
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 March 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.833318
This article is part of the Research Topic Emergence and re-emergence of plant diseases caused by Xanthomonas species View all 5 articles
 Denghui Chen1,2
Denghui Chen1,2 Xionghui Zhong2*
Xionghui Zhong2* Jian Cui2Hailong Li2Rui Han1,2Xiangqing Yue1,2
Jian Cui2Hailong Li2Rui Han1,2Xiangqing Yue1,2 Jianming Xie1*
Jianming Xie1* Jungen Kang2*
Jungen Kang2*
Black rot is a disease that has a severe impact on cabbage yield and quality in China. Xanthomonas campestris pv. campestris (Xcc) is the causal agent of black rot of Brassicaceae crops. So far, the whole genomic sequences of more than 30 Xcc isolates have been sequenced; however, little information about genomic variability and virulence has been reported. In this study, 12 Xcc isolates were isolated from diseased cabbage leaves in seven Chinese provinces and two municipalities from July 2019 to November 2020. Pathogenicity analysis showed that isolate GSXT20191014 was more aggressive than BJSJQ20200612 and HRIW 3811 on cabbage inbred line 1371. Both BJSJQ20200612 and GSXT20191014 were sequenced and comparatively analyzed. The results showed that BJSJQ20200612 and GSXT20191014 have a single circular chromosome comprising 5,115,975 and 4,975,682 bp, respectively. Compared to the other six sequenced strains, 26 and 47 variable genomic regions were found in BJSJQ2020061 and GSXT20191014 genomic sequences, respectively. The variable genomic regions could be responsible for the genetic variation in Xcc strains and have led to the differences in type III secreted effector repertoires, virulence factors and secreted proteins between these two strains. Among the identified secreted proteins, two copies of peptidase S8/S53 were found in GSXT20191014-specific chromosomal segments. The common effectors xopR, xopH, avrBs1, and xopAH are found in most Xcc genomes, but they are absent in the GSXT20191014 genome. Variations in the composition of exopolysaccharides (EPS) and lipopolysaccharides (LPS) may aid GSXT20191014 isolate infections to evade recognition by the host immune system. Our results revealed a direct correlation between genomic variability and Xcc virulence. We also developed several markers for detecting BJSJQ20200612 and GSXT20191014 isolates and further tested the rest of our other 10 isolates. Finally, the isolated Xcc strains were classified into three genetic subgroups by specific molecular markers and multilocus sequence typing (MLST) approach. BJSJQ20200612 and GSXT20191014 isolates were also classified into two subgroups of Xcc according to the core-genome-based phylogenetic tree. This study extended our understanding of Xcc genomic features and provided the foundation to further characterize the mechanisms for Xcc virulence and a clue for black rot resistance breeding.
Black rot of crucifers, caused by Xanthomonas campestris pv. campestris (Xcc), was first reported as a cabbage disease by Garman in 1890 in Kentucky, United States (Garman, 1890). Since then, the disease has been reported in all Brassicaceae-growing regions, and it is considered as one of the most destructive diseases of crucifers worldwide (Williams, 1980).
Black rot is a seed-borne disease. Xcc is favored by warm and humid conditions and Xcc can survive overwinter on plant debris, crucifer weeds, and wild relatives of cultivated brassicas (Cook et al., 1952; Schaad, 1981). These bacteria establish a systemic infection in susceptible hosts by penetrating the plant through the hydathodes at leaf margins or injuries. The typical symptom of black rot is the formation of V-shaped chlorotic yellow lesions along the leaf margins. Furthermore, the darkened veins appear along with the bacterial movement in the vascular system. Eventually, the affected tissues become necrotic and leaves fall prematurely, systemic infections cause stunted growth and the death of young plants. Black rot disease drastically reduces crop yields during the warm and wet seasons.
With the rapid development of DNA sequencing technologies, the increasing availability of genome data is enabling us to dig into bacterial genetic variability and pathogenesis. In 2002, the release of the genome sequence of ATCC 33913, the first genome sequence of Xcc, provided a profile of genetic information to explore the biological characteristics of Xcc (Ribeiro da Silva et al., 2002); however, a large repertoire of genes, including virulence factors and pathogenicity-related genes, have not been experimentally determined. Later, the genome sequence of Xcc 8004 became available, and comparative and functional genomic analyses provided valuable genetic information on Xcc pathogenicity. They found that three of unknown function genes (XC2055, XC2068 and XC2416) were located in Xcc 8004-specific chromosomal segments, which are responsible for the host-specific pathogenesis (Qian et al., 2005). The complete genome sequence of the Xcc B100 was established, where the updated annotated genome data was used to reconstruct the mechanistic model for xanthan biosynthesis (Vorhölter et al., 2008). In 2015, the draft genome sequences of strains CFBP 1869 and CFBP 5817 were determined; comparative genomics showed that core type III-secreted proteins were different between these two isolates (Bolot et al., 2015). As more genome sequences are released, we will gain further insight into interspecific genomic diversity and understanding of virulence variability between different Xcc isolates.
Successful infection of host tissue by bacteria often depends on the secreted bacterial virulence factors to disrupt the innate immunity. Virulence factors include bacterial toxins and adhesins that mediate bacterial attachment, cell surface-associated carbohydrates (polysaccharides and lipopolysaccharides) that protect a bacterium from hostile environments, and plant cell wall-degrading enzymes that may contribute to the pathogenicity of the bacterium (Büttner and Bonas, 2010; Li L. et al., 2019). Both EPS and LPS are surface-associated virulence factor that are major structural components of Xanthomonas spp. and offer protection from environmental stress (Büttner and Bonas, 2010). The type III secretion system is essential for most Gram-negative bacteria to establish infection (Puhar and Sansonetti, 2014). The core gene cluster of the Xcc type III secretion system consists of 25 genes and contains at least six operons (hrpA, hrpB, hrpC, hrpD, hrpE, and hrpF) (Weber et al., 2007; Li et al., 2014). P. syringae and Xanthomonastype III effectors play an important role in manipulating the host response. Pathogen effectors, like type III effectors, are major driving forces in plant pathogen co-evolution (Feng and Zhou, 2012). Pathogens may evolve new effectors or dislodge old effectors to escape host immunity. The difference in gene content, such as virulence factors, gene clusters in xanthan and LPS biosynthesis pathways, secreted proteins, and type III effectors, may contribute to genetic variability and differences in bacterial virulence.
Although, genetic diversity and population structure of the Xcc strains affecting cabbages in China has been revealed by MLST and Rep-PCR based genotyping (Chen et al., 2021), However, there is no detailed information on the whole genomic sequence of Xcc strains isolated from the main producing area of cabbage in China. Therefore, 12 Xcc virulent strains were isolated from seven Chinese provinces and two municipalities, and two isolates were chosen for whole-genome sequencing to compare their genetic diversity and pathogenicity. We conducted a comparative genomic analysis of BJSJQ20200612, GSXT20191014 and the known race strains including race 1 (HRIW 3811, B100, CFBP 1869), race 3 (ATCC 33913), race 4 (CFBP 5817) and race 9 (Xcc 8004) to understand the difference in genetic variability and virulence. Comparative genomic study clearly presented the discrepancy genes, variable genomic regions and strain specificities. The study revealed that an insight into the relationship between genetic diversity and pathogenic diversity in Xcc. It will lay the foundation for further study on Xcc virulence and provide the clue for black rot resistance breeding.
Xcc representative strains (HRIW 3811, HRIW 3849A, HRIW 5212, HRIW 1279A, HRIW 3880, HRIW 6181, and HRIW 8450A) were obtained from the School of Life Sciences, Wellesbourne Campus, University of Warwick, United Kingdom (HRIW). Xcc 8004 was obtained from Guangxi Key Laboratory of Biology for Crop Diseases and Insect Pests, Plant Protection Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, China. Another 12 strains (BJTZ20191009 and BJSJQ20200612, CQ20200923, HBXT20190705 and HBCL20191115, SXXY20191115, GSXT20191014 and GSNG20191014, JSNJ20191022, GDGZ20191212, ZJHZ20191121, and SDJN20201102) were collected from cabbages from Beijing, Chongqing, Hebei, Shaanxi, Gansu, Jiangsu, Guangdong, Zhejiang and Shandong provinces in China from July 2019 to November 2020. The isolates used in this study were obtained as described by Jensen et al. (2010), with minor modifications. Briefly, diseased leaves were collected from plants with V-shape lesions, and about 1 × 1 cm-leaf tissue segments were excised from lesion margins. The leaf segments were placed into 75% ethanol for 45 s, rinsed three times in sterile distilled water, and then ground with a glass mortar and pestle with sterile distilled water, and plated on the LB medium. LB plates were monitored for the presence of characteristic convex mucoid colonies after 2 days incubation at 28°C. Single yellow-pigmented colonies were transferred to another LB agar plate for purification. The typical pale yellow colonies of Xanthomonas were then transferred to new LB medium and incubated for 24 h at 28°C for further purification and identification of the species. For the long-term storage, the strains were typically incubated in LB liquid media at 28°C with shaking for 24 h. And then, 500 μL of the overnight culture was added to 500 μL of 50% glycerol in a 2 mL tube, and maintained at –80°C.
Pure cultures were grown on LB medium at 28°C for 48 h. Genomic DNA was extracted from 2-day-old bacterial colonies grown in nutrient agar. Bacterial strains were harvested from LB culture media, and genomic DNA was extracted using the DNeasy Plant Mini Kit (TIANGEN, Beijing of China) according to the manufacture instructions. The bacterial DNA was stored at –20°C for future use.
Sequence Characterized Amplified Region (SCAR) markers Xcc_48F/R and Xcc_53F/R were developed to amplify putative prolyl aminopeptidase and putative exported protein through whole-genome alignment (Rubel et al., 2019a). These primers were used for rapidly and accurately distinguishing Xcc from other pathovars. The two PCR assays were repeated independently for each strain. Primer names, sequences, and the size of the targeted product are listed in Supplementary Table 1. In addition to the 12 isolated strains, one known Xcc strain HRIW 3811 from cabbage and the Pseudomonas syringae pv. tomato DC3000 strain were used as the positive and negative controls, respectively. The PCR amplification was carried out using genomic DNA (gDNA) samples extracted from strains. PCR reaction volume for these primers was 10 μl including 5.0 μl of 1x Taq MasterMix (Biomed, China), 1 μl of each forward and reverse primer (10 pmol μl–1), 1 μl of bacterial DNA, and 2 μl of deionized water. PCR amplification conditions were standardized in a thermocycler with initial denaturation at 95°C for 5 min followed by 20 or 25 cycles of amplification with denaturation at 95°C for 30 s, annealing at different temperatures for 40 s, and elongation at 72°C for 5 min (Supplementary Table 1). A 5 μl aliquot of each amplified PCR product was loaded onto 1.5% agarose gels containing 1x TAE buffer, electrophoresed at 120 V for 30 min and checked for its respective size under UV transillumination. Confirmation of results was performed through repeated PCR amplifications. A DM2000 DNA marker (TransGen Biotech, China) was used to estimate the size of the PCR products.
Four housekeeping genes were chosen for MLST analysis based on previous studies (Fargier et al., 2011; Chen et al., 2021; Laala et al., 2021), including atpD (ATP synthase beta chain), gyrB (DNA gyrase subunit B), rpoD (RNA polymerase sigma-70 factor), and fyuA (TonB-dependent receptor). All PCR reaction volume for these primers was 50 μl including 25.0 μl of 2 × Primer STAR (Takara, China), 1 μl of each forward and reverse primer (10 pmol μl–1), 1 μl of bacterial DNA, and 22 μl of deionized water. Amplified products of these four housekeeping genes from our 12 isolated strains were purified and sequenced by the Beijing Genomics Institute (BGI, Beijing, China). The nucleotide and haplotype diversity analysis were performed using DNA Sequence polymorphism (DnaSP) software version 5.10 (Librado and Rozas, 2009). The phylogenetic tree was constructed with the four housekeeping gene concatenated sequences using Mega software version 6.06 based on the Maximum likelihood (ML) method (Tamura et al., 2013).
Whole-genome sequencing of BJSJQ20200612 and GSXT20191014 were performed by the Oxford Nanopore Technologies (ONT) method in Biomarker Technologies (Beijing, China) (Deamer et al., 2016; Jain et al., 2016; Magi et al., 2018). A DNA library with 19 and 20 kb inserts were constructed, respectively. The library sequencing was quantified by PromethION 48, and filtered subreads were assembled by CANU software (version 1.5) with default parameters (Koren et al., 2017). The error-corrected assembly was tested for possible circularity using Circlator software (version 1.5.5) (Hunt et al., 2015). The genomic circle map was constructed using Circos software (version 0.66) (Krzywinski et al., 2009).
Protein coding sequences (CDS) were predicted using the software Prodigal v2.6.3 (Hyatt et al., 2010). tRNA and rRNA genes were identified using tRNAscan-SE v2.0 (Chan and Lowe, 2019) and Infernal v1.1.3 (Nawrocki and Eddy, 2013), respectively. The functions of the predicted proteins were annotated based on a BLASTP search against universal databases, such as the Cluster of Orthologous Groups of proteins database (COG),1 and the Kyoto Encyclopedia of Genes and Genomes database (KEGG),2 and the exclusive database: virulence factor database (VFDB)3 (Chen et al., 2005). Putative signal peptides and transmembrane helices were predicted using SignalP v4.0 (Petersen et al., 2011) and TMHMM v2.0 (Krogh et al., 2001), respectively.
In order to design primers for specific detection of BJSJQ20200612 and GSXT20191014 isolates, we considered HRIW 3811 as a reference genome for designing primers by comparative whole-genome analyses of B100, CFBP 1869, ATCC 33913, CFBP 5817 and Xcc 8004, BJSJQ20200612 and GSXT20191014. The GenBank accession numbers are NZ_CP025750 for HRIW 3811, AM920689 for B100, NZ_CM002545 for CFBP 1869, AE008922 for ATCC 33913, NZ_CM002673 for CFBP 5817, NC_007086 for Xcc 8004, CP069085 for BJSJQ20200612, and CP069084 for GSXT20191014, respectively. The homology between sequence blocks among Xcc strains (shown by differential colors) was performed using mauve software (version 2.4.0). We developed specific primers to classify isolates from differential IS regions. The whole predicted protein sequences from BJSJQ20200612, GSXT20191014 and 33 completely sequenced or draft Xcc genomes were used to construct the phylogenetic tree. The core-genome was extracted from the whole genomes of Xcc strains using the USEARCH program (ver. 9.0). The concatenated amino acid sequences of the core-genome were aligned using MUSCLE v3.8.31 (Edgar, 2004). A core-genome-based phylogenetic tree was constructed from the concatenated sequences using the phyML version 3.0 by the maximum likelihood method (Guindon et al., 2005).
A genome-wide alignment among the sequences of B100, HRIW 3811, CFBP 1869, ATCC 33913, CFBP 5817, Xcc 8004, BJSJQ20200612, and GSXT20191014 was performed with Mauve multiple genome alignment tools. We identified several DNA fragments by comparative sequence alignments that were unique to either Xcc strain BJSJQ20200612 or Xcc strain GSXT20191014. Highly conserved regions were excluded when primers were designed. The variable genomic regions (> 800 bp) in the genomic sequence of BJSJQ20200612 and GSXT20191014 isolates compared to published strains were chosen to design the specific markers. Primers XccSJQ-12, XccSJQ-30, XccSJQ-31, XccSJQ-62 and XccXTZ-10, XccXTZ-16, XccXTZ-24, XccXTZ-28 were designed to detect BJSJQ20200612 and GSXT20191014 isolates, respectively (Supplementary Table 2). Among these designed primers, XccSJQ-12, XccSJQ-30, XccSJQ-31 and XccSJQ-62 anchored in the BJSJQ20200612 genomic sequence from 1,221,404 to 1,222,047 bp, 3,077,954–3,078,605 bp, 3,106,515–3,107,131 bp, 620,025–621,536 bp, respectively. XccXTZ-10, XccXTZ-16, XccXTZ-24 and XccXTZ-28 located at the position from 101,571 to 102,074 bp, 166,619–167,210 bp, 2,469,077–2,469,769 bp, 2,866,934–2,867,550 bp in GSXT20191014 genome, respectively. The PCR reaction mixture (10 μl) included 1 μl of gDNA, 10 pmol of each forward and reverse primer, and 7 μl of enzyme mixture. The conditions for PCR amplification for marker XccSJQ-62 was 95°C for 5 min followed by 25 cycles (95°C for 30 s, 67°C for 30 s, and 72°C for 60 s) and a final extension at 72°C for 10 min. The annealing temperature of XccSJQ-12, XccSJQ-30 and XccSJQ-31 was decreased to 65°C for 30 s. The annealing temperature of XccXTZ-10, XccXTZ-16, XccXTZ-24, and XccXTZ-28 was increased to 70°C for 30 s. The amplified PCR products were detected using gel electrophoresis (1.5% agarose gel).
Twelve isolated strains were finally selected for subsequent studies. For all biological assays, the strain HRIW 3811 was used as the positive control, and sterile water was used as the negative control. Strains preserved at –80°C were defrosted and grown in LB medium (5 ml) at 28°C for 24 h. The bacterial suspension was centrifuged for 5 min at 5,000 g, and the supernatant was discarded; then, the bacterial concentration was adjusted to 1 × 108CFU ml–1. Cabbage inbred line 1371 seeds were sown in plastic pots with sterilized substrate (peat: vermiculite = 2:1) and cultivated in a greenhouse at 24 ± 2°C with 16/8 h (light/dark). The seedlings with four fully expanded leaves were used for pathogen inoculation. For pathogenicity assays, the two youngest leaves were inoculated by dipping clipped secondary veins (with sterile scissors) into purified Xcc strain suspensions (1 × 108 CFU ml–1) (Vicente et al., 2001; Gu et al., 2008; Afrin et al., 2018a). Ten plants were inoculated per isolate; reference Xcc strain HRIW 3811 and water were used as the positive and negative controls. Inoculated plants were grown in a growth chamber at 24 ± 2°C with 16/8 h (light/dark) and 70–75% relative humidity. Plants were recorded for typical black rot symptoms with progressive V-shaped and yellow necrotic lesions with blackened veins after 5–14 days. The average lesion area/health area ratio from 20 leaves was measured with Adobe Photoshop 7.0.1. The method of pathogenicity testing on other brassica crops was same as above.
Twelve bacterial isolates were obtained from black rot diseased leaves (with V-shape lesions), which were harvested from cabbage-growing regions in Beijing, Chongqing, Hebei, Shaanxi, Gansu, Jiangsu, Guangdong, Zhejiang, and Shandong provinces in China from July 2019 to November 2020 (Figure 1A). Twelve of the purified bacterial isolates exhibited the typical yellow mucoid and glistening colonies and are suggestive of Xcc bacteria (Figure 1B). Xcc_48F/R and Xcc_53F/R were used to determine whether the isolates were the Xcc or other pathovars. Positive PCR amplification of 855 and 930 bp were observed in the 12 isolates and Xcc control (HRIW 3811), respectively (Figure 1C). The colonial morphology and PCR results indicated that the 12 isolates are Xcc pathovar.
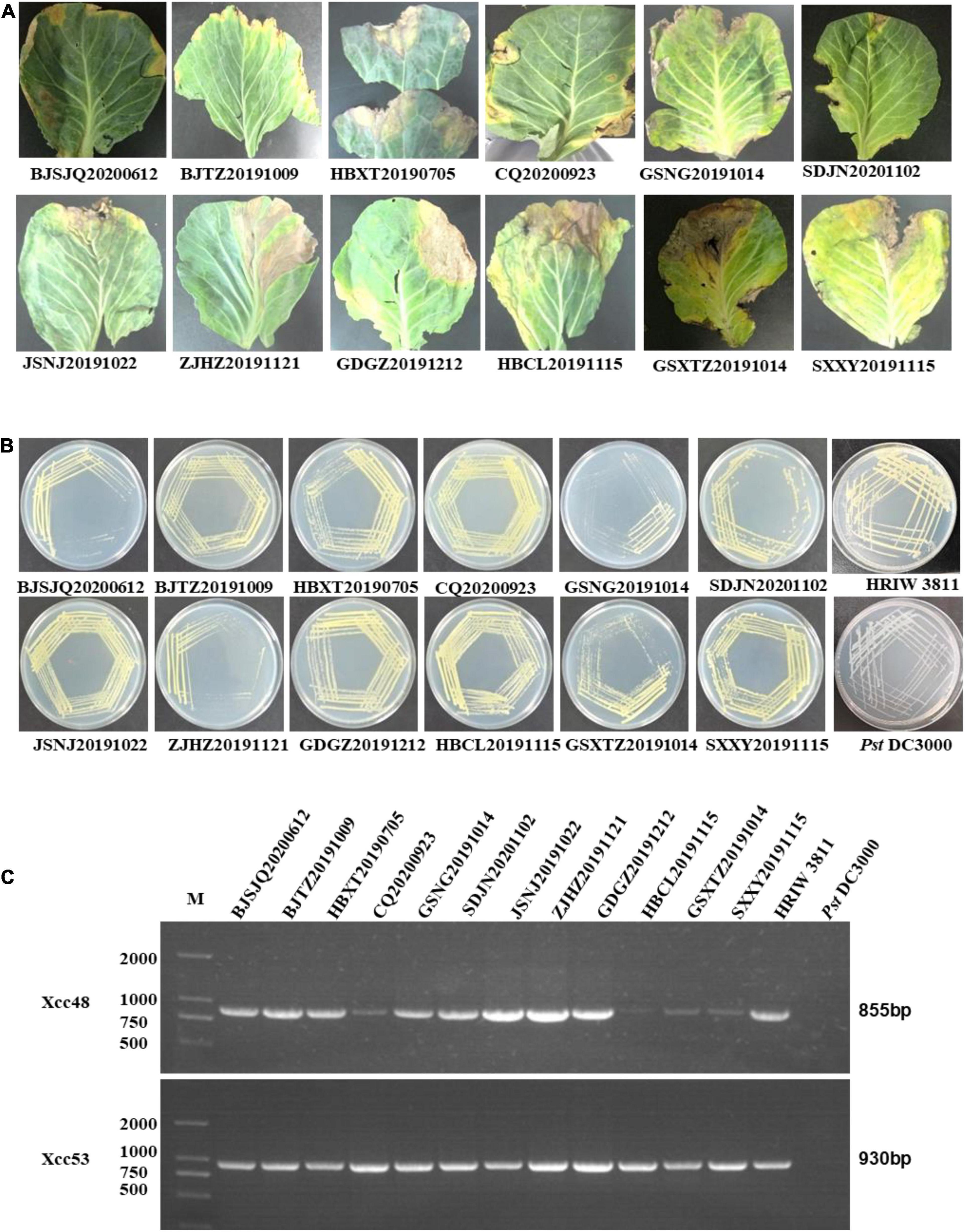
Figure 1. Purified colony morphology isolated from diseased cabbage leaf samples and specific molecular detection of Xcc in cabbage. (A) Diseased leaves collected in different cabbage growing regions of China. (B) Purified strain colonies growing on LB medium. (C) Detection of the isolated strains by Xcc-specific markers, where Xcc_48F/R and Xcc_53F/R were used to distinguish Xcc strains from other bacterial species. Isolated strains: BJTZ20191009 and BJSJQ20200612, CQ20200923, HBXT20190705 and HBCL20191115, SXXY20191115, GSXT20191014 and GSNG20191014, JSNJ20191022, GDGZ20191212, ZJHZ20191121 and SDJN20201102 were collected from Beijing, Chongqing, Hebei, Shaanxi, Gansu, Jiangsu, Guangdong, Zhejiang, and Shandong provinces in China. HRIW 3811 and Pseudomonas syringae pv. tomato DC3000 were used as the positive and negative control, respectively.
All 12 isolated strains were evaluated for their pathogenicity in cabbage by the leaf-clipping inoculation method. Seedlings inoculated with sterile water and HRIW 3811 were used as the negative and positive controls, respectively. The distinct V-shaped lesions were observed in all inoculated cabbages after 14 days (Figure 2A). Additionally, we also found that the isolated strains HBCL20191115, SXXY20191115 and GSXT20191014 induced more severe symptoms in cabbage inbred line 1371 than HRIW 3811, and the average lesion area/health area ratio (per leaf) caused by these three isolated strains were larger than that caused by the positive control (HRIW 3811 strain) (Figure 2B). However, the results showed that there was no difference in pathogenicity between the rest of the strains and HRIW 3811. Taken together, these results show that HBCL20191115, SXXY20191115 and GSXT20191014 (from the field) were more virulent than HRIW 3811 in cabbage inbred line 1371. Meanwhile, the virulence experiments were also tested in the Chinese cabbage, radish and broccoli; the pathogenicity results also showed that the isolate GSXT20191014 was more aggressive than BJSJQ20200612 and HRIW 3811 on these three brassica crops (Supplementary Figure 1).
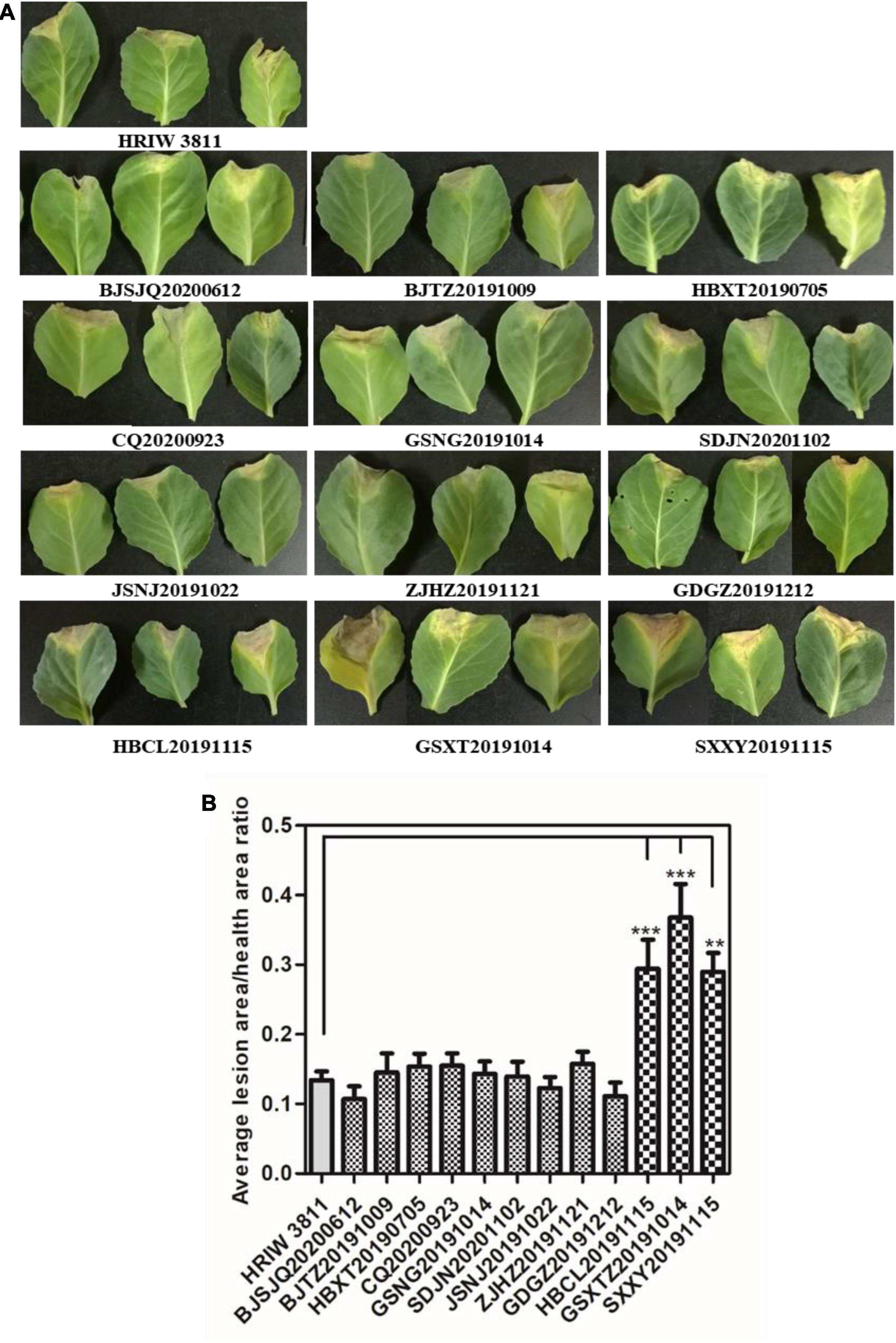
Figure 2. Pathogenicity analysis of isolated strains on cabbage. (A) All 12 isolated Xcc strains caused typical V-shaped lesions in the cabbage after leaf clipping inoculation. The average lesion area/health area ratio (per leaf) caused by three isolated strains (HBCL20191115, SXXY20191115, and GSXT20191014) were larger than that caused by the positive control (HRIW 3811 strain). Photos were taken at 7 d post-inoculation. (B) The average lesion area/health area ratio triggered by the isolated strains were bigger than that caused by HRIW 3811. Lesion and health area were measured using 20 leaves samples at 10 d post-inoculation (data are means ± SD; n > 3; **P < 0.01 and ***P < 0.001).
The length of these four housekeeping gene fragments (atpD, gyrB, rpoD, and fyuA) were 648, 705, 807, and 771 bp, and the total length of four housekeeping gene concatenated sequences was 2,931 bp. The number of polymorphic nucleotide sites among the strains varied from 13 to 107 (Supplementary Table 3). Phylogenetic analysis was performed by the concatenated sequences of four individual genes of 24 Xcc strains. Our isolated Xcc samples were distributed in three subgroups in the phylogenetic relationship (Figure 3). GDGZ20191212 and ZJHZ20191121 were distributed into subgroup I, which also consist of three race1 isolates CFBP1869, HRIW3811 and B100. Seven strains including BJSJQ20200612, BJTZ20191009, HBXT20190705, CQ20200923, GSNG20191014, SDJN20201102, and JSNJ20191022 were divided into the largest subgroup II, which has probably evolved from race 1. Interestingly, HBCL20191115, HBXT20190705 and GSXT20191014 were very closely related to race 2 (HRIW3849A), which were classified into subgroup III. Meanwhile, race 3 (ATCC33913), race 4 (CFBP5817), and race 9 (Xcc 8004) were divided into the same subgroup but in different clades. Taken together, our isolated Xcc strains were classified into three different genetic subgroups.
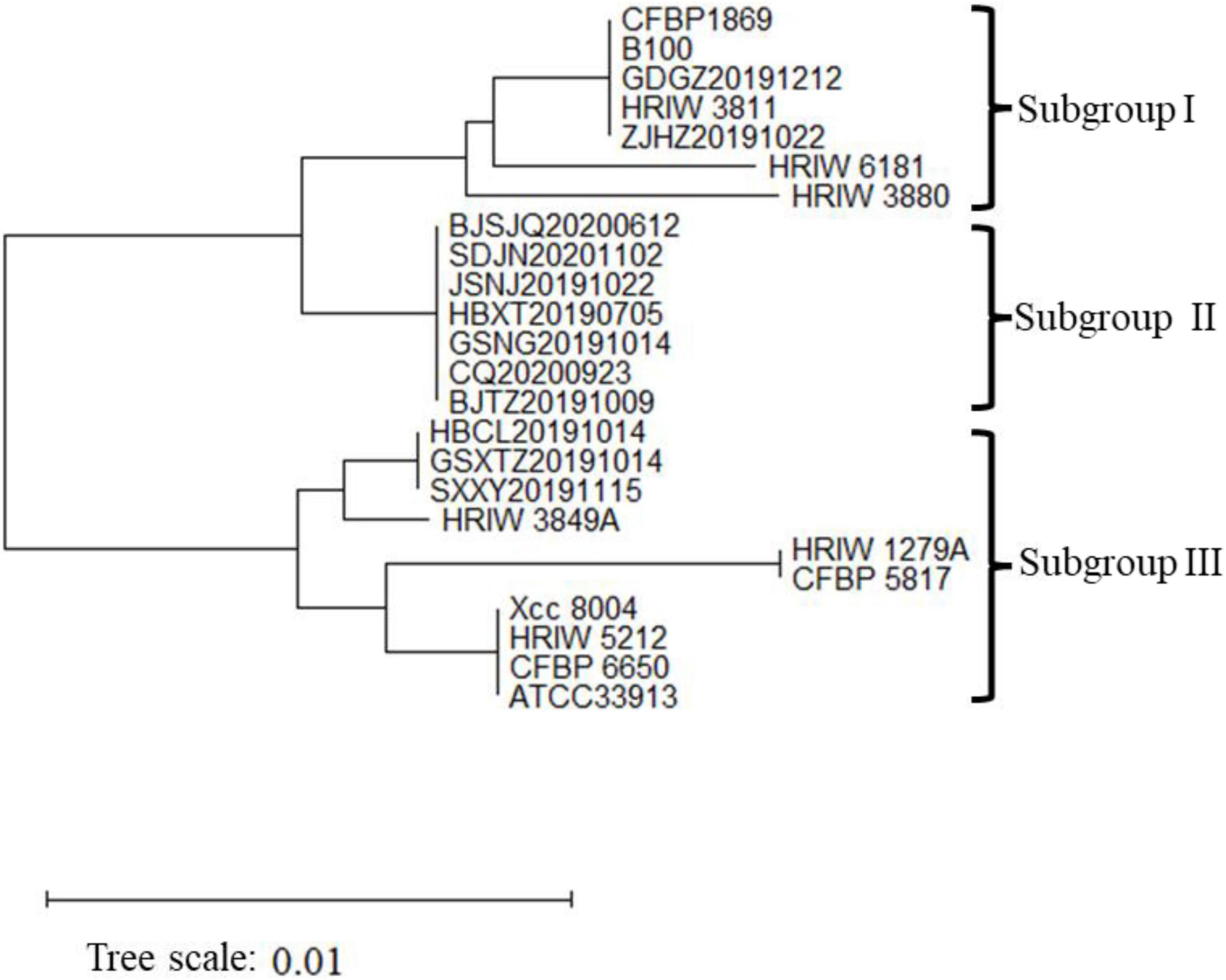
Figure 3. Neighbor-joining tree constructed with concatenated partial sequences of four housekeeping gene (atpD, gyrB, rpoD, and fyuA) of our 12 isolated strains and 12 different Xcc race strains. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The tree scale represents 0.01.
There were 43,522 and 35,039 raw sequences obtained from BJSJQ20200612 and GSXT20191014 isolates, respectively. We obtained 39,675 and 32,258 clean sequences after filtering out adapters and low quality and short segment (length < 2,000 bp) reads, respectively. Furthermore, we got the 158x and 140x genome coverages for isolates BJSJQ20200612 and GSXT20191014, respectively. The completeness of the genome sequences of BJSJQ20200612 and GSXT20191014 isolates is 99.67 and 99.55%, and there is no contamination in both isolates. The final assembly of the BJSJQ20200612 genome resides on a single circular chromosome with a size of 5,115,975 bp (Figure 4A). The genome of GSXT20191014 is composed of a single circular chromosome that is 4,975,682 bp in size without apparent autonomous plasmids (Figure 4B). The average G + C content of the genome is 64.99 and 65.29% of the BJSJQ20200612 and GSXT20191014 genomes, respectively, which is similar to HRIW 3811 (65.1%), B100 (65%), CFBP 1869 (65%), ATCC 33913 (65.1%), CFBP 5817 (65.2%), and Xcc 8004 (64.94%) (Table 1). The genome of GSXT20191014 possesses the highest G + C content in these sequenced Xcc strains. There were 4,382 and 4,184 predicted protein-coding genes in the genome of BJSJQ20200612 and GSXT20191014, respectively. Additionally, the BJSJQ20200612 genomic sequences had 181 RNA genes, including 56 tRNA genes, six rRNA genes (16S rRNA, 5S rRNA, and 23S rRNA), and 119 ncRNAs; whereas, the GSXT20191014 chromosome contained six rRNA genes, 57 tRNA genes, and 120 ncRNA genes (Table 1). The total repetitive sequence length is 49,353 bp and 61,786 bp in BJSJQ20200612 and GSXT20191014 isolates, accounting for 0.96 and 1.24% of the whole genome sequences, respectively. Functional categorization of the coding DNA sequences (CDSs) in BJSJQ20200612 and GSXT20191014 were analyzed using COG database. The results indicated that 32.25 and 33.14% predicted genes are related to metabolism, 23.06 and 23.49% of predicted genes are correlated with cellular processes and signaling, 16.76 and 16.87% are connected with information storage and processing. Furthermore, 27.95 and 26.51% of genes are poorly characterized in BJSJQ20200612 and GSXT20191014 genomes, respectively (Supplementary Table 4).
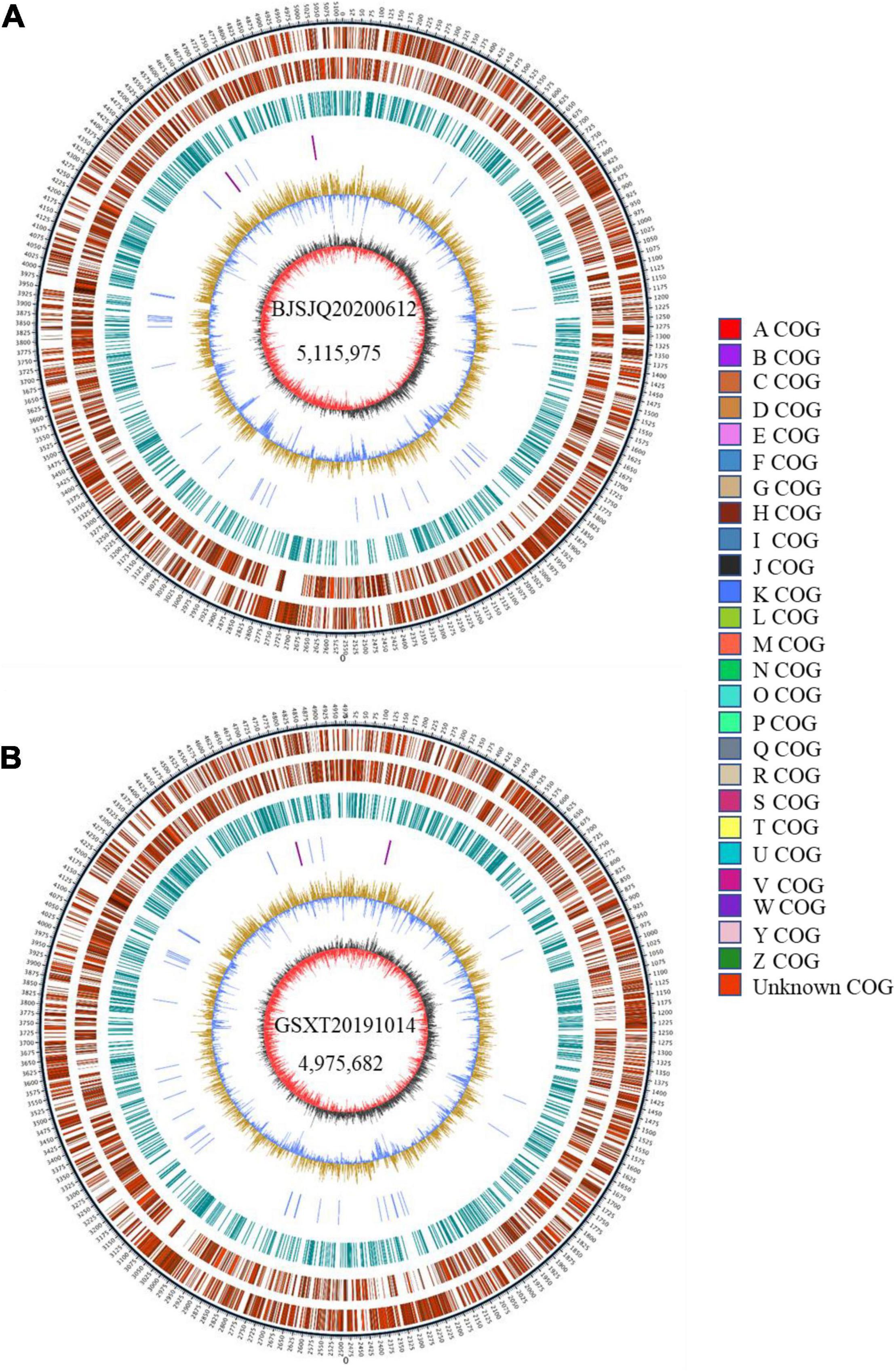
Figure 4. The Graphical circular map of the Xcc isolates BJSJQ20200612 and GSXT20191014. (A) Circular map of BJSJQ20200612. The genome of BJSJQ20200612 is composed of a single circular chromosome that is 5,115,975 bp in size with no apparent autonomous plasmids. (B) Circular map of GSXT20191014. The final assembly of the GSXT20191014 genome resides on a single circular chromosome with a size of 4,975,682 bp. Circles range from 1 to 7 (outer circle to inner circle). Circle 1: The size and position of the whole genome sequence, each scale represent 5 kb; circles 2 and 3: Genes on the positive and negative chains of the genome, respectively, and different colors represent different COG functional classification; circle 4: repeating sequence; circle 5: tRNA and rRNA genes; circle 6: GC content, the light yellow part indicates that the GC content in this region is higher than the average GC content in the genome, and the higher the peak value, the greater the difference between the region and the average GC content. The blue part indicates that the GC content in this region is lower than the average GC content in the genome; Circle 7: Is GC-skew, where dark gray represents the area where G content is greater than C, and red represents the area where C content is greater than G.
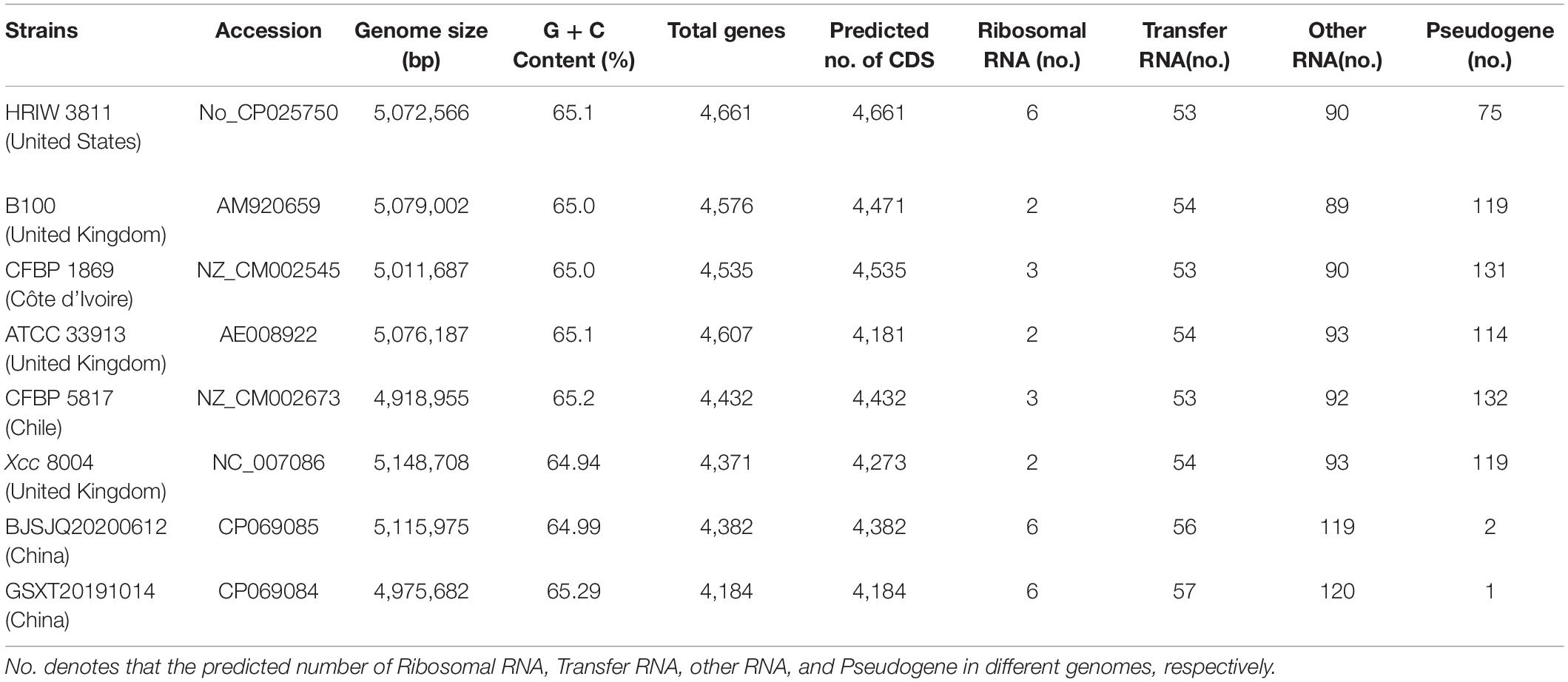
Table 1. Genome features of BJSJQ20200612 and GSXT20191014 and other sequenced Xcc strains.
To infer the phylogenetic relationships among Xcc strains, a phylogenetic tree was constructed using the concatenated amino acid sequences of genes in the core-genome (Figure 5). Unlike the phylogenetic tree based on housekeeping gene sequences, the phylogenetic tree based on the core-genome showed that Xcc strains have been more clearly separated into different phylogenetic lineages. It is generally accepted that closely related organisms share more orthologous genes, suggesting that evolutionary relationships among Xcc can be inferred by the presence/absence of orthologous genes. The results showed that BJSJQ20200612 was very closely related to isolate MAFF302021, isolated from Shiga, Japan. Interestingly, these two pathogens form a clade; these pathogens may have evolved from the same ancestor. Additionally, BJSJQ20200612 and MAFF302021 isolates shared a close relationship with HRIW 3811, B100 and CFBP 1869 sequences, which were commonly recognized as Xcc race 1. GSXT20191014 has the highest number of unique genes in this subgroup, formed a phylogenetic lineage clearly distinct from race 3 (ATCC33913), race 4 (CFBP5817), and race 9 (Xcc 8004) strains, which may reflect a different characters of GSXT20191014 from those of other Xcc. Interestingly, BJSJQ20200612 and GSXT20191014 isolates were also classified into two subgroups of Xcc according to the core-genome-based phylogenetic tree, which was consistent with subgrouping data performing by the MLST and specific molecular markers experiments.
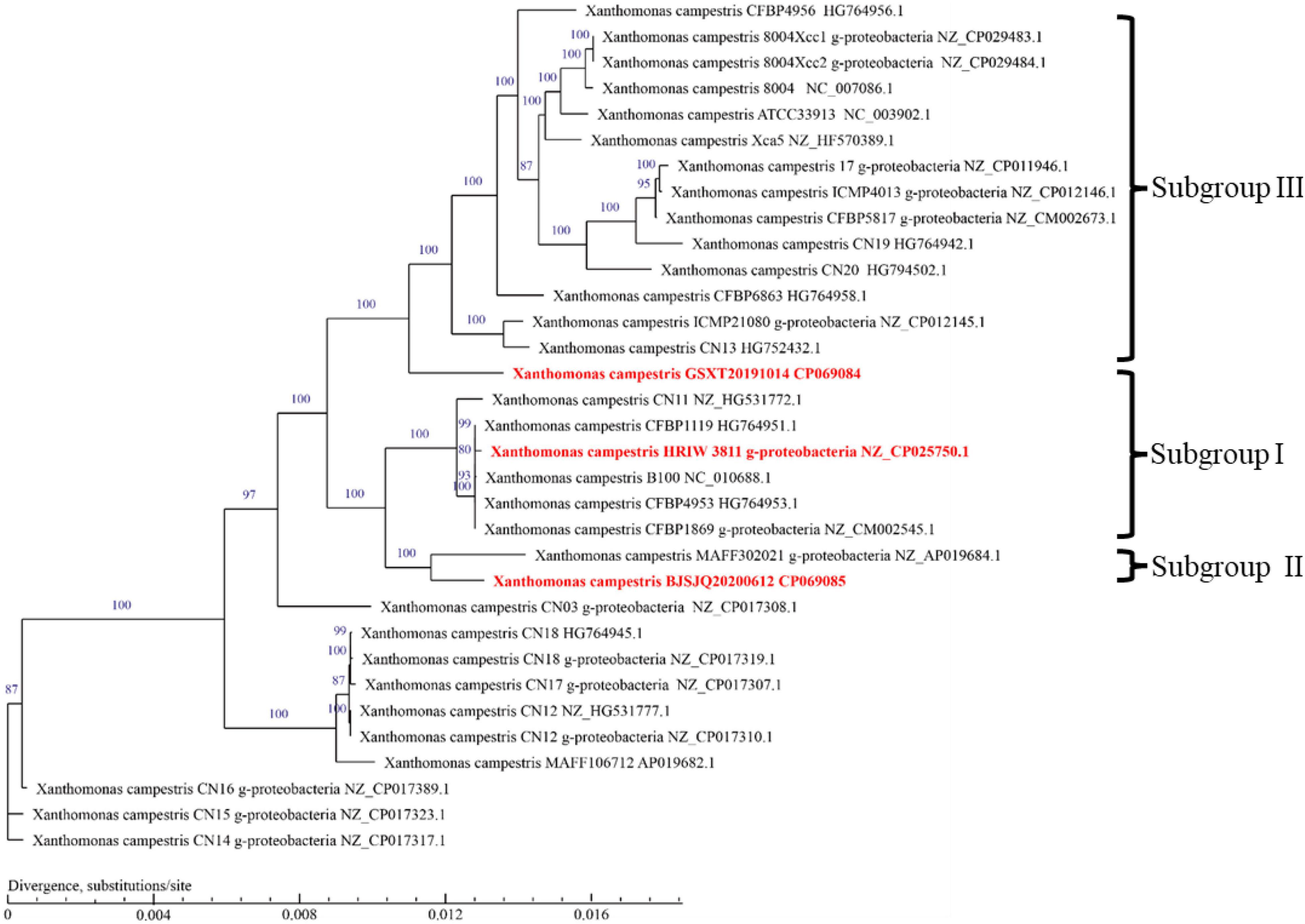
Figure 5. A phylogenetic tree with bootstrap values (1,000 bootstrap replications) reconstructed using the concatenated amino acid sequences of Xcc core-genome showing the relationships among Xcc strains. Strain names as described in GenBank are used in the tree and the type strains are highlighted in red bold. The bar indicates divergence, substitutions per site.
A genome-wide alignment among the sequences of BJSJQ20200612, GSXT20191014 and other sequenced Xcc strains was performed with Mauve multiple genome alignment tools (Figure 6A). XccSJQ-62 and XccXTZ-10 were chosen as the representative examples for the development of specific molecular markers for BJSJQ20200612 and GSXT20191014 isolates. An indel marker (XccXTZ-10) was designed according to a specific 35,024 bp insertion (101,595–136,619) in the genomic sequence of strain GSXT20191014 (Figure 6B). XccXTZ-10 amplified a 510 bp product in the GSXT20191014 isolate (Figure 6D). Equally, we found an 1,163 bp insertion at the BJSJQ20200612 genomic position 620,018–621,536, where a gene (IS5) encoding an IS5 family transposase was located and the sequence characterized amplified region primer set (SCAR; XccSJQ-62) was developed (Figure 6C). The PCR results showed a 1,512 bp product was specifically amplified in strain BJSJQ20200612, while a band size of 349 bp was observed in other strains (Figure 5D). Additionally, the PCR results showed that 644, 652, and 617 bp products were amplified in the BJSJQ20200612 genome using the XccSJQ-12, XccSJQ-30 and XccSJQ-31 markers. Meanwhile, XccXTZ-16, XccXTZ-24 and XccXTZ-28 markers amplified the 630, 693, and 617 bp products from GSXT20191014 genome, respectively (Supplementary Figure 2). A transposase mediated insertion and a large DNA fragment insertion were used to develop the molecular marker for bacteria identification. There are a variety of insertions in the genomic sequence of BJSJQ20200612 and GSXT20191014 isolates. Above-mentioned PCR data are consistent with the expected results according to our comparative comparison of B100, HRIW 3811, CFBP 1869, ATCC 33913, CFBP 5817, Xcc 8004, BJSJQ20200612, and GSXT20191014 genomes. Taken together, the developed markers (XccSJQ-12, XccSJQ-30, XccSJQ-31, XccSJQ-62 and XccXTZ-10, XccXTZ-16, XccXTZ-24, XccXTZ-28) could be used for distinguishing BJSJQ20200612 and GSXT20191014 isolates, respectively.
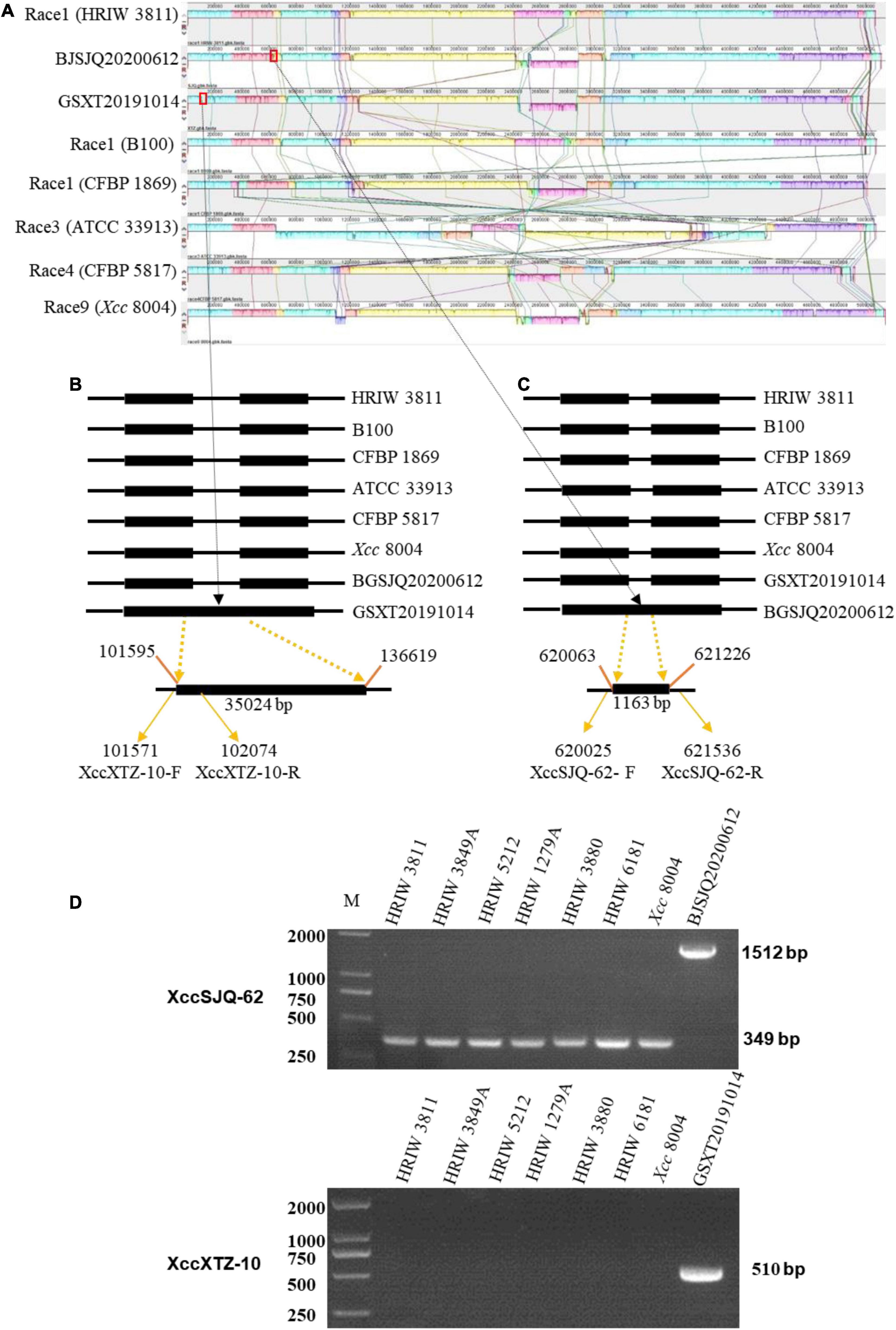
Figure 6. Multiple alignments of the whole genome sequence of Xcc strains by Mauve (version 2.4.0) and the development of molecular markers. (A) Alignment of the whole sequence of eight Xcc strains (B100, HRIW 3811, CFBP 1869, ATCC 33913, CFBP 5817, and Xcc 8004). HRIW 3811 was used as the reference genome. Each genome is laid out horizontally with homologous segments outlined as colored rectangular solid boxes. The syntenic blocks are connected among the strains with lines. The rectangular boxes (red outline) indicate the positions of inserted sequence in the genome. (B) An insertion sequence of BJSJQ20200612 isolate relative to other Xcc strains are set with the line diagram. A 35,024 bp large insertion was located from 101,595 to 136,619 of the genomic sequence. XccXTZ-10-F/R were the GSXT20191014-specific molecular markers. (C) An IS5 family transposase insertion in the GSXT20191014 isolate compared to other Xcc strains are shown with the line diagram. XccSJQ-62-F/R were the BJSJQ20200612-specific molecular marker. The GenBank accession numbers are NZ_CP025750 for HRIW 3811, AM920689 for B100, NZ_CM002545 for CFBP 1869, AE008922 for ATCC 33913, NZ_CM002673 for CFBP5817 and NC_007086 for Xcc 8004, respectively. (D) The usability of molecular markers XccSJQ-62 and XccXTZ-10 were tested by PCR using the genomic DNA of Xcc strains (HRIW 3811, HRIW 3849A, HRIW 5212, HRIW 1279A, HRIW 3880, HRIW 6181, and Xcc 8004). DNA concentration of all samples was 50 ng/μl. Lane M: 2k plus DNA ladder was used as the size marker. BJSJQ20200612 and GSXT20191014 were used as the positive control on the upper and lower panel, respectively.
Isolate-specific markers XccSJQ-12, XccSJQ-30, XccSJQ-31, XccSJQ-62 and XccXTZ-10, XccXTZ-16, XccXTZ-24, XccXTZ-28 were also used to determine other isolated strains. The PCR results indicated that the XccSJQ-12, XccSJQ-30, XccSJQ-31 and XccSJQ-62 primer sets amplified the 644, 652, 617, and 1,512 bp bands from the BJSJQ20200612 isolate and the isolates: BJTZ20191009, HBXT20190705, CQ20200923, GSNG20191014, SDJN20201102, and JSNJ20191022. However, there were no bands produced by markers (XccSJQ-12, XccSJQ-30 and XccSJQ-31) and a 349 bp product generated by marker XccSJQ-62 from the HBCL20191115, SXXY20191115, GSXT20191014, ZJHZ20191121, and GDGZ20191212 isolates. Likewise, a 510 bp band were specifically amplified by marker XccXTZ-10 and no band was observed by the primer sets XccXTZ-16, XccXTZ-24 and XccXTZ-28 from the GSXT20191014, HBCL20191115 and SXXY20191115 isolates (Figure 7). Taken together, we concluded that our isolated strains could be categorized into three subgroups according to eight isolate-specific markers: isolates ZJHZ20191121 and GDGZ20191212 belong to race 1 (HRIW 3811-like isolates); BJSJQ20200612, BJTZ20191009, HBXT20190705, CQ20200923, GSNG20191014, SDJN20210102, and JSNJ20191022 are classified as subgroup II, and isolates HBCL20191115, SXXY20191115 and GSXT20191014 are considered subgroup III.
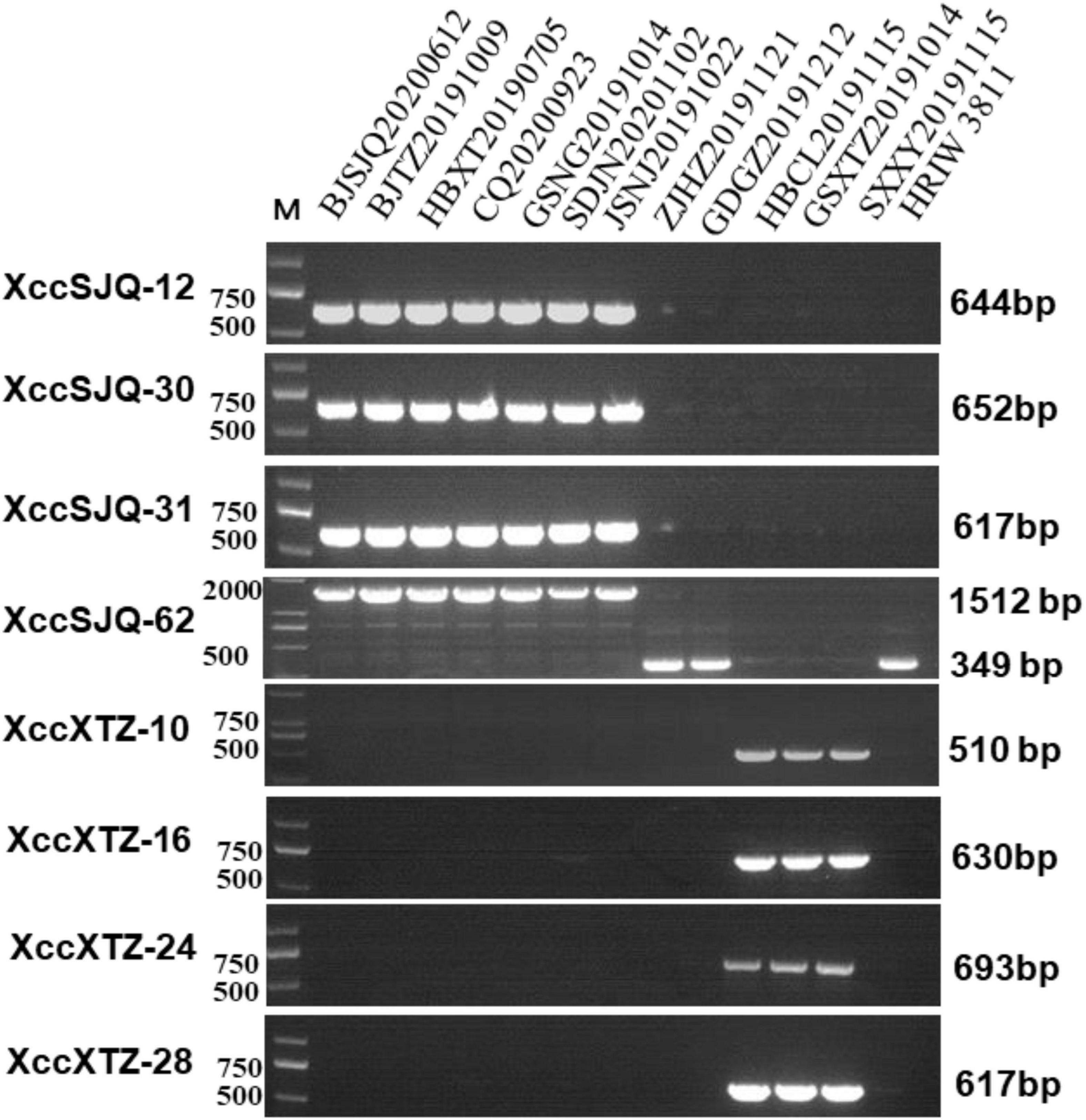
Figure 7. Subgrouping of the unknown Xcc strains by the specific molecular markers. The isolated strains were tested by specific molecular markers XccSJQ-12, XccSJQ-30, XccSJQ-31, XccSJQ-62, XccXTZ-10, XccXTZ-16, XccXTZ-24, and XccXTZ-28 for strain classification. DNA concentration of all samples was 50 ng/μl. Lane M: 2k plus DNA ladder was used as a size marker. Lane1–13: Xcc strains (BJSJQ20200612 and BJTZ20191009, HBXT20190705, CQ20200923, GSNG20191014, SDJN20201102, JSNJ20191022, ZJHZ20191121, GDGZ20191212, HBCL20191115, GSXT20191014, and SXXY20191115, and HRIW 3811, respectively).
In order to analyze the genomic variability of our isolated Xcc bacteria, we performed comparative genomic alignment analysis of BJSJQ20200612 and GSXT20191014 with five available complete genomes of Xcc isolates HRIW 3811, B100, CFBP 1869, ATCC 33913, CFBP 5817, and Xcc 8004 by Mauve software (version 2.4.0). We found 26 variable genomic regions (> 800 bp) in the genomic sequence of BJSJQ20200612 isolate compared to these five published strains. The length of variable genomic regions ranged from 884 bp to 31,905 bp. It should be noted that seven variable genomic regions larger than 5,000 bp, which are located at positions: 255,690–260,986, 1,199,954–1,223,227, 1,214,274–1,223,224, 2,500,995–2,510,117, 3,048,482–3,056,141, 3,074,557–3,080,689, and 3,086,987–3,118,892 bp of the genome sequence (Supplementary Table 5). In addition, 47 variable genomic regions (> 1,000 bp) were found in the genomic sequence of GSXT20191014; the size of the variable genomic regions ranged from 1,077 to 49,749 bp. There were five variable genomic regions larger than 5,000 bp, which are located at positions: 101,594–136,624, 1,244,066–1,252,479, 1,253,110–1,259,066, 2,450,290–2,500,038, and 2,505,237–2,520,734 bp (Supplementary Table 5). The variable genomic regions are rich in transposable elements. The IS3, IS4 and IS5 families are highly represented in both isolates (Supplementary Table 5). Most of these ISs are located near isolate-specific genes, which usually are associated with bacterial genome rearrangements.
A total of 820/830 known or putatively related genes encoding virulence factors were detected in the BJSJQ20200612 and GSXT20191014 genome, respectively (Supplementary Tables 6, 7). Furthermore, there were six and ten virulence factors identified inside the variable genomic regions of BJSJQ20200612 and GSXT20191014 genomes, respectively. These virulence factors, including toxin and surface-associated carbohydrates, such as zona occludens toxin, methyl-accepting chemotaxis protein (encoding by cheD gene), type IV secretion system component VirD4, were found in both isolates. Interestingly, the gene GE002595 (fhaB) encoding filamentous hemagglutinin (FHA) proteins were uniquely identified at the variable genomic regions in the GSXT20191014 genome. GE002595 has function in the two-partner secretion pathway (TPS), which is responsible for bacterial adherence. Meanwhile, it was important to point out that virB1 (GE000326) encoding type IV secretion system attachment mediating protein was also found at the variable genomic regions of the GSXT20191014 genome (Table 2).
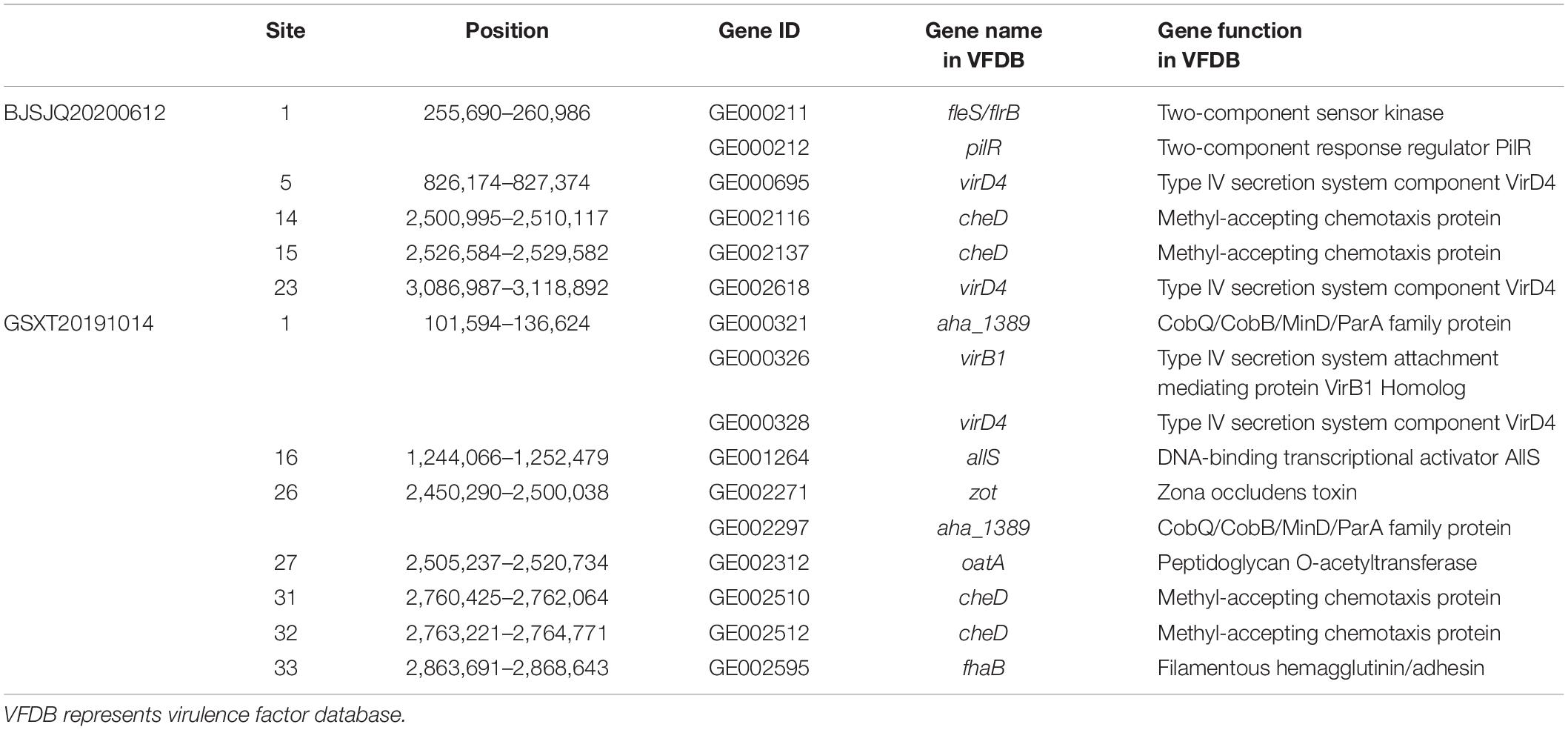
Table 2. The virulence factors in the variable genomic regions of BJSJQ20200612 and GSXT20191014 genomes according to the VFDB database.
A total of 665 and 632 proteins were predicted to be secreted in the BJSJQ20200612 and GSXT20191014 genomes by the SignalP and TMHMM software, respectively (Supplementary Table 8). Furthermore, the secreted proteins were also identified inside the variable genomic regions of BJSJQ20200612 and GSXT20191014. One lytic murein transglycosylase, one beta-propeller fold lactonase family protein and five hypothetical proteins were present in the variable genomic regions of the BJSJQ20200612 genome. Additionally, two copies of peptidase S8/S53 were found at site 1 and 26 of the variable genomic regions of the GSXT20191014 genome (Table 3). The gene GE002595 (fhaB) encoding FHA proteins were identified at the variable genomic regions in the GSXT20191014 genome.
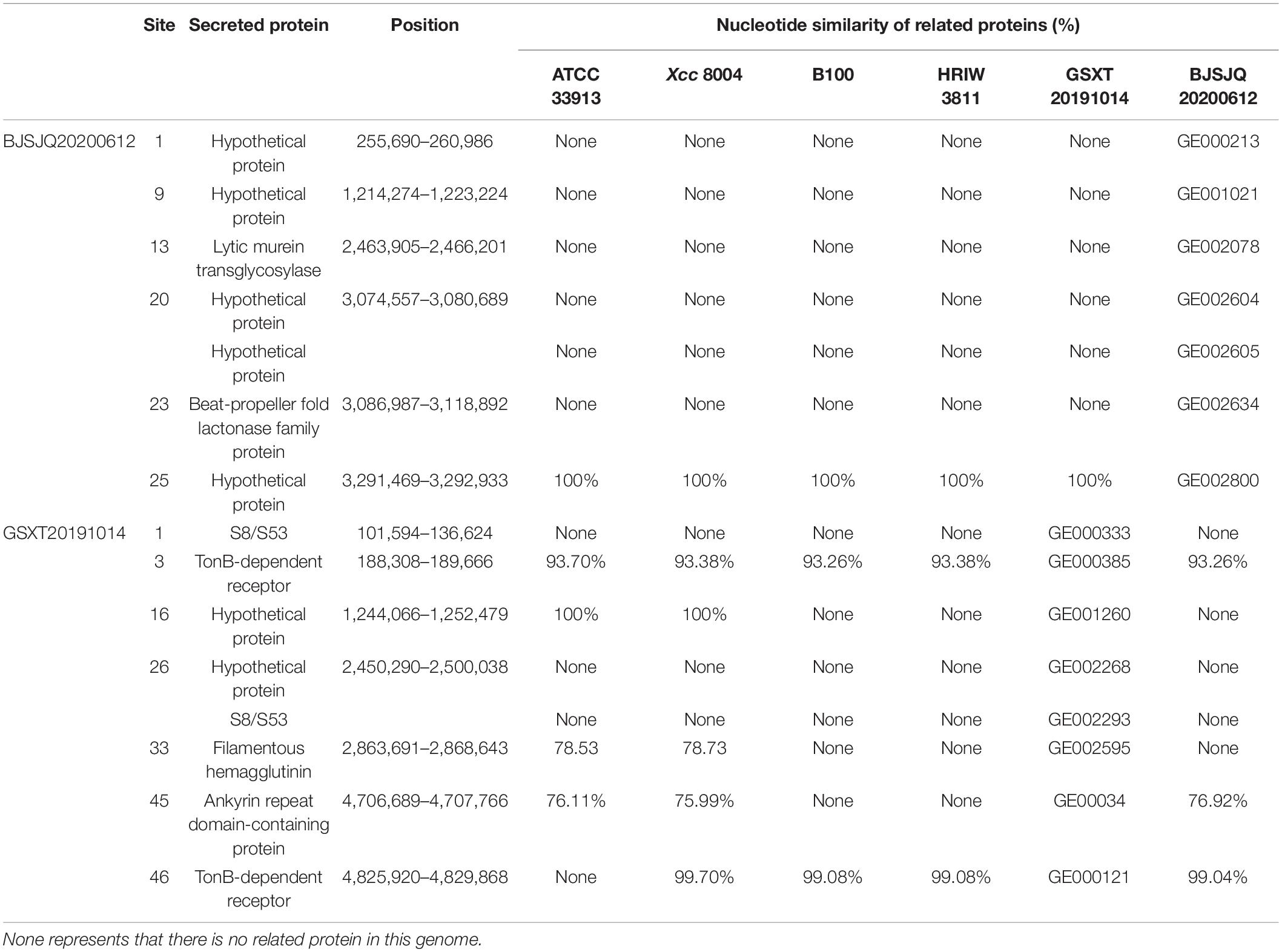
Table 3. The secreted proteins in the variable genomic regions of BJSJQ20200612 and GSXT20191014 genomes.
In Xanthomonas bacteria, the biosynthesis of EPS is regulated by the highly conserved gum gene cluster, which comprises 12 genes from gumB to gumM (Becker et al., 1998). Our results showed that there were few single-nucleotide polymorphisms (SNPs) distinguishing HRIW 3811 and BJSJQ20200612 isolates in these 12 gene sequences. However, more differences in these gene sequences were observed between HRIW 3811 and GSXT20191014 isolates (Figure 8A). LPS production is directly controlled by the wxc gene cluster, which consists of 15 genes (Vorholter et al., 2001). They were conserved between HRIW 3811 and BJSJQ20200612 isolates, only wxcV and wxzm genes showed low similarity, but 10 genes (wxcH, wxcL, wxcM, wxcN, wxcO, rmd, gmd, wxcE wxcD, and wxcC) were identical. However, five genes (wxcN, wxcV, rmd, gmd, and wxcE) in the GSXT20191014 genomic sequence showed less than 85% similarity compared to those in the HRIW 3811 isolate (Figure 8B). However, the differences in production of EPS or LPS need be further studied.

Figure 8. Comparative representation of the organization of Xcc gene clusters directing the synthesis of xanthan and lipopolysaccharide. The Xcc genes involved in the synthesis of xanthan are symbolized by shaded arrows that illustrate the similarities in size, order, and direction of transcription among these genes. Conventional gene names are given above the arrows of homologous genes. (A) Genes gumB to gumM are required for xanthan synthesis. The numbers within the arrows provide the numbers of the nucleotides deviating by SNPs or insertion-deletion events from the BJSJQ20200612 and GSXT20191014 gene sequences compared to the HRIW 3811. (B) The genes wxcH, wxcL, wxcM, wxcN, wxcO, rmd, gmd, wxcE, wxcD, and wxcC are required for lipopolysaccharide synthesis. The numbers within the arrows show the percentage of nucleotide similarity among BJSJQ20200612, GSXT20191014 and HRIW 3811.
The type III secretion system plays an important role in bacterial pathogenicity. BJSJQ20200612 and GSXT20191014 genomes contain a highly conserved type III gene cluster (including hrp and hrc) compared to that in the HRIW 3811 genome, covering 23,112 and 23,115 kb with 25 open reading frames (ORFs), respectively (Supplementary Table 9). However, secreted effectors via type III are different in these two genomes. The type III effectors were predicted and identified using the Xanthomonas resource.4 ATCC 33913 and Xcc 8004 contain 33 type III effector genes. Twenty-seven effector genes were present in the BJSJQ20200612 genome; and 23 effector genes were identified in GSXT20191014 genome. We identified 18 common effectors, these effector genes (avrBs2, xopN, xopQ, xopAY, xopAM, xopF, hpaW, hpaA, xopA, xopAL, xopP, xopZ, avrXccA2, xopAC, xopK, xopAG, xopL, and avrXccA1) are present in all six sequenced Xcc genomes (ATCC 33913, Xcc 8004, B100, HRIW 3811, BJSJQ20200612, and GSXT20191014) (Table 4). These 18 effector genes are possibly the core effector set for Xcc that cause black rot. Additionally, the effector repertoire is highly conserved in the BJSJQ20200612 and HRIW 3811 genomes except for xopJ. It is noteworthy that xopR, xopH, avrBs1, and xopAH are identified in ATCC 33913, Xcc 8004, B100, HRIW 3811, and BJSJQ20200612 genomes, but they are absent in the GSXT20191014 genome (Table 4).
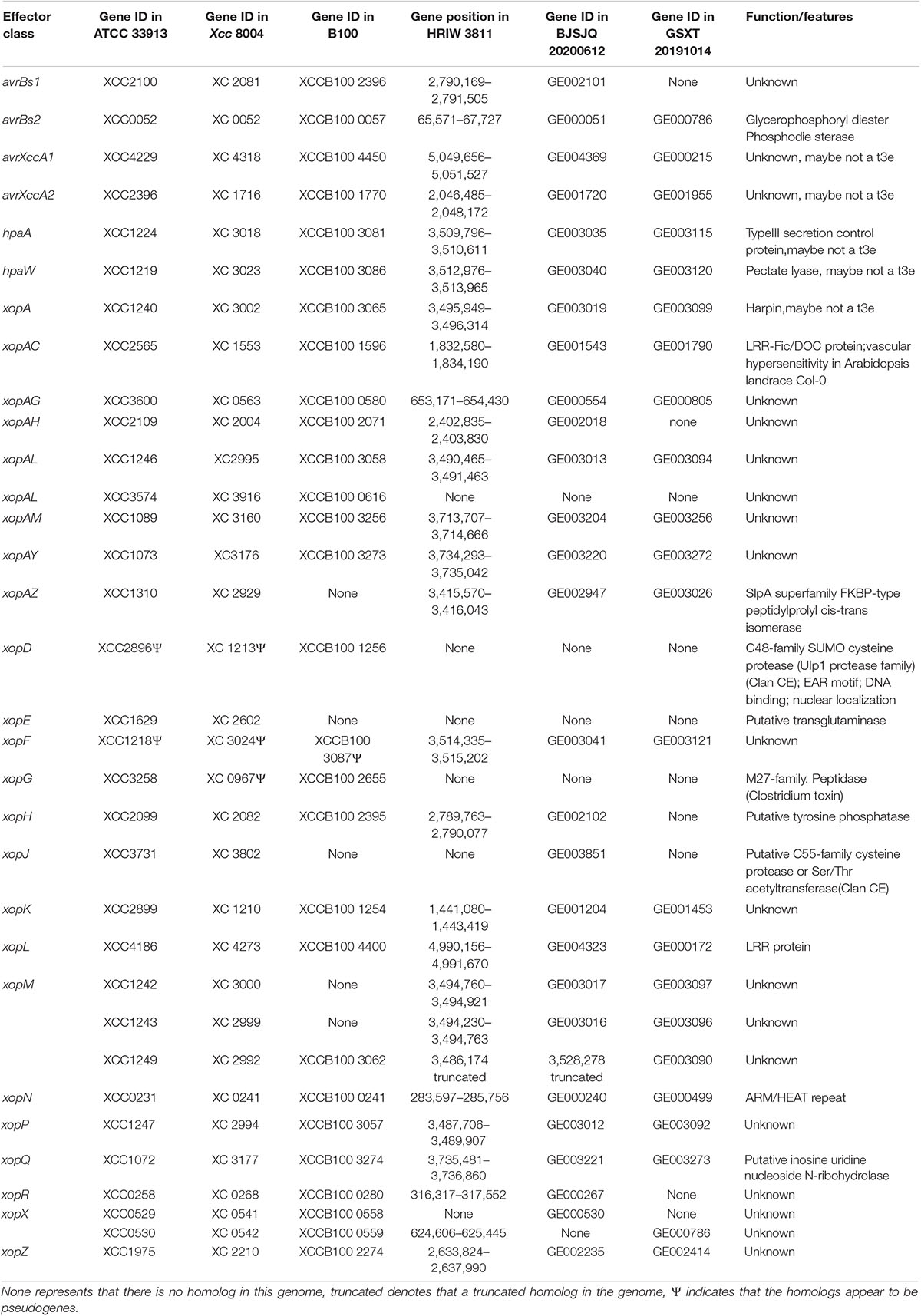
Table 4. Effector repertoire of ATCC 33913, Xcc 8004, B100, HRIW 3811, BJSJQ20200612 and GSXT20191014 genomes.
Taken together, the significant differences in the composition of above mentioned virulence factors may lead to GSXT20191014 isolate infections being more aggressive in cabbage inbred line 1371.
We sequenced the complete genome of BJSJQ20200612 and GSXT20191014 and compared them with HRIW 3811, B100, CFBP 1869, ATCC 33913, CFBP 5817, and Xcc 8004. Our results showed that large-scale genomic rearrangements were identified in both strains. These two genomes are quite different in the number of predicted CDSs, variable genomic regions, virulence factors (adhesion, secreted proteins, type III effectors, and the composition of EPS and LPS) (Tables 1–4 and Figure 8). These differences may contribute to the genomic sequence and virulence variations observed in Xcc strains.
It has been reported that genomic rearrangements of Xanthomonas genomes are often due to IS and transposons, which are responsible for bacterial pathogenicity and evolution (Ferreira et al., 2015). In this study, the pathogenicity of the GSXT20191014 isolate was more virulent than HRIW 3811 in cabbage inbred line 1371 and other brassica crops. By focusing on these variable genomic regions, we found that there were 26 and 47 variable genomic regions in the genomic sequence of BJSJQ2020061 and GSXT20191014, respectively (Supplementary Table 5). Most of the variable genomic regions possess IS family transposases, which are located at each end of insertion sequence and involved in the transposition of the insertion sequence, indicating that these DNA sequences may have been acquired through horizontal gene transfer (Garcia-Vallve et al., 2003; Ou et al., 2006; He et al., 2007). The horizontal gene transfer events that occurred in the genomic sequence of the GSXT20191014 were greater in scale than that in BJSJQ2020061.
Multiple strain-specific proteins were identified through comparative analysis of variable genomic regions in BJSJQ2020061 and GSXT20191014 genomes, especially virulence factors (adhesin, secreted proteins and Type III effectors) that may contribute to the differences in virulence. For example, the gene GE000326 encoding a type IV secretion system attachment mediating protein VirB1 homolog was found in the variable genomic regions of the GSXT20191014 genome (Table 2). VirB1 was reported to promote T-pilus assembly involving the attachment of Agrobacterium tumefaciens (Zupan et al., 2007). Here, GE000326 encoding VirB1 homolog was found in the variable genomic region may increase the attachment and virulence of GSXT20191014. The gene GE002595 (fhaB) encoding FHA proteins were specifically identified at the variable genomic regions of the GSXT20191014 genome. FHA, as the major adhesin of pathogens, is one of the most efficiently secreted proteins by means of the TPS pathway in Gram-negative bacteria, which is responsible for bacterial adherence (Clantin et al., 2004). Furthermore, it is still worth emphasizing that among the GSXT20191014 strain-specific genes, two copies of peptidase S8/S53 were found in the variable genomic regions of the GSXT20191014 genome (Table 3). Secreted proteins LtSP13 from Lasiodiplodia theobromae and Mp1 protein from Magnaporthiopsis poae contain conserved peptidase S8 domain known to be involved in plant pathogenesis (Sreedhar et al., 1999; Li Z. et al., 2019). Hence, it is possible that the secreted peptidase S8/S53 could be involved in strategies for establishing infection in cabbage. It was noteworthy that so many virulence factors with different functions were found at sites 1, 16, 26, and 33 of the variable genome regions in the GSXT20191014 genome (Tables 2, 3), meaning horizontal gene transfer introduced more pathogenic-related proteins in GSXT20191014. Taken together, many insertion events have occurred in the genome of GSXT20191014 during its evolution, which eventually made this segment of genome unique in GSXT20191014 and generated the variability in adhesion, secreted proteins and effector repertoires compared to other Xcc strains. And all these factors could contribute to the high pathogenicity of GSXT20191014.
Previous research showed that Xcc strains exhibit diversity in the physiological races even in the same subspecies (Vicente et al., 2001). The release of Xcc genomic data (Ribeiro da Silva et al., 2002; Qian et al., 2005; Vorhölter et al., 2008; Bolot et al., 2015), has led to the development of molecular markers from DNA polymorphic regions. Nou’s group in Korea have developed specific markers for Xcc strains HRIW 3811, HRIW 3849A, HRIW 5212, HRIW 1279A, and HRIW 3880, according to whole genome alignments, which could work more quickly, efficiently and reliably in the detection of specific Xcc strains (Afrin et al., 2018b,2019; Rubel et al., 2017, 2019b). Here, we also developed the specific molecular makers for BJSJQ20200612 and GSXT20191014, respectively, based on the variable genomic regions (Figure 6). These markers work efficiently in distinguishing the BJSJQ20200612 and GSXT20191014 genetic subgroups from other Xcc strains, revealing that the diversity and distribution of Xcc subgroups in the main cabbages-growing areas of China. The subgrouping results are consistent with that generated by MLST method. In addition, our findings suggest that the isolates from the same subgroups may have many similarities in disease aggressiveness in cabbage inbred line 1371, so accurate identification and recognition of Xcc in cabbages could help breeders rapidly distinguish the Xcc subgroups, aiding effective control measures.
In this study, three genetic subgroups of Xcc were found in our isolates (Figures 3, 5, 7). Notably, we obtained two race 1 isolates (GDGZ20191212 and ZJHZ20191121) from coastal provinces Guangdong and Zhejiang, which are in the south of China. Interestingly, HBCL20191115, SXXY20191115, and GSXT20191014 belonging to the subgroup III exhibited higher virulence than HRIW 3811 in cabbage inbred line 1371 and other brassica crops (Figure 2 and Supplementary Figure 1). Isolates SXXY20191115 and GSXT20191014 were found in the cabbages grown in northwest Shaanxi and Gansu provinces; and another isolate belongs to the same genetic subgroup was obtained from Changli County of the Hebei province. The rest of the isolates belonged to the genetic subgroup II strain, which was the dominant strain that has probably evolved from race 1 (Figure 7). We speculate that the Xcc are rapidly evolving as ISs were identified in the genomic sequences, which usually leads to rearrangement of genomic sequence and generation of more virulent strains.
At last, accurate identification and recognition of Xcc in cabbage could help breeders rapidly distinguish the Xcc subgroups, is a prerequisite in taking effective control measures. Ill-Sup Nou’s group found that the existence of race-specific resistance to Xcc races B100, HRIW 3849A, HRIW 5212, HRIW 3880, HRIW 6181, and HRIW 8450A in the cabbage germplasms (Afrin et al., 2018a). They also identified markers linked to Xcc races HRIW 6181 and HRIW 8450A resistance in cabbage (Hong et al., 2021). It seems that the broad spectrums of disease-resistant cabbage materials were shortage, the subgroup-specific resource could be found. So it was important to know if the isolates were from a group or another.
In conclusion, this is the first extensive analysis of the genetic diversity and distribution of Xcc based on whole genomic variation of Xcc strains in main cabbage growing areas of China, which could provide insight into for black rot-resistance breeding. The most effective way to control black rot wilt in brassica crops is to breed resistant varieties. In this study, our results showed that BJSJQ20200612 and GSXT20191014 genomes vary in the number of predicted CDSs, variable genomic regions, virulence factors (adhesion, secreted proteins, type III effectors, and the composition of EPS and LPS). These differences may contribute to the genomic sequence and virulence variations observed in Xcc strains. Meanwhile, our isolated Xcc strains were classified into three genetic subgroups according to MLST and our developed specific markers. However, it is important to notice that the pathogenicity experiment was done in controlled conditions and if conditions vary, the results might vary as well. In addition, there was only one cabbage inbred line 1371 tested and this might not be the case for all other cabbage lines. In the future, we aim to obtain more isolates to confirm the distribution and the virulence of Xcc in China. The pathogenicity test will be done in more cabbage inbred lines. At last, we aim to use these identified strains to screen and generate Xcc-resistant cabbage cultivars.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
XZ, JX, and JK conceived, designed the experiments, and revised the manuscript. DC, JC, and HL performed the experiments. RH and XY analyzed the data. DC and XZ wrote the manuscript. All the authors have read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (31972408) and the Science and Technology Innovation Capacity Building Projects of the Beijing Academy of Agriculture and Forestry Sciences (KJCX20210425, KJCX20200410, and KJCX20200113).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Beijing Vegetable Research Center, Beijing Academy of Agriculture and Forestry Sciences, Key Laboratory of Biology and Genetic Improvement of Horticultural Crops (North China), Ministry of Agriculture, Beijing, China for their completion of the work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.833318/full#supplementary-material
Afrin, K. S., Rahim, M. A., Jung, H. J., Park, J. I., Kim, H. T., and Nou, I. S. (2019). Development of Molecular Marker through Genome Realignment for Specific Detection of Xanthomonas campestris pv. campestris Race 5, a Pathogen of Black Rot Disease. J. Microbiol. Biotechnol. 29, 785–793. doi: 10.4014/jmb.1901.01050
Afrin, K. S., Rahim, M. A., Park, J.-I., Natarajan, S., Rubel, M., Kim, H., et al. (2018a). Screening of Cabbage (Brassica oleracea L.) Germplasm for Resistance to Black Rot. Plant Breed. Biotechnol. 6, 30–43. doi: 10.9787/pbb.2018.6.1.30
Afrin, K. S., Rahim, M. A., Rubel, M. H., Natarajan, S., Song, J.-Y., Kim, H.-T., et al. (2018b). Development of race-specific molecular marker for Xanthomonas campestris pv. campestris race 3, the causal agent of black rot of crucifers. Canad. J. Plant Sci. 98, 1119–1125. doi: 10.1139/cjps-2018-0035
Becker, A., Katzen, F., Pühler, A., and Ielpi, L. (1998). Xanthan gum biosynthesis and application: a biochemical/genetic perspective. Appl. Microbiol. Biotechnol. 50, 145–152. doi: 10.1007/s002530051269
Bolot, S., Cerutti, A., Carrere, S., Arlat, M., Saux, M. F., Portier, P., et al. (2015). Genome Sequences of the Race 1 and Race 4 Xanthomonas campestris pv. campestris Strains CFBP 1869 and CFBP 5817. Genome Announc. 3, e1023–e1015. doi: 10.1128/genomeA.01023-15
Büttner, D., and Bonas, U. (2010). Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 34, 107–133. doi: 10.1111/j.1574-6976.2009.00192.x
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chen, G., Kong, C., Yang, L., Zhuang, M., Zhang, Y., Wang, Y., et al. (2021). Genetic Diversity and Population Structure of the Xanthomonas campestris pv. campestris Strains Affecting Cabbages in China Revealed by MLST and Rep-PCR Based Genotyping. Plant Pathol. J. 37, 476–488. doi: 10.5423/PPJ.OA.06.2021.0088
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Clantin, B., Hodak, H., Willery, E., Locht, C., Jacob-Dubuisson, F., and Villeret, V. (2004). The crystal structure of filamentous hemagglutinin secretion domain and its implications for the two-partner secretion pathway. Proc. Natl. Acad. Sci. U S A. 101, 6194–6199. doi: 10.1073/pnas.0400291101
Cook, A., Walker, J., and Larson, R. (1952). Studies on the disease cycle of black rot of crucifers. Phytopathology 42:162.
Deamer, D., Akeson, M., and Branton, D. (2016). Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524. doi: 10.1038/nbt.3423
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fargier, E., Fischer-Le Saux, M., and Manceau, C. (2011). A multilocus sequence analysis of Xanthomonas campestris reveals a complex structure within crucifer-attacking pathovars of this species. Syst. Appl. Microbiol. 34, 156–165. doi: 10.1016/j.syapm.2010.09.001
Feng, F., and Zhou, J.-M. (2012). Plant–bacterial pathogen interactions mediated by type III effectors. Curr. Opin. Plant Biol. 15, 469–476. doi: 10.1016/j.pbi.2012.03.004
Ferreira, R. M., de Oliveira, A. C. P., Moreira, L. M., Belasque, J., Gourbeyre, E., Siguier, P., et al. (2015). A TALE of transposition: Tn3-like transposons play a major role in the spread of pathogenicity determinants of Xanthomonas citri and other xanthomonads. mBio 2015, e02505–e02514. doi: 10.1128/mBio.02505-14
Garcia-Vallve, S., Guzman, E., Montero, M. A., and Romeu, A. (2003). HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 31, 187–189. doi: 10.1093/nar/gkg004
Gu, Y., Mao, Y. W., Wang, C. G., Zhao, Q. C., Sun, D. L., and Song, W. Q. (2008). Cloning of differential expression fragments in cauliflower after Xanthomonas campestris inoculation. Biol. Plant. 52, 462–468. doi: 10.1007/s10535-008-0091-7
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559. doi: 10.1093/nar/gki352
He, Y.-Q., Zhang, L., Jiang, B.-L., Zhang, Z.-C., Xu, R.-Q., Tang, D.-J., et al. (2007). Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. campestris. Genome Biol. 8:R218. doi: 10.1186/gb-2007-8-10-r218
Hong, J.-E., Afrin, K., Rahim, M. A., Jung, H.-J., and Nou, I.-S. (2021). Inheritance of Black Rot Resistance and Development of Molecular Marker Linked to Xcc Races 6 and 7 Resistance in Cabbage. Plants 10:1940. doi: 10.3390/plants10091940
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17:239.
Jensen, B. D., Vicente, J. G., Manandhar, H. K., and Roberts, S. J. (2010). Occurrence and Diversity of Xanthomonas campestris pv. campestris in Vegetable Brassica Fields in Nepal. Plant Dis. 94, 298–305. doi: 10.1094/PDIS-94-3-0298
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Laala, S., Cesbron, S., Kerkoud, M., Valentini, F., Bouznad, Z., Jacques, M.-A., et al. (2021). Characterization of Xanthomonas campestris pv. campestris in Algeria. Phytopathol. Mediterr. 60, 51–62. doi: 10.36253/phyto-11726
Li, L., Yuan, L., Shi, Y., Xie, X., Chai, A., Wang, Q., et al. (2019). Comparative genomic analysis of Pectobacterium carotovorum subsp. brasiliense SX309 provides novel insights into its genetic and phenotypic features. BMC Genomics 20:486. doi: 10.1186/s12864-019-5831-x
Li, R. F., Lu, G. T., Li, L., Su, H. Z., Feng, G. F., Chen, Y., et al. (2014). Identification of a putative cognate sensor kinase for the two-component response regulator HrpG, a key regulator controlling the expression of the hrp genes in Xanthomonas campestris pv. campestris. Environ. Microbiol. 16, 2053–2071. doi: 10.1111/1462-2920.12207
Li, Z., Zhang, H., and Li, G. (2019). Characterization of phytotoxin and secreted proteins identifies of Lasiodiplodia theobromae, causes of peach gummosis. Fungal Biol. 123, 51–58. doi: 10.1016/j.funbio.2018.11.001
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Magi, A., Semeraro, R., Mingrino, A., Giusti, B., and D’Aurizio, R. (2018). Nanopore sequencing data analysis: state of the art, applications and challenges. Briefings Bioinformatics 19, 1256–1272. doi: 10.1093/bib/bbx062
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ou, H. Y., Chen, L. L., Lonnen, J., Chaudhuri, R. R., Thani, A. B., Smith, R., et al. (2006). A novel strategy for the identification of genomic islands by comparative analysis of the contents and contexts of tRNA sites in closely related bacteria. Nucleic Acids Res. 34:e3. doi: 10.1093/nar/gnj005
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Qian, W., Jia, Y., Ren, S., He, Y., Feng, J., Lu, L., et al. (2005). Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15, 757–767. doi: 10.1101/gr.3378705
Ribeiro da Silva, A., Ferro, J., Reinach, F., Farah, C., Furlan, L., et al. (2002). Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417, 459–463. doi: 10.1038/417459a
Rubel, M., Natarajan, S., Hossain, M., Nath, U., Afrin, K., Lee, J., et al. (2019a). Pathovar specific molecular detection of Xanthomonas campestris pv. campestris, the causal agent of black rot disease in cabbage. Canad. J. Plant Pathol. 41, 318–328. doi: 10.1128/mBio.00538-12
Rubel, M, Natarajan, S., Nath, U. K., Denison, M. I. J., Hee-Jeong, J., Hoy-Taek, K., et al. (2019b). Development of a marker for detection of Xanthomonas campestris pv. campestris races 1 and 2 in Brassica oleracea. Horticult. Environ. Biotechnol. 60, 511–517. doi: 10.1007/s13580-019-00143-7
Rubel, M. H., Robin, A. H. K., Natarajan, S., Vicente, J. G., Kim, H. T., Park, J. I., et al. (2017). Whole-Genome Re-Alignment Facilitates Development of Specific Molecular Markers for Races 1 and 4 of Xanthomonas campestris pv. campestris, the Cause of Black Rot Disease in Brassica oleracea. Int. J. Mol. Sci. 18:2523. doi: 10.3390/ijms18122523
Schaad, N. W. (1981). Cruciferous weeds as sources of inoculum of Xanthomonas campestris in black rot of crucifers. Phytopathology 71, 1215–1220. doi: 10.1111/j.1364-3703.2012.00833.x
Sreedhar, L., Kobayashi, D. Y., Bunting, T. E., Hillman, B. I., and Belanger, F. C. (1999). Fungal proteinase expression in the interaction of the plant pathogen Magnaporthe poae with its host. Gene 235, 121–129. doi: 10.1016/s0378-1119(99)00201-2
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Vicente, J. G., Conway, J., Roberts, S. J., and Taylor, J. D. (2001). Identification and Origin of Xanthomonas campestris pv. campestris Races and Related Pathovars. Phytopathology 91, 492–499. doi: 10.1094/PHYTO.2001.91.5.492
Vorholter, F. J., Niehaus, K., and Puhler, A. (2001). Lipopolysaccharide biosynthesis in Xanthomonas campestris pv. campestris: a cluster of 15 genes is involved in the biosynthesis of the LPS O-antigen and the LPS core. Mol. Genet. Genomics 266, 79–95. doi: 10.1007/s004380100521
Vorhölter, F.-J., Schneiker, S., Goesmann, A., Krause, L., Bekel, T., Kaiser, O., et al. (2008). The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J. Biotechnol. 134, 33–45. doi: 10.1016/j.jbiotec.2007.12.013
Weber, E., Berger, C., Bonas, U., and Koebnik, R. (2007). Refinement of the Xanthomonas campestris pv. vesicatoria hrpD and hrpE Operon Structure. Mol. Plant Microbe Interact. MPMI 20, 559–567. doi: 10.1094/MPMI-20-5-0559
Keywords: brassica oleracea, black rot, virulence, genomic comparisons, Xanthomonas campestris pv. campestris, IS family transposase, whole-genome sequencing, effectors
Citation: Chen D, Zhong X, Cui J, Li H, Han R, Yue X, Xie J and Kang J (2022) Comparative Genomic Analysis of Xanthomonas campestris pv. campestris Isolates BJSJQ20200612 and GSXT20191014 Provides Novel Insights Into Their Genetic Variability and Virulence. Front. Microbiol. 13:833318. doi: 10.3389/fmicb.2022.833318
Received: 11 December 2021; Accepted: 26 January 2022;
Published: 02 March 2022.
Edited by:
Marie-Agnès Jacques, INRA Centre Angers-Nantes Pays de la Loire, FranceReviewed by:
Alexander N. Ignatov, Peoples’ Friendship University of Russia, RussiaCopyright © 2022 Chen, Zhong, Cui, Li, Han, Yue, Xie and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xionghui Zhong, emhvbmd4aW9uZ2h1aUBuZXJjdi5vcmc=; Jianming Xie, eGllamlhbm1pbmdnc0AxMjYuY29t; Jungen Kang, a2FuZ2p1bmdlbkBuZXJjdi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.