 Guiliang Tan
Guiliang Tan Min Hu
Min Hu Xiangli Li3
Xiangli Li3 Ziqiang Pan
Ziqiang Pan Mei Li
Mei Li Lin Li
Lin Li Ziyi Zheng
Ziyi Zheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 25 February 2022
Sec. Food Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.841529
Koji making is a pre-fermentation stage in soy sauce manufacturing that impacts final product quality. Previous studies have provided valuable insights into the microbial species present in koji. However, changes in microbial community functional potential during koji-making are not well-known, nor are the associations among microbial populations and flavoring characteristics. In the present study, we investigated the succession of microbial communities, microbial community functional potential, metabolite profiles, and associations among microbial community members/functions with metabolites during koji making using shotgun metagenomic and metabolomic analyses. Firmicutes, Proteobacteria, and Ascomycota were identified as the most abundant microbial phyla in early koji making (0–12 h). Aspergillus (fungi) and Weissella (bacteria) exhibited marked abundance increases (0.98–38.45% and 0.31–30.41%, respectively) after 48 h of fermentation. Metabolite analysis revealed that aspartic acid, lysine, methyl acetate, isovaleraldehyde, and isoamyl alcohol concentrations increased ∼7-, 9-, 5-, 49-, and 10-fold after 48 h of fermentation. Metagenomic profiling demonstrated that koji communities were dominated by genes related to carbohydrate metabolism and amino acid metabolism, but functional profiles exhibited marked shifts after 24 h of fermentation. The abundances of genes within the categories of carbohydrate and amino acid metabolism all increased during koji making, except for pyruvate metabolism, glycolysis/gluconeogenesis, and the citrate cycle. Correlational analyses indicated that Aspergillus, Lactococcus, Enterococcus, Corynebacterium, and Kocuria abundances were positively correlated with 15 amino acid concentrations (all p < 0.05), while Weissella abundances were positively correlated with concentrations of volatile flavor compounds, including eight amino acids, phenylacetaldehyde, acetic acid, 2,3-butanediol, ethyl acetate, and ethanol (p < 0.05). These results provide valuable information for understanding the microbial-associated mechanisms of flavor formation during koji making.
Soy sauce is a traditional fermented soybean product that originated in China over 2,500 years ago (Feng et al., 2013). Soy sauce has distinctive, characteristic tastes and aromas and is consumed as an essential condiment in China and other Asian countries (Wei C. L. et al., 2013). Soy sauce production involves two fermentation stages: koji making and moromi fermentation. Koji is a solid-state fermentation of steamed whole or defatted soybeans and wheat flour using filamentous fungi (e.g., Aspergillus oryzae or Aspergillus sojae), with an incubation time from 26 h to 7 days (Wei C. L. et al., 2013). Moromi is a mixture of koji and a brine solution containing 18–22% NaCl that is allowed to spontaneously ferment for 3–6 months (Devanthi and Gkatzionis, 2019).
Koji making is the first step in soy sauce brewing and is an extremely important step in determining the quality of final products (Yan et al., 2013; Ding et al., 2019; Zhao et al., 2020). For example, most of the volatile compounds present in initial moromi fermentations might develop in the koji-making stage (Gao et al., 2010; Feng et al., 2013). During koji making, molds (e.g., Aspergillus) produce proteolytic enzymes that hydrolyze proteins into peptides and amino acids, in addition to amylases that also convert starches into simple sugars (Devanthi and Gkatzionis, 2019). Many metabolites have been identified in koji samples including volatile flavor compounds (VFCs), fatty acids, and lipids. Among these, aldehydes and alcohols are the major volatile compounds (Feng et al., 2013), and triacylglycerols are the most abundant fatty acids (Feng et al., 2014). In addition, koji making also provides enzymes (such as proteases and amylases) for the hydrolysis of raw materials in the subsequent brine fermentation, which then affects soy sauce chemical compositions, colors, and flavors (Wicklow et al., 2007).
Microorganisms play essential roles in flavor development during soy sauce fermentation (Devanthi and Gkatzionis, 2019; Mannaa et al., 2020). In particular, the bacterial genera Weissella, Staphylococcus, Tetragenococcus, and Bacillus; in addition to the fungal genera Aspergillus, Zygosaccharomyces, Candida, and Debaryomyces predominate throughout the moromi fermentation stage (Wei Q. Z. et al., 2013; Sulaiman et al., 2014; Han et al., 2020). However, few studies have evaluated the microbial community compositions and their contributions to flavor development during koji making. The non-sterile environment of koji making leads to contributions to flavoring from diverse microorganisms including Weissella, Staphylococcus, Lactobacillus, Streptococcus, Enterococcus, Kurthia, and Klebsiella (Tanaka et al., 2012; Wei Q. Z. et al., 2013; Yan et al., 2013). Among these, Weissella and Staphylococcus have been observed as the predominant bacterial genera in koji, while Aspergillus is the dominant fungal genus. In addition, less abundant fungal genera are present including Candida, Wickerhamomyces, Pichia, Geotrichum, and Trichosporon (Tanaka et al., 2012; Wei Q. Z. et al., 2013; Yan et al., 2013). Previous studies have provided valuable insights into the microbial communities present in koji, but changes in the functional potentials of microbial communities during koji making are not well known and correlations between microflora and metabolites are underexplored. The composition of microorganisms in soy sauce production (and in moromi fermentation) has been investigated primarily by 16S rRNA or ITS gene amplicon sequencing (Liang et al., 2019; Guo et al., 2020; Han et al., 2020; Liu et al., 2021; Qi et al., 2021), in addition to shotgun metagenomics (Sulaiman et al., 2014; Kim et al., 2021). A previous metagenomic study has shown that as fermentation progressed, microbial diversity decreased in the mid to late stages of moromi fermentation (Sulaiman et al., 2014; Kim et al., 2021), while inference of functional potential suggested characteristic profiles involved with heterotrophic fermentation of proteins and carbohydrates (Sulaiman et al., 2014). These studies have expanded our understanding of microbial community structure and functions in soy sauce moromi fermentation. However, the capacities of microbial communities for flavor generation during koji making requires further investigation.
In the present study, we investigated the microbial succession, community functional potential, and metabolite profiles present during koji making, while also evaluating correlations among microbial community composition/functions with metabolites using metagenomic and metabolomic analyses. These results provide a better understanding of the roles of microorganisms in koji making and flavor generation that can then be used to improve soy sauce product qualities.
Koji samples from high-salt liquid-state fermentations of soy sauce were collected from the Pearl River Bridge Biotechnology Co., Ltd (Zhongshan, Guangdong, China), which is one of the most famous food manufacturing companies in China. Prior to koji making, whole soybeans were steam-cooked and the steamed soybeans were mixed with wheat flour at a ratio of 3:1 (w/w), followed by cooling to 30°C and inoculation with 0.03% A. oryzae strain 3.042 as a spore starter (Feng et al., 2013). Subsequently, mixtures were fermented in a vessel (length: 8 m × width: 4 m × depth: 50 cm) at 28–35°C in a koji incubation room. Relative humidity was maintained at about 95% during koji making. The overall process of koji making is illustrated in Supplementary Figure 1. To investigate the succession of microbial communities and metabolites during the process, koji samples were periodically taken at 0–48 h. At each sampling time, 50 g of koji samples were randomly collected from three vessels, placed in 50 mL centrifuge tubes (Corning CentriStar, NY, United States), immediately transported on ice to the laboratory, and then stored at −20°C until subsequent DNA extraction and chemical analysis.
To measure sample pH, 10 g of koji sample was mixed with 100 mL of distilled water, and then centrifuged (9,000 × g, 10 min) at room temperature. The distilled water was heated to boiling and then cooled to room temperature before use. The supernatant pH was measured directly with a PB-10 pH meter (Sartorius, Göttingen, Germany). Free amino acid (FAA) contents were detected using ultra-HPLC tandem MS (UPLC-MS/MS; model 1290/6460; Agilent Technologies, Santa Clara, CA, United States). Briefly, samples were extracted with distilled water (pH 3.0) and the extracted solutions were purified using hexane (Merck, Germany). The FAAs were then separated on an ACQUITY UPLC BEH HILIC (2.1 × 100 mm, 1.7 μm; Waters Corp.) using ammonium formate-acetonitrile/ammonium formate-H2O (pH 3.0) as the mobile phase and detected with MS/MS using multiple reaction monitoring modes (Tan et al., 2020).
Volatile flavor compounds were analyzed as described previously (Feng et al., 2013), with minor modifications. Briefly, koji samples (2.5 g) were mixed with 0.5 g of NaCl and 20 μL of 2-methyl-3-heptanone (2 mg/L in methanol) as an internal standard in 15 mL amber SPME vials, followed by equilibration for 15 min with a thermostatic water bath at 55°C. VFCs were then extracted with SPME fiber (CAR/PDMS, 75 μm; Supelco Co., Bellefonte, PA, United States) at 55°C for 30 min. VFCs were analyzed using a GC-MS system (model 6890N/5975; Agilent Technologies, Santa Clara, CA, United States). The oven temperature gradient for GC-MS started at 33°C (2 min) and then increased at 5°C/min to 70°C, followed by increases at 10°C/min to 250°C. GC-MS settings included an injector temperature of 250°C and a run time of 30 min. Compounds were identified by comparison with mass spectral data from the NIST 14 mass spectra database. All extractions were conducted in triplicate. The concentrations of each volatile component in koji samples were quantified by comparing their peak areas to those of internal standard compounds on the total GC-MS ion chromatograms. All of the quantitative data represent mean values for triplicate measurements.
Total genomic DNA from koji samples (0.5 g) was extracted using an EZNA™ Mag-Bind Food DNA kit (Omega Bio-Tek, Inc., Norcross, GA, United States) according to the manufacturer’s instructions. Triplicate samples of extracted DNA from the same sample time were combined for downstream metagenomic sequencing. DNAs were then sheared into 300 bp fragments and sequenced on the Illumina HiSeqX-Ten platform (Illumina Inc., San Diego, CA, United States) to generate 2 × 150-bp paired-end sequencing reads. Sequencing was performed at the Novogene Bioinformatics Technology facility (Beijing, China).
Sequencing adapters were removed from reads that were then trimmed with Trimmomatic v 0.30 using a quality cutoff of 30, a sliding window of 6 bp, and a minimum length cutoff of 45 bp (Bolger et al., 2014). High quality-filtered reads were pooled and assembled with the IDBA-UD 1.1.1 assembler (Peng et al., 2012) to obtain contigs using a minimum k value of 60; a maximum k value of 120; incremented k-mer sizes of 10 for each iteration; a minimum multiplicity of 5 for filtering k-mers when building the graph; a seed k-mer size of 5 for alignment; and a minimum contig length of 1,000; while all other parameters were set to default values. Contigs shorter than 500 bp were excluded. Genes of the assembled contigs were predicted using the MetaGeneMark software program (Zhu et al., 2010) and genes shorter than 300 nt were removed. The CD-HIT software program (Fu et al., 2012) was used to remove redundant genes at a threshold of 95% nucleotide identity and ≥90% coverage, with the longest sequence in each gene cluster used for downstream analyses. Non-redundant gene sets were aligned to the National Center for Biotechnology Information (NCBI)-nr database using the DIAMOND aligner (Buchfink et al., 2015) and an e-value threshold of <e–5, followed by taxonomic profiling of genes with MEtaGenome ANalyzer (MEGAN) (Huson et al., 2007). Plant-associated genes were removed from the dataset to reduce the effects of plant genome contamination on the results. Thus, only bacterial- and fungal-affiliated genes were retained for downstream analysis. The filtered reads were subsequently mapped back to the microbial genes using Bowtie2 with default parameters, and the tags per million (TPM) values were calculated for each gene and their corresponding taxa to reduce the effects of sequencing depth and gene length on gene abundances. The taxonomic composition of koji metagenomes were then calculated by summing the TPM values for each lineage. To determine the relative abundances of genes in each sample, filtered reads were mapped back to the assembled microbial genes using Bowtie2 with default parameters. The functional profiles of the microbial communities were obtained by comparing the non-redundant microbial gene set against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) using KOBAS 3.0 with a threshold of e–5 (Xie et al., 2011). Gene abundances were normalized based on the Z scores of the TPM values for each stage.
All statistical analyses were conducted using the SPSS 18.0 Software package (SPSS Inc., Chicago, IL, United States). Differences in datasets were evaluated by conducting one-way ANOVA tests followed by least significant difference (LSD) tests. Differences were considered significant at p < 0.05. The unweighted pair group method with arithmetic mean analysis (UPGMA) hierarchical clustering was used to assess community compositional similarities based on Bray-Curtis distances with microbial abundance data within the R vegan package v.2.5-7 (Oksanen et al., 2019). Correlations among microbial compositional characteristics and chemical properties were estimated by Spearman’s correlation coefficients, and strong correlations were identified by values of | ρ| > 0.7 and a p < 0.05. Heatmap visualization was constructed in the R environment with the “vegan” package.
The metagenomic sequence data produced here are publicly available in the NCBI database under the BioProject accession PRJNA730347.
The pH slightly decreased during koji making from an initial value of 6.73 ± 0.06 to 6.33 ± 0.03 (Supplementary Figure 2). The contents of the 16 investigated amino acids all exhibited increased abundances with time (p < 0.05). In particular, arginine, aspartic acid, lysine, and phenylalanine were the predominant amino acid species at the end of fermentation (48 h), reaching concentrations of 12.44 ± 0.18 g/kg, 10.42 ± 0.03 g/kg, 10.10 ± 0.03 g/kg, and 8.11 ± 0.32 g/kg, respectively (Table 1). These amino acids might derive from the activity of the starter A. oryzae culture that hydrolyzes proteins into peptides and amino acids. In addition, a total of 66 VFCs were identified during koji making, including 12 esters, 10 alcohols, 10 aldehydes, 6 ketones, 4 acids, and 24 other compounds (Supplementary Table 1). The predominant volatile groups in the 48 h samples were aldehydes (31.22 ± 5.66%), alcohols (17.43 ± 1.31%), and esters (18.92 ± 3.90%), consistent with a previous study wherein the predominant volatile groups in koji samples were aldehydes and alcohols (Feng et al., 2013). The most abundant VFCs in the 48 h sample included isovaleraldehyde (with an average value of 939.75 μg/kg), 2,2,4,6,6-pentamethylheptane (470.53 μg/kg), methyl acetate (422.03 μg/kg), 1-octen-3-ol (385.46 μg/kg), and isoamyl alcohol (276.65 μg/kg) that exhibited approximately 49-, 3-, 5-, 1-, and 10-fold higher concentrations, respectively, compared to the initial 0 h sample (Supplementary Table 1). These results were consistent with those of a previous study wherein 3-methyl-butanal (isovaleraldehyde), 1-octen-3-ol, benzeneacetaldehyde, (E)-2-octenal, and benzaldehyde were the most abundant compounds (Feng et al., 2013). Among these, isovaleraldehyde is a branched short-chain aldehyde and is mainly produced from branched-chain amino acids via the Ehrlich pathway involving various fungal enzymes during fermentation (Chung et al., 2005). In addition, isovaleraldehyde was an important volatile compound in dry-cured ham products and fermented squid (Sabio et al., 1998; Huang L. D. et al., 2018). In addition, 1-octen-3-ol generated from lipid oxidation has been detected during soy sauce koji making (Feng et al., 2013) and is considered one of the most abundant compounds and important contributors to the sensory characteristics of koji.
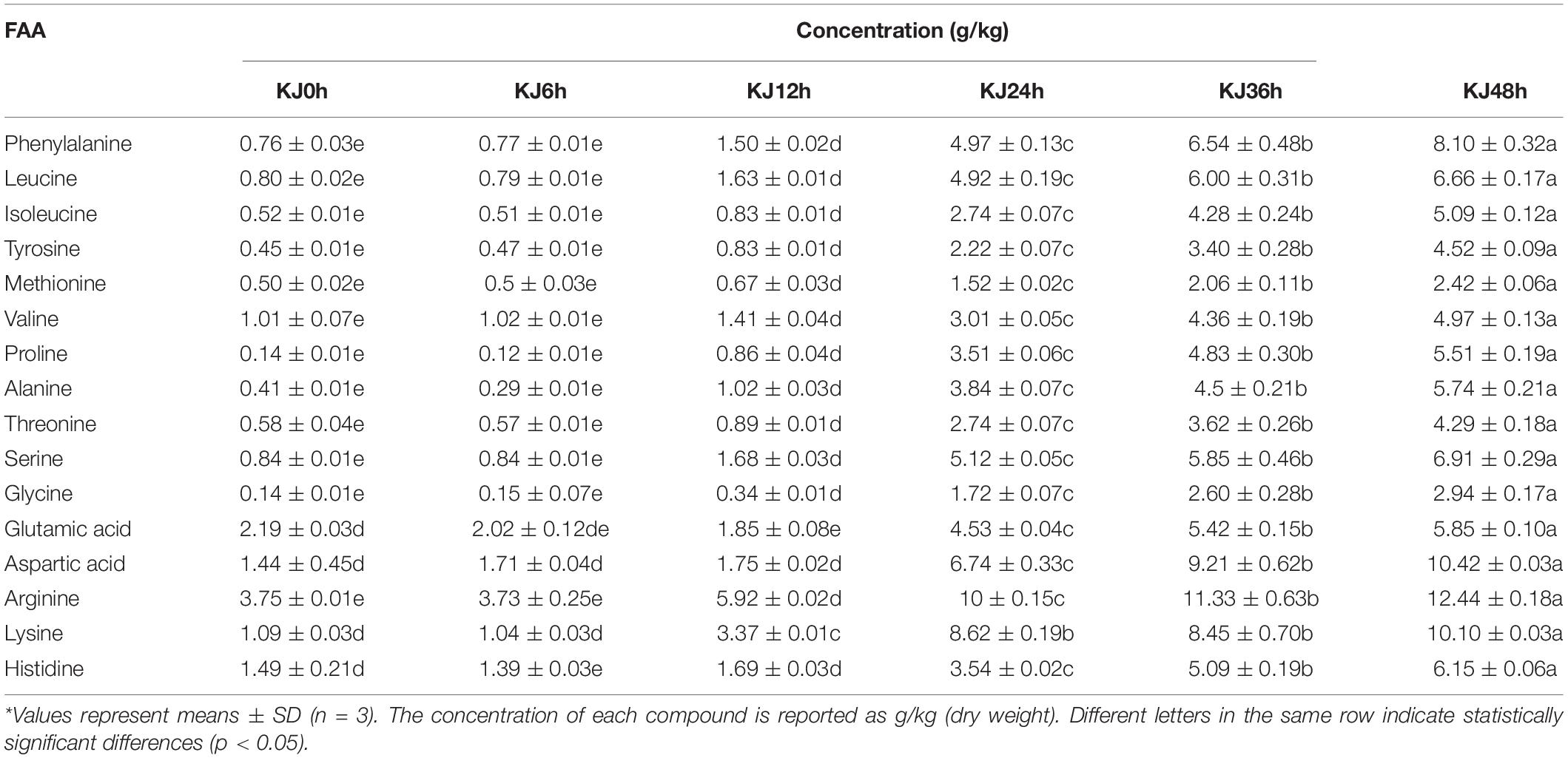
Table 1. Free amino acid (FAA) profiles of samples taken from koji making fermentations at six different stages.*
To identify the succession of microbial communities present in koji making, metagenomic sequencing of six koji samples was conducted at different stages of production. Several large metagenomic data sets were produced from the koji samples, resulting in an average of 15.04 million 2 × 150 bp paired-end reads for each sample, and a total of 25.79 Gbp of sequence data after quality filtering (Supplementary Table 2). A large fraction of the sequence reads were assembled in contigs ≥500 bp (i.e., 358,780 contigs comprising 353,846,420 bp), yielding an N50 (defined as the contig length above which 50% of all assembled data are included) of 883, with a maximum contig length of 511,628 bp, and a mean contig size of 986 bp. Read-mapping of the quality-filtered reads to the assemblies was successful overall, with an average alignment rate of 67.03 ± 15.65% for the six samples (Supplementary Table 2). Thus, the assembled contigs possessed the majority of the sequenced genetic information for the koji microbial communities. Bacterial-associated sequences decreased in relative abundance from 79.17 to 59.70% during koji making while fungus-affiliated sequences increased from 20.83 to 40.30% (Supplementary Table 2). Thus, the ratio of fungal to bacterial sequences within communities during Koji making increased across stages, from 0.26 to 0.68. Archaeal-affiliated sequences were not observed in the koji metagenomes. UPGMA demonstrated that koji samples from the first three stages (0–12 h) belonged to one cluster, whereas later stage communities grouped into another cluster (Figure 1). Firmicutes (61.86 ± 7.52% relative abundances in the later three stages), Ascomycota (33.57 ± 9.48% in the later three stages), Proteobacteria (46.55 ± 13.23% in the first three stages), and Basidiomycota (12.06 ± 3.60% in the first three stages) were the dominant organisms throughout the various fermentation stages (Supplementary Figure 3). The relative abundances of the bacterial phylum Proteobacteria were dramatically higher in the early to middle stages (from 0 to 24 h; decreasing from 56.91 to 4.92%) as were those for the fungal phylum Basidiomycota (decreasing from 14.44 to 0.52%), while the relative abundances of the bacterial phylum Firmicutes and the fungal phylum Ascomycota increased across the entire fermentation period (0–48 h), with relative abundances increasing from 19.84 to 56.78% and from 1.02 to 40.06%, respectively (Supplementary Figure 3). The bacterial genera Klebsiella, Lactobacillus, and Bradyrhizobium, in addition to the fungal genus Puccinia dominated the beginning stage of fermentation (0 h), with overall abundances of 34.09, 16.78, 16.75, and 14.32%, respectively, but transitioned to minor populations after 24 h of fermentation (Figure 1). In contrast, the fungal genus Aspergillus and the bacterial genus Weissella exhibited increased abundances after 24 h of fermentation and then became dominant in the later koji making stages (24–48 h), with abundances reaching approximately 38.45 and 30.41% after 48 h of incubation. As expected, Aspergillus exhibited the highest overall abundance since it is used as a starter for soy sauce production (Yan et al., 2013). Weissella is a typical lactic acid bacterium (LAB) that is isolated and detected in a variety of fermented foods, and plays an important role in flavor generation (e.g., via production of lactic acid, isoamyl acetate, and terpinyl acetate) (Fessard and Remize, 2017; Xiang et al., 2020). Weissella has also previously been observed as one of the most dominant bacterial genera in soy sauce koji making (Yan et al., 2013). Additionally, the abundances of Enterococcus increased during fermentation, representing 2.42% of the microbial community at 48 h, but only 0.01% at 0 h. Bacillus was detected at low abundances at 24 and 36 h, accounting for 1.17 and 3.98% of community totals, respectively, but then decreased to 0.33% at 48 h (Figure 1). Bacillus spp. are the dominant microorganisms in many fermented soybean products (Devanthi and Gkatzionis, 2019; He and Chung, 2020) and their primary contributions to these systems is the production of various hydrolytic enzymes like proteases, α-amylases, and β-glucosidases that hydrolyze macromolecules (Sanjukta et al., 2015). Bacillus are thought to contribute to flavor generation during soy sauce aging via amylase and protease activities (Wei C. L. et al., 2013; Liang et al., 2019), but can also reduce the concentrations of allergens and improve the nutritional value of soy products (Shi et al., 2017). At the species level, Klebsiella pneumoniae, Lactobacillus brevis, Bradyrhizobium sp., and Puccinia striiformis were the predominant species in the initial stage of fermentation (0–12 h), while A. oryzae, Weissella cibaria, Weissella confusa, Enterococcus italicus, and Bacillus subtilis abundances gradually increased and became the dominant species in the middle to late stages (24–48 h) (Supplementary Table 3). Changes in microbiota composition also reflected the role of microorganisms during koji making. At the beginning of fermentation, most bacteria (e.g., Klebsiella and Lactobacillus) originate from raw materials (Yan et al., 2013). As fermentation proceeds, microbial compositions change, leading to substantial increases in Aspergillus and Weissella abundances. Weissella spp. are frequently detected in many spontaneously fermented foods and can adapt to diverse environments (Fessard and Remize, 2017). A high abundance of Weissella populations is likely to be associated with the generation of antimicrobial substances (e.g., bacteriocins) that may inhibit the growth of other bacteria (Fessard and Remize, 2017).
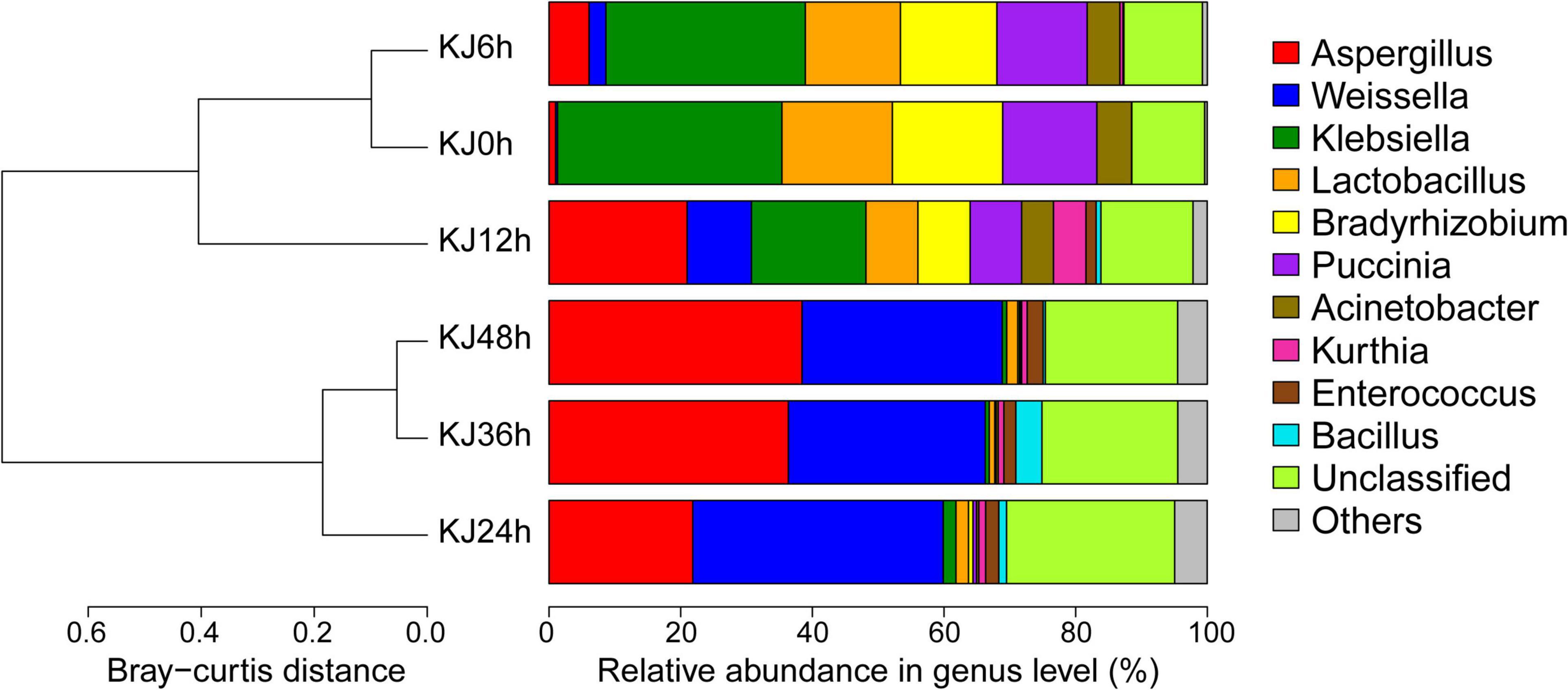
Figure 1. Taxonomic composition and community compositional clustering of koji metagenomes at the genus level among six fermentation stages spanning from 0 to 48 h. The 10-most abundant taxa are shown for all samples. “Others” comprise the less-abundant genus. Sequences that could not be assigned to known taxa were designated as “unclassified”. The cluster tree is based on the Bray-Curtis distance matrix of among-sample gene abundances.
Genes encoding metabolic functions dominated across the fermentation stages (increasing from 53.70 to 60.62%), followed by those involved in genetic information processing (decreasing from 23.34 to 17.21%), and environmental information processing (21.73–13.20%) based on KEGG annotations (Supplementary Figure 4). Gene abundances sharply changed at the 24 h fermentation point, then remained stable until the end of fermentation. The most represented metabolic gene sub-category was carbohydrate metabolism (relative abundance changes of 48.39–15.40%), followed by amino acid metabolism (1.05–9.68%), consistent with functional changes based on metagenomic analyses observed in soy sauce brines, sausage, and Chinese paocai (Sulaiman et al., 2014; Ferrocino et al., 2018; Liang et al., 2018; Kumar et al., 2019). Higher abundances of genes associated with carbohydrate and amino acid metabolism indicate that starches and proteins serve as critical flavor precursors. Within the carbohydrate metabolism category, genes involved in glycolysis/gluconeogenesis (ko00010, 9.52%), pyruvate metabolism (ko00620, average 9.49%), and the citrate cycle (TCA cycle) (ko00020, 9.17%) exhibited high abundances at the beginning of fermentation (e.g., in the 0, 6, and 12 h samples) that then decreased and remained stable as fermentation proceeded (Figure 2A and Supplementary Table 4). In the carbohydrate metabolism category, genes encoding dihydrolipoamide acetyltransferase (EC 2.3.1.12) within the glycolysis/gluconeogenesis pathway were detected in high abundance in the first two time points (0 and 6 h). Dihydrolipoamide acetyltransferase is a major component of the pyruvate dehydrogenase complex and is involved in pyruvic acid metabolism. The high observed abundances of genes encoding dihydrolipoamide acetyltransferases indicated a high activity of pyruvic acid biotransformation in the preliminary stage of koji making. The relative abundances then decreased throughout the fermentation process, although the abundances of other genes encoding L-lactate dehydrogenase (EC 1.1.1.27), pyruvate kinase (EC 2.7.1.40), and phosphoglycerate kinase (EC 2.7.2.3) increased from 0 to 24 h, but then slightly decreased (Figure 3). Decreases in the abundances of the gene encoding dihydrolipoamide acetyltransferase (EC 2.3.1.12), within the pyruvate metabolism category, were also observed. However, the relative abundances of genes encoding L-lactate dehydrogenase (EC 1.1.1.27), D-lactate dehydrogenase (EC 1.1.1.28), and acetyl-CoA hydrolase (EC 3.1.2.1) increased during the middle to late stages of fermentation (24–48 h) (Figure 3). Lactate dehydrogenase catalyzes the conversion of pyruvate to lactate with the oxidation of NADH to NAD+ (Schumann et al., 2002), while acetyl-CoA hydrolase catalyzes the production of acetate and corresponded to the increased concentrations of acetic acid that were measured during koji making (Supplementary Table 1). The above results suggest that the biotransformation of organic acids (e.g., lactate and acetate) in koji making derives from the metabolism of pyruvate and acetyl-CoA (from pyruvate), further suggesting that pyruvate is an important organic acid intermediate in the process. In addition, other genes related to carbohydrate metabolism also increased in abundance during fermentation. The abundances of genes involved in starch and sucrose metabolism (ko00500), in addition to amino sugar and nucleotide sugar metabolism (ko00520), increased from 0.08 to 2.50%, and from 0.07 to 1.89%, respectively (Figure 2A and Supplementary Table 4). In the middle to late stages of fermentation (24–48 h), starch and sucrose metabolic pathways (ko0500) were the most represented (average abundance of 2.63%), indicating that carbohydrates were likely used as an energy source during fermentation. Additional analysis of starch and sucrose metabolism revealed that the relative abundances of genes encoding alpha-amylase (EC 3.2.1.1) and glucoamylase (EC 3.2.1.3) steadily increased (Figure 3). Amylases and glucoamylases degrade starch molecules into simple sugars (e.g., glucose) that are useful energy sources for humans (Oguntoyinbo and Narbad, 2012; Tomasik and Horton, 2012). Starch and sucrose metabolism during fermentation are mainly related to Aspergillus taxa that are involved in the degradation of starch to glucose that is then utilized as a primary carbon source for microbial growth, while also contributing to the color and unique flavor profiles of fermentation products (Zhu et al., 2017). In contrast, genes involved in amino acid metabolism were present in low abundances prior to 12 h of fermentation, but then dramatically increased and remained stable at high abundances during the later stages (Figure 2B). The most abundant genes in the last three stages of fermentation were primarily associated with phenylalanine, tyrosine, and tryptophan biosynthesis (ko00400, average abundance of 0.65% in the last three stages); lysine biosynthesis (ko00300, 0.99%); glycine, serine, and threonine metabolism (ko00260, 1.00%); cysteine and methionine metabolism (ko00270, 0.93%); and alanine, aspartate and glutamate metabolism (ko00250, 1.06%) (Supplementary Table 4). These observations suggest that the koji microbiome exhibited a high potential for amino acid metabolism (biosynthesis), consistent with high FAA contents detected in the samples of this study (Table 1). Further, amino acids can be converted to various acids, alcohols (e.g., isoamyl alcohol), aldehydes (e.g., isovaleraldehyde, 2-methylbutyraldehyde, benzaldehyde, and phenylacetaldehyde), and esters (e.g., methyl acetate and ethyl acetate) (Supplementary Table 1), all of which may contribute to the flavor development of koji. In particular, increases in the relative abundances of genes (0.03–0.43%) were observed related to valine, leucine, and isoleucine degradation (ko00280) during koji making. This metabolic pathway involves the three branched-chain amino acids (BCAA; isoleucine, leucine, and valine) that have been reported as precursors to volatile compounds like acids, alcohols, and esters (Marilley and Casey, 2004; Smit et al., 2005).
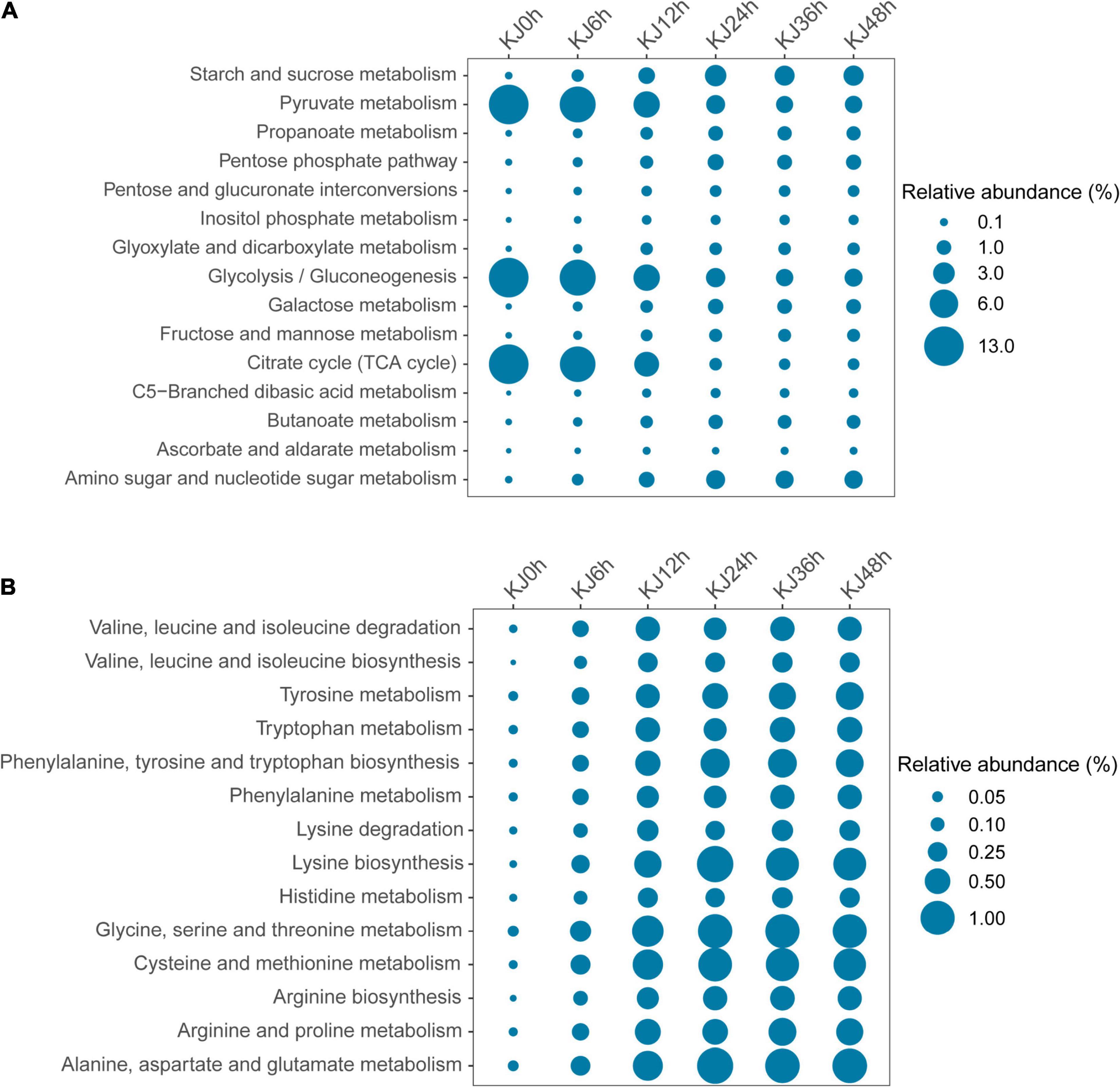
Figure 2. Functional profiling of carbohydrate metabolism (A) and amino acid metabolism (B) genes from whole-shotgun metagenome data that were classified at level 3 of the Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation database.
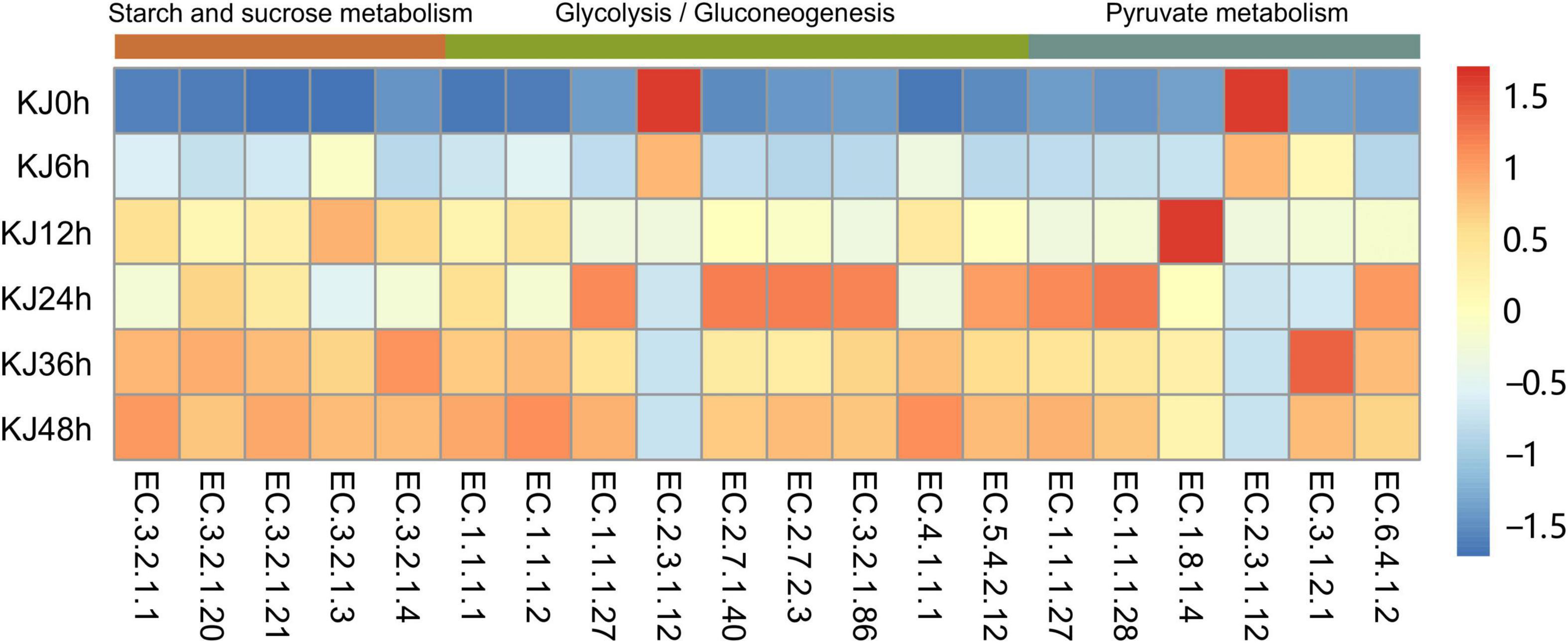
Figure 3. Changes in enzyme-encoding gene abundances among fermentation stages based on annotations against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database within the carbohydrate metabolism category (e.g., starch and sucrose metabolism, glycolysis/gluconeogenesis, and pyruvate metabolism). Gene abundances were normalized by calculating Z scores of the tags per million (TPM) values for each fermentation stage. Heatmap values range from +1.5 to −1.5 and represent high abundance to low abundance levels.
Correlations between microbial populations and flavor profiles have been reported for moromi fermentation, wherein Lactococcus and Weissella abundances were positively correlated with oxalic acid concentrations (Liu et al., 2021). However, these relationships have not been evaluated for the koji making process. Here, the abundances of Aspergillus, Kocuria, Corynebacterium, Enterococcus, and Lactococcus were positively correlated with the concentrations of all amino acids (except for glutamic acid) (all p < 0.05) (Figure 4), indicating potential roles of these genera in the production and transformation of amino acids in koji making. In addition, the abundances of Acinetobacter, Puccinia, Bradyrhizobium, Klebsiella, and Lactobacillus were negatively associated with the contents of the metabolites mentioned above and positively with 2-ethylfuran (all p < 0.05). A. oryzae is a common starter that is inoculated into soy sauce koji and is capable of secreting proteases to hydrolyze proteins and promote the production of peptides and amino acids (Vishwanatha et al., 2009; Gao et al., 2019). Further, Enterococcus is one of the main producers of methyl and ethyl esters due to its observed esterase and lipase activities in food fermentation (Jin et al., 2019). Moreover, Enterococcus abundances have also been positively correlated with the concentrations of 16 amino acids in the fermented soybean food sufu (Huang X. N. et al., 2018). This study also demonstrated the positive correlation of Enterococcus abundances to concentrations of all 16 amino acids that were analyzed, with 15 exhibiting significantly positive correlations to abundances (all p < 0.05).
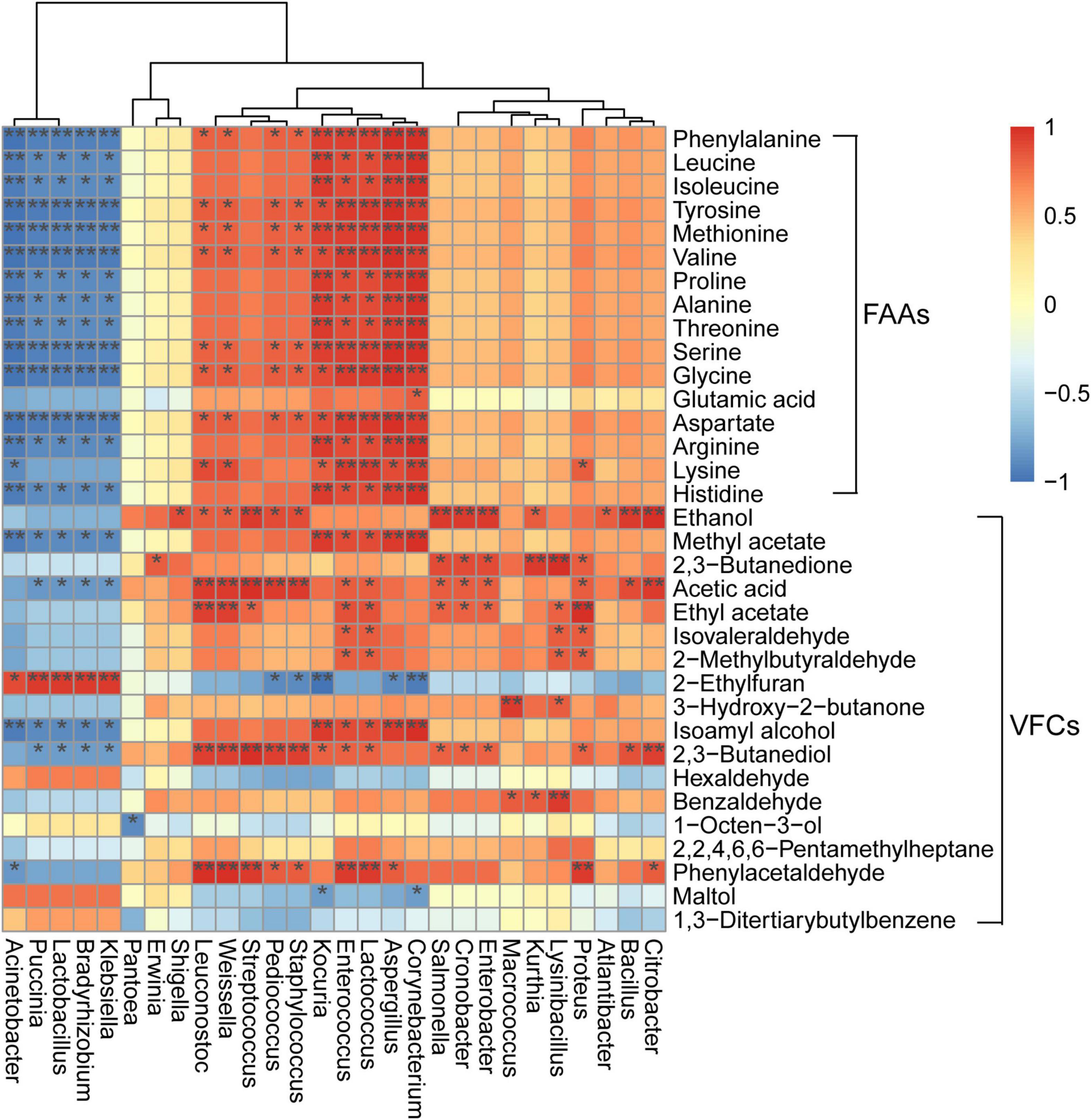
Figure 4. Heatmap of correlations among microbial genera and metabolites. Correlation strength (based on Spearman’s r-value) and correlation significance values are shown as shaded colors (red, positive correlation; blue, negative correlation). Heatmap values range from +1.0 to −1.0. Values above/below zero represent positive/negative correlations, respectively, between genera and parameters. *p < 0.05, **p < 0.01.
The abundances of the low-abundance genus Corynebacterium (<1% relative abundance at each stage) were also positively correlated to the concentrations of all 16 amino acids and two VFCs (methyl acetate and isoamyl alcohol) (all p < 0.05), suggesting a possible role in flavor generation. Weissella was positively correlated to the concentrations of eight amino acids (phenylalanine, tyrosine, methionine, valine, serine, glycine, aspartate, and lysine), phenylacetaldehyde, acetic acid, 2,3-butanediol, ethyl acetate, and ethanol (all p < 0.05). Additionally, the abundances of other genera (such as Streptococcus, Bacillus, Cronobacter, Pediococcus, and Kurthia) also exhibited positive relationships with ethanol concentrations (all p < 0.05) (Figure 4), indicating that these genera, may contribute to the production of ethanol. It is worth noting that these correlational results between microbial populations and metabolites in koji were from fermentation that used whole soybeans. However, different soybean materials may influence microbial communities and the associations between microorganism abundances and metabolites. Therefore, further studies are needed to evaluate these interactions in koji.
In this study, changes in microbial community compositions, genetic functions, and metabolites were investigated during koji making by coupling shotgun metagenomics and metabolomics. To the best of our knowledge, this is the first study to comprehensively evaluate the functional potentials of microbial communities and correlations between microbiota and metabolites during koji making. Aspergillus and Weissella were identified as the most abundant microbial taxa during koji making. In addition, functional profiling analysis indicated that metabolic functions of the koji microbiome exhibited drastic shifts after 24 h of fermentation, wherein the abundances of genes related to pyruvate metabolism, glycolysis/gluconeogenesis, and the TCA cycle greatly decreased, while the abundances increased for other functional genes associated with carbohydrate and amino acid metabolism. Among the latter category, genes that were enriched in later stages of fermentation included those involved in starch and sucrose metabolism; amino sugar and nucleotide sugar metabolism; alanine, aspartate, and glutamate metabolism; glycine, serine, and threonine metabolism; lysine biosynthesis; and cysteine and methionine metabolism. Correlational analyses indicated that the abundances of Aspergillus, Kocuria, Enterococcus, Lactococcus, and Corynebacterium were all positively correlated with the concentrations of all amino acids (except for glutamic acid), while Weissella abundances were positively associated with the concentrations of eight amino acids, phenylacetaldehyde, acetic acid, 2,3-butanediol, ethyl acetate, and ethanol. Overall, this study provides novel insights into the roles of microbial communities in the generation of metabolites during koji making. A better understanding of the microbial taxonomic and functionality within koji can help optimize product quality and further improve the flavor of soy sauce products. Lastly, Aspergillus and Weissella populations were frequently detected in similar high abundance during koji making, but little is known about their potential inter-species interactions. Future studies should be conducted to investigate potential coexistence relationships between these two species during soy sauce manufacturing.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
GT and MH conceived and designed the experiments in addition to writing the manuscript. GT and YW conducted the experiments and data analyses. XiL, XuL, and ZP performed most of the experiments, while ML, LL, and ZZ supervised the execution of the experiments. All authors read and approved the final manuscript.
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (Grant Nos. 2020A1515011308, 2214050006757, and 2020A1515011577), the Major Projects in Key Fields of Colleges and Universities of Guangdong Province (Grant No. 2020ZDZX3027), the National Science Foundation of China (Grant No. 41977138), General University Project of Guangdong Provincial Department of Education (Grant Nos. 2021KCXTD070 and 2021ZDZX4072), the Construction Project of Teaching Quality and Teaching Reform in Guangdong Province (Grant No. SJD202001), the Key Project of Social Welfare and Basic Research of Zhongshan City (Grant No. 2020B2010), and start-up funds at the Zhongshan Institute, University of Electronic Science and Technology of China (Grant No. 419YKQN12).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to Ecogene Biotech Co., Ltd. (Shenzhen, China) for assistance with bioinformatics analysis of metagenomic data.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.841529/full#supplementary-material
Supplementary Figure 1 | Koji making overview and sampling times.
Supplementary Figure 2 | pH changes during koji making.
Supplementary Figure 3 | Taxonomic composition of koji metagenomes at the phylum level showing changes in microbial communities across six fermentation stages (0–48 h). Sequences that could not be classified to known taxa were designated as “unclassified”.
Supplementary Figure 4 | Functional profiles produced from whole shotgun metagenome-derived ORFs annotated at level 2 of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Chung, H. Y., Fung, P. K., and Kim, J. S. (2005). Aroma impact components in commercial plain sufu. J. Agric. Food Chem. 53, 1684–1691. doi: 10.1021/jf048617d
Devanthi, P. V. P., and Gkatzionis, K. (2019). Soy sauce fermentation: microorganisms, aroma formation, and process modification. Food Res. Int. 120, 364–374. doi: 10.1016/j.foodres.2019.03.010
Ding, C. F., Meng, M., Jiang, Y. Y., and Hou, L. H. (2019). Improvement of the quality of soy sauce by reducing enzyme activity in Aspergillus oryzae. Food Chem. 292, 81–89. doi: 10.1016/j.foodchem.2019.04.052
Feng, Y. Z., Chen, Z. Y., Liu, N., Zhao, H. F., Cui, C., and Zhao, M. M. (2014). Changes in fatty acid composition and lipid profile during koji fermentation and their relationships with soy sauce flavour. Food Chem. 158, 438–444. doi: 10.1016/j.foodchem.2014.02.147
Feng, Y. Z., Cui, C., Zhao, H. F., Gao, X. L., Zhao, M. M., and Sun, W. Z. (2013). Effect of koji fermentation on generation of volatile compounds in soy sauce production. Int. J. Food Sci. Technol. 48, 609–619. doi: 10.1111/ijfs.12006
Ferrocino, I., Bellio, A., Giordano, M., Macori, G., Romano, A., Rantsiou, K., et al. (2018). Shotgun metagenomics and volatilome profile of the microbiota of fermented sausages. Appl. Environ. Microbiol. 84, e2110–e2117. doi: 10.1128/aem.02120-17
Fessard, A., and Remize, F. (2017). Why are Weissella spp. not used as commercial starter cultures for food fermentation? Fermentation 3:38. doi: 10.3390/fermentation3030038
Fu, L. M., Niu, B. F., Zhu, Z. W., Wu, S. T., and Li, W. Z. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, X. L., Cui, C., Zhao, H. F., Zhao, M. M., Yang, L., and Ren, J. Y. (2010). Changes in volatile aroma compounds of traditional Chinese-type soy sauce during moromi fermentation and heat treatment. Food Sci. Biotechnol. 19, 889–898. doi: 10.1007/s10068-010-0126-7
Gao, X. L., Yin, Y. Y., Yan, J. K., Zhang, J. K., Ma, H. L., and Zhou, C. S. (2019). Separation, biochemical characterization and salt-tolerant mechanisms of alkaline protease from Aspergillus oryzae. J. Sci. Food Agric. 99, 3359–3366. doi: 10.1002/jsfa.9553
Guo, J., Luo, W., Fan, J., Suyama, T. K., and Zhang, W. X. (2020). Co-inoculation of Staphylococcus piscifermentans and salt-tolerant yeasts inhibited biogenic amines formation during soy sauce fermentation. Food Res. Int. 137:109436. doi: 10.1016/j.foodres.2020.109436
Han, D. M., Chun, B. H., Feng, T., Kim, H. M., and Jeon, C. O. (2020). Dynamics of microbial communities and metabolites in ganjang, a traditional Korean fermented soy sauce, during fermentation. Food Microbiol. 92:103591. doi: 10.1016/j.fm.2020.103591
He, W. M., and Chung, H. Y. (2020). Exploring core functional microbiota related with flavor compounds involved in the fermentation of a natural fermented plain sufu (Chinese fermented soybean curd). Food Microbiol. 90:103408. doi: 10.1016/j.fm.2019.103408
Huang, L. D., Wu, Z. F., Chen, X. Q., Weng, P. F., and Zhang, X. (2018). Characterization of flavour and volatile compounds of fermented squid using electronic nose and HPMS in combination with GC-MS. Int. J. Food Prop. 21, 760–770. doi: 10.1080/10942912.2018.1454466
Huang, X. N., Yu, S. Z., Han, B. Z., and Chen, J. Y. (2018). Bacterial community succession and metabolite changes during sufu fermentation. LWT 97, 537–545. doi: 10.1016/j.lwt.2018.07.041
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jin, Y., Li, D. Y., Ai, M., Tang, Q. X., Huang, J., Ding, X. F., et al. (2019). Correlation between volatile profiles and microbial communities: a metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 121, 422–432. doi: 10.1016/j.foodres.2019.03.021
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim, K. H., Chun, B. H., Kim, J., and Jeon, C. O. (2021). Identification of biogenic amine-producing microbes during fermentation of ganjang, a Korean traditional soy sauce, through metagenomic and metatranscriptomic analyses. Food Control 121:107681. doi: 10.1016/j.foodcont.2020.107681
Kumar, J., Sharma, N., Kaushal, G., Samurailatpam, S., Sahoo, D., Rai, A. K., et al. (2019). Metagenomic insights into the taxonomic and functional features of kinema, a traditional fermented soybean product of Sikkim Himalaya. Front. Microbiol. 10:1744. doi: 10.3389/fmicb.2019.01744
Liang, H. P., Chen, H. Y., Zhang, W. X., Yu, C. X., Ji, C. F., and Lin, X. P. (2018). Investigation on microbial diversity of industrial Zhacai paocai during fermentation using high-throughput sequencing and their functional characterization. LWT 91, 460–466. doi: 10.1016/j.lwt.2018.01.088
Liang, R., Huang, J., Wu, X. M., Xu, Y., Fan, J., Wu, C. D., et al. (2019). Characterizing the metabolites and the microbial communities of the soy sauce mash affected by temperature and hydrostatic pressure. Food Res. Int. 123, 801–808. doi: 10.1016/j.foodres.2019.06.002
Liu, X. Y., Qian, M., Shen, Y. X., Qin, X., Huang, H. C., Yang, H., et al. (2021). An high-throughput sequencing approach to the preliminary analysis of bacterial communities associated with changes in amino acid nitrogen, organic acid and reducing sugar contents during soy sauce fermentation. Food Chem. 349:129131. doi: 10.1016/j.foodchem.2021.129131
Mannaa, M., Seo, Y. S., and Park, I. (2020). Addition of coriander during fermentation of Korean soy sauce (gangjang) causes significant shift in microbial composition and reduction in biogenic amine levels. Foods 9:1346. doi: 10.3390/foods9101346
Marilley, L., and Casey, M. G. (2004). Flavours of cheese products: metabolic pathways, analytical tools and identification of producing strains. Int. J. Food Microbiol. 90, 139–159. doi: 10.1016/s0168-1605(03)00304-0
Oguntoyinbo, F. A., and Narbad, A. (2012). Molecular characterization of lactic acid bacteria and in situ amylase expression during traditional fermentation of cereal foods. Food Microbiol. 31, 254–262. doi: 10.1016/j.fm.2012.03.004
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., Mcglinn, D., et al. (2019). Vegan: Community Ecology Package. R package Version 2.5–6.2019.
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Qi, Q., Huang, J., Zhou, R., Yang, M., Zhang, L., Peng, C., et al. (2021). Characterizing microbial community and metabolites of Cantonese soy sauce. Food Biosci. 40:100872. doi: 10.1016/j.fbio.2020.100872
Sabio, E., Vidal-Aragón, M. C., Bernalte, M. J., and Gata, J. L. (1998). Volatile compounds present in six types of dry-cured ham from south European countries. Food Chem. 61, 493–503. doi: 10.1016/S0308-8146(97)00079-4
Sanjukta, S., Rai, A. K., Muhammed, A., Jeyaram, K., and Talukdar, N. C. (2015). Enhancement of antioxidant properties of two soybean varieties of Sikkim Himalayan region by proteolytic Bacillus subtilis fermentation. J. Funct. Foods 14, 650–658. doi: 10.1016/j.jff.2015.02.033
Schumann, G., Bonora, R., Ceriotti, F., Clerc-Renaud, P., Ferrero, C. A., Férard, G., et al. (2002). IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37°C. Part 3. reference procedure for the measurement of catalytic concentration of lactate dehydrogenase. Clin. Chem. Lab. Med. 40, 643–648. doi: 10.1515/CCLM.2002.111
Shi, C. Y., Zhang, Y., Lu, Z. Q., and Wang, Y. Z. (2017). Solid-state fermentation of corn-soybean meal mixed feed with Bacillus subtilis and Enterococcus faecium for degrading antinutritional factors and enhancing nutritional value. J. Anim. Sci. Biotechnol. 8:50. doi: 10.1186/s40104-017-0184-2
Smit, G., Smit, B. A., and Engels, W. J. M. (2005). Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol. Rev. 29, 591–610. doi: 10.1016/j.fmrre.2005.04.002
Sulaiman, J., Gan, H. M., Yin, W. F., and Chan, K. G. (2014). Microbial succession and the functional potential during the fermentation of Chinese soy sauce brine. Front. Microbiol. 5:556. doi: 10.3389/fmicb.2014.00556
Tan, G. L., Hu, M., Li, X. Y., Pan, Z. Q., Li, M., Li, L., et al. (2020). High-throughput sequencing and metabolomics reveal differences in bacterial diversity and metabolites between red and white sufu. Front. Microbiol. 11:758. doi: 10.3389/fmicb.2020.00758
Tanaka, Y., Watanabe, J., and Mogi, Y. (2012). Monitoring of the microbial communities involved in the soy sauce manufacturing process by PCR-denaturing gradient gel electrophoresis. Food Microbiol. 31, 100–106. doi: 10.1016/j.fm.2012.02.005
Tomasik, P., and Horton, D. (2012). “Chapter 2 - Enzymatic conversions of starch,” in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton (Cambridge, MA: Academic Press), 59–436. doi: 10.1016/B978-0-12-396523-3.00001-4
Vishwanatha, K. S., Rao, A. G. A., and Singh, S. A. (2009). Characterisation of acid protease expressed from Aspergillus oryzae MTCC 5341. Food Chem. 114, 402–407. doi: 10.1016/j.foodchem.2008.09.070
Wei, C. L., Chao, S. H., Tsai, W. B., Lee, P. S., Tsau, N. H., Chen, J. S., et al. (2013). Analysis of bacterial diversity during the fermentation of inyu, a high-temperature fermented soy sauce, using nested PCR-denaturing gradient gel electrophoresis and the plate count method. Food Microbiol. 33, 252–261. doi: 10.1016/j.fm.2012.10.001
Wei, Q. Z., Wang, H. B., Chen, Z. X., Lv, Z. J., Xie, Y. F., and Lu, F. P. (2013). Profiling of dynamic changes in the microbial community during the soy sauce fermentation process. Appl. Microbiol. Biotechnol. 97, 9111–9119. doi: 10.1007/s00253-013-5146-9
Wicklow, D. T., Mcalpin, C. E., and Yeoh, Q. L. (2007). Diversity of Aspergillus oryzae genotypes (RFLP) isolated from traditional soy sauce production within Malaysia and Southeast Asia. Mycoscience 48, 373–380. doi: 10.1007/S10267-007-0383-3
Xiang, W. L., Zhang, N. D., Lu, Y., Zhao, Q. H., Xu, Q., Rao, Y., et al. (2020). Effect of Weissella cibaria co-inoculation on the quality of Sichuan pickle fermented by Lactobacillus plantarum. LWT 121:108975. doi: 10.1016/j.lwt.2019.108975
Xie, C., Mao, X. Z., Huang, J. J., Ding, Y., Wu, J. M., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Yan, Y. Z., Qian, Y. L., Ji, F. D., Chen, J. Y., and Han, B. Z. (2013). Microbial composition during Chinese soy sauce koji-making based on culture dependent and independent methods. Food Microbiol. 34, 189–195. doi: 10.1016/j.fm.2012.12.009
Zhao, G. Z., Liu, C., Li, S., Wang, X. W., and Yao, Y. P. (2020). Exploring the flavor formation mechanism under osmotic conditions during soy sauce fermentation in Aspergillus oryzae by proteomic analysis. Food and Funct. 11, 640–648. doi: 10.1039/C9FO02314C
Zhu, W. H., Lomsadze, A., and Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132–e132. doi: 10.1093/nar/gkq275
Keywords: koji making, metagenome, microbial community structure, metabolites, functional potential
Citation: Tan G, Hu M, Li X, Li X, Pan Z, Li M, Li L, Wang Y and Zheng Z (2022) Microbial Community and Metabolite Dynamics During Soy Sauce Koji Making. Front. Microbiol. 13:841529. doi: 10.3389/fmicb.2022.841529
Received: 22 December 2021; Accepted: 03 February 2022;
Published: 25 February 2022.
Edited by:
Lin Lin, Jiangsu University, ChinaReviewed by:
Marko Verce, Catholic University of Louvain, Belgium Saenjum, Chiang Mai University, Thailand
Saenjum, Chiang Mai University, ThailandCopyright © 2022 Tan, Hu, Li, Li, Pan, Li, Li, Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Hu, aHVtaW5Ac29pbC5nZC5jbg==; Yi Wang, d2FuZ3lpQHpzYy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.