase family (Özgen and Schmidt, 2019). We therefore took the conservative approach of focusing only on the genes identified by both IMG and pplacer as Methylobacterium for the remainder of our analyses.
A separate analysis was carried out to assess the relative abundances of all Methylobacterium reads within each metagenome in the JGI IMG database. The “Phylogenetic Distribution: Genomes v. Metagenomes” tool was used; this tool conducts a BLAST search to find reads in a set of metagenomes that have similarity to a query genome (JGI IMG, 2017). 18 query genomes were used: Methylobacterium radiotolerans JCM 2831, Methylobacterium pseudosasiacola BL36, M. extorquens PA1, Methylobacterium sp. AMS5, Methylobacterium sp. Leaf88, Methylobacterium gosipiicola Gh-105, Methylobacterium indicum SE3.6, M. indicum SE2.11, Methylobacterium aquaticum MA-22A, M. platani JCM 14648, Methylobacterium variabile 16961, Methylobacterium tarhaniae DSM 25844, M. aquaticum DSM 16371, Methylobacterium sp. WSM2598, Methylobacterium sp. 4–46, M. nodulans ORS 2060, Microvirga flocculans ATCC BAA-817, and Enterovirga rhinocerotis DSM 25903 (genome accessions in Supplementary Table 1). A total of 33,967 metagenomes were searched; this search set comprised all metagenomes published in IMG as of January 10, 2021 that were returned by the following 6 search terms:
• GOLD Ecosystem = Host-Associated and GOLD Ecosystem Category = Plant
• GOLD Ecosystem = Host-Associated and GOLD Ecosystem Category = Human
• GOLD Ecosystem = Host-Associated and GOLD Ecosystem Category = Mammals
• GOLD Ecosystem = Environmental and GOLD Ecosystem Category = Terrestrial
• GOLD Ecosystem = Environmental and GOLD Ecosystem Category = Aquatic
• GOLD Ecosystem = Engineered
Metagenomes with a published gene count of 0 were excluded from analysis, but otherwise no attempt was made to sample evenly across ecosystems, or to screen metagenomes by size, assembly status, etc., as the goal was to be as complete as possible in the search of available data. Due to computational constraints of IMG, metagenomes were searched in groups of 5,000 or fewer and 1 genome at a time, and results were combined. The output of each search consisted of a data table including identity (and metadata) of each metagenome sample where the genome was found, and the number of reads that hit to the genome at the 90% similarity level.
Results
Genetic Capacity for Metabolism of Methoxylated Aromatic Compounds Is Present Primarily in Two Clades of Methylobacterium
To investigate the possibility of aromatic catabolism among Methylobacterium strains, we first searched 131 published genomes for genes encoding four key enzymes known to be involved in the degradation pathway of methoxylated aromatic compounds: vanA (vanillate monooxygenase alpha subunit, KO:K03862); pcaG (protocatechuate 3,4-dioxygenase alpha subunit, KO:K00448); pobA (p-hydroxybenzoate 3-monooxygenase, KO:K00481); and ech (trans-feruloyl-CoA hydratase/vanillin synthase, KO:K18383; we searched also for IMG Term Object ID 8865, vanillin synthase, and used genome synteny; see Section “Material and Methods”). We found that all four genes are indeed present within the genus Methylobacterium. They are primarily limited to two closely related clades, the aquaticum clade (C1) and the nodulans clade (C2); furthermore, they are present in nearly all the genomes we surveyed in these clades (Figure 1, Supplementary Table 1, and Supplementary Figure 2). However, there are exceptions. Of the genomes searched, Methylobacterium sp. AMS5 in the extorquens clade (clade B) and M. pseudosasiacola in the radiotolerans clade (clade A) also carry some aromatic catabolism genes (Figure 1).
Two members of clade C2 (sp. 4–46 and sp. WSM2598) appear to lack vanA but carry ech (Figure 1). It is possible that the ech homologs annotated as trans-feruloyl-CoA hydratase/vanillin synthase in fact carry out a different reaction in these organisms (Lohans et al., 2017); however, they are located adjacent other genes annotated as vanillin dehydrogenase (see below), which catalyzes the conversion of vanillin to vanillate. This suggests that Methylobacterium sp. 4–46 and sp. WSM2598 may have previously had the capacity to utilize the product of these reactions but have since lost vanA. Among the closest sister taxa to Methylobacterium, E. rhinocerotis has none of the four genes of interest, and M. flocculans has only pobA and pcaH.
While the absence of a gene in a draft-status genome is not always an indication that the gene is not there, several of the genomes without aromatic catabolism genes, including M. sp. 4–46 and M. extorquens AM1 and PA1, are complete (Vuilleumier et al., 2009; Marx et al., 2012) (Supplementary Table 1). The substantial number of genomes we observed without any evidence of aromatic catabolism genes suggested a broad phylogenetic pattern: the capacity to metabolize several lignin-derived aromatic compounds was likely acquired by clade C early in its divergence within the Methylobacterium, but members of other clades have acquired the genes more recently by horizontal gene transfer. We sought further support for this evolutionary hypothesis by analyzing the phylogenies of the genes themselves, examining the genomic contexts of vanA and pcaG, and assaying the ability of several strains to grow on methoxylated aromatic compounds in the lab.
Phylogeny of Aromatic Catabolism Genes Supports Ancestral Origins in M. aquaticum and M. nodulans Clades and Horizontal Acquisition by Two Other Species
The phylogenies of pobA, pcaG, and vanA were largely congruent with the phylogeny of conserved housekeeping genes within the Methylobacterium but not between Methylobacterium and their closest relatives, supporting the hypothesis that the four genes are ancestral to clade C but were not necessarily acquired together (Figure 2). Phylogeny also supported our hypothesis of the function of the vanA gene in Methylobacterium species: although vanA homologs, members of the Rieske [2Fe-2S] oxygenases, are known to carry out a variety of reactions (Kweon et al., 2008; Özgen and Schmidt, 2019), and Methylobacterium species have several such genes, only the genes annotated as vanA cluster with genes that have been characterized as vanillate monooxygenases in known lignin-degrading bacteria (Supplementary Figure 3). The ech phylogeny was the most difficult to interpret, as the Methylobacterium separated into three clades (Figure 2). It is possible that not all gene homologs in the phylogeny code for enzymes specific to ferulate; further biochemical and genetic work will be necessary to be certain. Notably, Methylobacterium sp. AMS5 and M. pseudosasiacola were more closely related to each other than to any other species in both their pobA and pcaG sequences, despite their phylogenetic distance from each other at the genome level. And while the pobA sequences of those two strains fell within those of other Methylobacterium, their pcaG sequences clustered with the Sphingomonas, further support not only for the hypothesis that these two disparate species both acquired the beta-ketoadipate pathway via horizontal gene transfer, but that they may have acquired it from the same donor or that one served as the donor to the other.
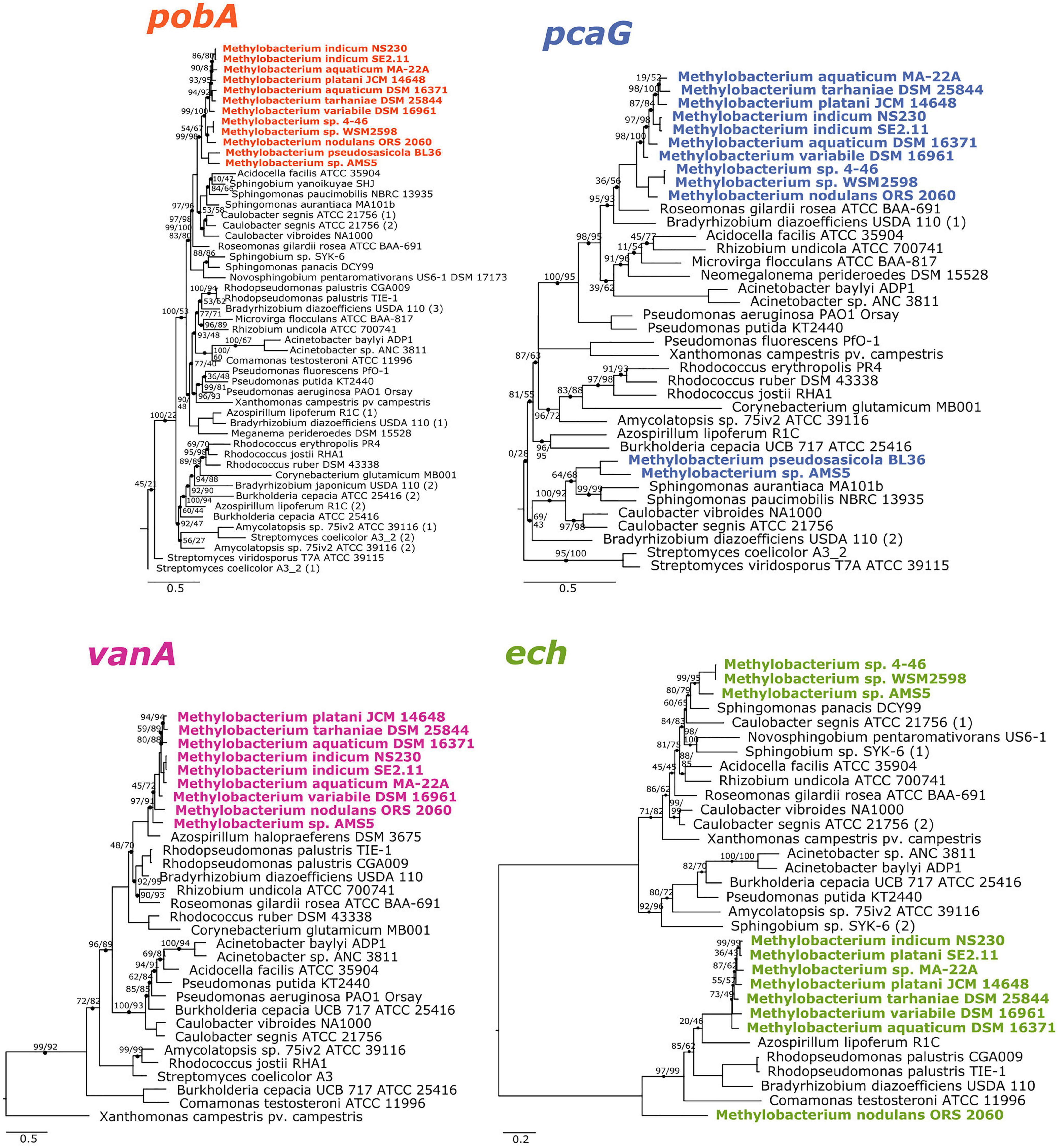
Figure 2. Phylogenies of aromatic catabolism genes suggest that all four are ancestral to the M. aquaticum/nodulans clades and have been horizontally acquired by two other Methylobacterium species. For all organisms in Figure 1, the sequences of pobA (p-hydroxybenzoate 3-monooxygenase), pcaH (protocatechuate 3,4-dioxygenase beta subunit), vanA (vanillate monooxygenase subunit A), and ech (trans-feruloyl-CoA hydratase/vanillin synthase) were aligned and phylogenies constructed using maximum likelihood, with homologs from archaeal organisms as outgroups (not shown). Accession numbers for all genes are given in Supplementary Tables 1, 2. Branch labels indicate bootstrap support. Comparison of these phylogenies and that in Figure 1 suggest that all four genes are restricted to and prevalent in the M. aquaticum and M. nodulans clades, with a few losses (vanA by Methylobacterium sp. 4–46 and WSM2598) and some gains by horizontal gene transfer (in M. pseudosasiacola and Methylobacterium sp. AMS-5).
Gene Synteny in Aromatic Catabolism Gene Neighborhoods Is Consistent With Phylogeny
We further examined the relationships of vanA and pcaG across species by comparing patterns of synteny within the genome neighborhood for each (Figure 3). In all strains with pcaG, the gene was located within a cluster of beta-ketoadipate pathway genes, and five of these genes were present in all strains in the same order. Strikingly, we found no genes annotated as pcaI and pcaJ (beta-ketoadipate:succinyl-CoA transferase) or pcaF (encoding beta-ketoadipyl-CoA thiolase) within these operons, which are necessary for the final steps of the beta-ketoadipate pathway (Harwood and Parales, 1996; Buchan et al., 2004), leaving it initially unclear whether and how these organisms might be able to metabolize vanillic acid and related compounds. However, almost all strains contained an additional gene between pcaG and pcaB, annotated as an uncharacterized conserved protein DUF849 (COG3246, beta-keto acid cleavage enzyme). In all Methylobacterium strains where vanA was found, it was accompanied by the other two genes required to confer the ability for the demethoxylation of vanillate: vanB (encoding the other subunit of vanillate monooxygenase) and vanR (an AraC family transcriptional regulator). Some strains also had genes encoding a Major Facilitator Superfamily transporter specific to protocatechuate (pcaK) or vanillate (vanK) (Figure 3).

Figure 3. Genomic context of aromatic catabolism genes in Methylobacterium species supports a common evolutionary origin within the M. aquaticum/nodulans clades and separate origin in M. pseudosasiacola and Methylobacterium sp. AMS-5. Genomic regions (from fully assembled genomes) or scaffolds (from partially assembled genomes) showing the neighborhoods of pcaG and vanA were aligned at the genes of interest. Gene phylogenies are excerpted from Figure 2. Genes of interest and their operons are shown in white with gene names labeled. Within each of the two alignments, homologous regions shared across multiple genomes are shown in the same color to facilitate comparison among genomes. In most cases, members of the M. aquaticum clade share some synteny in the regions surrounding the van and pca gene clusters, with the degree of synteny roughly paralleling their phylogenetic distance. Exceptions indicate likely cases of horizontal gene transfer, in agreement with figure.
The genomic contexts of pcaG and vanA (Figure 3), as well as pobA and ech (Supplementary Figure 4), exhibited patterns that agreed overall with the phylogenies described above. Closely related strains shared common genes in the regions near the genes of interest, with differences among the strains increasing with increasing distance from the genes of interest. Also consistent with phylogeny, Methylobacterium sp. AMS5 and M. pseudosasiacola shared no commonalities with members of clade C or with each other in the neighborhoods surrounding any of the genes of interest. In addition, these were the only two organisms in which pobA and pobR were located adjacent to the pca gene cluster; this contrasts with their arrangement in the other Methylobacterium and in most previously described organisms, where the pca genes are not co-located with any of the other genes of interest in this study (Supplementary Tables 1, 2). However, PCA, the reaction product of the enzyme encoded by pobA, is the substrate for the gene encoded by pcaGH; the proximity of the genes therefore suggests that if these two organisms gained the pca genes via horizontal transfer, the pob genes may have been transferred simultaneously as part of a catabolic island. We found further support for the catabolic island hypothesis in the proximity of vanA and ech to the pca genes in Methylobacterium sp. AMS5.
Methylobacterium sp. AMS5 Carries a Catabolic Island Conferring the Ability to Degrade Lignin-Derived Aromatic Compounds, in an M. extorquens-Like Genome
The co-localization of all four aromatic catabolism genes of interest in the genome of Methylobacterium sp. AMS5 prompted us to examine the region more closely. AMS5 was originally isolated in 2011 from the stem of a hypernodulating strain of soybean and, like other organisms in the group not formally described, is currently named as a Methylobacterium (Anda et al., 2011), but is very closely related to the model organism M. extorquens PA1 and therefore likely to belong to the Methylorubrum group (Figure 1). We aligned the two genomes, as well as that of another clade member, Methylorubrum zatmanii PSBB041, at the region where the aromatic catabolism genes are located (Figure 4). The comparison revealed that the region appears to be prone to the insertion and excision of mobile genetic elements.
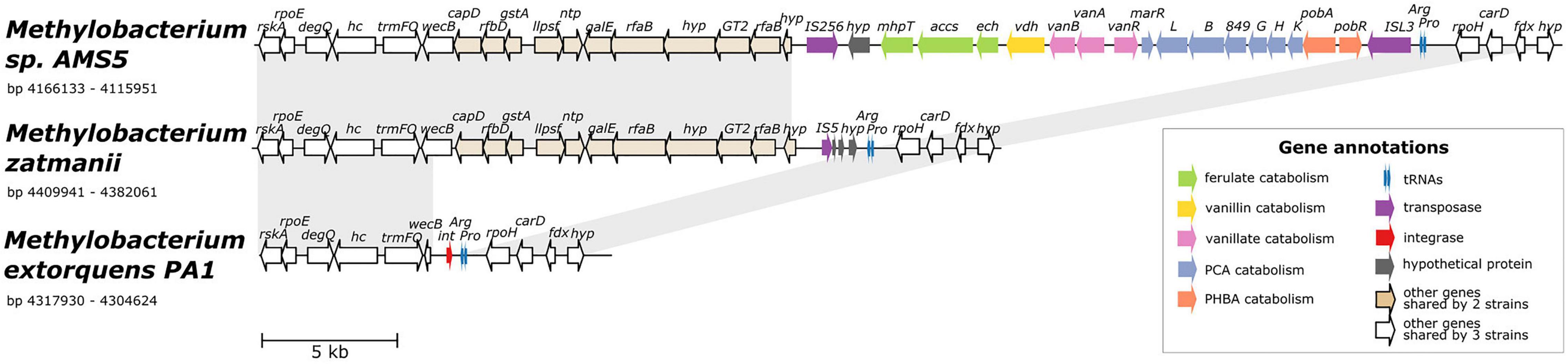
Figure 4. Alignment of three genomes from the extorquens clade reveals that Methylobacterium sp. AMS-5 harbors a catabolic island conferring the ability to degrade several aromatic substrates, in a genomic region prone to insertions and deletions. The genome of Methylobacterium sp. AMS5 is shown aligned with that of two close relatives, M. zatmanii PSBB041 and M. extorquens PA1. Genes are color-coded to indicate either function or commonality between genomes, as indicated by the key. Light gray shading between pairs of species connects regions shared by both. See Supplementary Table 3 for full annotations corresponding to the short gene codes.
At the locus where M. zatmanii carries a gene encoding a putative IS5-type transposase (possibly non-functional due to a frameshift with an internal stop codon), AMS5 contained an additional 22-kb region: a catabolic island containing genes that appear to confer the full ability to degrade ferulate, vanillate, vanillin, p-hydroxybenzoate, and protocatechuate, flanked by two different transposase genes from the MULE superfamily. On the 3′ side of this region, the two strains share 11 genes (14 kb), many related to nucleotide sugar metabolism, that are also not present in PA1 (Figure 4), or in any of the other Methylobacterium species surveyed in this study. The corresponding locus in the PA1 genome carries only a putative site-specific integrase/recombinase, and this gene is present only in members of the extorquens clade (clade B). The gene adjacent to the integrase encodes UDP-N-acetylglucosamine 2-epimerase (wecB); the sequence appears to be complete in M. zatmanii (1,137 bp) but truncated in PA1 (missing 912 bp from the 3′ end). In all three genomes, the region of variability lies immediately at the 3′ end of the genes for tRNA-Pro and tRNA-Arg. Due to the extensive genome rearrangement that is common within the Methylobacterium (Vuilleumier et al., 2009; Lee and Marx, 2012), we were unable to identify a syntenic region in species of clades A or C for comparison.
The presence of the truncated wecB gene in PA1 suggests that the region of sugar metabolism genes may originally have been present but were excised, whereas the arrangement of genes in the catabolic island suggest that the island was acquired into a M. zatmanii-like genomic background. The origins of the catabolic island itself are uncertain; as shown above (Figure 2), there appear to be diverse phylogenetic origins represented among the different genes within the pathway, and BLAST search for the entire region in the NCBI Nucleotide database found no other organisms outside of the Methylobacterium containing all genes in the same order. We could detect no genes in the region relating to replication or conjugation as might be expected in an integrative and conjugative element (ICE) (Wozniak and Waldor, 2010). Furthermore, no significant difference in tetranucleotide composition or in GC content was found between the inserted region (GC% = 71.4) and the full genome (GC% = 71.1) (Supplementary Figure 5), indicating that the catabolic island was likely either transferred from an unknown closely-related organism, or has been present in the genome long enough for amelioration (Lawrence and Ochman, 1997).
Genome Content Predicts Ability to Grow on Aromatic Compounds in Most Methylobacterium Strains
We next wanted to assess whether Methylobacterium strains carrying genes for the catabolism of methoxylated aromatic compounds could indeed use those compounds as growth substrates. We chose 8 species from the M. aquaticum/nodulans clades, shown in Figure 5 (and Supplementary Table 1). All of these strains carry all four genes of interest except Methylobacterium sp. 4–46, which lacks vanA and so was predicted not to grow on vanillate or ferulate (Figure 1). We conducted a series of growth experiments on defined mineral medium with substrates of each of the four gene products of interest, as well as several other substrates for context: methanol (a simple C1 substrate), succinate (a known growth substrate for all the strains tested), glucose (used by only some Methylobacterium species), benzoic acid [another aromatic acid with a degradation pathway separate from the beta-ketoadipate pathway (Moreno and Rojo, 2008)], and Kraft lignin (a soluble form of polymeric lignin). For most strains, growth on PHBA, PCA, vanillate, and ferulate was as expected for all strains given their genome content: that is, organisms with pobA grew on PHBA, those with pcaG grew on PCA, those with vanA grew on vanillate, and those with ech grew on ferulate.
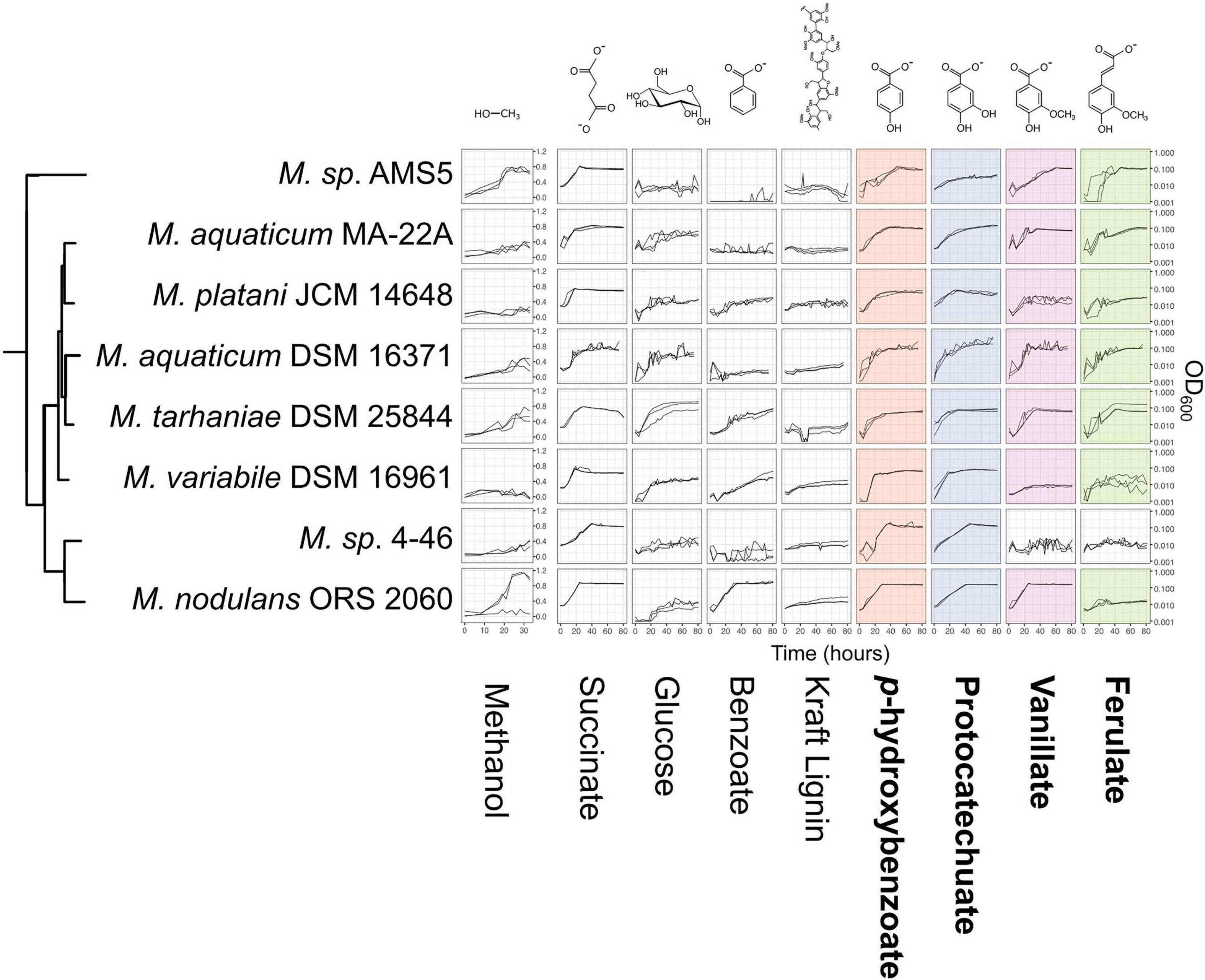
Figure 5. Growth of Methylobacterium species on aromatic compounds is predicted by genome content in most cases. Methylobacterium species were grown in defined mineral medium with a single carbon substrate; all conditions contained 16 mM of carbon. Substrates corresponding to the four degradation pathways featured in this study are outlined labeled in bold type, and presence of the corresponding genes in each strain’s genome is indicated by colored shading (as in Figure 1). Lines show growth curves from three biological replicates. Note that the y-axis for methanol is on a different scale, as these cultures were grown in different vessels. There were only 3 cases (out of 32) in which organisms had the genes associated with degradation of a given substrate but showed little or no growth. There were no cases in which organisms grew on substrates for which they did not have the associated genes. Source data are available in Supplementary Tables 4, 5.
There were three exceptions: M. variabile did not grow on vanillate or ferulate, and M. nodulans did not grow on ferulate. Furthermore, M variabile growth on vanillate was only modest (Figure 5 and Supplementary Tables 3, 4). However, because a number of factors may lead to lack of growth, further tests in different culture conditions would be necessary to further explore these strains’ ability to grow on these substrates. For example, all strains grew on succinate, indicating that the cultures were viable and capable of growth on the medium provided; however, M. variabile and M. platani did not show substantial growth on methanol within 30 h, possibly indicating that conditions were not optimal for methylotrophic growth, or simply that a longer incubation time was needed. Growth on benzoate was observed for M. platani, M. tarhaniae, M. variabile, and M. nodulans but not for other species, and growth on Kraft lignin was negligible for all species in the 80 h tested.
Non-methylotrophs Excrete Formaldehyde During Vanillate Growth, Whereas Methylobacterium Do Not
We were especially interested to ask whether methylotrophs are different from non-methylotrophs in their ability to cope with the formaldehyde produced during vanillate metabolism. As mentioned above, it has been recognized by multiple researchers that the formaldehyde released by vanillate monooxygenase is a burden to organisms growing on vanillate, and we have previously observed formaldehyde accumulation during vanillate utilization by P. putida KT2440 (Lee et al., 2021). We therefore measured formaldehyde in the medium during vanillate metabolism by P. putida and Rhodococcus jostii RHA1, two well-studied lignin-degrading bacteria that, unlike Methylobacterium, do not utilize C1 compounds for growth (Hibi et al., 2005; Ahmad et al., 2011; Sainsbury et al., 2013; Otani et al., 2014; Ravi et al., 2017). We assayed each strain on vanillate as well as on PCA as a control compound, as PCA is the direct product of vanillate demethylation and therefore involves the same metabolism except for the effect of formaldehyde. In both strains, when growth occurred on vanillate, formaldehyde accumulated in the medium concomitant with the increase in optical density, peaking at concentrations of 0.94 mM (Figure 6A and Supplementary Table 5). When cultures entered stationary phase (approximately 10 h for P. putida, 15 h for R. jostii), formaldehyde concentrations began to decrease, ultimately returning to below the detection limit, as might be expected from known formaldehyde detoxification pathways. No formaldehyde was detected at any time during growth on PCA. Moreover, growth on PCA was faster: stationary phase was reached at 7 h for P. putida and 10 h for R. jostii. These results suggest that while both P. putida and R. jostii can oxidize formaldehyde, degradation is slower than production.
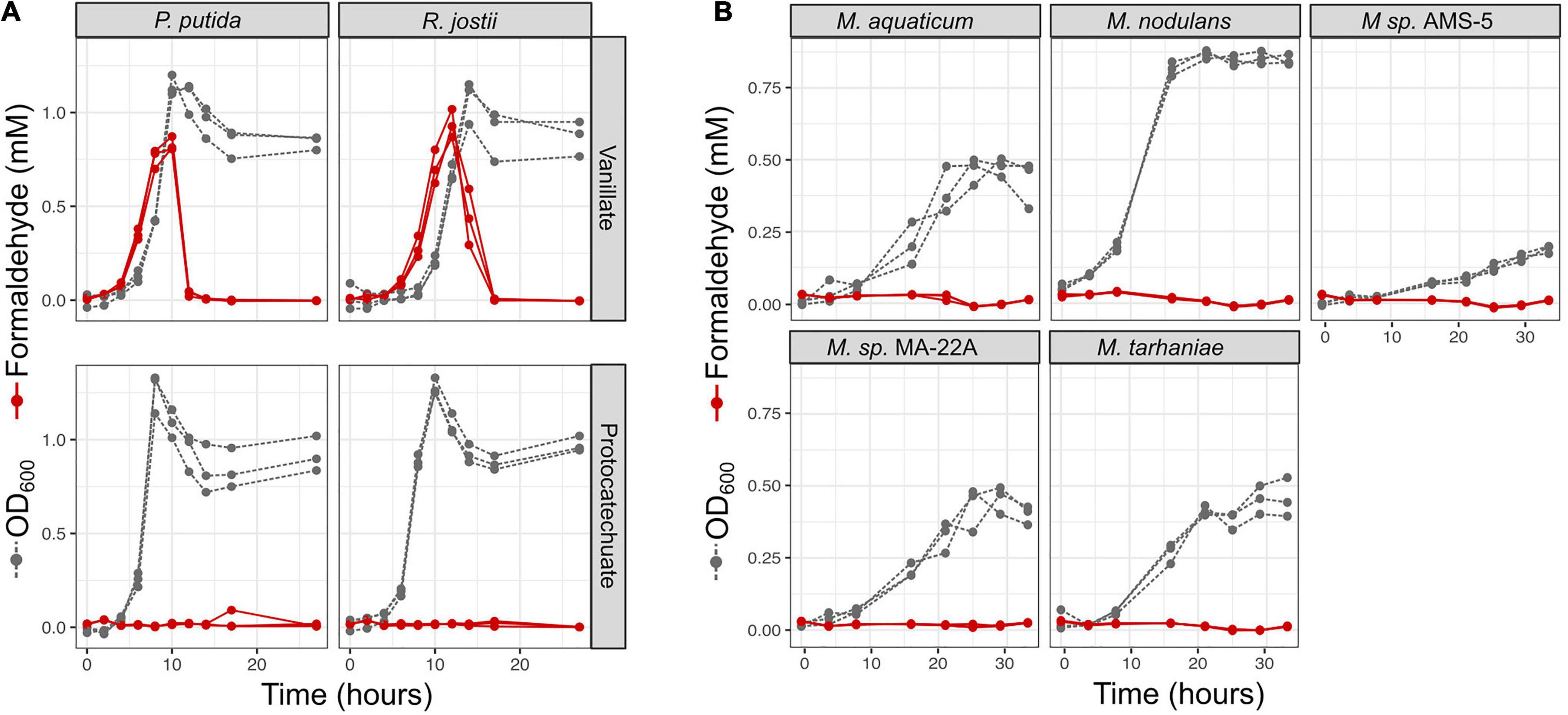
Figure 6. Non-methylotrophic lignin degraders Pseudomonas putida and Rhodococcus jostii produce formaldehyde when growing on methoxy-substituted aromatic compounds such as vanillate; Methylobacterium do not. All organisms were grown in defined mineral medium with vanillate or protocatechuate (PCA) as a sole carbon source; growth was assayed by optical density at 600 nm (gray symbols and dashed lines) and formaldehyde in the medium was measured by a colorimetric assay (red symbols and solid lines). Each line represents one biological replicate. (A) P. putida and R. jostii accumulate formaldehyde transiently in the medium when growing on vanillate, but not on the non-methoxylated compound PCA, and growth is slower on vanillate. (B) All Methylobacterium species tested grew on vanillate without producing measurable formaldehyde. Source data are available in Supplementary Table 6.
We conducted a similar experiment on the five strains of Methylobacterium showing the greatest growth on vanillate in our laboratory conditions: M. aquaticum, M. nodulans, M. tarhaniae, Methylobacterium sp. AMS5, and M. aquaticum MA-22A. We incubated all cultures with vanillate for 33 h, long enough for AMS5 to show marked growth and all other species to consume all the substrate and reach stationary phase. No detectable formaldehyde was produced at any time (Figure 6B and Supplementary Table 5). These results suggest that Methylobacterium species are able to consume the formaldehyde generated from vanillate demethylation as rapidly as it is produced, likely via the same pathways by which the formaldehyde generated from methanol is used for energy generation and biosynthesis (Marx et al., 2005; Crowther et al., 2008).
Methylobacterium-Derived vanA Reads in Published Metagenomes Are Predominantly From the M. nodulans Cluster, and Most Numerous in Soil
Aside from the environments in which they were isolated, there exists scant information on the ecological niches of the aromatic-degrading Methylobacterium clades—for instance, whether they are more likely than the other Methylobacterium to inhabit ecosystems rich in lignin, such as soil or the rhizosphere. We therefore sought to learn more about the prevalence and abundance of vanA-carrying Methylobacterium species in the environment by querying the publicly available metagenome datasets on the JGI IMG/M database. The distinctiveness of the Methylobacterium genes within the phylogeny of known vanA sequences (Figure 2) makes it possible to deduce phylogeny from DNA sequence and vanA is present as a single copy in most genomes in which it is found.
Our study set comprised 1,651 metagenomes, with a total assembled gene count of 5.60 × 109; we retrieved 317,816 scaffolds carrying vanA, of which 348 had Methylobacterium or Methylorubrum as their IMG Phylogenetic Lineage Assignment (a frequency of 0.11%) (Supplementary Tables 6–8). We then used pplacer (Matsen et al., 2010) (RRID:SCR_004737) for phylogenetic placement of those reads onto a reference tree of vanA genes from known Methylobacterium, and found that 182 of them actually clustered with the Methylobacterium; the others fell outside the Methylobacterium and were therefore omitted from further study (Supplementary Figure 1). Of those 182 Methylobacterium vanA, 114 (63%) clustered with the M. nodulans vanA gene; 31 genes (17%) with Methylobacterium sp. AMS5, and 37 genes (20%) fell into the M. aquaticum-like clade (Figure 7). Overall, they were most commonly found in samples associated with soil (43%) and roots (40%); only 10% were found in leaf samples.
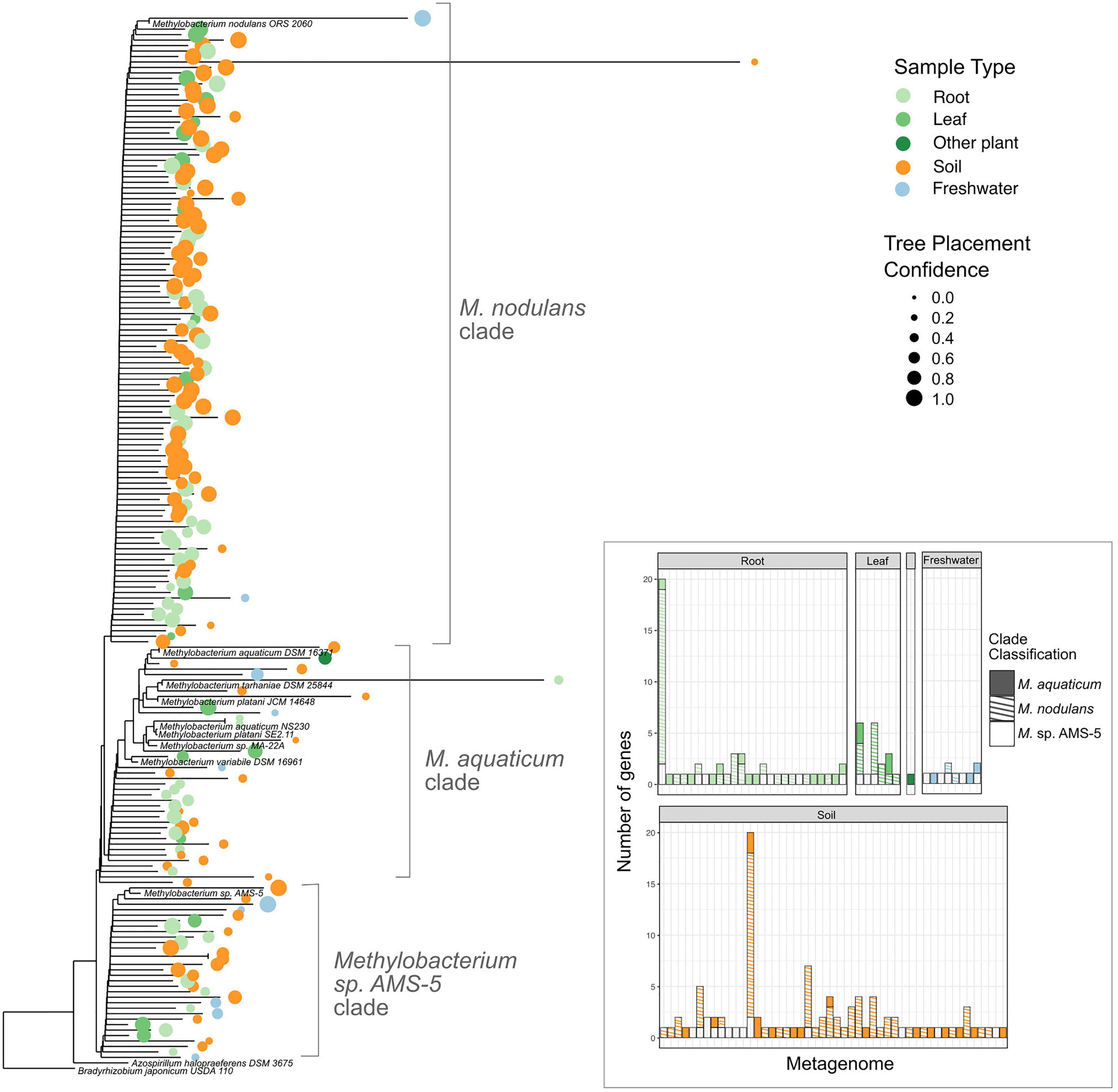
Figure 7. Among Methylobacterium vanA sequences in published metagenomes, M. nodulans-like sequences are the most abundant. Sequences of 182 Methylobacterium-associated vanA gene fragments found in published metagenomes were added using pplacer to a phylogeny of full-length vanA genes sequenced from reference genomes. 166 genes that were classified by IMG as Methylobacterium but clustered outside of the genus are not shown. Colored dots indicate the ecosystem type from which the metagenome sample originated, with size scaled to the likelihood weight ratio of the pplacer classification as a measure of confidence in the placement. Inset bar plot: abundance of Methylobacterium vanA genes, shaded by clade and colored by sample type, in each metagenome in which they were found. Each bar represents one metagenome (genome IDs not shown).
Methylobacterium Metagenome Reads Show Ecological Differences Among Clades
To place these findings in context, we carried out a survey for Methylobacterium metagenome reads across all ecosystem types. We used the Phylogenetic Distribution of Genes in Metagenome tool (Markowitz et al., 2012; JGI IMG, 2017; Chen et al., 2019) to query all published metagenomes for reads with 90% BLAST similarity to each of 18 genomes from the Methylobacteriaceae. We observed a pattern in the samples that had BLAST hits to the query genomes suggesting ecological differences among the clades (Figure 8, Supplementary Figure 6, and Supplementary Tables 9–11). For example, the hits to query genomes from clades A and B were generally found with higher frequency in metagenomes from leaf samples than from soil (for Methylobacterium sp. Leaf88 and Methylobacterium gossipiicola, respectively, 4 and 10% of hits were from the soil, whereas 76 and 76% of were from leaf samples). In contrast, hits to query genomes from clade C2 were found primarily in soil and rarely in leaf samples (for M. nodulans, Methylobacterium sp. 4–46, and Methylobacterium sp. WSM2598, respectively, 74, 73, and 49% of hits were from the soil, whereas 2, 1, and 4% were from leaf samples). Clade C1 differed from the others in that there were fewer hits to these query genomes (between 8 and 71 hits per genome, in contrast to 96–487 hits for clades A–B and 104–523 hits for clade C2), and those hits more evenly distributed across all the sample types surveyed. These results point to ecological differences between the two clades of Methylobacterium capable of aromatic catabolism (clades C1 and C2). However, in general they agree with the survey of Methylobacterium vanA metagenome reads, suggesting that these two clades are more likely to be found in soil and root environments, and less likely to be found in the phyllosphere, than other clades of Methylobacterium.
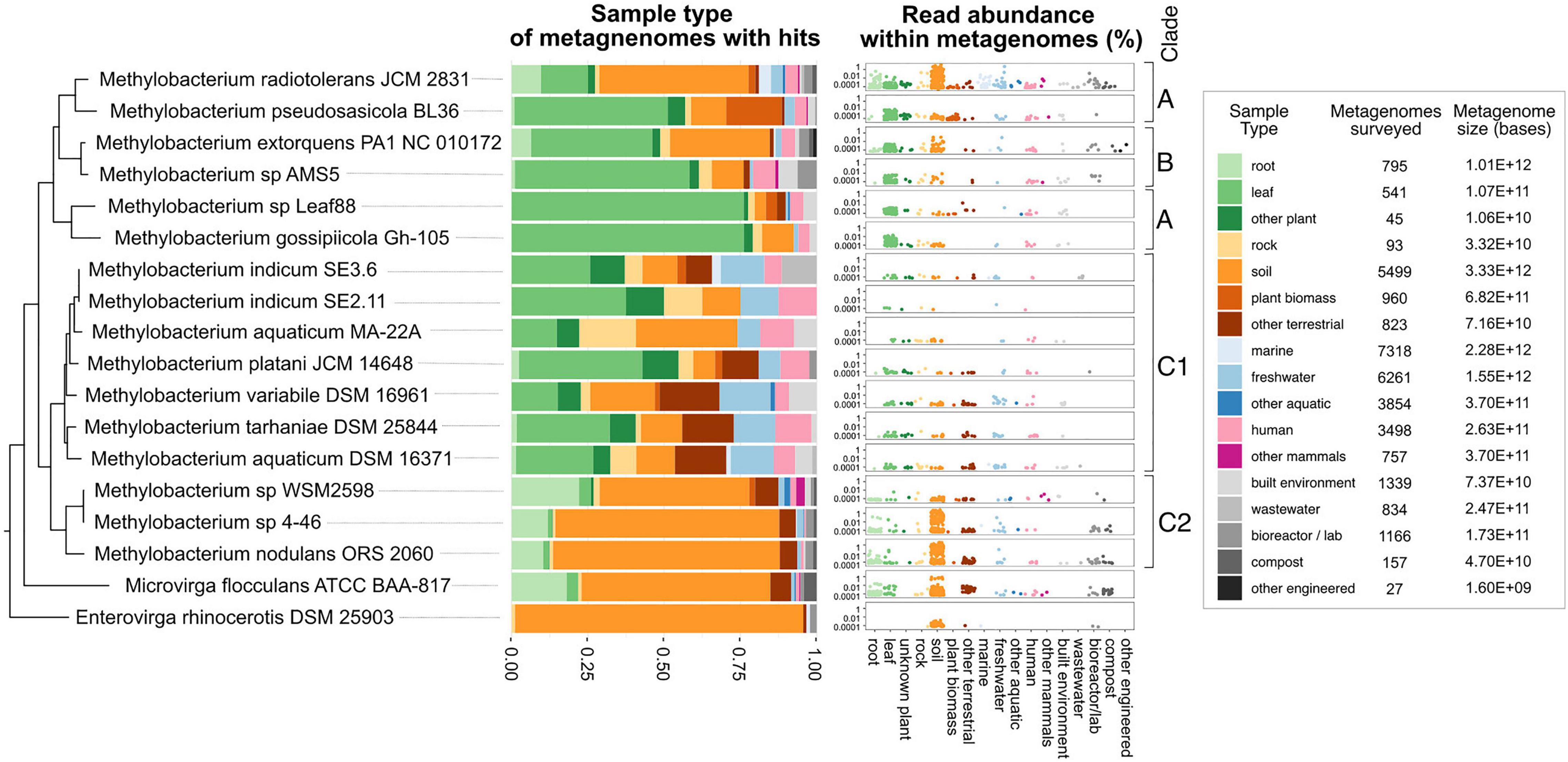
Figure 8. Methylobacterium strains vary substantially in their distribution among ecosystems; not all strains are phyllosphere-associated. Published reads from 33,967 metagenomes were surveyed for BLAST hits with 90% or greater similarity to the genomes of 18 strains from the Methylobacteriaceae, using the Phylogenetic Distribution of Genes in Metagenome tool in IMG. Each metagenome was classified into one of 17 ecological sample types. For each strain, the barplot shows the distribution of BLAST hits among those sample types. In the dotplots, each dot represents one metagenome, with height indicating the abundance of reads matching the strain’s genome within that metagenome. Points are color-coded, and clustered along the x-axis, by sample type. For context, the number of metagenomes, and total number of bases surveyed (in all metagenomes) is provided for each sample type. Phylogeny of the rpoB gene of the target species (as in Supplementary Figure 2) is shown at left.
Discussion
We have provided the first description of the methoxydotrophic growth of Methylobacterium on lignin-derived methoxylated aromatic acids. Methylobacterium species with this capability can use vanillate as a sole carbon substrate, without the transient formaldehyde accumulation in the environment observed in vanillate degraders that do not grow on C1 compounds. The genes encoding aromatic catabolism are found in almost all genome-sequenced members of clades C1 and C2 (those related to M. aquaticum and M. nodulans); they were largely absent in clades A and B (the M. radiotolerans and M. extorquens clades), but acquisition by horizontal gene transfer appears to have taken place in some species of those clades.
These findings shed new light on the ecology and evolution of the Methylobacterium. Methylobacterium have traditionally been of interest due to their ability to use single-carbon compounds as a sole source of carbon and energy; indeed, as their name suggests, methylotrophy has been one of the criteria used for classification. However, most work thus far has focused on methanol, methylamine, or formate as growth substrates; to our knowledge, ours is the first study to address whether methoxylated aromatic compounds might serve as a source for those single-carbon compounds. In fact, very few studies have addressed the possibility of Methylobacterium species using aromatic compounds as growth substrates at all.
As organisms known for their association with plants, it is possible that Methylobacterium play a role in the bacterial lignin-degrading community. In the environment, these organisms may depend upon aromatic acids released by the action of other lignin-degrading organisms, which have been found to be a prominent component of leaf litter leachate (Kuiters, 1990). Alternatively, Methylobacterium may encounter methoxylated aromatic acids primarily in plant root exudates (Zhalnina et al., 2018). In addition to acting as a growth substrate, aromatic acids play an important role in plant-microbe signaling: vanillate, ferulate, p-hydroxybenzoate, and protocatechuate all influence on the process and productivity of root nodulation by other members of the Rhizobiales (Seneviratne and Jayasinghearachchi, 2003; Mandal et al., 2010). This is significant in light of the fact that the members of the M. nodulans clade were, as the species name suggests, isolated from root and stem nodules of their plant hosts (Fleischman and Kramer, 1998; Jourand, 2004); and that we found this clade to be the most abundant among the Methylobacterium vanA genes found in the environment. Furthermore, Methylobacterium sp. AMS5 was isolated in a study on soybean epiphytes that are particularly responsive to host nodulation phenotype (Anda et al., 2011). It is likely that at least part of the importance of aromatic catabolism in Methylobacterium is to facilitate the relationships of these plant-associated organisms with their hosts. A link between aromatic catabolism and plant-microbe symbioses could help to explain our finding that vanA sequences related to those from M. nodulans and Methylobacterium sp. AMS5 are dominant among the Methylobacterium vanA sequences found in the environment despite the fact that there are few genome-sequenced representatives from those groups.
Perhaps the most compelling results from this study are the new insights into the evolution of the Methylobacterium group. One element is the absence of the vanA gene in Methylobacterium sp. 4–46 and sp. WSM298, the only two species we found in clade C to lack the capacity for methoxydotrophic growth. These species do carry genes for transforming ferulate to vanillin (ech) and vanillin to vanillate (vdh), which may be remnants from predecessors that were able to metabolize vanillate but lost the capability. Notably, Methylobacterium sp. 4–46 is also one of very few Methylobacterium species reported not to be capable of methylotrophic growth (Green and Ardley, 2018). Similarly to other strains described in Alessa et al. (2021), we have found that Methylobacterium sp. 4–46 is indeed capable of growth on methanol, but only in the presence of LaCl3 (Supplementary Figure 7 and Supplementary Table 12). This suggests the involvement of a XoxF-type methanol dehydrogenase (Skovran and Martinez-Gomez, 2015), and possibly a different role for methanol oxidation in this organism’s ecology. Given our hypothesis that methanol oxidation and vanillate demethylation require the same pathways for metabolizing the formaldehyde produced, it is possible that the loss of vanA in Methylobacterium sp. 4–46 might be related to its different style of methylotrophy. We have also observed that this and several non-Methylobacterium lignin-degrading species possess the genes to use PCA (the aromatic product of vanillate demethylation) but not the methoxy group (Figure 1). Yet we have found no species in which the reverse is true, although it would theoretically be possible for a methylotroph with van genes but no pca genes to carry out methoxydotrophic growth by utilizing only the methoxy group of vanillate.
The other unexpected evolutionary finding relates to the acquisition of aromatic catabolism genes by horizontal transfer in two Methylobacterium species from outside of clade C. The discovery of a catabolic island in Methylobacterium sp. AMS5 is itself not unusual; Methylobacterium and Methylorubrum species have long been recognized to carry an abundance of IS (insertion sequence) elements, and it has been postulated that the associated genome rearrangements and horizontal gene transfer associated are important mechanisms of evolution in the group (Schmid-Appert et al., 1997; Vuilleumier et al., 2009; Nayak and Marx, 2015). Relevant to the present study is the prior observation that diverse features of the genomic background—and not necessarily those predicted by phylogeny—influence whether a newly introduced set of genes is immediately useful to the recipient organism (Michener et al., 2014, 2016). Are there particular features of the M. pseudosasiacola and Methylobacterium sp. AMS5 genomes that allowed them to acquire the capacity for the degradation of methoxylated aromatics when no other known members of their clades did, or is the maintenance of this genomic capability the result of selective pressure specific to their ecological niche? Further work on the ecology of AMS5 and the biology of aromatic catabolism in Methylobacterium is necessary to address these questions.
This study has benefited from the wealth of knowledge that already exists on pathways for the degradation of lignin-derived aromatics in other microorganisms, and on methylotrophic metabolism in Methylobacterium, to deduce the likely fate of the methoxy group from vanillate. We found that in most cases, the gene annotations in IMG enabled us to correctly predict the substrates each strain could grow on (the exceptions were cases of no growth, and we cannot rule out the possibility that growth could occur under different conditions). However, we did find some novel features of the Methylobacterium pathways: almost all the strains we studied appear have no homologs of pcaI, pcaJ, or pcaF encoded within the pca gene cluster. It is possible that the functions of these genes are carried out by homologs located elsewhere in the genome, as has been found in some other organisms (Parke, 1997). A second possibility is raised by a previous study that carried out enzymatic screening and active site modeling on the DUF849 family of genes (Bastard et al., 2014): several of the DUF849 genes found in these Methylobacterium gene clusters were classified as beta-keto acid cleavage enzymes (G4 BKACE) predicted to act on beta-ketoadipate, raising the possibility that they might carry out the pcaI/pcaJ function and thus constitute a novel variant of the already diverse family of beta-ketoadipate pathway configurations (Parke, 1997; Buchan et al., 2004).
Our survey of where methoxydotrophic vanillate-utilizing Methylobacterium might be found in the environment suggests that they are more prominent in soil than in leaf environments. This is a notable given that reports on the Methylobacterium often focus on the phyllosphere as a characteristic habitat (e.g., Iguchi et al., 2015; Alessa et al., 2021). Our findings relating to Methylobacterium-associated vanA genes and Methylobacterium reads in metagenomes are subject to numerous caveats, including variability in extraction and sequencing methods and metadata annotation in public datasets, and the limitations in our ability to deduce taxonomy from short sequencing reads. Moreover, the methods we used are unable to capture the role of the horizontal gene transfer that is likely important for conferring aromatic catabolism to organisms such as Methylobacterium sp. AMS5. To gain true insight into the ecology of aromatic catabolism in this group will require more targeted sequencing efforts, as well as isolation and cultivation. However, this work provides a solid motivation for future studies, by demonstrating that Methylobacterium have diverse ecological patterns across their phylogeny, and that part of that diversity may be a soil-associated niche for aromatic-degrading strains.
Data Availability Statement
The original contributions presented in the study are included in the article or Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
JL: conceptualization, methodology, investigation, data curation, writing – original draft, and visualization. SS: methodology, resources, and writing – review and editing. CM: methodology, resources, writing – review and editing, supervision, and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by grants from the United States Department of Energy Genomic Science Program in Systems Biology for Energy and Environment, awards DE-SC0012627 and DE-SC0019436, and from the United States National Science Foundation Dimensions of Biodiversity Program, award DEB-1831838.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Nicholas Shevalier, Alyssa Baugh, and Tomislav Ticak for their assistance with experiments and support during the scientific process. We thank Armando MacDonald for technical assistance with measurements, Akio Tani for providing the M. aquaticum MA-22A culture, Andrea Lubbe and Trent Northen for the P. putida and R. jostii cultures, and José de la Torre for assistance with calculating genome signatures. We are thankful to Dipti Nayak, Mete Yuksel, Elizabeth Winters, Brittany Baker, and Tomislav Ticak for their valuable feedback on the manuscript. A preprint of this manuscript has been posted on the bioRxiv server with the doi: 10.1101/712836 (Lee et al., 2022).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.849573/full#supplementary-material
Supplementary Figure 1 | IMG phylogenetic lineage annotations do not match pplacer phylogenetic placement of vanA genes.
Supplementary Figure 2 | Phylogeny of all Methylobacterium and Methylorubrum strains included in this study, based on the rpoB gene.
Supplementary Figure 3 | Phylogeny of vanillate monooxygenase (vanA) genes with other COG 4638 (phenylpropionate dioxygenase or related ring-hydroxylating dioxygenase, large terminal subunit) genes from M. nodulans, M. indicum NS230, M. aquaticum DSM 16371, M. pseudosasiacola, and M. extorquens PA1.
Supplementary Figure 4 | Genomic context of aromatic catabolism genes pobA and ech in Methylobacterium species.
Supplementary Figure 5 | Methylobacterium sp. AMS5 catabolic island has similar GC content and tetranucleotide composition to the rest of the genome.
Supplementary Figure 6 | Ecological distribution of metagenomes surveyed for Methylobacterium reads, for comparison with Figure 8.
Supplementary Figure 7 | Methylobacterium sp. 4–46 grows on methanol only when lanthanum is provided.
Supplementary Table 1 | Methylobacterium strains included in this study, and locus tags of genes analyzed.
Supplementary Table 2 | Reference organisms included in Figures 1, 2, and locus tags of genes analyzed.
Supplementary Table 3 | Growth data for Methylobacterium strains on diverse compounds, shown in Figure 5. Time, incubation time in hours. Species, abbreviated strain name. Substrate, carbon substrate. Well, location of the replicate in a multiwell culture plate. OD, optical density at 600 nm.
Supplementary Table 4 | Growth data for Methylobacterium strains on methanol, shown in Figure 5. Time, incubation time in hours. Species, abbreviated strain name. Substrate, carbon substrate. Rep, replicate designation (A, B, or C). OD, optical density at 600 nm.
Supplementary Table 5 | Full OD and formaldehyde data from growth experiments on PCA and vanillate shown in Figure 6.
Supplementary Table 6 | Summary of metagenomes surveyed for Methylobacterium vanA (Figure 7).
Supplementary Table 7 | All metagenomes in which Methylobacterium vanA genes (shown in Figure 7) were found.
Supplementary Table 8 | All metagenome scaffolds containing vanA genes and identified as Methylobacterium, shown in Figure 7.
Supplementary Table 9 | Metagenomes surveyed for BLAST hits to each of 18 Methylobacteriaceae query genomes.
Supplementary Table 10 | Metagenomes in which BLAST hits with 90% similarity to a Methylobacteriaceae query genome were found.
Supplementary Table 11 | Summary of results from BLAST search of metagenomes for hits to each query Methylobacteriaceae genome, by sample type.
Supplementary Table 12 | Growth of Methylobacterium sp. 4–46 with and without lanthanum, shown in Supplementary Figure 7. Time, incubation time in hours. OD, optical density at 600 nm. La, with or without lanthanum (25 μM LaCl3). Rep, replicate designation (A, B, or C).
Supplementary File 1 | atpD gene alignment (from Figure 1).
Supplementary File 2 | recA gene alignment (from Figure 1).
Supplementary File 3 | rpoB gene alignment (from Figure 1).
Supplementary File 4 | 16S rRNA gene alignment (from Figure 1).
Supplementary File 5 | ech gene alignment (from Figure 2).
Supplementary File 6 | pcaG gene alignment (from Figure 2).
Supplementary File 7 | pobA gene alignment (from Figure 2).
Supplementary File 8 | vanA gene alignment (from Figure 2).
Supplementary File 9 | rpoB gene alignment, all Methylobacterium surveyed (from Supplementary Figure 3).
Supplementary File 10 | Alignment of COG 4638 homologs (from Supplementary Figure 4).
References
Ahmad, M., Roberts, J. N., Hardiman, E. M., Singh, R., Eltis, L. D., and Bugg, T. D. H. (2011). Identification of DypB from Rhodococcus jostii RHA1 as a Lignin Peroxidase. Biochemistry 50, 5096–5107. doi: 10.1021/bi101892z
Alessa, O., Ogura, Y., Fujitani, Y., Takami, H., Hayashi, T., Sahin, N., et al. (2021). Comprehensive comparative genomics and phenotyping of Methylobacterium species. Front. Microbiol. 12:2852. doi: 10.3389/fmicb.2021.740610
Anda, M., Ikeda, S., Eda, S., Okubo, T., Sato, S., Tabata, S., et al. (2011). Isolation and genetic characterization of Aurantimonas and Methylobacterium strains from stems of hypernodulated soybeans. Microbes Environ. 26, 172–180. doi: 10.1264/jsme2.ME10203
Bastard, K., Smith, A. A. T., Vergne-Vaxelaire, C., Perret, A., Zaparucha, A., De Melo-Minardi, R., et al. (2014). Revealing the hidden functional diversity of an enzyme family. Nat. Chem. Biol. 10, 42–49. doi: 10.1038/nchembio.1387
Brunel, F., and Davison, J. (1988). Cloning and sequencing of Pseudomonas genes encoding vanillate demethylase. J. Bacteriol. 170, 4924–4930. doi: 10.1128/jb.170.10.4924-4930.1988
Buchan, A., Neidle, E. L., and Moran, M. A. (2004). Diverse organization of genes of the β-ketoadipate pathway in members of the marine Roseobacter lineage. Appl. Environ. Microbiol. 70, 1658–1668. doi: 10.1128/AEM.70.3.1658-1668.2004
Chen, H.-P., Chow, M., Liu, C.-C., Lau, A., Liu, J., and Eltis, L. D. (2012). Vanillin catabolism in Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 78, 586–588. doi: 10.1128/AEM.06876-11
Chen, I.-M. A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., et al. (2019). IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. doi: 10.1093/nar/gky901
Chen, N. H., Djoko, K. Y., Veyrier, F. J., and McEwan, A. G. (2016). Formaldehyde stress responses in bacterial pathogens. Front. Microbiol. 7:257. doi: 10.3389/fmicb.2016.00257
Chistoserdova, L. (2011). Modularity of methylotrophy, revisited. Environ. Microbiol. 13, 2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x
Chistoserdova, L., and Lidstrom, P. M. E. (2013). “Aerobic methylotrophic prokaryotes,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 267–285. doi: 10.1007/978-3-642-30141-4_68
Compton, B. J., and Purdy, W. C. (1980). Fluoral-P, a member of a selective family of reagents for aldehydes. Anal. Chim. Acta 119, 349–357. doi: 10.1016/S0003-2670(01)93636-0
Crowther, G. J., Kosály, G., and Lidstrom, M. E. (2008). Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J. Bacteriol. 190, 5057–5062. doi: 10.1128/JB.00228-08
Delaney, N. F., Kaczmarek, M. E., Ward, L. M., Swanson, P. K., Lee, M.-C., and Marx, C. J. (2013). Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS One 8:e62957. doi: 10.1371/journal.pone.0062957
Díaz-García, L., Huang, S., Spröer, C., Sierra-Ramírez, R., Bunk, B., Overmann, J., et al. (2021). Dilution-to-stimulation/extinction method: a combination enrichment strategy to develop a minimal and versatile lignocellulolytic bacterial consortium. Appl. Environ. Microbiol. 87, e02427–20. doi: 10.1128/AEM.02427-20
Eddy, S. R. (2007). HMMER - biosequence analysis using profile hidden Markov models. Available online at: http://hmmer.org (accessed April 14, 2019).
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fleischman, D., and Kramer, D. (1998). Photosynthetic rhizobia. Biochim. Biophys. Acta Bioeng. 1364, 17–36. doi: 10.1016/S0005-2728(98)00011-5
Gaunt, M. W., Turner, S. L., Rigottier-Gois, L., Lloyd-Macgilp, S. A., and Young, J. P. (2001). Phylogenies of atpD and recA support the small subunit rRNA-based classification of rhizobia. Int. J. Syst. Evol. Microbiol. 51, 2037–2048. doi: 10.1099/00207713-51-6-2037
Green, P. N., and Ardley, J. K. (2018). Review of the genus Methylobacterium and closely related organisms: a proposal that some Methylobacterium species be reclassified into a new genus, Methylorubrum gen. nov. Int. J. Syst. Evol. Microbiol. 68, 2727–2748. doi: 10.1099/ijsem.0.002856
Gumerov, V. M., Ortega, D. R., Adebali, O., Ulrich, L. E., and Zhulin, I. B. (2020). MiST 3.0: an updated microbial signal transduction database with an emphasis on chemosensory systems. Nucleic Acids Res. 48, D459–D464. doi: 10.1093/nar/gkz988
Harwood, C. S., and Parales, R. E. (1996). The beta-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50, 553–590. doi: 10.1146/annurev.micro.50.1.553
Hibi, M., Sonoki, T., and Mori, H. (2005). Functional coupling between vanillate-O-demethylase and formaldehyde detoxification pathway. FEMS Microbiol. Lett. 253, 237–242. doi: 10.1016/j.femsle.2005.09.036
Hördt, A., López, M. G., Meier-Kolthoff, J. P., Schleuning, M., Weinhold, L.-M., Tindall, B. J., et al. (2020). Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 11:468. doi: 10.3389/fmicb.2020.00468
Huntemann, M., Ivanova, N. N., Mavromatis, K., Tripp, H. J., Paez-Espino, D., Tennessen, K., et al. (2016). The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Stand. Genom. Sci. 11:17. doi: 10.1186/s40793-016-0138-x
Iguchi, H., Yurimoto, H., and Sakai, Y. (2015). Interactions of methylotrophs with plants and other heterotrophic bacteria. Microorganisms 3, 137–151. doi: 10.3390/microorganisms3020137
Inkscape Team (2003). Inkscape: Draw Freely. Available online at: https://inkscape.org/ (accessed April 14, 2019).
Ito, H., and Iizuka, H. (1971). Taxonomic studies on a radio-resistant Pseudomonas. Agric. Biol. Chem. 35, 1566–1571. doi: 10.1080/00021369.1971.10860119
JGI IMG (2017). Phylogenetic Distribution of Genes in Metagenome (User Guide). Available online at: https://img.jgi.doe.gov/docs/MetagPhyloDist.pdf (accessed May 18, 2021).
Jourand, P. (2004). Methylobacterium nodulans sp. nov., for a group of aerobic, facultatively methylotrophic, legume root-nodule-forming and nitrogen-fixing bacteria. Int. J. Syst. Evol. Biol. 54, 2269–2273. doi: 10.1099/ijs.0.02902-0
Karlin, S. (2001). Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 9, 335–343. doi: 10.1016/S0966-842X(01)02079-0
Kato, N., Yurimoto, H., and Thauer, R. K. (2006). The physiological role of the ribulose monophosphate pathway in bacteria and archaea. Biosci. Biotechnol. Biochem. 70, 10–21. doi: 10.1271/bbb.70.10
Kelly, D. P., McDonald, I. R., and Wood, A. P. (2014). “The family Methylobacteriaceae,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 313–340. doi: 10.1007/978-3-642-30197-1_256
Kuiters, A. T. (1990). Role of phenolic substances from decomposing forest litter in plant–soil interactions. Acta Bot. Neerl. 39, 329–348. doi: 10.1111/j.1438-8677.1990.tb01412.x
Kweon, O., Kim, S.-J., Baek, S., Chae, J.-C., Adjei, M. D., Baek, D.-H., et al. (2008). A new classification system for bacterial Rieske non-heme iron aromatic ring-hydroxylating oxygenases. BMC Biochem. 9:11. doi: 10.1186/1471-2091-9-11
Lawrence, J. G., and Ochman, H. (1997). Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 44, 383–397. doi: 10.1007/PL00006158
Leducq, J.-B., Seyer-Lamontagne, É, Condrain-Morel, D., Bourret, G., Sneddon, D., Foster, J. A., et al. (2022). Fine-scale adaptations to environmental variation and growth strategies drive phyllosphere Methylobacterium diversity. mBio 13:e03175-21. doi: 10.1128/mbio.03175-21
Lee, J. A., Baugh, A. C., Shevalier, N. J., Strand, B., Stolyar, S., and Marx, C. J. (2021). Cross-feeding of a toxic metabolite in a synthetic lignocellulose-degrading microbial community. Microorganisms 9:321. doi: 10.3390/microorganisms9020321
Lee, J. A., Stolyar, S., and Marx, C. J. (2022). Aerobic methoxydotrophy: growth on methoxylated aromatic compounds by Methylobacterium. bioRxiv [Preprint]. doi: 10.1101/712836
Lee, M.-C., and Marx, C. J. (2012). Repeated, selection-driven genome reduction of accessory genes in experimental populations. PLOS Genet. 8:e1002651. doi: 10.1371/journal.pgen.1002651
Lee, M.-C., Chou, H.-H., and Marx, C. J. (2009). Asymmetric, bimodal trade-offs during adaptation of Methylobacterium to distinct growth substrates. Evolution 63, 2816–2830. doi: 10.1111/j.1558-5646.2009.00757.x
Lehmann, J., and Kleber, M. (2015). The contentious nature of soil organic matter. Nature 528, 60–68. doi: 10.1038/nature16069
Lessmeier, L., Hoefener, M., and Wendisch, V. F. (2013). Formaldehyde degradation in Corynebacterium glutamicum involves acetaldehyde dehydrogenase and mycothiol-dependent formaldehyde dehydrogenase. Microbiology 159, 2651–2662. doi: 10.1099/mic.0.072413-0
Lloyd, M. K., Trembath-Reichert, E., Dawson, K. S., Feakins, S. J., Mastalerz, M., Orphan, V. J., et al. (2021). Methoxyl stable isotopic constraints on the origins and limits of coal-bed methane. Science 374, 894–897. doi: 10.1126/science.abg0241
Lohans, C. T., Wang, D. Y., Wang, J., Hamed, R. B., and Schofield, C. J. (2017). Crotonases: nature’s exceedingly convertible catalysts. ACS Catal. 7, 6587–6599. doi: 10.1021/acscatal.7b01699
Mandal, S. M., Chakraborty, D., and Dey, S. (2010). Phenolic acids act as signaling molecules in plant-microbe symbioses. Plant Signal. Behav. 5, 359–368. doi: 10.4161/psb.5.4.10871
Markowitz, V. M., Chen, I.-M. A., Chu, K., Szeto, E., Palaniappan, K., Grechkin, Y., et al. (2012). IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res. 40, D123–D129. doi: 10.1093/nar/gkr975
Marx, C. J., Bringel, F., Chistoserdova, L., Moulin, L., Farhan Ul Haque, M., Fleischman, D. E., et al. (2012). Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 194, 4746–4748. doi: 10.1128/JB.01009-12
Marx, C. J., Chistoserdova, L., and Lidstrom, M. E. (2003). Formaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J. Bacteriol. 185, 7160–7168. doi: 10.1128/JB.185.23.7160-7168.2003
Marx, C. J., Van Dien, S. J., and Lidstrom, M. E. (2005). Flux analysis uncovers key role of functional redundancy in formaldehyde metabolism. PLOS Biol. 3:e16. doi: 10.1371/journal.pbio.0030016
Masai, E., Katayama, Y., and Fukuda, M. (2007). Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci. Biotechnol. Biochem. 71, 1–15. doi: 10.1271/bbb.60437
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11:538. doi: 10.1186/1471-2105-11-538
Mayumi, D., Mochimaru, H., Tamaki, H., Yamamoto, K., Yoshioka, H., Suzuki, Y., et al. (2016). Methane production from coal by a single methanogen. Science 354, 222–225. doi: 10.1126/science.aaf8821
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Menna, P., Barcellos, F. G., and Hungria, M. (2009). Phylogeny and taxonomy of a diverse collection of Bradyrhizobium strains based on multilocus sequence analysis of the 16S rRNA gene, ITS region and glnII, recA, atpD and dnaK genes. Int. J. Syst. Evol. Microbiol. 59, 2934–2950. doi: 10.1099/ijs.0.009779-0
Merkens, H., Beckers, G., Wirtz, A., and Burkovski, A. (2005). Vanillate metabolism in Corynebacterium glutamicum. Curr. Microbiol. 51, 59–65. doi: 10.1007/s00284-005-4531-8
Michener, J. K., Vuilleumier, S., Bringel, F., and Marx, C. J. (2014). Phylogeny poorly predicts the utility of a challenging horizontally transferred gene in Methylobacterium strains. J. Bacteriol. 196, 2101–2107. doi: 10.1128/JB.00034-14
Michener, J. K., Vuilleumier, S., Bringel, F., and Marx, C. J. (2016). Transfer of a catabolic pathway for chloromethane in Methylobacterium strains highlights different limitations for growth with chloromethane or with dichloromethane. Front. Microbiol. 7:1116. doi: 10.3389/fmicb.2016.01116
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), (New Orleans: IEEE), 1–8.
Mitsui, R., Kusano, Y., Yurimoto, H., Sakai, Y., Kato, N., and Tanaka, M. (2003). Formaldehyde fixation contributes to detoxification for growth of a nonmethylotroph, Burkholderia cepacia TM1, on vanillic acid. Appl. Environ. Microbiol. 69, 6128–6132. doi: 10.1128/AEM.69.10.6128-6132.2003
Moreno, R., and Rojo, F. (2008). The target for the Pseudomonas putida Crc global regulator in the benzoate degradation pathway is the BenR transcriptional regulator. J. Bacteriol. 190, 1539–1545. doi: 10.1128/JB.01604-07
Nash, T. (1953). The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J. 55, 416–421. doi: 10.1042/bj0550416
Nayak, D. D., and Marx, C. J. (2015). Experimental horizontal gene transfer of methylamine dehydrogenase mimics prevalent exchange in nature and overcomes the methylamine growth constraints posed by the sub-optimal N-methylglutamate pathway. Microorganisms 3, 60–79. doi: 10.3390/microorganisms3010060
Ortega, D. R. (2020). GeneHood. California: Caltech Jensen Lab. Available online at: https://genehood.io/.
Otani, H., Lee, Y.-E., Casabon, I., and Eltis, L. D. (2014). Characterization of p-Hydroxycinnamate Catabolism in a Soil Actinobacterium. J. Bacteriol. 196, 4293–4303. doi: 10.1128/JB.02247-14
Özgen, F. F., and Schmidt, S. (2019). “Rieske non-heme iron dioxygenases: applications and future perspectives,” in Biocatalysis: Enzymatic Basics and Applications, eds Q. Husain and M. F. Ullah (Cham: Springer International Publishing), 57–82. doi: 10.1007/978-3-030-25023-2_4
Pagès, H., Aboyoun, P., Gentleman, R., and DebbRoy, S. (2019). Biostrings: Efficient manipulation of biological strings. R package version 2.62.0. Available online at: http://bioconductor.org/packages/release/bioc/html/Biostrings.html (accessed April 27, 2019).
Parke, D. (1997). Acquisition, reorganization, and merger of genes: novel management of the β-ketoadipate pathway in Agrobacterium tumefaciens. FEMS Microbiol. Lett. 146, 3–12. doi: 10.1111/j.1574-6968.1997.tb10164.x
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ragauskas, A. J., Beckham, G. T., Biddy, M. J., Chandra, R., Chen, F., Davis, M. F., et al. (2014). Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. doi: 10.1126/science.1246843
Ravi, K., García-Hidalgo, J., Gorwa-Grauslund, M. F., and Lidén, G. (2017). Conversion of lignin model compounds by Pseudomonas putida KT2440 and isolates from compost. Appl. Microbiol. Biotechnol. 101, 5059–5070. doi: 10.1007/s00253-017-8211-y
Roca, A., Rodríguez-Herva, J. J., and Ramos, J. L. (2009). Redundancy of enzymes for formaldehyde detoxification in Pseudomonas putida. J. Bacteriol. 191, 3367–3374. doi: 10.1128/JB.00076-09
Ruiz-Dueñas, F. J., and Martínez, ÁT. (2009). Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb. Biotechnol. 2, 164–177. doi: 10.1111/j.1751-7915.2008.00078.x
Sainsbury, P. D., Hardiman, E. M., Ahmad, M., Otani, H., Seghezzi, N., Eltis, L. D., et al. (2013). Breaking down lignin to high-value chemicals: the conversion of lignocellulose to vanillin in a gene deletion mutant of Rhodococcus jostii RHA1. ACS Chem. Biol. 8, 2151–2156. doi: 10.1021/cb400505a
Schmid-Appert, M., Zoller, K., Traber, H., Vuilleumier, S., and Leisinger, T. (1997). Association of newly discovered IS elements with the dichloromethane utilization genes of methylotrophic bacteria. Microbiology 143, 2557–2567. doi: 10.1099/00221287-143-8-2557
Seneviratne, G., and Jayasinghearachchi, H. S. (2003). Phenolic acids: possible agents of modifying N2-fixing symbiosis through rhizobial alteration? Plant Soil 252, 385–395. doi: 10.1023/A:1024725511783
Skovran, E., and Martinez-Gomez, N. C. (2015). Just add lanthanides. Science 348, 862–863. doi: 10.1126/science.aaa9091
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sudtachat, N., Ito, N., Itakura, M., Masuda, S., Eda, S., Mitsui, H., et al. (2009). Aerobic vanillate degradation and C1 compound metabolism in Bradyrhizobium japonicum. Appl. Environ. Microbiol. 75, 5012–5017. doi: 10.1128/AEM.00755-09
Vanholme, R., Demedts, B., Morreel, K., Ralph, J., and Boerjan, W. (2010). Lignin biosynthesis and structure. Plant Physiol. 153, 895–905. doi: 10.1104/pp.110.155119
Ventorino, V., Sannino, F., Piccolo, A., Cafaro, V., Carotenuto, R., and Pepe, O. (2014). Methylobacterium populi VP2: plant growth-promoting bacterium isolated from a highly polluted environment for polycyclic aromatic hydrocarbon (PAH) biodegradation. Sci. World J. 2014:931793. doi: 10.1155/2014/931793
Vorholt, J. A., Chistoserdova, L., Lidstrom, M. E., and Thauer, R. K. (1998). The NADP-dependent methylene tetrahydromethanopterin dehydrogenase in Methylobacterium extorquens AM1. J. Bacteriol. 180, 5351–5356. doi: 10.1128/JB.180.20.5351-5356.1998
Vuilleumier, S., Chistoserdova, L., Lee, M.-C., Bringel, F., Lajus, A., Zhou, Y., et al. (2009). Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS One 4:e5584. doi: 10.1371/journal.pone.0005584
Welte, C. U. (2016). A microbial route from coal to gas. Science 354, 184–184. doi: 10.1126/science.aai8101
Wilhelm, R. C., Singh, R., Eltis, L. D., and Mohn, W. W. (2019). Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 13, 413–429. doi: 10.1038/s41396-018-0279-6
Wozniak, R. A. F., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563. doi: 10.1038/nrmicro2382
Zhalnina, K., Louie, K. B., Hao, Z., Mansoori, N., da Rocha, U. N., Shi, S., et al. (2018). Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 3, 470–480. doi: 10.1038/s41564-018-0129-3
Keywords: methylotrophy, lignin degradation, aromatic catabolism, Methylobacterium, formaldehyde
Citation: Lee JA, Stolyar S and Marx CJ (2022) Aerobic Methoxydotrophy: Growth on Methoxylated Aromatic Compounds by Methylobacteriaceae. Front. Microbiol. 13:849573. doi: 10.3389/fmicb.2022.849573
Received: 06 January 2022; Accepted: 16 February 2022;
Published: 11 March 2022.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology (RAS), RussiaReviewed by:
Yasuyoshi Sakai, Kyoto University, JapanMarina G. Kalyuzhanaya, San Diego State University, United States
Copyright © 2022 Lee, Stolyar and Marx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica A. Lee, amVzc2ljYS5hLmxlZUBuYXNhLmdvdg==; Christopher J. Marx, Y21hcnhAdWlkYWhvLmVkdQ==