 Muhammad Ali
Muhammad Ali Tong Gu
Tong Gu Xun Yu
Xun Yu Anum Bashir
Anum Bashir Zhiyong Wang
Zhiyong Wang Xiaowen Sun
Xiaowen Sun Naeem Mahmood Ashraf
Naeem Mahmood Ashraf Lin Li
Lin Li- 1State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan, China
- 2Department of Biotechnology, COMSATS University Islamabad, Abbottabad, Pakistan
- 3Department of Biochemistry and Biotechnology, University of Gujrat, Gujrat, Pakistan
Nematicidal potential of the common plant pathogen Pseudomonas syringae has been recently identified against Caenorhabditis elegans. The current study was designed to investigate the detailed genetic mechanism of the bacterial pathogenicity by applying comparative genomics, transcriptomics, mutant library screening, and protein expression. Results showed that P. syringae strain MB03 could kill C. elegans in the liquid assay by gut colonization. The genome of P. syringae MB03 was sequenced and comparative analysis including multi locus sequence typing, and genome-to-genome distance placed MB03 in phylogroup II of P. syringae. Furthermore, comparative genomics of MB03 with nematicidal strains of Pseudomonas aeruginosa (PAO1 and PA14) predicted 115 potential virulence factors in MB03. However, genes for previously reported nematicidal metabolites, such as phenazine, pyochelin, and pyrrolnitrin, were found absent in the MB03 genome. Transcriptomics analysis showed that the growth phase of the pathogen considerably affected the expression of virulence factors, as genes for the flagellum, glutamate ABC transporter, phoP/phoQ, fleS/fleR, type VI secretion system, and serralysin were highly up-regulated when stationary phase MB03 cells interacted with C. elegans. Additionally, screening of a transposon insertion mutant library led to the identification of other nematicidal genes such as acnA, gltP, oprD, and zapE. Finally, the nematicidal activity of selected proteins was confirmed by heterologous expression in Escherichia coli.
Introduction
Caenorhabditis elegans has been widely utilized to study host–pathogen interaction. Pathogenicity mechanisms of numerous bacterial species of Bacillus (Geng et al., 2016), Burkholderia (Lee et al., 2011; Paiva et al., 2013), Pseudomonas (Dubern et al., 2015; Nandi et al., 2015; Ali et al., 2016), Salmonella (Alegado et al., 2011), Staphylococcus (Begun et al., 2005; Irazoqui et al., 2010) and Yersinia (Darby et al., 2002; Atkinson et al., 2011) have been reported against C. elegans. Bacterial species have acquired different mechanisms to kill C. elegans such as slow killing due to infection and colonization (Ali et al., 2016), biofilm formation (Darby et al., 2002; Atkinson et al., 2011), fast killing due to diffusible metabolites (Kirienko et al., 2015; Nandi et al., 2015), and secretion of proteins (Luo et al., 2012; Geng et al., 2016; Zhang et al., 2016). Several species of the genus Pseudomonas, including P. aeruginosa, P. chlororaphis, P. fluorescens, P. putida, and P. protegens have been reported for their pathogenicity against animals (Feinbaum et al., 2012; Burlinson et al., 2013; Fernandez et al., 2015; Wei et al., 2017). Among these species, P. aeruginosa PA14 has been reported as multi-host pathogen, capable of infecting nematodes, Drosophila melanogaster, and plants (Plotnikova et al., 2000; Kim et al., 2008). The interaction of P. aeruginosa with C. elegans has been well-characterized (Cezairliyan et al., 2013; Kirienko et al., 2013). The killing of C. elegans by the strains of P. aeruginosa has been attributed due to various mechanisms such as lethal paralysis, agar-based fast killing, liquid killing, and red death, along with gut colonization (Darby et al., 1999; Mahajan-Miklos et al., 1999; Tan et al., 1999b; Gallagher and Manoil, 2001; Zaborin et al., 2009; Cezairliyan et al., 2013; Kirienko et al., 2013, 2015; Ray et al., 2015). It has been established that change in the physical form of the killing assay (from agar-based killing to liquid-based killing) altered the killing mechanism; for instance, agar-based killing was mediated by the phenazines (Cezairliyan et al., 2013) and gut colonization (Tan et al., 1999a), whereas liquid killing was facilitated by pyoverdine (Kirienko et al., 2013).
Pseudomonas syringae is an important plant pathogen that causes foliar necroses in host plants and a hypersensitive reaction in non-hosts. Depending upon host range and host-pathogen interaction, this bacterial species was sub-divided into 50 pathovars (Hirano and Upper, 2000). More recently the species has been further divided into 60 pathovars (Gomila et al., 2017) and 13 phylogroups (Berge et al., 2014). The strains of P. syringae have been isolated from various agricultural and other environmental sites preferably in proximity to water cycle (Morris et al., 2007, 2008). Among 13 phylogroups, most of the strains in the phylogroup I were found from the damaged tissues of the plants. The phylogroup I also contained strains isolated from some other environmental samples (non-agricultural). The phyogroup II was the most diverse group as it contained strains from all known habitats (Berge et al., 2014). The released genomes of the different P. syringae pathovars, such as P. syringae pv. syringae B728a (Feil et al., 2005), P. syringae pv. tomato DC3000 (Buell et al., 2003), P. syringae CC1557 (Hockett et al., 2014), and P. syringae pv. syringae HS191 (Ravindran et al., 2015), have shown notable variations in terms of number and function of genes related to the bacterial pathogenicity. These strains not only have the common virulence factors but also have host-specific ones (O’Brien et al., 2011).
Pseudomonas syringae has been generally annotated as the “most notorious plant pathogen” (Mansfield et al., 2012), however, little information is available regarding its interaction with C. elegans (Ali et al., 2016; Dorati et al., 2018; Manan et al., 2018; Bashir et al., 2020). Previous attempts to investigate its pathogenic potential against C. elegans showed that the strain B728a and DC3000 did not possess nematicidal activity on NGM medium (Burlinson et al., 2013). The assay conditions are among the major factors that shape the outcome of the host–pathogen interaction (Burlinson et al., 2013). On these grounds, the pathogenicity of P. syringae MB03 against C. elegans was re-investigated under various assay conditions, and an obvious shift in the bacterial behavior was observed (from non-pathogenic to pathogenic) (Ali et al., 2016). Moreover, we have previously investigated the pathogenic behavior of P. syringae MB03 under nutrient-rich conditions and found that on PG medium, P. syringae MB03 could colonize the gut of C. elegans (Ali et al., 2016). In contrast, the killing of C. elegans by P. aeruginosa on PG medium (Cezairliyan et al., 2013) and in a liquid assay (Kirienko et al., 2013) was mediated by toxin secretion, rather than by gut colonization. Recently, Nif3-family protein from P. syringae MB03 was predicted as nematicidal factor and the purified protein was able to kill C. elegans and Meloidogyne incognita (Manan et al., 2018). These observations raised the possibility that P. syringae MB03 might be capable of killing nematodes because of multifactor mechanism.
This study was designed to investigate the key virulence factors of the P. syringae MB03 required for pathogenicity against C. elegans in the liquid killing assay. Comparative genomics, transcriptomics, and transposon insertion mutant library screening were performed to predict and identify key virulence factors. Further, the selected nematicidal genes were heterologously expressed in Escherichia coli to evaluate their toxicity. Collectively from these experiments, various species-common and strain-specific novel virulence factors belonging to P. syringae were identified.
Materials and Methods
Strains and Growth Conditions
A previously isolated laboratory isolate P. syringae MB03 (Li et al., 2012) was used to study bacterial pathogenicity against C. elegans. This strain has been deposited to the China Culture Collection with accession number CCTCC M2014114. P. syringae MB03 was cultured at 28°C and E. coli (DH5α, BL21, TOP10) strains were cultured at 37°C for routine growth. The C. elegans wild-type N2 (Bristol) was used as a model host in all bioassays. C. elegans N2 wildtype was obtained from Caenorhabditis Genetics Center College of Biological Sciences, University of Minnesota, United States. The worms were maintained on NGM medium having E. coli OP50 as food source at 20°C (Stiernagle, 2006). Synchronization of the population of worms was performed as reported previously (Stiernagle, 2006). The synchronized population of L4 larval stage worms was used in all assays.
Nematode Killing Assay
Pseudomonas syringae MB03 was grown overnight in LB medium. Log phase bacterial cells were washed with M9 buffer and appropriate dilutions were made in S medium (Stiernagle, 2006). The assay was carried out in a 96 well plate. Each well contained 150 μl of bacterial cell suspension, 5 μl of 8 mM 5-fluorodeoxyuridine (FUdR, 0.2 mM final concentration), 40 μl S medium, and 30–40 C. elegans wild-type L4 stage.
Plant Infection Assay
Fresh seedlings of wheat plants were used in the infection assay as previously reported (Uppalapati et al., 2008). Briefly, the bacterial strain was grown overnight in LB medium, and log-phase cells were harvested. Wheat plants were grown under optimum environmental conditions and healthy leaves of approximately similar size were selected for infection assay. Leaves were surface sterilized with 1% sodium hypochlorite solution. After rinsing with sterilized water, leaves were dipped into 100 ml of the bacterial suspension of P. syringae MB03 with a cell density of 0.05OD600. Leaves were placed on wet plates and sealed to avoid contamination. For the determination of infection, leaves were regularly observed for the appearance of lesions.
Gut Colonization
To express red fluorescent protein (RFP), the plasmid pMCh-23 (Berry et al., 2012) was introduced into P. syringae MB03. Fluorescent protein-expressing P. syringae cells were grown overnight under optimum conditions, repeatedly washed with S medium and appropriate cell dilutions were prepared. L4 synchronized worms were washed with M9 buffer. The assay was performed in a 96 well plate, and each well contained 150 μl of RFP expressing P. syringae cell suspension, 5 μl of 8 mM 5-fluorodeoxyuridine (FUdR, 0.2-mM final concentration), 40 μl S medium, and 30–40 L4 worms.
Genome Sequencing
To increase the understanding of the virulence mechanism of this bacterial strain, whole-genome shotgun sequencing of P. syringae MB03 was performed using Illumina technology. Quality trimming of 150-nucleotide (nt) paired-end reads was produced from a 500-bp genomic library (5.7 Mb, 175-fold coverage). For de novo assembly of the genome, SOAPdenovo software was used. Open reading frame calling and annotation was performed by using Glimmer software (Delcher et al., 2007). Blastall software was used to assign putative roles to proteins. The KEGG automatic annotation server (KAAS) was used to determine metabolic pathways (Kanehisa et al., 2016). The analysis generated 76 contigs, where the largest contig size was 549643 bp. The genome sequence was deposited at DDBJ/EMBL/GenBank under accession LAGV00000000. The version described in this article is version LAGV01000000. The genome sequence was also deposited to Integrated Microbial Genome (Genome submission ID 60303).
Phylogeny and Comparative Genomics
Multi loci sequence analysis (MLSA) using seven housekeeping genes including gyrB, gapA, fruK, pgi, rpoD, anB, and gltA was performed to further classify P. syringae MB03 as reported previously (Sarkar and Guttman, 2004). The P. syringae MB03 genome was compared with other strains of P. syringae with the help of Mauve (Darling et al., 2004). GC skew was generated by submitting the draft genome of P. syringae MB03 to the CGView Server (Grant and Stothard, 2008). To distribute genes of P. syringae MB03 into auxiliary and core genome, the genome of P. syringae MB03 was compared with the previously determined core genome of P. syringae species (Baltrus et al., 2011). For this purpose, in silico genome subtraction was performed using mGenomeSubtractor with default settings (H value > 0.64) (Shao et al., 2010). The EDGAR online tool was used to determine the strain-specific genes (Blom et al., 2009). In addition, the amino acid identity matrix and genome-to-genome distance were also generated at EDGAR. Genomic islands were predicted by IslandViewer3 (Dhillon et al., 2013). Among the genome sequences used in this study for the comparison with P. syringae MB03, complete genome sequences of strains P. syringae B728a, CC1557, DC3000, HS191 are available at NCBI. In case of strains B64 and SM, only draft genome sequences are available at NCBI.
Pathogenomic Analysis
To identify nematicidal proteins and virulence factors, protein databases were searched for nematicidal proteins, and their homologs were determined in P. syringae MB03 genome. Previously, the nematicidal proteins of P. aeruginosa strain PA14 and strain PAO1 have been identified by screening the transposon insertion mutant libraries (Feinbaum et al., 2012; Dubern et al., 2015). The protein sequences of these virulence factors were used to identify their homologs in P. syringae MB03. Moreover, amino acid sequences of virulence factors of P. syringae pv. syringae B728a, P. syringae pv. tomato DC3000, P. aeruginosa PA14 and P. aeruginosa PA01 were obtained from the virulence factor database (VFDB) (Chen et al., 2012) and their homologs were also identified in P. syringae MB03 by applying BLASTp. Sequences of type III secretion system effectors were obtained from Pseudomonas syringae Genome Resources Home Page1 and again BLASTp was used to identify the presence of genes of effector proteins in the genome P. syringae MB03. To investigate insertion or deletion of genomic sequences, the mauve suite was used and the genome of P. syringae MB03 was compared with the previously reported genomes of P. syringae pv. syringae B728a, P. syringae pv. syringae HS191, P. syringae pv. syringae B64, and P. syringae pv. syringae SM.
Bioinformatics Analysis of Hypothetical Proteins and Transcriptional Regulators
The sequences of selected hypothetical proteins of P. syringae MB03 were retrieved from the NCBI database. Physiochemical properties, including molecular weight, aliphatic index, stability, and isoelectric points of hypothetical proteins, were identified by ProtParam server (Gasteiger et al., 2005). To explore the sub-cellular localization of hypothetical proteins, three different servers SignalP 4.1 (Petersen et al., 2011), CELLO (Yu et al., 2004), and TMHMM 2.0 (Krogh et al., 2001) were used. SignalP 4.1 is designed to identify a leader sequence at the N-terminus of a protein to provide an idea about the localization of the protein in the cell. Similarly, CELLO identifies the localization of proteins by support vector mechanics (SVM) based on the n-peptide composition. Conversely, TMHMM predicts the localization of the protein in the membrane by determining the transmembrane helical structures in the protein. Virulence of hypothetical proteins was predicted using the VICMpred server (Saha and Raghava, 2006). HPIDB 2.0 (Ammari et al., 2016) was used to predict hypothetical proteins, which are potentially involved in host-pathogen interaction. HPIDB is a database of experimentally characterized proteins, which are involved in host–pathogen interactions. Sequence homology-based search in HPIDB was used to predict proteins involved in host–pathogen interaction. To elucidate the 3D structures of hypothetical proteins suspected to be involved in host-pathogen interaction, I-TASSER (Yang et al., 2015) server was employed. Predicted 3D models for each protein by the server were further sorted based on the C-score and refined using the 3Drefine algorithm (Bhattacharya et al., 2016). Finally, transcription regulator binding sites were predicted by using CollecTF (Kılıç et al., 2016) using a homology based method, and their target genes were predicted.
RNA Sequencing and Transcriptional Analysis
RNA sequencing of the P. syringae MB03 cells was performed to determine the change in the bacterial gene expression during the host–pathogen interactions. Interaction of host-pathogen was studied during two different growth phases of bacterial culture [(I) exponential phase and (II) stationary phase]. For this purpose, a single pure colony of P. syringae MB03 was cultured in LB broth. The overnight culture was used to further inoculate two different groups (namely, exponential phase group and stationary phase group) which were incubated at 28°C with shaking at 160 rpm for 12 h. Bacterial cells were harvested by centrifugation and washed repeatedly with S buffer. Finally, pre-fasting L4 C. elegans were exposed to bacterial cells in NGM broth for 5 h. After a specified time of interaction, worms were removed by centrifugation and washed repeatedly to obtain bacterial cells. This resulted in the fabrication of the exponential phase group. The pellets of bacterial cells were used to extract total RNA (P. syringae MB03 exposed to C. elegans as treatment and P. syringae MB03 incubated without C. elegans as control). For the stationary phase group, the entire sample treatment was the same, except the bacterial cells were grown for 24 h so that the cells might enter the stationary phase of growth. After 24 h growth, bacterial cells were exposed to pre-fasting L4 worms for 5 h. Worms were removed after a specified time interval. The bacterial cell pellet was obtained by repeated washing of worms. A bacterial cell pallet was used to isolate total RNA from the stationary phase bacterial cells for transcriptomics (P. syringae MB03 exposed to C. elegans as treatment and P. syringae MB03 incubated without C. elegans as control).
The overall methodology for transcriptomics was followed as described previously (Abbasi et al., 2021). Cellular RNA was isolated by Trizol method (Sigma-Aldrich, St. Louis, MO, United States). RNA samples were run on agarose gel for qualitative analysis. Further Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States) was used to quantify RNA samples and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States) was used to check integrity of the samples. Before library construction, rRNA was removed using Illumina rRNA depletion kit. Random hexamers were used for the preparation of cDNA. After RNA end repair and tail addition, sequencing adapters were connected. PCR enrichment was performed to obtain final cDNA library. Finally, HiSeq/MiSeq sequencing was performed. Clean reads were obtained by removing reads containing adapter sequence and low-quality reads. Clean reads were mapped with the genome sequence of P. syringae MB03 and with reference genomes including P. syringae B728a, P. syringae DC3000 using Bowtie 2 (Langmead and Salzberg, 2012). The enrichment analysis of GO and KEGG pathways was performed by using webserver for gene ontology (GO) and KEGG database.
Construction and Screening of the Transposon Insertion Mutant Library
For the construction of transposon insertion mutant library, donor E. coli MB266 (S17-λpir harboring pUT mini-Tn5 Km2) and recipient cells (P. syringae MB03) were grown overnight in LB at 37 and 28°C, respectively. One ml from each culture was subjected to centrifugation. Bacterial pellets of donor cells and recipient cells were washed and re-suspended in 1ml of LB. One hundred μl of each re-suspended culture was mixed and spread on LB agar plate. After 24 h of incubation, bacterial cells were scraped from the agar and suspended in 1 ml phosphate-buffered saline (PBS). Appropriate dilutions of the bacterial growth were spread on MG agar plate (to inhibit the growth of donor strains) supplemented with 50 mg/ml kanamycin (to select the plasmid transformed cells of P. syringae MB03) and incubated at 28°C for 48 h.
Screening of the Transposon Mutant Library for the Loss of Virulence
For the screening of P. syringae MB03 attenuated mutants, the liquid killing assay was conducted. Briefly, mutants of P. syringae MB03 were grown for 24 h at 28°C. The cells were washed and diluted in S medium supplemented with 5 μg/ml cholesterol. Cell density was adjusted at 0.6OD600 and 150 μL of cell suspension was added into the assay. Worms were synchronized and 30–50 L4 synchronized worms were added in the assay as described previously (Powell and Ausubel, 2008). For the inhibition of egg lying, 5-fluorodeoxyuridine (FUdR) was added at a concentration of 50 μg/ml (Meisel et al., 2014). The assay was set in a 96 well-plate incubated at 25°C. Mutants were screened based upon their pathogenicity against worms compared with that of the wild-type P. syringae MB03. Genes harboring transposons were identified by HiTAIL PCR, sequencing and the subsequent DNA alignment revealed the insertion position of the transposon.
Heterologous Expression of Candidate Nematicidal Genes
The genes of potential nematicidal proteins were selected from the results of three different approaches; (1) de novo RNA sequencing, (2) comparative genomics, and (3) prediction based upon an online web tool, VirulentPred (Garg and Gupta, 2008). Selected genes were amplified through PCR and were inserted into the pTrcHis series (Invitrogen, Waltham, MA, United States). Newly prepared vectors were cloned into E. coli DH5α. The recombinant strains were grown over LB agar containing ampicillin as selection marker. Further verification of cloned genes was performed by PCR and gene sequencing. After final verification, vectors were cloned into E. coli strain TOP10 or E. coli strain JM109 for protein expression. The His-tag labeled proteins were induced by adding IPTG to the bacterial growth medium, and the proteins were purified with a Ni-NTA affinity column. After dialysis, SDS PAGE was run for the quantitative analysis of proteins and Bradford assay was used for the quantification of proteins (Bradford, 1976). Recombinant cells and purified proteins were used to determine nematicidal activities.
Results
Pathogenicity of Pseudomonas syringae MB03 Against Caenorhabditis elegans
The killing assay showed that the P. syringae MB03 could kill C. elegans in the liquid assay. The percent mortality increased with an increase in cell inoculum (Figure 1A). Interestingly, P. syringae MB03 was found to be capable of gut colonization in this liquid-based killing assay. An obvious distention in the anterior and posterior parts of the gut of worms was observed due to bacterial colonization (Figure 1B). Previously, it was reported that gut colonization of C. elegans by P. aeruginosa was observed during agar-based slow killing (Tan et al., 1999a) whereas liquid killing was not mediated by gut colonization (Kirienko et al., 2013).
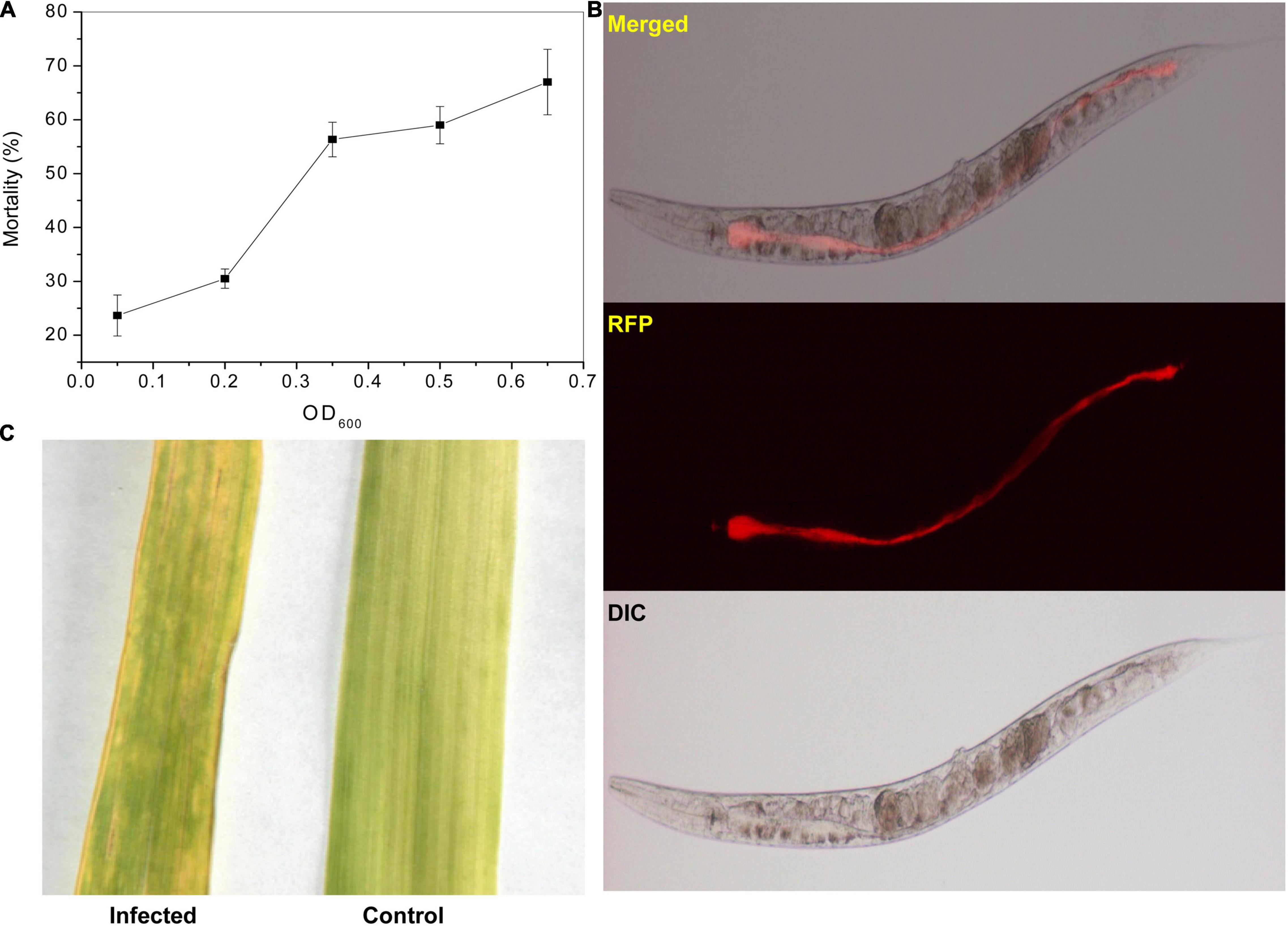
Figure 1. Pathogenicity of P. syringae MB03 against C. elegans and wheat leaves. The impact of the initial cell density of MB03 on killing C. elegans is shown in figure. (A) Assay was performed in a 96 well plate. L4 synchronized worms (30–40) were placed in each assay well. The fraction of dead worms was determined after 72 h. The worms were considered dead if no response was shown to touch. Figure shows increase in worm mortality with an increase in initial cell density of P. syringae MB03. (B) Gut colonization of C. elegans by P. syringae MB03 is shown. The red fluorescence protein (RFP) expressing vector was cloned into P. syringae MB03. The worms were fed on RFP expressing P. syringae MB03 and worms were investigated after 48 h to observe distention of anterior and posterior parts of the gut. The red fluorescence is depicting colonization of the host gut by bacterial cells of P. syringae MB03 harboring pMCh-23 plasmid. (C) Infection of the wheat leaf by P. syringae MB03. The bacterial strain was grown overnight, and a cell suspension made in MgSO4 was used to infect leaves. Deterioration in the leaf infected with P. syringae MB03 is obvious.
Comparative Genomics for the Identification of Unique Features of Pseudomonas syringae MB03
Genomic Organization of Pseudomonas syringae MB03 Is Highly Similar to the Strains of Phylogroup II
The de novo gene prediction showed that the 5.77 Mb genome of P. syringae MB03 contains 5,026 genes with an average gene length of 930 bp. The general features of the P. syringae MB03 genome are described in Supplementary Tables 1, 2. Previously, strains of P. syringae have been classified into 13 different phylogroups based on multi-locus sequence typing (Berge et al., 2014). All the strains within the same phylogroup showed less than 5% genetic distance (Berge et al., 2014). Results of “Multi Locus Sequence Analysis” (MLSA) showed a common ancestry among strains MB03, DSM50255, and B64. Based on the results of MLSA, MB03 was placed in P. syringae phylogroup II (Figure 2A). The strains of phylogroup II, contained very few Type three secretion system genes and were found very active for the ice nucleation damage on plants (Berge et al., 2014). We have previously reported ice nucleation activity of P. syringae MB03 (Li et al., 2012). Moreover, genome-to-genome distance (Figure 2B) and an amino acid identity matrix (AAI) were generated (Figure 2C) to further deepen the phylogenetics of MB03. The homologs of MB03 showed the highest AAI to the strains B64 and DSM50255 followed by SM. The results of genomic distance analysis coincided with the results of MLSA and AAI. The strains of phylogroup II such as B64 and SM are known for their potential to infect monocot plants, such as wheat (Dudnik and Dudler, 2013a). Therefore, it was hypothesized that MB03 would be able to infect wheat because it had been predicted as a member of phylogroup II. The pathogenicity of MB03 in wheat was assessed, and the strain could infect the wheat leaf (Figure 1C). Based on these results, MB03 was placed in P. syringae phylogroup II (Figure 2A).
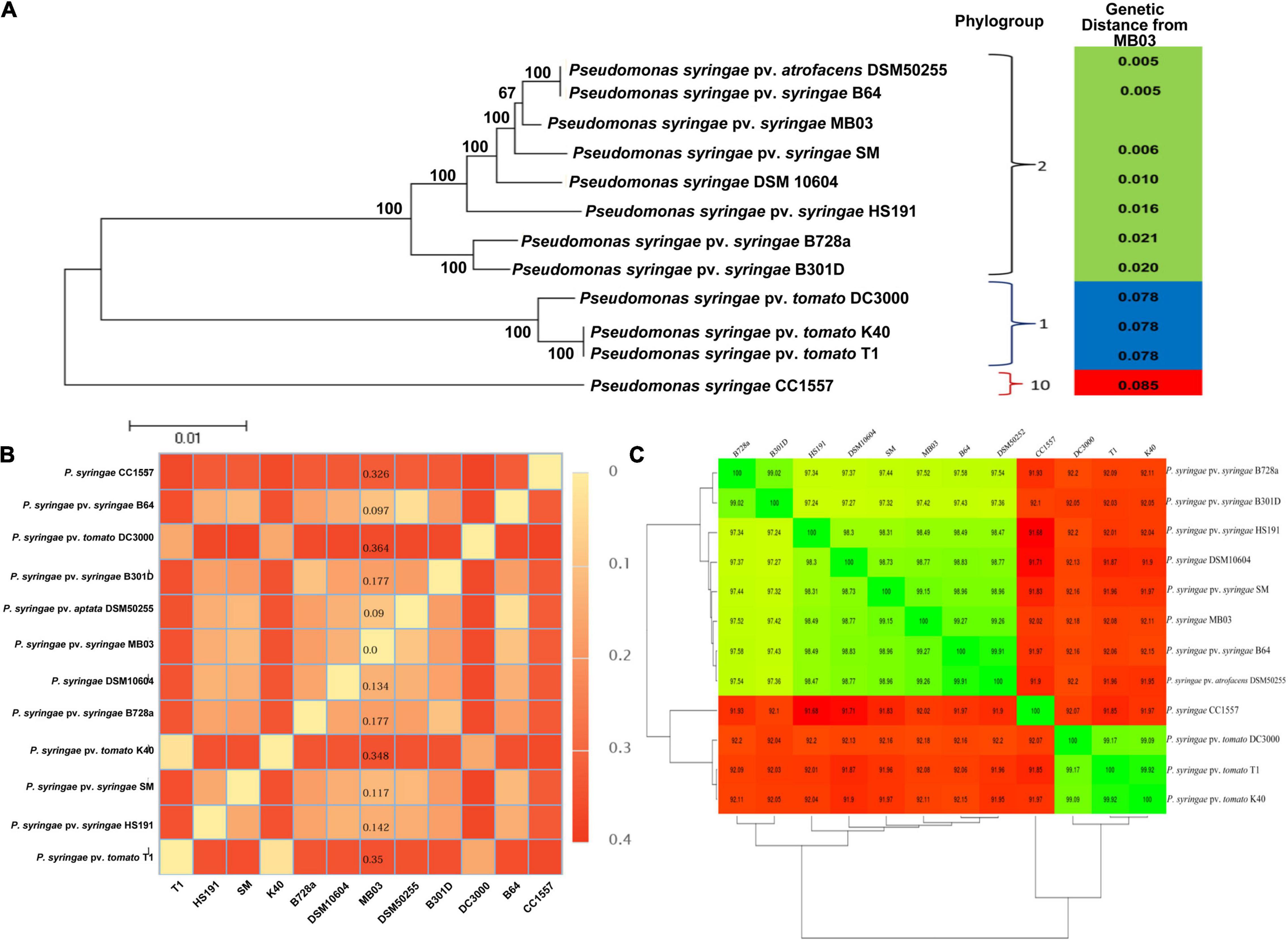
Figure 2. Phylogenetics of P. syringae MB03. Phylogenetic analysis of P. syringae MB03 was performed based on MLSA using seven housekeeping genes (A). The tree was constructed by the neighbor-joining method by MEGA5 and bootstrap values are shown at internal nodes. Based on seven genes, the genetic distance of MB03 from other strains was determined, shown in the right panel. Strain MB03 was grouped in phylogroup II of P. syringae species. Genome-to-genome distance of strain MB03 with other strains is also shown (B). Least distance of P. syringae MB03 was observed with P. syringae DSM50255, B64 and SM. The mean amino acid identity matrix is shown for selected strains (C). The numerical value within a box shows the percent of the mean amino acid identity of homologs of two strains. Red and green colors are used to show percent similarity.
The GC skew of MB03 was generated using the CGView server. All the predicted ORFs of MB03 were compared with the reference strains (P. syringae B728a, DC3000 and CC1557); the opacity of the line is representing the degree of similarity (Supplementary Figure 1). The genome alignment of MB03, SM and B64 showed noticeable similarity in terms of evolutionary rearrangements (as shown by colored block in Supplementary Figure 2).
The Strain-Specific Gene Content of MB03 Is Exceeded by the Hypothetical Proteins
Previously, strains of P. syringae were considered harmless to C. elegans (Burlinson et al., 2013). Contrary to this, P. syringae MB03 could kill C. elegans. This raised the possibility of the presence of strain-specific genes in MB03 strain which could involve in host infection. The distinction between the core and dispensable genomes provides better insight into the phenotypes and virulence mechanisms of a strain (Pallen and Wren, 2007). Formerly, the core genome of P. syringae was determined (40% identity over 40% length), and the analysis resulted in the determination of 3397 genes as the core genome of P. syringae species (Baltrus et al., 2011). When the P. syringae MB03 genome was compared (using 40% identity over 40% length as selection criteria) with the previously determined core genome by Baltrus et al. (2011), homologs of 3346 genes (out of 3397 genes) were found in the genome of strain MB03. For the determination of species-common genes and strain-specific genes, P. syringae MB03 was compared with DC3000, CC1557, B728a, HS191, B64, and SM (Table 1). The highest number of strain-specific genes was found when P. syringae MB03 was compared with DC3000. Moreover, 156 genes of P. syringae MB03 showed no homologs in the six tested strains; hence, these genes were specified as unique genes (Supplementary Table 3). Among these unique genes, 135 were predicted as hypothetical proteins. Moreover, four transcriptional regulators (VT47_00425, VT47_00955, VT47_13240, and VT47_22400) were only found in P. syringae MB03. Among these regulators, VT47_00420, VT47_00425, and VT47_22400 were found on a predicted genomic island.

Table 1. Strain-specific genes of P. syringae MB03 compared with other reference strains.
Predicted Genomic Islands of Pseudomonas syringae MB03 Have the Potential to Amend the Pool of Virulence Factors
Genomic islands evolve due to horizontal gene transfer and sometimes play a vital role in bacterial lifestyle (O’Brien et al., 2011). Regarding P. syringae species, two candidate pathogenicity islands of P. syringae B728a have been reported in PAI DB2. The hrp pathogenicity island of B728a (related to Type III secretion system) contains most of the hop effectors of this strain (Psyr_1175 to Psyr_1239). When compared with MB03, integrase, transposase, hopX1, avrB3 and two hypothetical proteins were absent. Alternatively, several additional unique proteins were found in MB03. The rest of the island was similar between the two strains (Supplementary Figure 3). The other candidate pathogenicity island of B728a was not observed in MB03.
When analyzed at Islandviewver3, 24 genomic islands were observed (Supplementary Table 4 and Supplementary Figure 4). To avoid false-positive results, only those islands were analyzed that were present within a single contig. One of these islands contained genes for a Type VI secretion system and hop proteins. This cluster (VT47_11230–VT47_11300) was not present in other P. syringae strains. Another important island (VT47_00420−VT47_00465) contained many strain-specific genes. The gene VT47_00420 encodes a LasR transcriptional regulator that is absent in the genome of HS191, B728a, DC3000, B64, SM, and CC1557. According to the KEGG pathway and previous literature reports, the homologs of LasR transcriptional regulators have a well-defined role in bacterial pathogenicity (Feinbaum et al., 2012). LasR controls the expression of hundreds of genes including quorum sensing, secreted virulence factors, and secondary metabolites (Gilbert et al., 2009).
Transcriptomics for Determining Bacterial Response During Host–Pathogen Interaction
The Transcriptional Response of the Pathogen Varied Depending Upon Its Growth Phase
Pseudomonas syringae MB03 cells were exposed to the model host C. elegans during the exponential phase, as well as the stationary phase of bacterial growth, to analyze the transcriptome of MB03 cells. Various genes were differentially expressed depending on the growth phase of the pathogen (Figure 3). In total, 76 and 138 genes showed log2 ≥ 2-fold differential expression when the host–pathogen interaction took place during the exponential and stationary phase, respectively (Supplementary Table 5). Among these genes, only 11 genes overlapped and showed similar patterns of expression regardless of the growth phase. Up-regulated genes of interest included atoE (V47_15130) for short-chain fatty acid degradation; hisJ (VT47_18480), which is a part of an ABC transporter for histidine transport; and gltI (VT47_18645), which might be correlated to a shift in nutrient availability. Additionally, pntA (VT47_24115) and scoB (VT47_15125) were up-regulated, whereas scoP (V47_01435), queD (VT47_04670), rpsT (VT47_03545), and two hypothetical proteins (VT47_13300, VT47_14685) were down-regulated during host–pathogen interaction regardless of the bacterial growth phase (Supplementary Table 5).
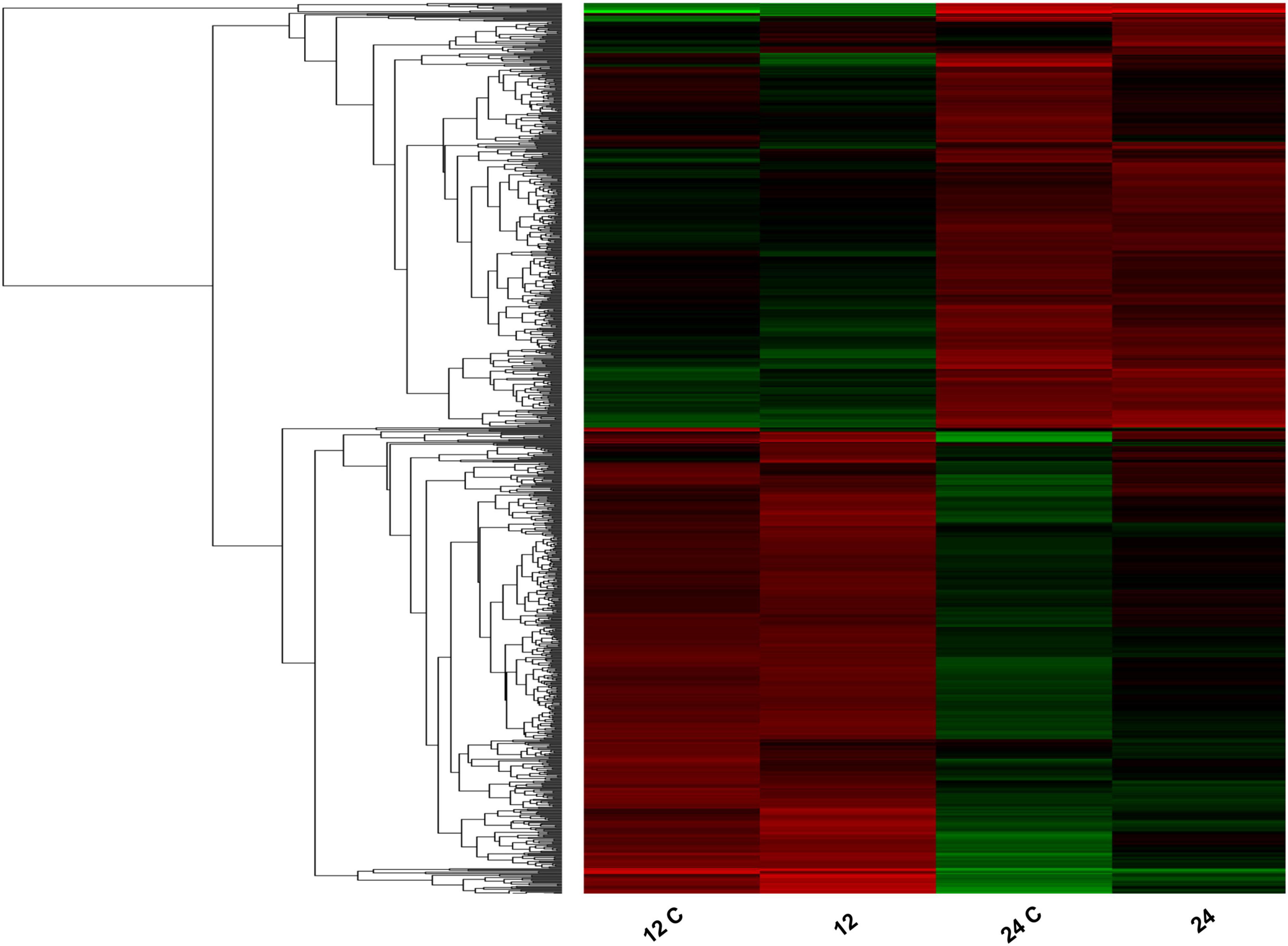
Figure 3. Heatmap showing differential expression of bacterial genes in response to C. elegans. Total RNA was extracted from the bacterial cells with/without the host from two different phases of bacterial growth (exponential phase represented as 12 and 12 C whereas stationary phase represented as 24 and 24 C). Control samples of 12 h growth and 24 h growth are represented as 12 and 24, respectively whereas bacterial cells cultured with C. elegans are shown as 12 C and 24 C. Red bars show up-regulation and green bars show downregulation of genes. The intensity of the color is directly proportional to the magnitude of gene expression. Results of transcriptomics were cross verified by performing qRT-PCR on selected genes.
Genes for the utilization of D-galactonate (VT47_09385, VT47_09390, and VT47_09395) were only up-regulated in the exponential phase cells. Another important difference in the gene expression pattern was observed in the case of ABC transporters primarily related to amino acids. In bacterial two-component systems, phoP/phoQ genes were up-regulated when interaction was studied with exponentially growing bacterial cells. This system has been documented to trigger various bacterial virulence factor during environmental stress and host pathogen interaction (Alegado and Tan, 2008; Gellatly et al., 2012). Similarly, genes for tricarboxylate transport (tctA VT47_18945, tctB VT47_18940, tctC VT47_18935) and amino acid uptake and metabolism (aatP VT47_18660, aatM VT47_18655, aatQ VT47_18650, aatJ VT47_18645) were up-regulated during the exponential phase interaction. It has been reported that most of hosts enforce amino acid starvation to the invading bacteria. In response, bacterial pathogens manipulate the host metabolism to overcome nutrient depletion (Zhang and Rubin, 2013). Most genes related to flagellar assembly were up-regulated when bacterial cells in the stationary phase were used in host-pathogen interaction (Supplementary Table 5). Previously, flagellum has been reported for its role in the adhesion and penetration of pathogen into host tissue (Haiko and Westerlund-Wikstrom, 2013; Coloma-Rivero et al., 2020). Moreover, in our previous study, genes related to flagellum were found up-regulated during host-pathogen interaction (Ali et al., 2016).
Among the genes present on the predicted genomic islands, five genes showed highly differentiated expression when MB03 was exposed to C. elegans (Supplementary Table 6). All these genes were part of the auxiliary genome of P. syringae. Among these genes, VT47_06210 was responsible for the conversion of nitroalkane into nitrite, whereas VT47_06205 was involved in nitric oxide detoxification. Previously, nitric oxide detoxification genes have been reported in certain strains of P. syringae (Marcelletti et al., 2011). Analysis of the genome organization revealed the presence of one transcriptional regulator (VT47_06200) just upstream of these genes. This transcriptional regulator was annotated as norR, which has also been reported for nitric oxide detoxification.
Up Regulation of Bacterial Two-Component Systems Might Help the Bacterial Strain in Sensing the Host Environment
According to COG, 282 genes of MB03 were grouped under the signal transduction category (Supplementary Table 2). Among two-component systems (TCS), phoQ of phoQ/phoP was up-regulated during host-pathogen interaction regardless of the growth phase of the bacterial pathogen. Previously, up regulation of this system has been reported in P. aeruginosa cells after interaction with human bronchial epithelial cells, and mutation in the phoQ gene resulted in reduced virulence (Gellatly et al., 2012). This system has also been known to confer antimicrobial resistance to the bacterial cells. Previously, it was reported that the phoP mutant of Salmonella enterica was unable to colonize the gut of C. elegans (Aballay et al., 2000).
Other TCS related to bacterial pathogenicity include fleS/fleR, which regulates flagellum biosynthesis along with rpoN sigma factor (Dasgupta et al., 2003). Previously, the synthesis of flagellum and transcription of biosynthetic genes have been divided into four phases. The FleS/FleR system was reported to control expression of genes required for transcription of class III genes of the flagellum (Dasgupta et al., 2003). Down regulation of this TCS during the exponential phase interaction and up-regulation during the stationary phase interaction highlighted its role in the late phase of MB03 infection of C. elegans (Supplementary Table 5). Interestingly, the same transcriptional pattern was observed for rpoN. The genes related to flagellum synthesis and functioning were also up-regulated during the stationary phase interaction.
The KdpD/KdpE TCS has been reported not only to regulate intracellular potassium concentration but also to play an important role in bacterial virulence (Freeman et al., 2013). Bacterial species such as enterohemorrhagic Escherichia coli, Francisella tularensis, and Salmonella typhimurium having mutated kdpD/kdpE genes, showed significantly attenuated virulence when studied in different host cells (Alegado et al., 2011; Njoroge et al., 2012). Accordingly, kdpD mutants of P. aeruginosa and S. typhimurium species were attenuated in killing C. elegans (Alegado et al., 2011; Feinbaum et al., 2012). A fully functional kdpD gene was required for colonization of the host gut by bacterial cells. In this study, no significant change was observed in the expression of the kdpD/kdpE two component system. However, the kdpB gene was significantly up-regulated during the stationary phase interaction. Previously, it was reported that a kdpB mutant of P. aeruginosa PAO1 was attenuated in its virulence against C. elegans (Dubern et al., 2015). Another important two-component system is gacA/gacS, which is well-studied for its role in bacterial virulence. However, in our transcriptomics data, no notable change was observed in the expression of this system.
Genes of Type III and VI Secretion Systems Were Differentially Expressed During Host–Pathogen Interaction
In the case of P. syringae pathovars, the Type III secretion system and its effectors (TTE) are among the major factors in its plant host determination (Lindeberg et al., 2006). To date, more than 50 TTEs have been identified in P. syringae strains (Baltrus et al., 2011). Their sequences were obtained to identify homologs in strain MB03 by applying BLASTp, and a total of 10 effectors were identified (Supplementary Table 7). Five effectors (AvrE, HopAA, HopAH, HopI, and HopM), which are found in almost all of the strains (Baltrus et al., 2011), were also present in MB03. Previously, it was reported that members of P. syringae genomogroup II contain fewer effectors compared to other groups. P. syringae B64, which was isolated from the wheat plant, contained 10 effector proteins (Dudnik and Dudler, 2013b), and out of those, six are common between B64 and MB03 (HopZ3 and five core effectors including AvrE1, HopAA1 HopI1 HopM1, and HopAH1).
Although most of the genes related to the Type III secretion system and effectors showed no change in their transcription levels, hrcN (VT47_05965) and hrcQa (VT47_05955) were up-regulated when stationary phase MB03 cells were exposed to the worms. Both of these proteins are among the core proteins of Type III secretion system and share significant similarities with flagellar proteins (Van Gijsegem et al., 1995). Other than the well-known motility function of the flagellum, secretion of proteins especially virulence factors has also been well-documented (Young et al., 1999).
Type VI Secretion System
Among bacterial secretion systems, the Type VI secretion system plays an important role in the host-microbe interaction and virulence (Mougous et al., 2006; Jiang et al., 2014). However, its role in P. syringae is still not well-characterized. Recently, an effort was made to analyze the distribution of the Type VI secretion system in P. syringae species by in silico characterization (Sarris et al., 2010). Although we observed two Hcp secretion islands (HSI) clusters in MB03 (VT47_23900–VT47_23835 and VT47_11235–VT47_11300), these were not identical to the clusters found in DC3000 in terms of gene number and function. The Type VI secretion system of B728a is comprised of the HSI-I cluster, Ppka locus, and certain hcp/vgrG genes. The Ppka and Pppa were observed in many strains of P. syringae such as B728a, DC3000 and T1 (Sarris et al., 2010). Compared with B728a, the Ppka locus along with homologs for genes Psyr_0101 and Psyr_1935 were absent in MB03. The ppkA and pppA are regulatory genes, and both work antagonistically. The homolog of PpkA locus was not found in MB03. However, this is not unusual as many other bacterial strains with missing PpkA loci have been reported (Sarris et al., 2010). Except for one gene, impM (VT47_23885), the entire cluster was up-regulated during stationary phase host-pathogen interaction. Two Rhs element Vgr proteins (Valine-glycine repeat protein, an essential component of secretion machinery) (VT47_23935 and VT47_23990) were also up-regulated.
Moreover, MB03 contained another contiguous cluster (Supplementary Figure 5) encoding for a Type VI secretion system (VT47_11230–VT47_11300). This cluster of MB03 was absent in strain B728a; however, homologs of genes of this cluster were found in B64, and SM. The ClpB/V, DotU, and IcmF are among the core components of T6SS, and the VT47_11270 loci of MB03 encodes a ClpB protein, which can provide energy to the secretion system. Interestingly, this cluster was accompanied by two transcriptional regulators: one σ54 dependent (VT47_11275) within the cluster and another LysR family protein (VT47_11320) downstream of the cluster. Previously, a σ54 dependent transcriptional regulator was reported to play an important role in the regulation of the T6SS clusters (Bernard et al., 2011). Among the genes of HSI-II, impA, which encodes a secretion protein, was up-regulated during the stationary phase interaction. There is no direct evidence to explain the role of these Type VI effector proteins; however, it appears that these proteins might have important roles in the animal pathogenicity of P. syringae MB03.
Genes Related to Locomotion and Adhesion Were Up Regulated During Host–Pathogen Interaction
The flagellum has a well-defined role in host colonization, locomotion, protein secretion, and chemotaxis, and all these functions help in pathogenesis (Young et al., 1999; Dasgupta et al., 2003). Mutations in alginate biosynthesis genes of P. syringae pathovars lead to a significant decrease in plant pathogenesis (Dorati et al., 2018). In the current study, alg44, algE, and algD were upregulated during the exponential phase interaction, whereas algD, algE, algF, algL, and alg8 were upregulated during the stationary phase interaction. Interestingly, the genes related to the flagellum were down-regulated during the exponential phase interaction and up-regulated during the stationary phase interaction (Supplementary Table 5). In P. aeruginosa, transcription of flagellum-related genes is controlled by fleQ, vfr, and other sigma factors (Dasgupta et al., 2003). Gene fleQ, which regulates transcription of class 1 flagellum genes, was also down-regulated during the exponential phase interaction. On the other hand, almost all the genes for the flagellum were up-regulated when the stationary phase MB03 was cultured with C. elegans. Animal pathogens have been well-reported for protein secretions through the flagellum (Fretin et al., 2005; Hautefort et al., 2008). Late expression of the flagella-related genes in MB03 infection is in accordance with a previous report where flagellum-related genes were expressed in the late phase of infection (Hautefort et al., 2008). Up regulation of flagellum-related genes during host–pathogen interaction might be related to protein secretion and motility. Possibly, this up-regulation occurred after infection by the pathogen as it was previously observed in the case of the Salmonella strain (Hautefort et al., 2008). Additionally, the involvement of flagellar genes in the killing of C. elegans has also been reported in Burkholderia pseudomallei (O’Quinn et al., 2001).
Homologs of Nematicidal Genes of Pseudomonas aeruginosa in MB03 and Their Transcriptional Response
Virulence factors of different pathogens, such as P. aeruginosa, have been previously characterized using C. elegans as a model organism; a total of 170 nematicidal genes in P. aeruginosa PA14 (Feinbaum et al., 2012) and 68 in P. aeruginosa PAO1 have been identified (Dubern et al., 2015). To further evaluate the nematicidal potential of MB03, its proteins were compared with the reported virulence factors of P. aeruginosa PA14 and PAO1. For this purpose, in silico subtraction was used, and conserved proteins were retrieved (H-value ≥ 0.64). This revealed 87 and 30 homologs of PA14 and PAO1, respectively, in MB03 (Supplementary Table 8). Among these, 10 homologs showed significant variation in their transcriptional profiles (Table 2) upon host-pathogen interaction. Six homolog genes were up regulated during the stationary phase interaction: lysR transcriptional regulator (VT47_13020), isovaleryl-CoA dehydrogenase (VT47_11890), putative acyl-CoA carboxylase alpha chain (VT47_11875), acyl-CoA carboxyltransferase beta chain (VT47_11885), prpC (VT47_09990), and PrpB (VT47_09985). On the other hand, crfX (VT47_10030), (VT47_19750) and hypothetical protein (VT47_10035) were down regulated (Table 2). Regarding nematicidal genes of PAO1, homologs of 30 genes were found in MB03, as well as in the core genome of P. syringae (H value ≥ 0.64). Among these 30 homologs, one named kdpB was markedly up regulated.
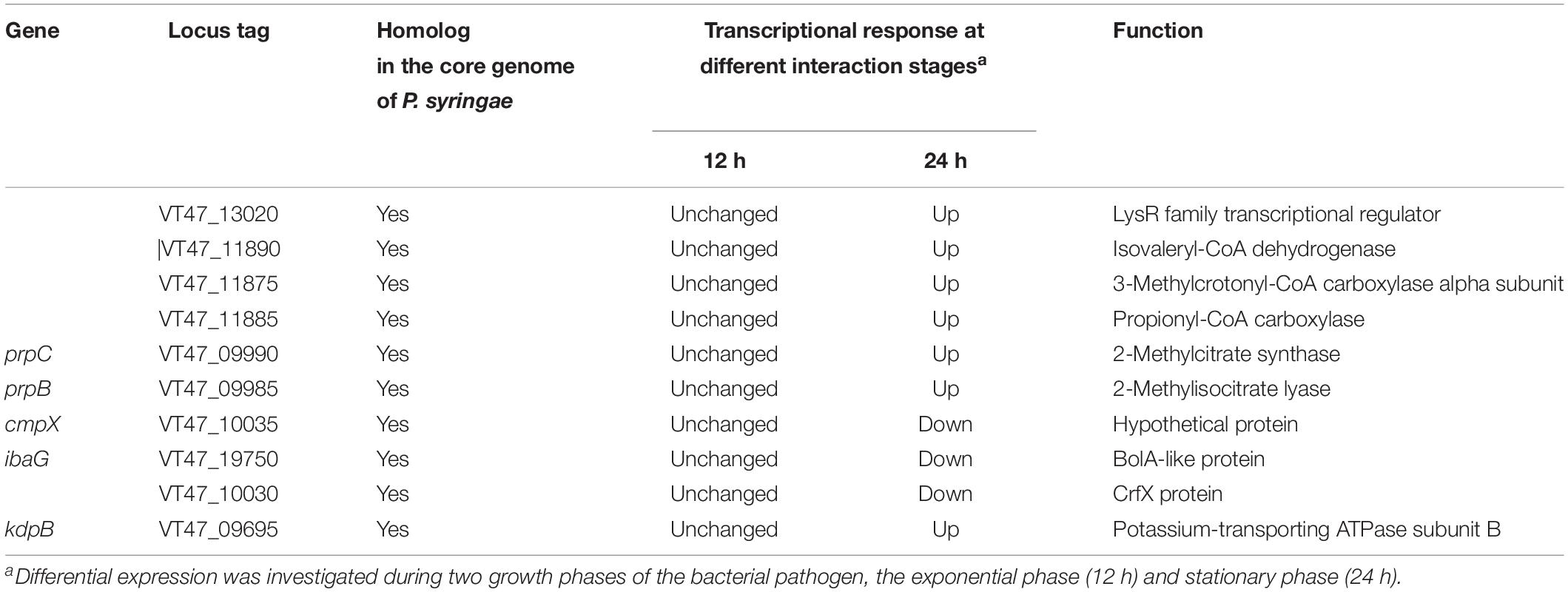
Table 2. Homologs of P. aeruginosa PA14 and PAO1 nematicidal genes in P. syringae MB03 showing differential transcriptional response.
Up Regulation of ABC Transporters Provide Insight Into Nutritional Availability
Comparative genomics revealed the presence of six ABC transporter genes of MB03 (VT47_07640, VT47_08280, VT47_08285, VT47_12125, VT47_13510, VT47_20470) homologs, which were previously demonstrated to be essential for full bacterial virulence of P. aeruginosa against C. elegans (Feinbaum et al., 2012; Dubern et al., 2015). The results of the transcriptomics showed up-regulation of different genes related to ABC transporters (Table 3). Glutamate/aspartate transporters encoded by gltI, gltJ, gltK, and gltL, were highly up-regulated when MB03 cells were exposed to C. elegans. This system binds and transports glutamate and aspartate amino acids to the bacterial cell. It has been proposed that the release of these amino acids indicated disruption of host cells and that bacterial cells used these as sources of carbon and nitrogen (Hassel et al., 2014).
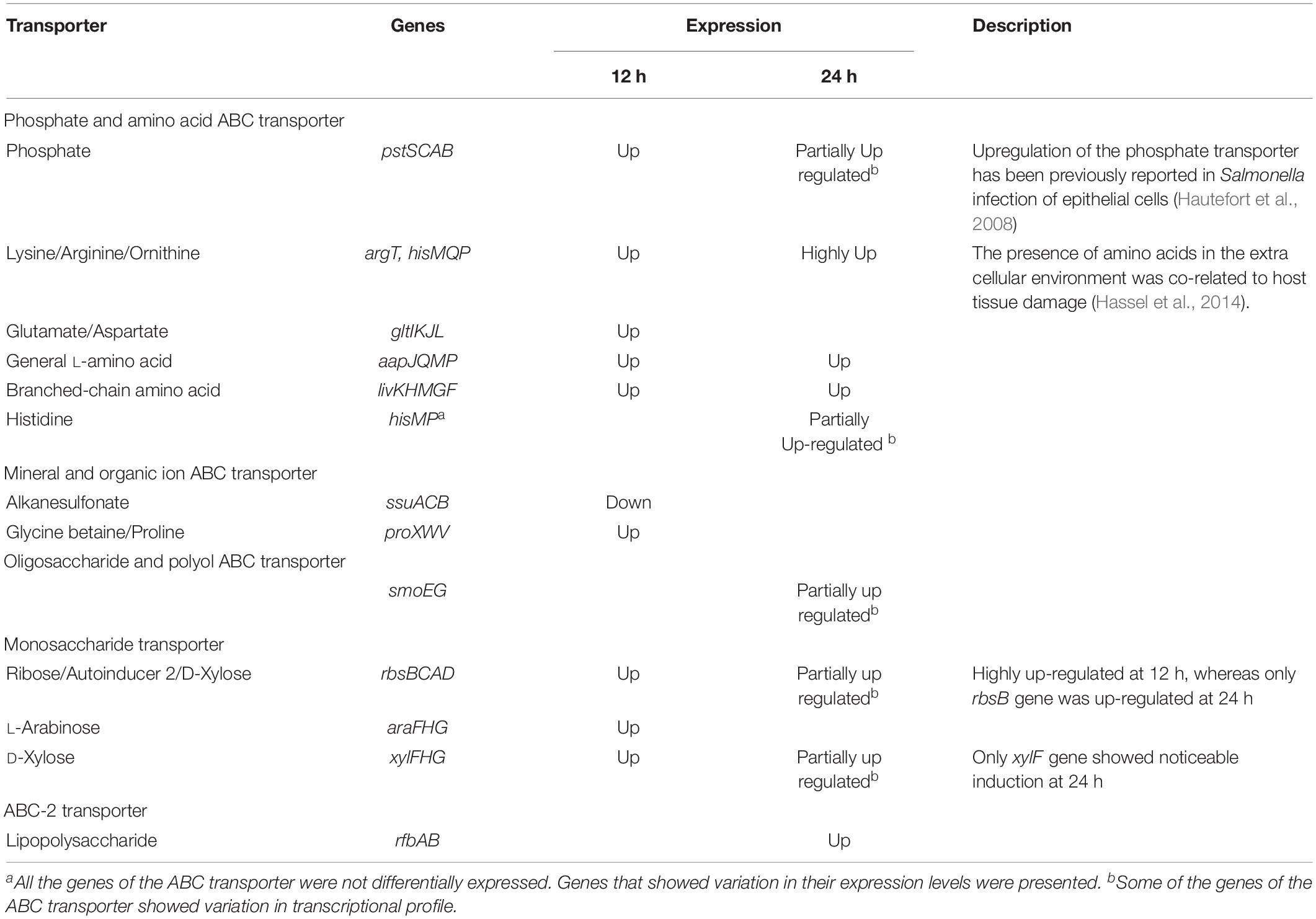
Table 3. Transcriptional profile of ABC transporters during the exponential and the stationary phase interactions.
Also, glycine betaine/proline transporter was up-regulated during the exponential phase interaction (Table 3). This has also been reported for osmoprotection in P. aeruginosa (Wargo, 2013). Moreover, glycine betaine uptake by proXVWZ ABC transporter was found to be vital for the growth and survival of Mycobacterium tuberculosis in human macrophages (Price et al., 2008).
Various Transcriptional Regulators Showed Significant Differential Expression During Host–Pathogen Interaction
Eight transcriptional regulators showed significant change in their expression profiles when P. syringae MB03 interacted with C. elegans (Table 4). Out of these transcriptional regulators, the homolog of one gene (LysR family, VT47_13020) was reported as essential for full virulence of P. aeruginosa PA14 against C. elegans (Feinbaum et al., 2012). The LysR family transcriptional regulators are well-documented for their impact on bacterial pathogenicity by regulating numerous virulence factors, helping in bacterial adhesion and adaptation to a hostile environment (Xiao et al., 2006). Two other regulators, araC family, and betI, were differentially expressed when P. syringae MB03 interacted with C. elegans. Among these regulators, the AraC family transcriptional regulator was up-regulated during the exponential and stationary phase interactions. This regulator is located upstream of glycine betaine/proline ABC transporter genes (proXVW) in the genome of P. syringae MB03. Genes for betaine transport were also up-regulated during the exponential phase interaction. Transcriptional factors related to the AraC family have been reported for carbon metabolism, bacterial pathogenicity, and stress response (Gallegos et al., 1997).
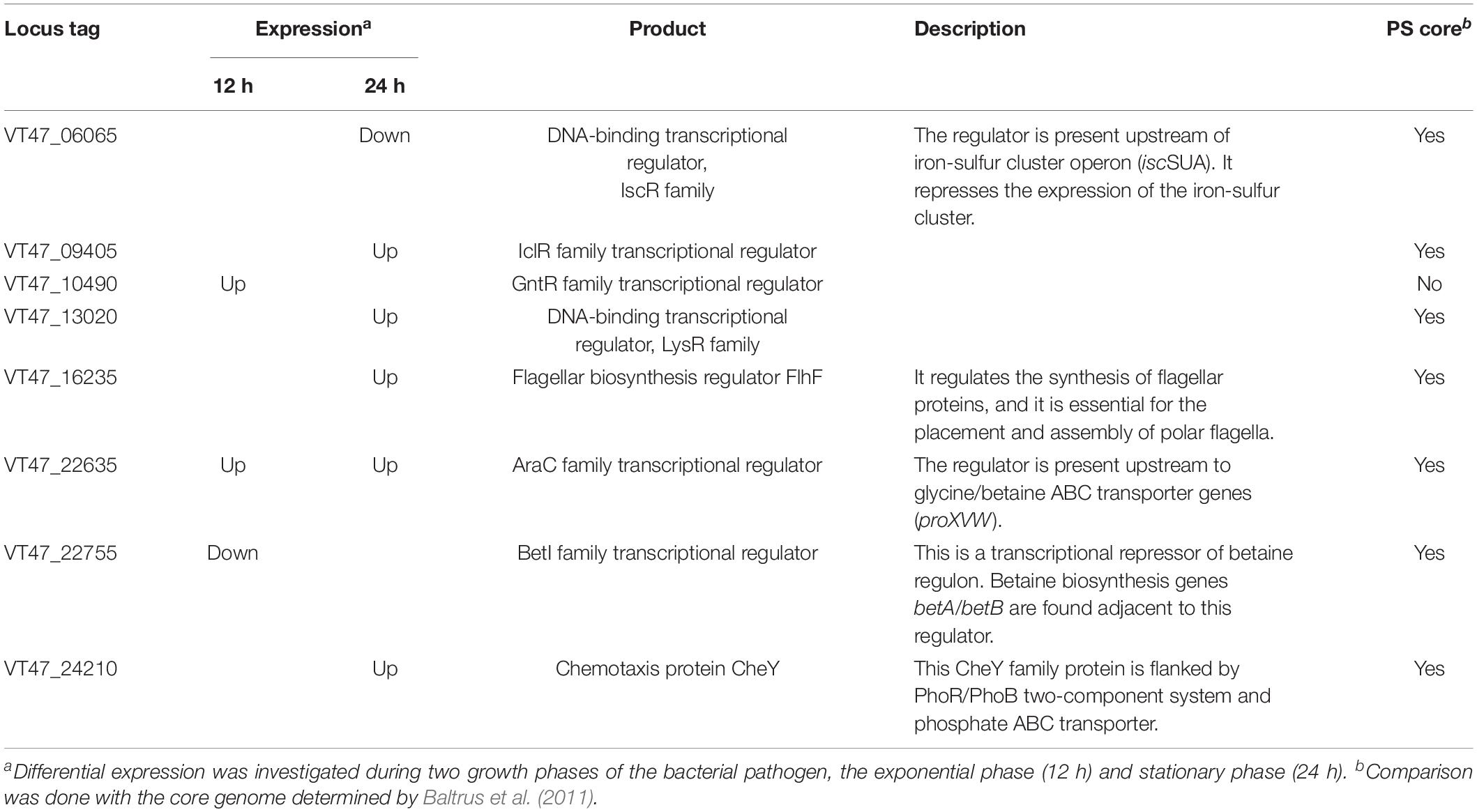
Table 4. Expression profile of selected transcriptional regulators and their function.
Screening of a Mutant Library Revealed Nematicidal Genes of Pseudomonas syringae MB03
The construction of a transposon insertion mutant library resulted in 1265 mutants of P. syringae MB03. The killing assay was performed to screen for mutants with attenuated virulence. For comparison, P. syringae wildtype strain MB03 was used, and all the worms died after 7–8 days when exposed to the wild-type strain. Primary screening resulted in the identification of 12 mutants with attenuated virulence. The second round of screening was performed which resulted in the identification of seven mutants (Table 5). The worms survived for at least 10 days when exposed to these attenuated mutants. These seven mutant genes were assessed for their homologs in the previously reported virulence factors of Gram-negative bacteria, especially virulence factors of P. aeruginosa and P. syringae (Feinbaum et al., 2012; Dubern et al., 2015). For this purpose, VFDB was also searched; however, homologs of these seven genes have not been previously reported for their virulence against C. elegans.
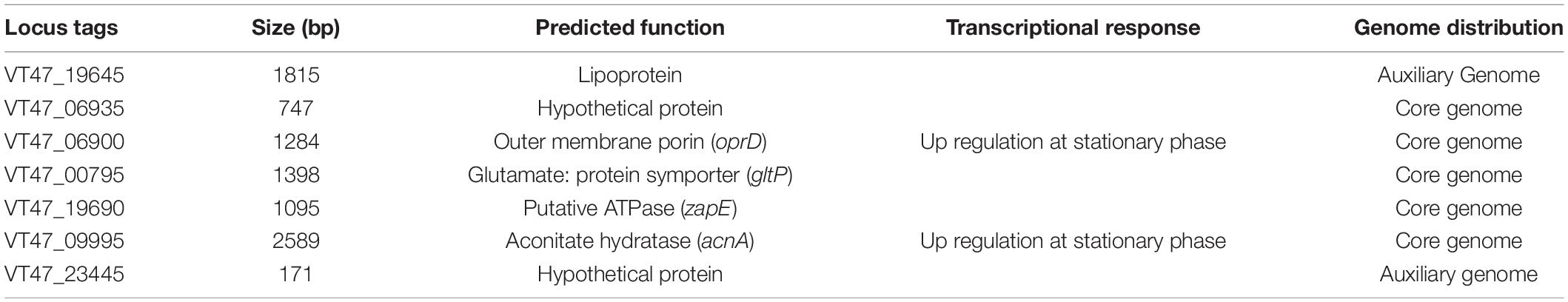
Table 5. Nematicidal genes identified by transposon insertion mutant library screening.
Among these mutants, VT47_19690 was identical to zapE of E. coli strain K-12 and a hypothetical protein (PA4438) of P. aeruginosa PA14. Recently, the ZapE protein has been reported for its role in cell division, and the protein is required for bacterial infection (Marteyn et al., 2014). The importance of cell division has also been elucidated in the P. aeruginosa – C. elegans model, where a mutation in the minD gene resulted in attenuated virulence (Feinbaum et al., 2012). Another mutant strain ΔacnA (VT47_09995) showed attenuated virulence. The protein can interconvert citrate and isocitrate thereby facilitating a shift in different metabolic pathways, including the citrate cycle and glyoxylate cycle. Similarly, the mutation in the oprD (V47_06900) gene resulted in attenuated pathogenicity against C. elegans. The gene oprD encodes porin which forms a channel in the bacterial cell membrane. Different functions have been associated with membrane porins, including resistance against antimicrobials, nutrient uptake, and adhesion (Tamber et al., 2006; Muller et al., 2011). In addition, the porin of P. aeruginosa may also possess protease activity (Yoshihara et al., 1996).
In silico Characterization of Selected Hypothetical Proteins and Transcriptional Regulators
The hypothetical proteins and transcriptional regulators, which showed differential expression during transcriptomics analysis were selected for in silico characterization. Molecular weight, isoelectric point, aliphatic index, and stability of the hypothetical proteins were predicted by Protparam software. Among 25 hypothetical proteins, 12 were found to be unstable (Supplementary Table 9). Signal peptides were predicted at the N-terminus of 7 hypothetical proteins, indicating their localization in periplasmic space, outer or inner membrane. This notion was further validated by CELLO and TMHMM results. Seven hypothetical proteins were predicted in the periplasmic space while four were associated with the outer and inner membranes. The proteins on the outer membrane could be potentially involved in the host-pathogen interaction, and therefore, 3D structures of these were predicted using I-TASSER. Interestingly, one hypothetical protein (Accession No: KZL40945) was predicted to be involved in host–pathogen interaction by the VICMpred server. This was also supported by our experimental data from the transposon insertion mutant library screening. Moreover, this protein is predicted as a transcriptional regulator based on homology modeling by the HPIDB server. Similarly, uncharacterized transcriptional regulators which showed highly differential expression were selected to predict target genes (Supplementary Table 10).
Heterologous Expression of Predicted Virulence Factors
Proteases were expressed in E. coli, and their impact on virulence acquisition by living cells was investigated. For this purpose, the fraction of killed worms was determined during the interaction of recombinant E. coli strains and C. elegans. The screening resulted in the identification of potential proteases and esterases (Table 6 and Figure 4). Among these enzymes, VT47_14740 showed 54% identity with metalloproteinase serralysin (AFX62372) of S. marcescens (Paiva et al., 2013). VT47_14210 also showed 34% identity with metalloproteinase serralysin (AFX62372). The protein VT47_17780 had 33% identity with a serine protease precursor (JX667979) of S. marcescens (Paiva et al., 2013). However, the results showed high up-regulation of a metallo-protease serralysin (VT47_14740) during the stationary phase interaction (Supplementary Table 5). Recently, a serralysin-like protein has been reported to play an important role in the pathogenicity of Serratia marcescens against insects (Pineda-Castellanos et al., 2015).
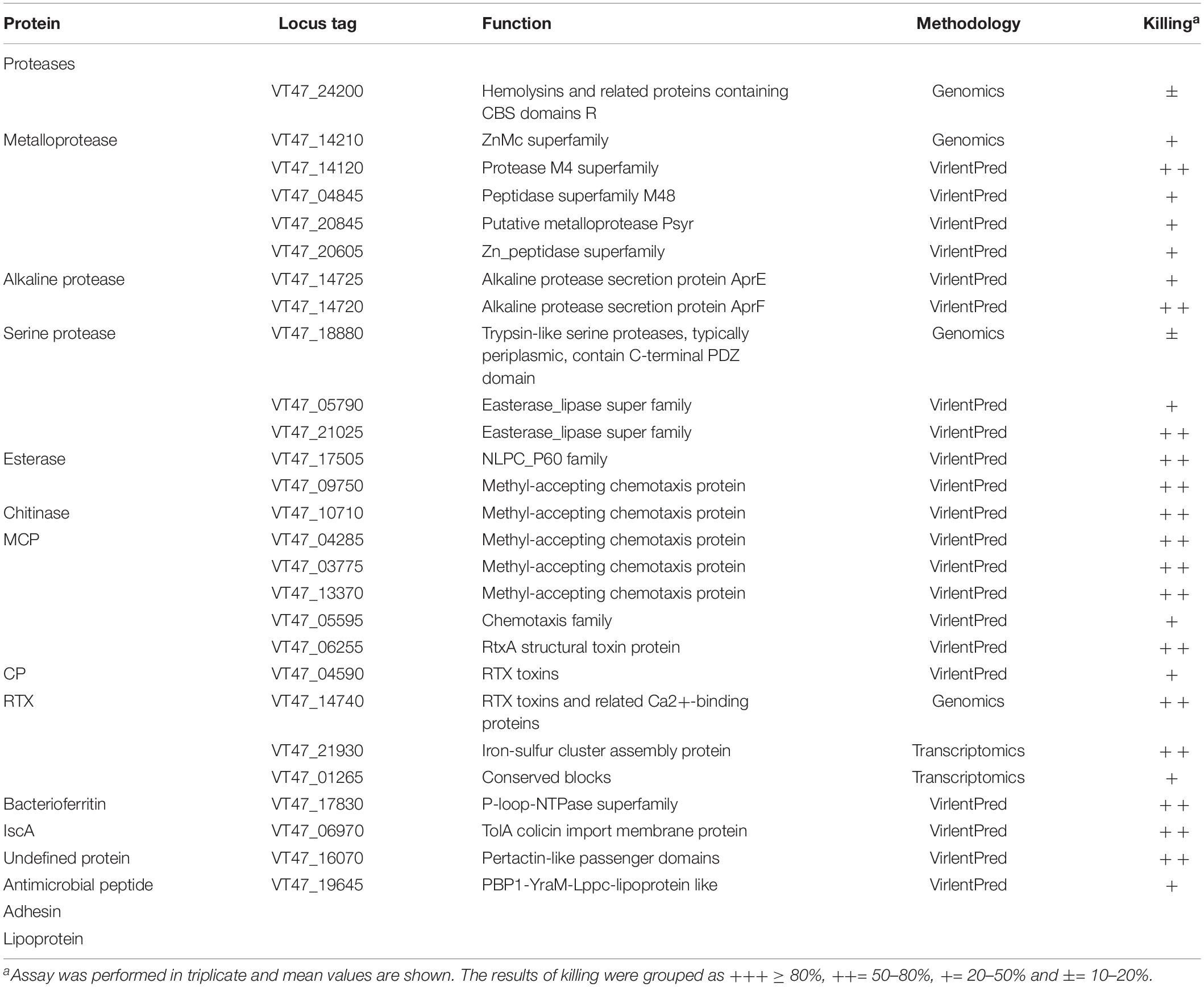
Table 6. Heterologous expression of proteins and nematicidal activity of recombinant E. coli cells.
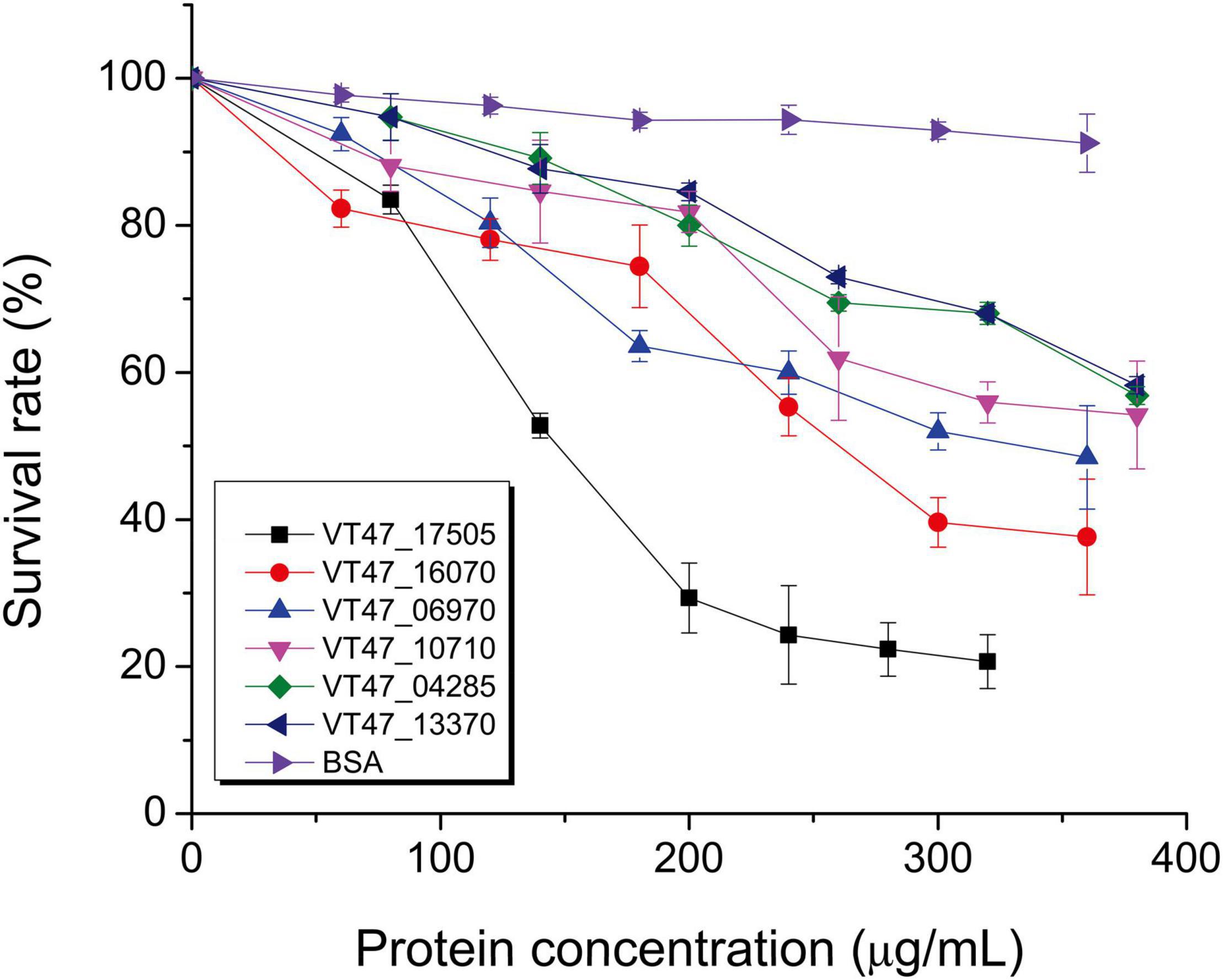
Figure 4. Nematicidal activity of purified bacterial proteins. Potential nematicidal proteins of P. syringae MB03 were expressed in E. coli TOP10 or JM109 and purified by affinity column chromatography for the liquid killing assay. The killing of worms was determined after 3 days. Worms that did not respond to the touch were considered dead. Bovine serum albumin (BSA) was used as a control. The experiment was performed in triplicate and mean values are represented. On X-axis, concentrations of purified proteins are shown. On Y-axis, survival of C. elegans is shown. NCBI accession numbers of the genes are shown in text box (VT47_17505 esterase, VT47_16070 undefined protein, VT47_06970 membrane protein, VT47_10710 chitinase, VT47_04285 MCP, VT47_13370 MCP). The details and selection criteria of these proteins are provided in Table 6.
Discussion
Pseudomonas syringae has been traditionally recognized as a plant pathogen; however, notable killing activity against the animal model C. elegans has been recently demonstrated for a P. syringae wild-type strain MB03 (Ali et al., 2016; Bashir et al., 2020, 2021). Various killing mechanisms of P. syringae MB03 such as gut colonization of host (Ali et al., 2016), production of secondary metabolites (Bashir et al., 2020), and nematicidal proteins (Manan et al., 2018) have been discovered. For instance, strain MB03 was capable of gut colonization under nutrient-rich conditions (Ali et al., 2016) and secreted pyoverdine under an iron-deficient environment which mediated host killing (Bashir et al., 2020). In the current study, comparative genomics, transcriptomics, and transposon insertion mutant library analyses were applied for genome-wide identification of common and unique virulence factors required for the nematicidal activity of P. syringae MB03 against C. elegans. The results indicated that contrary to the P. aeruginosa liquid-based killing mechanism, P. syringae MB03 colonized the gut of the worm, and lethality followed an infection-like course. When analyzed at the genetic level, we identified some unique potential nematicidal virulence factors, as well as factors for locomotion, nutrient acquisition, adhesion to host, and protein secretion that could play important roles in nematode killing.
Classification of potential virulence factors into strain-specific, auxiliary and core genome showed that most of the virulence factors were from the core genome P. syringae. In the case of genes showing high differential expression (log2 ≥ 2) during host–pathogen interaction, approximately 70% of the genes were part of the core genome. The fraction of the core genome also outnumbered the auxiliary genome during the determination of nematicidal homologs of P. aeruginosa and from mutant library screening. It should be noted that approximately 66% of genes of MB03 contribute to the core genome, and the remaining 34% of the genome is comprised of auxiliary and strain-specific genes. Hence, it can be concluded that the distribution of virulence factors was not biased toward the core genome or auxiliary genome. The core genome of P. syringae, which was employed in comparative genomics, was determined by considering three diverse phylogroups of the P. syringae species (Baltrus et al., 2011). Moreover, most strain-specific genes were hypothesized to reside on genomic islands, and indeed, 66 strain-specific genes (42%) were found on the predicted genomic island (Supplementary Table 4).
Previously, near-complete transposon insertion mutant libraries were constructed to identify virulence factors of P. aeruginosa strains PAO1 and PA14 (Feinbaum et al., 2012; Dubern et al., 2015). These genome-wide studies revealed various genes related to nematicidal activity including enzymes, secondary metabolites, and two-component systems, etc. Comparative genomics was applied to identify the homologs of those genes in P. syringae MB03. The analysis resulted in the identification of 115 candidate genes that may assist pathogens during host-pathogen interaction.
Transcriptomics provided information about a variety of candidate nematicidal genes of P. syringae MB03, and the results showed a notable influence of the growth phase on the expression of bacterial genes during host–pathogen interaction. Moreover, the expression of some of the genes was in accordance with previous reports. For example, certain MB03 homolog genes of nematicidal proteins of P. aeruginosa showed differential expression (Table 2; Feinbaum et al., 2012; Dubern et al., 2015). It is worth noting that these P. aeruginosa homologs showed differential expression during the stationary phase interaction. Similarly, the expression of two-component signal transduction systems can be justified based on previous reports showing their role in bacterial pathogenicity. Up regulation of the phoQ/phoP and fleS/fleR two-component systems during the stationary phase, interaction might be correlated to bacterial infection and colonization of C. elegans (Alegado and Tan, 2008; Gellatly et al., 2012; Ali et al., 2016).
Different mechanisms have been proposed by which bacterial species kill C. elegans. The most common pathogenicity mechanisms include toxin secretion, gut colonization, and persistent infection (Cezairliyan et al., 2013). In the case of toxin-mediated killing, various metabolites such as pyoverdine, phenazine, pyochelin, pyrrolnitrin have been identified in the Pseudomonas – C. elegans infection model (Cezairliyan et al., 2013; Kirienko et al., 2013). Comparative genomics revealed that MB03 only possessed genes for pyoverdine. In addition, agar-based pathogenicity of P. aeruginosa was found to be dependent upon quorum sensing (Feinbaum et al., 2012), a mechanism by which population-dependent genes are regulated. The strain B728a harbors two mechanisms for quorum sensing, ahlI-ahlR and hdtS (Feil et al., 2005). Similar quorum sensing-related genes were observed in B64, SM, and HS191 (Dudnik and Dudler, 2013a,b; Ravindran et al., 2015). However, only the hdtS (VT47_24855) homolog was found in MB03, and no significant change in the expression of hdtS was observed. Hence, hdtS did not appear to play a significant role in the killing of C. elegans.
Siderophores are low-molecular-weight iron chelators that are secreted outside the cell. In DC3000, three different iron chelators, including pyoverdine, pyochelin, and yersiniabactin, have been reported. However, only pyoverdine and achromobactin were found in MB03. Recently, it has been reported that pyoverdine alone was sufficient to kill C. elegans (Kirienko et al., 2015). Interestingly, in a liquid killing assay, mutations in pyochelin biosynthesis genes showed no effects on the virulence of P. aeruginosa against C. elegans, whereas mutations in pvdA, pvdD, pvdE, pvdF, and pvdP resulted in decreased killing efficacy (Kirienko et al., 2013). A recent study conducted on P. syringae MB03 demonstrated the role of pyoverdine in the killing of C. elegans (Bashir et al., 2020).
Other than secreted metabolites, bacterial enzymes, and proteins capable of host degradation also play a vital role in bacterial invasion and infection (Matsumoto, 2004; Yoon et al., 2018). Similarly, signaling molecules such as cyclic-di-GMP secreted by V. cholerae attracted C. elegans toward pathogen (Angeloni et al., 2020). In the case of enzymes, alkaline proteases, serine proteases, metalloproteinase, and neutral protease with nematicidal activities have been identified in Brevibacillus laterosporus, Bacillus sp., Pseudoalteromonas tunicate, Serratia sp., and Stenotrophomonas maltophilia (Qiuhong et al., 2006; Paiva et al., 2013; Salikin et al., 2021). Extracellular secreted proteases might assist bacterial infection by degrading the outer proteinaceous membrane of the cuticle (Cox et al., 1981). These proteases help bacterial strains in nutrient acquisition, resistance against host defense by modulating host proteins, and colonization of host by tissue invasion and damage (Matsumoto, 2004). In the current study, some bacterial enzymes were expressed in E. coli, and some of the proteases of P. syringae MB03 were found to be toxic against C. elegans (Table 6).
Gut colonization is another mechanism by which pathogens kill C. elegans (White et al., 2015; Ali et al., 2016). It is one of the mechanisms used by P. aeruginosa to kill C. elegans in the slow killing assay (Mahajan-Miklos et al., 1999). P. syringae also kills C. elegans by gut colonization (Ali et al., 2016). Gut colonization in the liquid killing assay (Figure 1) is contrary to P. aeruginosa, which implies this killing mechanism in the liquid killing assay (Kirienko et al., 2013, 2015).
The integrated utilization of various techniques resulted in the genome-wide prediction of bacterial virulence factors that required lethal host–pathogen interaction. For instance, transcriptomics and transposon insertion mutant library identified novel genes required for successful infection.
Conclusion
In summary, P. syringae MB03 can kill C. elegans in the liquid assay via gut colonization. Comparative genomics revealed 156 unique genes and 115 potential nematicidal genes in the MB03 genome. The current study was performed using draft genome sequence of P. syringae MB03 hence, there is a possibility of identification more genes related to the virulence. Transcriptomics analysis showed that a variety of virulence genes were highly up-regulated. Furthermore, seven nematicidal genes were identified via screening of transposon insertion mutant library and bioassays. Total 27 nematicidal enzymes/proteins were identified based upon the activity of heterologously expressing strains of E. coli. The pathogenicity appeared to be a combinatorial action of various genes, including regulatory genes (signal transduction system and transcriptional regulators), genes related to locomotion (flagella proteins), and genes for nutrient acquisition (different metabolic proteins and catabolic enzymes).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
LL: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review and editing. MA: formal analysis, investigation, and writing – original draft. TG, XY, AB, ZW, XS, and NA: validation and visualization. All authors: contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 32170124, 31770108, and 31570123) and a grant from the National Basic Research Program of China (973 Program, grant 2013CB127504).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to Dr. Teresa De Kievit, Department of Microbiology, University of Manitoba, MB, Canada for providing vector pMCh-23.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.826962/full#supplementary-material
Footnotes
References
Aballay, A., Yorgey, P., and Ausubel, F. M. (2000). Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr. Biol. 10, 1539–1542. doi: 10.1016/S0960-9822(00)00830-7
Abbasi, A. Z., Bilal, M., Khurshid, G., Yiotis, C., Zeb, I., Hussain, J., et al. (2021). Expression of cyanobacterial genes enhanced CO2 assimilation and biomass production in transgenic Arabidopsis thaliana. PeerJ. 9:e11860. doi: 10.7717/peerj.11860
Alegado, R. A., Chin, C.-Y., Monack, D. M., and Tan, M. -W. (2011). The two-component sensor kinase KdpD is required for Salmonella typhimurium colonization of Caenorhabditis elegans and survival in macrophages. Cell. Microbiol. 13, 1618–1637. doi: 10.1111/j.1462-5822.2011.01645.x
Alegado, R. A., and Tan, M.-W. (2008). Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell. Microbiol. 10, 1259–1273. doi: 10.1111/j.1462-5822.2008.01124.x
Ali, M., Sun, Y., Xie, L., Yu, H., Bashir, A., and Li, L. (2016). The pathogenicity of Pseudomonas syringae MB03 against Caenorhabditis elegans and the transcriptional response of nematicidal genes upon different nutritional conditions. Front. Microbiol. 7:805. doi: 10.3389/fmicb.2016.00805
Ammari, M. G., Gresham, C. R., Mccarthy, F. M., and Nanduri, B. (2016). HPIDB 2.0: a curated database for host–pathogen interactions. Database 2016:baw103. doi: 10.1093/database/baw103
Angeloni, J., Dong, Y., Wang, Z., and Cao, M. (2020). Bacterial second messenger 3’, 5’-cyclic diguanylate attracts Caenorhabditis elegans and suppresses its immunity. Commun. Biol. 3, 1–11. doi: 10.1038/s42003-020-01436-9
Atkinson, S., Goldstone, R. J., Joshua, G. W., Chang, C. Y., Patrick, H. L., Camara, M., et al. (2011). Biofilm development on Caenorhabditis elegans by Yersinia is facilitated by quorum sensing-dependent repression of type III secretion. PLoS Pathog. 7:e1001250. doi: 10.1371/journal.ppat.1001250
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukhtar, M. S., Cherkis, K., et al. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7:e1002132. doi: 10.1371/journal.ppat.1002132
Bashir, A., Sun, Y., Yu, X., Sun, X., and Li, L. (2021). Nematicidal effects of 2-methyl-aconitate isomerase from the phytopathogen Pseudomonas syringae MB03 on the model nematode Caenorhabditis elegans. J. Inverteb. Pathol. 185:107669. doi: 10.1016/j.jip.2021.107669
Bashir, A., Tian, T., Yu, X., Meng, C., Ali, M., and Li, L. (2020). Pyoverdine-mediated killing of Caenorhabditis elegans by Pseudomonas syringae MB03 and the role of iron in Its pathogenicity. Int. J. Mol. Sci. 21:2198. doi: 10.3390/ijms21062198
Begun, J., Sifri, C. D., Goldman, S., Calderwood, S. B., and Ausubel, F. M. (2005). Staphylococcus aureus virulence factors identified by using a high-throughput caenorhabditis elegans-killing model. Infect. Immunity 73, 872–877. doi: 10.1128/IAI.73.2.872-877.2005
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C., et al. (2014). A user’s guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PLoS One 9:e105547. doi: 10.1371/journal.pone.0105547
Bernard, C. S., Brunet, Y. R., Gavioli, M., Lloubès, R., and Cascales, E. (2011). Regulation of type VI secretion gene clusters by σ54 and cognate enhancer binding proteins. J. Bacteriol. 193, 2158–2167. doi: 10.1128/JB.00029-11
Berry, C. L., Brassinga, A. K. C., Donald, L. J., Fernando, W. G. D., Loewen, P. C., and De Kievit, T. R. (2012). Chemical and biological characterization of sclerosin, an antifungal lipopeptide. Can. J. Microbiol. 58, 1027–1034. doi: 10.1139/w2012-079
Bhattacharya, D., Nowotny, J., Cao, R., and Cheng, J. (2016). 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 44, W406–W409. doi: 10.1093/nar/gkw336
Blom, J., Albaum, S. P., Doppmeier, D., Puehler, A., Vorhoelter, F.-J., Zakrzewski, M., et al. (2009). EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 10:154. doi: 10.1186/1471-2105-10-154
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Buell, C. R., Joardar, V., Lindeberg, M., Selengut, J., Paulsen, I. T., Gwinn, M. L., et al. (2003). The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 100, 10181–10186. doi: 10.1073/pnas.1731982100
Burlinson, P., Studholme, D., Cambray-Young, J., Heavens, D., Rathjen, J., Hodgkin, J., et al. (2013). Pseudomonas fluorescens NZI7 repels grazing by C. elegans, a natural predator. ISME J. 7, 1126–1138. doi: 10.1038/ismej.2013.9
Cezairliyan, B., Vinayavekhin, N., Grenfell-Lee, D., Yuen, G. J., Saghatelian, A., and Ausubel, F. M. (2013). Identification of Pseudomonas aeruginosa phenazines that kill Caenorhabditis elegans. PLoS Pathog. 9:e1003101. doi: 10.1371/journal.ppat.1003101
Chen, L., Xiong, Z., Sun, L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–D645. doi: 10.1093/nar/gkr989
Coloma-Rivero, R. F., Gómez, L., Alvarez, F., Saitz, W., Del Canto, F., Céspedes, S., et al. (2020). The role of the flagellar protein FlgJ in the virulence of Brucella abortus. Front. Cell. Infect. Microbiol. 10:178. doi: 10.3389/fcimb.2020.00178
Cox, G. N., Kusch, M., and Edgar, R. S. (1981). Cuticle of Caenorhabditis elegans: its isolation and partial characterization. J. Cell Biol. 90, 7–17. doi: 10.1083/jcb.90.1.7
Darby, C., Cosma, C. L., Thomas, J. H., and Manoil, C. (1999). Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 96, 15202–15207. doi: 10.1073/pnas.96.26.15202
Darby, C., Hsu, J. W., Ghori, N., and Falkow, S. (2002). Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature 417, 243–244. doi: 10.1038/417243a
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dasgupta, N., Wolfgang, M. C., Goodman, A. L., Arora, S. K., Jyot, J., Lory, S., et al. (2003). A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol. Microbiol. 50, 809–824. doi: 10.1046/j.1365-2958.2003.03740.x
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Dhillon, B. K., Chiu, T. A., Laird, M. R., Langille, M. G. I., and Brinkman, F. S. L. (2013). IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res. 41, W129–W132. doi: 10.1093/nar/gkt394
Dorati, F., Barrett, G. A., Sanchez-Contreras, M., Arseneault, T., José, M. S., Studholme, D. J., et al. (2018). Coping with environmental eukaryotes; identification of Pseudomonas syringae genes during the interaction with alternative hosts or predators. Microorganisms 6:32. doi: 10.3390/microorganisms6020032
Dubern, J.-F., Cigana, C., De Simone, M., Lazenby, J., Juhas, M., Schwager, S., et al. (2015). Integrated whole-genome screening for Pseudomonas aeruginosa virulence genes using multiple disease models reveals that pathogenicity is host specific. Environ. Microbiol. 17, 4379–4393. doi: 10.1111/1462-2920.12863
Dudnik, A., and Dudler, R. (2013a). High-quality draft genome sequence of Pseudomonas syringae pv. syringae strain SM, isolated from wheat. Genome Announc. 1, e00610–e00613. doi: 10.1128/genomeA.00610-13
Dudnik, A., and Dudler, R. (2013b). Non contiguous-finished genome sequence of Pseudomonas syringae pathovar syringae strain B64 isolated from wheat. Stand. Genomic Sci. 8, 420–429. doi: 10.4056/sigs.3997732
Feil, H., Feil, W. S., Chain, P., Larimer, F., Dibartolo, G., Copeland, A., et al. (2005). Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 102, 11064–11069. doi: 10.1073/pnas.0504930102
Feinbaum, R. L., Urbach, J. M., Liberati, N. T., Djonovic, S., Adonizio, A., Carvunis, A.-R., et al. (2012). Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLoS Pathog. 8:e1002813. doi: 10.1371/journal.ppat.1002813
Fernandez, M., Porcel, M., De La Torre, J., Molina-Henares, M. A., Daddaoua, A., Llamas, M. A., et al. (2015). Analysis of the pathogenic potential of nosocomial Pseudomonas putida strains. Front. Microbiol. 6:871. doi: 10.3389/fmicb.2015.00871
Freeman, Z. N., Dorus, S., and Waterfield, N. R. (2013). The KdpD/KdpE two-component system: integrating K+ homeostasis and virulence. PLoS Pathog. 9:e1003201. doi: 10.1371/journal.ppat.1003201
Fretin, D., Fauconnier, A., Kohler, S., Halling, S., Leonard, S., Nijskens, C., et al. (2005). The sheathed flagellum of Brucella melitensis is involved in persistence in a murine model of infection. Cell. Microbiol. 7, 687–698. doi: 10.1111/j.1462-5822.2005.00502.x
Gallagher, L. A., and Manoil, C. (2001). Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J. Bacteriol. 183, 6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001
Gallegos, M. T., Schleif, R., Bairoch, A., Hofmann, K., and Ramos, J. L. (1997). Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61, 393–410. doi: 10.1128/mmbr.61.4.393-410.1997
Garg, A., and Gupta, D. (2008). VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 9:62. doi: 10.1186/1471-2105-9-62
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S. E., Wilkins, M. R., Appel, R. D., et al. (2005). Protein Identification and Analysis Tools on the ExPASy Server. Germany: Springer. doi: 10.1385/1-59259-890-0:571
Gellatly, S. L., Needham, B., Madera, L., Trent, M. S., and Hancock, R. E. W. (2012). The Pseudomonas aeruginosa PhoP-PhoQ two-component regulatory system is induced upon interaction with epithelial cells and controls cytotoxicity and inflammation. Infect. Immunity 80, 3122–3131. doi: 10.1128/IAI.00382-12
Geng, C., Nie, X., Tang, Z., Zhang, Y., Lin, J., Sun, M., et al. (2016). A novel serine protease, Sep1, from Bacillus firmus DS-1 has nematicidal activity and degrades multiple intestinal-associated nematode proteins. Sci. Rep. 6:25012. doi: 10.1038/srep25012
Gilbert, K. B., Kim, T. H., Gupta, R., Greenberg, E. P., and Schuster, M. (2009). Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol. Microbiol. 73, 1072–1085. doi: 10.1111/j.1365-2958.2009.06832.x
Gomila, M., Busquets, A., Mulet, M., García-Valdés, E., and Lalucat, J. (2017). Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front. Microbiol. 8:2422. doi: 10.3389/fmicb.2017.02422
Grant, J. R., and Stothard, P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Haiko, J., and Westerlund-Wikstrom, B. (2013). The role of the bacterial flagellum in adhesion and virulence. Biology 2, 1242–1267. doi: 10.3390/biology2041242
Hassel, B., Dahlberg, D., Mariussen, E., Goverud, I. L., Antal, E.-A., Tonjum, T., et al. (2014). Brain infection with Staphylococcus aureus leads to high extracellular levels of glutamate, aspartate, gamma-aminobutyric acid, and zinc. J. Neurosci. Res. 92, 1792–1800. doi: 10.1002/jnr.23444
Hautefort, I., Thompson, A., Eriksson-Ygberg, S., Parker, M. L., Lucchini, S., Danino, V., et al. (2008). During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell. Microbiol. 10, 958–984. doi: 10.1111/j.1462-5822.2007.01099.x
Hirano, S. S., and Upper, C. D. (2000). Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae - a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 64, 624–653. doi: 10.1128/MMBR.64.3.624-653.2000
Hockett, K. L., Nishimura, M. T., Karlsrud, E., Dougherty, K., and Baltrus, D. A. (2014). Pseudomonas syringae CC1557: a highly virulent strain with an unusually small type III effector repertoire that includes a novel effector. Mol. Plant Microbe Interact. 27, 923–932. doi: 10.1094/MPMI-11-13-0354-R
Irazoqui, J. E., Troemel, E. R., Feinbaum, R. L., Luhachack, L. G., Cezairliyan, B. O., and Ausubel, F. M. (2010). Distinct pathogenesis and host responses during Infection of C. elegans by P. aeruginosa and S. aureus. PloS Pathog. 6:e1000982. doi: 10.1371/journal.ppat.1000982
Jiang, F., Waterfield, N. R., Yang, J., Yang, G., and Jin, Q. (2014). A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe 15, 600–610. doi: 10.1016/j.chom.2014.04.010
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: kEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kılıç, S., Sagitova, D. M., Wolfish, S., Bely, B., Courtot, M., Ciufo, S., et al. (2016). From data repositories to submission portals: rethinking the role of domain-specific databases in CollecTF. Database 2016:baw055. doi: 10.1093/database/baw055
Kim, S.-H., Park, S.-Y., Heo, Y.-J., and Cho, Y.-H. (2008). Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor. HudA. Infect. Immunity 76, 4152–4162. doi: 10.1128/IAI.01637-07
Kirienko, N. V., Ausubel, F. M., and Ruvkun, G. (2015). Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 112, 1821–1826. doi: 10.1073/pnas.1424954112
Kirienko, N. V., Kirienko, D. R., Larkins-Ford, J., Wahlby, C., Ruvkun, G., and Ausubel, F. M. (2013). Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe 13, 406–416. doi: 10.1016/j.chom.2013.03.003
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, S. H., Ooi, S. K., Mahadi, N. M., Tan, M. W., and Nathan, S. (2011). Complete killing of Caenorhabditis elegans by Burkholderia pseudomallei is dependent on prolonged direct association with the viable pathogen. PLoS One 6:e16707. doi: 10.1371/journal.pone.0016707
Li, Q., Yan, Q., Chen, J., He, Y., Wang, J., Zhang, H., et al. (2012). Molecular characterization of an ice nucleation protein variant (InaQ) from Pseudomonas syringae and the analysis of its transmembrane transport activity in Escherichia coli. Int. J. Biol. Sci 8, 1097–1108. doi: 10.7150/ijbs.4524
Lindeberg, M., Cartinhour, S., Myers, C. R., Schechter, L. M., Schneider, D. J., and Collmer, A. (2006). Closing the circle on the discovery of genes encoding Hrp regulon members and type III secretion system effectors in the genomes of three model Pseudomonas syringae strains. Mol. Plant Microbe Interact. 19, 1151–1158. doi: 10.1094/MPMI-19-1151
Luo, X., Chen, L., Huang, Q., Zheng, J., Zhou, W., Peng, D., et al. (2012). Bacillus thuringiensis metalloproteinase Bmp1 functions as a nematicidal virulence factor. Appl. Environ. Microbiol. 79, 460–468. doi: 10.1128/AEM.02551-12
Mahajan-Miklos, S., Tan, M. W., Rahme, L. G., and Ausubel, F. M. (1999). Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96, 47–56. doi: 10.1016/S0092-8674(00)80958-7
Manan, A., Bazai, Z., Fan, J., Yu, H., and Li, L. (2018). The Nif3-family protein YqfO03 from Pseudomonas syringae MB03 has multiple nematicidal activities against Caenorhabditis elegans and Meloidogyne incognita. Int. J. Mol. Sci. 19:3915. doi: 10.3390/ijms19123915
Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, P., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. doi: 10.1111/j.1364-3703.2012.00804.x
Marcelletti, S., Ferrante, P., Petriccione, M., Firrao, G., and Scortichini, M. (2011). Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS One 6:e27297. doi: 10.1371/journal.pone.0027297
Marteyn, B. S., Karimova, G., Fenton, A. K., Gazi, A. D., West, N., Touqui, L., et al. (2014). ZapE is a novel cell division protein interacting with FtsZ and modulating the Z-ring dynamics. Mbio 5, e22–e14. doi: 10.1128/mBio.00022-14
Matsumoto, K. (2004). Role of bacterial proteases in pseudomonal and serratial keratitis. Biol. Chem. 385, 1007–1016. doi: 10.1515/BC.2004.131
Meisel, Joshua D, Panda, O., Mahanti, P., and Schroeder, Frank C, and Kim, Dennis H (2014). Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell 159, 267–280. doi: 10.1016/j.cell.2014.09.011
Morris, C. E., Kinkel, L. L., Xiao, K., Prior, P., and Sands, D. C. (2007). Surprising niche for the plant pathogen Pseudomonas syringae. Infect. Genet. Evol. 7, 84–92. doi: 10.1016/j.meegid.2006.05.002
Morris, C. E., Sands, D. C., Vinatzer, B. A., Glaux, C., Guilbaud, C., Buffiere, A., et al. (2008). The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2, 321–334. doi: 10.1038/ismej.2007.113
Mougous, J. D., Cuff, M. E., Raunser, S., Shen, A., Zhou, M., Gifford, C. A., et al. (2006). A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312, 1526–1530. doi: 10.1126/science.1128393
Muller, C., Plesiat, P., and Jeannot, K. (2011). A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and beta-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 55, 1211–1221. doi: 10.1128/AAC.01252-10
Nandi, M., Selin, C., Brassinga, A. K. C., Belmonte, M. F., Fernando, W. G. D., Loewen, P. C., et al. (2015). Pyrrolnitrin and hydrogen cyanide production by Pseudomonas chlororaphis strain PA23 exhibits nematicidal and repellent activity against Caenorhabditis elegans. PLoS One 10:e0123184. doi: 10.1371/journal.pone.0123184
Njoroge, J. W., Nguyen, Y., Curtis, M. M., Moreira, C. G., and Sperandio, V. (2012). Virulence meets metabolism: cra and KdpE gene regulation in Enterohemorrhagic Escherichia coli. Mbio 3, e00280–12. doi: 10.1128/mBio.00280-12
O’Brien, H. E., Thakur, S., and Guttman, D. S. (2011). Evolution of plant pathogenesis in Pseudomonas syringae: a genomics perspective. Annu. Rev. Phytopathol. 49, 269–289. doi: 10.1146/annurev-phyto-072910-095242
O’Quinn, A. L., Wiegand, E. M., and Jeddeloh, J. A. (2001). Burkholderia pseudomallei kills the nematode Caenorhabditis elegans using an endotoxin-mediated paralysis. Cell. Microbiol. 3, 381–393. doi: 10.1046/j.1462-5822.2001.00118.x
Paiva, G., Proenca, D. N., Francisco, R., Verissimo, P., Santos, S. S., Fonseca, L., et al. (2013). Nematicidal bacteria associated to pinewood nematode produce extracellular proteases. PLoS One 8:e79705. doi: 10.1371/journal.pone.0079705
Pallen, M. J., and Wren, B. W. (2007). Bacterial pathogenomics. Nature 449, 835–842. doi: 10.1038/nature06248
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pineda-Castellanos, M. L., Rodriguez-Segura, Z., Villalobos, F. J., Hernandez, L., Lina, L., and Nunez-Valdez, M. E. (2015). Pathogenicity of Isolates of Serratia marcescens towards larvae of the scarab Phyllophaga blanchardi (coleoptera). Pathogens 4, 210–228. doi: 10.3390/pathogens4020210
Plotnikova, J. M., Rahme, L. G., and Ausubel, F. M. (2000). Pathogenesis of the human opportunistic pathogen Pseudomonas aeruginosa PA14 in Arabidopsis. Plant Physiol. 124, 1766–1774. doi: 10.1104/pp.124.4.1766
Powell, J. R., and Ausubel, F. A. (2008). “Models of Caenorhabditis elegans infection by bacterial and fungal pathogens” in Innate Immunity. eds J. Ewbank and E. Vivier (United States: Humana Press Inc). 403–427. doi: 10.1007/978-1-59745-570-1_24
Price, C. T. D., Bukka, A., Cynamon, M., and Graham, J. E. (2008). Glycine betaine uptake by the ProXVWZ ABC transporter contributes to the ability of Mycobacterium tuberculosis to initiate growth in human macrophages. J. Bacteriol. 190, 3955–3961. doi: 10.1128/JB.01476-07
Qiuhong, N., Xiaowei, H., Baoyu, T., Jinkui, Y., Jiang, L., Lin, Z., et al. (2006). Bacillus sp. B16 kills nematodes with a serine protease identified as a pathogenic factor. Appl. Microbiol. Biotechnol. 69, 722–730. doi: 10.1007/s00253-005-0019-5
Ravindran, A., Jalan, N., Yuan, J. S., Wang, N., and Gross, D. C. (2015). Comparative genomics of Pseudomonas syringae pv. syringae strains B301D and HS191 and insights into intrapathovar traits associated with plant pathogenesis. MicrobiologyOpen 4, 553–573. doi: 10.1002/mbo3.261
Ray, A., Rentas, C., Caldwell, G. A., and Caldwell, K. A. (2015). Phenazine derivatives cause proteotoxicity and stress in C. elegans. Neurosci. Lett. 584, 23–27. doi: 10.1016/j.neulet.2014.09.055
Saha, S., and Raghava, G. (2006). VICMpred: an SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genomics Proteomics Bioinform. 4, 42–47. doi: 10.1016/S1672-0229(06)60015-6
Salikin, N. H., Dubois, M., Nappi, J., Lebhar, H., Marquis, C., and Egan, S. (2021). Novel nematode-killing protein-1 (Nkp-1) from a marine epiphytic bacterium Pseudoalteromonas tunicata. Biomedicines 9:1586. doi: 10.3390/biomedicines9111586
Sarkar, S. F., and Guttman, D. S. (2004). Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol. 70, 1999–2012. doi: 10.1128/AEM.70.4.1999-2012.2004
Sarris, P. F., Skandalis, N., Kokkinidis, M., and Panopoulos, N. J. (2010). In silico analysis reveals multiple putative type VI secretion systems and effector proteins in Pseudomonas syringae pathovars. Mol. Plant Pathol. 11, 795–804. doi: 10.1111/j.1364-3703.2010.00644.x
Shao, Y., He, X., Harrison, E. M., Tai, C., Ou, H.-Y., Rajakumar, K., et al. (2010). mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 38, W194–W200. doi: 10.1093/nar/gkq326
Tamber, S., Ochs, M. M., and Hancock, R. E. (2006). Role of the novel OprD family of porins in nutrient uptake in Pseudomonas aeruginosa. J. Bacteriol. 188, 45–54. doi: 10.1128/JB.188.1.45-54.2006
Tan, M. W., Mahajan-Miklos, S., and Ausubel, F. M. (1999a). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 96, 715–720. doi: 10.1073/pnas.96.2.715
Tan, M. W., Rahme, L. G., Sternberg, J. A., Tompkins, R. G., and Ausubel, F. M. (1999b). Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 96, 2408–2413. doi: 10.1073/pnas.96.5.2408
Uppalapati, S. R., Ishiga, Y., Wangdi, T., Urbanczyk-Wochniak, E., Ishiga, T., Mysore, K. S., et al. (2008). Pathogenicity of Pseudomonas syringae pv. tomato on tomato seedlings: phenotypic and gene expression analyses of the virulence function of coronatine. Mol. Plant Microbe Interact. 21, 383–395. doi: 10.1094/MPMI-21-4-0383
Van Gijsegem, F., Gough, C., Zischek, C., Niqueux, E., Arlat, M., Genin, S., et al. (1995). The hrp gene locus of Pseudomonas solanacearum, which controls the production of a type III secretion system, encodes eight proteins related to components of the bacterial flagellar biogenesis complex. Mol. Microbiol. 15, 1095–1114. doi: 10.1111/j.1365-2958.1995.tb02284.x
Wargo, M. J. (2013). Homeostasis and catabolism of choline and glycine betaine: lessons from Pseudomonas aeruginosa. Appl. Environ. Microbiol. 79, 2112–2120. doi: 10.1128/AEM.03565-12
Wei, J.-Z., Siehl, D. L., Hou, Z., Rosen, B., Oral, J., Taylor, C. G., et al. (2017). An enterotoxin-like binary protein from Pseudomonas protegens with potent nematicidal activity. Appl. Environ. Microbiol. 83, e00942–17. doi: 10.1128/AEM.00942-17
White, C. V., Darby, B. J., Breeden, R. J., and Herman, M. A. (2015). A Stenotrophomonas maltophilia strain evades a major Caenorhabditis elegans defense pathway. Infect. Immunity 84, 524–536. doi: 10.1128/IAI.00711-15
Xiao, G., Deziel, E., He, J., Lepine, F., Lesic, B., Castonguay, M. H., et al. (2006). MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol. Microbiol. 62, 1689–1699. doi: 10.1111/j.1365-2958.2006.05462.x
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yoon, S. J., Park, Y. J., Kim, J. S., Lee, S., Lee, S. H., Choi, S., et al. (2018). Pseudomonas syringae evades phagocytosis by animal cells via type III effector-mediated regulation of actin filament plasticity. Environ. Microbiol. 20, 3980–3991. doi: 10.1111/1462-2920.14426
Yoshihara, E., Gotoh, N., Nishino, T., and Nakae, T. (1996). Protein D2 porin of the Pseudomonas aeruginosa outer membrane bears the protease activity. FEBS Lett. 394, 179–182. doi: 10.1016/0014-5793(96)00945-3
Young, G. M., Schmiel, D. H., and Miller, V. L. (1999). A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. U. S. A. 96, 6456–6461. doi: 10.1073/pnas.96.11.6456
Yu, C. S., Lin, C. J., and Hwang, J. K. (2004). Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13, 1402–1406. doi: 10.1110/ps.03479604
Zaborin, A., Romanowski, K., Gerdes, S., Holbrook, C., Lepine, F., Long, J., et al. (2009). Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc. Natl. Acad. Sci. U. S. A. 106, 6327–6332. doi: 10.1073/pnas.0813199106
Zhang, F., Peng, D., Cheng, C., Zhou, W., Ju, S., Wan, D., et al. (2016). Bacillus thuringiensis crystal protein Cry6Aa triggers Caenorhabditis elegans necrosis pathway mediated by aspartic protease (ASP-1). PLoS Pathog. 12:e1005389. doi: 10.1371/journal.ppat.1005389
Keywords: Pseudomonas syringae, Caenorhabditis elegans, pathogenomics, transcriptomics, transposon mutant library, gut colonization, nematicidal activity
Citation: Ali M, Gu T, Yu X, Bashir A, Wang Z, Sun X, Ashraf NM and Li L (2022) Identification of the Genes of the Plant Pathogen Pseudomonas syringae MB03 Required for the Nematicidal Activity Against Caenorhabditis elegans Through an Integrated Approach. Front. Microbiol. 13:826962. doi: 10.3389/fmicb.2022.826962
Received: 01 December 2021; Accepted: 11 February 2022;
Published: 09 March 2022.
Edited by:
Imen Nouioui, German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), GermanyReviewed by:
Petar Pujic, Centre National de la Recherche Scientifique (CNRS), FranceHilal Ay, Ondokuz Mayıs University, Turkey
Copyright © 2022 Ali, Gu, Yu, Bashir, Wang, Sun, Ashraf and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Li, bGlsaW5AbWFpbC5oemF1LmVkdS5jbg==