 Lynise C. Pillay
Lynise C. Pillay Lucpah Nekati
Lucpah Nekati Phuti J. Makhwitine
Phuti J. Makhwitine Sizwe I. Ndlovu
Sizwe I. Ndlovu- Discipline of Medical Microbiology, School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
The discovery of silent biosynthetic gene clusters (BGCs) in fungi provides unlimited prospects to harness the secondary metabolites encoded by gene clusters for various applications, including pharmaceuticals. Amplifying these prospects is the new interest in exploring fungi living in the extremes, such as those associated with plants (fungal endophytes). Fungal species in endosymbiosis relationship with plants are recognized as the future factories of clinically relevant agents since discovering that they can produce similar metabolites as their plant host. The endophytes produce these compounds in natural environments as a defense mechanism against pathogens that infect the plant host or as a strategy for mitigating competitors. The signaling cascades leading to the expression of silent biosynthetic gene clusters in the natural environment remain unknown. Lack of knowledge on regulatory circuits of biosynthetic gene clusters limits the ability to exploit them in the laboratory. They are often silent and require tailor-designed strategies for activation. Epigenetic modification using small molecular compounds that alter the chromatin network, leading to the changes in secondary metabolites profile, has achieved considerable success. This review aims to comprehensively analyze the secondary metabolite profiles expressed after treatment with various epigenetic modifiers. We first describe the regulatory circuits governing the expression of secondary metabolites in fungi. Following this, we provide a detailed review of the small molecular modifiers, their mechanism(s) of action, and the diverse chemistries resulting from epigenetic modification. We further show that genetic deletion or epigenetic inhibition of histone deacetylases does not always lead to the overexpression or induction of silent secondary metabolites. Instead, the response is more complex and often leads to differential expression of secondary metabolites. Finally, we propose using this strategy as an initial screening tool to dereplicate promising fungal species.
Introduction
Endophytic fungi have recently attracted attention in screening programs after discovering that they can produce similar metabolites to their plant hosts (Deepika et al., 2016). Endophytic microorganisms colonize the intracellular and intercellular regions of healthy plant tissues without damaging the host plant (Rodriguez et al., 2009; Nair and Padmavathy, 2014). In this relationship, they produce secondary metabolites that play an essential role in defending the plant host from pathogens, thus protecting its habitat (Hardoim et al., 2015; Deepika et al., 2016). Secondary metabolism is not directly involved in the producing organism’s growth, development, or reproduction. Still, it may act as chemical signals during cell-to-cell communications in natural environments (Netzker et al., 2015). Interestingly, the produced secondary metabolites exhibit sought-after properties in the pharmaceutical industries such as antibacterial, antifungal, anticancer, and other bioactivities (Tyurin et al., 2018).
Secondary metabolites (SMs) are low molecular weight substances encoded by contiguous gene assembles termed biosynthetic gene clusters (BGCs), which encode enzymes responsible for a stepwise assembly of complex bioactive molecules (Pfannenstiel and Keller, 2019). Many of the fungal-derived secondary metabolites, such as cyclosporines, statins, and penicillins were discovered in the early drug screening programs (Harvey et al., 2018; Ding et al., 2020). However, the search for active molecules from microorganisms declined due to a continuous rediscovery of known metabolites (Jackson et al., 2018). As a result, pharmaceutical companies shifted their investment interests to synthetic drug development routes (Jackson et al., 2018).
Recent advances in genome sequencing technologies have revealed that fungi are host to many novel BGCs other than those previously identified in natural product screening programs (Macheleidt et al., 2016). Fungi, more especially those from unexplored environments such as fungal endophytes, are promising alternative sources of novel chemistries that will provide potent drugs that have the potential to combat the growing challenge of antibiotic resistance and the emergence of new multidrug-resistant pathogens (Phukan et al., 2018). However, the problem is that most of these BGCs do not produce appreciable concentrations of metabolites during cultivation in standard laboratory conditions and are regarded as “silent” or “cryptic” (Xu et al., 2017; Pfannenstiel and Keller, 2019). Therefore, there is a discrepancy between fungi’ secondary metabolite production potential and the currently known fungal metabolites in the clinical pipeline. For this reason, there is a fast-growing research interest aimed at identifying new approaches to activating the silent secondary metabolites encoding genes (Macheleidt et al., 2016).
Successful activation of silent BGCs requires a proper understanding of the regulatory circuits governing the secondary metabolites gene clusters. The signaling cues that trigger the expression of the silent or transiently expressed BGCs in natural environments remain unknown (Reen et al., 2015). Several strategies developed to awaken the ‘silent’ genes have shown to significantly influence the profile of secondary metabolites produced (VanderMolen et al., 2013; Hewage et al., 2014; Nielsen and Larsen, 2015; Begani et al., 2018; Scherlach and Hertweck, 2021). The activation of these genes can be achieved at the genome, transcriptome, proteome, or metabolome levels (Scherlach and Hertweck, 2009; Cichewicz, 2010; Ochi and Hosaka, 2013; Begani et al., 2018; Scherlach and Hertweck, 2021). There are several exciting reviews on these strategies, such as those noted by Rutledge and Challis (2015), which categorizes these approaches into pleiotropic and pathway-specific.
Several BGC awakening strategies (based on culturing techniques and genetic manipulation) proposed to date have varying advantages and disadvantages. The culture-based strategies include the variation of growth conditions, also known as one strain many compounds (OSMAC) and co-culturing (Pan et al., 2019). In the OSMAC strategy, growth conditions such as temperature, media, cultivation time, etc., can be systematically varied to simulate the changing signaling cues in the environment (Bode et al., 2002; Guzmán-Chávez et al., 2018). Different studies have shown that small changes in cultivation conditions and media composition (OSMAC approach) completely shift the metabolic profile of various filamentous fungi (Bode et al., 2002; Romano et al., 2018; González-Menéndez et al., 2019; Toghueo et al., 2020). The second culture-based approach involves co-culturing with microorganisms such as bacteria, where crosstalk between the co-cultivated organisms induces the production of metabolites encoded by the silent BGCs (Bertrand et al., 2014; Rutledge and Challis, 2015; Pfannenstiel and Keller, 2019). The challenge with the culture-based approaches is that they are laborious, requiring scientists to set up several experiments encompassing different conditions (Romano et al., 2018).
Several genetic manipulation strategies have been developed to awaken the silent biosynthetic gene clusters in fungi (Jiang et al., 2021). One of these strategies involves engineering the transcription and translation machinery, which is achieved by introducing mutational changes in RNA polymerase and proteins, thus increasing BGC expression (Rutledge and Challis, 2015; Pfannenstiel and Keller, 2019). Another genetic approach includes manipulating global regulators to activate more than one pathway to discover multiple metabolites (Rutledge and Challis, 2015; Baral et al., 2018). In a review by Rutledge and Challis (2015), they discussed the four pathway-specific approaches, including (1) Manipulating pathway-specific regulators to increase the expression of BGCs. This approach requires prior knowledge of genes that encode putative pathway-specific transcription factors (Rutledge and Challis, 2015). (2) Reporter-guided mutant selection method allows for the visualization of mutant expressing a target BGC (Rutledge and Challis, 2015). (3) Refactoring by using constitutive or readily inducible promoters, replacing natural promoters (Rutledge and Challis, 2015; Fan et al., 2017; Baral et al., 2018). (4) After the BGC of interest has been identified, the genes are activated and expressed in a heterologous host. The heterologous expression approach simplifies metabolite identification workflows (Rutledge and Challis, 2015; Fan et al., 2017; Harvey et al., 2018). Refactoring is seen as the best approach since fungi are typically resistant to genetic manipulation, and heterologous expression might provide a better alternative for expressing fungal biosynthetic genes (Keller, 2019). However, there are limited successes achieved through these approaches. Although these genetic approaches have shown successes in different fungal species, their wide application in fungi remains limited. The limitations are due to fungi’s complex genetic system and the lack of genetic tools such as selection markers for application in filamentous fungi (Jiang et al., 2021). It appears that non-genetic strategies are still valuable, especially for novel fungal isolates with no previous genetic information.
Application of small molecular compounds that epigenetically modifies the chromatin leading to the induction of silent fungal BGCs have shown considerable successes (Strauss and Reyes-Dominguez, 2011; Akone et al., 2016). Despite the attention given to this approach, there are a few reviews (Cichewicz, 2010; Pettit, 2011; Poças-Fonseca et al., 2020; Toghueo et al., 2020) that consolidate the findings thus far. This review aims to analyze the successes achieved using this strategy thus far. This analysis will be achieved by exploring the secondary metabolite profiles expressed after treatment with various epigenetic modifiers. We first describe the regulatory circuits governing the expression of secondary metabolites in fungi. The relationship between the plants and fungal endophytes is presented as an example of an environmental niche with dynamic interactions and a high potential for novel chemistries. Following this, we provide a detailed review of the small molecular modifiers, their mechanism(s) of action, and the diverse chemistries resulting from epigenetic modification. We further show that genetic deletion or epigenetic inhibition of histone deacetylases does not always lead to the overexpression or induction of silent secondary metabolites. Instead, the response is more complex and often leads to differential expression of secondary metabolites. Finally, we propose using this strategy as an initial screening tool to dereplicate promising fungal species.
Endosymbiotic Relationship Between Fungal Endosymbionts and their Plant Hosts
Endophytic fungi belong to mitosporic and meiosporic ascomycetes that asymptomatically colonize the internal tissues of the plant host (Jia et al., 2016). Endophytic fungi have a significant biological diversity, as most plants host one or more endophytic fungus species (Jia et al., 2016; Rana et al., 2019). The host plant secretes compounds as a defense system to protect itself from colonization (Jia et al., 2016). In response, the endophytic fungi secrete the matching detoxifying enzymes such as cellulases, lactase, xylanase, and protease, to decompose these plant protective compounds, thus allowing these endophytes to penetrate through the plant defense systems (Jia et al., 2016; Khare et al., 2018). Once the endophytic fungi colonize the inner tissues of the plant, fungi assume a latent state either for the whole lifetime of the plant host (neutralism) or for an extended period (mutualism or antagonism) (Jia et al., 2016).
There are several benefits for the plant host resulting from this symbiotic relationship with fungal endophytes. The benefits include induced plant growth, increased resistance to disease, and the stimulation of secondary metabolites production (Alvin et al., 2014; Fadiji and Babalola, 2020). Fungal endophytes produce secondary metabolites in response to external stimuli such as changes in nutritional needs or pathogenic threats (Alvin et al., 2014; Macheleidt et al., 2016). These secondary metabolites may serve as defense molecules mediating nutrient acquisition and species-species communication (Strobel, 2003; Berg and Hallmann, 2006; Macheleidt et al., 2016). Secondary metabolites produced by fungal endophytes can trigger a defense mechanism by the plant host against specific plant pathogens (Kloepper and Ryu, 2006; Alvin et al., 2014). Also, fungal endophytes can encourage increased cell apoptosis when pathogens infect the plant host. This response can occur in several ways, such as producing phytohormones, siderophores, etc. (Berg and Hallmann, 2006; Alvin et al., 2014). Other benefits include a natural resistance to soil contaminants, the ability to degrade xenobiotics, and their action as vectors to present degradative traits to plants (Ryan et al., 2008). The host plant also provides many benefits to the development and survival of the endophytic fungi (Fadiji and Babalola, 2020).
The host plant’s genetic background determines its endophyte population structure as substrates produced by the host plant impact the colonization and distribution of the endophytic fungi (Card et al., 2016; Jia et al., 2016). These factors indicate that the host plant’s fitness affects the fitness of endophytic fungi (Jia et al., 2016). The symbiotic relationship between the plant and its endophytes proves that endophytic bioactive compounds are likely to possess reduced cell toxicity since the secondary metabolites produced during this interaction do not harm the eukaryotic host system (Jia et al., 2016; Fadiji and Babalola, 2020). This suggests that active secondary metabolites produced by endophytic fungi in this symbiotic relationship are potentially selectively toxic to invading agents but not their plant host. Selective toxicity is an important criterion when developing pharmaceutical drugs, as potential drugs may not adversely affect recipient cells (Strobel, 2003; Alvin et al., 2014).
Biosynthetic Gene Clusters in Fungal Species and their Regulation
Organization of the Biosynthetic Gene Clusters in Fungal Species
Secondary metabolites are produced by contiguous gene assembles termed biosynthetic gene clusters (BGCs), which encode enzymes responsible for a stepwise assembly of complex bioactive molecules (Xu et al., 2017; Tyurin et al., 2018). The clustering of the biosynthetic genes is suggested to ensure coordinated regulation of secondary metabolism (Shwab et al., 2007). Fungal BGCs characteristically exhibit modularity of enzymatic domains. They can exceed 100 kb in size, a feature that creates bottlenecks when using molecular biology techniques to capture their activities through heterologous expression (Reen et al., 2015). Classes of BGCs include non-ribosomal peptides (NRPs), polyketides (PKs), ribosomally synthesized and post-translationally modified peptides (RiPPs), terpenoids, saccharides, and hybrid compounds being the most widely characterized (Alvin et al., 2014; Reen et al., 2015; Macheleidt et al., 2016). The NRPs are characterized by three main domains, (1) the adenylation (A) domain responsible for amino acid recognition, (2) the peptidyl carrier protein (PCP) responsible for activation as well as (3) a condensation (C) domain responsible for bond formation. Typical PKs also consists of three domains which include (1) an acyltransferase (AT) which acts as the gatekeeper for substrate specificity, selecting and activating the monomers and the intermediate acyl chain, (2) a ketosynthase (KS) which catalyzes C-C bond formation via the Claisen condensation in the elongation of the polyketide chain, and (3) the thioesterase (TE) domain which is responsible for the termination of the growing chain The structural details of the fungal biosynthetic gene clusters will not be covered in details in this review, but the reader is referred to an excellent review by Guzmán-Chávez et al. (2018). The fungal BGCs represent a potential source for new scaffolds to discover novel antimicrobial compounds (Tyurin et al., 2018).
Fungal biosynthetic genes tend to position proximal to the telomeres in the genome in heterochromatin regions (Brosch et al., 2008). Biosynthetic genes are often transcriptionally controlled by epigenetic regulation through histone deacetylation and DNA methylation (Pettit, 2011). The changes in biotic and abiotic conditions in the natural environments trigger the fungal chromatin-based induction of silent secondary metabolites biosynthetic gene clusters. Essentially, these triggers are environmental factors and nutrients which strongly influence the production of secondary metabolites by fungi (Zutz et al., 2013). However, fungal fermentations on artificially defined media in the laboratory tend to be poor surrogates to mimic the endophytic environmental conditions. This often results in the induction of only a subset of biosynthetic pathways encoding secondary metabolites thus limiting the full biosynthetic prospects of fungi (Williams et al., 2008). Understanding the regulatory processes will assist in realizing the full potential of endophytic fungi.
Genetic Regulation of Fungal Biosynthetic Gene Clusters Encoding Secondary Metabolite at a Chromatin Level
Secondary metabolism is not immediately essential for the organism but produced secondary metabolites may influence the organism’s competitiveness in natural environments, hence the tight regulation of these biosynthetic genes (Gacek and Strauss, 2012). Biosynthetic gene clusters encoding secondary metabolites are typically activated in response to environmental stimuli, and this expression depends on the developmental stage of the fungus (Keller, 2019). In fungi, the chromatin structure has emerged as a fundamental regulatory circuit of cellular metabolism, with the chromatin-modifying enzymes playing an essential role in fungal secondary metabolism (Brosch et al., 2008; Nützmann et al., 2013).
The Structure of the Chromatin and Its Role in the Control of Gene Expression
The chromatin is composed of DNA (∼147 base pairs of double-stranded DNA) cross-linked to an octamer of histone proteins, H2A, H2B, H3, and H4 (Aghcheh and Kubicek, 2015; Sun et al., 2021). The tightly packed DNA and structural nuclear proteins like histones and non-histone chromatin-associated proteins together form a nucleosome (Nützmann et al., 2013). Linker histones, mainly H1, bind to the nucleosome at the entry and exit site. It does not make up the histone’ “’bead” but sits on top of the structure, which assists the chromatin in folding into higher-order structures (Fyodorov et al., 2018).
The chromatin acts as a natural substrate for the transcriptional machinery and regulatory proteins that replicate and repair DNA and chromosome segregation (Gacek and Strauss, 2012; Nützmann et al., 2013). Histones H2A, H2B, H3, and H4 exist as dimers and are the four core proteins (Fyodorov et al., 2018; Sun et al., 2021). They have a basic N-terminal domain and a histone-fold C-terminal domain (Caterino and Hayes, 2011). Histones H3 and H4 are almost identical in plants and animals, while H2A and H2B histones differ from species to species (Fyodorov et al., 2018). Histone H2A is well characterized and is found at sites with specific transcriptional control, chromatin remodeling, and when DNA repair is required (Giaimo et al., 2019). The H3 and H4 histones have long tails extending from the nucleosome, representing sites where pathways for signal transduction affect the chromatin structure. Moreover, numerous other minor histone variants play a specialized role in regulating the chromatin (Martire and Banaszynski, 2020).
Histones are substrates for several post-translational modifications (PTMs), including methylation, acetylation, phosphorylation, ubiquitination, SUMOylation, citrullination, and ADP-ribosylation that can modify their interaction with nuclear proteins and DNA (Bannister and Kouzarides, 2011; Aghcheh and Kubicek, 2015). Post-translational modifications are central in defining the state of the chromatin, which can either be loosely (euchromatin) or tightly (heterochromatin) packaged (Figure 1; Studt et al., 2013). These modifications are likely to control the interaction of histones with transcriptional activators and repressors (Shwab et al., 2007). Histone acetylation and DNA methylation are the most commonly occurring of all post-translational modifications (Studt et al., 2013). When the histones are highly acetylated, they are loosely bound to DNA and renders the chromatin transcriptionally accessible (Okada and Seyedsayamdost, 2017). However, it has been shown that the post-translational modification of histones through either methylation, acetylation and/or phosphorylation can lead to chromatin rearrangements that can improve selective accessibility of DNA by some transcription factors (Bannister and Kouzarides, 2011; Fischer et al., 2016).
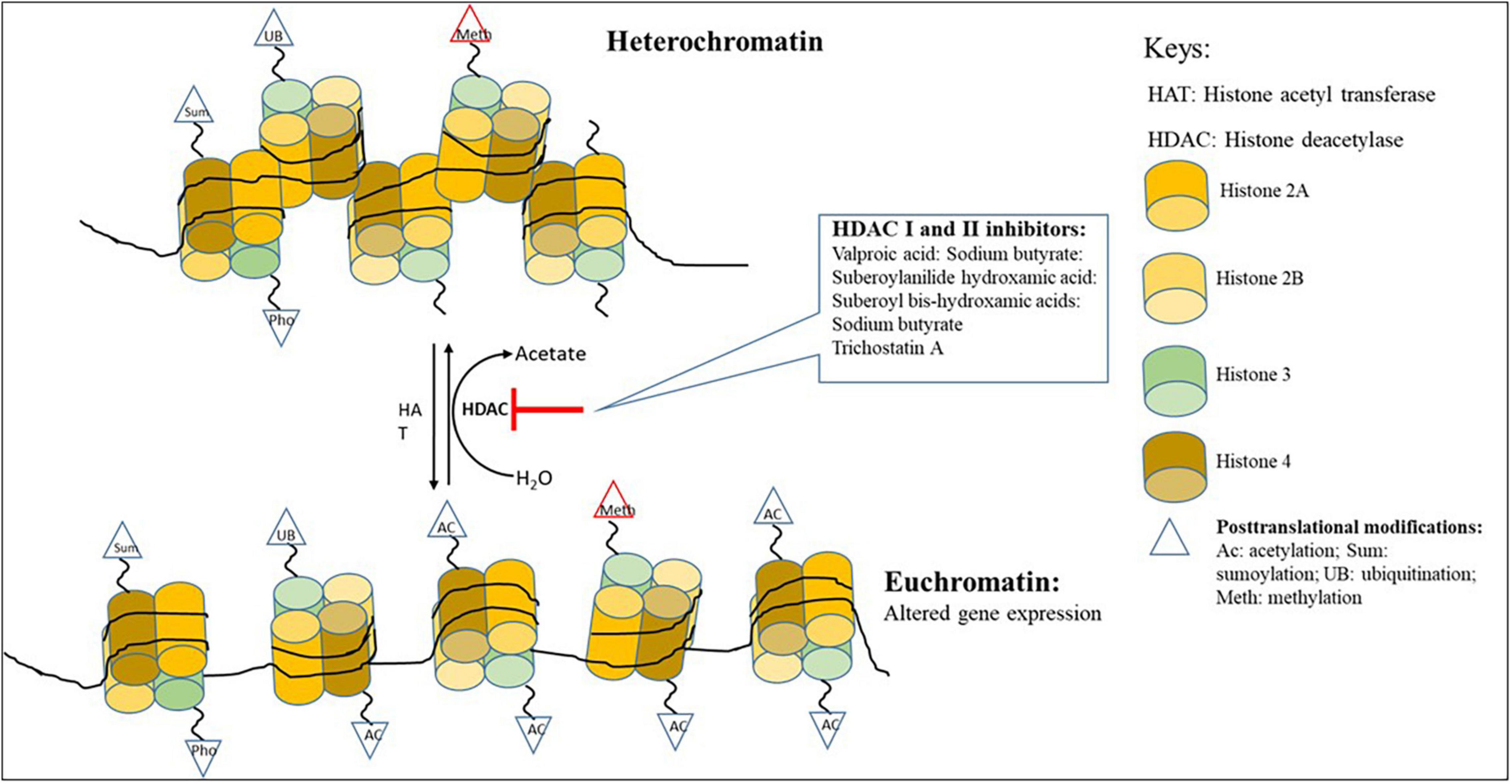
Figure 1. Chromatin remodeling showing the changing of heterochromatin to euchromatin with the addition of HDAC inhibitors.
Changes in the chromatin structure are essential in the functioning of the cell and may lead to short and long alterations in the transcriptional activities of the cell. These changes are governed by intrinsic cellular and extrinsic environmental factors (Brosch et al., 2008). They play a central role in maintaining cellular integrity, differentiation, development, and metabolism (Brosch et al., 2008). The intrinsic cellular factors are mediated by enzymes such as histone deacetylases (HDACs) and histone acetyltransferases (HATs) which are responsible for eliminating, establishing, and preserving local chromatin modification that either expresses or represses gene transcription (Albright et al., 2015). Histone binding proteins, histone acetyltransferase, and methyltransferases can regulate the expression of secondary metabolite BGCs in filamentous fungi (Gacek and Strauss, 2012). Two main groups of these small-molecule epigenetic modifiers: HDAC inhibitors and DNMT inhibitors, play an influential role as elicitors in producing secondary metabolites (Okada and Seyedsayamdost, 2017). The HATs and HDACs have antagonistic action on histone acetylation. HATs are responsible for acetylation, which is linked to gene transcription, and HDACs involved in deacetylation is associated with gene silencing (Bannister and Kouzarides, 2011; Studt et al., 2013).
Histone deacetylases (HDACs) are categorized into three families (1) zinc-dependent (classical) HDACs (e.g., class 1, class 2, HOS3-like HDACs in fungi, and class 4 found in other Eukaryotes), (2) nicotinamide adenine dinucleotide (NAD+)-dependent SIR-like HDACs (Sirtuins), and (3) HD2-like enzymes (found exclusively in plants) (Brosch et al., 2008; Lamoth et al., 2015). Classical HDACs are particularly interesting as they play a pivotal role in the regulation of secondary metabolism. Classical HDACs can be further classified into class1 or RPD3-type HDACs (RPD3; RPDA in A. nidulans, Hos1, Hos2; HosA in A. nidulans) and class 2 or HDA1-type (HDA1; HdaA in A. nidulans) (Tribus et al., 2010; Lamoth et al., 2015). Deleting deacetylases or chemical inhibition of deacetylase using small molecular elicitors that inactivate deacetylases’ activity often leads to increased production of known or novel secondary metabolites in several fungi (Gacek and Strauss, 2012). The HDACs and HATs are instrumental in transitioning between heterochromatin and euchromatin (Fischer et al., 2016).
Global Regulation of Biosynthetic Gene Clusters in Fungal Species
The discovery of LaeA (loss of aflR expression) in Aspergillus species, a nuclear methyltransferase protein, has provided insights into regulating secondary metabolite production in filamentous fungi (Bok and Keller, 2004). Sequence analysis of a gene encoding LaeA protein showed that this protein is similar to histone and arginine methyltransferases (Chiang et al., 2009). Furthermore, in a study by Perrin et al. (2007), they compared the transcriptional profile of the wild type, ΔlaeA mutant, and complemented strains of A. fumigatus. They showed that laeA was involved in the transcriptional control of 13 of the 22 secondary metabolite gene clusters of this species. Seven of these gene clusters controlled by laeA were located in subtelomeric regions. The location of laeA regulated genes and its sequence similarity to methyltransferases provided strong evidence that laeA might be involved in chromatin remodeling (Fox and Howlett, 2008). Apart from regulating secondary metabolite gene clusters in fungi, LaeA is also a critical factor in fungal morphology and development (Calvo et al., 2002; Bok and Keller, 2004). Global manipulation of transcription factors allows for simultaneous targeting of several secondary metabolite gene clusters (Bok and Keller, 2004). This approach has been demonstrated in Aspergillus spp. through the overexpression of laeA, which activated or enhanced the production of several known bioactive compounds (Bok and Keller, 2004). Furthermore, LaeA, alongside the highly conserved velvet complex proteins, is believed to play a role in fungal pathogenicity (Fischer et al., 2016).
The LaeA/LAE1 protein is directly involved in the transcription of various secondary metabolite gene clusters through direct interaction with the velvet domain family (Aghcheh and Kubicek, 2015). It forms a heterotrimeric with the VelB and VelA of the velvet family proteins (VelB-VelA-LaeA complex), that coordinate the light signal-dependent fungal development linked to secondary metabolism (Fischer et al., 2016; Chang et al., 2019). The presence of a methyltransferase domain in LaeA is associated with the regulation of secondary metabolite gene cluster expression through the epigenetic modification of the chromatin structure (Perrin et al., 2007). The LaeA protein is thought to facilitate the repression of the heterochromatin via association with either heterochromatin-associated methylases or deacetylases (Shwab and Keller, 2008; Kosalková et al., 2009).
LaeA in chromatin remodeling is a global regulator whose role is mainly facilitated by histone methylation (Wu and Yu, 2015). Deletion of laeA blocked the expression of sterigmatocystin, while its overexpression triggered an increased gene transcription of certain products such as penicillin and lovastatin in various Aspergillus spp. (Bok and Keller, 2004; Wu and Yu, 2015). Deletion of hdaA, a methyltransferase from Aspergillus that is a homologous of LaeA histone HDAC, increased secondary metabolite production (Okada and Seyedsayamdost, 2017). Further evidence indicated that the deletion of hdaA, in the fungal endophyte, Calcarisporium arbuscular led to the activation of ten compounds. Four of these were novel structures, including three tricyclic diterpenes and a novel meroterpenoid (Mao et al., 2015; Ding et al., 2020). LaeA protein is thus considered a global regulator of secondary metabolism whose levels modify the profile of secondary metabolites production (Lim et al., 2012).
Bok et al. (2009) hypothesized that in addition to LaeA protein, other chromatin-modifying proteins such as the members of the COMPASS (complex associated with Set1) also play a vital role in regulating the biosynthetic gene clusters. They identified CclA from A. nidulans, an ortholog of Bre2 in Saccharomyces cerevisiae. This gene was deleted together with a gene encoding a fatty acid synthase (stcJ) previously identified in A. nidulans, which is required for sterigmatocystin production (Bok and Keller, 2004; Bok et al., 2006, 2009). The double mutant strain could not produce sterigmatocytin. However, it was able to produce two known compounds (A. nidulans austinol and dehydroaustinol) and six aromatic compounds not previously identified in this fungus, and they were identified as monodictyphenone, emodin and four emodin analogs. All the compounds produced shared a similar aromatic non-reduced polyketide structure (Bok et al., 2009). The authors targeted ten unknown non-reduced polyketide clusters and were able to link polyketides responsible for producing all the identified compounds (Bok et al., 2009).
Chromatin Remodeling Alters the Profile of Fungal Secondary Metabolites
Following this discovery that the disruption of histone deacetylase activity led to the transcriptional activation of gene clusters encoding for sterigmatocystin and penicillin, Shwab et al. (2007) showed that histone-modifying enzymes influence secondary metabolism. They found that HDAC mutants of A. nidulans generated by the deletion of hdaA bypassed the regulation by laeA. The genetic depletion of hdaA resulted in an increased expression of subtelomeric secondary metabolites, sterigmatocystin and penicillin (Shwab et al., 2007; Gacek and Strauss, 2012). These results provided strong evidence that hdaA mediate the expression of subtelomeric secondary metabolite gene clusters in Aspergillus. The role of hdaA as a global suppresser of biosynthetic genes was postulated to be a common occurrence since this HDAC is conserved in filamentous fungi (Shwab et al., 2007; Mao et al., 2015). Indeed, deletion of hdaA in filamentous fungi such as Calcarisporium arbuscular (Mao et al., 2015), Penicillium chrysogenum Fes1701 (Ding et al., 2020) resulted in a pleiotropic activation of secondary metabolites. Although this gene is an obvious target for activating secondary metabolite production, its deletion often results in other development defects such as slower growth, shorter mycelia, and defective sporulation (Mao et al., 2015). For example, in a study by Niu et al. (2015), they observed a decrease in conidia production upon the deletion of a putative histone deacetylase gene, hid1 in Pestalotiopsis microspore NK17. The deletion of this HDAC gene resulted in a twofold increase pestalotiollide B yield. These observations are expected since HDACs are directly or indirectly involved in the development, proliferation, differentiation, and cell death (Kim and Bae, 2011; Niu et al., 2015).
The results observed after genetic depletion of HDAC suggested that pharmacological depletion using chemical HDAC inhibitors (known as an epigenetic modification) could provide means of regulating or increasing secondary metabolite production without genetic deletions (Shwab et al., 2007; Zutz et al., 2014). This strategy has since been popular in inducing a wide variety of novel secondary metabolites that are otherwise transiently expressed by fungi or have never been detected due to silencing of the encoding biosynthetic gene (Shwab et al., 2007). Interestingly, fungal species are primed for chromatin remodeling since their biosynthetic genes group together in locations that tend to be near the telomeres of their chromosomes (Toghueo et al., 2020). This localization of biosynthetic gene clusters near telomeres of the chromosomes might be a setup to facilitate efficiently coordinated regulation through chromatin remodeling within the subtelomeric regions (Brosch et al., 2008).
Epigenetic modification of the chromatin structure has shown to be a powerful tool in activating silent genes (Reyes-Dominguez et al., 2010; Strauss and Reyes-Dominguez, 2011; Chávez et al., 2015). In a vast number of cases, it has also led to the production of novel bioactive molecules. There are three levels involved in epigenetic regulation (i) DNA methylation, essential for normal development and differentiation. Transcription is inhibited by the covalent addition of a methyl group at the 5-C of the cytosine ring resulting in a 5-methylcytosine (5-mC) which extends into the major groove of DNA (Aghcheh and Kubicek, 2015); (ii) chromatin remodeling by histone modification where histones can undergo post-translational modifications (PTMs), which alter the interaction with DNA and nuclear proteins (Bannister and Kouzarides, 2011; Aghcheh and Kubicek, 2015); and (iii) RNA interference where non-coding RNA (ncRNA), which are non-translated into proteins, regulate transcriptional and post-transcriptional gene expression (Aghcheh and Kubicek, 2015). Two groups of ncRNA have been identified; short ncRNA (subcategorized into microRNA, short interfering RNA, and piwi-interacting RNA) and long ncRNA (Aghcheh and Kubicek, 2015). Short interfering RNA binds to specific target messenger RNA and inhibits translation by inducing its degradation, thus silencing post-transcriptional modification (Aghcheh and Kubicek, 2015; Collemare and Seidl, 2019).
This review will focus on epigenetic modification of the chromatin employing small molecule effectors. In a study by Nützmann et al. (2013), they demonstrated, using Aspergillus nidulans, that chemical inhibitors can lead to the upregulation of secondary metabolite gene clusters, with the possible alteration in the production of secondary metabolites of the targeted activation or inactivation of specific chromatin modifiers (König et al., 2013). The use of HDAC inhibitors like valproic acid and suberoylanilide hydroxamic acid (SAHA) led to the increased production of secondary metabolites in fungi. This inhibition of the HDACs has been attributed to the hyperacetylation of chromatin, which subsequently opens the chromatin (Beau et al., 2012; Takahashi et al., 2016). Several studies show the effects of treatment of fungi in culture media with these small molecule inhibitors and the changes in the expression profile of SM gene clusters (Cichewicz, 2010; Collemare and Seidl, 2019). The profile of metabolites detected in the broths (Williams et al., 2008; Chung et al., 2013) has allowed for the isolation of previously unknown compounds such as nygerone A from Aspergillus niger (Henrikson et al., 2009) and new sesquiterpenoids from Aspergillus sydowii (Chung et al., 2013). Since this strategy does not require genome sequencing, it may be suitable as a tool for initial metabolite screening of fungi from extreme environments, in combination with OSMAC strategies. However, there is no significant alteration of the metabolic profile observed after treatment in some cases, indicating that this strategy may not be applicable in all fungal species. For example, fungi such as Alternaria brassicicola and Diheretospora chlamydosporia were resistant to HDAC inhibitors (Brosch et al., 2008). The resistance may probably be due to that other filamentous fungus producing HDAC inhibitors allowing the fungus to escape inhibition.
Mechanisms of Action of Small Molecular Compounds, Target Sites, Advantages, and Disadvantages
Epigenetic modifiers or epidrugs are natural or synthetic small molecular compounds targeting the epigenetic marks or enzymes with epigenetic activity leading to epigenetic alterations (Poças-Fonseca et al., 2020). Many of these compounds act by inhibiting enzyme machinery essential for transferring methyl, acetyl, or alkyl groups to DNA or histones (Table 1). In the past two decades, vast and diverse compounds have been produced by treating fungi with epigenetic modifiers (Williams et al., 2008; Linnakoski et al., 2018; Toghueo et al., 2020). Chromatin remodeling using epigenetic modifiers such as histone deacetylase (HDAC) and DNA methyltransferase (DNMT) inhibitors presents a straightforward and low-cost approach to uncovering the regulatory circuit in fungal biosynthetic genomes. The fungal biosynthetic genes hold a promise as a reservoir for novel chemistries that could be developed into pharmaceuticals (Gacek and Strauss, 2012).
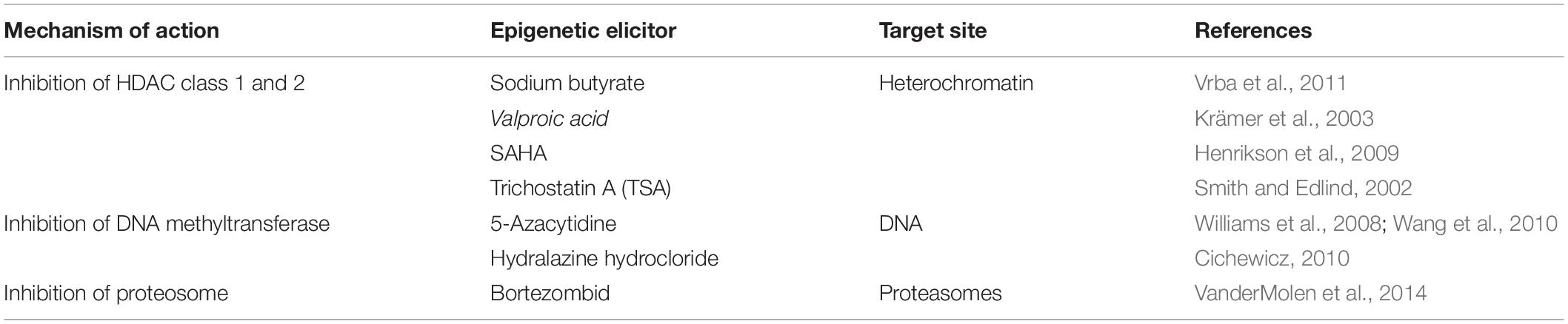
Table 1. Small molecular compounds and their mechanism of action.
Histone deacetylases inhibitors are structurally classified into four classes: hydroxamates, cyclic peptides, aliphatic acids, and benzamides b (reviewed by Kim and Bae, 2011). HDAC inhibitors alter gene expression patterns and endorse changes in non-histone proteins occurring at the post-translational level (Shwab et al., 2007; Williams et al., 2008). Trichostatin A (TSA), its derivative, suberoylanilide hydroxamic acid (SAHA), and sodium butyrate are frequently used as HDAC inhibitors in filamentous fungi. Both TSA and SAHA present a hydroxamic group that binds to the zinc ion of class 1 and 2 classical HDAC’s active sites, thus preventing their activities. Sodium butyrate inhibits the histone deacetylase activity, leading to differentiation in eukaryotic cells (Zutz et al., 2014; Poças-Fonseca et al., 2020). Class 1 and class 2 classical HDAC inhibitor, TSA, and other HDAC chemical inhibitors such as SAHA, sodium butyrate, and valproic acid have been shown to enhance the chemical diversity of secondary metabolites produced by fungi from the genera Clonostachys, Diatrype, and Verticillium (Williams et al., 2008). Valproic acid is also frequently used, and it inhibits class 1 classical HDACs and also induces proteosomal degradation of class2 classical HDACs (Zutz et al., 2014, 2016).
DNA methyltransferases (DNMTs) are a conserved family of cytosine methylases that play an essential role in maintaining DNA methylation patterns, transcriptional activation, and silencing (Lyko, 2018). DNMT inhibitors: 5-azacytidine (5-AZA) and decitabine (5-AZA-2′-deoxycytidine) are synthetic analogs of cytidine presenting nitrogen instead of carbon in the “5” of the pyrimidine ring. This ligand is incorporated in the DNA and less into RNA, preventing the proper transferring of the methyl group by the DNMT (Zutz et al., 2014). Inhibition by DNMT inhibitors results in passive demethylation through consecutive DNA replication cycles. In the presence of DNMT, DNMTs remain bound to the DNA and are degraded by the proteasome pathway (Poças-Fonseca et al., 2020).
In Table 2, we present a comprehensive summary of the application of HDAC and DNMT inhibitors in various fungal species. We show that a variety of chemical diversity, instead of silenced in untreated fungal species, can be produced upon treatment with different pan inhibitors. The use of pan inhibitors for inducing BGC expression is a vital tool for screening fungal environmental isolates without any prior knowledge of their genetic information. However, treatment with small molecular compounds often lead to a pleiotropic activation, making it difficult to predict which secondary metabolites will be produced and to link the producing gene to a particular phenotype. Although the approach of using small molecular epigenetic modifiers yields diverse chemistries, many studies do not assess the activity profiles of these compounds. This challenge can be attributed to limited access to standardized in vitro methods for activity testing in different fungal natural products laboratories. This highlights a need for collaborations and the adoption of a standard workflow to discover secondary metabolites from natural resources such as fungi.
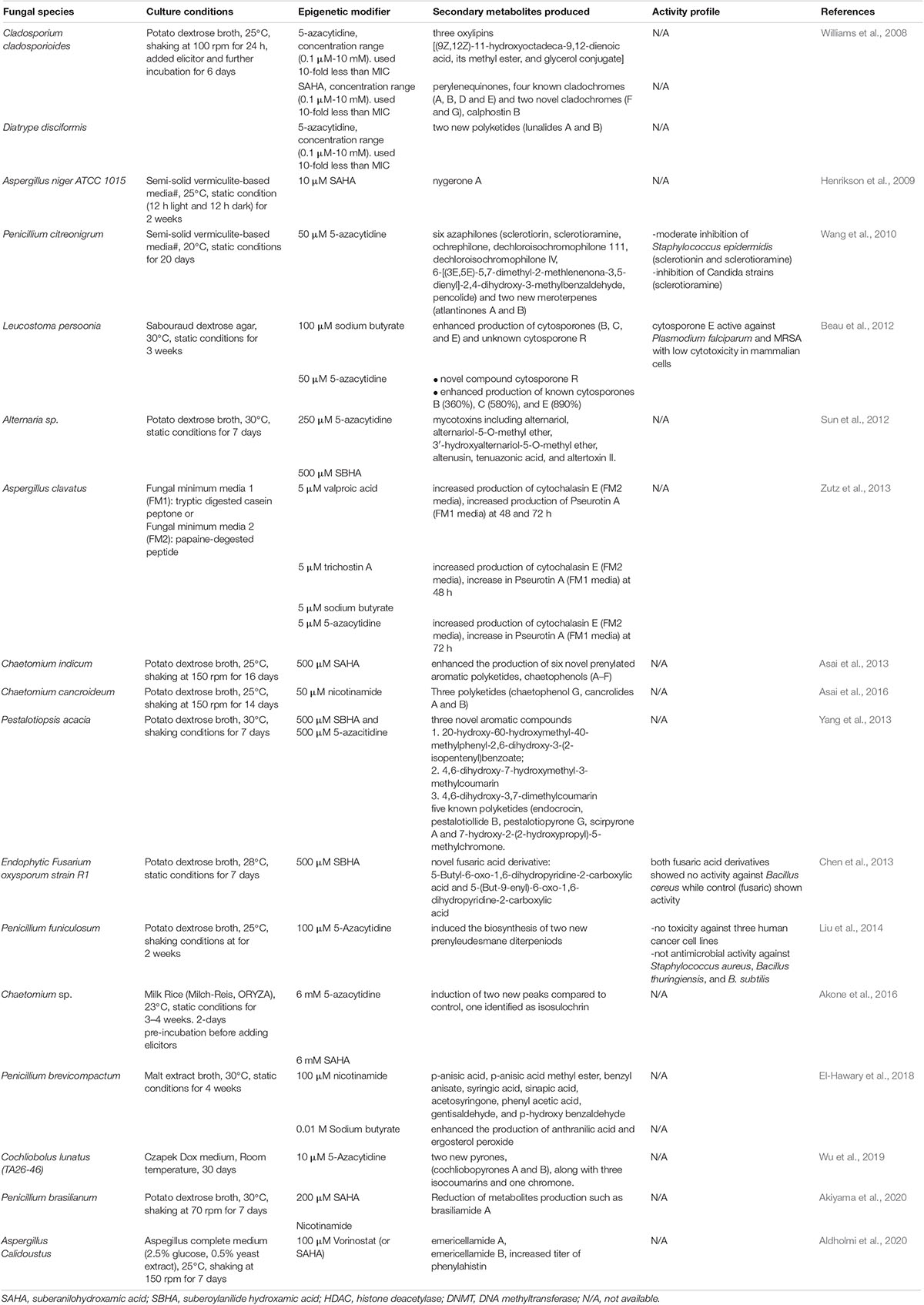
Table 2. Epigenetic induction of fungal biosynthetic gene clusters encoding secondary metabolites using small molecular compounds.
Response to Epigenetic Treatment is Complex at a Metabolome Level
Histone post-translational modifications (PTMs) such as acetylation and methylation play a profound role in chromatin-based regulation of secondary metabolite (SM) gene clusters (Gacek-Matthews et al., 2016). However, the exact molecular mechanism mediated by histone PTMs leading to the activation or repression of SM gene clusters is not yet fully understood. Studies in the model organism, Aspergillus nidulans, revealed that SM gene clusters are under chromatin-based regulatory control (Shwab et al., 2007). Subsequent studies in HDAC inhibition and SAGA-complex mutants showed the importance of acetylation in the expression of these SM gene clusters (Gacek and Strauss, 2012). Consequently, genetic deletion of, HDACs in various fungal species such as Aspergillus fumigatus, Aspergillus oryzae, Fusarium fujikuroi, Penicillium oryzae, Fusarium asiaticum, P. chrysogenum, and A. nidulans have resulted in increased expression of known gene clusters and expression of novel clusters that are repressed in the control strain (Albright et al., 2015; Collemare and Seidl, 2019).
Generally, acetylation of histones (e.g., H3K27ac in Neurospora crassa) is associated with transcriptional activation of SM gene clusters (Ferraro et al., 2021; Zhang et al., 2021). Although the exact mechanism of this activation is not known, there are various proposed mechanisms suggested by observations of histone marks in different genetic analysis studies (Gacek-Matthews et al., 2016; Ferraro et al., 2021). The earlier proposed mechanism is that histone marks such as acetylation of histones neutralizes the positive charge of the ε-amino group of lysine in the nucleosome, thus weakening the interaction between the core histones with negatively charged DNA and subsequent chromatic accessibility (Gacek-Matthews et al., 2016). Another possibility is that the acetylation of lysines alters the interaction of regulatory proteins with histones (Brosch et al., 2008) or recruit chromatin-modifying enzymes that promote or inhibit transcription of chromatin-regulated gene clusters (Gacek-Matthews et al., 2016).
In addition to the specific modification site, the complex combination of distinct histone modifications creates a pattern that is likely recognized and interpreted as a modification signal by regulatory factors (Brosch et al., 2008; Pidroni et al., 2018). For example, increased SM production has been associated with the global increase of H3K14ac and SM gene cluster-specific H3K9ac (Nützmann et al., 2013; Collemare and Seidl, 2019). However, in a study by Gacek-Matthews et al. (2016), they reported that histone marks associated with activation were observed in some transcribed clusters of A. nidulans, but this was not true for most of the transcribed gene clusters. These observations suggest that chromatin-based regulation is more complex and depends on many interacting factors. Indeed, the histone modifications do not occur independently but are interrelated, thus influencing the biological outcome (Brosch et al., 2008). In a study by Rea et al. (2000), the effect of functional interdependence was clear where the phosphorylation of Serine 10 in H3 was linked to facilitating the acetylation of H3k9 and H3K4. They also observed that H3K9 methylation inhibited phosphorylation, thus suggesting that chromatin networks are rather regulated by dynamic mechanisms challenging to elucidate in vitro (Brosch et al., 2008).
The complex regulatory nature of the chromatin has been observed also in genetic depletion studies probing the role of HDACs in secondary metabolite production. Evidence from these studies suggest that while the deletion of HDACs may lead to the activation of previously silenced biosynthetic gene clusters, a subset of genes may also be repressed by the genetic deletion of HDACs (Brosch et al., 2008; Pfannenstiel and Keller, 2019). Evidence of this differential response was first shown by Albright et al. (2015), where they used untargeted metabolomics to profile >1000 small molecules following genetic or chemical reductions in histone deacetylase activity. They found that A. nidulans could increase its capacity to produce more than 61 compounds, including a lipopeptide aldehyde, fellutamides known as a proteasome inhibitor by ∼100-fold upon inhibition of a single HDAC, RpdA. Interestingly, there was a simultaneous decrease in the expression of 47 compounds by >100-fold. Further evidence of differential expression has been recently reported by Ding et al. (2020), where deletion of HdaA from an endophytic fungus, Penicillium chrysogenum FES1701. These researchers observed that in the HdaA mutant strain, the BGC encoding the meleagrin/roquefortine was upregulated while the chrysogine encoding BGC was downregulated. Interestingly, a similar pattern has been observed upon treatment of fungal species with HDAC inhibitors. Magotra et al. (2017) reported a 10-fold upregulation of fumiquinazoline C when Aspergillus fumigatus (GA-L7), an endophyte of Grewia asiatica L. was treated with valproic acid, a class I HDAC inhibitor that also induces the proteosomal degradation of class II HDACs (Krämer et al., 2003). In the same study, a reduction was observed in the production of seven other metabolites produced under normal conditions. These observations provide evidence that the overall response to HDAC inhibition at the level of secondary metabolome is more complex than the currently assumed global increase in the abundance of secondary metabolites (Albright et al., 2015).
Future Perspectives
This review highlights the influence of epigenetic modifiers on rearranging the chromatin network, thus ensuring fungal ability to produce secondary metabolites. Epigenetic modification using small molecular modulators offers a convenient tool for the initial screening of novel fungal isolates such as those from the less explored environments. The discovery that endophytic fungi can produce similar metabolites to their host plants places the endophytes as attractive candidates for future drug discoveries. Moreover, bioprospecting plant endophytes instead of plants provide a sustainable alternative as plants are prone to extinction due to overharvesting. Over recent years there has been a remarkably rapid development of new techniques for biosynthetic gene clusters used in drug discovery. Low molecular weight molecules such as 5-AZA, SAHA, and valproic acid can induce secondary metabolite production in fungi by displaying chromatin remodeling. This method provides a considerable potential for inducing a vast number of bioactive compounds. It has been found that each low molecular weight molecule can induce different patterns of secondary metabolite production in the same fungal culture species. These findings can assist in optimizing secondary metabolite production using epigenetic modifiers as an economical, non-invasive approach. However, future studies need to establish the exact mechanism by which these small molecular inhibitors facilitate the changes in the chromatin network. Establishing the mechanism of action will enable a more targeted approach that results in a specific expression of compounds of interest.
Author Contributions
SIN conceptualized, critically reviewed, and added value to the overall manuscript. LCP and SIN wrote the manuscript, gathered literature, and made necessary edits after critical reviews. LN and PJM were involved in the literature collection and contributed to writing the section on small molecular modifiers. All authors contributed to the article and approved the submitted version.
Funding
LCP would like to thank the University of KwaZulu-Natal, College of Health Science Scholarship for funding her studies. SIN was funded by the National Research Foundation (NRF) grant number 129778.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aghcheh, R. K., and Kubicek, C. P. (2015). Epigenetics as an emerging tool for improvement of fungal strains used in biotechnology. Appl. Microbiol. Biotechnol. 99, 6167–6181. doi: 10.1007/s00253-015-6763-2
Akiyama, D. Y., Rocha, M. C., Costa, J. H., Malavazi, I., and Fill, T. P. (2020). The histone deacetylase clr3 regulates secondary metabolite production and growth under oxidative stress conditions in Penicillium brasilianum. bioRxiv [Preprint]. doi: 10.1101/2020.05.01.072108
Akone, S. H., Mandi, A., Kurtan, T., Hartmann, R., Lin, W., Daletos, G., et al. (2016). Inducing secondary metabolite production by the endophytic fungus Chaetomium sp. through fungal-bacterial co-culture and epigenetic modification. Tetrahedron 72, 6340–6347.
Albright, J. C., Henke, M. T., Soukup, A. A., McClure, R. A., Thomson, R. J., Keller, N. P., et al. (2015). Large-scale metabolomics reveals a complex response of Aspergillus nidulans to epigenetic perturbation. ACS Chem. Biol. 10, 1535–1541. doi: 10.1021/acschembio.5b00025
Aldholmi, M., Wilkinson, B., and Ganesan, A. (2020). Epigenetic modulation of secondary metabolite profiles in Aspergillus calidoustus and Aspergillus westerdijkiae through histone deacetylase (HDAC) inhibition by vorinostat. J. Antibiot. 73, 410–413. doi: 10.1038/s41429-020-0286-5
Asai, T., Morita, S., Taniguchi, T., Monde, K., and Oshima, Y. (2016). Epigenetic stimulation of polyketide production in Chaetomium cancroideum by an NAD(+)-dependent HDAC inhibitor. Org. Biomol. Chem. 14, 646–651. doi: 10.1039/c5ob01595b
Asai, T., Yamamoto, T., Shirata, N., Taniguchi, T., Monde, K., Fujii, I., et al. (2013). Structurally diverse chaetophenol productions induced by chemically mediated epigenetic manipulation of fungal gene expression. Org. Lett. 15, 3346–3349. doi: 10.1021/ol401386w
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Baral, B., Akhgari, A., and Metsä-Ketelä, M. (2018). Activation of microbial secondary metabolic pathways: avenues and challenges. Synth. Syst. Biotechnol. 3, 163–178. doi: 10.1016/j.synbio.2018.09.001
Beau, J., Mahid, N., Burda, W. N., Harrington, L., Shaw, L. N., Mutka, T., et al. (2012). Epigenetic tailoring for the production of anti-infective cytosporones from the marine fungus Leucostoma persoonii. Mar. Drugs 10, 762–774. doi: 10.3390/md10040762
Begani, J., Lakhani, J., and Harwani, D. (2018). Current strategies to induce secondary metabolites from microbial biosynthetic cryptic gene clusters. Ann. Microbiol. 68, 419–432. doi: 10.1007/s13213-018-1351-1
Berg, G., and Hallmann, J. (2006). “Control of Plant Pathogenic Fungi with Bacterial Endophytes,” in Microbial Root Endophytes. Soil Biology, vol 9, eds B. J. E. Schulz, C. J. C. Boyle, and T. N. Sieber (Berlin: Springer), doi: 10.1007/3-540-33526-9_4
Bertrand, S., Bohni, N., Schnee, S., Schumpp, O., Gindro, K., and Wolfender, J. L. (2014). Metabolite induction via microorganism co-culture: a potential way to enhance chemical diversity for drug discovery. Biotechnol. Adv. 32, 1180–1204. doi: 10.1016/j.biotechadv.2014.03.001
Bode, H. B., Bethe, B., Höfs, R., and Zeeck, A. (2002). Big effects from small changes: possible ways to explore nature’s chemical diversity. Chem. Biochem. 3, 619–627. doi: 10.1002/1439-7633(20020703)3:7<619::AID-CBIC619<3.0.CO;2-9
Bok, J. W., Chiang, Y. M., Szewczyk, E., Reyes-Dominguez, Y., Davidson, A. D., Sanchez, J. F., et al. (2009). Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 5, 462–464. doi: 10.1038/nchembio.177
Bok, J. W., and Keller, N. P. (2004). LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3, 527–535. doi: 10.1128/EC.3.2.527-535.2004
Bok, J. W., Noordermeer, D., Kale, S. P., and Keller, N. P. (2006). Secondary metabolic gene cluster silencing in Aspergillus nidulans. Mol. Microbiol. 61, 1636–1645. doi: 10.1111/j.1365-2958.2006.05330.x
Brosch, G., Loidl, P., and Graessle, S. (2008). Histone modifications and chromatin dynamics: a focus on filamentous fungi. FEMS Microbiol. Rev. 32, 409–439. doi: 10.1111/j.1574-6976.2007.00100.x
Calvo, A. M., Wilson, R. A., Bok, J. W., and Keller, N. P. (2002). Relationship between secondary metabolism and fungal development. Microbiol. Mol Biol. Rev. 66, 447–459. doi: 10.1128/MMBR.66.3.447-459.2002
Card, S., Johnson, L., Teasdale, S., and Caradus, J. (2016). Deciphering endophyte behaviour: the link between endophyte biology and efficacious biological control agents. FEMS Microbiol. Ecol. 92:fiw114. doi: 10.1093/femsec/fiw114
Caterino, T. L., and Hayes, J. J. (2011). Structure of the H1 C-terminal domain and function in chromatin condensation. Biochem. Cell Biol. 89, 35–44. doi: 10.1139/O10-024
Chang, Z., Yadav, V., Lee, S. C., and Heitman, J. (2019). Epigenetic mechanisms of drug resistance in fungi. Fungal Genet. Biol. 132:103253. doi: 10.1016/j.fgb.2019.103253
Chávez, R., Fierro, F., García-Rico, R. O., and Vaca, I. (2015). Filamentous fungi from extreme environments as a promising source of novel bioactive secondary metabolites. Front. Microbiol. 6:903. doi: 10.3389/fmicb.2015.00903
Chen, H. J., Awakawa, T., Sun, J. Y., Wakimoto, T., and Abe, I. (2013). Epigenetic modifier-induced biosynthesis of novel fusaric acid derivatives in endophytic fungi from Datura stramonium L. Nat. Prod. Bioprospect. 3, 20–23. doi: 10.1007/s13659-013-0010-2
Chiang, Y. M., Lee, K. H., Sanchez, J. F., Keller, N. P., and Wang, C. C. (2009). Unlocking fungal cryptic natural products. Nat. Prod. Commun. 4, 1505–1510.
Chung, Y. M., Wei, C. K., Chuang, D. W., El-Shazly, M., Hsieh, C. T., Asai, T., et al. (2013). An epigenetic modifier enhances the production of anti-diabetic and anti-inflammatory sesquiterpenoids from Aspergillus sydowii. Bioorg. Med. Chem. 21, 3866–3872. doi: 10.1016/j.bmc.2013.04.004
Cichewicz, R. H. (2010). Epigenome manipulation as a pathway to new natural product scaffolds and their congeners. Nat. Prod. Rep. 27, 11–22. doi: 10.1039/b920860g
Collemare, J., and Seidl, M. F. (2019). Chromatin-dependent regulation of secondary metabolite biosynthesis in fungi: is the picture complete? FEMS Microbiol. Rev. 43, 591–607. doi: 10.1093/femsre/fuz018
Deepika, V. B., Murali, T. S., and Satyamoorthy, K. (2016). Modulation of genetic clusters for synthesis of bioactive molecules in fungal endophytes: a review. Microbiol. Res. 182, 125–140. doi: 10.1016/j.micres.2015.10.009
Ding, Z., Zhou, H., Wang, X., Huang, H., Wang, H., Zhang, R., et al. (2020). Deletion of the Histone Deacetylase HdaA in Endophytic Fungus Penicillium chrysogenum Fes1701 Induces the Complex Response of Multiple Bioactive Secondary Metabolite Production and Relevant Gene Cluster Expression. Molecules 25:3657. doi: 10.3390/molecules25163657
El-Hawary, S. S., Sayed, A. M., Mohammed, R., Hassan, H. M., Zaki, M. A., Rateb, M. E., et al. (2018). Epigenetic Modifiers Induce Bioactive Phenolic Metabolites in the Marine-Derived Fungus Penicillium brevicompactum. Mar. Drugs 16:253. doi: 10.3390/md16080253
Fadiji, A. E., and Babalola, O. O. (2020). Elucidating Mechanisms of Endophytes Used in Plant Protection and Other Bioactivities with Multifunctional Prospects. Front. Bioeng. Biotechnol. 8:467. doi: 10.3389/fbioe.2020.00467
Fan, A., Mi, W., Liu, Z., Zeng, G., Zhang, P., Hu, Y., et al. (2017). Deletion of a Histone Acetyltransferase Leads to the Pleiotropic Activation of Natural Products in Metarhizium robertsii. Org. Lett. 19, 1686–1689. doi: 10.1021/acs.orglett.7b00476
Ferraro, A. R., Ameri, A. J., Lu, Z., Kamei, M., Schmitz, R. J., and Lewis, Z. A. (2021). Chromatin accessibility profiling in Neurospora crassa reveals molecular features associated with accessible and inaccessible chromatin. BMC Genomics 22:459. doi: 10.1186/s12864-021-07774-0
Fischer, J., Schroeckh, V., and Brakhage, A. A. (2016). “Awakening of fungal secondary metabolite gene clusters,” in Gene Expression Systems in Fungi: Advancements and Applications, eds M. Schmoll and C. Dattenböck (Cham: Springer), 253–273.
Fox, E. M., and Howlett, B. J. (2008). Secondary metabolism: regulation and role in fungi biology. Curr. Opin. Microbiol. 11, 481–487.
Fyodorov, D. V., Zhou, B. R., Skoultchi, A. I., and Bai, Y. (2018). Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 19, 192–206. doi: 10.1038/nrm.2017.94
Gacek, A., and Strauss, J. (2012). The chromatin code of fungal secondary metabolite gene clusters. Appl. Microbiol. Biotechnol. 95, 1389–1404. doi: 10.1007/s00253-012-4208-8
Gacek-Matthews, A., Berger, H., Sasaki, T., Wittstein, K., Gruber, C., Lewis, Z. A., et al. (2016). KdmB, a Jumonji Histone H3 Demethylase, Regulates Genome-Wide H3K4 Trimethylation and Is Required for Normal Induction of Secondary Metabolism in Aspergillus nidulans. PLoS Genet. 12:e1006222. doi: 10.1371/journal.pgen.1006222
Giaimo, B. D., Ferrante, F., Herchenröther, A., Hake, S. B., and Borggrefe, T. (2019). The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 12:37. doi: 10.1186/s13072-019-0274-9
González-Menéndez, V., Crespo, G., Toro, C., Martín, J., de Pedro, N., Tormo, J. R., et al. (2019). Extending the Metabolite Diversity of the Endophyte Dimorphosporicola tragani. Metabolites 9:197. doi: 10.3390/metabo9100197
Guzmán-Chávez, F., Zwahlen, R. D., Bovenberg, R., and Driessen, A. (2018). Engineering of the Filamentous Fungus Penicillium chrysogenum as Cell Factory for Natural Products. Front. Microbial. 9:2768. doi: 10.3389/fmicb.2018.02768
Hardoim, P. R., van Overbeek, L. S., Berg, G., Pirttilä, A. M., Compant, S., Campisano, A., et al. (2015). The Hidden World within Plants: ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol. Mol. Biol. Rev. 79, 293–320. doi: 10.1128/MMBR.00050-14
Harvey, C. J. B., Tang, M., Schlecht, U., Horecka, J., Fischer, C. R., Lin, H. C., et al. (2018). HEx: a heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 4:5459. doi: 10.1126/sciadv.aar5459
Henrikson, J. C., Hoover, A. R., Joyner, P. M., and Cichewicz, R. H. (2009). A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Org Biomol. Chem. 7, 435–438. doi: 10.1039/b819208a
Hewage, R. T., Aree, T., Mahidol, C., Ruchirawat, S., and Kittakoop, P. (2014). One strain-many compounds (OSMAC) method for production of polyketides, azaphilones, and an isochromanone using the endophytic fungus Dothideomycete sp. Phytochemistry 108, 87–94. doi: 10.1016/j.phytochem.2014.09.013
Jackson, N., Czaplewski, L., and Piddock, L. J. V. (2018). Discovery and development of new antibacterial drugs: learning from experience? J. Antimicrob. Chemother. 73, 1452–1459. doi: 10.1093/jac/dky019
Jia, M., Chen, L., Xin, H. L., Zheng, C. J., Rahman, K., Han, T., et al. (2016). A Friendly Relationship between Endophytic Fungi and Medicinal Plants: a Systematic Review. Front. Microbiol. 7:906. doi: 10.3389/fmicb.2016.00906
Jiang, C., Lv, G., Tu, Y., Cheng, X., Duan, Y., Zeng, B., et al. (2021). Applications of CRISPR/Cas9 in the Synthesis of Secondary Metabolites in Filamentous Fungi. Front. Microbiol. 12:638096. doi: 10.3389/fmicb.2021.638096
Keller, N. P. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180. doi: 10.1038/s41579-018-0121-1
Khare, E., Mishra, J., and Arora, N. K. (2018). Multifaceted Interactions Between Endophytes and Plant: developments and Prospects. Front. Microbiol. 9:2732. doi: 10.3389/fmicb.2018.02732
Kim, H. J., and Bae, S. C. (2011). Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 3, 166–179.
Kloepper, J. W., and Ryu, C. M. (2006). “Bacterial Endophytes as Elicitors of Induced Systemic Resistance,” in Microbial Root Endophytes, eds B. J. E. Schulz, C. J. C. Boyle, and T. N. Sieber (Berlin: Springer), 33–52. doi: 10.1007/3-540-33526-9_3
König, C. C., Scherlach, K., Schroeckh, V., Horn, F., Nietzsche, S., Brakhage, A. A., et al. (2013). Bacterium induces cryptic meroterpenoid pathway in the pathogenic fungus Aspergillus fumigatus. Chem. Biochem. 14, 938–942. doi: 10.1002/cbic.201300070
Kosalková, K., García-Estrada, C., Ullán, R. V., Godio, R. P., Feltrer, R., Teijeira, F., et al. (2009). The global regulator LaeA controls penicillin biosynthesis, pigmentation and sporulation, but not roquefortine C synthesis in Penicillium chrysogenum. Biochemie 91, 214–225. doi: 10.1016/j.biochi.2008.09.004
Krämer, O. H., Zhu, P., Ostendorff, H. P., Golebiewski, M., Tiefenbach, J., Peters, M. A., et al. (2003). The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 22, 3411–3420. doi: 10.1093/emboj/cdg315
Lamoth, F., Juvvadi, P. R., and Steinbach, W. J. (2015). Histone deacetylase inhibition as an alternative strategy against invasive aspergillosis. Front. Mcrobiol. 6:96. doi: 10.3389/fmicb.2015.00096
Lim, F. Y., Sanchez, J. F., Wang, C. C., and Keller, N. P. (2012). Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods Enzymol. 517, 303–324. doi: 10.1016/B978-0-12-404634-4.00015-2
Linnakoski, R., Reshamwala, D., Veteli, P., Cortina-Escribano, M., Vanhanen, H., and Marjomäki, V. (2018). Antiviral Agents From Fungi: diversity, Mechanisms and Potential Applications. Front. Microbiol. 9:2325. doi: 10.3389/fmicb.2018.02325
Liu, D. Z., Liang, B. W., Li, X. F., and Liu, Q. (2014). Induced production of new diterpenoids in the fungus Penicillium funiculosum. Nat. Prod. Commun. 9, 607–608.
Alvin, A., Miller, K. I, and Neilan, B. A. (2014). Exploring the potential of endophytes from medicinal plants as sources of antimicrobial compounds. Microbiol. Res. 169, 483–495.
Lyko, F. (2018). The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92. doi: 10.1038/nrg.2017.80
Macheleidt, J., Mattern, D. J., Fischer, J., Netzker, T., Weber, J., Schroeckh, V., et al. (2016). Regulation and Role of Fungal Secondary Metabolites. Annu. Rev. Genet. 50, 371–392. doi: 10.1146/annurev-genet-120215-035203
Magotra, A., Kumar, M., Kushwaha, M., Awasthi, P., Raina, C., Gupta, A. P., et al. (2017). Epigenetic modifier induced enhancement of fumiquinazoline C production in Aspergillus fumigatus (GA-L7): an endophytic fungus from Grewia asiatica L. AMB Express 7:43. doi: 10.1186/s13568-017-0343-z
Mao, X. M., Xu, W., Li, D., Yin, W. B., Chooi, Y. H., Li, Y. Q., et al. (2015). Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. Engl. 54, 7592–7596. doi: 10.1002/anie.201502452
Martire, S., and Banaszynski, L. A. (2020). The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 21, 522–541. doi: 10.1038/s41580-020-0262-8
Nair, D. N., and Padmavathy, S. (2014). Impact of endophytic microorganisms on plants, environment and humans. Sci. World J. 2014:250693. doi: 10.1155/2014/250693
Netzker, T., Fischer, J., Weber, J., Mattern, D. J., König, C. C., Valiante, V., et al. (2015). Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front. Microbiol. 6:299. doi: 10.3389/fmicb.2015.00299
Nielsen, K. F., and Larsen, T. O. (2015). The importance of mass spectrometric dereplication in fungal secondary metabolite analysis. Front. Microbiol. 6:71. doi: 10.3389/fmicb.2015.00071
Niu, X., Hao, X., Hong, Z., Chen, L., Yu, X., and Zhu, X. (2015). A Putative Histone Deacetylase Modulates the Biosynthesis of Pestalotiollide B and Conidiation in Pestalotiopsis microspora. J. Microbiol. Biotechnol. 25, 579–588. doi: 10.4014/jmb.1409.09067
Nützmann, H. W., Fischer, J., Scherlach, K., Hertweck, C., and Brakhage, A. A. (2013). Distinct amino acids of histone H3 control secondary metabolism in Aspergillus nidulans. Appl. Environ. Microbiol. 79, 6102–6109. doi: 10.1128/AEM.01578-13
Ochi, K., and Hosaka, T. (2013). New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol. 97, 87–98. doi: 10.1007/s00253-012-4551-9
Okada, B. K., and Seyedsayamdost, M. R. (2017). Antibiotic dialogues: induction of silent biosynthetic gene clusters by exogenous small molecules. FEMS Microbiol. Res. 41, 19–33. doi: 10.1093/femsre/fuw035
Pan, R., Bai, X., Chen, J., Zhang, H., and Wang, H. (2019). Exploring Structural Diversity of Microbe Secondary Metabolites Using OSMAC Strategy: a Literature Review. Front. Microbiol. 10:294. doi: 10.3389/fmicb.2019.00294
Perrin, R. M., Fedorova, N. D. D., Bok, J. W., Cramer, R. A., Wortman, J. R., Kim, H. S., et al. (2007). Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 3:e50. doi: 10.1371/journal.ppat.0030050
Pettit, R. K. (2011). Small-molecule elicitation of microbial secondary metabolites. Microb. Biotechnol. 4, 471–478. doi: 10.1111/j.1751-7915.2010.00196.x
Pfannenstiel, B. T., and Keller, N. P. (2019). On top of biosynthetic gene clusters: how epigenetic machinery influences secondary metabolism in fungi. Biotechnol. Adv. 37:107345. doi: 10.1016/j.biotechadv.2019.02.001
Phukan, H., Mitra, P. K., and Saikia, M. (2018). Comparative study of Endophytic fungal metabolite isolated from black turmeric (Curcuma caesia roxb) in ROS associated Caenorhabditis elegans model system. World J. Pharm. Res. 4, 79–82.
Pidroni, A., Faber, B., Brosch, G., Bauer, I., and Graessle, S. (2018). A Class 1 Histone Deacetylase as Major Regulator of Secondary Metabolite Production in Aspergillus nidulans. Front. Microbiol. 9:2212. doi: 10.3389/fmicb.2018.02212
Poças-Fonseca, M. J., Cabral, C. G., and Manfrão-Netto, J. (2020). Epigenetic manipulation of filamentous fungi for biotechnological applications: a systematic review. Biotechnol. Lett. 42, 885–904. doi: 10.1007/s10529-020-02871-8
Rana, K. L., Kour, D., Sheikh, I., Yadav, N., Yadav, A. N., Kumar, V., et al. (2019). “Biodiversity of Endophytic Fungi from Diverse Niches and Their Biotechnological Applications,” in Advances in Endophytic Fungal Research. Fungal Biology, ed. B. Singh (Cham: Springer), doi: 10.1007/978-3-030-03589-1_6
Rea, S., Eisenhaber, F., O’Carroll, D., Strahl, B. D., Sun, Z. W., Schmid, M., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. doi: 10.1038/35020506
Reen, J., Romano, S., Dobson, A. D. W., and O’Gara, F. (2015). The sound of Silence: activating Silent Biosynthetic Gene Clusters in Marine Microorganisms. Mar. Drugs 13, 4754–4783. doi: 10.3390/md13084754
Reyes-Dominguez, Y., Bok, J. W., Berger, H., Shwab, E. K., Basheer, A., Gallmetzer, A., et al. (2010). Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol. Microbiol. 76, 1376–1386. doi: 10.1111/j.1365-2958.2010.07051.x
Rodriguez, R. J., White, J. F. Jr., Arnold, A. E., and Redman, R. S. (2009). Fungal endophytes: diversity and functional roles. New Phytol. 182, 314–330. doi: 10.1111/j.1469-8137.2009.02773.x
Romano, S., Jackson, S. A., Patry, S., and Dobson, A. D. W. (2018). Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 16:244. doi: 10.3390/md16070244
Rutledge, P. J., and Challis, G. L. (2015). Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 13, 509–523. doi: 10.1038/nrmicro3496
Ryan, R. P., Germaine, K., Franks, A., Ryan, D. J., and Dowling, D. N. (2008). Bacterial endophytes: recent developments and applications. FEMS Microbiol. Lett. 278, 1–9. doi: 10.1111/j.1574-6968.2007.00918.x
Scherlach, K., and Hertweck, C. (2009). Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 7, 1753–1760. doi: 10.1039/b821578b
Scherlach, K., and Hertweck, C. (2021). Mining and unearthing hidden biosynthetic potential. Nat. Commun. 12:3864. doi: 10.1038/s41467-021-24133-5
Shwab, E. K., Bok, J. W., Tribus, M., Galehr, J., Graessle, S., and Keller, N. P. (2007). Histone Deacetylase Activity Regulates Chemical Diversity in Aspergillus. Eukaryot. Cell 6, 1656–1664. doi: 10.1128/EC.00186-07
Shwab, E. K., and Keller, N. P. (2008). Regulation of secondary metabolite production in filamentous ascomycetes. Mycol. Res. 112, 225–230. doi: 10.1016/j.mycres.2007.08.021
Smith, W. L., and Edlind, T. D. (2002). Histone deacetylase inhibitors enhance Candida albicans sensitivity to azoles and related antifungals: correlation with reduction in CDR and ERG upregulation. Antimicrob. Agents Chemother. 46, 3532–3539. doi: 10.1128/AAC.46.11.3532-3539.2002
Strauss, J., and Reyes-Dominguez, Y. (2011). Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet. Biol. 48, 62–69. doi: 10.1016/j.fgb.2010.07.009
Studt, L., Schmidt, F. J., Jahn, L., Sieber, C. M. K., Connolly, L. R., Niehaus, E.-M., et al. (2013). Two histone deacetylases, FfHda1 and FfHda2, are important for Fusarium fujikuroi secondary metabolism and virulence. Appl. Environ. Microbiol. 79, 7719–7734. doi: 10.1128/AEM.01557-13
Sun, J., Awakawa, T., Noguchi, H., and Abe, I. (2012). Induced production of mycotoxins in an endophytic fungus from the medicinal plant Datura stramonium L. Bioorganic Med. Chem. Lett. 22, 6397–6400. doi: 10.1016/j.bmcl.2012.08.063
Sun, R., Wen, M., Wu, L., Lan, H., Yuan, J., Wang, S., et al. (2021). The Fungi-specific histone Acetyltransferase Rtt109 mediates morphogenesis, Aflatoxin synthesis and pathogenicity in Aspergillus flavus by acetylating H3K9. IMA Fungus 12:9. doi: 10.1186/s43008-021-00060-4
Takahashi, J. A., Gomes, D. C., Lyra, F. H., and dos Santos, G. F. (2016). “Modulation of fungal secondary metabolites biosynthesis by chemical epigenetics,” in Fungi: Applications Management Strategies, eds S. K. Deshmukh, J. K. Misra, and J. P. Tewari (Boca Raton: CRC Press), 117–133.
Toghueo, R., Sahal, D., and Boyom, F. F. (2020). Recent advances in inducing endophytic fungal specialized metabolites using small molecule elicitors including epigenetic modifiers. Phytochemistry 174:112338. doi: 10.1016/j.phytochem.2020.1123
Tribus, M., Bauer, I., Galehr, J., Rieser, G., Trojer, P., Brosch, G., et al. (2010). A novel motif in fungal class 1 histone deacetylases is essential for growth and development of Aspergillus. Mol. Biol. Cell 21, 345–353. doi: 10.1091/mbc.e09-08-0750
Tyurin, A. P., Alferova, V. A., and Korshun, V. A. (2018). Chemical Elicitors of Antibiotic Biosynthesis in Actinomycetes. Microorganisms 6:52. doi: 10.3390/microorganisms6020052
VanderMolen, K. M., Darveaux, B. A., Chen, W. L., Swanson, S. M., Pearce, C. J., and Oberlies, N. H. (2014). Epigenetic Manipulation of a Filamentous Fungus by the Proteasome-Inhibitor Bortezomib Induces the Production of an Additional Secondary Metabolite. RSC Adv. 4, 18329–18335. doi: 10.1039/C4RA00274A
VanderMolen, K. M., Raja, H. A., El-Elimat, T., and Oberlies, N. H. (2013). Evaluation of culture media for the production of secondary metabolites in a natural products screening program. AMB Express 3:71. doi: 10.1186/2191-0855-3-71
Vrba, J., Trtkova, K., and Ulrichova, J. (2011). HDAC inhibitors sodium butyrate and sodium valproate do not affect human ncor1 and ncor2 gene expression in HL-60 cells. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. 155, 259–262. doi: 10.5507/bp.2011.033
Wang, X., Sena Filho, J. G., Hoover, A. R., King, J. B., Ellis, T. K., Powell, D. R., et al. (2010). Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an atlantic-forest-soil-derived Penicillium citreonigrum. J. Nat. Prod. 73, 942–948. doi: 10.1021/np100142h
Williams, R. B., Henrikson, J. C., Hoover, A. R., Lee, A. E., and Cichewicz, R. H. (2008). Epigenetic remodeling of the fungal secondary metabolome. Org. Biomol. Chem. 6, 1895–1897. doi: 10.1039/b804701d
Wu, J. S., Shi, X. H., Zhang, Y. H., Yu, J. Y., Fu, X. M., Li, X., et al. (2019). Co-cultivation With 5-Azacytidine Induced New Metabolites From the Zoanthid-Derived Fungus Cochliobolus lunatus. Front. Chem. 7:763. doi: 10.3389/fchem.2019.00763
Wu, M. Y., and Yu, J. H. (2015). “Epigenetics of fungal secondary metabolism related genes,” in Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites, eds S. Zeilinger, J. F. Martín, and C. García-Estrada (New York: Springer), 29–42.
Xu, F., Nazari, B., Moon, K., Bushin, L. B., and Seyedsayamdost, M. R. (2017). Discovery of a Cryptic Antifungal Compound from Streptomyces albus J1074 Using High-Throughput Elicitor Screens. J. Am. Chem. Soc. 139, 9203–9212. doi: 10.1021/jacs.7b02716
Yang, X. L., Awakawa, T., Wakimoto, T., and Abe, I. (2013). Induced production of novel prenyldepside and coumarins in endophytic fungi Pestalotiopsis acacia. Tetrahedron Lett. 54, 5814–5817.
Zhang, W., Huang, J., and Cook, D. E. (2021). Histone modification dynamics at H3K27 are associated with altered transcription of in planta induced genes in Magnaporthe oryzae. PLoS Genet. 17:e1009376. doi: 10.1371/journal.pgen.1009376
Zutz, C., Bacher, M., Parich, A., Kluger, B., Gacek-Matthews, A., Schuhmacher, R., et al. (2016). Valproic Acid Induces Antimicrobial Compound Production in Doratomyces microspores. Front. Microbiol. 7:510. doi: 10.3389/fmicb.2016.00510
Zutz, C., Bandian, D., Neumayer, B., Speringer, F., Gorfer, M., Wagner, M., et al. (2014). Fungi treated with small chemicals exhibit increased antimicrobial activity against facultative bacterial and yeast pathogens. BioMed Res. Int. 2014:540292. doi: 10.1155/2014/540292
Keywords: endophytic fungi, biosynthetic gene cluster, histone deacetylases, epigenetic modifiers, chromatin network, secondary metabolites
Citation: Pillay LC, Nekati L, Makhwitine PJ and Ndlovu SI (2022) Epigenetic Activation of Silent Biosynthetic Gene Clusters in Endophytic Fungi Using Small Molecular Modifiers. Front. Microbiol. 13:815008. doi: 10.3389/fmicb.2022.815008
Received: 14 November 2021; Accepted: 19 January 2022;
Published: 14 February 2022.
Edited by:
Vijay K. Sharma, Agricultural Research Organization (ARO), IsraelReviewed by:
Rufin Toghueo, Institute of Natural Resources and Agrobiology of Salamanca, Spanish National Research Council (CSIC), SpainJuan F. Martin, Universidad de León, Spain
Copyright © 2022 Pillay, Nekati, Makhwitine and Ndlovu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sizwe I. Ndlovu, bWhsb25nb3M5QHVrem4uYWMuemE=