 Prabha Liyanapathiranage
Prabha Liyanapathiranage Naama Wagner2†
Naama Wagner2† Oren Avram
Oren Avram Tal Pupko
Tal Pupko Neha Potnis
Neha Potnis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 14 April 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.840308
This article is part of the Research TopicPopulation and Comparative Genomics of Plant Pathogenic BacteriaView all 16 articles
The type VI secretion system (T6SS) present in many Gram-negative bacteria is a contact-dependent apparatus that can directly deliver secreted effectors or toxins into diverse neighboring cellular targets including both prokaryotic and eukaryotic organisms. Recent reverse genetics studies with T6 core gene loci have indicated the importance of functional T6SS toward overall competitive fitness in various pathogenic Xanthomonas spp. To understand the contribution of T6SS toward ecology and evolution of Xanthomonas spp., we explored the distribution of the three distinguishable T6SS clusters, i3*, i3***, and i4, in approximately 1,740 Xanthomonas genomes, along with their conservation, genetic organization, and their evolutionary patterns in this genus. Screening genomes for core genes of each T6 cluster indicated that 40% of the sequenced strains possess two T6 clusters, with combinations of i3*** and i3* or i3*** and i4. A few strains of Xanthomonas citri, Xanthomonas phaseoli, and Xanthomonas cissicola were the exception, possessing a unique combination of i3* and i4. The findings also indicated clade-specific distribution of T6SS clusters. Phylogenetic analysis demonstrated that T6SS clusters i3* and i3*** were probably acquired by the ancestor of the genus Xanthomonas, followed by gain or loss of individual clusters upon diversification into subsequent clades. T6 i4 cluster has been acquired in recent independent events by group 2 xanthomonads followed by its spread via horizontal dissemination across distinct clades across groups 1 and 2 xanthomonads. We also noted reshuffling of the entire core T6 loci, as well as T6SS spike complex components, hcp and vgrG, among different species. Our findings indicate that gain or loss events of specific T6SS clusters across Xanthomonas phylogeny have not been random.
The genus Xanthomonas belongs to the gamma subdivision of the phylum Proteobacteria (Jun et al., 2010). It consists of more than 35 species, some of which cause economically important diseases in more than 400 host plants (Timilsina et al., 2020). Even though a wide host range is observed with Xanthomonas spp., individual strains can be tightly restricted to a particular host or host group. Hence, the Xanthomonas spp. are further differentiated into pathovars based on this characteristic (White et al., 2009; An et al., 2020). To date, Xanthomonas spp. have been isolated from host plants belonging to 124 monocotyledonous and 268 dicotyledonous species (White et al., 2009; Kyrova et al., 2020). In terms of host tissue specificity, Xanthomonas spp. can either invade the vascular tissues or survive in the intercellular spaces of the mesophyll parenchyma tissue (An et al., 2020). To successfully adapt to the hostile host environment, Xanthomonas spp. usually depend on several factors that are important for their invasion. These factors are responsible for various functions, including adherence to the plant surface, invasion to the intercellular space of the host tissue, manipulation of plant cellular processes, acquisition of nutrients, counteracting plant defense responses, and competing with other microbes that occupy the same niche (Büttner and Bonas, 2010). Identification of the adaptation factors that contribute to host–pathogen interactions is crucial in implementing future management strategies.
To date, more than 1,700 Xanthomonas genomes are publicly available in the National Center for Biotechnology Information (NCBI) database. With the availability of such a large amount of genomic data, comparative genomic studies enabled the identification of many potential virulence and fitness strategies contributing to the adaptation to a variety of monocot/dicot hosts and the establishment of successful vascular or non-vascular modes of infection (Jacques et al., 2016; An et al., 2020). These strategies include various secretion systems, by which pathogenic bacteria translocate proteins/DNA directly into the host cell or into the extracellular milieu.
In Gram-negative bacteria, 10 different secretion systems have been identified, but only six of them (I–VI) have been described in the genus Xanthomonas (Meuskens et al., 2019; Alvarez-Martinez et al., 2020; Palmer et al., 2020). Of these six systems, types II, III, and IV have been extensively studied in the genus Xanthomonas. In contrast, the role, distribution, and evolutionary history of the most recently identified type VI secretion system (T6SS) has been described only in few studies (Bayer-Santos et al., 2018; Bansal et al., 2019, 2020; Bayer-Santos et al., 2019; Ceseti et al., 2019; Alvarez-Martinez et al., 2020; Choi et al., 2020; Zhu et al., 2020; Montenegro Benavides et al., 2021). Most of the aforementioned studies used only representative Xanthomonas spp., to derive conclusions regarding the distribution of the T6SS clusters in the whole genus Xanthomonas. Yet, the evolutionary history of multiple T6SS clusters, their distribution across the genus, and their association with particular pathogen lifestyle all remain unclear.
T6SS is present in approximately 25% of Gram-negative bacteria, mainly within the phylum Proteobacteria and the classes α-, β-, and γ-proteobacteria, including pathogenic, beneficial, and commensal bacteria, indicating possible functions of this system unrelated to pathogenicity (Boyer et al., 2009; Durand et al., 2014). T6SS is composed of 15 to 23 different proteins, including 13 core structural proteins that assemble into an injectisome-like structure (Shyntum et al., 2014). Core components are named as Tss (for type VI secretion) A-M and are usually encoded in the same gene cluster (Zoued et al., 2014). In addition to the core genes, most T6 clusters also include Tag (type VI–associated genes), which encode accessory proteins, suggested to be important for the transcriptional or posttranscriptional regulation and assembly of the T6SS. Finally, T6SS includes T6 effectors and their associated immunity proteins (Shalom et al., 2007; Boyer et al., 2009; Silverman et al., 2011; Coulthurst, 2013). This Tss nomenclature is now widely adopted by many, while common names, such as Hcp, VgrG, and ClpV, are also in use.
T6SS is comprised of three major complexes: the transmembrane complex, the baseplate, and the tail. Together, they form a larger machine that extends from the bacterial cytoplasm, across the bacterial cell wall and into the target cell (Silverman et al., 2012; Coulthurst, 2013; Durand et al., 2015; Nguyen et al., 2018; and Hernandez et al., 2020). The transmembrane complex made from TssM, TssJ, and TssL acts as a docking site for the cytoplasmic baseplate-like structure made from TssE, TssF, TssG, and TssK. It also serves as an adaptor between the transmembrane complex and the tail. The internal tube of the tail component is made of Hcp tubes that are assembled into the central VgrG–PAAR spike and is enclosed by a contractile sheath composed of TssB and TssC. Secretion of effector proteins through the T6SS is a one-step process, independent of the Sec or Tat secretion machinery, which is mediated through contraction of the sheath to propel the Hcp–VgrG–PAAR complex toward the target cell and deliver a cocktail of effectors in each contraction. Unlike other secretion systems, after each firing event, the T6 tube disassembles, and Hcp is lost to the extracellular milieu. AAA+ClpV (TssH) ATPase is recruited to the contracted sheath to disassemble the complex and to recycle the sheath components for the next round of firing. Apart from being core components of the T6SS, Hcp (hemolysin coregulated protein) and VgrG (valine-glycine repeat protein G) have also been identified as specialized effectors important in antibacterial activity in several pathogen systems (Lien and Lai, 2017; Ma et al., 2017). Effectors that get delivered via the T6SS are categorized as either “cargo” or “specialized.” Cargo effectors interact with the Hcp–VgrG–PAAR complex non-covalently, whereas specialized effectors are covalently linked to these proteins. T6 effectors and their antitoxins or immunity proteins may be encoded either within the T6 gene cluster, which encodes the core genes, or scattered in other parts of the bacterial genome (Hernandez et al., 2020).
Depending on the 13 core proteins, T6 clusters have been separated into four subtypes (T6SSi–iv). The majority of the proteobacteria have T6 clusters belonging to the subtype T6SSi, and because of the diversity observed in this subgroup, it has been further subdivided into five phylogenetic clades (i1–i5) (Boyer et al., 2009). Bacteria belonging to the family Xanthomonadales harbor three such clades: i1, i3, and i4, whereas bacteria within genus Xanthomonas harbor only the i3 and i4 clades (Bayer-Santos et al., 2019; Alvarez-Martinez et al., 2020). Clade i3 was further subdivided into distinct subclades: i3*, i3**, and i3*** based on the phylogenetic analysis conducted using tssC gene (Bayer-Santos et al., 2019). Species within the genus Xanthomonas harbor subclades i3* (e.g., Xanthomonas citri), i3*** (e.g., Xanthomonas oryzae), or both i3* and i3*** (e.g., Xanthomonas perforans), but not i3**.
The phylogenetic-based division to clades and subclades is also reflected in the set of genes included within the T6SS gene cluster and its genome organization. For example, all Xanthomonas species within the i3 clade lack tssJ, a core component in the outer membrane complex of T6 machinery (Alvarez-Martinez et al., 2020). In addition, i3* and i3*** differ in their genetic architecture, for example, i3* cluster in Xanthomonas euvesicatoria 85-10 is approximately 40 kb, whereas the i3*** cluster is approximately 20 kb. This difference in cluster size is due to the presence of different genes encoding hypothetical proteins and transcriptional regulators in each cluster (Boyer et al., 2009; Abendroth et al., 2017; Alvarez-Martinez et al., 2020).
Similar to observations with other bacteria, some Xanthomonas spp. encode multiple T6 copies: either both i3* and i3*** or one copy from each subclade together with the i4 cluster (Bingle et al., 2008; Alvarez-Martinez et al., 2020). These multiple copies of T6 gene clusters are believed to be acquired by independent horizontal gene transfer events, suggesting different roles played by the T6 clusters in different bacteria (Bingle et al., 2008; Blondel et al., 2009; Boyer et al., 2009). In X. oryzae pv. oryzae, one T6 cluster is distantly related to the phaseoli clade, whereas the second cluster forms a monophyletic clade with i3*** cluster of X. euvesicatoria and X. perforans, implicating a recent common origin (Montenegro Benavides et al., 2021).
The goal of this study is to harness a large set of publicly available Xanthomonas genomes spanning the entire genus Xanthomonas with the genomes of multiple important pathogenic and non-pathogenic or commensal species and study the conservation, genetic organization, and evolutionary patterns of multiple T6SS clusters. We hypothesize that the acquisition of the multiple T6SS clusters occurred as independent events during the adaptation of Xanthomonas species to specific hosts, as they diverged into various clades. Specific cases of loss or gain events happened in certain clades as an adaptation to vascular or non-vascular lifestyles. We examined the phylogenetic distribution of T6SS clusters and systematically assessed the acquisition and loss events among different species along with the reshuffling of entire loci or individual genes, to obtain insights into the significance of T6SS in host adaptation of xanthomonads.
All Xanthomonas RefSeq genomes available in NCBI on January 16, 2022, were downloaded. This included a total of 1,740 genomes of 39 Xanthomonas species (see full list of species and accession numbers in Supplementary Table 1). For each of these genomes, protein translation of open reading frames was extracted using Prodigal (Hyatt et al., 2010).
To find the core genes of T6-i3*, T6-i3***, and T6-i4 clusters in each of the Xanthomonas genomes, we first established datasets of core genes of each of the three clusters. The core genes of T6-i3* and T6-i3*** were taken from X. euvesicatoria str. 85-10 (accession NC_007508.1), whereas the core genes of T6-i4 were taken from X. oryzae pv. oryzae PXO99A (accession NC_010717.2). The core gene loci of all three clusters included TssA, TssB, TssF, TssG, TssK, TssL (T6-i4 included both TssL1 and TssL2), and TssM. T6-i3* and T6-i3*** also included TssC and TssD. T6-i3*** and T6-i4 also included TssE. T6-i3* also included TssH, T6-i3*** also included ImpE-like protein TagF, and T6-i4 also included TssJ and ClpV.
The core genes of T6-i3*, T6-i3***, and T6-i4 clusters were searched for in each of the Xanthomonas genomes using BLASTx (Altschul et al., 1990). We used the core gene sequences described above as query and the protein sequences of each of the genomes as databases. The commands used were makeblastdb -in {dataset} -dbtype prot -out {dataset_db} to create the BLAST database, and blastx -db {dataset_db} -query {query} -outfmt 6 -out {out} to run BLASTx. “dataset” was the protein FASTA file of a certain Xanthomonas genome, and “query” was a DNA FASTA file containing the core genes of a certain T6 system. It was repeated for each genome and system combination. For each of the core genes in the query, the best BLAST hit in the genome was considered, if it had an E-value lower than 10−10 and identity of at least 50% to the core gene, and a coverage of at least 30% (i.e. that the length of the aligned region is at least 30% of the query gene length). In case a certain gene in the genome was found to be a hit matching our criteria for genes of more than one T6 system, for example, TssA of T6-i3* and TssA of T6-i3***, the hit was saved only for the gene of the system it had the highest identity to. ClpV shares domains with ClpB, so to avoid classifying ClpB genes as ClpV; we blasted ClpB versus the genomes, and protein sequences that showed a higher sequence similarity to ClpB than to ClpV were discarded. Then, for each genome, the presence of each T6 system was evaluated. If all the core genes of a specific system were found in the genome, we considered this system to be full in this genome. Otherwise, if it had at least one core gene, we considered it partial. Gene Neighborhood tool was used to identify gene synteny and cluster organization (Chen et al., 2021).
Following the identification of core genes, as described previously, the corresponding protein sequences of each gene were aligned using MAFFT (v.7.471) with the L-ins-I option (Katoh et al., 2002). Phylogenetic trees under the maximum likelihood (ML) criterion were reconstructed using the IQ-TREE multicore version 2.1.2 COVID edition (Minh et al., 2020). The “–m MFP” function was used for the model selection, whereas “–merge rclusterf” was used for partition finding. Branch support was determined using 1,000 bootstraps and 1,000 SH-aLRT bootstrap replicates (-B 1000, -alrt 1000). Nodes with ≥80% support with the SH-aLRT and >80% with normal bootstrap supports were considered well supported (Davis et al., 2021). Trees were visualized using FigTree v1.4.4 (Rambaut et al., 2018).
To identify and visualize possible conflicting signals, which would suggest possible recombination events and evolutionary relationships within sequence data, phylogenetic networks were inferred using SplitsTree4 (version 4.17.0) (Huson and Bryant, 2006). Concatenated T6 core genes from each cluster were used to generate SplitsTree files, for a selected subset of genomes using the automated multilocus sequence analysis pipeline (automlsa2) (Davis et al., 2021). Resultant splits.nex files were visualized using the SplitsTree4 (version 4.17.0), with the rooted equal angle algorithm (Gambette and Huson, 2008). Recombination events were identified by the branches that form parallelograms (Joseph and Forsythe, 2012).
To reconstruct the species phylogeny, we conducted a multilocus sequence analysis (MLSA) using 12 housekeeping genes: atpD (ATP synthase β chain), dnaK (70-kDa heat shock protein), efp (elongation factor P), fusA (translation elongation factor aEF-2), fyuA (transmembrane protein; Ton-B–dependent transporter), gapA (glyceraldehyde 3-phosphate dehydrogenase), glnA (glutamine synthetase I), gltA (citrate (Si)-synthase gene), gyrB (DNA gyrase β subunit), lacF (sugar ABC transporter permease), lepA (GTP-binding protein), and rpoD (RNA polymerase sigma 70 factor). These genes were previously used for MLSA (Young et al., 2008; Fischer-Le Saux et al., 2015; Timilsina et al., 2015). We created a multiple sequence alignment (MSA) for each of these 12 genes using MAFFT (Katoh et al., 2002; Katoh and Standley, 2013) and then concatenated these MSAs. The concatenated MSA was used as input for RaxML (Stamatakis, 2014) to create the phylogenetic tree.
We used GLOOME to analyze the gain and loss probabilities for three T6 clusters during evolution of the entire Xanthomonas genus (Cohen et al., 2010). GLOOME analyzes phyletic patterns based on presence and absence of individual core genes of the cluster and accurately infers branch-specific and site-specific gain and loss events. We generated the input file of phyletic pattern based on the BLASTx results described above, in which “1” indicates presence and “0” indicates absence. The MLSA tree was used as input tree. Cladogram was drawn with FigTree v1.4.4 (Rambaut et al., 2018). Gain and loss events were added manually based on the GLOOME output files.
For the reconstruction of the core genome phylogeny Roary was used (Page et al., 2015). All the genomes downloaded from the NCBI GenBank in the fna format were first annotated using the Prokka (v1.14.5) software tool (Seemann, 2014). After annotating the genome to determine the location and attributes of the genes contained in them, the resultant gff file in the GFF3 format was used as input file to perform the pan-genome analysis using Roary. Roary extracted the coding sequences from the input GFF3 files, converted them into protein sequences, removed partial clusters, and created preclusters. Next Roary performed BLASTP with 95% sequence identity, and sequences were clustered with Markov cluster algorithm. As the next step, Roary generated clusters and inferred orthologs. Finally, Roary identified core genes and generated a concatenated core gene alignment that was used to generate maximum likelihood phylogeny using RAxML (v 8.2.12) (Stamatakis, 2014).
In this study, we analyzed 1,740 complete genome sequences, belonging to 39 Xanthomonas spp. retrieved from the NCBI RefSeq with a large proportion of them from species, X. oryzae (396 genomes), X. citri (195 genomes), X. phaseoli (97 genomes), X. perforans (151 genomes), and X. arboricola (136 genomes). This collection of genomes also included commensal Xanthomonas spp., isolated from environment (rain) as well as isolated from diverse plant species (Potnis et al., unpublished). Based on core gene sequence identity of T6SS clusters, presence of complete T6 i3*** was identified in 832 genomes; T6 i3* in 710 genomes and 497 genomes had the complete T6 i4 cluster (Table 1). Presence of only a single T6 cluster, T6 i3* was identified in 23% of Xanthomonas genomes, the majority belonging to X. citri, X. phaseoli, X. vesicatoria, X. axonopodis, and few strains belonging to X. arboricola, X. campestris, X. pisi, X. dyei, and X. cucurbitae, whereas that of T6SS i3*** alone in X. vasicola and that of T6 i4 alone in X. oryzae, X. translucens, X. fragarie, couple of X. hortorum strains and X. bromi. Some X. oryzae strains contain both T6 i3*** and T6 i4. The T6SS makeup in X. citri is highly variable, with majority of strains containing T6 i3* alone, 14 strains with T6 i3* and T6 i4, whereas a couple of them lack T6 clusters. X. phaseoli also shows variability in genetic makeup of T6 clusters, with 40 strains with T6 i3* alone, 52 strains with T6 i3* and T6 i3***, and 2 strains containing T6 i3* and T6 i4. Eighty-nine percent of the X. oryzae strains possess both T6 i3*** and T6 i4, and 2.77% contain T6 i4 alone. X. euvesicatoria and X. perforans contain both T6 i3* and T6 i3*** clusters. Xanthomonas albilineans, Xanthomonas sacchari, X. cannabis, and majority of X. arboricola strains lack T6 cluster. Xanthomonas maliensis strains contain all three T6 clusters. Approximately 40% of the currently classified X. campestris strains contain either i3* or i3*** complete clusters, with 13% of them containing both i3* and i3*** clusters (Table 1).
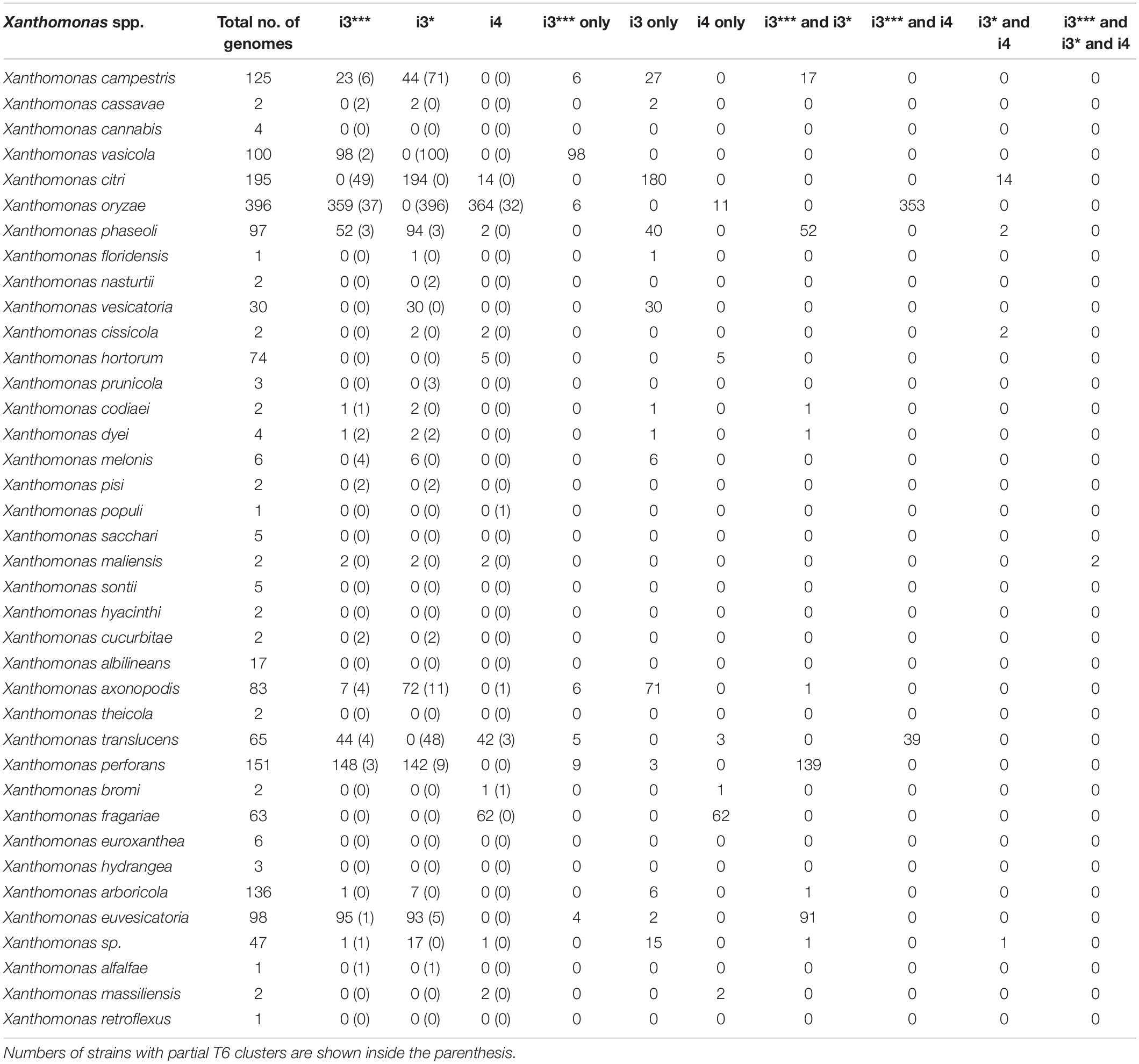
Table 1. Analysis of T6 cluster distribution in genus Xanthomonas.
Among early-branching strains, also known as group 1 xanthomonads, X. translucens and few strains belonging to unclassified species contain T6 clusters. Of 65 sequenced X. translucens strains, 44 strains contain intact T6 i3*** cluster, and 42 strains contain intact i4 cluster, with 39 strains containing both i3*** and i4 clusters (Table 1). While Xanthomonas hyancinthi genomes available in RefSeq do not contain any intact T6 cluster, X. hyacinthi DSM 19077 contains intact T6 i3*** cluster, with core genes of this cluster showing 100% nucleotide sequence identity to those from Luteibacter rhizovicinus DSM 16549 strain. This strain did not get included in Table 1 as it lacks RefSeq record. L. rhizovicinus is a gamma proteobacterium in the order Xanthomonadales isolated from the rhizosphere of barley (Hordeum vulgare) (Johansen et al., 2005). This observation indicates the possible independent acquisitions of the T6 clusters by individual strains within the early-branching clade from species belonging to closely related genera within Xanthomonadaceae family.
The size of each T6 cluster was also compared in this study. Most of the species with complete i3* cluster were 49–60 kbp whereas the i3* cluster in X. cucurbitae CFBP 2542 was approximately 70 kbp (Supplementary Figure 1). Shorter i3*** clusters with 23–28 kbps in size were identified in the analyzed genomes, whereas determining the size of the i4 cluster was not possible because of the presence of the i4 cluster in two loci.
Phylogeny based on concatenated sequences of 12 housekeeping genes (MLSA) was compared with that of T6 i3***, i3*, and i4 core gene phylogenies including conserved effector genes, vgrG and hcp (Figure 1). Incongruencies among MLSA phylogeny and that of T6 i3* were noted for the phylogenetic placements of clades belonging to species X. perforans and X. euvesicatoria. X. euvesicatoria is nestled between two X. perforans clades, whereas X. euvesicatoria–related pathovars (those pathogenic on other hosts such as alfalfa, and citrus) form a separate clade based on the MLSA phylogeny. T6 i3* phylogeny with hcp and vgrG sequences reshuffled some X. perforans strains among the two clades. Incongruencies between MLSA phylogeny and T6 i4 cluster phylogeny with hcp and vgrG sequences were observed in case of X. oryzae, X. translucens, and X. fragariae species. Each of these species forms a distinct clade in the MLSA phylogeny. However, based on T6 i4 phylogeny, X. oryzae was highly diverse, with strains dispersed throughout the phylogeny. Some X. translucens and X. fragarie strains carry T6 i4 loci that are closely related to X. oryzae. X. translucens strain ART-Xtg2 clustered with X. bromi based on T6 i4 phylogeny (Figures 1A,B). No apparent incongruencies were observed when i3*** phylogeny was compared with that of the MLSA phylogeny.
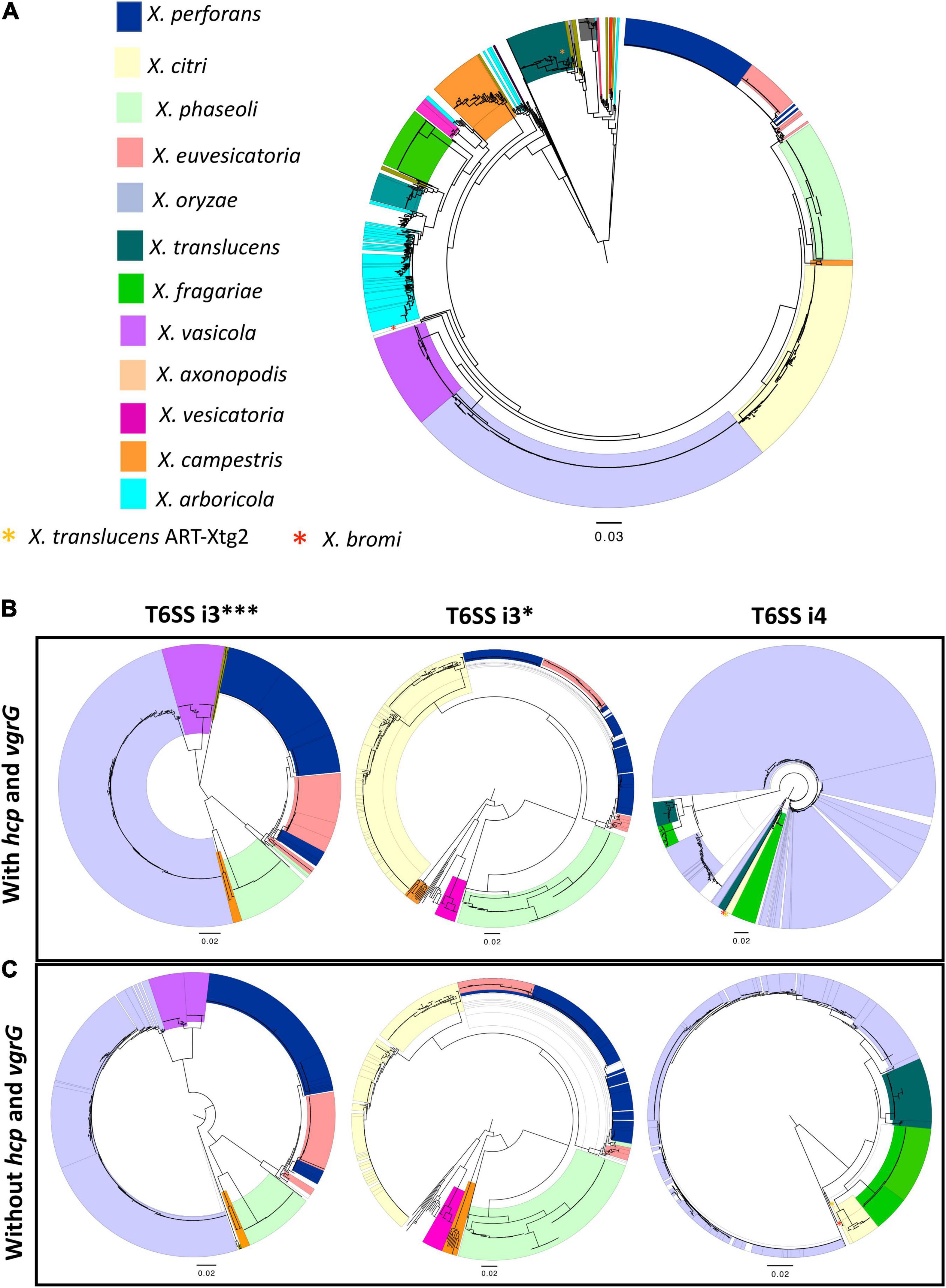
Figure 1. Distribution of T6 clusters in the genus Xanthomonas. (A) Relationship among 1,577 Xanthomonas genomes, multilocus sequence analysis (MLSA) phylogeny of Xanthomonas spp. based on the concatenation of partial sequences of housekeeping genes atpD, dnaK, efP, fusA, fyuA, gapA, glnA, gltA, gyrB, lacF, lepA, and rpoD. (B) T6 core gene phylogeny of Xanthomonas spp. based on the concatenation of T6 core genes tssA, tssB, tssC, tssD (hcp), tssE, tssF, tssG, tssH, tssI (vgrG), tssJ (only in i4 cluster), tssK, tssL, and tssM. (C) T6SS core gene phylogeny of i3***, i3*, and i4 clusters in the absence of hcp and vgrG core genes. (B,C) T6 core gene phylogenies were generated using the automated multilocus sequence analysis pipeline (automlsa2). Midpoint rooted phylogenetic trees in maximum likelihood (ML) criterion were reconstructed using the IQ-TREE and branch support was determined using 1,000 bootstraps and 1,000 SH-aLRT bootstrap replicates.
Hcp and VgrG are the components of T6SS spike complex and play a role in secretion of effectors by covalently linking to the effectors. To investigate if hcp and vgrG contribute to the incongruencies observed among the above phylogeny comparisons, these two gene sequences were removed from the concatenated T6 core gene query file, and the phylogenies of T6SS core genes without hcp and vgrG sequences were reconstructed for the T6 clusters (Figure 1C). Phylogeny of core T6SS i3*** genes without hcp and vgrG showed X. euvesicatoria–related pathovars that form a separate clade in MLSA phylogeny, now clustering together with pepper pathogenic X. euvesicatoria strains. Likewise, some X. perforans strains that formed a distinct clade nested between X. euvesicatoria and X. euvesicatoria–related pathovars in MLSA phylogeny from the other X. perforans clade were now clustering together in phylogeny based on T6SS i3*** genes without hcp and vgrG. A distantly related species, X. maliensis, was observed closely related to X. euvesicatoria species complex based on T6 i3*** phylogeny with or without hcp and vgrG genes. Interestingly, comparison of i3* phylogeny without hcp and vgrG to that with hcp and vgrG showed change in the placement of some X. perforans strains in separate clades. The incongruencies observed with placement of X. oryzae, and X. fragarie observed in T6 i4 phylogeny with vgrG genes in comparison to MLSA phylogeny were resolved in the phylogeny constructed without vgrG genes (Figure 1). X. oryzae was observed as highly diverse species in phylogeny with vgrG and formed one distinct clade in T6 i4 phylogeny without vgrG, indicating a high degree of variation in vgrG genes in this species in addition to a gene flow with other species such as X. translucens and X. fragarie. Multiple copies of vgrG are presently dispersed throughout the genome of X. oryzae (Supplementary Figure 2). Group 1 species, X. translucens, was found to cluster with group 2 species, X. oryzae based on core gene T6 i4 phylogeny without vgrG genes, indicating a shared evolutionary path for T6 i4 cluster in these two species. A strain of X. translucens, ART-Xtg2, was an exception, which is more closely related to X. bromi, a group 2 species, based on T6 i4 core gene phylogeny with or without vgrG genes, suggesting shared evolutionary patterns for T6 i4 cluster (Figure 1).
Given our observation of incongruencies among MLSA phylogeny and T6SS core gene phylogeny without hcp, vgrG in case of clusters i3* and i3***, we hypothesized that there is genetic exchange of T6 core genes among different strains belonging to different species. We constructed phylogenetic networks using SplitsTree focusing on representatives from individual species. SplitsTree can demonstrate conflicting signals for phylogenetic placements as reticulation. Upon examining the split decomposition tree generated for T6 i3* without hcp and vgrG, we observed reticulation at the base of the branches connecting unusual recombinant strains thought to be hybrid between X. perforans and X. euvesicatoria (NI1, NI38), strains belonging to X. euvesicatoria sister species (CFBP 6369, F1, LMG12749, LMG 495, CFBP 3836, CFBP3371, DAR26930), commensal Xanthomonas strains (CFBP7921, CFBP7922), some strains belonging to X. phaseoli (A1962, LMG12749), X. axonopodis (LMG26789), and X. euvesicatoria 85-10 and X. perforans 91-118 (Figure 2A). Another evident reticulated event was identified to involve X. citri (LMG859, Bagalkot, LMG7439, 12609, NCPPB3660, NCPPB1495, NCPPB1056), X. axonopodis (LMG753, LMG9049, LMG9050, LMG558), and X. campestris strains (LMG954, LMG940, LMG939, LMG9044), indicating flow of genetic information between them with respect to T6 i3* core genes. In a split decomposition tree inferred for T6 i3*** without hcp and vgrG, a highly reticulated network encompassing above identified X. perforans, X. euvesicatoria, X. euvesicatoria–related pathovars, and some strains of X. phaseoli was observed (Figure 2B).
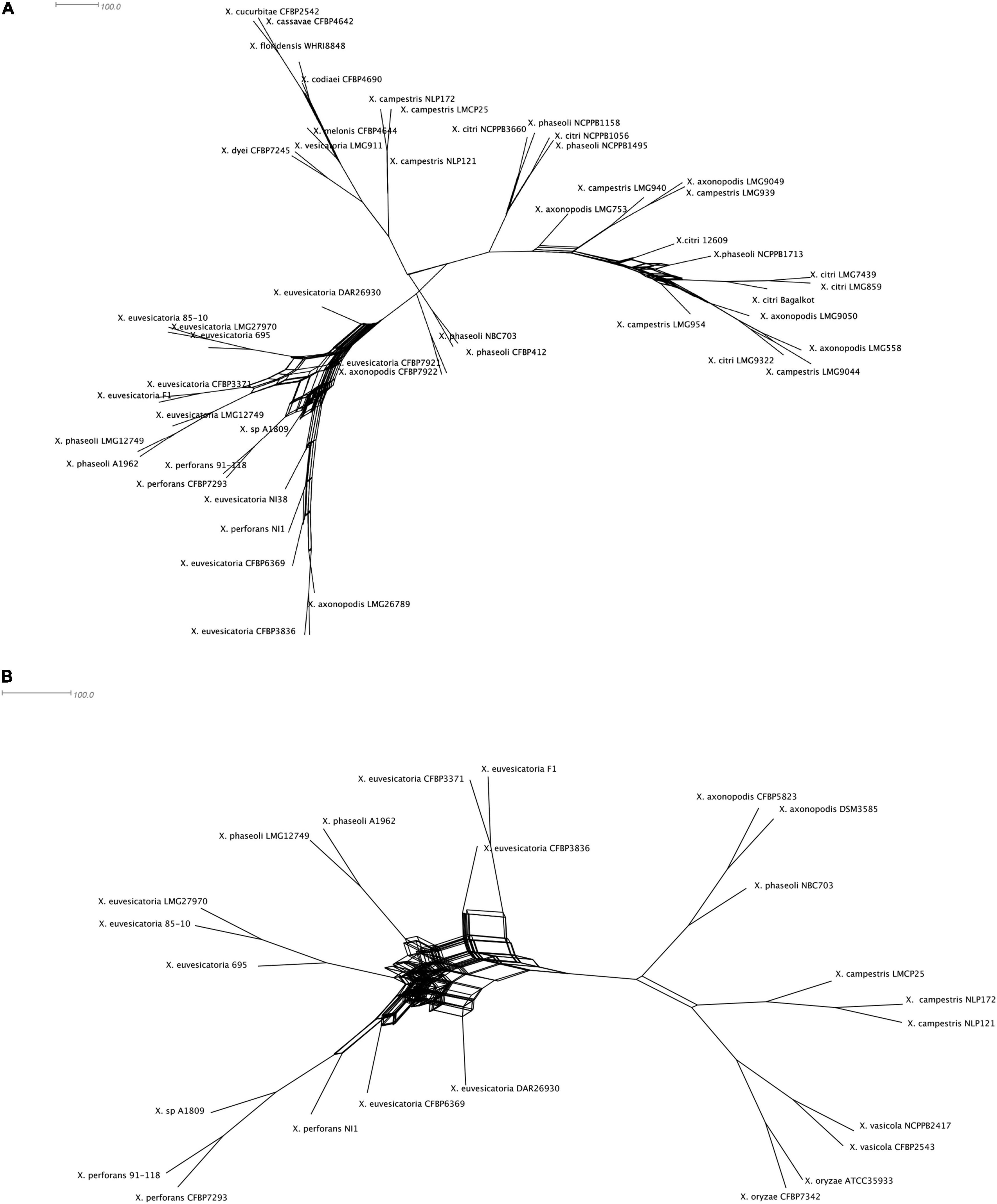
Figure 2. Split decomposition tree of the selected Xanthomonas spp. demonstrating incongruencies between the MLSA-based phylogeny and (A) T6 i3* without hcp and vgrG and (B) T6 i3*** without hcp and vgrG. The figure was drawn to scale using Splitstree4. The formation of parallel lines indicates conflicting phylogeny or possible recombination events.
Gene gain and loss evolutionary dynamics among bacterial species can be inferred by contrasting the reference species phylogeny to that of individual genes (Kettler et al., 2007; Cohen and Pupko, 2011; Iranzo et al., 2019). We next analyzed the evolutionary origin and distribution of multiple T6SS clusters using such an approach, as implemented in GLOOME (Cohen et al., 2010). For the reference species tree, we used a phylogenetic tree generated based on a concatenated MSA of 12 housekeeping gene sequences: atpD, dnaK, efP, fusA, fyuA, gapA, glnA, gltA, gyrB, lacF, lepA, and rpoD. A profile of presence and absence of core genes of individual T6 clusters was used as input, and the output is a probabilistic mapping of events over the reference tree (Figure 3). We additionally added clade information based on core genome phylogeny presented by Merda et al. (2017) (Supplementary Figure 4). Our results clearly present a pattern of multiple gain and loss events for all clusters (Figure 3 and Supplementary Figure 4).
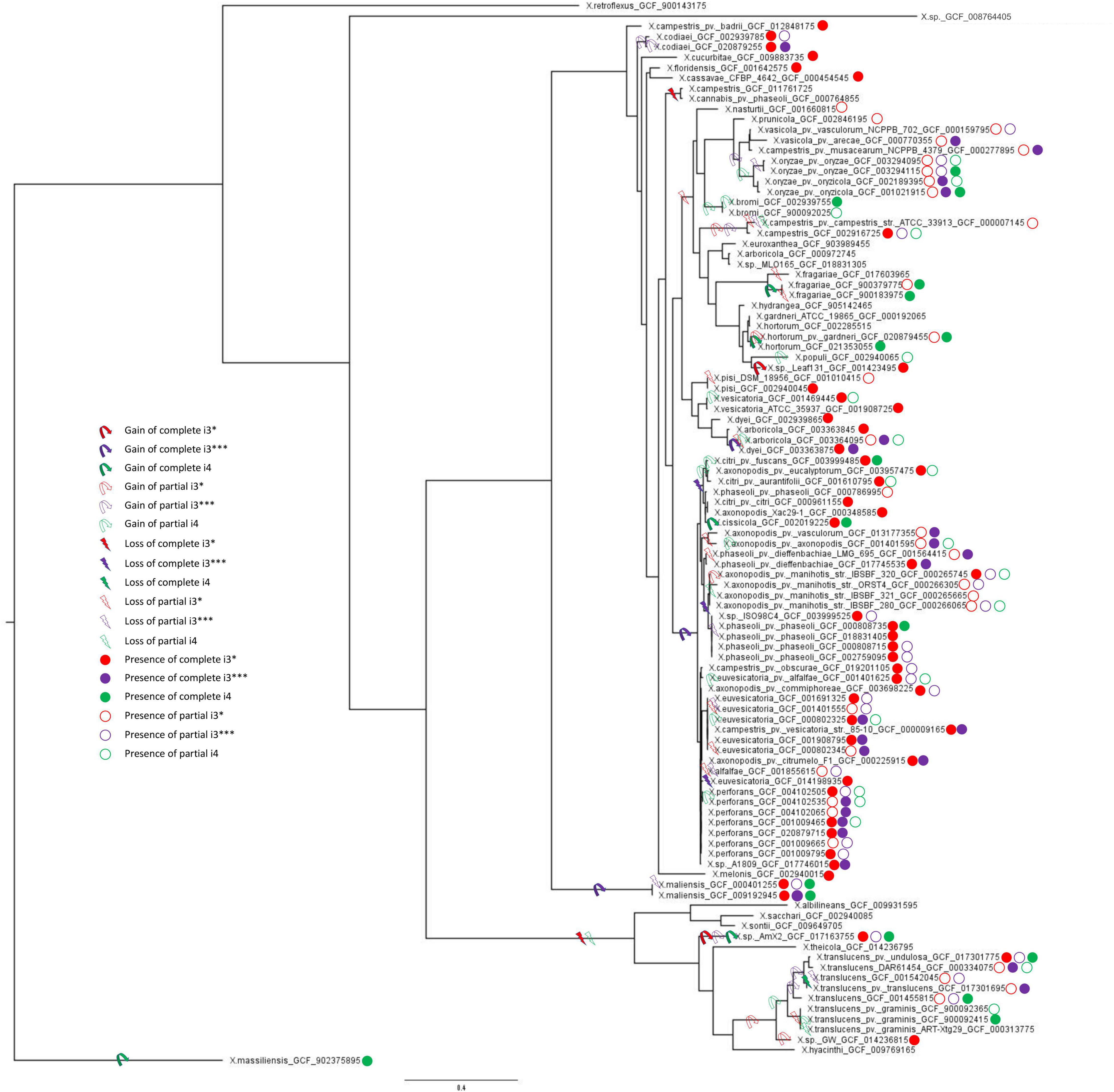
Figure 3. Multilocus sequence analysis phylogenetic tree and T6SS clusters presence/absence and gain/loss prediction for the genus Xanthomonas. Gain and loss of individual T6SS genes were mapped onto the MLSA phylogeny of 1,740 Xanthomonas spp. From each species, one genome with each phyletic pattern was included; that is, if two or more genomes from the same species had identical presence/absence profile of T6SS genes, only a single representative was included in the analysis. The tree was rooted using X. massiliensis as outgroup. The presence of complete and partial T6 clusters (i3*, i3***, and i4) in all the strains was taken from Table 1. Ancestral gain and loss events were mapped onto the tree based on the output of GLOOME. For all the predicted events, the probability was higher than 0.5.
The i3* cluster is present in the majority of group 2 xanthomonads, and in few X. translucens strains belonging to group 1. GLOOME predicted a strong signal for multiple loss events of the i3* cluster in both groups 1 and 2 Xanthomonas spp. For example, in group 1, the bacteria in which this cluster is present are nested among multiple strains in which the cluster is absent. Thus, a likely scenario, is that the T6 i3* cluster was acquired by lateral gene transfer event by the ancestor of the X. translucens clade, after its divergence from the clade that includes X. albilineans, Xanthomonas sontii, and X. sacchari (Figure 3). The i3* cluster is present in many but not all group 2 xanthomonads. The phyletic pattern analysis suggests that it experienced multiple independent loss events, for example, in the lineage leading to the clade that includes X. cannabis and also in the lineage leading to a clade that includes X. arboricola 3307 (Supplementary Figure 4). The presence of the i3* cluster in X. sp. leaf 131, a commensal Xanthomonas, suggests a recent independent acquisition of the cluster in that strain (Figure 3). X. oryzae, X. vasicola, X. prunicola, X. bromi, and Xanthomonas nasturtii have recently lost T6 i3*, whereas some strains belonging to these species have retained a partial cluster. These species belong to clade B based on Merda et al., 2017 core genome phylogeny (Supplementary Figure 4). In addition, several strains of X. phaseoli, X. axonopodis, and X. euvesicatoria have experienced recent partial loss of this cluster (Figure 3).
Our analysis suggests that the T6 i4 cluster was independently acquired in X. oryzae, X. translucens, X. fragarie, and in some strains of X. hortorum, X. citri, X. phaseoli, and X. maliensis. These multiple gain events are also supported by a SplitsTree analysis that indicated a gene flow of core genes of T6 i4 cluster among X. oryzae and X. translucens, as well as among X. citri and X. hortorum, suggesting more recent spread of this cluster with repeated events of horizontal dissemination into distinct clades across Xanthomonas phylogeny (Figure 2).
T6 i3*** cluster was likely gained by the ancestor of X. axonopodis complex, containing species euvesicatoria, citri, phaseoli, and perforans, and was subsequently lost by some species within this clade, as indicated by the presence of partial clusters in strains of these species. An independent gain is inferred to have occurred in the lineage leading to species X. campestris (clade D, as shown in Supplementary Figure 4), a conclusion that is corroborated by the SplitsTree analysis that indicated a gene flow of core genes of this cluster among X. axonopodis species complex (clade B) and X. campestris. The presence of the i3*** cluster in X. maliensis sharing higher similarity to that of X. euvesicatoria compared with X. campestris might suggest a recent independent acquisition of this cluster into X. maliensis. Another independent acquisition of this cluster was also identified in X. oryzae pv. oryzicola strains. Presence of intact T6 i3*** cluster in some X. translucens strains and partial in some X. translucens strains might indicate its presence in the ancestor of group 1. It is likely that the availability of more genomes of species belonging to group 1 might be able to resolve the phylogenetic inference of evolutionary patterns of the three clusters. Nevertheless, this analysis highlights that all three types of clusters experienced numerous transfer and loss events along the evolution of the analyzed Xanthomonas genomes.
Presence of more than one T6SS cluster was detected in X. axonopodis complex (i3* and i3***) and in X. oryzae (i3*** and i4). Strong evidence of acquisition of both i3* and i3*** by the ancestors of X. axonopodis species complex was obtained in aforementioned GLOOME analysis. We also observed genetic exchange of core T6 genes among different species of X. axonopodis in the SplitsTree analysis. Evolutionary history of X. axonopodis complex has been recently well studied, where it has been proposed that the first step of generalist diversification into the X. axonopodis subgroups was followed by the second step of ecology-driven specialization that favored the emergence of novel pathovars (Mhedbi-Hajri et al., 2013). Given our prior work that suggested the potential role of core gene tssM of cluster T6 i3*, in overall epiphytic fitness in X. perforans and during initial stages of disease development (Liyanapathiranage et al., 2021), we hypothesized that during diversification into X. axonopodis subgroups, a combination of specific T6SS clusters in individual species provided significant advantage during the process of niche adaptation onto specific hosts. We closely examined the patterns of the evolutionary history of the i3* and i3*** T6 clusters in the X. axonopodis complex subspecies groups and the lifestyle of these pathogens. When the distribution of the T6 clusters was examined according to the subspecies group they belong to, it was observed that strains belonging to the same subgroup have a similar distribution of the T6 clusters except for X. phaseoli pv. dieffenbachiae LMG 695 in subgroup 9.4. Strains belonging to subgroup 9.2 and X. phaseoli pv. dieffenbachiae LMG 695 in subgroup 9.4 possess both complete clusters, whereas strains belonging to subgroups 9.4, 9.5, and 9.6 have complete i3* clusters. Subgroup 9.3 contains only a single complete i3*** cluster, along with an incomplete i3* cluster with one to three core genes (Table 2). Interestingly, 9.3 is the only subgroup that has strains pathogenic only on monocot plants. X. axonopodis pv. axonopodis belonging to subgroup 9.3 are pathogenic on Axonopus scoparius, whereas X. axonopodis pv. vasculorum are pathogenic on Saccharum officinarum. All the other subgroups consist of strains that are pathogenic on dicot plants, except for X. axonopodis pv. allii (9.2) that is pathogenic on Allium sp.
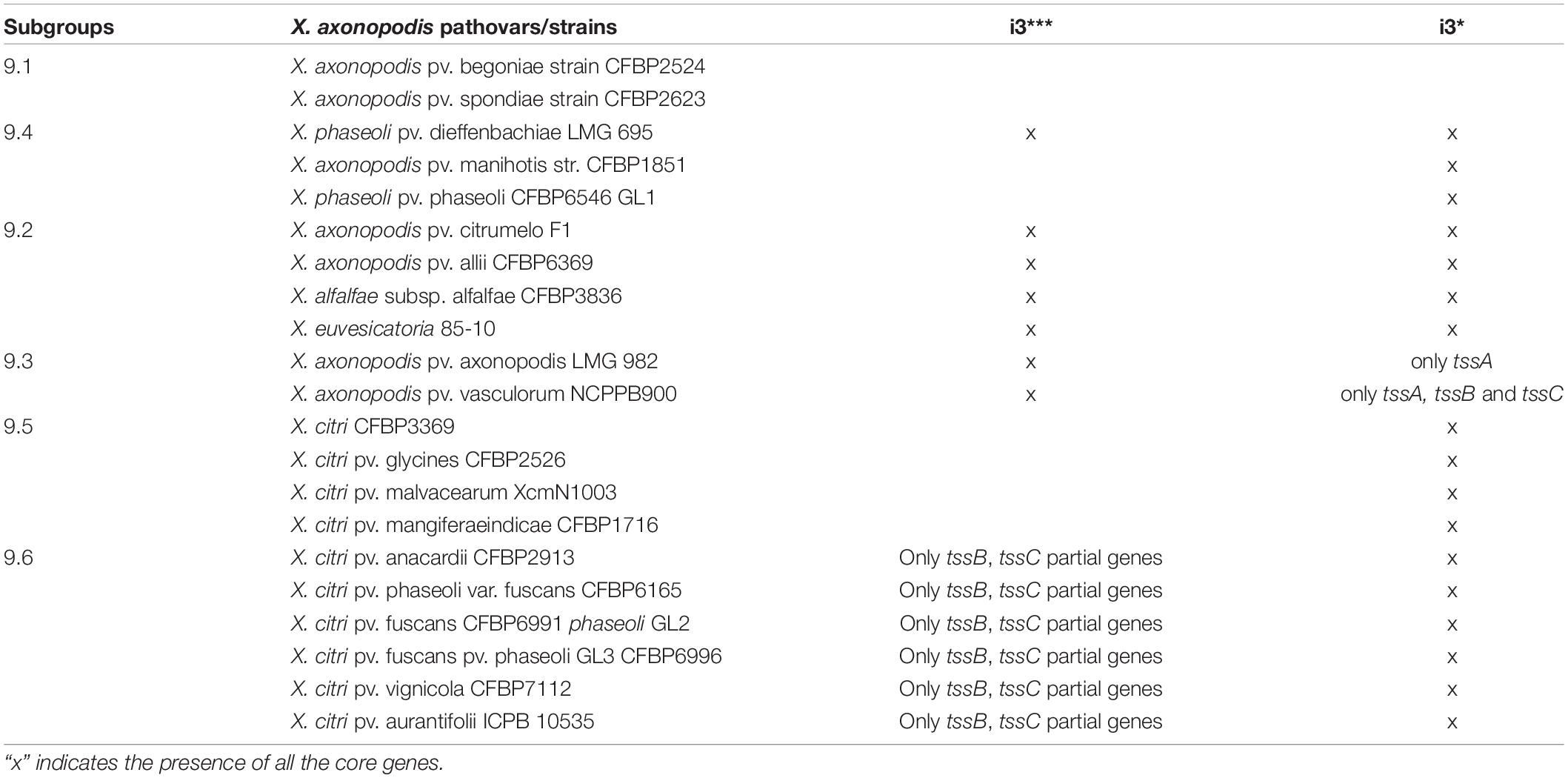
Table 2. Distribution of the i3* and i3*** clusters in the X. axonopodis complex.
To determine if the T6 clusters follow the same evolutionary history as the species belonging to the X. axonopodis complex, a phylogeny developed based on the T6 core genes in i3*/i3*** clusters was compared with the phylogenies generated based on the core genome of strains with either i3* or i3*** core genes encoded in their genomes. Incongruencies were observed when comparing core genome phylogeny and phylogeny based on i3* or i3*** core genes (Figure 4). Core genome phylogeny clustered subgroups 9.2 and 9.4 in a single clade, whereas i3*** phylogeny clustered subgroups 9.4 and 9.3 in one clade, and i3* phylogeny indicates an early divergence of the subgroup 9.4. Clustering of subgroups 9.5 and 9.6 shows the same arrangement when core genome phylogeny was compared with the i3* phylogeny. These differences in the phylogeny hint at a possible evolution pattern in T6 clusters independent of its core genome evolution, thus further suggesting that T6 might have played a major role in host adaptation.
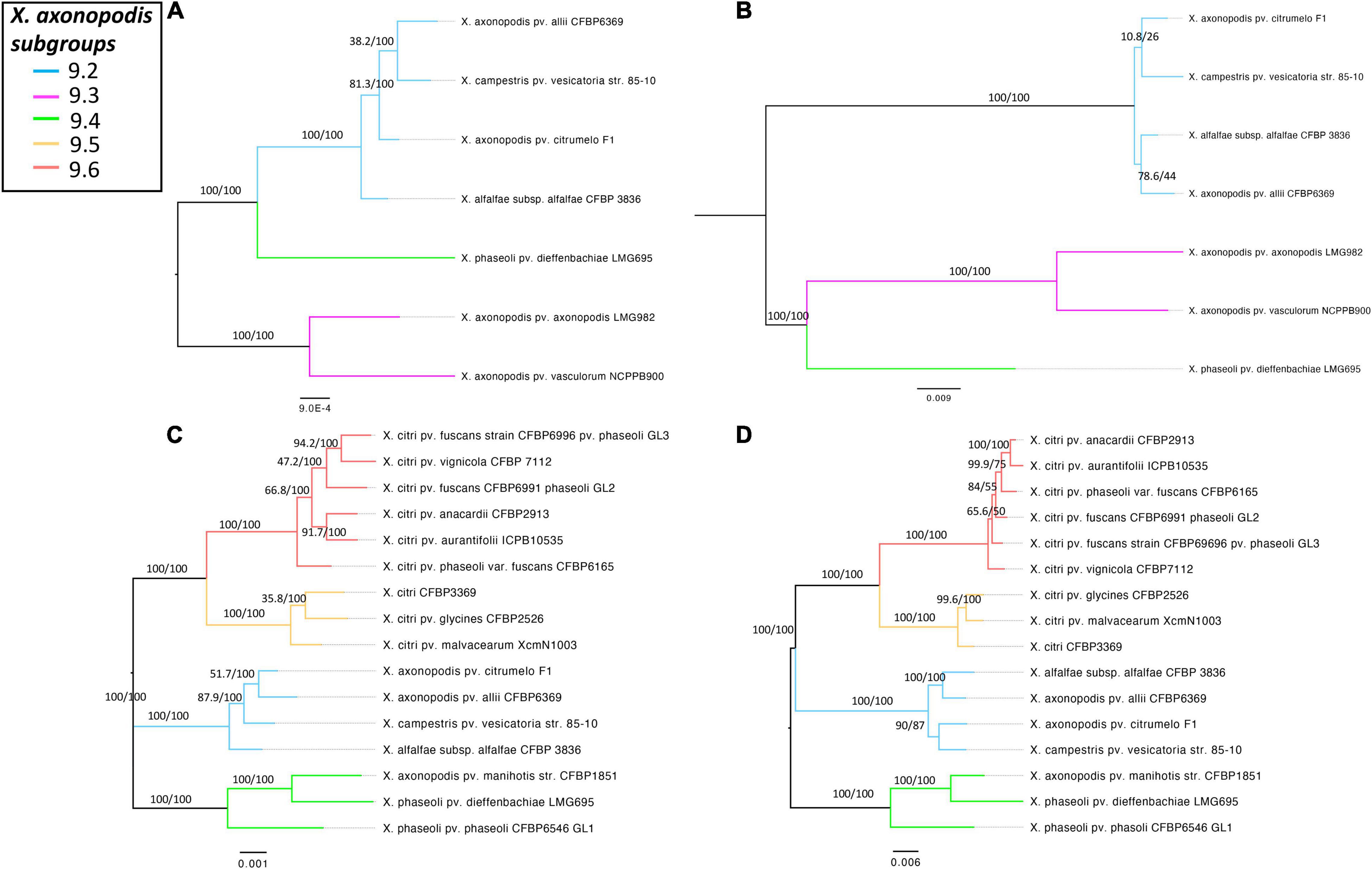
Figure 4. Comparison of the core genome phylogeny of X. axonopodis subspecies with T6 i3***/i3* core genome phylogeny to T6 i3*/i3*** core gene phylogeny. (A) Core genome phylogeny for the strains with i3***. (B) T6SS i3*** phylogeny. (C) Core genome phylogeny for the strains with i3*. (D) T6SS i3* phylogeny. Core genome phylogenies were developed based on the core alignment of nucleotide sequences using the Roary pipeline (Page et al., 2015), whereas T6 phylogenies were generated using the automated multilocus sequence analysis pipeline (automlsa2). Midpoint rooted phylogenetic tree in maximum likelihood (ML) criterion was developed using the IQ-TREE, and branch support was determined using 1,000 bootstraps and 1,000 SH-aLRT bootstrap replicates.
We further assessed the distribution of T6 clusters according to vascular and non-vascular mode of colonization as well as preferred dicot versus monocot host. Most of the dicotyledonous species infecting Xanthomonas contain T6 i3*, whereas most of the monocotyledonous host plants infecting Xanthomonas contain T6 i3*** cluster (Table 3). Absence of i3* cluster was noted in the genomes of species infecting monocots. Furthermore, the T6 cluster distribution was analyzed depending on their lifestyles of vascular versus non-vascular mode. We observed presence of i4 cluster and its association with the ability of the majority of those strains to colonize vascular tissues. Notable examples of the presence of i4 cluster and demonstrated vascular colonization are mentioned below. Presence of i4 cluster was noted in only some strains of species, X. citri, more specifically, X. citri pv. fuscans and X. citri pv. glycines, of which X. citri pv. fuscans has been demonstrated to colonize vascular tissues (Table 3). While all X. citri strains including vascular colonizers contain intact i3* cluster, loss of i3* was seen in X. oryzae strains. Presence of intact i4 cluster was observed in 393 strains out of 396 sequenced genomes of X. oryzae. Vascular mode of infection has been demonstrated in pathovar oryzae, whereas pathovar oryzicola colonizes non-vascular tissues during infection. A partial i4 cluster was observed in some oryzicola strains. Another vascular pathogen, X. fragariae, encodes an intact i4 cluster, similar to X. oryzae, with absence of any other intact T6 cluster (Table 3). Other known vascular colonizers, X. phaseoli pv. manihotis and pv. dieffenbachiae, were found to lack complete i4 cluster, although a partial cluster was noted in some pathovar dieffenbachiae strains. Two X. phaseoli pv. phaseoli strains contain intact i4 clusters, strains of which are known to colonize both vascular and mesophyll tissues. X. campestris, X. arboricola pv. pruni, and X. hortorum pv. vitians can be considered as exception to the observation of association of intact i4 cluster with vascular mode of pathogenesis (Table 3). Only one sequenced strain of pathovar vitians contains intact i4 cluster. One strain of X. hortorum pv. gardneri was found to contain intact i4 cluster. While X. hortorum pv. gardneri is not known to colonize vascular tissues, Bernal et al. (2021) recently showed colonization of hydathodes upon inoculation of tomato plants with X. hortorum pv. gardneri. X. translucens strains show diversity in their preferences to colonize vascular and non-vascular tissues. However, our above observation of association of intact i4 cluster with vascular pathogenesis does not hold true with X. translucens species complex, which contains strains known to colonize vascular as well as non-vascular tissues. Some X. translucens strains despite being vascular lack T6 cluster (Table 3).
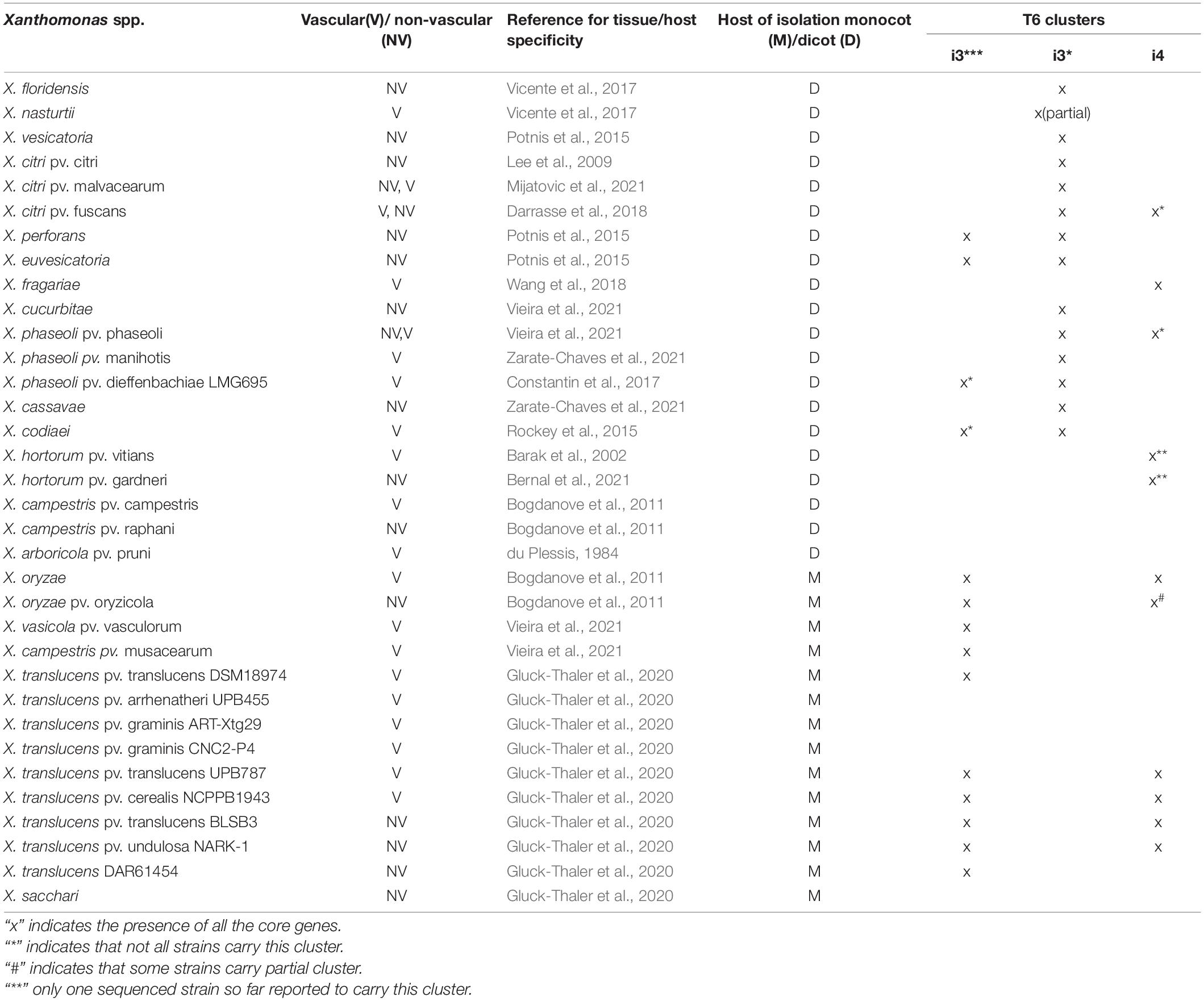
Table 3. Distribution of the T6 clusters according to the lifestyle of Xanthomonas spp.
Next, we expanded our analysis of distribution of T6SS clusters to non-pathogenic or environmental isolates. This could provide added clarification to its role in Xanthomonas strains regardless of their pathogenicity. Thirteen strains collected from rainwater and 16 non-pathogenic Xanthomonas collected from tomato, beans, citrus, pepper, and rice seed or phyllosphere were included in a core genome phylogenetic analysis (Supplementary Table 2). Among the non-pathogenic Xanthomonas species, X. maliensis strains, early-branching strain X. translucens F5, and X. floridensis WHRI 8848 contain i3* cluster encoded in their genome. Interestingly, X. maliensis contains all three clusters, i3*, i3***, and i4 clusters, whereas other non-pathogenic rice leaf–associated Xanthomonas species such as X. sontii and X. sacchari lack any T6 clusters. Similarly, non-pathogenic X. floridensis WHRI 8848 isolated from watercress has a complete i3* cluster, whereas watercress pathogenic X. nasturtii WHRI 8853 only has a single i3* core gene with flanking regions similar to the X. floridensis, suggesting recent loss (Supplementary Figure 1). A commensal Xanthomonas strain F5 isolated from pepper belonging to group 1 contains complete i3* cluster, with core genes showing 95% to 100% identity to the genes from non-pathogenic Xanthomonas strains SI, SS, and GW isolated from perennial ryegrass seed (Supplementary Table 3). These strains showed antagonistic activity against key fungal pathogens. Li et al. (2020) hypothesized the involvement of unique secondary metabolites in this antagonistic activity. Our results suggest that T6SS may also play a role in this antagonism to some extent.
Among the Xanthomonas strains collected from the rainwater, two X. arboricola strains (4461 and 3793) possess a T6 i3* cluster (Supplementary Figure 4). Other X. arboricola strains including both known pathogens and commensals contain a partial tssB gene along with potential regulatory genes encoding serine/threonine protein kinase and RNA polymerase sigma factor and conserved synteny for i3* cluster, indicating recent loss.
In this study, we conducted a comprehensive analysis to study the distribution and the evolutionary patterns of multiple T6 clusters in the 1,740 genomes spanning the entire genus Xanthomonas. Complete T6 clusters, partial clusters, and even single T6 genes belonging to i3*, i3***, and i4 subgroups of T6 secretion system are dispersed throughout the genus. Our data indicate that T6SS cluster genes have been inherited both vertically and horizontally throughout the genus phylogeny. Like other genera, Xanthomonas species contain multiple copies of T6 clusters, with presence of either i3* + i3*** or i3* + i4 combinations being frequently observed. X. maliensis, associated with healthy rice seeds or leaves in Mali (Triplett et al., 2015), was the only species identified carrying all three clusters, i3*, i3***, and i4. Being an early branching species within group 2 xanthomonads, conservation of all three clusters in this species and the genomic context is important to infer evolutionary scenarios explaining gain/loss events of the three types of T6 clusters. Simultaneous maintenance of all three clusters in the species is surprising, given the energy investment in these large clusters. Sequencing multiple strains of this species might help address whether presence of all three clusters may represent random flicker or a sign of positive selection for acquisition in the evolutionary history. If the latter is true, this species would represent an ideal candidate for studying differential regulation of each cluster and how the environment influences this regulation.
The majority of the species belonging to X. axonopodis complex possess both i3* and i3*** clusters, with exception of X. citri that lacks cluster i3***. The loss of i3*, observed in clade A strains and strains of X. oryzae, and X. vasicola from clade B, was accompanied by gain of i4 cluster. Some strains belonging to X. citri and X. phaseoli, which possess both i3 and i4 clusters in 1% to 5% of the strains are an exception to the rule of two i3 clusters in X. axonopodis complex and to the observation of replacement of i3 by i4. The identification of two X. phaseoli strains with the i4 cluster was an unusual observation as the X. phaseoli strains were previously only known to harbor i3* and i3*** clusters (Montenegro Benavides et al., 2021). This highlights the importance of the availability of genomes of multiple hundreds of strains of each species for understanding intraspecific diversity. It is unclear whether the loss of i3* and subsequent gain of i4 cluster in species such as X. oryzae, and X. vasicola, was in response to selection pressure from the environment including resident microflora.
The role of i3* has been suggested not only in competition against resident flora, more specifically, resistance to predation by the bacterivorous amoeba, Dictyostelium discoideum, but also in adaptation to the phyllosphere non-vascular environment (Bayer-Santos et al., 2018; Liyanapathiranage et al., 2021). Loss of i3* in lineages containing X. oryzae and X. vasicola, both vascular pathogens, might be in response to the vascular lifestyle. In the rice pathogen X. oryzae pv. oryzicola, T6SS i4 has been identified for its role in the interbacterial competition, whereas in X. oryzae pv. oryzae, it was identified for its role in virulence (Choi et al., 2020; Zhu et al., 2020; Montenegro Benavides et al., 2021).
As it has been identified that presence of all the core T6SS genes is required for the functionality of the T6SS (Boyer et al., 2009), it is intriguing to identify some strains that lack single T6 genes and raise the questions about the possible function of that apparently inactive T6 cluster. In our analysis, we observed the T6SS locus lacking clpV gene in some of the X. oryzae strains, and interestingly, in previous studies on the distribution of the T6 clusters in Campylobacter jejuni, T6SS locus lacking the clpV (tssH) gene has been noted (Bönemann et al., 2009; Bleumink-Pluym et al., 2013). Even though ClpV is required for the recycling of the TssB/TssC tubular sheath, in Vibrio cholerae, a clpV mutant was still effective in killing Escherichia coli, thus indicating the possibility of function of a clpV-related ClpB family of ATPase encoded from elsewhere compensating for the lack of clpV in the T6SS (Pietrosiuk et al., 2011; Zheng et al., 2011).
The distribution of T6SS among Xanthomonas species does not correlate with the host preference. Four Xanthomonas spp. that cause bacterial leaf spot in tomato and pepper show the diverse distribution of T6 clusters. X. euvesicatoria and X. perforans have both i3* and i3*** clusters; whereas X. vesicatoria has i3*cluster, X. hortorum pv. gardneri does not have T6 clusters encoded in its genome, with the exception of a single strain carrying i4 cluster (Potnis et al., 2011). Even the three species that have the i3* cluster show differences in cluster organization (Supplementary Figure 3), with different set of putative effectors. Another example could be of rice-associated strains. An extensive diversity in the T6 cluster distribution was observed in the rice-associated xanthomonads, ranging from absence of T6 clusters in non-pathogenic strains, X. sontii (Bansal et al., 2019) and X. sacchari R1 (Fang et al., 2015), to presence of i4 alone or i3*** and i4 clusters in pathogenic X. oryzae and X. oryzicola. Another such example is of watercress-associated strains. A non-pathogenic strain, X. floridensis WHRI 8848 isolated from the diseased leaves of watercress (Nasturtium officinale) (Vicente et al., 2017), contains intact i3* cluster, but a pathogenic strain, X. nasturtii WHRI 8853, lacks a functional cluster and encodes only a single partial T6SS gene (tssA), indicating a recent loss of the cluster with similar flanking regions.
The distribution of T6 clusters does not depend on host of isolation or pathogenic status. We observed presence of intact T6 clusters in non-pathogenic or commensal strains, even those recovered from rainwater. This observation highlights the role of T6 clusters and associated effectors in conferring fitness during host colonization and also possibly outside the host environment, in conferring traits such as nutrient acquisition, or bacterial communication. While host environment has been suggested to contribute to induction of T6 clusters in pathogenic bacteria, commensal strains would be an ideal background to understand how T6SS is regulated in absence of network of other virulence factors, although constitutive expression of T6SS cannot be ruled out as observed in V. cholerae non-pathogenic strains (Lopez et al., 2020).
Based on the probability of gain/loss events during Xanthomonas evolution, as well as information gathered by analysis of flanking regions of T6 clusters and identities of core genes, the data obtained in this study indicate that T6SS was already present in the ancestors of the major xanthomonad lineages. When we mapped the presence of various T6 clusters onto the MLSA tree, we inferred numerous gain events via horizontal gene transfer as well as many loss events. We hypothesize that such evolutionary dynamics is driven by selective pressure related to adaptation to specific environments. In case of T6 i3* cluster, a conserved flanking region across group 2 xanthomonads indicate vertical acquisition of T6 clusters by a common ancestor in a single acquisition event. However, T6 i3* cluster experienced subsequent loss in several lineages, including those in X. arboricola, X. oryzae, and X. hortorum, and has left signatures within genomes, such as partial cluster, or presence of tRNA indicating a rearrangement event. This loss was also followed by gain of T6 i4 cluster in some vascular pathogenic species such as X. oryzae and X. fragarie. T6 i4 gain events were identified as independent events across the phylogeny. Recent gain of the i4 cluster via horizontal transfer events in multiple species was further confirmed by the fact that flanking regions of the i4 cluster show extreme heterogeneity.
Comparing T6 cluster core gene phylogenies to that of housekeeping genes suggested gene flow of core genes among different species of xanthomonads. This is in agreement with previous findings of T6SS gene clusters as recombination hotspots in X. euvesicatoria species complex (Newberry et al., 2019), which suggests reshuffling of alleles of core genes of T6 clusters. In addition to core genes, we also observed reticulations in phylogenetic network, indicating frequent exchange of Hcp and VgrG among Xanthomonas species. Such events of reshuffling of VgrG and Hcp have been observed as a strong force in diversification of effector-immunity proteins in many genera, such as Agrobacterium and Vibrio (Salomon et al., 2015; Thomas et al., 2017; Wu et al., 2021). Although it is true that poorly resolved phylogenies or phylogenies of closely related strains with low branch support could appear as reticulations in the network, reticulations observed specifically for distantly related species suggest that recombination could be a highly likely explanation in those cases.
Species belonging to the X. axonopodis complex are pathogenic on a wide range of crops (Vauterin et al., 1995) and are classified into six groups named 9.1 to 9.6 within X. axonopodis (Rademaker et al., 2005; Young et al., 2008; Hajri et al., 2009; Bui Thi Ngoc et al., 2010; Mhedbi-Hajri et al., 2013). According to the core genome phylogeny and i3* or i3*** core genes phylogeny in this current study, we observe the same pattern in the way each strain categorizes into the subgroupings as explained in the previous literature, suggesting acquisition of these clusters by the ancestor of X. axonopodis. Mhedbi-Hajri et al. (2013) speculated that the first step of generalist diversification was followed by a second step of ecology-driven specialization that probably occurred in the past two centuries and favored the emergence of novel pathovars. This second event of host specialization has occurred likely through agricultural intensification and expansion that facilitated genetic exchanges of virulence-associated genes. All of these pathovars are restricted to one of the six subgroups and most of them formed monophyletic (e.g., citri, mangiferaeindicae, malvacearum, begonia, and manihotis) clusters, whereas some strains were polyphyletic (e.g., dieffenbachiae, glycines, and phaseoli) (Hajri et al., 2009; Mhedbi-Hajri et al., 2013). Some strains that belong to the same pathovar can be found in phylogenetically distant clusters. For example, X. axonopodis strains that infect legumes can be found in subgroups 9.2, 9.4, and 9.6; mango Mangifera indica–infecting strains were found in subgroups 9.5 and 9.6, and citrus-infecting strains were identified in clusters 9.2, 9.5, and 9.6. Similarly, X. axonopodis strains that are pathogenic on phylogenetically distant hosts are clustered in the same subgroups, as seen with subgroup 9.5, where citrus-, mango-, legume-, and cotton-infecting X. axonopodis strains are grouped together. An independent rapid evolution in the subgroup 9.3 compared with the other subgroups might also be reflected in its loss of i3* cluster during niche adaptation. X. phaseoli pv. dieffenbachiae is polyphyletic and found in subgroups 9.6 (A- Dieffenbachia sp.), 9.2 (B- Philodendron sp.) and 9.4 (C- Anthurium sp.) according to the rpoD sequences analysis and Rep-PCR, this grouping is also dependent on the host of isolation (Hajri et al., 2009; Mhedbi-Hajri et al., 2013). Our observation of heterogeneity of T6 cluster distribution in the subgroup 9.4 suggests preferences in specific T6 clusters during niche adaptation of different host species. This phylogenetic analysis and T6SS cluster distributions suggest that T6SS cluster conservation and associated effector alleles might have played a role during host specialization in the X. axonopodis strains in addition to the already suggested factors such as methyl-accepting chemotaxis proteins and type III effectors (Hajri et al., 2009; Mhedbi-Hajri et al., 2013).
In conclusion, our findings point to the scenario in which the different T6 clusters are often gained and lost during the evolution of Xanthomonas species, with some species encoding two clusters, whereas others, not a single one. We hypothesize that the majority of gain and loss events are not random, but rather reflect adaptations and specialization to specific environments. The factors involved in niche adaptation have been of interest in case of plant pathogenic bacteria, and a new paradigm has suggested involvement of multiple genetic determinants underlying this process with small to major effect genes during the process of adaptation of a pathogen to a host type or to a particular tissue (Bogdanove et al., 2011; Huang et al., 2015; Jacques et al., 2016). Here, we explored association of T6 clusters and preference of Xanthomonas species toward dicot or monocot hosts and vascular or non-vascular mode of infection. As not all species have been studied for their breadth of host range or tissue colonization, the associations evaluated here are based on current literature. We noted the absence of i3* cluster in monocot-infecting pathogens. The gain/loss patterns also indicated loss of i3* followed by gain of i4 cluster in vascular pathogenic species in group 2 xanthomonads, such as oryzae and fragarie. But this observation was not true in case of X. translucens species complex and X. campestris pv. campestris, X. vasicola. In brief, there does not seem to be association of specific T6 cluster for a lifestyle of Xanthomonas when evaluating the entire genus as a whole. However, functional characterization using T6SS-defective mutants of Xanthomonas has suggested importance of T6SS in overall fitness during host colonization, as well as its role in competing with the resident flora. As pathogen fitness criteria vary depending on the niche that pathogen experiences, involvement of T6 cluster/s and its role in regulatory network may vary depending on the host environment. Thus, it is likely that associations that we have observed with gain/loss patterns for a specific T6 cluster may be valid for specific clades that experience similar host environments or that harness similar virulence factor repertoires to coordinate the process of colonization. It is also possible that secreted effectors or toxins may be more important in specialization to the host environment rather than association of a specific T6 cluster/s.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
NP conceived the project. PL, NW, and NP conducted the comparative genome analyses and SplitsTree analysis, generated the core T6 gene and core genome phylogenies, and wrote the manuscript. OA conducted the Gloome analysis. OA, NP, NW, and TP interpreted the Gloome analysis results. NW, OA, and TP edited the manuscript. All authors approved final manuscript.
This research was supported in part with funds from the NSF-CAREER award to NP, award, IOS-1942956 from the Plant Biotic Interactions Program of the National Science Foundation (NSF). OA and NW were supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. OA was also supported by a fellowship from the Dalia and Eli Hurvitz Foundation LTD. This work was made possible in part by a grant of high-performance computing resources and technical support from the Alabama Supercomputer Authority.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.840308/full#supplementary-material
Abendroth, U., Adlung, N., Otto, A., Grüneisen, B., Becher, D., and Bonas, U. (2017). Identification of new protein-coding genes with a potential role in the virulence of the plant pathogen Xanthomonas euvesicatoria. BMC Genomics 18:625. doi: 10.1186/s12864-017-4041-7
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alvarez-Martinez, C. E., Sgro, G. G., Araujo, G. G., Paiva, M. R. N., Matsuyama, B. Y., Guzzo, C. R., et al. (2020). Secrete or perish: the role of secretion systems in Xanthomonas biology. Comput. Struct. Biotechnol. J. 19, 279–302. doi: 10.1016/j.csbj.2020.12.020
An, S.-Q., Potnis, N., Dow, M., Vorhölter, F.-J., He, Y.-Q., Becker, A., et al. (2020). Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 44, 1–32. doi: 10.1093/femsre/fuz024
Bansal, K., Kumar, S., and Patil, P. B. (2020). Phylogenomic insights into diversity and evolution of nonpathogenic Xanthomonas strains associated with citrus. mSphere 5, e00087–20. doi: 10.1128/mSphere.00087-20
Bansal, K., Midha, S., Kumar, S., Kaur, A., Sonti, R. V., and Patil, P. B. (2019). Ecological and evolutionary insights into pathogenic and non-pathogenic rice associated Xanthomonas. bioRxiv [Preprint]. doi: 10.1101/453373
Barak, J. D., Koike, S. T., and Gilbertson, R. L. (2002). Movement of Xanthomonas campestris pv. vitians in the stems of lettuce and seed contamination. Plant Pathol. 51, 506–512.
Bayer-Santos, E., Ceseti, L., de, M., Farah, C. S., and Alvarez-Martinez, C. E. (2019). Distribution, function and regulation of type 6 secretion systems of Xanthomonadales. Front. Microbiol. 10:1635. doi: 10.3389/fmicb.2019.01635
Bayer-Santos, E., Lima, L. D. P., Ceseti, L. M., Ratagami, C. Y., de Santana, E. S., da Silva, A. M., et al. (2018). Xanthomonas citri T6SS mediates resistance to Dictyostelium predation and is regulated by an ECF σ factor and cognate Ser/Thr kinase. Environ. Microbiol. 20, 1562–1575. doi: 10.1111/1462-2920.14085
Bernal, E., Deblais, L., Rajashekara, G., and Francis, D. M. (2021). Bioluminescent Xanthomonas hortorum pv. gardneri as a tool to quantify bacteria in planta, screen germplasm, and identify infection routes on leaf surfaces. Front. Plant Sci. 12:667351. doi: 10.3389/fpls.2021.667351
Bingle, L. E., Bailey, C. M., and Pallen, M. J. (2008). Type VI secretion: a beginner’s guide. Curr. Opin. Microbiol. 11, 3–8. doi: 10.1016/j.mib.2008.01.006
Bleumink-Pluym, N. M. C., van Alphen, L. B., Bouwman, L. I., Wösten, M. M., and van Putten, J. P. (2013). Identification of a functional type VI secretion system in campylobacter jejuni conferring capsule polysaccharide sensitive cytotoxicity. PLoS Pathog. 9:e1003393. doi: 10.1371/journal.ppat.1003393
Blondel, C. J., Jiménez, J. C., Contreras, I., and Santiviago, C. A. (2009). Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC Genomics 10:354. doi: 10.1186/1471-2164-10-354
Bogdanove, A. J., Koebnik, R., Lu, H., Furutani, A., White, F. F., Salzberg, S. L., et al. (2011). Two new complete genome sequences offer insights into host and tissue specificity of plant pathogenic Xanthomonas spp. J. Bacteriol. 193, 5450–5464. doi: 10.1128/JB.05262-11
Bönemann, G., Pietrosiuk, A., Diemand, A., Zentgraf, H., and Mogk, A. (2009). Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 28, 315–325. doi: 10.1038/emboj.2008.269
Boyer, F., Fichant, G., Berthod, J., Vandenbrouck, Y., and Attree, I. (2009). Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 10:104. doi: 10.1186/1471-2164-10-104
Bui Thi Ngoc, L., Vernière, C., Jouen, E., Ah-You, N., Lefeuvre, P., Chiroleu, F., et al. (2010). Amplified fragment length polymorphism and multilocus sequence analysis-based genotypic relatedness among pathogenic variants of Xanthomonas citri pv. citri and Xanthomonas campestris pv. bilvae. Int. J. Syst. Evol. Microbiol. 60, 515–525. doi: 10.1099/ijs.0.009514-0
Büttner, D., and Bonas, U. (2010). Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 34, 107–133. doi: 10.1111/j.1574-6976.2009.00192.x
Ceseti, L. M., de Santana, E. S., Ratagami, C. Y., Barreiros, Y., Lima, L. D. P., Dunger, G., et al. (2019). The Xanthomonas citri pv. citri Type VI secretion system is induced during epiphytic colonization of citrus. Curr. Microbiol. 76, 1105–1111. doi: 10.1007/s00284-019-01735-3
Chen, I.-M. A., Chu, K., Palaniappan, K., Ratner, A., Huang, J., Huntemann, M., et al. (2021). The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49, D751–D763. doi: 10.1093/nar/gkaa939
Choi, Y., Kim, N., Mannaa, M., Kim, H., Park, J., Jung, H., et al. (2020). Characterization of Type VI secretion system in Xanthomonas oryzae pv. oryzae and its role in virulence to rice. Plant Pathol. J. 36, 289–296. doi: 10.5423/PPJ.NT.02.2020.0026
Cohen, O., and Pupko, T. (2011). Inference of gain and loss events from phyletic patterns using stochastic mapping and maximum parsimony a simulation study. Genome Biol. Evol. 3, 1265–1275. doi: 10.1093/gbe/evr101
Cohen, O., Ashkenazy, H., Belinky, F., Huchon, D., and Pupko, T. (2010). GLOOME: gain loss mapping engine. Bioinformatics 26, 2914–2915. doi: 10.1093/bioinformatics/btq549
Constantin, E. C., Haegeman, A., Van Vaerenbergh, J., Baeyen, S., Van Malderghem, C., Maes, M., et al. (2017). Pathogenicity and virulence gene content of Xanthomonas strains infecting Araceae, formerly known as Xanthomonas axonopodis pv. dieffenbachiae. Plant Pathol. 66, 1539–1554. doi: 10.1111/ppa.12694
Coulthurst, S. J. (2013). The Type VI secretion system – a widespread and versatile cell targeting system. Res. Microbiol. 164, 640–654. doi: 10.1016/j.resmic.2013.03.017
Darrasse, A., Barret, M., Cesbron, S., Compant, S., and Jacques, M.-A. (2018). Niches and routes of transmission of Xanthomonas citri pv. fuscans to bean seeds. Plant Soil 422, 115–128.
Davis, E. W., Okrent, R. A., Manning, V. A., and Trippe, K. M. (2021). Unexpected distribution of the 4-formylaminooxyvinylglycine (FVG) biosynthetic pathway in Pseudomonas and beyond. PLoS One 16:e0247348. doi: 10.1371/journal.pone.0247348
du Plessis, H. J. (1984). Scanning electron microscopy of Xanthomonas campestris pv. pruni in plum petioles and buds. J. Phytopathol. 109, 277–284.
Durand, E., Cambillau, C., Cascales, E., and Journet, L. (2014). VgrG, Tae, Tle, and beyond: the versatile arsenal of Type VI secretion effectors. Trends Microbiol. 22, 498–507. doi: 10.1016/j.tim.2014.06.004
Durand, E., Nguyen, V. S., Zoued, A., Logger, L., Péhau-Arnaudet, G., Aschtgen, M.-S., et al. (2015). Biogenesis and structure of a type VI secretion membrane core complex. Nature 523, 555–560. doi: 10.1038/nature14667
Fang, Y., Lin, H., Wu, L., Ren, D., Ye, W., Dong, G., et al. (2015). Genome sequence of Xanthomonas sacchari R1, a biocontrol bacterium isolated from the rice seed. J. Biotechnol. 206, 77–78. doi: 10.1016/j.jbiotec.2015.04.014
Fischer-Le Saux, M., Bonneau, S., Essakhi, S., Manceau, C., and Jacques, M.-A. (2015). Aggressive emerging pathovars of Xanthomonas arboricola represent widespread epidemic clones distinct from poorly pathogenic strains, as revealed by multilocus sequence typing. Appl. Environ. Microbiol. 81, 4651–4668. doi: 10.1128/AEM.00050-15
Gambette, P., and Huson, D. H. (2008). Improved layout of phylogenetic networks. IEEE/ACM Trans. Comput. Biol. Bioinformatics 5, 472–479. doi: 10.1109/tcbb.2007.1046
Gluck-Thaler, E., Cerutti, A., Perez-Quintero, A. L., Butchacas, J., Roman-Reyna, V., Madhavan, V. N., et al. (2020). Repeated gain and loss of a single gene modulates the evolution of vascular plant pathogen lifestyles. Sci. Adv. 6:eabc4516. doi: 10.1126/sciadv.abc4516
Hajri, A., Brin, C., Hunault, G., Lardeux, F., Lemaire, C., Manceau, C., et al. (2009). A «Repertoire for repertoire» hypothesis: repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS One 4:e6632. doi: 10.1371/journal.pone.0006632
Hernandez, R. E., Gallegos-Monterrosa, R., and Coulthurst, S. J. (2020). Type VI secretion system effector proteins: effective weapons for bacterial competitiveness. Cell. Microbiol. 22:e13241. doi: 10.1111/cmi.13241
Huang, C.-L., Pu, P.-H., Huang, H.-J., Sung, H.-M., Liaw, H.-J., Chen, Y. M., et al. (2015). Ecological genomics in Xanthomonas: the nature of genetic adaptation with homologous recombination and host shifts. BMC Genomics 16:188. doi: 10.1186/s12864-015-1369-8
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Iranzo, J., Wolf, Y. I., Koonin, E. V., and Sela, I. (2019). Gene gain and loss push prokaryotes beyond the homologous recombination barrier and accelerate genome sequence divergence. Nat. Commun. 10:5376. doi: 10.1038/s41467-019-13429-2
Jacques, M.-A., Arlat, M., Boulanger, A., Boureau, T., Carrere, S., Cesbron, S., et al. (2016). Uisng ecology, physeiology, and genomics to understand host specificity in Xanthomonas. Annu. Rev. Phytopathol. 54, 163–187. doi: 10.1146/annurev-phyto-080615-100147
Johansen, J. E., Binnerup, S. J., Kroer, N., and Mølbak, L. (2005). Luteibacter rhizovicinus gen. nov., sp. nov., a yellow-pigmented gammaproteobacterium isolated from the rhizosphere of barley (Hordeum vulgare L.). Int. J. Syst. Evol. Microbiol. 55, 2285–2291. doi: 10.1099/ijs.0.63497-0
Joseph, S., and Forsythe, S. (2012). Insights into the emergent bacterial pathogen cronobacter spp., generated by multilocus sequence typing and analysis. Front. Microbiol. 3:397. doi: 10.3389/fmicb.2012.00397
Jun, S.-R., Sims, G. E., Wu, G. A., and Kim, S.-H. (2010). Whole-proteome phylogeny of prokaryotes by feature frequency profiles: an alignment-free method with optimal feature resolution. Proc. Natl. Acad. Sci. U. S. A. 107, 133–138. doi: 10.1073/pnas.0913033107
Katoh, M., Miyata, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kettler, G. C., Martiny, A. C., Huang, K., Zucker, J., Coleman, M. L., Rodrigue, S., et al. (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 3:e231. doi: 10.1371/journal.pgen.0030231
Kyrova, E. I., Dzhalilov, F. S., and Ignatov, A. N. (2020). The role of epiphytic populations in pathogenesis of the genus Xanthomonas bacteria. Bio Web Conf. 23:03010. doi: 10.1051/bioconf/20202303010
Lee, I.-J., Kim, K.-W., Hyun, J. W., Lee, Y. H., and Park, E.-W. (2009). Comparative ultrastructure of nonwounded mexican lime and yuzu leaves infected with the citrus canker bacterium Xanthomonas citri pv. citri. Microscopy Res. Technique 72, 507–516. doi: 10.1002/jemt.20707
Li, T., Mann, R., Sawbridge, T., Kaur, J., Auer, D., and Spangenberg, G. (2020). Novel Xanthomonas species from the perennial ryegrass seed microbiome - Assessing the bioprotection activity of non-pathogenic relatives of pathogens. Front. Microbiol. 11:1991. doi: 10.3389/fmicb.2020.01991
Lien, Y.-W., and Lai, E.-M. (2017). Type VI secretion effectors: methodologies and biology. Front. Cell Infect. Microbiol. 7:254. doi: 10.3389/fcimb.2017.00254
Liyanapathiranage, P., Jones, J. B., and Potnis, N. (2021). A mutation of a single core gene, tssM, of Type VI secretion system of Xanthomonas perforans influences virulence, epiphytic survival and transmission during pathogenesis on tomato. Phytopathology. doi: 10.1094/PHYTO-02-21-0069-R [Epub ahead of print].
Lopez, J., Ly, P. M., and Feldman, M. F. (2020). The tip of the VgrG spike is essential to functional type VI secretion system assembly in Acinetobacter baumannii. mBio 11, e02761–19. doi: 10.1128/mBio.02761-19
Ma, J., Pan, Z., Huang, J., Sun, M., Lu, C., and Yao, H. (2017). The Hcp proteins fused with diverse extended-toxin domains represent a novel pattern of antibacterial effectors in type VI secretion systems. Virulence 8, 1189–1202. doi: 10.1080/21505594.2017.1279374
Merda, D., Briand, M., Bosis, E., Rousseau, C., Portier, P., Barret, M., et al. (2017). Ancestral acquisitions, gene flow and multiple evolutionary trajectories of the type three secretion system and effectors in Xanthomonas plant pathogens. Mol. Ecol. 26, 5939–5952. doi: 10.1111/mec.14343
Meuskens, I., Saragliadis, A., Leo, J. C., and Linke, D. (2019). Type V secretion systems: an overview of passenger domain functions. Front. Microbiol. 10:1163. doi: 10.3389/fmicb.2019.01163
Mhedbi-Hajri, N., Hajri, A., Boureau, T., Darrasse, A., Durand, K., Brin, C., et al. (2013). Evolutionary history of the plant pathogenic bacterium Xanthomonas axonopodis. PLoS One 8:e58474. doi: 10.1371/journal.pone.0058474
Mijatovic, J., Severns, P. M., Kemerait, R. C., Walcott, R. R., and Kvitko, B. (2021). Patterns of seed-to-seedling transmission of Xanthomonas citri pv. malvacearum, the causal agent of cotton bacterial blight. Phytopathology 111, 2176–2184. doi: 10.1094/PHYTO-02-21-0057-R
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534.
Montenegro Benavides, N. A., Alvarez, B. A., Arrieta-Ortiz, M. L., Rodriguez-R, L. M., Botero, D., Tabima, J. F., et al. (2021). The type VI secretion system of Xanthomonas phaseoli pv. manihotis is involved in virulence and in vitro motility. BMC Microbiol. 21:14. doi: 10.1186/s12866-020-02066-1
Newberry, E. A., Bhandari, R., Minsavage, G. V., Timilsina, S., Jibrin, M. O., Kemble, J., et al. (2019). Independent evolution with the gene flux originating from multiple xanthomonas species explains genomic heterogeneity in Xanthomonas perforans. Appl. Environ. Microbiol. 85, e00885–19. doi: 10.1128/AEM.00885-19
Nguyen, V. S., Douzi, B., Durand, E., Roussel, A., Cascales, E., and Cambillau, C. (2018). Towards a complete structural deciphering of Type VI secretion system. Curr. Opin. Struct. Biol. 49, 77–84. doi: 10.1016/j.sbi.2018.01.007
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palmer, T., Finney, A. J., Saha, C. K., Atkinson, G. C., and Sargent, F. (2020). A holin/peptidoglycan hydrolase-dependent protein secretion system. Mol. Microbiol. 115, 345–355. doi: 10.1111/mmi.14599
Pietrosiuk, A., Lenherr, E. D., Falk, S., Bönemann, G., Kopp, J., Zentgraf, H., et al. (2011). Molecular basis for the unique role of the AAA+ chaperone ClpV in type VI protein secretion. J. Biol. Chem. 286, 30010–30021. doi: 10.1074/jbc.M111.253377
Potnis, N., Krasileva, K., Chow, V., Almeida, N. F., Patil, P. B., Ryan, R. P., et al. (2011). Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 12:146. doi: 10.1186/1471-2164-12-146
Potnis, N., Timilsina, S., Strayer, A., Shantharaj, D., Barak, J. D., Paret, M. L., et al. (2015). Bacterial spot of tomato and pepper: diverse Xanthomonas species with a wide variety of virulence factors posing a worldwide challenge. Mol. Plant Pathol. 16, 907–920. doi: 10.1111/mpp.12244
Rademaker, J. L. W., Louws, F. J., Schultz, M. H., Rossbach, U., Vauterin, L., Swings, J., et al. (2005). A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology 95, 1098–1111. doi: 10.1094/PHYTO-95-1098
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rockey, W., Potnis, N., Hong, J. C., Timilsina, S., Vallad, G. E., Jones, J. B., et al. (2015). Multilocus sequence analysis reveals genetic diversity in Xanthomonads associated with poinsettia production. Plant Dis. 99, 874–882. doi: 10.1094/PDIS-08-14-0867-RE
Salomon, D., Klimko, J. A., Trudgian, D. C., Kinch, L. N., Grishin, N. V., Mirzaei, H., et al. (2015). Type VI secretion system toxins horizontally shared between marine bacteria. PLoS Pathog. 11:e1005128. doi: 10.1371/journal.ppat.1005128
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shalom, G., Shaw, J. G., and Thomas, M. S. (2007). In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153, 2689–2699. doi: 10.1099/mic.0.2007/006585-0
Shyntum, D., Venter, S., Moleleki, L., Toth, I., and Coutinho, T. (2014). Comparative genomics of type VI secretion systems in strains of Pantoea ananatis from different environments. BMC Genomics 15:163. doi: 10.1186/1471-2164-15-163
Silverman, J. M., Austin, L. S., Hsu, F., Hicks, K. G., Hood, R. D., and Mougous, J. D. (2011). Separate inputs modulate phosphorylation-dependent and -independent type VI secretion activation: posttranslational regulation of type VI secretion. Mol. Microbiol. 82, 1277–1290. doi: 10.1111/j.1365-2958.2011.07889.x
Silverman, J. M., Brunet, Y. R., Cascales, E., and Mougous, J. D. (2012). Structure and Regulation of the Type VI Secretion System. Ann. Rev. Microbiol. 66, 453–472. doi: 10.1146/annurev-micro-121809-151619
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Thomas, J., Watve, S. S., Ratcliff, W. C., and Hammer, B. K. (2017). Horizontal gene transfer of functional type VI killing genes by natural transformation. mBio 8, e00654–17. doi: 10.1128/mBio.00654-17
Timilsina, S., Jibrin, M. O., Potnis, N., Minsavage, G. V., Kebede, M., Schwartz, A., et al. (2015). Multilocus sequence analysis of xanthomonads causing bacterial spot of tomato and pepper plants reveals strains generated by recombination among species and recent global spread of Xanthomonas gardneri. Appl. Environ. Microbiol. 81, 1520–1529. doi: 10.1128/AEM.03000-14
Timilsina, S., Potnis, N., Newberry, E. A., Liyanapathiranage, P., Iruegas-Bocardo, F., White, F. F., et al. (2020). Xanthomonas diversity, virulence and plant–pathogen interactions. Nat. Rev. Microbiol. 18, 415–427. doi: 10.1038/s41579-020-0361-8
Triplett, L. R., Verdier, V., Campillo, T., Van Malderghem, C., Cleenwerck, I., Maes, M., et al. (2015). Characterization of a novel clade of Xanthomonas isolated from rice leaves in Mali and proposal of Xanthomonas maliensis sp. nov. Antonie van Leeuwenhoek 107, 869–881. doi: 10.1007/s10482-015-0379-5
Vauterin, L., Hoste, B., Kersters, K., and Swings, J. Y. (1995). Reclassification of Xanthomonas. Int. J. Syst. Evol. Microbiol. 45, 472–489. doi: 10.1099/00207713-45-3-472
Vicente, J. G., Rothwell, S., Holub, E. B., and Studholme, D. J. (2017). Pathogenic, phenotypic and molecular characterisation of Xanthomonas nasturtii sp. nov. and Xanthomonas floridensis sp. nov., new species of Xanthomonas associated with watercress production in Florida. Int. J. Syst. Evol. Microbiol. 67, 3645–3654. doi: 10.1099/ijsem.0.002189
Vieira, P. S., Bonfim, I. M., Araujo, E. A., Melo, R. R., Lima, A. R., Fessel, M. R., et al. (2021). Xyloglucan processing machinery in Xanthomonas pathogens and its role in the transcriptional activation of virulence factors. Nat. Commun. 12:4049. doi: 10.1038/s41467-021-24277-4
Wang, H., McTavish, C., and Turecheck, W. W. (2018). Colonization and movement of Xanthomonas fragariae in strawberry tissues. Phytopathology 108, 681–690. doi: 10.1094/PHYTO-10-17-0356-R
White, F. F., Potnis, N., Jones, J. B., and Koebnik, R. (2009). The type III effectors of Xanthomonas. Mol. Plant Pathol. 10, 749–766. doi: 10.1111/j.1364-3703.2009.00590.x
Wu, C. F., Weisberg, A. J., Davis, E. II., Chou, L., Khan, S., Lai, E.-M., et al. (2021). Diversification of the type VI secretion system in Agrobacteria. mBio 12:e0192721. doi: 10.1128/mBio.01927-21
Young, J. M., Park, D.-C., Shearman, H. M., and Fargier, E. (2008). A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 31, 366–377. doi: 10.1016/j.syapm.2008.06.004
Zarate-Chaves, C. A., de la Cruz, D. G., Verdier, V., Lopez, C. E., and Bernal, A. (2021). Cassava diseases caused by Xanthomonas phaseoli pv. manihotis and Xanthomonas cassavae. Mol. Plant Pathol. 22, 1520–1537. doi: 10.1111/mpp.13094
Zheng, J., Ho, B., and Mekalanos, J. J. (2011). Genetic analysis of anti-amoebae and anti-bacterial activities of the type VI secretion system in Vibrio cholerae. PLoS One 6:e23876. doi: 10.1371/journal.pone.0023876
Zhu, P.-C., Li, Y.-M., Yang, X., Zou, H.-F., Zhu, X.-L., Niu, X.-N., et al. (2020). Type VI secretion system is not required for virulence on rice but for inter-bacterial competition in Xanthomonas oryzae pv. oryzicola. Res. Microbiol. 171, 64–73. doi: 10.1016/j.resmic.2019.10.004
Keywords: T6SS, Xanthomonas, evolution, non-pathogenic, phylogenetics
Citation: Liyanapathiranage P, Wagner N, Avram O, Pupko T and Potnis N (2022) Phylogenetic Distribution and Evolution of Type VI Secretion System in the Genus Xanthomonas. Front. Microbiol. 13:840308. doi: 10.3389/fmicb.2022.840308
Received: 21 December 2021; Accepted: 10 February 2022;
Published: 14 April 2022.
Edited by:
Chih-Horng Kuo, Institute of Plant and Microbial Biology, Academia Sinica, TaiwanReviewed by:
Alexandra J. Weisberg, Oregon State University, United StatesCopyright © 2022 Liyanapathiranage, Wagner, Avram, Pupko and Potnis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neha Potnis, bnpwMDAyNEBhdWJ1cm4uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.