 Katelyn Boase
Katelyn Boase Carolina González
Carolina González Eva Vergara
Eva Vergara Gonzalo Neira
Gonzalo Neira David Holmes
David Holmes Elizabeth Watkin
Elizabeth Watkin- 1Curtin Medical School, Curtin University, Perth, WA, Australia
- 2Center for Bioinformatics and Genome Biology, Centro Ciencia & Vida, Santiago, Chile
- 3Facultad de Medicina y Ciencias, Universidad San Sebastián, Santiago, Chile
Acidihalobacter is a genus of acidophilic, gram-negative bacteria known for its ability to oxidize pyrite minerals in the presence of elevated chloride ions, a capability rare in other iron-sulfur oxidizing acidophiles. Previous research involving Acidihalobacter spp. has focused on their applicability in saline biomining operations and their genetic arsenal that allows them to cope with chloride, metal and oxidative stress. However, an understanding of the molecular adaptations that enable Acidihalobacter spp. to thrive under both acid and chloride stress is needed to provide a more comprehensive understanding of how this genus can thrive in such extreme biomining conditions. Currently, four genomes of the Acidihalobacter genus have been sequenced: Acidihalobacter prosperus DSM 5130T, Acidihalobacter yilgarnensis DSM 105917T, Acidihalobacter aeolianus DSM 14174T, and Acidihalobacter ferrooxydans DSM 14175T. Phylogenetic analysis shows that the Acidihalobacter genus roots to the Chromatiales class consisting of mostly halophilic microorganisms. In this study, we aim to advance our knowledge of the genetic repertoire of the Acidihalobacter genus that has enabled it to cope with acidic stress. We provide evidence of gene gain events that are hypothesized to help the Acidihalobacter genus cope with acid stress. Potential acid tolerance mechanisms that were found in the Acidihalobacter genomes include multiple potassium transporters, chloride/proton antiporters, glutamate decarboxylase system, arginine decarboxylase system, urease system, slp genes, squalene synthesis, and hopanoid synthesis. Some of these genes are hypothesized to have entered the Acidihalobacter via vertical decent from an inferred non-acidophilic ancestor, however, horizontal gene transfer (HGT) from other acidophilic lineages is probably responsible for the introduction of many acid resistance genes.
Introduction
Environments that exhibit both acidic and saline conditions are relatively rare. There are few studies aimed at understanding acid stress responses in halophiles and these typically show that halophiles are unable to grow in low pH environments (Bowers and Wiegel, 2011; Moran-Reyna and Coker, 2014; He et al., 2016; Zammit and Watkin, 2016). For example, it has been hypothesized that chloride ions, present in saline conditions, can disrupt the reversed membrane potential of acidophiles making them potentially sensitive to acidification of their cytoplasm followed by subsequent cellular death (Suzuki et al., 1999).
Acidihalobacter is one of a limited number of genera of bacteria that has been shown to be an extreme acidophile (pH ≤ 3.0) and halophile (Minegishi, 2013; Zammit and Watkin, 2016) and is thus a polyextremophile. Currently four organisms belonging to the Acidihalobacter genus have been isolated and cultivated in laboratory conditions: Acidihalobacter prosperus DSM 5130T from the geothermally heated seafloor at Porto di Levante, Vulcano, Italy; Acidihalobacter aeolianus DSM 14174T, and Acidihalobacter ferrooxydans DSM 14175T from the hydrothermal pools at the Aeolian Islands, Vulcano Italy and Acidihalobacter yilgarnensis DSM 105917T from an acidic saline lake drain in Western Australia (Huber and Stetter, 1989; Simmons and Norris, 2002; Zammit et al., 2009). Several studies have been conducted on the Acidihalobacter genus, understanding its response to chloride ions, osmotic stress, metal stress and oxidative stress (Dopson et al., 2016; Khaleque et al., 2018, 2019, 2020) but there has been limited focus on their mechanisms of survival at extremely low pH. The Acidihalobacter are interesting not only because they are deeply intriguing polyextremophiles but also because they are among the few organisms that can be used for copper recovery (bioleaching) from chalcopyrite (Khaleque et al., 2017a). Extreme acidophiles capable of oxidizing iron and sulfur have been of interest to the scientific and industrial communities regarding their biotechnological and biomining applications (Johnson and Schippers, 2017; Gumulya et al., 2018) and their potential in protein engineering (Parashar and Satyanarayana, 2018).
Acidophilic microorganisms are found in all three domains of life. When describing acid stress responses in microorganisms, it is useful to make a distinction between “extreme” acidophiles (pH opt < 3.0) and “moderate” acidophiles (pH opt between 3 and 5). Both share multiple acid tolerance mechanisms, however, extreme acidophiles have additional adaptations absent in moderate acidophiles (reviewed in Baker-Austin and Dopson, 2007). Both categories of acidophiles thrive in acidic conditions by maintaining a circumneutral cytoplasmic pH despite a large proton gradient compared to their environment. In contrast, another class of acidophiles, the so-called “amateur” acidophiles, e.g., Helicobacter pylori, survive by neutralizing their acidic environment (Foster, 2004; Baker-Austin and Dopson, 2007; Slonczewski et al., 2009; Quatrini and Barrie Johnson, 2016). The maintenance of a circumneutral cytoplasmic pH is achieved through multiple mechanisms including the generation of a reversed membrane potential, creating a positive charge, referred to as the Donnan potential. This chemi-osmotic barrier effectively repels positive ions from entering the cell, preventing the acidification of their cytoplasm and has been defined as the first line of defense (Vergara et al., 2020). The use of potassium transporters, importing K+ ions, are thought to be the most likely mechanism acidophiles use to generate this membrane potential (Cholo et al., 2015; Buetti-Dinh et al., 2016; Christel et al., 2018). The potassium channel transporters Kdp, Trk, and Kch have been identified in extreme acidophiles (Cholo et al., 2015; Buetti-Dinh et al., 2016; Christel et al., 2018; Vergara et al., 2020). Extreme acidophiles are also hypothesized to have membranes that are less permeable to protons through membrane adaptations (Yamauchi et al., 1993; Macalady et al., 2004; Chao et al., 2008; Mykytczuk et al., 2010). Hopanoids, spermidine, and starvation-inducible membrane-altering lipoproteins are all associated with conferring a higher resistance to proton permeability (Alexander and St John, 1994; Hommais et al., 2004; Jones et al., 2012). Second line of defense mechanisms (Vergara et al., 2020) describe processes that consume or expel protons that have entered the cell including buffering reactions, for example the glutamate decarboxylase system, the arginine decarboxylase system, spermidine synthesis, the urease system and Na+/H+ antiporters (Hommais et al., 2004; Richard and Foster, 2004; Zhao and Houry, 2010; Feehily and Karatzas, 2013; Mangold et al., 2013; Marcus and Scott, 2016). These mechanisms are commonly found in both extreme and moderate acidophiles. To deepen our understanding of acid stress response in the Acidihalobacter genus, a genome-wide comparison study was undertaken to generate an inventory of predicted acid resistance genes. Using phylogenomic approaches, we suggest possible events, including gene gain/loss and gene duplication, that led to the evolution of the acidophilic Acidihalobacter from an inferred non-acidophilic ancestor.
Materials and Methods
Genomes and Phylogenetic Analysis
The four currently published Acidihalobacter assembly sequences (GCF_000754095.2, GCF_001753245.1, GCF_001753165.1 and GCF_001975725.1) as well as the outgroup Halothiobacillus neapolitanus c2 (GCF_000024765.1) were downloaded from the National Center for Biotechnology Information (NCBI) GenBank genomic database in October 2019 (Pruitt et al., 2012). Phylogenetic analysis of four Acidihalobacter genomes in the context of the Chromatiales order was conducted using 52 NCBI reference proteomes of the Chromatiales order available from the GenBank database (Supplementary Table 1) in October 2019. PhyloPhlAn3 was used to construct a phylogenetic tree of the Chromatiales proteomes, using the Phylophlan database and diversity set to low (set of 400 conserved proteins) (Asnicar et al., 2020). Diamond (Buchfink et al., 2015) was used for mapping the database to the proteomes in study, MAFFT (Katoh and Standley, 2013) for the multiple sequence alignment and the maximum likelihood tree was constructed with IQTREE (Nguyen et al., 2015), using 1000 replicates, with the best suited evolutionary model proposed by IQTREE. The final tree was visualized using iTOL.1
Prediction of Mobile Genetic Elements and Genome Islands
Mobile genetic elements (MGEs) were predicted and classified using TnpPred and ISsaga (Siguier et al., 2006; Varani et al., 2011; Riadi et al., 2012). Horizontal gene transfer events were predicted by HGTector (Waack et al., 2006; Zhu et al., 2014). Genome context of mobile elements and genome islands were analyzed using STRING, MAUVE, and ARTEMIS (Darling et al., 2010; Carver et al., 2012; Szklarczyk et al., 2019). These bioinformatic tools were used to predict genes involved in mechanisms of HGT (see Supplementary Information 1).
Identification of Genes Related to Low pH Resistance
Genes and mechanisms involved in acidic resistance were identified through previous research (Vergara et al., 2020; González-Rosales et al., 2022). Identification of these genes in the Acidihalobacter genus genomes and the outgroup was done using BlastP comparison with a minimum E-value cutoff of 1e–5. Synteny blocks were predicted and visualized using MAUVE Progressive alignment (Darling et al., 2010). Genome contexts were visualized using Artemis (Carver et al., 2012). Acid tolerance proteins found in the Acidihalobacter genomes and H. neapolitanus were back-blast to assess orthologous proteins in other organisms. To assess the relatedness of these proteins, phylogenetic trees were made as previously described in section 2.1 using MAFFT, IQTREE, and iTOL.
Mapping Evolutionary Events
To infer branch-site-specific evolutionary events across genomes of the Acidihalobacter genus, a conserved markers tree of 400 proteins was constructed between the Acidihalobacter genomes and using H. neapolitanus c2 as outgroup using Phylophlan3 (Asnicar et al., 2020). The presence of genes predicted to be involved in acid resistance were mapped onto each branch of the phylogenomic tree to model gene gain and loss events. Inference of evolutionary events were based on parsimony criteria.
Results and Discussion
Genomic Features of the Acidihalobacter Genus
Acidihalobacter spp. are aerobic, chemolithotrophic, mesophilic, halotolerant acidophiles with pH optimums between 1.9 and 2.75. The four publicly available Acidihalobacter genomes were analyzed together with the H. neapolitanus outgroup genome (Table 1). A. yilgarnensis is the only complete genome of the Acidihalobacter, with the other three being high quality permanent draft genomes. GC% content between the Acidihalobacter genomes varies between 59.9 and 64.4%. Acidihalobacter genomes are larger than the H. neapolitanus c2 outgroup genome with a range of 0.78–0.99 Mb.
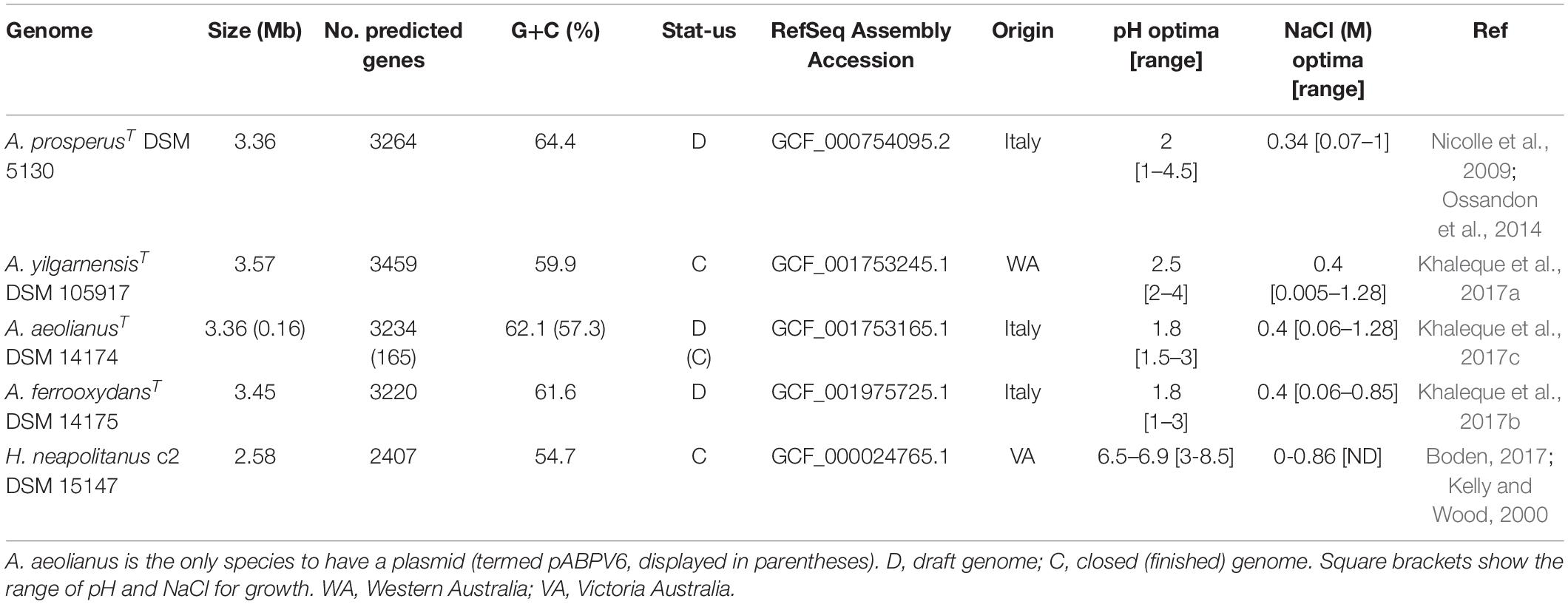
Table 1. Genomic information of the Acidihalobacter spp. genomes and Halothiobacillus neapolitanus genome used in the study.
Phylogeny of Acidihalobacter Within the Chromatiales Order
To understand the recent evolutionary history of the Acidihalobacter genus, we compared the relative chloride and acid tolerance of microbes in the Chromatiales order to that of the Acidihalobacter spp. Using the complete reference genomes in the Chromatiales order, a phylogenetic tree was constructed, and literature was reviewed to identify the chloride and acid tolerances of the genomes used (Figure 1). Most microbes in the Chromatiales order were halotolerant and either neutrophilic or alkaliphilic. The Acidihalobacter microbes were the only microbes in the Chromatiales order that tolerate pH environments below four. This is an indication that Acidihalobacter has likely evolved from a halophilic past and has gained the ability to cope with acidic stress.
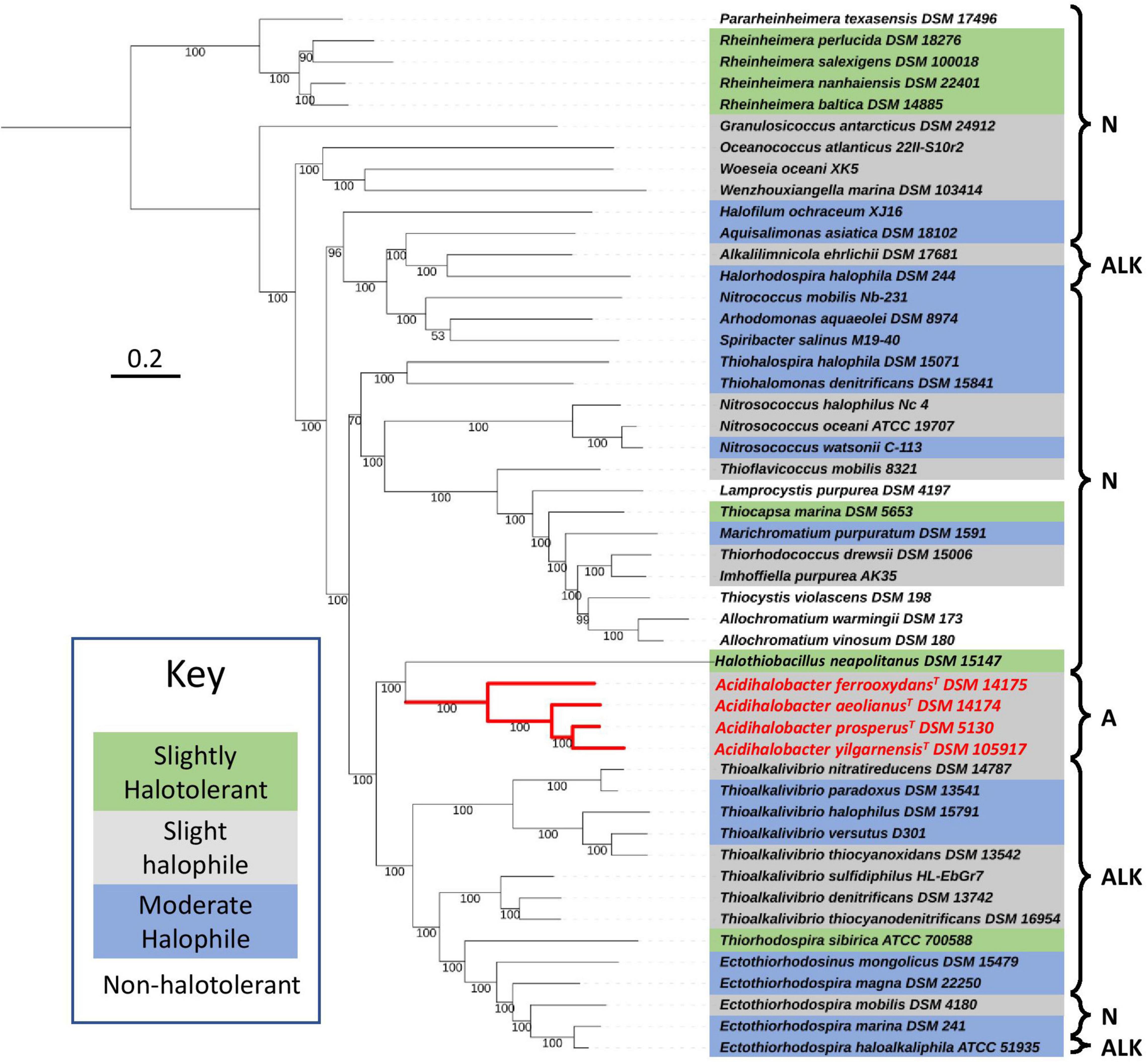
Figure 1. Phylogenomic tree constructed, using 400 protein markers of 52 representative complete genomes of the Chromatiales order, using the Phylophlan3 program. Gray highlights slightly halophilic genomes (optimum growth 2–5% w/v NaCl), blue highlights moderately halophilic genomes (optimum growth 5–20% w/v NaCl), green highlights slightly halotolerant genomes (no NaCl required to grow however optimum growth in 2–8% w/v NaCl) and no highlight represents non-halophiles. A, acidophiles (optimum growth < pH 5); N, neutrophiles (optimum growth between pH 5 and 9); and ALK, alkaliphiles (optimum growth > pH 9). Red lines indicate the phylogenetic relationships between the acidophiles. The sequences were aligned with MAFFT and the concatenated alignment was used to construct the phylogenetic tree with IQTREE and LG+R3 as the best-suited evolutionary model. Scale bar indicates 0.2 substitutions per amino acid. See Supplementary Table 1 for the full information on the microorganisms used in the tree.
Acid Resistance: First Line of Defense
“First line of defense” (Vergara et al., 2020) mechanisms are involved in reducing the influx of protons into the cytoplasm of acidophiles. Potassium pumps are primarily responsible for this by creating the reversed membrane potential, limiting proton influx (reviewed in Quatrini and Barrie Johnson, 2016). Membrane alterations that limit proton permeability across the cellular membrane also fall under the “first line of defense” mechanisms and include hopanoid synthesis, Slp proteins and porin alterations, as described in the following sections.
Membrane Potential and Potassium Transporters
Potassium transport systems are found in many bacteria as potassium is the major intracellular cation responsible for osmotic regulation, activating intracellular enzymes, regulating internal pH and as a secondary messenger (Epstein, 2003; Korolev, 2021; Raven, 2021). It is widely accepted that potassium transporters are regulated by ion concentrations and turgor pressure (Epstein, 2003). In terms of acid tolerance in extreme acidophiles, potassium transporters are hypothesized to be responsible for the reversed membrane potential, which is the primary mechanism reducing proton influx (Baker-Austin and Dopson, 2007). In halophiles, it has been shown that potassium transport systems aid in osmotic stress and typically halophiles have multiple copies of potassium transport genes (Epstein, 2003; Kraegeloh et al., 2005; Mongodin et al., 2005).
The Trk potassium system symports K+ and H+ into the cell and has been identified in plants, Bacteria and Archaea (reviewed in Epstein, 2003; Pandey and Mahiwal, 2020). In prokaryotes the most studied Trk system is in Escherichia coli and consists of TrkH and TrkG, the multimeric functional transporters; TrkA, the cytoplasmic regulating subunit; and TrkE (also known as sapD), the ATP-binding component of the Trk system (Epstein, 2003; reviewed in Stautz et al., 2021). In E. coli, the genes involved in the Trk system are distributed throughout the genome, however in halophilic organisms trkA and trkH have been found clustered together (Nakamura et al., 1998; Kraegeloh et al., 2005). Many acidophile genomes lack trkHG, and only contain the trkA homolog (exception Alicyclobacillus ferrooxidans) (Rivera-Araya et al., 2020). In Acidihalobacter spp. and H. neapolitanus c2 the TrkAH potassium transporter system was present. The system is in a highly conserved syntenic region in all genomes and the genes are arranged contiguously (Supplementary Figure 1). A second copy of trkH was also found in the Acidihalobacter genomes directly downstream of the first trkH copy. The genomic arrangement of the Trk system in Acidihalobacter and H. neapolitanus c2 is very similar to that found in Vibrio alinolyticus (a halophile) with fmt (methionyl-tRNA formyltransferase) and rsmB (a ribosomal RNA small subunit methyltransferase B, also identified as fmu in other bacteria) being present upstream of trkA (Supplementary Figure 1) (Nakamura et al., 1998). Three extra genes were found between rsmB and trkA in Acidihalobacter and H. neapolitanus, a predicted gene that produces a protein with an unknown function, a histidine kinase, and a sigma-54 RNA polymerase holoenzyme. Both the histidine kinase and σ-54 proteins are predicted to be nitrogen regulated and involved in nitrogen assimilation (ntrY and atoC also known as ntrX). Trk proteins have been identified in the genomes of other acidophiles such as Leptospirillum (Vergara et al., 2020), however, information about how the Trk system is arranged in their genome is lacking. No other case of a Trk system with two adjacent TrkH proteins has been published. Multiple copies of trkA and trkH have been found in the same region in Salinibacter ruber, however, they are not juxtaposed (López-Pérez et al., 2013). Whether this alteration results in a more efficient adaptation to acidity or salinity remains to be explored. Evidence of HGT in the genome surrounding the Trk system in Acidihalobacter was not found, and phylogenetic analysis of the protein sequences and their best BlastP hits show the Acidihalobacter TrkH proteins clustering with other TrkH proteins found in other organisms in the same Chromatiales order (Supplementary Figure 2). In light of the Trk system being in a highly conserved region of the genome in both Acidihalobacter and H. neapolitanus c2 and the Trk proteins all clustering with Trk proteins in the Chromatiales order, vertical descent of the Trk system in Acidihalobacter is likely. Whether the alteration of the gene structure in Acidihalobacter and H. neapolitanus when compared to Trk systems in other halophiles results in differential control or efficiency of Trk in these bacteria remains to be investigated.
Kdp is a high-affinity potassium transport system known to be involved in potassium homeostasis and the osmotic stress response in many bacteria, encompassing halophiles and non-halophiles, and is responsible for the active influx of potassium ions in environments with low potassium concentrations (Epstein, 2003; Strahl and Greie, 2008; Kixmüller et al., 2011; Price-Whelan et al., 2013). The Kdp system is also present in many acidophiles and has been found to be actively transcribed in acidic environments (Bakker et al., 1987; Acuña et al., 2013; Cholo et al., 2015; Rivera-Araya et al., 2019, 2020; Vergara et al., 2020). In E. coli the Kdp system consists of two operons, one encompassing the structural genes kdpFABC, and the other encompassing the regulatory genes kdpDE (Walderhaug et al., 1992). These two operons overlap in E. coli, however, different genetic arrangements of the system are found in other bacteria (Walderhaug et al., 1992; Epstein, 2003). KdpF is involved in stabilizing the Kdp complex but is not essential. KdpA is responsible for binding and translocating the K+ and KdpB and KdpC are responsible for the ATP hydrolysis (Hesse et al., 1984; Gaßel et al., 1998, 1999; Dorus et al., 2001; Reviewed in Epstein, 2003). The kdpDE operon is a two-component response regulator responsible for regulating the Kdp complex as KdpD is a histidine kinase and KdpE is a response regulator (Walderhaug et al., 1992; Reviewed in Epstein, 2003). The Kdp system was only identified in the genome of A. aeolianus. The gene arrangement of the system was similar to that of E. coli (Supplementary Figure 3A) (Walderhaug et al., 1992). Both carbonic anhydrase (can) and the sigma-54 regulator (σ-54) have been identified in the same genomic context and previously have been indicated to be involved in acid tolerance of other microorganisms (Sachs et al., 2006; Bury-Moneì et al., 2008; Mitra et al., 2012, 2014). HGTector analysis predicts KdpA, KdpB, KdpC, KdpD, KdpE, and the sigma-54 transcription regulator to have been gained through HGT from a Bacteria, and the can from a Proteobacteria. The sigma-54 regulator, a hypothetical zinc-containing protein and the can upstream of the Kdp system all have significant similarity to their corresponding proteins in multiple Acidithiobacillus spp. (Acidithiobacillus ferriphilus and Acidithiobacillus ferrivorans) as well as the closely related Ambacidithiobacillus sulfuriphilus and Fervidacidithiobacillus caldus acidophiles. The lack of kdp genes in all other Acidihalobacter genomes and the outgroup suggests it has been acquired via HGT, as well as the genes and gene contexts having significant similarity to species in the Burkholderiales order. We propose that the Kdp system in A. aeolianus was a result of HGT and has the potential to aid acid tolerance in low potassium environments.
Kch is a voltage-gated membrane potassium transporter commonly found in plants but has also been identified in E. coli, although its biological role is currently not understood (Reviewed in Epstein, 2003; Lundbäck et al., 2009). In eukaryotes, it is recognized as a voltage-gated ion channel that is associated with membrane potential regulation (Capera et al., 2019). The Kch protein was identified in all Acidihalobacter genomes and was not identified in H. neapolitanus. Best-hit analysis of the Kch proteins to the NCBI BlastP non-redundant database revealed that the Kch proteins in Acidihalobacter have significant similarity (46–53%) to Kch proteins from the acidophiles, F. caldus and Am. sulfuriphilus. putA (a bifunctional proline dehydrogenase/L-glutamate gamma-semialdehyde dehydrogenase) was found closely associated with kch in A. yilgarnensis and A. prosperus and is known to be involved in osmotolerance. Phylogenetic analysis, however, shows the Kch proteins from Acidihalobacter clustering closely to Kch proteins from other Chromatiales, and not the acidophiles Kch proteins (see Supplementary Figure 4). As Kch proteins are present, and cluster closely to the Chromatiales proteins, it is likely the Kch protein is a result of vertical decent in the Acidihalobacter genomes. The proximity of the kch to other osmotic tolerance genes may point to this area of the genome being important to both acid and osmotolerance in Acidihalobacter.
Spermidine Biosynthesis and Associated Genes
Spermidine is a positively charged membrane associated aliphatic, polycation polyamine that is possibly involved in acid and osmotic stress responses in E. coli and has been shown to reduce the outer membrane permeability caused by porins (Samartzidou et al., 2003). Spermidine biosynthesis pathways vary between bacterial species. Arginine is the precursor which can either be converted to citrulline or ornithine (via ArcB and RocF, respectively) which is then converted to putrescine (via SpeC) and subsequently converted to Spermidine via SpeE. An alternative pathway also includes arginine conversion to agmatine (AguA), which can be either converted directly to putrescine via SpeB, or to an intermediate CPT by AguA. The full spermidine biosynthesis pathway identified in Acidihalobacter is summarized in Figure 2. Both speE and speH (involved in the production of dSAM important in the conversion of putrescine to spermidine) were identified in all Acidihalobacter genomes and had significant similarity (66–68%) to SpeE and (73–79%) SpeH proteins in the closely related sister clade Thioalkalivibrio genus and other bacteria in the Chromatiales order. speE is found in the same genomic context as the urease accessory genes. speH is in a separate part of the genome that is also conserved between the Acidihalobacter genomes. Genes involved in osmotic tolerance are found upstream of speH including yhfA (a protein belonging to the osmotically inducible OsmC superfamily). As spermidine has been shown to be involved in both acid and osmotic stress responses, it is likely that spermidine synthesis in Acidihalobacter has been acquired via vertical descent from a common ancestor considering the presence of spermidine genes in other organisms from Chromatiales order and has possibly been lost in the H. neapolitanus outgroup genome.
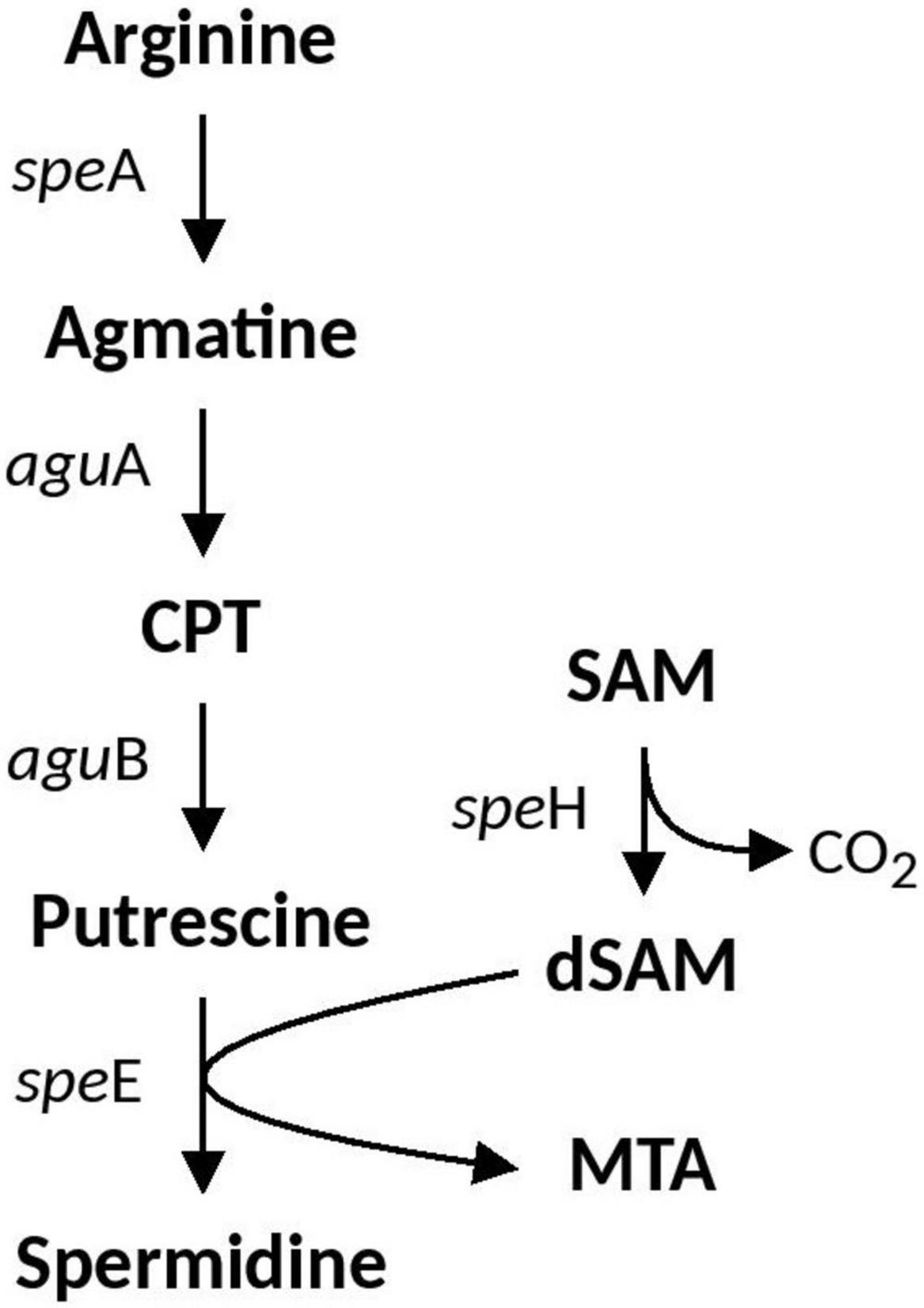
Figure 2. Spermidine biosynthesis pathway identified in Acidihalobacter. CPT, N-carbamoylputrescine; SAM, S-Adenosyl methionine; dSAM, decarboxylated-Adenosyl methionine; MTA, methylthioadenosine.
Hopanoid Biosynthesis
Hopanoid biosynthesis is hypothesized to be involved in regulating the fluidity and permeability of bacterial membranes, aiding in stress responses involved in acidity, temperature and salinity (Belin et al., 2018). Squalene is the precursor to hopanoid biosynthesis and the genes involved in it (hpnDE and hpnC) were predicted in all Acidihalobacter genomes, and were not identified in the H. neapolitanus genome. hpnDEC all had high similarity to orthologous proteins in the Thioalkalivibrio genus (>90%). A. ferrooxydans was the only genome that had the complete set of genes required to produce bacteriohopanetetrol (BHT) (hpnFHG) (the full hopanoid biosynthesis pathway can be seen in Figure 3). HpnF and HpnH both had high similarity to proteins from the Nitrosomonas genus (61.5 and 74–68%, respectively) and HpnH also had high similarity to proteins from microbes in the Methylococcaceae family (70–67%). hpnH, ispH, hpnF, hpG, and hpnA are all in a region of the genome with no synteny with the other Acidihalobacter genomes. An assembly gap was detected in the genome adjacent to hpnA, thus the full genomic context and evidence of HGT could not be determined.
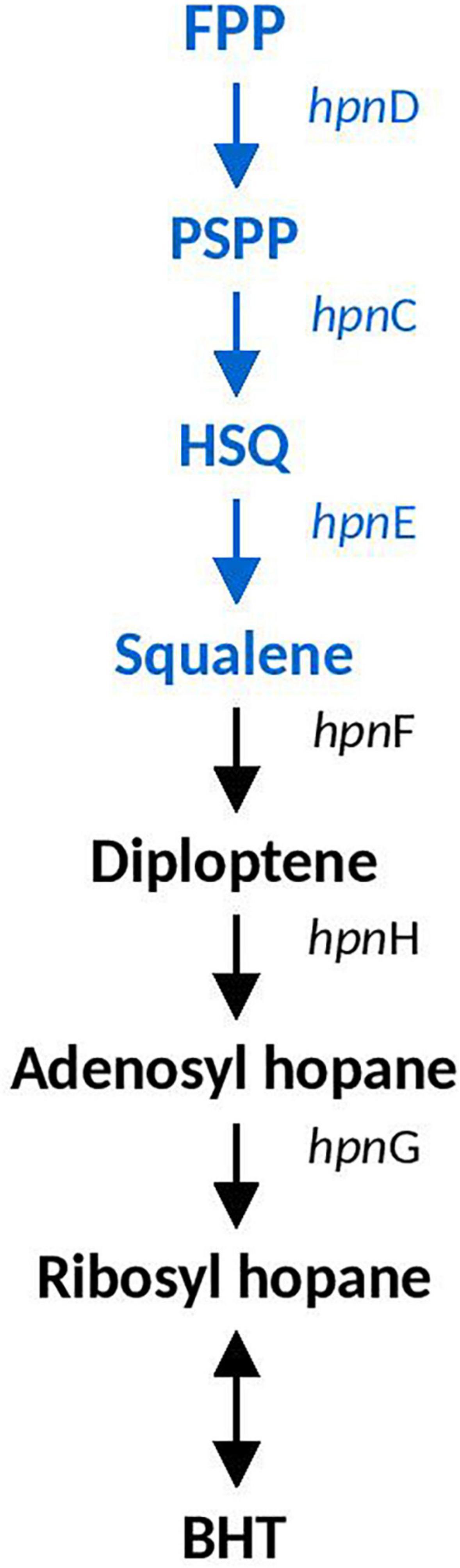
Figure 3. Hopanoid biosynthesis pathway identified in Acidihalobacter. Blue text indicates pathway identified in all Acidihalobacter genomes, black text indicates hopanoid biosynthesis pathway found in A. ferrooxydans. HSQ, hydroxysqualene; BHT, bacteriohopanetetrol.
Starvation Lipoprotein Slp
The outer membrane “starvation lipoprotein” gene slp is associated with the protection of E. coli during carbon starvation and metabolites that are toxic in low pH environments (namely organic acids) (Alexander and St John, 1994; Hommais et al., 2004; Mates et al., 2007). slp is located in the “acid fitness island” of E. coli and is co-expressed with other acid resistance genes. Two copies of slp were identified in all species of Acidihalobacter, excluding A. ferrooxydans, which only had one copy, and was absent in the outgroup. Slp copies contained a characteristic lipobox motif (Zückert, 2014; Vergara et al., 2020) with an Alanine amino acid residue in the position +2, suggesting an exportation of Slp (Supplementary Figure 5). Both copies, named slp-1 and slp-2, are adjacent to each other in A. yilgarnensis, A. prosperus and A. aeolianus, and the area of the genome is conserved between the three species (see Supplementary Figure 6). A cardiolipin synthase (cls) gene was found upstream of slp in all Acidihalobacter genomes and is involved in the structural organization of membranes and is associated with osmoregulation and acidic resistance (Romantsov et al., 2009; MacGilvray et al., 2012). The cardiolipin synthase presented higher similarity with Planctomycetes microorganisms according BlastP results. engB, a gene associated with the slp and glutamate decarboxylase genes in E. coli, was also identified in the same genomic context as Acidihalobacters slp (Mates et al., 2007). NhaP, a K+(Na+)/H+ antiporter, and lysR, a transcriptional regulator, were identified downstream of slp in A. ferrooxydans and both are associated with acid stress response of Vibrio cholerae (Kovacikova et al., 2010; Ante et al., 2015; Mourin et al., 2019). Phylogenetic analysis of the proteins shows the Slp-1 protein clustering with Slp proteins of Acidithiobacillus, and Slp-2 clustered with Chromatiales Slp proteins (see Figure 4). HGT events signals were identified for two genes upstream of slp-1 (AOU99095.1–AOU99096.1) by HGTector, suggesting Proteobacteria donor. This information, in addition to the genomic arrangement and phylogenetic analysis, allow us to hypothesize that genomic segment of Acidihalobacter including slp-1, cardiolipin synthase and genes upstream were gained by HGT events from a Proteobacteria donor, such as the extreme acidophile Acidithiobacillaceae; meanwhile slp-2 copies correspond to an ancestral gene from Chromatiales.

Figure 4. Unrooted phylogenetic tree of the predicted Acidihalobacter Slp amino acid sequences and their best hits from the NCBI non-redundant database, with collapsed branches by genera when possible (showing in parenthesis the number of leaves inside each). Acidihalobacter Slp proteins are colored red, Chromatiales proteins are colored green and other acidophiles proteins are colored blue. The time scale bar represents 1 amino acid substitution per site.
Acid Resistance Mechanisms: Second Line of Defense
“Second line of defense” (Vergara et al., 2020) mechanisms are involved in removing excess protons that have passed the cellular membrane and have entered the cytoplasm. Such mechanisms consist of cytoplasmic buffering systems and proton antiporters, pumping protons out of the cell. The glutamate decarboxylase system, the arginine decarboxylase system, the urease system, carbonic anhydrase and the ClcA antiporters found in Acidihalobacter are described below.
Glutamate Decarboxylase System
The glutamate decarboxylase system has been identified as a major mechanism in the acid stress response of E. coli (reviewed in Foster, 2004). Glutamate is transported into the cell by GadC (an amino acid permease), and GadA or GadB (the glutamate decarboxylase isozymes) replaces the a-carboxyl group of glutamate with H+, creating y-amino butyric acid (GABA) and CO2 removing a proton from the cytoplasm in the process. GABA is subsequently removed from the cell by GadC. The Gad decarboxylase system has also been identified in extreme acidophiles such as Leptospirillum spp. and F. caldus (Mangold et al., 2013; Vergara et al., 2020). GadA and GadC were both identified in all Acidihalobacter genomes but was missing in the H. neapolitanus genome. gadA and gadC in A. aeolianus and A. ferrooxydans were separated by a protein with no predicted putative domains. This hypothetical protein was predicted to be 248 amino acids and 7 transmembrane domains by HMMER, and has matches to ArcD, an arginine/ornithine antiporter in the HMMER database. In all genomes, best-hit analysis of GadA and GadC proteins showed 100% coverage and high amino acid percentage identity (ranging from 75 to 85.12%) to orthologs in Salinisphaera sp. LB1 (a haloacidophile), Mangrovitalea sediminis (a halophile), and Acidithiobacillus spp. (including At. thiooxidans, At. Ferridurans, and At. ferrivorans). This is an indication of a possible gene gain event in Acidihalobacter (see Supplementary Figures 7A,B).
Arginine Decarboxylase System
The Arginine decarboxylase system is another acid stress tolerance system identified in E. coli that works in the same way as the glutamate decarboxylase system (reviewed in Richard and Foster, 2004). Arginine decarboxylase, encoded by the adiA, replaces the a-carboxyl group of arginine with a proton, producing CO2 and agmatine, which is removed from the cell via antiporter AdiC. All Acidihalobacter genomes had hits to adiA, however, no hit to adiC was identified (no arginine decarboxylase genes were found in the H. neapolitanus genome). The complete arginine synthesis pathway was identified (argABCDEFGHO), thus Acidihalobacter may be capable of producing its own arginine, to be used in the arginine decarboxylase system. Best hits of the Acidihalobacter AdiA proteins to the BlastP non-redundant database indicated significant similarity to AdiA proteins of other Chromatiales organisms, suggesting vertical descent.
Urease System
One of the key mechanisms used by the enteric pathogen H. pylori to combat acid stress is the production of a highly expressed urease enzyme (Marcus and Scott, 2016). Urease converts urea to ammonia which is protonated to NH4+, neutralizing its environment. Urea import in H. pylori is controlled by UreI, a proton gated inner membrane urea channel, found to be activated by low pH (Bury-Moné et al., 2001; McNulty et al., 2013). Further buffering capacity is achieved by carbonic anhydrase which converts the carbon dioxide produced as a by-product of the urease activity into carbonic acid, which has a higher buffering capacity compared to ammonium (Marcus et al., 2005). The urease enzyme is coded for by ureA, ureB, and ureC (urease subunit gamma, beta, and alpha, respectively) and ureD (an ortholog of ureH), ureE, ureF, and ureG all of which are accessory proteins necessary for the expression of a functional urease enzyme (Akada et al., 2000). In H. pylori the regulation in the expression of the urease complex is dependent on the cellular pH and environmental pH, conditions sensed by the ArsRS and FlgRS two-component system. Urease is also found widely in soil bacteria, cyanobacteria and the extreme acidophile Ferrovum (Ullrich et al., 2016a) and studied in halophilic alkaliphiles in the production of biocement (Burbank et al., 2012; Dhami et al., 2013; Stabnikov et al., 2013; Veaudor et al., 2019). In the previously mentioned cases the urease activity is largely related to nitrogen assimilation and microbially induced calcite precipitation (Burbank et al., 2012). In cyanobacteria urea transport is controlled by the urea transport genes urtABCDE (Veaudor et al., 2019).
In all the Acidihalobacter genomes, ureA, ureB, ureC, ureD, ureE, ureF, and ureG were detected (ureC in A. yilganensis has multiple frameshift mutations resulting in a truncated version of UreC). These genes were not detected in the outgroup H. neapolitanus c2. The gene cluster structure of the urease system was unique in the Acidihalobacter genomes with a cyanase (cynS) gene between ureA and ureB. Cyanase is an enzyme that converts cyanate to carbamate, which spontaneously converts to ammonia and carbon dioxide (OCN– + HCO3– + 2H+ ↔ NH3 + 2CO2), directly consuming two protons as well as more protons consumed via the spontaneous conversion of ammonia to ammonium (NH3 + H+ ↔ NH4+) (Anderson and Little, 1986). Reporting of cynS incorporated into the urease operon in other genomes has not been found and could possibly be a novel alteration in Acidihalobacter allowing for further buffering capacity. As previously stated, H. pylori contains arsR, a pH activated transcriptional regulator, responsible for the transcription of the urease operon in low pH environments (Pflock et al., 2005). The arsR transcriptional regulator was found in A. yilgarnensis, A. prosperus, and A. aeolianus. This regulator was found upstream of the urease operon and also encompasses the genes adiA and speE (Figure 5). SpeE is a polyamine aminopropyltransferase in E. coli that is involved in the production of spermidine and is found to be involved in the acid tolerance of E. coli, possibly through the regulation of a wide range of acid tolerance genes including the glutamate decarboxylase system and the arginine decarboxylase system (Rhee et al., 2007). Whether or not the urease operon, adiA and speE are controlled by the arsR transcriptional regulator remains to be seen and must be tested further. The only urease transport system detected was urtABCDE in A. ferrooxydans and two copies in H. nepolitanus c2. In the case of H. pylori, gastrointestinal urea concentrations are very low (∼3 mM), and thus requires the active transport of urea into its cell for the urease system to be effective at combating acid stress (Scott et al., 2000). Environmental urea typically ranges from 0 to 13uM in concentration, over 2 orders of magnitude lower than the case of H. pylori; thus, it is unclear if the urease system found in A. yilgarnensis, A. prosperus and A. aeolianus would be effective enough in combating acid stress if no active urea transport system is found. A. ferrooxydans may be able to use the urease system as an acid resistance mechanism more effectively than the other member of Acidihalobacter due to the presence of the active urease transporter system in its genome. Further proteomic studies would be needed to validate this hypothesis. Due to the lack of the urease gene cluster in the outgroup and the similarity of the Acidihalobacter and Betaproteobacteria urease gene clusters, we speculate that the urease system was acquired via HGT in an ancestral Acidihalobacter.
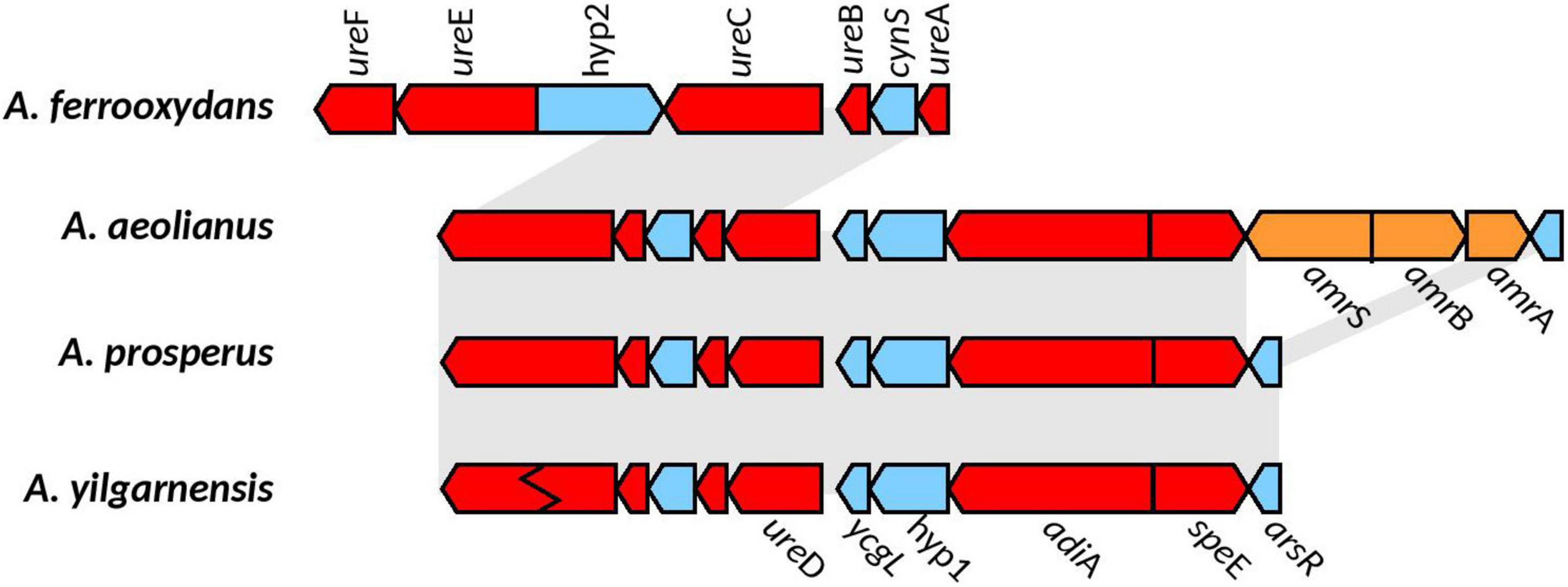
Figure 5. Genomic context of the urease gene cluster in the Acidihalobacter spp. genomes. Gray background shows synteny between genomes; blue genes are found in all genomes; red shows genes hypothesized to be involved in acid and osmotic tolerance; genes in orange correspond to unique genes (only found in their respective genomes). Ziz-zag pattern indicates a frameshift mutation in the gene. Hyp1, hypothetical 1; Hyp2, hypothetical 2; cynS, cynanate hydratase; ycgL, protein YcgL; arsR, arsenic resistance transcriptional regulator; amrS, AmmeMemoRadiSam system radical SAM enzyme; amrB, AmmeMedoRadiSam system protein B; amrA, AmmeMedoRadiSam system protein A.
Carbonic Anhydrase
Carbonic anhydrase (CA) is a proton-consuming enzyme that has been found to be upregulated in response to acid stress in H. pylori and the lack of the enzyme delays growth of H. pylori in acidic conditions (Sachs et al., 2006; Bury-Moneì et al., 2008). CAs are found in the cytoplasm and thus would directly contribute to minimizing the effect of protons leaking into the cytoplasm. CA is also known to catalyze the dissolution of carbonate minerals and in Aspergillus fumigatus is upregulated in potassium limited environments to increase the dissolution of potassium minerals to obtain potassium (Xiao et al., 2012; Sun et al., 2013). CA have also been found in acidophiles, but conclusions about their involvement in acid tolerance is not clear (Levicán et al., 2008; Li et al., 2012; Acuña et al., 2013). HGTector predicted the CA gene to have been acquired in the A. aeolianus and the A. yilgarnensis genomes from a Proteobacteria. The proximity of the CA to the Kdp system in A. aeolianus, as well as its clustering with CA proteins from the extreme acidophile class Acidithiobacillia may indicate an involvement in acid tolerance. Although, best BlastP hit analysis had many top hits to CAs of the extreme acidophile class Acidithiobacillia, phylogenetic analysis reveals clustering of the Acidihalobacter CAs to both Acidithiobacillia and Chromatiales CAs (see Supplementary Figure 8). This makes it unclear as to whether the horizontal gene transfer occurred from the acidophiles to the Acidihalobacter genomes, and subsequent loss in A. prosperus and A. ferrooxydans, or if the HGT occurred from an early Acidihalobacter common ancestor to the acidophiles.
ClcA Antiporters
Voltage gated ClC-type Cl–/H+ transporters found in E. coli are thought to be important in acid resistance systems. These proteins are hypothesized to function as a “electrical shunt for an outwardly directed virtual proton pump, linked to amino acid decarboxylation” (Iyer et al., 2002). Although chloride regulation is important in halophiles, no studies have indicated ClcA proteins to be involved in osmotic tolerance. Multiple predicted ClcA proteins were identified in the Acidihalobacter genomes, two in A. yilgarnensis, three in A. prosperus and A. aeolianus, four in A. ferrooxydans and one in the outgroup. Phylogenetic analysis of the ClcA proteins reveal five distinct clades of ClcA proteins which were named ClcA-1, ClcA-2, ClcA-3, ClcA-4, and ClcA-5 (see Supplementary Figure 9). clcA-1 and clcA-3 are located in the same area of the genome in A. yilgarnensis, A. prosperus, and A. aeolianus. Evidence of the loss of clcA-3 in A. yilgarnensis is evident as genomic synteny is conserved between these genomes (see Figure 6A). Both clcA-1 and clcA-3 are hypothesized to have been inherited via HGT, as phylogenetic analysis reveals clcA-1 clustering closely to ClcA proteins from Acidithiobacillaceae and clcA-3 clustering with Sneathiella spp., Symmachiella spp., Desulfobacteraceae spp., and Thiomicrospira spp. The genes surrounding clcA-1 and clcA-3 may also be involved in both acid and osmotic tolerance; pstACS (proton buffering), yggTUS (osmotic stress signaling), proH (associated with the production of proline as a compatible solute), and pilU and pilT (involved in cell adhesion, colonization, biofilm maturation and twitching) (Ito et al., 2009; Chen et al., 2015). pilG was identified to be upregulated in Acidihalobacter during increasing osmotic stress, thus these proteins may also play a role in osmotic tolerance, although no direct evidence is available of this mechanism in other bacteria (Dopson et al., 2016). This area of the genome may be important to acidic and osmotic tolerance in the Acidihalobacter, given that many of the genes in the same context are possibly involved in both osmotic and acidic tolerance. A. ferrooxydans is the only Acidihalobacter that had a second copy of the clcA-1 and is hypothesized to have been acquired via gene duplication of the original clcA-1 gene and has undergone further diversification. The second copy of clcA-1 in A. ferrooxydans is directly upstream of the hypothesized original clcA-1 gene and clusters closely with the original clcA-1 proteins (Figure 6B). clcA-2 was only identified in A. ferrooxydans and was in an area of the genome with no synteny between any other Acidihalobacter genomes (Figure 6A). Phylogenetic analysis indicates the clcA-2 protein also clustered closely with ClcA proteins in Acidithiobacillaceae, and is hypothesized to have been acquired via HGT from said Acidithiobacillaceae ClcA-4 clusters closely to ClcA proteins from other Chromatiales ClcA proteins, and is in a conserved region of the genome across all the Acidihalobacter genomes (see Supplementary Figure 10), thus we suspect that clcA-4 was acquired via vertical descent, and subsequently lost in H. neapolitanus. ClcA-5 was the only ClcA protein found in the outgroup H. neapolitanus c2 genome, however, is hypothesized to not be orthologous to any of the ClcA proteins found in the Acidihalobacter genomes as it was not phylogenetically related to any of the Acidihalobacter ClcA proteins. We hypothesize that the multiple copies of clcA in Acidihalobacter may point to its usefulness in the acidic response of Acidihalobacter. The abundance of chloride ions in Acidihalobacter environments could possibly allow for the ClcA mechanism of exporting H+ in exchange for Cl– is an effective and energetically favored mechanism, due to the abundance of chloride in Acidihalobacter environment. Although this mechanism is found in other acidophiles, it may be more useful for a haloacidophile like Acidihalobacter as it is adapted to a high chloride environment.
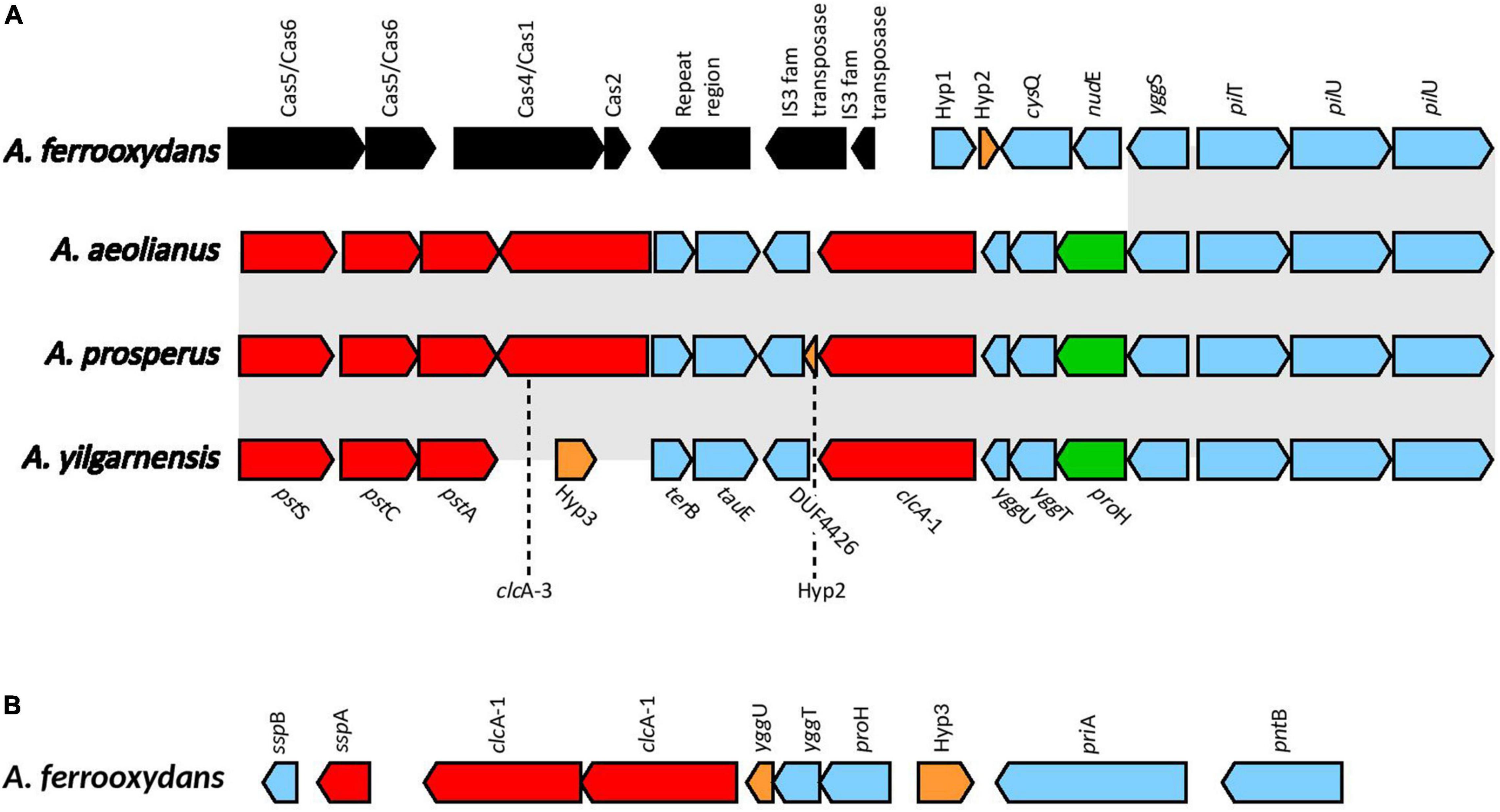
Figure 6. (A) The genomic context of clcA-1 in A. yilgarnensis, A. prosperus and A. aeolianus. (B) The genomic context of the clcA-1 genes in A. ferrooxydans. Genes in red; involved in acid tolerance, in green; involved in osmotolerance, in blue; others, in black; genes associated with HGT. Hyp, hypothetical proteins with no predicted putative domains; cysQ, 3′(2’),5′-bisphosphate nucleotidase; nudE, ADP compounds hydrolase; yggS, Pyridoxal phosphate homeostasis protein; pilT, type IV pilus twitching motility protein; pilU, type IV pilus ATPase; terB, tellurite resistance protein; tauE, sulfite exporter; DUF4426, domain of unknown function 4426; yggU, UPF0235 protein YggU; yggT, uncharacterized protein; proH, pyrroline-5-carboxylate reductase 1; sspB, stringent starvation protein B; sspA, stringent starvation protein A; priA, primosomal protein N′; pntB, NAD(P) transhydrogenase subunit beta.
Acid Tolerance Genes Absent in the Acidihalobacter Genus
Multiple genes that are hypothesized to be involved in acid tolerance were not identified in the Acidihalobacter genomes (the complete list of genes queried against Acidihalobacter can be found in the Supplementary Table 2). Acidihalobacter lacked the lysine, ornithine and agmatine decarboxylase systems. Regulatory genes involved in the glutamate decarboxylase system were absent, including ybaS, gadX, gadW, and gadE. Alternative regulatory genes could be responsible for the glutamate decarboxylase system in Acidihalobacter, and further experimental data may shed light on such genes. Multiple hopanoid biosynthesis genes that produce alternative hopanoid proteins were either absent in the Acidihalobacter genomes, or lacked essential intermediate genes. Genes involved in producing general stress proteins were not covered in this study as they are not specialized acid tolerance cellular mechanisms.
Model of Acidihalobacter Acid Resistance
A model explaining the acid resistance mechanisms identified in Acidihalobacter is proposed in Figure 7. We propose the major mechanisms aiding in the acid resistance of the Acidihalobacter spp. to be a combination of potassium transporters, multiple cytoplasmic buffering systems and cellular membrane alterations. The potassium transporters including TrkAH and Kch are likely involved in generating a reversed membrane potential, repelling protons from entering the cell in Acidihalobacter spp. A. aeolianus has the additional Kdp potassium transport system, possibly aiding it in maintaining a reversed membrane potential when exposed environments low in potassium ions. Acidihalobacter has multiple cytoplasmic buffering systems including the glutamate decarboxylase system, the urease system, and carbonic anhydrase. These buffering systems likely contribute to removing any protons that have leaked through the membrane and thus aid in maintaining a circumneutral intracellular pH. The urease system is reported as a strong buffering system, allowing H. pylori to survive in the acidity of the stomach. The effectiveness of this system in Acidihalobacter is unknown, however, as only A. ferrooxydans has a urea transport system. The glutamate decarboxylase system is another well-known buffering system used by enteric bacteria that have transient exposure to the acidic conditions of the stomach. It is evident in the study that the glutamate decarboxylase system is present in Acidihalobacter spp. and was likely acquired from acidophilic bacteria. Interestingly, glutamate is also commonly accumulated as an osmoprotectant in halophilic bacteria (Dinnbier et al., 1988; Csonka et al., 1994; Kang and Hwang, 2018), therefore the import of glutamate by the glutamate decarboxylase system may also aid during osmotic stress. Membrane alterations also likely play a role in the acid resistance of Acidihalobacter spp. including Slp lipoprotein, spermidine and BHT production in A. ferrooxydans. Multiple Slp lipoproteins identified in Acidihalobacter likely provide resistance to organic acids, that are detrimental to many chemolithotrophic acidophiles, including Acidihalobacter. This area of the genome has also uncovered many genes involved in outer membrane proteins, that may be important to assess for their function in Acidihalobacter response to acidic and osmotic stress. BHTs produced in A. ferrooxydans likely aid in its resistance to acidic conditions as hopanoids have been identified in acid mine drainage and other acidic environments (Jones et al., 2012). Although the mechanism of spermidine in acid tolerance is not fully understood, Acidihalobacter spp. have the genetic potential to produce spermidine, and may aid in its acid tolerance. Multiple ClcA proteins identified in Acidihalobacter likely exchange chloride for the removal of protons, which may be especially effective in Acidihalobacter as opposed to other acidophiles sensitive to chloride ions.
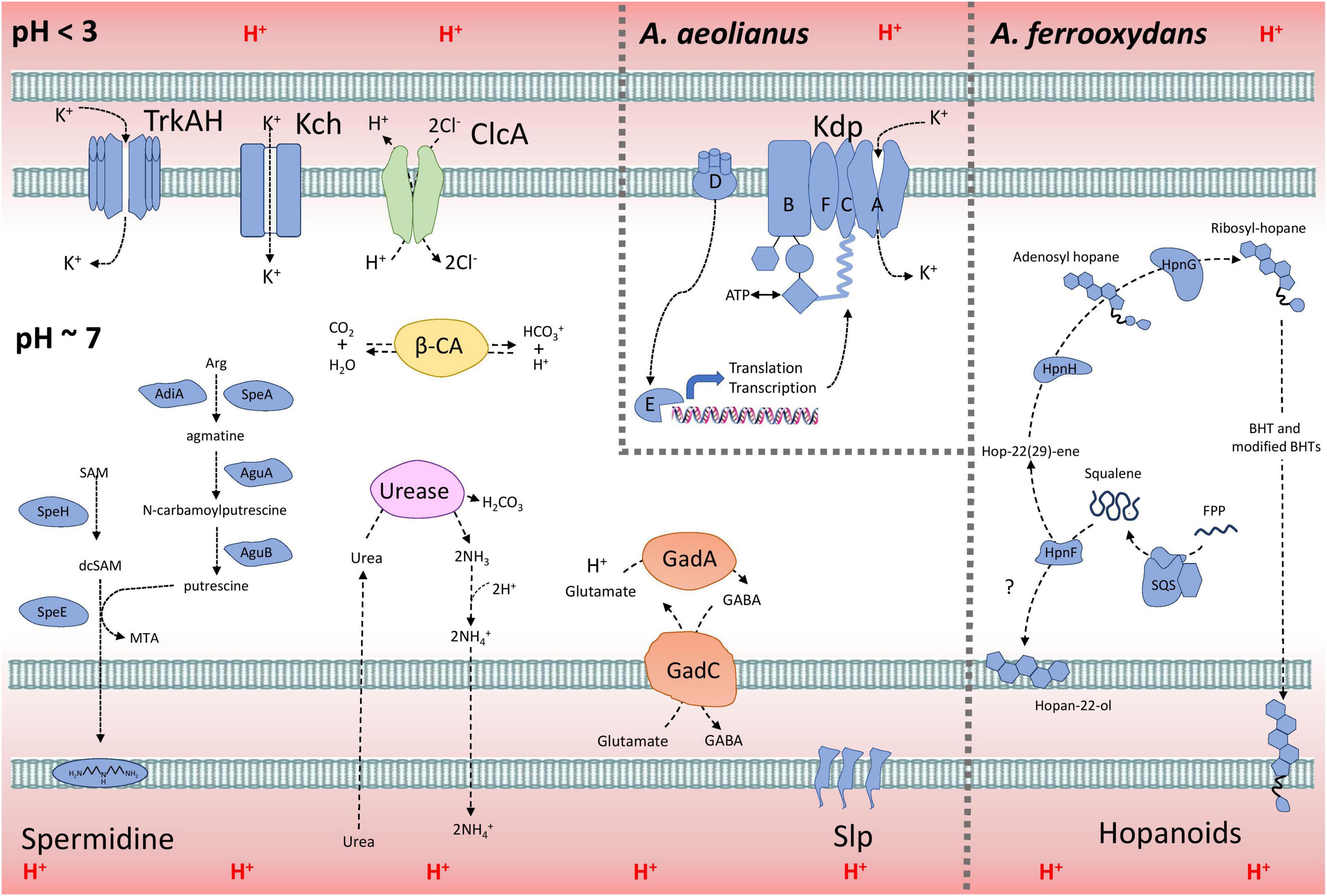
Figure 7. The model of acid resistance in the Acidihalobacter spp. First line of defense mechanisms are displayed in blue and second line of defense mechanisms are displayed in other colors (green = ClcA; yellow = β-CA; purple = urease system; and orange = Gad system).
Phylogenetic Distribution of Acid Resistance Genes and Their Inferred Evolutionary Trajectories
The hypothesized evolutionary trajectory leading to the Acidihalobacter genus becoming acidophilic from an inferred neutrophilic ancestor is summarized in Figure 8, displaying gene gain and gene loss events and gene inheritance through vertical descent. Many genes involved in acid resistance, were hypothesized to be in the last inferred common ancestor including the potassium transporters (trkAH, kch) and membrane alterations (hpnJCDE, ispH and speEH). It is evident that the accumulation of potassium via potassium pumps is vital when responding to both osmotic and acidic stress. In this study we hypothesize the TrkAH low affinity potassium transporter to have been acquired via vertical descent from a common halophilic ancestor. Although the Kch proteins have Best-BlastP hits to many Kch proteins from the Acidithiobacillaceae family, phylogenetic analysis reveals the Acidihalobacter spp. Kch proteins cluster closely with Kch proteins from organisms of the Chromatiales class. Thus, we inferred the Kch protein to have also been inherited via vertical descent, and subsequently lost in the outgroup. The Kdp high affinity potassium transport system was hypothesized to have been acquired in the A. aeolianus genome via HGT likely from Betaproteobacteria, according to the phylogenetic tree (Supplementary Figure 3B). A. ferrooxydans BHT synthesis genes were likely obtained in the genome via both vertical descent and HGT. HpnCDE are involved in squalene synthesis, an early intermediate product of BHT production. These protein sequences were phylogenetically related to the same proteins in other Chromatiales species, indicating they were likely vertically inherited. Squalene is known to be associated with halophilic archaea’s membranes, integrating with cellular lipids, helping them to pack closer together, thus is supports HpnCDE likely being inherited from a halophilic common ancestor (Gilmore et al., 2013). Hopanoids are also associated with saline stress, however, the remainder of the BHT synthesis genes (hpnFGH) had best-BlastP hits to non-Chromatiales proteins and due to the lack of these genes in the other Acidihalobacter genomes, as well as the outgroup, we hypothesize these genes were gained via HGT in the A. ferrooxydans genome. All the typical “first line of defense” mechanisms described in extreme acidophiles, in the case of Acidihalobacter, seem to originate from a neutrophilic-halophilic ancestry. When it comes to second line of defense mechanisms, it is clear in this study that Acidihalobacter has likely gained two cytoplasmic buffering systems through HGT. The glutamate decarboxylase system (gadAC) was acquired via HGT into the Acidihalobacter genome from a possible acidophilic microbe. Urease (ureABCDEFG) genes were also acquired into the Acidihalobacter genome via HGT, but we hypothesize from a non-acidophilic counterpart. Although not acquired from an acidophile, the urease system is known to be a strong buffering system for non-acidophilic enteric bacteria such as H. pylori, thus may be able to contribute to acid tolerance in Acidihalobacter. Urease was identified in the extreme acidophile Ferrovum sp. JA12, and it was also noted in the study that urease has not been identified in any other iron oxidizing acidophiles (Ullrich et al., 2016b). As mentioned previously, it is unclear how effective the urease system is in A. yilgarnensis, A. prosperus, and A. aeolianus, as no urea transporter was identified in these genomes. ClcA proteins in the Acidihalobacter genomes were hypothesized to have independent evolutionary histories. clcA-1 was likely acquired via HGT in the Acidihalobacter genomes from an acidophilic counterpart and underwent a duplication event in A. ferrooxydans. clcA-2 we hypothesize was also acquired via HGT in A. ferrooxydans independently. clcA-3 was likely acquired via HGT, however, from a non-acidophilic counterpart. clcA-4 was the only ClcA protein to have been vertically inherited and subsequently lost in the outgroup as it is phylogenetically related to other ClcA proteins from Chromatiales species. Although ClcA proteins are recognized in the acid stress responses of acidophiles, they have also been identified in some halophilic bacteria, although the function of ClcA proteins in halophiles has not been discussed (John et al., 2019). The evolutionary origin of carbonic anhydrase remains unclear. Acidihalobacter has been identified in shared environments with acidophilic microorganisms that we hypothesize had HGT events between species namely members of the Acidithiobacillaceae family (Norris et al., 2020; Abramov et al., 2021). Both Acidihalobacter and Acidithiobacillaceae species have been isolated from the acidified sulfide-rich seawater sites from the islands of Vulcano and Milos (Simmons and Norris, 2002; Norris et al., 2020). Other environments that have identified Acidihalobacter and other acidophiles inhabiting the same ecological niche include arid soils in the Atacama desert Chile (Neilson et al., 2012) and acidic hypersaline river sediments of Western Australia (Lu et al., 2016). The evidence of professional acidophiles and Acidihalobacter supports environmental scenarios for HGT events to have occurred between these bacteria. These environmental niches hypothetically provide the ideal conditions for acidophiles and halophiles to live in close proximity and also apply the selection pressure for existing halophiles and acidophiles alike to share genetic capabilities. We hypothesize Acidihalobacter is an example of a halophile gaining acid tolerance mechanisms, however, the opposite case may still be discovered in other bacteria. The cellular mechanisms for microorganisms to thrive in saline and acidophilic conditions independently is well established. Understanding what combination of these cellular mechanisms Acidihalobacter has, that allows it to cope in both extreme acidity and saline conditions will give insight into the unique capabilities of haloacidophiles. The current understanding of Acidihalobacter as a halophile, is that it utilizes potassium pumps, periplasmic glucans and multiple compatible solutes to regulate its internal osmotic pressure to its saline environment. It is proposed by Khaleque et al. (2019) that initially potassium accumulation and the production of periplasmic glucans are the primary mechanisms used to respond to initial saline stress, and as salinity increases, the accumulation and production of compatible solutes would take priority. Identifying the genetic capabilities Acidihalobacter spp. possess that allows the genus to thrive in acidic conditions is vital to understand its evolution as a polyextremophile.
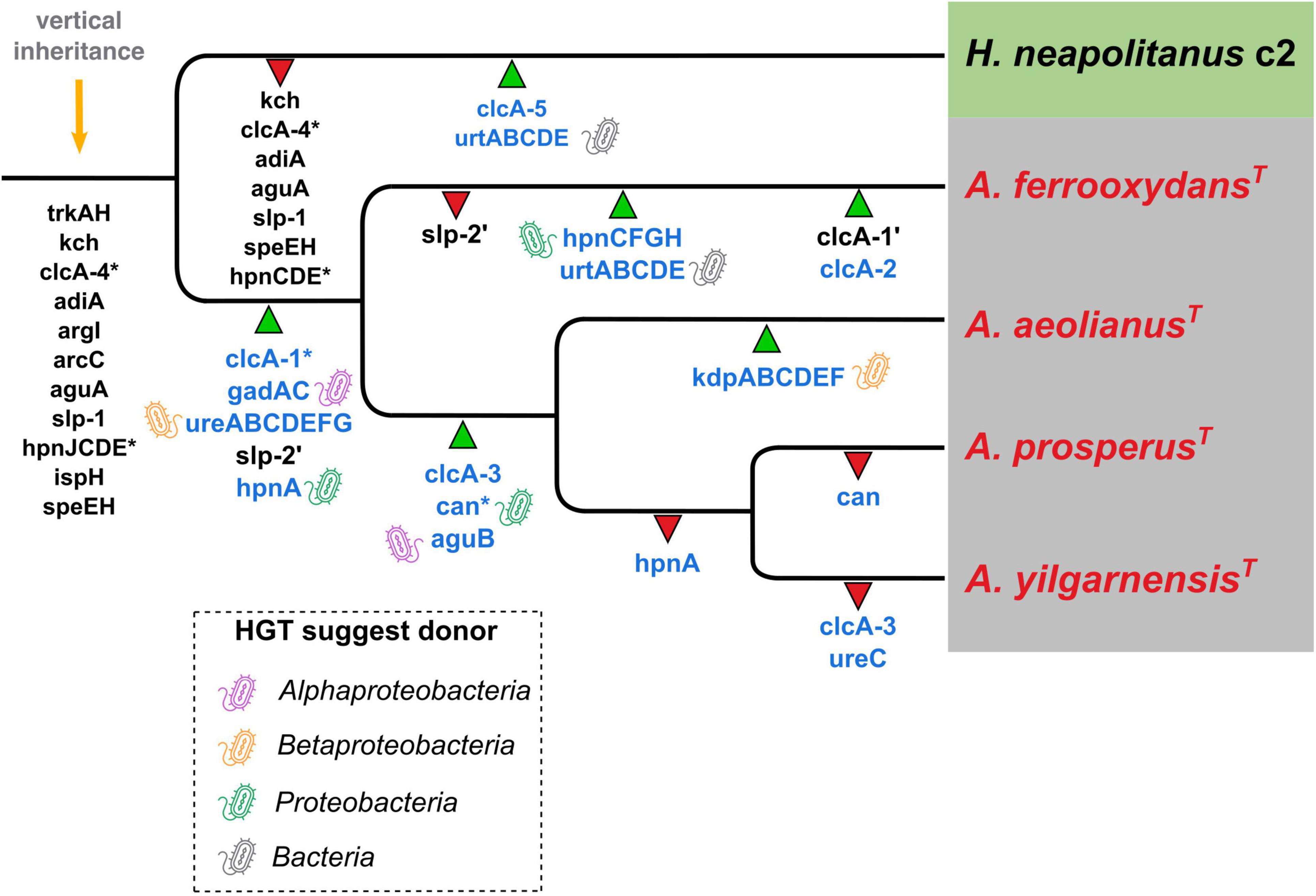
Figure 8. Inferred phylogenetic distribution of acid resistance genes. Green triangles represent gene gain events (the ’ in slp and clcA genes represent a duplication event) and red triangles represent gene loss events. Gene names in blue indicate hypothesized HGT events (* marks the uncertain origin for that gene) and the bacteria-icon represents the putative origin of those events. Orange arrow = predicted genes inherited by vertical descent.
It is evident that Acidihalobacter is a genus rooted in a class of bacteria that are mostly halophilic/halotolerant neutrophiles and alkaliphiles. Acidihalobacter is also the only genus in the Chromatiales order that has been found to be acidophilic. We hypothesize that the ancestor of Acidihalobacter was a halophile that subsequently gained the genetic capability to thrive in acidic conditions. This research has identified some of these hypothesized genes involved in acid tolerance and uncovered evidence of HGT events that may have been involved in Acidihalobacter becoming acidophilic. The main mechanisms identified that likely are involved in acid tolerance in Acidihalobacter spp. include the potassium transporters TrkAH, Kch, and Kdp, membrane-associated alterations Slp, spermidine and BHTs, cytoplasmic buffering mechanisms, glutamate decarboxylase, urease and carbonic anhydrase, and the antiporters ClcA’s. This study can inform further experimentation studying the acid tolerance of Acidihalobacter.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: NCBI GCF_000754095.2, GCF_001753245.1, GCF_001753165.1 GCF_001975725.1, and GCF_000024765.1.
Author Contributions
DH and EW conceived the study. KB carried out the research. All authors contributed to data collection, analysis, and manuscript preparation and read and approved the final manuscript.
Funding
This work was supported by Fondecyt 1181717 (DH), ANID FONDECYT 3190792 (CG), and Centro Ciencia & Vida, FB210008, Financiamiento Basal para Centros Científicos y Tecnológicos de Excelencia de ANID.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Diego Cortez for his insightful input. KB acknowledges support through an Australian Government Research Training Program Scholarship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.848410/full#supplementary-material
Footnote
References
Abramov, S. M., Straub, D., Tejada, J., Grimm, L., Schädler, F., Bulaev, A., et al. (2021). Biogeochemical niches of fe-cycling communities influencing heavy metal transport along the rio tinto, Spain. Appl. Environ. Microbiol. 88:e0229021. doi: 10.1128/AEM.02290-21
Acuña, L. G., Cárdenas, J. P., Covarrubias, P. C., Haristoy, J. J., Flores, R., Nuñez, H., et al. (2013). Architecture and repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus. PLoS One 8:e78237. doi: 10.1371/journal.pone.0078237
Akada, J. K., Shirai, M., Takeuchi, H., Tsuda, M., and Nakazawa, T. (2000). Identification of the urease operon in Helicobacter pylori and its control by mRNA decay in response to pH. Mol. Microbiol. 36, 1071–1084. doi: 10.1046/j.1365-2958.2000.01918.x
Alexander, D. M., and St John, A. C. (1994). Characterization of the carbon starvation-inducible and stationary phase-inducible gene slp encoding an outer membrane lipoprotein in Escherichia coli. Mol. Microbiol. 11, 1059–1071. doi: 10.1111/j.1365-2958.1994.tb00383.x
Anderson, P. M., and Little, R. M. (1986). Kinetic properties of cyanase. Biochemistry 25, 1621–1626. doi: 10.1021/bi00355a026
Ante, V. M., Bina, X. R., and Bina, J. E. (2015). The LysR-type regulator LeuO regulates the acid tolerance response in Vibrio cholerae. Microbiology 161, 2434–2443. doi: 10.1099/mic.0.000194
Asnicar, F., Thomas, A. M., Beghini, F., Mengoni, C., Manara, S., Manghi, P., et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11:2500. doi: 10.1038/s41467-020-16366-7
Baker-Austin, C., and Dopson, M. (2007). Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 15, 165–171. doi: 10.1016/j.tim.2007.02.005
Bakker, E. P., Borchard, A., Michels, M., Altendorf, K., and Siebers, A. (1987). High-affinity potassium uptake system in Bacillus acidocaldarius showing immunological cross-reactivity with the Kdp system from Escherichia coli. J. Bacteriol. 169, 4342–4348. doi: 10.1128/jb.169.9.4342-4348.1987
Belin, B. J., Busset, N., Giraud, E., Molinaro, A., Silipo, A., and Newman, D. K. (2018). Hopanoid lipids: from membranes to plant–bacteria interactions. Nat. Rev. Microbiol. 16, 304–315. doi: 10.1038/nrmicro.2017.173
Boden, R. (2017). Reclassification of Halothiobacillus hydrothermalis and Halothiobacillus halophilus to Guyparkeria gen. nov. in the Thioalkalibacteraceae fam. nov., with emended descriptions of the genus Halothiobacillus and family Halothiobacillaceae. Int. J. Syst. Evol. Microbiol. 67, 3919–3928. doi: 10.1099/ijsem.0.002222
Bowers, K. J., and Wiegel, J. (2011). Temperature and pH optima of extremely halophilic archaea: a mini-review. Extremophiles 15, 119–128. doi: 10.1007/s00792-010-0347-y
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Buetti-Dinh, A., Dethlefsen, O., Friedman, R., and Dopson, M. (2016). Transcriptomic analysis reveals how a lack of potassium ions increases Sulfolobus acidocaldarius sensitivity to pH changes. Microbiology 162, 1422–1434. doi: 10.1099/mic.0.000314
Burbank, M. B., Weaver, T. J., Williams, B. C., and Crawford, R. L. (2012). Urease activity of ureolytic bacteria isolated from six soils in which calcite was precipitated by indigenous bacteria. Geomicrobiol. J. 29, 389–395. doi: 10.1080/01490451.2011.575913
Bury-Moneì, S., Mendz, G. L., Ball, G. E., Thibonnier, M., Stingl, K., Ecobichon, C., et al. (2008). Roles of α and β carbonic anhydrases of Helicobacter pylori in the urease-dependent response to acidity and in colonization of the murine gastric mucosa. Infect. Immunity 76, 497–509. doi: 10.1128/iai.00993-07
Bury-Moné, S., Skouloubris, S., Labigne, A., and De Reuse, H. (2001). The Helicobacter pylori UreI protein: role in adaptation to acidity and identification of residues essential for its activity and for acid activation. Mol. Microbiol. 42, 1021–1034. doi: 10.1046/j.1365-2958.2001.02689.x
Capera, J., Serrano-Novillo, C., Navarro-Pérez, M., Cassinelli, S., and Felipe, A. (2019). The potassium channel odyssey: mechanisms of traffic and membrane arrangement. Int. J. Mol. Sci. 20:734. doi: 10.3390/ijms20030734
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Chao, J., Wang, W., Xiao, S., and Liu, X. (2008). Response of Acidithiobacillus ferrooxidans ATCC 23270 gene expression to acid stress. World J. Microbiol. Biotechnol. 24, 2103–2109. doi: 10.1007/s11274-008-9715-5
Chen, L.-X., Hu, M., Huang, L.-N., Hua, Z.-S., Kuang, J.-L., Li, S.-J., et al. (2015). Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 9, 1579–1592. doi: 10.1038/ismej.2014.245
Cholo, M. C., van Rensburg, E. J., Osman, A. G., and Anderson, R. (2015). Expression of the Genes encoding the Trk and Kdp potassium transport systems of Mycobacterium tuberculosis during growth in vitro. Biomed Res. Int. 2015:608682. doi: 10.1155/2015/608682
Christel, S., Herold, M., Bellenberg, S., El Hajjami, M., Buetti-Dinh, A., Pivkin, I. V., et al. (2018). Multi-omics reveals the lifestyle of the acidophilic, mineral-oxidizing model species Leptospirillum ferriphilum. Appl. Environ. Microbiol. 84:e02091–17. doi: 10.1128/AEM.02091-17
Csonka, L. N., Ikeda, T. P., Fletcher, S. A., and Kustu, S. (1994). The accumulation of glutamate is necessary for optimal growth of Salmonella typhimurium in media of high osmolality but not induction of the proU operon. J. Bacteriol. 176, 6324–6333. doi: 10.1128/jb.176.20.6324-6333.1994
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Dhami, N. K., Reddy, M. S., and Mukherjee, A. (2013). Biomineralization of calcium carbonates and their engineered applications: a review. Front. Microbiol. 4:314. doi: 10.3389/fmicb.2013.00314
Dinnbier, U., Limpinsel, E., Schmid, R., and Bakker, E. P. (1988). Transient accumulation of potassium glutamate and its replacement by trehalose during adaptation of growing cells of Escherichia coli K-12 to elevated sodium chloride concentrations. Arch. Microbiol. 150, 348–357. doi: 10.1007/BF00408306
Dopson, M., Holmes, D. S., Lazcano, M., McCredden, T. J., Bryan, C. G., Mulroney, K. T., et al. (2016). Multiple osmotic stress responses in result in tolerance to chloride ions. Front. Microbiol. 7:2132. doi: 10.3389/fmicb.2016.02132
Dorus, S., Mimura, H., and Epstein, W. (2001). Substrate-binding clusters of the k -transporting Kdp ATPase of Escherichia coli investigated by amber suppression scanning mutagenesis. J. Biol. Chem. 276, 9590–9598. doi: 10.1074/jbc.m009365200
Epstein, W. (2003). The roles and regulation of potassium in bacteria. Prog. Nucleic Acid Res. Mol. Biol. 75, 293–320. doi: 10.1016/s0079-6603(03)75008-9
Feehily, C., and Karatzas, K. A. G. (2013). Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 114, 11–24. doi: 10.1111/j.1365-2672.2012.05434.x
Foster, J. W. (2004). Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2, 898–907. doi: 10.1038/nrmicro1021
Gaßel, M., Möllenkamp, T., Puppe, W., and Altendorf, K. (1999). The KdpF subunit is part of the K -translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J. Biol. Chem. 274, 37901–37907. doi: 10.1074/jbc.274.53.37901
Gaßel, M., Siebers, A., Epstein, W., and Altendorf, K. (1998). Assembly of the Kdp complex, the multi-subunit K -transport ATPase of Escherichia coli. Biochim. Biophys. Acta BBA Biomembranes 1415, 77–84. doi: 10.1016/s0005-2736(98)00179-5
Gilmore, S. F., Yao, A. I., Tietel, Z., Kind, T., Facciotti, M. T., and Parikh, A. N. (2013). Role of squalene in the organization of monolayers derived from lipid extracts of Halobacterium salinarum. Langmuir 29, 7922–7930. doi: 10.1021/la401412t
González-Rosales, C., Vergara, E., Dopson, M., Valdés, J. H., and Holmes, D. S. (2022). Integrative genomics shed light on evolutionary forces shaping the acidithiobacillia class acidophilic lifestyle. Front. Microbiol. 12:822229. doi: 10.3389/fmicb.2021.822229
Gumulya, Y., Boxall, N. J., Khaleque, H. N., Santala, V., Carlson, R. P., and Kaksonen, A. H. (2018). In a quest for engineering acidophiles for biomining applications: challenges and opportunities. Genes 9:116. doi: 10.3390/genes9020116
He, G., Wu, C., Huang, J., and Zhou, R. (2016). Acid tolerance response of Tetragenococcus halophilus: a combined physiological and proteomic analysis. Process Biochem. 51, 213–219. doi: 10.1016/j.procbio.2015.11.035
Hesse, J. E., Wieczorek, L., Altendorf, K., Reicin, A. S., Dorus, E., and Epstein, W. (1984). Sequence homology between two membrane transport ATPases, the Kdp-ATPase of Escherichia coli and the Ca2 -ATPase of sarcoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 81, 4746–4750. doi: 10.1073/pnas.81.15.4746
Hommais, F., Krin, E., Coppée, J.-Y., Lacroix, C., Yeramian, E., Danchin, A., et al. (2004). GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology 150, 61–72. doi: 10.1099/mic.0.26659-0
Huber, H., and Stetter, K. O. (1989). Thiobacillus prosperus sp. nov., represents a new group of halotolerant metal-mobilizing bacteria isolated from a marine geothermal field. Arch. Microbiol. 151, 479–485. doi: 10.1007/bf00454862
Ito, T., Uozumi, N., Nakamura, T., Takayama, S., Matsuda, N., Aiba, H., et al. (2009). The implication of YggT of Escherichia coli in osmotic regulation. Biosci. Biotechnol. Biochem. 73, 2698–2704. doi: 10.1271/bbb.90558
Iyer, R., Iverson, T. M., Accardi, A., and Miller, C. (2002). A biological role for prokaryotic ClC chloride channels. Nature 419, 715–718. doi: 10.1038/nature01000
John, J., Siva, V., Richa, K., Arya, A., and Kumar, A. (2019). Life in high salt concentrations with changing environmental conditions: insights from genomic and phenotypic analysis of Salinivibrio sp. Microorganisms 7:577. doi: 10.3390/microorganisms7110577
Johnson, D. B., and Schippers, A. (2017). Editorial: recent advances in acidophile microbiology: fundamentals and applications. Front. Microbiol. 8:428. doi: 10.3389/fmicb.2017.00428
Jones, D. S., Albrecht, H. L., Dawson, K. S., Schaperdoth, I., Freeman, K. H., Pi, Y., et al. (2012). Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 6, 158–170. doi: 10.1038/ismej.2011.75
Kang, Y., and Hwang, I. (2018). Glutamate uptake is important for osmoregulation and survival in the rice pathogen Burkholderia glumae. PLoS One 13:e0190431. doi: 10.1371/journal.pone.0190431
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kelly, D. P., and Wood, A. P. (2000). Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 50 Pt 2, 511–516. doi: 10.1099/00207713-50-2-511
Khaleque, H. N., Corbett, M. K., Ramsay, J. P., Kaksonen, A. H., Boxall, N. J., and Watkin, E. L. J. (2017a). Complete genome sequence of Acidihalobacter prosperus strain F5, an extremely acidophilic, iron- and sulfur-oxidizing halophile with potential industrial applicability in saline water bioleaching of chalcopyrite. J. Biotechnol. 262, 56–59. doi: 10.1016/j.jbiotec.2017.10.001
Khaleque, H. N., Fathollazadeh, H., González, C., Shafique, R., Kaksonen, A. H., Holmes, D. S., et al. (2020). Unlocking survival mechanisms for metal and oxidative stress in the extremely acidophilic, halotolerant acidihalobacter Genus. Genes 11:1392. doi: 10.3390/genes11121392
Khaleque, H. N., González, C., Shafique, R., Kaksonen, A. H., Holmes, D. S., and Watkin, E. L. J. (2019). Uncovering the mechanisms of halotolerance in the extremely acidophilic members of the acidihalobacter genus through comparative genome analysis. Front. Microbiol. 10:155. doi: 10.3389/fmicb.2019.00155
Khaleque, H. N., Ramsay, J. P., Murphy, R. J. T., Kaksonen, A. H., Boxall, N. J., and Watkin, E. L. J. (2017c). Draft genome sequence of the acidophilic, halotolerant, and iron/sulfur-oxidizing Acidihalobacter prosperus DSM 14174 (Strain V6). Genome Announc. 5:e01469–16. doi: 10.1128/genomeA.01469-16
Khaleque, H. N., Ramsay, J. P., Murphy, R. J. T., Kaksonen, A. H., Boxall, N. J., and Watkin, E. L. J. (2017b). Draft genome sequence of acidihalobacter ferrooxidans DSM 14175 (Strain V8), a new iron- and sulfur-oxidizing, halotolerant, acidophilic species. Genome Announcements 5:e00413–17. doi: 10.1128/genomea.00413-17
Khaleque, H. N., Shafique, R., Kaksonen, A. H., Boxall, N. J., and Watkin, E. L. J. (2018). Quantitative proteomics using SWATH-MS identifies mechanisms of chloride tolerance in the halophilic acidophile Acidihalobacter prosperus DSM 14174. Res. Microbiol. 169, 638–648. doi: 10.1016/j.resmic.2018.07.002
Kixmüller, D., Strahl, H., Wende, A., and Greie, J.-C. (2011). Archaeal transcriptional regulation of the prokaryotic KdpFABC complex mediating K uptake in H. salinarum. Extremophiles 15, 643–652. doi: 10.1007/s00792-011-0395-y
Korolev, N. (2021). How potassium came to be the dominant biological cation: of metabolism, chemiosmosis, and cation selectivity since the beginnings of life. BioEssays 43:2000108. doi: 10.1002/bies.202000108
Kovacikova, G., Lin, W., and Skorupski, K. (2010). The LysR-type virulence activator AphB regulates the expression of Genes in Vibrio cholerae in response to low pH and Anaerobiosis. Journal of Bacteriology 192, 4181–4191. doi: 10.1128/jb.00193-10
Kraegeloh, A., Amendt, B., and Kunte, H. J. (2005). Potassium transport in a halophilic member of the bacteria domain: identification and characterization of the K uptake systems TrkH and TrkI from Halomonas elongata DSM 2581 T. J. Bacteriol. 187, 1036–1043. doi: 10.1128/jb.187.3.1036-1043.2005
Levicán, G., Ugalde, J. A., Ehrenfeld, N., Maass, A., and Parada, P. (2008). Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations. BMC Genomics 9:581. doi: 10.1186/1471-2164-9-581
Li, L., Fu, M.-L., Zhao, Y.-H., and Zhu, Y.-T. (2012). Characterization of carbonic anhydrase II from Chlorella vulgaris in bio-CO2 capture. Environ. Sci. Pollut. Res. 19, 4227–4232. doi: 10.1007/s11356-012-1077-8
López-Pérez, M., Ghai, R., Leon, M. J., Rodríguez-Olmos, Á, Copa-Patiño, J. L., Soliveri, J., et al. (2013). Genomes of “Spiribacter”, a streamlined, successful halophilic bacterium. BMC Genomics 14:787. doi: 10.1186/1471-2164-14-787
Lu, S., Peiffer, S., Lazar, C. S., Oldham, C., Neu, T. R., Ciobota, V., et al. (2016). Extremophile microbiomes in acidic and hypersaline river sediments of Western Australia. Environ. Microbiol. Rep. 8, 58–67. doi: 10.1111/1758-2229.12351
Lundbäck, A.-K., Müller, S. A., Engel, A., and Hebert, H. (2009). Assembly of Kch, a putative potassium channel from Escherichia coli. J. Struct. Biol. 168, 288–293. doi: 10.1016/j.jsb.2009.07.018
Macalady, J. L., Vestling, M. M., Baumler, D., Boekelheide, N., Kaspar, C. W., and Banfield, J. F. (2004). Tetraether-linked membrane monolayers in Ferroplasma spp: a key to survival in acid. Extremophiles 8, 411–419. doi: 10.1007/s00792-004-0404-5
MacGilvray, M. E., Lapek, J. D., Friedman, A. E., and Quivey, R. G. (2012). Cardiolipin biosynthesis in Streptococcus mutans is regulated in response to external pH. Microbiology 158, 2133–2143. doi: 10.1099/mic.0.057273-0
Mangold, S., Rao Jonna, V., and Dopson, M. (2013). Response of Acidithiobacillus caldus toward suboptimal pH conditions. Extremophiles 17, 689–696. doi: 10.1007/s00792-013-0553-5
Marcus, E. A., and Scott, D. R. (2016). “Gastric colonization by H. pylori,” in Helicobacter pylori, 1st Edn, ed. N. Kim (Singapore: Springer), 23–34. doi: 10.1007/978-981-287-706-2_2
Marcus, E. A., Moshfegh, A. P., Sachs, G., and Scott, D. R. (2005). The periplasmic α-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 187, 729–738. doi: 10.1128/jb.187.2.729-738.2005
Mates, A. K., Sayed, A. K., and Foster, J. W. (2007). Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low ph and mediate a cell density-dependent acid resistance. J. Bacteriol. 189, 2759–2768. doi: 10.1128/jb.01490-06
McNulty, R., Ulmschneider, J. P., Luecke, H., and Ulmschneider, M. B. (2013). Mechanisms of molecular transport through the urea channel of Helicobacter pylori. Nat. Commun. 4:2900. doi: 10.1038/ncomms3900
Minegishi, H. (2013). “Halophilic, acidophilic, and haloacidophilic prokaryotes,” in Polyextremophiles. Cellular Origin, Life in Extreme Habitats and Astrobiology, Vol. 27, eds J. Seckbach, A. Oren, and H. Stan-Lotter (Dordrecht: Springer), 201–213. doi: 10.1007/978-94-007-6488-0_7
Mitra, A., Fay, P. A., Morgan, J. K., Vendura, K. W., Versaggi, S. L., and Riordan, J. T. (2012). Sigma factor n, liaison to an ntrc and rpos dependent regulatory pathway controlling acid resistance and the lee in enterohemorrhagic Escherichia coli. PLoS One 7:e46288. doi: 10.1371/journal.pone.0046288
Mitra, A., Fay, P. A., Vendura, K. W., Alla, Z., Carroll, R. K., Shaw, L. N., et al. (2014). σ N −dependent control of acid resistance and the locus of enterocyte effacement in enterohemorrhagic E scherichia coli is activated by acetyl phosphate in a manner requiring flagellar regulator FlhDC and the σ S antagonist FliZ. Microbiol. Open 3, 497–512. doi: 10.1002/mbo3.183
Mongodin, E. F., Nelson, K. E., Daugherty, S., Deboy, R. T., Wister, J., Khouri, H., et al. (2005). The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. U.S.A. 102, 18147–18152. doi: 10.1073/pnas.0509073102
Moran-Reyna, A., and Coker, J. A. (2014). The effects of extremes of pH on the growth and transcriptomic profiles of three haloarchaea. F1000Research 3:168. doi: 10.12688/f1000research.4789.2
Mourin, M., Wai, A., O’Neil, J., Hausner, G., and Dibrov, P. (2019). Physiological, structural, and functional analysis of the paralogous cation–proton antiporters of nhap type from Vibrio cholerae. Int. J. Mol. Sci. 20:2572. doi: 10.3390/ijms20102572
Mykytczuk, N. C. S., Trevors, J. T., Ferroni, G. D., and Leduc, L. G. (2010). Cytoplasmic membrane fluidity and fatty acid composition of Acidithiobacillus ferrooxidans in response to pH stress. Extremophiles 14, 427–441. doi: 10.1007/s00792-010-0319-2
Nakamura, T., Yamamuro, N., Stumpe, S., Unemoto, T., and Bakker, E. P. (1998). Cloning of the trkAH gene cluster and characterization of the Trk K(+)-uptake system of Vibrio alginolyticus. Microbiology 144 (Pt 8), 2281–2289. doi: 10.1099/00221287-144-8-2281
Neilson, J. W., Quade, J., Ortiz, M., Nelson, W. M., Legatzki, A., Tian, F., et al. (2012). Life at the hyperarid margin: novel bacterial diversity in arid soils of the Atacama Desert, Chile. Extremophiles 16, 553–566. doi: 10.1007/s00792-012-0454-z
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nicolle, J. L. C., Le, C., Nicolle, J., Simmons, S., Bathe, S., and Norris, P. R. (2009). Ferrous iron oxidation and rusticyanin in halotolerant, acidophilic “Thiobacillus prosperus.”. Microbiology 155, 1302–1309. doi: 10.1099/mic.0.023192-0
Norris, P. R., Davis-Belmar, C. S., Calvo-Bado, L. A., and Ogden, T. J. (2020). Salt-tolerant Acidihalobacter and Acidithiobacillus species from Vulcano (Italy) and Milos (Greece). Extremophiles 24, 593–602. doi: 10.1007/s00792-020-01178-w
Ossandon, F. J., Cárdenas, J. P., Corbett, M., Quatrini, R., Holmes, D. S., and Watkin, E. (2014). Draft genome sequence of the iron-oxidizing, acidophilic, and halotolerant “Thiobacillus prosperus” type strain DSM 5130. Genome Announc. 2:e01042–14. doi: 10.1128/genomeA.01042-14
Pandey, G. K., and Mahiwal, S. (2020). “Potassium homeostasis,” in Role of Potassium in Plants, eds G. K. Pandey and S. Mahiwal New York, NY: Springer International Publishing, 11–18. doi: 10.1007/978-3-030-45953-6_2
Parashar, D., and Satyanarayana, T. (2018). An insight into ameliorating production, catalytic efficiency, thermostability and starch saccharification of acid-stable α-amylases from acidophiles. Front. Bioeng. Biotechnol. 6:125. doi: 10.3389/fbioe.2018.00125
Pflock, M., Kennard, S., Delany, I., Scarlato, V., and Beier, D. (2005). Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73, 6437–6445. doi: 10.1128/IAI.73.10.6437-6445.2005
Price-Whelan, A., Poon, C. K., Benson, M. A., Eidem, T. T., Roux, C. M., Boyd, J. M., et al. (2013). Transcriptional profiling of Staphylococcus aureus during growth in 2 M NaCl leads to clarification of physiological roles for Kdp and Ktr K+ uptake systems. MBio 4:e00407–13. doi: 10.1128/mBio.00407-13
Pruitt, K. D., Tatusova, T., Brown, G. R., and Maglott, D. R. (2012). NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–D135. doi: 10.1093/nar/gkr1079
Quatrini, R., and Barrie Johnson, D. (2016). Acidophiles: Life in Extremely Acidic Environments. Norfolk: Caister Academic Press.
Raven, J. A. (2021). Origin of the roles of potassium in biology. Bioessays 43:e2000302. doi: 10.1002/bies.202000302
Rhee, H. J., Kim, E.-J., and Lee, J. K. (2007). Physiological polyamines: simple primordial stress molecules. J. Cell. Mol. Med. 11, 685–703. doi: 10.1111/j.1582-4934.2007.00077.x
Riadi, G., Medina-Moenne, C., and Holmes, D. S. (2012). TnpPred: a web service for the robust prediction of prokaryotic transposases. Comp. Funct. Genomics 2012:678761. doi: 10.1155/2012/678761
Richard, H., and Foster, J. W. (2004). Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 186, 6032–6041. doi: 10.1128/JB.186.18.6032-6041.2004
Rivera-Araya, J., Huynh, N. D., Kaszuba, M., Chávez, R., Schlömann, M., and Levicán, G. (2020). Mechanisms of NaCl-tolerance in acidophilic iron-oxidizing bacteria and archaea: comparative genomic predictions and insights. Hydrometallurgy 194:105334. doi: 10.1016/j.hydromet.2020.105334
Rivera-Araya, J., Pollender, A., Huynh, D., Schlömann, M., Chávez, R., and Levicán, G. (2019). Osmotic imbalance, cytoplasm acidification and oxidative stress induction support the high toxicity of chloride in acidophilic bacteria. Front. Microbiol. 10:2455. doi: 10.3389/fmicb.2019.02455
Romantsov, T., Guan, Z., and Wood, J. M. (2009). Cardiolipin and the osmotic stress responses of bacteria. Biochim. Biophys. Acta 1788, 2092–2100. doi: 10.1016/j.bbamem.2009.06.010
Sachs, G., Kraut, J. A., Wen, Y., Feng, J., and Scott, D. R. (2006). Urea transport in bacteria: acid acclimation by gastric Helicobacter spp. J. Membr. Biol. 212, 71–82. doi: 10.1007/s00232-006-0867-7
Samartzidou, H., Mehrazin, M., Xu, Z., Benedik, M. J., and Delcour, A. H. (2003). Cadaverine inhibition of porin plays a role in cell survival at Acidic pH. J. Bacteriol. 185, 13–19. doi: 10.1128/jb.185.1.13-19.2003
Scott, D. R., Marcus, E. A., Weeks, D. L., Lee, A., Melchers, K., and Sachs, G. (2000). Expression of the Helicobacter pylori ureI gene is required for acidic pH activation of cytoplasmic urease. Infect. Immun. 68, 470–477. doi: 10.1128/IAI.68.2.470-477.2000
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Simmons, S., and Norris, R. (2002). Acidophiles of saline water at thermal vents of Vulcano, Italy. Extremophiles 6, 201–207. doi: 10.1007/s007920100242
Slonczewski, J. L., Fujisawa, M., Dopson, M., and Krulwich, T. A. (2009). Cytoplasmic pH measurement and homeostasis in bacteria and archaea. Adv. Microb. Physiol. 55, 1–79, 317. doi: 10.1016/S0065-2911(09)05501-5
Stabnikov, V., Chu, J., Ivanov, V., and Li, Y. (2013). Halotolerant, alkaliphilic urease-producing bacteria from different climate zones and their application for biocementation of sand. World J. Microbiol. Biotechnol. 29, 1453–1460. doi: 10.1007/s11274-013-1309-1
Stautz, J., Hellmich, Y., Fuss, M. F., Silberberg, J. M., Devlin, J. R., Stockbridge, R. B., et al. (2021). Molecular mechanisms for bacterial potassium homeostasis. J. Mol. Biol. 433:166968. doi: 10.1016/j.jmb.2021.166968
Strahl, H., and Greie, J.-C. (2008). The extremely halophilic archaeon Halobacterium salinarum R1 responds to potassium limitation by expression of the K+-transporting KdpFABC P-type ATPase and by a decrease in intracellular K+. Extremophiles 12, 741–752. doi: 10.1007/s00792-008-0177-3
Sun, L., Xiao, L., Xiao, B., Wang, W., Pan, C., Wang, S., et al. (2013). Differences in the gene expressive quantities of carbonic anhydrase and cysteine synthase in the weathering of potassium-bearing minerals by Aspergillus niger. Sci. China Earth Sci. 56, 2135–2140. doi: 10.1007/s11430-013-4704-4
Suzuki, I., Lee, D., Mackay, B., Harahuc, L., and Oh, J. K. (1999). Effect of various ions, pH, and osmotic pressure on oxidation of elemental sulfur by Thiobacillus thiooxidans. Appl. Environ. Microbiol. 65, 5163–5168. doi: 10.1128/AEM.65.11.5163-5168.1999
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Ullrich, S. R., González, C., Poehlein, A., Tischler, J. S., Daniel, R., Schlömann, M., et al. (2016a). Gene Loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile “ferrovum.”. Front. Microbiol. 7:797. doi: 10.3389/fmicb.2016.00797
Ullrich, S. R., Poehlein, A., Tischler, J. S., González, C., Ossandon, F. J., Daniel, R., et al. (2016b). Genome analysis of the biotechnologically relevant acidophilic iron oxidising strain ja12 indicates phylogenetic and metabolic diversity within the novel genus “ferrovum.”. PLoS One 11:e0146832. doi: 10.1371/journal.pone.0146832
Varani, A. M., Siguier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30. doi: 10.1186/gb-2011-12-3-r30
Veaudor, T., Cassier-Chauvat, C., and Chauvat, F. (2019). Genomics of urea transport and catabolism in cyanobacteria: biotechnological implications. Front. Microbiol. 10:2052. doi: 10.3389/fmicb.2019.02052
Vergara, E., Neira, G., González, C., Cortez, D., Dopson, M., and Holmes, D. S. (2020). Evolution of predicted acid resistance mechanisms in the extremely acidophilic Leptospirillum Genus. Genes 11:389. doi: 10.3390/genes11040389
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Walderhaug, M. O., Polarek, J. W., Voelkner, P., Daniel, J. M., Hesse, J. E., Altendorf, K., et al. (1992). KdpD and KdpE, proteins that control expression of the kdpABC operon, are members of the two-component sensor-effector class of regulators. J. Bacteriol. 174, 2152–2159. doi: 10.1128/jb.174.7.2152-2159.1992
Xiao, B., Lian, B., Sun, L., and Shao, W. (2012). Gene transcription response to weathering of K-bearing minerals by Aspergillus fumigatus. Chem. Geol. 306–307, 1–9. doi: 10.1016/j.chemgeo.2012.02.014
Yamauchi, K., Doi, K., Yoshida, Y., and Kinoshita, M. (1993). Archaebacterial lipids: highly proton-impermeable membranes from 1,2-diphytanyl-sn-glycero-3-phosphocholine. Biochim. Biophys. Acta 1146, 178–182. doi: 10.1016/0005-2736(93)90353-2
Zammit, C. M., and Watkin, E. L. J. (2016). “Adaptation to extreme acidity and osmotic stress,” in Acidophiles: Life in Extremely Acidic Environments, eds R. Quatrini and D. B. Johnson (Poole: Caister Academic Press), 49–62. doi: 10.21775/9781910190333.03
Zammit, C. M., Mutch, L. A., Watling, H. R., and Watkin, E. L. J. (2009). The characterization of salt tolerance in biomining microorganisms and the search for novel salt tolerant strains. Adv. Materials Res. 71-73, 283–286. doi: 10.4028/www.scientific.net/amr.71-73.283
Zhao, B., and Houry, W. A. (2010). Acid stress response in enteropathogenic gammaproteobacteria: an aptitude for survival. Biochem. Cell Biol. 88, 301–314. doi: 10.1139/o09-182
Zhu, Q., Kosoy, M., and Dittmar, K. (2014). HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genomics 15:717. doi: 10.1186/1471-2164-15-717
Keywords: polyextremophile, extreme acidophile, acid resistance, genome evolution, phylogenomics, potassium transporters, urease, chloride/proton antiporters
Citation: Boase K, González C, Vergara E, Neira G, Holmes D and Watkin E (2022) Prediction and Inferred Evolution of Acid Tolerance Genes in the Biotechnologically Important Acidihalobacter Genus. Front. Microbiol. 13:848410. doi: 10.3389/fmicb.2022.848410
Received: 04 January 2022; Accepted: 28 February 2022;
Published: 18 April 2022.
Edited by:
Claudia P. Saavedra, Andres Bello University, ChileReviewed by:
Mario Tello, University of Santiago, ChileDaniel Aguayo, Universidad Andres Bello, Santiago, Chile
Copyright © 2022 Boase, González, Vergara, Neira, Holmes and Watkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Holmes, ZHNob2xtZXMyMDAwQHlhaG9vLmNvbQ==; Elizabeth Watkin, RS5XYXRraW5AY3VydGluLmVkdS5hdQ==
†These authors share first authorship