 Michela Ruinelli1
Michela Ruinelli1 Jochen Blom
Jochen Blom Theo H. M. Smits
Theo H. M. Smits Joël F. Pothier
Joël F. Pothier- 1Environmental Genomics and Systems Biology Research Group, Institute for Natural Resources Sciences, Zurich University of Applied Sciences (ZHAW), Wädenswil, Switzerland
- 2Bioinformatics and Systems Biology, Justus-Liebig-University Giessen, Giessen, Germany
Members of the Pseudomonas syringae species complex cause symptoms that are ranging from leaf spots to cankers on a multitude of plant species, including some of the genus Prunus. To date, a total of two species of the P. syringae species complex and six different pathovars have been associated with diseases on Prunus spp., which were shown to belong to different phylogenetic units (phylogroups, PG) based on sequence similarity of housekeeping genes or whole genomes, suggesting that virulence to Prunus spp. may be the result of convergent pathoadaptation. In this study, a comparative genomics approach was used to determine genes significantly associated with strains isolated from Prunus spp. across a phylogeny of 97 strains belonging to the P. syringae species complex. Our study revealed the presence of a set of orthologous proteins which were significantly associated with strains isolated from Prunus spp. than in strains isolated from other hosts or from non-agricultural environments. Among them, the type III effector HopAY predicted to encode for a C58 cysteine protease was found to be highly associated with strains isolated from Prunus spp. and revealed patterns supporting co-evolution and host adaptation.
Introduction
Members of the Pseudomonas syringae species complex are responsible for the development of plant disease-causing blights, spots, specks, galls, and cankers on a wide range of economically important plant species including both herbaceous and woody hosts. Strains belonging to the P. syringae species complex have also been isolated from non-agricultural habitats, and therefore, their persistence and transmission is probably linked to the water cycle (Morris et al., 2008). Despite the economic and ecological importance of this bacterium, the taxonomy and nomenclature of strains belonging to the P. syringae species complex is quite confusing and remains largely unsettled (Palleroni, 2005; Gomila et al., 2017). Within the P. syringae species complex, at least nine independent species have been determined based on phenotypical and molecular characteristics while more than 60 pathovars have been defined based on the host range (Dye et al., 1980; Palleroni, 2005; Young, 2010). DNA–DNA hybridization experiments among strains belonging to 48 different pathovars of P. syringae revealed the existence of nine different genomospecies (Gardan et al., 1999), which were later reflected by the so-called phylogroups (PG) obtained based on sequence similarity of housekeeping genes (Sarkar and Guttman, 2004; Hwang et al., 2005; Sarkar et al., 2006; Parkinson et al., 2011). With the inclusion of strains isolated from non-agricultural environments, a total of 13 PG were defined (Berge et al., 2014).
Many studies have been performed in the last decades with the intent to investigate and determine factors related to pathogenicity of P. syringae strains. The presence of the hypersensitive reaction and pathogenicity (hrp)/hypersensitive reaction and conserved (hrc) cluster was shown to be essential for pathogenicity of P. syringae pv. phaseolicola on bean and for triggering hypersensitive response (HR) on non-host plants, such as tobacco and tomato (Lindgren et al., 1986, 1988; Bogdanove et al., 1996). A homologous region with similar function was found also in other plant pathogens (Beer et al., 1991; Bonas et al., 1991; Bogdanove et al., 1996) and was later shown to encode for a type III secretion system (T3SS) with homology to the virulence protein secretion system (Yop) of animal-pathogenic Yersinia spp. (Gough et al., 1992). In P. syringae, the T3SS encodes for a protein apparatus which is responsible for the delivery of virulence-related factors, so-called type III effectors (T3E), into the plant cell (Wei et al., 2000). T3E generally act by promoting pathogenicity or by suppressing host immune defense but constitute a double-edge sword since T3E can also be recognized by specific plant resistance proteins which in turn trigger host immune system (Mackey et al., 2002; Shao et al., 2003; Xiang et al., 2008). However, many T3E have been shown to be functionally redundant thus decreasing the selective pressure on the host to evolve resistance proteins against single T3E (Kvitko et al., 2009). This observation suggested that the compatible interaction between P. syringae and its host is defined by the totality of its T3E repertoire (Lindeberg et al., 2012).
With the advent of affordable next-generation sequencing technologies, many complete and draft genome sequences of strains belonging to the P. syringae species complex have become available. Comparative genomics studies within different pathovars of the P. syringae species complex also revealed that adaptation to woody hosts was reflected by the presence of genes involved in the degradation of woody plant species-related compounds like the pentose sugar xylose and aromatic compounds, such as toluene and catechol (Green et al., 2010; Bartoli et al., 2015; Caballo-Ponce et al., 2016; Nowell et al., 2016; Hulin et al., 2020). Many studies have focused on the determination of the T3E repertoire of strains isolated from different hosts (Lindeberg et al., 2012; Ruinelli et al., 2019) and it is only recently that a few of them reported the convergent acquisition of T3E in strains adapted to the same host (Hulin et al., 2018; Newberry et al., 2019; Moreno-Pérez et al., 2020) or that strain differences in T3E alleles could be linked to host specificity (Zembek et al., 2018; Jayaraman et al., 2020). These findings underline the importance of whole genome-based comparisons to investigate factors involved in the host–pathogen interactions, which indeed are more complex than initially thought.
The plant genus Prunus includes economically important stone fruit trees, such as sweet cherry (Prunus avium), sour cherry (Prunus cerasus), and peach (Prunus persica), which in 2018 accounted for 11.6% of the total fruit orchard area in Europe (Eurostat, 2018). Even more important for the European market are almond trees (Prunus amygdalus) which in 2018 occupied as single species 22.6% of the total area dedicated to growing fruits (Eurostat, 2018). Bacterial canker on Prunus spp. caused by members of the P. syringae species complex affects all aboveground organs of the tree causing heavy yield reduction (up to 75%) and can lead to death of the whole tree, especially in young orchards (Crosse, 1966; Spotts et al., 2010; Hulin et al., 2020). Typical symptoms visible on trunks and branches include sunken, dark brown dieback, and cankers, which are sometimes accompanied by gummy leaks (Puławska et al., 2017). Blossom wilting and browning is mainly visible on highly susceptible varieties and constitute an important source of secondary infection. In addition, necrotic spots can be observed on leaves and on fruits which then lose their commercial values (Puławska et al., 2017). Within the P. syringae species complex, three different PG contain two Pseudomonas species and six P. syringae pathovars, which were found in association with diseases on Prunus spp.
Bacterial canker of sweet and sour cherry is mainly caused by strains belonging to P. syringae pv. morsprunorum race 1 and P. syringae pv. morsprunorum race 2 (Crosse, 1959; Crosse and Garrett, 1963; Freigoun and Crosse, 1975; Ruinelli et al., 2019). Despite being classified as races of the same pathovar, phylogenetic analysis based on sequence similarity of four housekeeping genes or of core genome of 2,085 coding sequences revealed that strains of the P. syringae pv. morsprunorum race 1 belong to PG3, whereas strains of the P. syringae pv. morsprunorum race 2 cluster within PG1 (Nowell et al., 2016; Ruinelli et al., 2019), underlying the need for clarification of the nomenclature of members of the P. syringae species complex.
Bacterial dieback of peach is caused by P. syringae pv. persicae (PG1; Young, 1987) which is also causes disease on nectarine and is weakly pathogenic to plum but not causing disease on apricot and cherry (Young, 1987). Due to its limited distribution in Europe, P. syringae pv. persicae was classified as quarantine organism from the European and Mediterranean Plant Protection Organization (EPPO, 2005) and as recommended regulated non-quarantine pest in the EU plant health regulation in force since December 2019 (Picard et al., 2018). Strains belonging to the P. syringae pv. avii (PG1) were isolated from wild cherry trees (Prunus avium) affected by bacterial canker in France and were shown to be only weakly pathogenic to peach, plum, and apricot (Ménard et al., 2003). Pseudomonas amygdali and P. syringae pv. cerasicola, both belonging to PG3, are the causal agents of the bacterial hyperplastic canker of almond (P. amygdalus; Psallidas, 1997) and bacterial gall of ornamental cherry (Prunus × yedoensis; Kamiunten et al., 2000), respectively. A few years ago, a new species belonging to PG2, namely, Pseudomonas cerasi (Kałużna et al., 2016), was found to be responsible for the development of bacterial canker on cherry trees in Poland and more recently on pear tree in South Korea (Choi et al., 2020).
In addition, symptoms of bacterial canker on Prunus spp. are also caused by strains of P. syringae pv. syringae belonging to PG2 (Crosse and Garrett, 1966). However, in contrast to all above-mentioned pathovars which have been specifically found in association with plant species belonging to the genus Prunus, strains of P. syringae pv. syringae display a broader host range and are responsible for diseases on many other woody and herbaceous hosts (Cazorla et al., 1998; Garibaldi et al., 2007; Zhou et al., 2012; Popović et al., 2015; Ivanović et al., 2017).
In this study, a comparative genomics approach was used to investigate factors potentially involved in the adaptation of P. syringae to plant species belonging to the Prunus genus. Our study revealed the presence of a set of orthologous proteins, which were significantly more present in strains isolated from Prunus spp. than in strains isolated from other hosts or environments. Among them, the T3E HopAY, potentially encoding for a C58 cysteine protease was found to be highly associated with strains isolated from Prunus spp. and revealed patterns supporting co-evolution and host adaptation.
Materials and Methods
Phylogenomics
For comparative genomics purpose, the whole genomes data of 97 strains belonging to the P. syringae species complex, together with one P. fluorescens (strain Pf0-1) and one P. putida (strain KT2440) were used (Table 1). A total of 20 genomes were complete and 79 were draft. The selected set of P. syringae genomes consisted of strains isolated from plants (n = 81) as well as strains isolated from non-agricultural environments (n = 15) and represents 11 of the 13 PG defined by Berge et al. (2014). Plant-associated strains were isolated from over 30 different plant species comprising Prunus spp. (n = 20), Actinidia chinensis (n = 4), Solanum lycopersicum (n = 4), Corylus avellana (n = 5), Cucumis spp. (n = 5), Aesculus hippocastanum (n = 3), Triticum aestivum (n = 3), Hordeum vulgare (n = 3), Phaseolus vulgaris (n = 2), Olea europaea (n = 2), Glycine max (n = 2), Nicotiana sp. (n = 2), Pyrus sp. (n = 2), and other herbaceous and woody hosts (n = 22). Non-annotated genomes retrieved from the NCBI database were annotated using a command line annotation pipeline based on HMMer against an EDGAR based database of Pseudomonas ortholog groups followed by reference genome annotation and a comparison to the Swiss-Prot and RefSeq databases for genes that had no high-quality hit in previous steps (Linke et al., 2011).
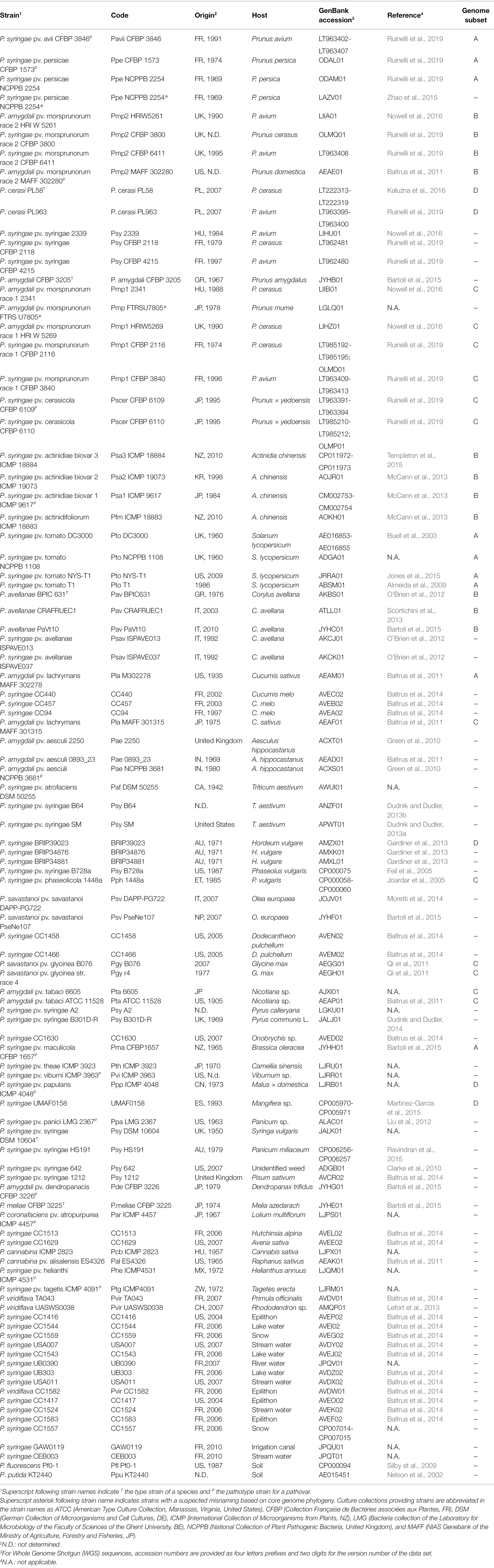
Table 1. List of strains used for this study.
The core genome phylogenetic relationships were obtained using EDGAR 2.2 (Blom et al., 2016) as previously described (Ruinelli et al., 2019).
Comparative Genomics, Gene Sets Calculation, and Identification of Prunus-Associated Genes
Based on the core genome phylogeny, four subsets of genomes were defined (subsets A–D; Table 1) to be used in comparative genomics. Within each of the subsets, the sets of orthologous proteins present in Prunus-associated strains but absent in their phylogenetically closely related non-Prunus-associated strains were determined using EDGAR 2.2 (Blom et al., 2016). The protein sequences (n = 1,058) resulting for each of the subsets (subsets A–D; Figure 1) were used as large query against each other using standalone BLAST v.2.2.29+ (Camacho et al., 2009). All BLASTP hits having identity and coverage higher or equal to 70% were considered as ortholog and displayed in a Venn diagram. Orthologous proteins shared among each combination of subsets (n = 52) were checked for orthologs in the whole set of genomes (n = 97) using EDGAR 2.2.
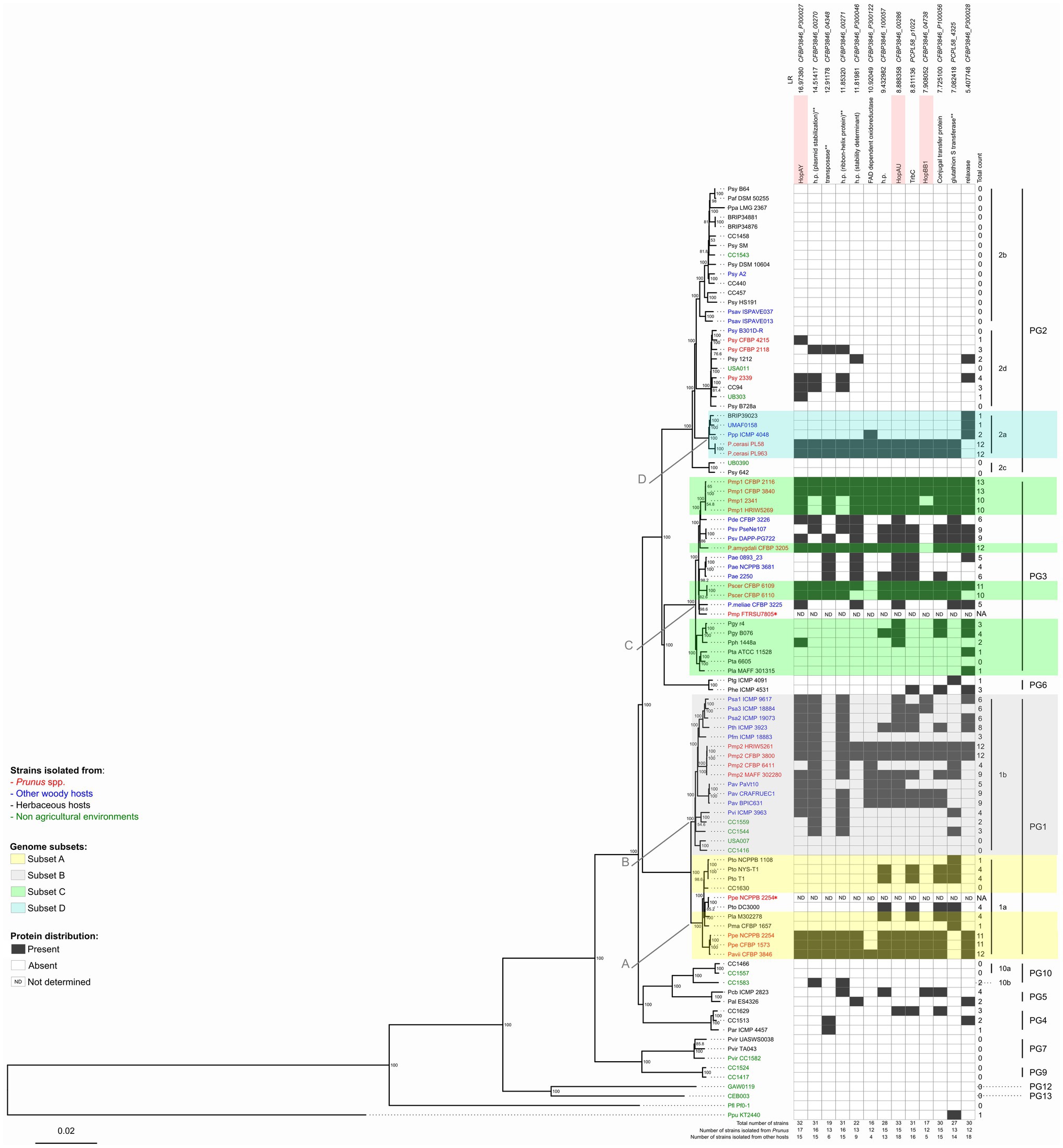
Figure 1. Neighbor-joining (NJ) phylogeny based on the core genome of the Pseudomonas syringae species complex and distribution profile of 13 proteins considered to be significantly associated with Prunus spp. among these strains. A set of 99 genomes of strains belonging to the P. syringae species complex as well as one Pseudomonas fluorescens and one Pseudomonas putida strains were used for this analysis (Table 1). The tree was built using EDGAR 2.2 (Blom et al., 2016) out of a core genome of 1,344 CDS giving a total alignment of 5,36,722 amino acids per genome. Percent bootstrap (bt) support values calculated for 500 reiterations are indicated near nodes. Only bt values over 51 are displayed. The strain names refer to the code field from Table 1. Phylogroups (PG) and clades are indicated on the right. Strains isolated from Prunus spp. are indicated in red, from other woody hosts in blue, from herbaceous hosts in black, and from non-agricultural environments in green. Strain names followed by an asterisk (*) indicate strains which were excluded from further comparative analyses due to a presumed misnaming of the genome. Genome subsets (A-D) used to determine the correlation between gene presence and Prunus spp. association are indicated with color highlights. Arrows indicate the node at which Prunus-associated strains are diverging from the non-Prunus-associated strains within the same genome subset. Protein orthologs were retrieved out of these 97 genomes using EDGAR 2.2 (Blom et al., 2016). Black squares indicate presence of the protein based on FIGURE 1the orthology criteria of EDGAR 2.2. Proteins highlighted in pink are involved in virulence based on their annotation. Protein descriptions followed by two asterisks (**) indicate that orthologs were also found using online TBLASTN analysis against 13 additional Pseudomonas species closely related to the P. syringae species complex as reported in Supplementary Figure 4. The proteins are ordered by decreasing significance of the likelihood ratio (LR) statistic when exceeding the p ≤ 0.05 threshold of 5.36. This order is not indicative of any physical proximity. Locus tags and LR statistic are reported over each considered protein; h.p.: hypothetical protein; NA: not applicable; and ND: not determined.
Using the core genome phylogeny as a reference, associations were identified between the presence/absence of each orthologous protein in the analyzed genomes (n = 99) and the discrete binary trait designated “Prunus spp. isolate” or “other host/environment isolate” using BayesTraits v.3.0.5 (Pagel, 1994; Barker and Pagel, 2005; Pagel and Meade, 2006). The goodness of fit of the dependent versus independent model was compared with a likelihood ratio (LR) test by using a Perl script to run both models (available from https://github.com/reubwn/bayestraits-wrapper; Nowell et al., 2016). The LR test was conducted for the 52 genes that occurred in either greater than six or fewer than 92 strains, resulting in a total of 49 LRs. A null LR distribution model was constructed by randomly permuting a total of 100 times either the gene occurrence data for each of the 52 tested genes, the binary trait designation or both variables, in each case calculating a new LR statistic (Nowell et al., 2016). The null distribution was then used to derive the p-value thresholds. The proteins considered to be significantly more present in Prunus-associated strains were also used as online TBLASTN queries against nucleotide databases from 13 additional Pseudomonas species closely related to P. syringae (Mulet et al., 2010; Lalucat et al., 2020; Supplementary Figure 4). The presence of an ortholog in any of these 13 closely related Pseudomonas species was then reported when at least one TBLASTN hit having identity and coverage higher or equal to 70% was detected.
HopAY and HopAR Ortholog Retrieval and Phylogenetic Analysis
The bidirectional best hits protein orthology criteria used in EDGAR 2.2 in the previous step is mostly designed to determine the presence of a complete and probably functional ortholog protein among different genomes. However, in order, to investigate the evolution of a gene within different strains it is also important to differentiate between absence or inactivation of that gene. For this purpose, the hopAY reference sequence (GenBank accession number CP000059.1; locus tag: PSPPH_A0129) was derived from the T3E database (PPI, 2010) and used as online BLASTN query against all genomes selected for comparative genomics (n = 97). The resulting nucleotide sequence was translated using the ExPASy translate tool (ExPASy) and the longest open reading frame corresponding to the reference HopAY sequence (GenBank accession number: AAZ37994.1) was used for alignment. Deviations between the BLASTN hit and the identified protein were investigated in comparison to the reference hopAY gene for the possibility of pseudogenization due to frameshift or insertion of a stop codon in the correct reading frame. DNA and amino acid sequences were aligned using ClustalW, while MEGA 6.0 was used to generate neighbor-joining (NJ) phylogeny using the Jones–Taylor–Thornton model with the gamma parameter set at 2.25 and bootstrap values after 1,000 repeats as suggested elsewhere (Lindeberg et al., 2005). A similar method was used twith hopAR (GenBank accession number AJ870974.1 positions 17,471–18,274) and hopAU (GenBank accession number LT963409.1; locus tag: CFBP3840_01698).
Comparison of the Arabidopsis PBS Resistance Protein Among Different Plant Species
The T3E HopAR1 (formerly AvrPphB) from P. syringae pv. phaseolicola belongs to the same family of C58 protease as HopAY and has been shown to proteolytically cleave the serine/threonine protein kinase PBS1 in Arabidopsis. The amino acid sequence of PBS1 from Prunus persica (GenBank accession number XP_007225732) was used to perform a TBLASTN search in the Transcriptome Shotgun Assembly Sequence (TSA) and Protein NCBI databases of the plants associated with strains possessing a full-length or truncated HopAY.
The transcribed mRNA sequences retrieved from the TSA were translated using the ExPASy translate tool and the obtained amino acid sequences were aligned to the PBS1 amino acid sequence retrieved from the NCBI protein database of 22 additional plant species (Supplementary Table 1) using ClustalW on the MEGA 6.0 software. To clarify the phylogenetic relationships among the PBS1 proteins of different plants, a maximum likelihood phylogeny was reconstructed using the Jones–Taylor–Thornton model with the gamma parameter set at 2.25 and bootstrap values of 1,000.
Data Availability
The data sets analyzed for this study are available at the NCBI GenBank/DDJ/EMBL database under the accession detailed in Table 1.
Results
Phylogenomics
In order to clarify the exact phylogenetic position of the Prunus-associated strains in the data set within the P. syringae species complex and to define suitable strains and subgroups for comparative genomics (Table 1), a core genome-based phylogeny was generated for the selected set of genomes using EDGAR 2.2 (Blom et al., 2016). The obtained tree was generated based on the concatenated and aligned amino acid sequences of 1,344 genes consisting of a total length of 536,722 amino acids (Figure 1).
The main clustering obtained from the core genome phylogeny reflects the PG previously defined by Multi Locus Sequence Analysis (MLSA; Sarkar and Guttman, 2004; Hwang et al., 2005; Sarkar et al., 2006) and single locus phylogeny (Parkinson et al., 2011; Berge et al., 2014). However, our analysis revealed that two genomes obtained from the Whole Genome Shotgun (WGS) NCBI database which were supposed to represent strains isolated from Prunus spp. did not cluster as expected based on previous work (Parkinson et al., 2011). Indeed, the sequence with the GenBank WGS accession prefix LAZV01 which is supposed to represent P. syringae pv. persicae strain NCPPB 2254 and should cluster close to P. syringae pv. avii (Parkinson et al., 2011) was found to be clustering really close to the complete genome of P. syringae pv. tomato DC3000 and quite distant from the two other P. syringae pv. persicae genomes generated previously (Ruinelli et al., 2019). Additionally, the sequence with accession number LGLQ01 which was deposited in the NCBI database as P. amygdali pv. morsprunorum strain FTRSU7805 clustered closer to P. syringae pv. cerasicola and Pseudomonas meliae than to other strains of P. syringae pv. morsprunorum race 1. This observation was supported by the calculation of the average nucleotide identity (ANI) values among the suspected strains, their observed phylogenetically closely related strains and their supposed closely related strains (Supplementary Figure 1). Considering these facts, the sequences with the WGS accession prefixes LAZV01 and LGLQ01 were not included in further comparative genomics analysis.
Correlation Between Genes Presence and Prunus spp. Association
The number of orthologous proteins present in Prunus-associated strains but absent in non-Prunus-associated strains retrieved for each of the compared genome subsets (subsets A–D; Table 1; Figure 1) ranged from 41 (PG3, genome subset D) to 758 (PG2a, genome subset C; Figure 2). This considerable difference could be because Prunus-associated strains within PG3 belonged to different pathovars and species (P. syringae pv. cerasicola, P. syringae pv. morsprunorum race 1 and P. amygdali), whereas within PG2a only strains of P. cerasi have been described to date as being associated with Prunus diseases. Among P. syringae pv. avii and P. syringae pv. persicae (PG1a, genome subset A), a relatively high number of orthologous proteins were retrieved (n = 249), whereas only 70 orthologous proteins were found within strains of the genome subset B (PG1b; Figure 2).
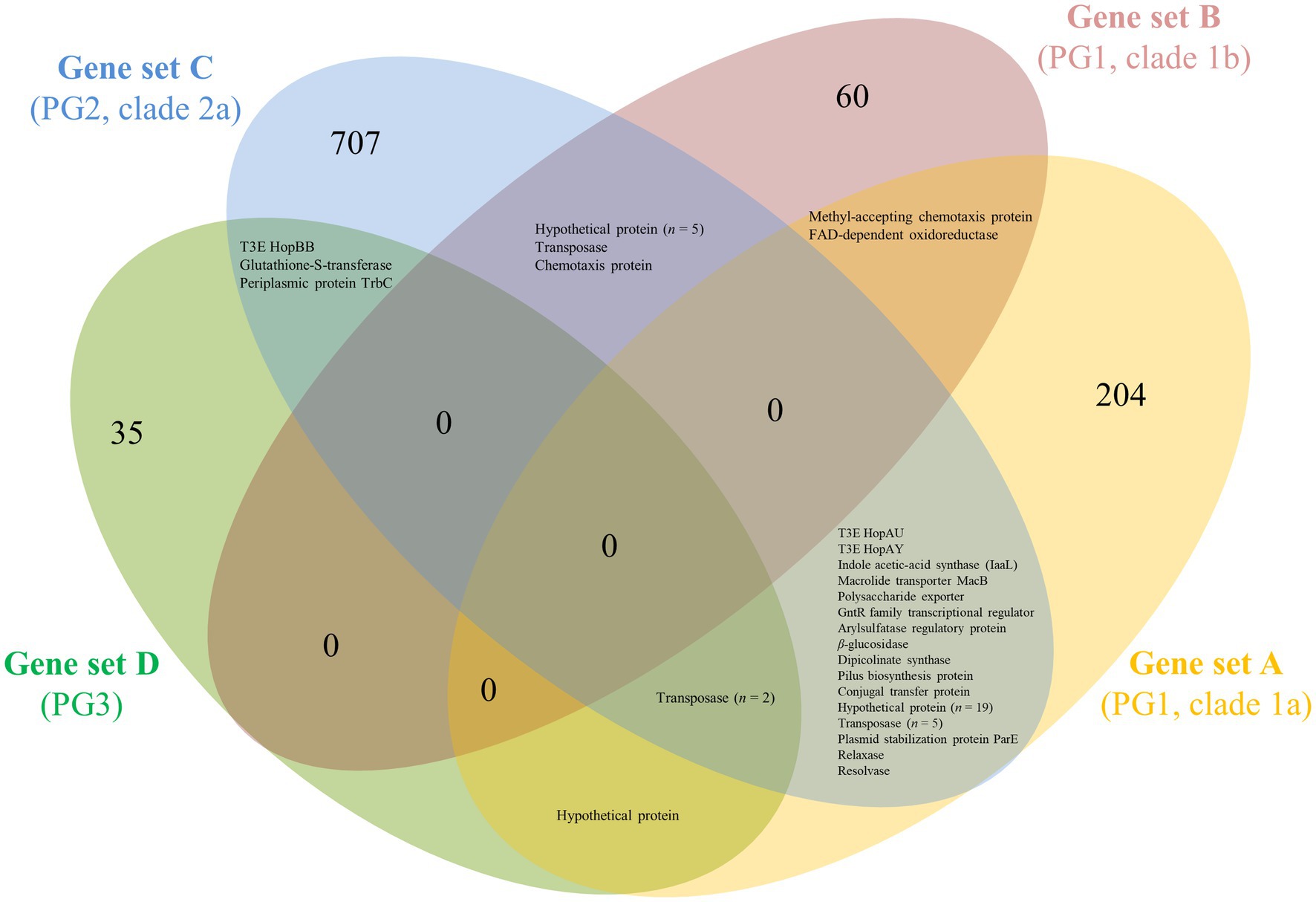
Figure 2. Venn diagram showing groups of ortholog proteins within Prunus-associated Pseudomonas syringae strains belonging to different PG but absent from phylogenetically closely related non-Prunus-associated strains. Subsets of genomes to be compared were defined based on core genome phylogeny within PG1a (subset A), PG1b (subset B), PG2a (subset C), and PG3 (subset D; see Figure 1). For each subset of genomes, the core genome of the Prunus-associated strains was calculated, and at the same time, all ortholog proteins found in non-Prunus-associated strains within the same subset were discarded using EDGAR 2.2 (Blom et al., 2016). The protein sequences resulting for each of the subsets (gene sets A–D) were used as BLASTP query against each other using standalone BLAST v.2.2.29+ (Camacho et al., 2009) and considered as ortholog if identity and coverage were higher or equal to 60%.
To verify which proteins were shared among Prunus spp. associated strains belonging to different PG, the proteins retrieved for each genome subset (n = 1,058) were compared for orthology using BLASTP and the results obtained for each possible combination represented in a Venn diagram (Figure 2). A total of 52 proteins were found to be shared at least between two genome subsets but no protein was found to be shared among all Prunus-associated members of the P. syringae species complex. Each protein was checked for distribution across all the initially selected set of genomes (n = 97). None of the analyzed proteins (n = 52) was found exclusively in Prunus-associated strains but 19 of them were found to be significantly more abundant in Prunus-associated strains than in non-Prunus-associated strains (likelihood ratio statistic exceeding the p ≤ 0.05 threshold of 5.36; Figure 1; Table 2; Supplementary Figure 2). Out of these, only proteins present in at least 60% of the Prunus spp. isolated strains were finally considered, giving a total of 13 proteins (Figure 1; Table 2). Strains isolated from Prunus spp. belonging to PG1a, PG1b, PG2a, and PG3 possessed a similar distribution profile with exception of the P. syringae pv. morsprunorum race 2 strain CFBP 6411 (PG1b) and strains from PG2d which were more divergent (Figure 1).
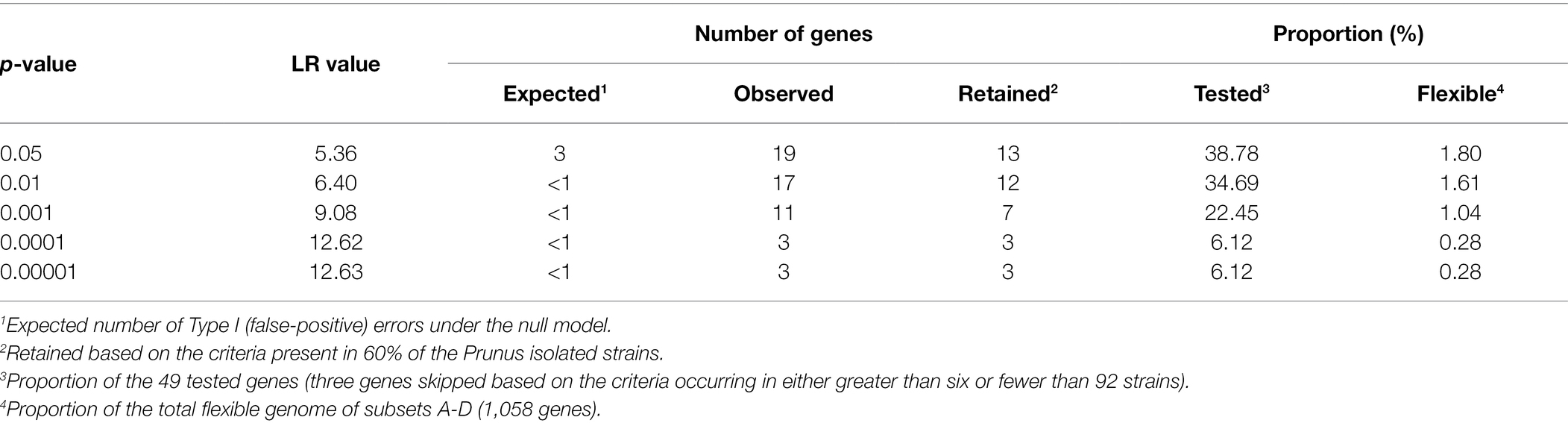
Table 2. Number of genes significantly more present in Prunus spp. isolated strains.
A third of the analyzed proteins were hypothetical proteins (n = 4) and also a third were located potentially on plasmids (n = 4) when complete genomes were available (Table 1; Supplementary Figure 3). However, three known virulence factors were found to be significantly more present in Prunus spp. associated members of the P. syringae species complex, namely, three T3E (HopAY, HopAU, and HopBB; Figure 1). These three known virulence factors were only reported in the species P. syringae during ortholog analysis within 13 additional Pseudomonas species closely related to the P. syringae species complex (Supplementary Figure 4).
The T3E HopAY was the protein with the highest LR statistic and the most abundant in Prunus spp. associated strains (89%) if compared to all other considered proteins (n = 12) and it was found only in 19% of strains isolated from other hosts or from non-agricultural environment (Figure 1). Within strains of the PG2 (n = 31) only six strains harbored HopAY of which four were isolated from Prunus spp. (Figure 1).
A similar distribution was observed for the T3E HopAU, which was present in 80% of Prunus-associated strains and 23% of strains isolated from other hosts. Out of the 32 strains possessing HopAY, 27 also possessed HopAU (Figure 1; Supplementary Figure 5). The T3E HopBB was present in only 6% of non-Prunus isolated strains but its abundance was also lower in strains isolated from Prunus (63%, Figure 3).
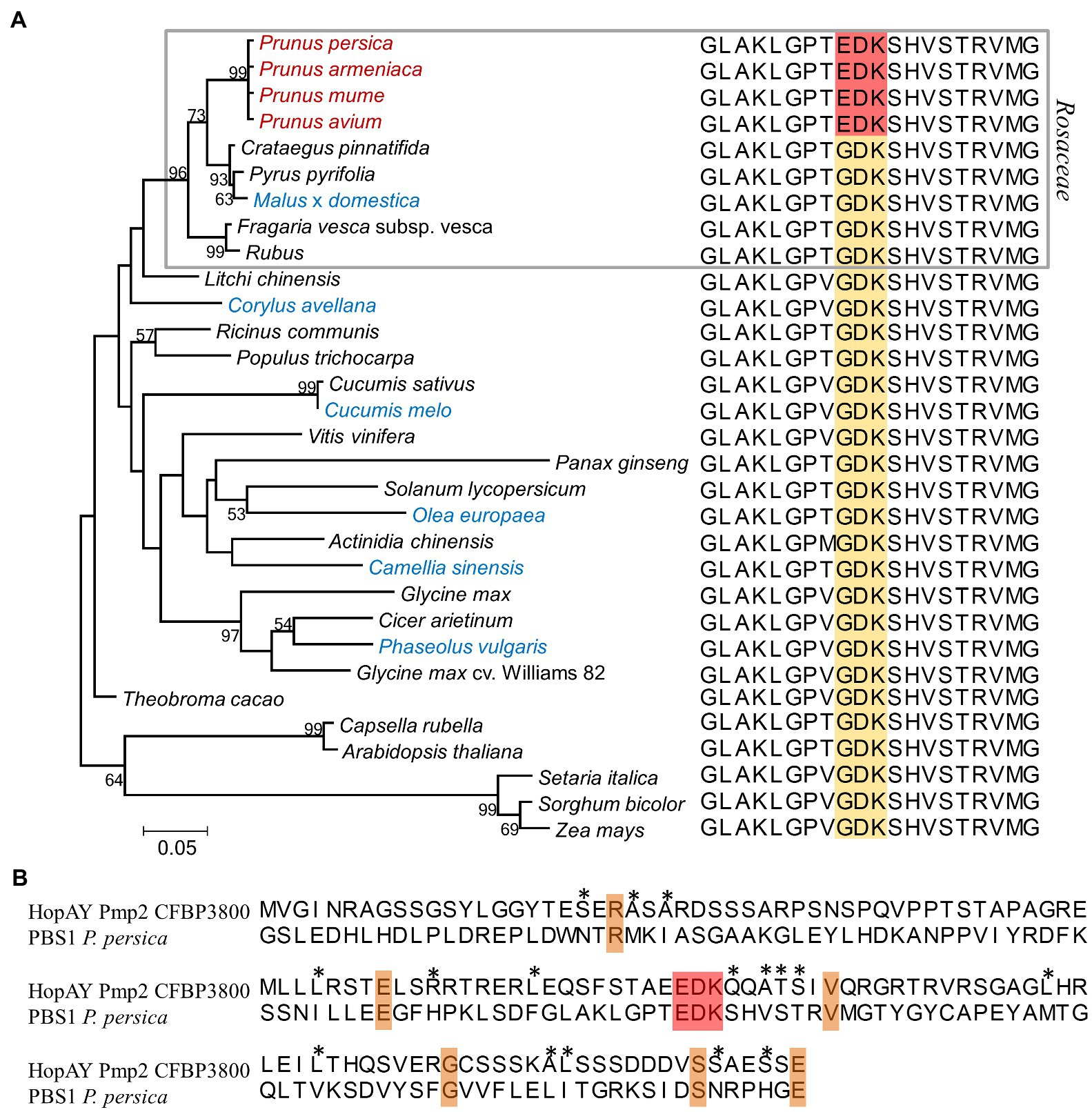
Figure 3. Comparison of the serine/threonine protein kinase PBS1 among different plant species (A) and relation to the HopAY sequence (B). (A) Maximum likelihood phylogeny of the PBS1 protein among different plant species and relative PBS1 sequence stretch corresponding to the amino acids 233–252 of the Arabidopsis thaliana PBS1 sequence (NCBI Acc. Nr. NP_196820) containing the HopAR1 cleavage site GDK (brown block). The corresponding EDK region in the PBS1 sequence of the Prunus spp. is highlighted in red. In the phylogeny Prunus spp. members are reported in red, whereas plants other than Prunus spp. from which Pseudomonas syringae strains possessing the HopAY gene were isolated are indicated in blue. The gray block indicates species belonging to the Rosaceae family. The evolutionary distances were computed using the Jones–Taylor–Thornton model matrix-based with a gamma distribution (shape parameter = 2.25). Percent bootstrap (bt) support values calculated for 1,000 reiterations are indicated near node. Only bt values over 51 are displayed. All ambiguous positions were removed for each sequence pair giving a total of 598 positions. Alignments were obtained using ClustalW and phylogenetic analysis was performed with MEGA 6.0. (B) Alignment of HopAY and PBS1 around the EDK region. The first 133 amino acids (aa) of the HopAY sequence of Pseudomonas syringae pv. morsprunorum race 2 CFBP 3800 (Pmp2 CFBP3800) were aligned to the aa 170–302 of the Prunus persica PBS1 sequence (NCBI Acc. Nr. XP_007225732). The EDK motif is highlighted in red, identical residues are highlighted in orange, whereas residues sharing similar side chain properties at a specific position are indicated by asterisks.
HopAY is predicted to belong to the same class of C58 peptidases like the well-characterized T3E HopAR (formerly AvrPphB). HopBB has been shown to interact with regulators of the jasmonic acid hormone signaling pathway in Arabidopsis (Yang et al., 2017), whereas HopAU was recently shown to activate plant immunity by interacting with a calcium-sensing receptor in Nicotiana benthamiana and in kiwifruit (Zhang et al., 2022).
Sequence Comparison of HopAY and HopAR, a Very Well-Characterized C58 Cysteine Protease in Pseudomonas syringae
The T3E HopAY showed the strongest level association with Prunus isolated strains in respect to all other genes (n = 13) analyzed in this study (Figure 1) and belongs to the C58 cysteine proteases family. Another well-studied and characterized T3E encoding for a C58 peptidase is HopAR which was initially identified in P. syringae pv. phaseolicola as being responsible for elicitation of HR in bean (Jenner et al., 1991; Puri et al., 1997). Orthologs of hopAR were retrieved from 10 out of 19 strains isolated from Prunus spp. and in 13 strains isolated from other hosts (Supplementary Figure 5). Around 15 strains, including nine strains isolated from Prunus spp., possessed both hopAY and hopAR orthologs. The target of HopAR in Arabidopsis is the serine/threonine protein kinase AVRPPHB SUSCEPTIBLE 1 (PBS1) and the ability of HopAR to cleave PBS1 is related to the presence in PBS1 of the Glycine (G241)-Aspartate (D242)-Lysine (K243) motif which is also found at the autocleavage site of HopAR (Shao et al., 2003). Mutations in the amino acids G241, D242, and K243 of PBS1 in Arabidopsis reduced the proteolytic activity of HopAR by 90, 75, and 15%, respectively (Shao et al., 2003). The cleavage of PBS1 by HopAR induces a conformational change of PBS1 causing the exposition of a particular motif (SEMPH) which is sensed by the resistance protein RESISTANCE TO PSEUDOMONAS SYRINGAE 5 (RPS5) in Arabidopsis, leading to HR (Ade et al., 2007; Qi et al., 2012, 2014). In addition, the determination of the crystal structure of HopAR1 revealed the presence of a catalytic triad composed by a cysteine (C98), histidine (H212), and aspartate (D227) which has been shown to be essential for catalysis (Zhu et al., 2004). As already noticed by Zumaquero et al. (2010), the amino acid sequence similarity between HopAY and HopAR is very limited (68% query coverage and 27% identity; Supplementary Figure 6). Nevertheless, motifs corresponding to the catalytic triad were identified also on HopAY and localized at C156, H265, and D280 using the HopAY reference present in the T3E database (PPI, 2010; NCBI locus tag: PSPPH_A0129), whereas no motif corresponding to the cleavage site of HopAR (GDK) was found in the HopAY sequence (Figure 3B). Secondary structure prediction revealed a conserved pattern of α-helices and β-sheets between HopAR and HopAY as well as other members of the C58 proteases (Zhu et al., 2004). Alignment of the PBS1 protein sequence from different plant species (n = 31) revealed that the protein kinase PBS1 is quite conserved among different plant families (Qi et al., 2014). However, we noticed that members of the Prunus spp. (n = 4) possess an EDK motif instead of the GDK motif essential for HopAR cleavage in PBS1, which was in contrast conserved in all other plant species included in the comparison (n = 27; Figure 3A). The alignment of the PBS1 sequence of P. persica with HopAY revealed that the same EDK motif was found also within the N-terminal half of HopAY (E76, D77, and K78) followed by a stretch of four amino acids with the same physical properties (Figure 3B). In addition, all PBS1 sequences analyzed in this study with exception of PBS1 of Arabidopsis thaliana and Capsella rubella were also lacking the SEMPH motif, which was shown to be essential for RPS5 mediated resistance in Arabidopsis (Qi et al., 2014).
Sequence Comparison of HopAY Among Different Members of the Pseudomonas syringae Species Complex
In order to determine the evolutionary relationships of hopAY within different strains of the P. syringae species complex, a BLASTN search was performed using the hopAY sequence of P. syringae pv. phaseolicola 1448a (PPI; NCBI locus tag: PSPPH_A0129) against the set of genomes selected for comparative genomics (n = 97; Table 1). The BLASTN analysis revealed the presence of 43 hopAY sequences in a total of 36 strains. In addition to the strains retrieved by the protein-based ortholog search (n = 32; Figure 1), a hopAY ortholog was present in the horse chestnut-associated P. amygdali pv. aesculi strains 2250, 0893_23, and NCPPB 3681 as well as in the apple tree pathogen P. syringae pv. papulans ICMP 4048. With exception of strain HRIW5269, all other P. syringae pv. morsprunorum race 1 strains analyzed in this study (n = 3) were possessing more than one copy of hopAY. In the genomes of P. syringae pv. avii strain CFBP 3846, P. amygdali CFBP 3205, and P. syringae pv. dendropanacis CFBP 3226, two copies of hopAY were found as well.
Sequence analysis revealed that the retrieved hopAY sequences (n = 43) could be divided into five major groups based on the insertion–deletion (indel) scheme affecting this gene (Figure 4). The indel group 1 (n = 25) consisted of sequences with no insertions or deletions if compared to the reference hopAY sequence available in the hop database and were mostly retrieved from genomes of strains isolated from Prunus spp. (n = 16). Sequences belonging to the indel group 2 (n = 3) were affected by a probable transposase insertion leading to a 41-bp deletion at the 5′ end (Figure 4) and were retrieved only from Pseudomonas avellanae strains. Indel groups 3, 4, and 5 displayed an additive indel profile. In fact, the indel group 3 (n = 2) displayed a 4-bp deletion at position 66–70 which was shared also from groups 4 (n = 6) and 5 (n = 2). A 1-bp deletion located at position 737 was also present in sequences of groups 4 and 5, whereas group 5 was additionally having a 12-bp deletion at position 149–160. Sequences of the indel group 4 were retrieved only from strains of the PG3 and mostly isolated from Prunus spp. (n = 5), namely, P. syringae pv. cerasicola and P. syringae pv. morsprunorum race 1. Based on the complete genomes previously sequenced using PacBio (Ruinelli et al., 2019), it was possible to determine that all hopAY of the indel group 4 were located on the chromosome, whereas the hopAY of indel group 1 were located on both chromosome and plasmids. Sequences of the indel group 5 were retrieved from two P. syringae pv. aesculi strains isolated in Europe, whereas the P. syringae pv. aesculi isolated in India displayed an additional resolvase insertion within hopAY (Figure 4). In addition to the above-described groups, four sequences displayed unique indel profiles varying from transposase insertions (Psa ICMP 18884) to 1-bp deletions (CC94; Figure 4).
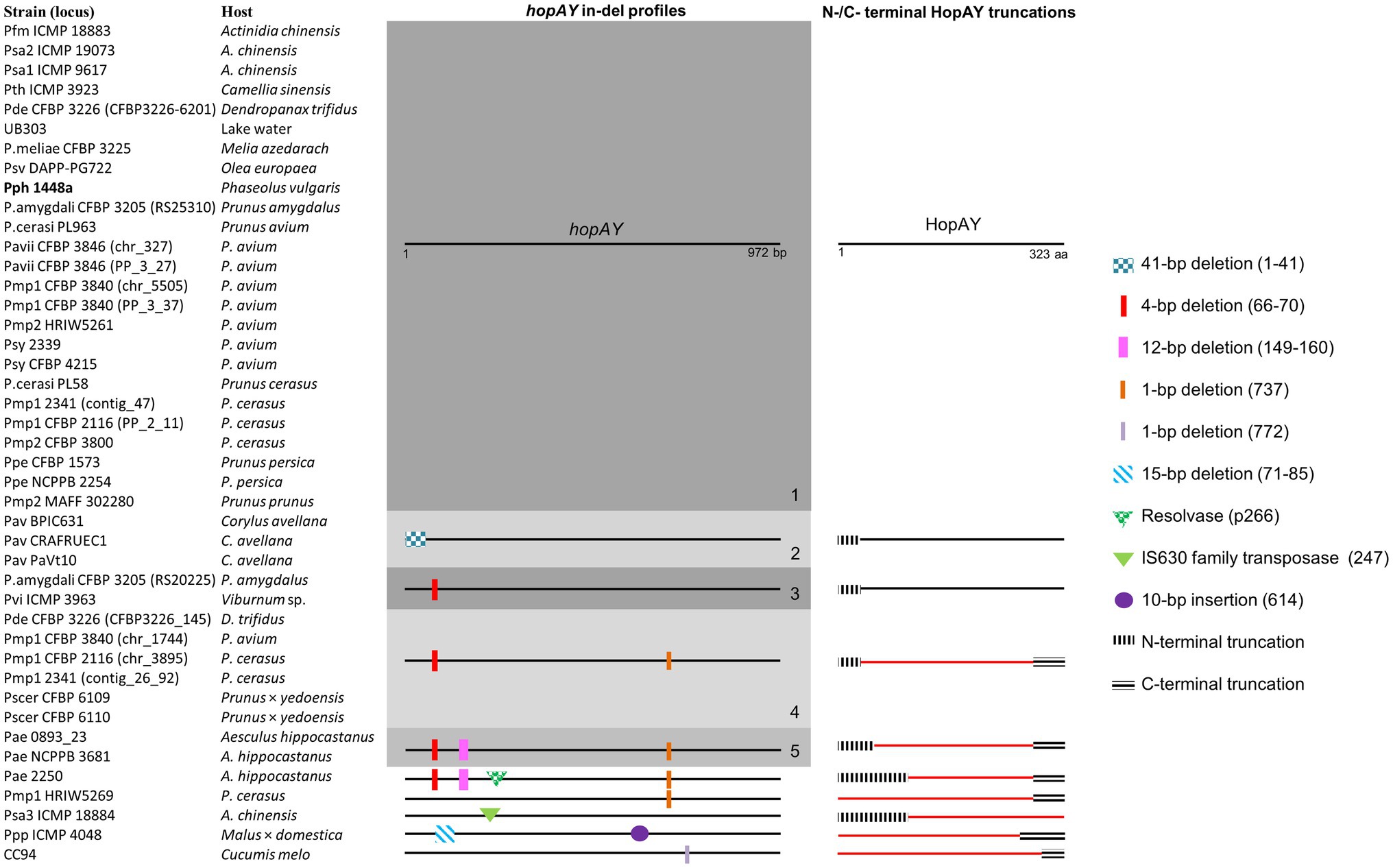
Figure 4. Insertion–deletion profiles of hopAY retrieved by BLASTN and corresponding HopAY truncation scheme. Five major groups (1–5) were defined based on conserved indel mutations. Numbers in bracket indicate the position of the insertion or deletion based on the reference hopAY sequence from Pseudomonas syringae pv. phaseolicola 1448a (indicated in bold) available in the Hop database (PPI). HopAY proteins with a truncation affecting the catalytic domains characteristic for C58 cysteine protease and thus considered as non-functional are represented in red. The strain names used refer to the code field from Table 1. Figure is not to scale.
Alignment of HopAY sequences retrieved from the BLASTN search (n = 43) revealed that sequences belonging to the previously described indel groups 4 and 5 as well as four of five additional sequences with unique indel profiles (Figure 4) were missing both H256 and D280 due to the introduction of a premature stop codon (Figure 4). On the other side, the transposase insertion within hopAY of P. syringae pv. actinidiae ICMP 18884 led to a N-terminal truncation deleting the C156 motif. In addition, the HopAY from P. amygdali CFBP 3205 belonging to the indel group 1 possessed a tyrosine instead of the expected H256. With exception of both P. syringae pv. cerasicola strains, the other three strains isolated from Prunus belonging to the indel group 4 thus possess an inactivated HopAY and at least another copy of hopAY encoding a full-length protein. The N-terminal truncations observed in the P. avellanae strains (indel group 2) and in the sequences of the indel group 3 did not affect the catalytic triad of HopAY and thus it was not possible to determine if the derived protein would be functional or not (Figure 4).
Phylogeny of HopAY
The NJ phylogeny obtained from the 43 retrieved HopAY sequences did not reflect the phylogeny obtained from the core genome of the 36 strains possessing a hopAY ortholog (Figures 5A,B). In particular, the HopAY sequence of Prunus-associated strains belonging to PG1a, PG1b, PG2a, and PG3 cluster closer to each other than to strains isolated from other hosts belonging to the same PG.
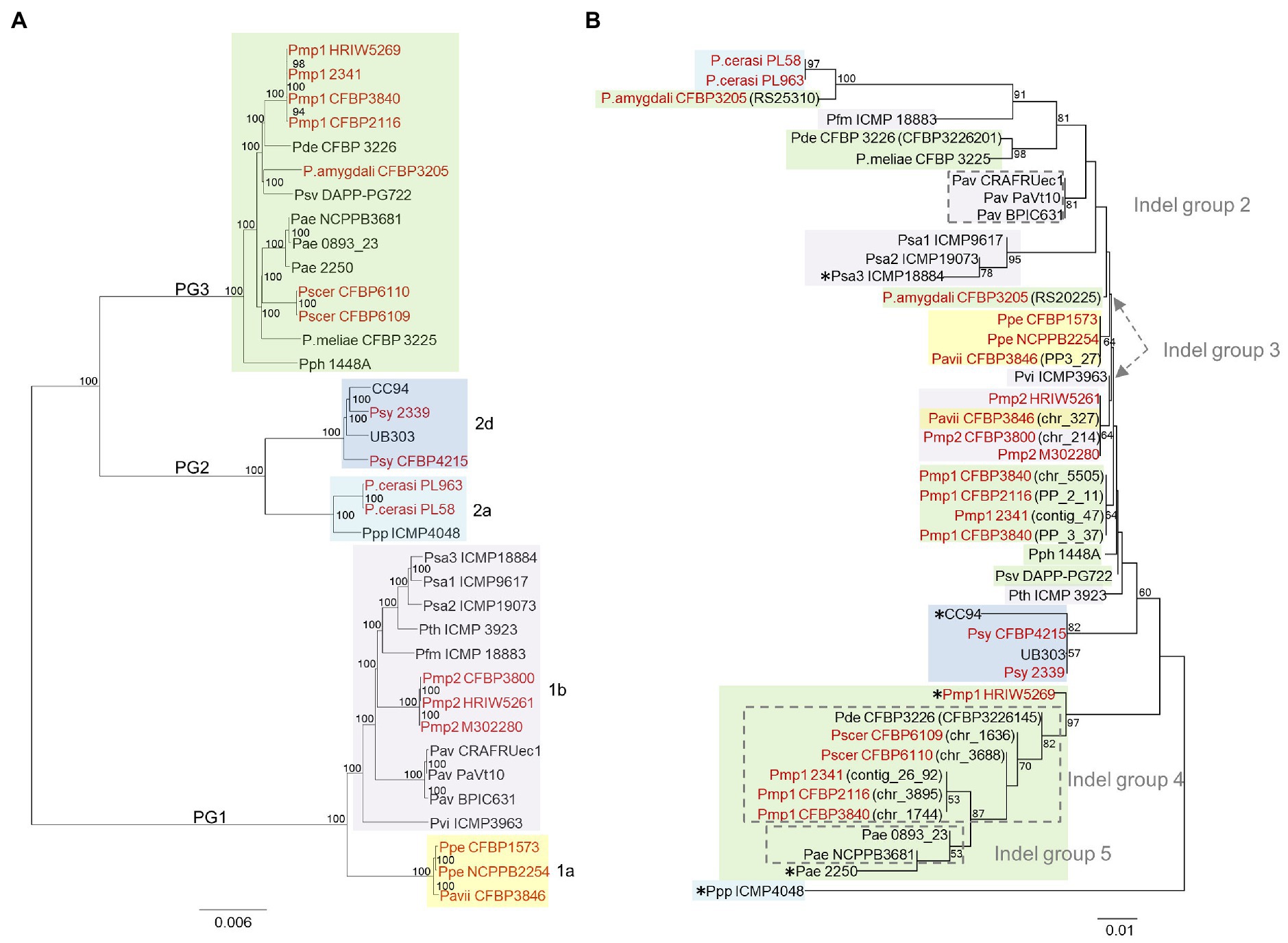
Figure 5. Comparison of the neighbor-joining phylogeny based on the core genome (A) and on HopAY (B). (A) The core genome of the 36 strains possessing an hopAY ortholog based on the BLASTN search was determined using EDGAR 2.2 (Blom et al., 2016) out of a core genome of 2,511 CDS giving a total alignment of 872,675 amino acids per genome. The strain names used refer to the code field from Table 1. Phylogroups and clades are indicated on the left and on the right, respectively. (B) The phylogeny of the 43 retrieved HopAY sequences was computed using the Jones–Taylor–Thornton model matrix-based with a gamma distribution (shape parameter = 2.25). Percent bootstrap (bt) support values calculated for 1,000 reiterations are indicated near nodes. Only bt values over 51 are displayed. All ambiguous positions were removed for each sequence pair giving a total of 323 positions in the final data set. Evolutionary analyses were conducted in MEGA 6.0. Indel groups (gray dashed lines boxes) refer to Figure 4. Asterisks (*) indicate strains with unique indel profiles. If nothing stated, sequences belong to indel group 1. For strains possessing multiple copies of HopAY, the locus tag is indicated in brackets. Strains isolated from Prunus spp. are highlighted in red. PG and clades are indicated with the same color code as used on the left in panel (A).
For example, HopAY sequences from P. syringae pv. morsprunorum race 2 strains belonging to clade PG1b cluster closer to Prunus isolated strains of PG1a than to strains of the PG1b, namely, P. syringae pv. actinidiae and P. avellanae (Figure 5B). In addition, protein sequences from strains of the PG2d form a monophyletic cluster, which is distantly related to strains of the PG2a clade (Figure 5B). Within PG2a, the proteins from Prunus-associated P. cerasi strains are more closely related to the full-length protein of the PG3 strain P. amygdali CFBP 3205, also isolated from Prunus, than to that of P. syringae pv. papulans strain ICMP 4048 (PG2a). HopAY sequences belonging to the indel groups 4 and 5, which were all retrieved from members of the PG3, form a clearly separated cluster together with the proteins from two strains with unique indel profile, being P. syringae pv. morsprunorum race 1 HRIW5269 and P. syringae pv. aesculi strain 2250 (Figure 5B).
Discussion
The development of effective measures to control plant diseases would be facilitated by a founded knowledge on the pathogen biology as well as on mechanisms involved in the plant–pathogen interactions. Diseases caused by members of the P. syringae species complex on species belonging to the Prunus genus are responsible for relevant yield losses, particularly in young orchards (Puławska et al., 2017). To date, a total of two species and six pathovars belonging to three different PG of the P. syringae species complex have been found in association with diseases of species within the Prunus genus. Despite their economic importance, not much is known about the evolution and adaptation strategies of members belonging to the P. syringae species complex toward Prunus spp. In this study, a whole-genome comparison approach was used aiming to identify genetic traits shared among these phylogenetically distantly related pathovars and species that could give insights into the evolutionary aspects related to the adaptation toward Prunus spp. hosts.
From the core genome-based phylogeny obtained in this study, it was evident that the pathoadaptation toward Prunus spp. is not the result of a single evolutionary event but have evolved independently at least three times in the evolutionary history of the P. syringae species complex. This convergent pathoadaptation in distantly related strains leading to virulence on the same host is not unique for the P. syringae—Prunus spp. pathosystem within the P. syringae species complex (Morris et al., 2019). In fact, phylogenetically distantly related members of the P. syringae species complex were also found to have converged onto hazelnut (Wang et al., 2007).
Wang et al. (2007) explained the occurrence of convergent pathoadaptation toward a specific host is not only by the independent acquisition of genes necessary for a successful association but also by the specific loss or inactivation of genes resulting in the same host range limitation. The predominant evolutionary force driving such events in the P. syringae species complex is horizontal gene transfer (HGT), which allows the transfer (gain or loss) of genes between closely and distantly related strains within relatively short evolutionary periods (Nowell et al., 2014). Based on HGT, genes having a selective advantage can be easily accumulated leading to new pathovars or lineages which can adapt to new ecological niches and hosts.
The comparative genomic analysis performed in this study revealed a strong correlation between the presence of the T3E HopAY and the association of members of the P. syringae species complex with hosts belonging to the genus Prunus. The gene hopAY was claimed to be significantly associated with the woody host niche (Nowell et al., 2016), something that we also noticed if considering the hopAY orthologs. However, our analysis highlighted the importance of considering not only the gene sequence but also the protein sequence to correctly interpret T3E profiles. A few studies recently took this also into consideration and showed that T3E alleles were linked to host specificity (Zembek et al., 2018; Jayaraman et al., 2020).
Unlike many T3E which have no known function, HopAY is a putative member of the C58 cysteine protease family which is characterized by the presence of an invariant catalytic triad composed by a Cysteine (C), a Histidine (H), and an Aspartate (D) which are essential for catalysis (Shao et al., 2002). Based on that knowledge, it was possible to determine that half of the hopAY sequences retrieved based on DNA orthology were encoding for proteins missing at least one of those essential amino acids (C/H/D) and thus would not be functional. Inactivated HopAY were found also in Prunus-associated strains but most of them were shown to possess an additional hopAY encoding for a full-length protein possessing the C/H/D catalytic triad, often located on a plasmid. The evolutionary dynamics observed within the retrieved HopAY sequences suggests that this protein may be of selective disadvantage on certain hosts and therefore mutated at higher rate than other T3E, like already observed for other T3E families (Baltrus et al., 2011). The phylogeny obtained based on HopAY did not reflect the core genome-based phylogeny, thus excluding a vertical pattern of inheritance and further support the importance of HGT as adaptive force in the evolution of the P. syringae species complex. In addition, it revealed that the HopAY sequence present in many Prunus spp. associated strains belonging to PG1a, PG1b, and PG3 was nearly identical, supporting the theory of convergent pathoadaptation of these strains.
HopAR (former AvrPphB), another T3E of the C58 cysteine protease family, was subject of many molecular studies in the last decades. These studies revealed that HopAR targets the protein kinase PBS1 in Arabidopsis due to the presence of a particular recognition motif (GDK) which was also found in the sequence of HopAR (Shao et al., 2003). Cleavage of PBS1 by HopAR could result in increased virulence or lead to resistance in Arabidopsis plants lacking or possessing the resistance protein RPS5, respectively (Ade et al., 2007). The PBS1 protein is quite conserved among different plant species representing a good target for T3Es. In contrast to all other considered plant species, including other members of the Rosaceae family, the PBS1 sequence found in Prunus spp. lacked the GDK motif necessary for HopAR cleavage and possessed instead an EDK motif which was found also in the N-terminal half of HopAY. The N-terminal part of members of the C58 cysteine protease family is known to be involved in substrate specificity as shown for HopAR and for the DKM motif of Y4zC, a putative T3E of Rhizobium (Zhu et al., 2004). Based on this observation, we speculate that HopAY could act in a similar way as HopAR but specifically evolved to cleave the PBS1 ortholog of Prunus spp., thus explaining why HopAY is significantly associated with strains adapted to this group of hosts. This hypothesis is supported by the fact that strains isolated from other hosts, such as Corylus avellana and Aesculus hippocastanum, both harboring a GDK motif in the PBS1 sequence, possessed a truncated or non-functional HopAY, respectively. In addition, Zumaquero et al. (2010) showed that knocking out HopAY does not affect pathogenicity of P. syringae pv. phaseolicola 1448a on bean, whose PBS1 protein also possesses a GDK motif. Of course, it could also be hypothesized that the PBS1 protein in Prunus has evolved to be cleaved by HopAY to trigger resistance by action of a third unknown resistance protein (similarly to RPS5). However, pathogenicity tests using wild-type strains revealed no direct correlation with presence or absence of hopAY (Ruinelli et al., 2019). Therefore, this suggests that Prunus spp. does not possess a recognition system for HopAY. At the same time, based on that data, HopAY does not seem to be the determinant factor for pathogenicity but it could still play a role interfering with plant immune response. In order to confirm this hypothesis, additional experiments are needed to show that HopAY is a functional protease able to cleave PBS1 from Prunus spp. but the comparative genomic analysis conducted here already provided evidence for sequence correlation between HopAY and its putative cognate target in Prunus spp.
This study identifies traits supporting the adaptation between members of the P. syringae species complex with species belonging to the Prunus genus. It also revealed that most of the mutations affecting hopAY were short insertions or deletions that would not be detected by regular PCR and gel electrophoresis, a method that was often used to determine T3E profiles of P. syringae and other plant pathogens before the advent of next-generation sequencing technologies (Escalon et al., 2013; Ferrante and Scortichini, 2015). Besides highlighting the biases linked to DNA-based T3E profiling, this study also underlines the importance of integrating host genomic data to correctly interpret the relevance of genomic traits found in the pathogen.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TS and JP conceptualized the study with the assistance of MR. MR designed the methodology and carried out the experiments. MR and JP analyzed the data with the assistance from JB and TS, contributed to the data visualization, prepared the original draft with assistance from JB and TS for review and editing, and curated the data. JB and JP helped with software. All the authors revised the final version of the manuscript, while JP acted as the corresponding author. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Swiss Secretariat for Education, Research and Innovation, grant number SBFI C12.0099, within the European research network COST Action FA1104 “Sustainable production of high-quality cherries for the European market” and in part by the European Union Seventh Framework (FP7/2007–2013) under the grant agreement no. 613678 (DROPSA). TS and JP were supported by the Department of Life Sciences and Facility Management of ZHAW. The EDGAR platform is financially supported by the BMF grant FKZ 031A533 within the de.NBI network. Open access funding provided by University Library, Zurich University of Applied Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.804681/full#supplementary-material
References
Ade, J., DeYoung, B. J., Golstein, C., and Innes, R. W. (2007). Indirect activation of a plant nucleotide binding site–leucine-rich repeat protein by a bacterial protease. Proc. Natl. Acad. Sci. U. S. A. 104, 2531–2536. doi: 10.1073/pnas.0608779104
Almeida, N. F., Yan, S., Lindeberg, M., Studholme, D. J., Schneider, D. J., Condon, B., et al. (2009). A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of Pseudomonas syringae pv. tomato DC3000. Mol. Plant-Microbe Interact. 22, 52–62. doi: 10.1094/mpmi-22-1-0052
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukhtar, M. S., Cherkis, K., et al. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7:e1002132. doi: 10.1371/journal.ppat.1002132
Baltrus, D. A., Yourstone, S., Lind, A., Guilbaud, C., Sands, D. C., Jones, C. D., et al. (2014). Draft genome sequences of a phylogenetically diverse suite of Pseudomonas syringae strains from multiple source populations. Genome Announc. 2, e01195–e01213. doi: 10.1128/genomeA.01195-13
Barker, D., and Pagel, M. (2005). Predicting functional gene links from phylogenetic-statistical analyses of whole genomes. PLoS Comput. Biol. 1:e3. doi: 10.1371/journal.pcbi.0010003
Bartoli, C., Lamichhane, J. R., Berge, O., Guilbaud, C., Varvaro, L., Balestra, G. M., et al. (2015). A framework to gauge the epidemic potential of plant pathogens in environmental reservoirs: the example of kiwifruit canker. Mol. Plant Pathol. 16, 137–149. doi: 10.1111/mpp.12167
Beer, S.V., Bauer, D.W., Jiang, X.H., Laby, R.J., Sneath, B.J., Wei, Z.-M., et al. (1991). “The hrp gene cluster of Erwinia amylovora,” in Advances in Molecular Genetics of Plant-Microbe Interactions, Vol. 1. Proceedings of the 5th International Symposium on the Molecular Genetics of Plant-Microbe Interactions, Interlaken, Switzerland. (eds.) H. Hennecke and D.P.S. Verma. September 9–14, 1990. Dordrecht: Springer Netherlands, 53–60.
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C., et al. (2014). A user's guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PLoS One 9:e105547. doi: 10.1371/journal.pone.0105547
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bogdanove, A. J., Beer, S. V., Bonas, U., Boucher, C. A., Collmer, A., Coplin, D. L., et al. (1996). Unified nomenclature for broadly conserved hrp genes of phytopathogenic bacteria. Mol. Microbiol. 20, 681–683. doi: 10.1046/j.1365-2958.1996.5731077.x
Bonas, U., Schulte, R., Fenselau, F., Minsavage, G. V., Staskawicz, B. J., and Stall, R. E. (1991). Isolation of a gene cluster from Xanthomonas campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol. Plant-Microbe Interact. 4, 81–88. doi: 10.1094/MPMI-4-081
Buell, C. R., Joardar, V., Lindeberg, M., Selengut, J., Paulsen, I. T., Gwinn, M. L., et al. (2003). The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 100, 10181–10186. doi: 10.1073/pnas.1731982100
Caballo-Ponce, E., van Dillewijn, P., Wittich, R. M., and Ramos, C. (2016). WHOP, a genomic region associated with woody hosts in the Pseudomonas syringae complex contributes to the virulence and fitness of Pseudomonas savastanoi pv. savastanoi in olive plants. Mol. Plant-Microbe Interact. 30, 113–126. doi: 10.1094/MPMI-11-16-0233-R
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cazorla, F. M., Torés, J. A., Olalla, L., Pérez-García, A., Farré, J. M., and de Vicente, A. (1998). Bacterial apical necrosis of mango in southern Spain: a disease caused by Pseudomonas syringae pv. syringae. Phytopathology 88, 614–620. doi: 10.1094/PHYTO.1998.88.7.614
Choi, O., Kang, B., Lee, Y., Kim, S., Oh, J., Kim, H., et al. (2020). Bacterial shoot blight caused by Pseudomonas cerasi, a new pathogen of pear tree. Aust. Plant Dis. Notes 15:24. doi: 10.1007/s13314-020-00393-w
Clarke, C. R., Cai, R., Studholme, D. J., Guttman, D. S., and Vinatzer, B. A. (2010). Pseudomonas syringae strains naturally lacking the classical P. syringae hrp/hrc locus are common leaf colonizers equipped with an atypical type III secretion system. Mol. Plant-Microbe Interact. 23, 198–210. doi: 10.1094/mpmi-23-2-0198
Crosse, J. E. (1959). Bacterial canker of stone-fruits: IV. Investigation of a method for measuring the inoculum potential of cherry trees. Ann. Appl. Biol. 47, 306–317. doi: 10.1111/j.1744-7348.1959.tb02546.x
Crosse, J. (1966). Epidemiological relations of the pseudomonad pathogens of deciduous fruit trees. Annu. Rev. Phytopathol. 4, 291–310. doi: 10.1146/annurev.py.04.090166.001451
Crosse, J. E., and Garrett, C. M. E. (1963). Studies on the bacteriophagy of Pseudomonas morsprunorum, Ps. syringae and related organisms. J. Appl. Microbiol. 26, 159–177. doi: 10.1111/j.1365-2672.1963.tb04764.x
Crosse, J. E., and Garrett, C. M. E. (1966). Bacterial canker of stone-fruits: infection experiments with Pseudomonas morsprunorum and P. syringae. Ann. Appl. Biol. 58, 31–41. doi: 10.1111/j.1744-7348.1966.tb05068.x
Dudnik, A., and Dudler, R. (2013a). High-quality draft genome sequence of Pseudomonas syringae pv. syringae strain SM, isolated from wheat. Genome Announc. 1, e00610–e00613. doi: 10.1128/genomeA.00610-13
Dudnik, A., and Dudler, R. (2013b). Non contiguous-finished genome sequence of Pseudomonas syringae pathovar syringae strain B64 isolated from wheat. Stand. Genomic Sci. 8, 420–429. doi: 10.4056/sigs.3997732
Dudnik, A., and Dudler, R. (2014). Genome and transcriptome sequences of Pseudomonas syringae pv. syringae B301D-R. Genome Announc. 2, e00306–e003314. doi: 10.1128/genomeA.00306-14
Dye, D., Bradbury, J., Goto, M., Hayward, A., Lelliott, R., and Schroth, M. N. (1980). International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Rev. Plant Pathol. 59, 153–168. doi: 10.1079/cabireviews/19801362818
EPPO (2005). PM 7/43 (1) Pseudomonas syringae pv. persicae. EPPO Bull. 35, 285–287. doi: 10.1111/j.1365-2338.2005.00823.x
Escalon, A., Javegny, S., Vernière, C., Noël, L. D., Vital, K., Poussier, S., et al. (2013). Variations in type III effector repertoires, pathological phenotypes and host range of Xanthomonas citri pv. citri pathotypes. Mol. Plant Pathol. 14, 483–496. doi: 10.1111/mpp.12019
Eurostat (2018). Database—Eurostat [Online]. Available at: https://ec.europa.eu/eurostat/databrowser/view/APRO_CPSH1 (Accessed January 13, 2022).
Feil, H., Feil, W. S., Chain, P., Larimer, F., DiBartolo, G., Copeland, A., et al. (2005). Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 102, 11064–11069. doi: 10.1073/pnas.0504930102
Ferrante, P., and Scortichini, M. (2015). Redefining the global populations of Pseudomonas syringae pv. actinidiae based on pathogenic, molecular and phenotypic characteristics. Plant Pathol. 64, 51–62. doi: 10.1111/ppa.12236
Freigoun, S. O., and Crosse, J. E. (1975). Host relations and distribution of a physiological and pathological variant of Pseudomonas morsprunorum. Ann. Appl. Biol. 81, 317–330. doi: 10.1111/j.1744-7348.1975.tb01647.x
Gardan, L., Shafik, H., Belouin, S., Broch, R., Grimont, F., and Grimont, P. A. D. (1999). DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int. J. Syst. Evol. Microbiol. 49, 469–478. doi: 10.1099/00207713-49-2-469
Gardiner, D. M., Stiller, J., Covarelli, L., Lindeberg, M., Shivas, R. G., and Manners, J. M. (2013). Genome sequences of Pseudomonas spp. isolated from cereal crops. Genome Announc. 1, e00209–e00213. doi: 10.1128/genomeA.00209-13
Garibaldi, A., Minuto, A., Scortichini, M., and Gullino, M. L. (2007). First report of syringae leaf spot caused by Pseudomonas syringae pv. syringae on tomato in Italy. Plant Dis. 91:1518. doi: 10.1094/PDIS-91-11-1518B
Gomila, M., Busquets, A., Mulet, M., García-Valdés, E., and Lalucat, J. (2017). Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front. Microbiol. 8:2422. doi: 10.3389/fmicb.2017.02422
Gough, C. L., Genin, S., Zischek, C., and Boucher, C. A. (1992). Hrp genes of Pseudomonas solanacearum are homologous to pathogenicity determinants of animal pathogenic bacteria and are conserved among plant pathogenic bacteria. Mol. Plant-Microbe Interact. 5, 384–389. doi: 10.1094/MPMI-5-384
Green, S., Studholme, D. J., Laue, B. E., Dorati, F., Lovell, H., Arnold, D., et al. (2010). Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum. PLoS One 5:e10224. doi: 10.1371/journal.pone.0010224
Hulin, M. T., Armitage, A. D., Vicente, J. G., Holub, E. B., Baxter, L., Bates, H. J., et al. (2018). Comparative genomics of Pseudomonas syringae reveals convergent gene gain and loss associated with specialization onto cherry (Prunus avium). New Phytol. 219, 672–696. doi: 10.1111/nph.15182
Hulin, M. T., Jackson, R. W., Harrison, R. J., and Mansfield, J. W. (2020). Cherry picking by pseudomonads: after a century of research on canker, genomics provides insights into the evolution of pathogenicity towards stone fruits. Plant Pathol. 69, 962–978. doi: 10.1111/ppa.13189
Hwang, M. S. H., Morgan, R. L., Sarkar, S. F., Wang, P. W., and Guttman, D. S. (2005). Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl. Environ. Microbiol. 71, 5182–5191. doi: 10.1128/aem.71.9.5182-5191.2005
Ivanović, Ž., Perović, T., Popović, T., Blagojević, J., Trkulja, N., and Hrnčić, S. (2017). Characterization of Pseudomonas syringae pv. syringae, causal agent of citrus blast of mandarin in Montenegro. Plant Pathol. J. 33, 21–33. doi: 10.5423/PPJ.OA.08.2016.0161
Jayaraman, J., Yoon, M., Applegate, E. R., Stroud, E. A., and Templeton, M. D. (2020). AvrE1 and HopR1 from Pseudomonas syringae pv. actinidiae are additively required for full virulence on kiwifruit. Mol. Plant Pathol. 21, 1467–1480. doi: 10.1111/mpp.12989
Jenner, C., Hitchin, E., Mansfield, J., Walters, K., Betteridge, P., Teverson, D., et al. (1991). Gene-for-gene interactions between Pseudomonas syringae pv. phaseolicola and Phaseolus. Mol. Plant-Microbe Interact. 4, 553–562. doi: 10.1094/MPMI-4-553
Joardar, V., Lindeberg, M., Jackson, R. W., Selengut, J., Dodson, R., Brinkac, L. M., et al. (2005). Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J. Bacteriol. 187, 6488–6498. doi: 10.1128/JB.187.18.6488-6498.2005
Jones, L. A., Saha, S., Collmer, A., Smart, C. D., and Lindeberg, M. (2015). Genome-assisted development of a diagnostic protocol for distinguishing high virulence Pseudomonas syringae pv. tomato strains. Plant Dis. 99, 527–534. doi: 10.1094/pdis-08-14-0833-re
Kałużna, M., Willems, A., Pothier, J. F., Ruinelli, M., Sobiczewski, P., and Puławska, J. (2016). Pseudomonas cerasi sp. nov. (non griffin, 1911) isolated from diseased tissue of cherry. Syst. Appl. Microbiol. 39, 370–377. doi: 10.1016/j.syapm.2016.05.005
Kamiunten, H., Nakao, T., and Oshida, S. (2000). Pseudomonas syringae pv. cerasicola, pv. nov., the causal agent of bacterial gall of cherry tree. J. Gen. Plant Pathol. 66, 219–224. doi: 10.1007/PL00012949
Kvitko, B. H., Park, D. H., Velásquez, A. C., Wei, C.-F., Russell, A. B., Martin, G. B., et al. (2009). Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5:e1000388. doi: 10.1371/journal.ppat.1000388
Lalucat, J., Mulet, M., Gomila, M., and García-Valdés, E. (2020). Genomics in bacterial taxonomy: impact on the genus Pseudomonas. Gene 11:139. doi: 10.3390/genes11020139
Lefort, F., Calmin, G., Crovadore, J., Osteras, M., and Farinelli, L. (2013). Whole-genome shotgun sequence of Pseudomonas viridiflava, a bacterium species pathogenic to Ararabidopsis thaliana. Genome Announc. 1, e00116–e00212. doi: 10.1128/genomeA.00116-12
Lindeberg, M., Cunnac, S., and Collmer, A. (2012). Pseudomonas syringae type III effector repertoires: last words in endless arguments. Trends Microbiol. 20, 199–208. doi: 10.1016/j.tim.2012.01.003
Lindeberg, M., Stavrinides, J., Chang, J. H., Alfano, J. R., Collmer, A., Dangl, J. L., et al. (2005). Proposed guidelines for a unified nomenclature and phylogenetic analysis of type III Hop effector proteins in the plant pathogen Pseudomonas syringae. Mol. Plant-Microbe Interact. 18, 275–282. doi: 10.1094/MPMI-18-0275
Lindgren, P. B., Panopoulos, N. J., Staskawicz, B. J., and Dahlbeck, D. (1988). Genes required for pathogenicity and hypersensitivity are conserved and interchangeable among pathovars of Pseudomonas syringae. Mol. Gen. Genet. 211, 499–506. doi: 10.1007/bf00425707
Lindgren, P. B., Peet, R. C., and Panopoulos, N. J. (1986). Gene cluster of Pseudomonas syringae pv. "phaseolicola" controls pathogenicity of bean plants and hypersensitivity of nonhost plants. J. Bacteriol. 168, 512–522. doi: 10.1128/jb.168.2.512-522.1986
Linke, B., Giegerich, R., and Goesmann, A. (2011). Conveyor: a workflow engine for bioinformatic analyses. Bioinformatics 27, 903–911. doi: 10.1093/bioinformatics/btr040
Liu, H., Qiu, H., Zhao, W., Cui, Z., Ibrahim, M., Jin, G., et al. (2012). Genome sequence of the plant pathogen Pseudomonas syringae pv. panici LMG 2367. J. Bacteriol. 194, 5693–5694. doi: 10.1128/jb.01267-12
Mackey, D., Holt Iii, B. F., Wiig, A., and Dangl, J. L. (2002). RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108, 743–754. doi: 10.1016/S0092-8674(02)00661-X
Martínez-García, P. M., Rodríguez-Palenzuela, P., Arrebola, E., Carrión, V. J., Gutiérrez-Barranquero, J. A., Pérez-García, A., et al. (2015). Bioinformatics analysis of the complete genome sequence of the mango tree pathogen Pseudomonas syringae pv. syringae UMAF0158 reveals traits relevant to virulence and epiphytic lifestyle. PLoS One 10:e0136101. doi: 10.1371/journal.pone.0136101
McCann, H. C., Rikkerink, E. H. A., Bertels, F., Fiers, M., Lu, A., Rees-George, J., et al. (2013). Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 9:e1003503. doi: 10.1371/journal.ppat.1003503
Ménard, M., Sutra, L., Luisetti, J., Prunier, J. P., and Gardan, L. (2003). Pseudomonas syringae pv. avii (pv. nov.), the causal agent of bacterial canker of wild cherries (Prunus avium) in France. Eur. J. Plant Pathol. 109, 565–576. doi: 10.1023/A:1024786201793
Moreno-Pérez, A., Pintado, A., Murillo, J., Caballo-Ponce, E., Tegli, S., Moretti, C., et al. (2020). Host range determinants of Pseudomonas savastanoi pathovars of woody hosts revealed by comparative genomics and cross-pathogenicity tests. Front. Plant Sci. 11:973. doi: 10.3389/fpls.2020.00973
Moretti, C., Cortese, C., Passos da Silva, D., Venturi, V., Ramos, C., Firrao, G., et al. (2014). Draft genome sequence of Pseudomonas savastanoi pv. savastanoi strain DAPP-PG 722, isolated in Italy from an olive plant affected by knot disease. Genome Announc. 2, e00864–e00914. doi: 10.1128/genomeA.00864-14
Morris, C. E., Lamichhane, J. R., Nikolić, I., Stanković, S., and Moury, B. (2019). The overlapping continuum of host range among strains in the Pseudomonas syringae complex. Phytopathol. Res. 1:4. doi: 10.1186/s42483-018-0010-6
Morris, C. E., Sands, D. C., Vinatzer, B. A., Glaux, C., Guilbaud, C., Buffière, A., et al. (2008). The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2, 321–334. doi: 10.1038/ismej.2007.113
Mulet, M., Lalucat, J., and García-Valdés, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x
Nelson, K. E., Weinel, C., Paulsen, I. T., Dodson, R. J., Hilbert, H., Martins dos Santos, V. A., et al. (2002). Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4, 799–808. doi: 10.1046/j.1462-2920.2002.00366.x
Newberry, E. A., Ebrahim, M., Timilsina, S., Zlatković, N., Obradović, A., Bull, C. T., et al. (2019). Inference of convergent gene acquisition among Pseudomonas syringae strains isolated from watermelon, cantaloupe, and squash. Front. Microbiol. 10:270. doi: 10.3389/fmicb.2019.00270
Nowell, R. W., Green, S., Laue, B. E., and Sharp, P. M. (2014). The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol. Evol. 6, 1514–1529. doi: 10.1093/gbe/evu123
Nowell, R. W., Laue, B. E., Sharp, P. M., and Green, S. (2016). Comparative genomics reveals genes significantly associated with woody hosts in the plant pathogen Pseudomonas syringae. Mol. Plant Pathol. 17, 1409–1424. doi: 10.1111/mpp.12423
O’Brien, H. E., Thakur, S., Gong, Y., Fung, P., Zhang, J., Yuan, L., et al. (2012). Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol. 12:141. doi: 10.1186/1471-2180-12-141
Pagel, M. (1994). Detecting correlated evolution on phylogenies: a general method for the comparative analysis of discrete characters. Proc. R. Soc. Lond. Ser. B Biol. Sci. 255, 37–45. doi: 10.1098/rspb.1994.0006
Pagel, M., and Meade, A. (2006). Bayesian analysis of correlated evolution of discrete characters by reversible-jump Markov chain Monte Carlo. Am. Nat. 167, 808–825. doi: 10.1086/503444
Palleroni, N. J. (2005). “Genus I. Pseudomonas” in Bergey's Manual of Systematic Bacteriology. The Proteobacteria, Part C: The Gammaproteobacteria. eds. D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (Dordrecht: Springer, Netherlands), 323–379.
Parkinson, N., Bryant, R., Bew, J., and Elphinstone, J. (2011). Rapid phylogenetic identification of members of the Pseudomonas syringae species complex using the rpoD locus. Plant Pathol. 60, 338–344. doi: 10.1111/j.1365-3059.2010.02366.x
Picard, C., Afonso, T., Benko-Beloglavec, A., Karadjova, O., Matthews-Berry, S., Paunovic, S. A., et al. (2018). Recommended regulated non-quarantine pests (RNQPs), associated thresholds and risk management measures in the European and Mediterranean region. EPPO Bull. 48, 552–568. doi: 10.1111/epp.12500
Popović, T., Ivanović, Ž., Trkulja, N., Milosavljević, A., and Ignjatov, M. (2015). First report of Pseudomonas syringae pv. syringae on pea (Pisum sativum) in Serbia. Plant Dis. 99:724. doi: 10.1094/PDIS-11-14-1212-PDN
PPI (2010). Pseudomonas syringae genome resources home page [Online]. Available at: www.pseudomonas-syringae.org (Accessed January 23, 2017).
Psallidas, P. G. (1997). Hyperplastic canker—a perennial disease of almond caused by Pseudomonas amygdali. EPPO Bull. 27, 511–517. doi: 10.1111/j.1365-2338.1997.tb00675.x
Puławska, J., Gétaz, M., Kałużna, M., Kuzmanović, N., Obradović, A., Pothier, J. F., et al. (2017). “Bacterial diseases” in Cherries: Botany, Production and Uses. eds. J. Quero-García, A. Iezzoni, J. Puławska, and G. Lang (Wallingford, United Kingdom: CAB International), 365–385.
Puri, N., Jenner, C., Bennett, M., Stewart, R., Mansfield, J., Lyons, N., et al. (1997). Expression of avrPphB, an avirulence gene from Pseudomonas syringae pv. phaseolicola, and the delivery of signals causing the hypersensitive reaction in bean. Mol. Plant-Microbe Interact. 10, 247–256. doi: 10.1094/MPMI.1997.10.2.247
Qi, D., DeYoung, B. J., and Innes, R. W. (2012). Structure-function analysis of the coiled-coil and leucine-rich repeat domains of the RPS5 disease resistance protein. Plant Physiol. 158, 1819–1832. doi: 10.1104/pp.112.194035
Qi, D., Dubiella, U., Kim, S. H., Sloss, D. I., Dowen, R. H., Dixon, J. E., et al. (2014). Recognition of the protein kinase AVRPPHB SUSCEPTIBLE1 by the disease resistance protein RESISTANCE TO PSEUDOMONAS SYRINGAE5 is dependent on S-acylation and an exposed loop in AVRPPHB SUSCEPTIBLE1. Plant Physiol. 164, 340–351. doi: 10.1104/pp.113.227686
Qi, M., Wang, D., Bradley, C. A., and Zhao, Y. (2011). Genome sequence analyses of Pseudomonas savastanoi pv. glycinea and subtractive hybridization-based comparative genomics with nine pseudomonads. PLoS One 6:e16451. doi: 10.1371/journal.pone.0016451
Ravindran, A., Jalan, N., Yuan, J. S., Wang, N., and Gross, D. C. (2015). Comparative genomics of Pseudomonas syringae pv. syringae strains B301D and HS191 and insights into intrapathovar traits associated with plant pathogenesis. Microbiology 4, 553–573. doi: 10.1002/mbo3.261
Ruinelli, M., Blom, J., Smits, T. H. M., and Pothier, J. F. (2019). Comparative genomics and pathogenicity potential of members of the Pseudomonas syringae species complex on Prunus spp. BMC Genomics 20:172. doi: 10.1186/s12864-019-5555-y
Sarkar, S. F., Gordon, J. S., Martin, G. B., and Guttman, D. S. (2006). Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics 174, 1041–1056. doi: 10.1534/genetics.106.060996
Sarkar, S. F., and Guttman, D. S. (2004). Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol. 70, 1999–2012. doi: 10.1128/aem.70.4.1999-2012.2004
Scortichini, M., Marcelletti, S., Ferrante, P., and Firrao, G. (2013). A Genomic redefinition of Pseudomonas avellanae species. PLoS One 8:e75794. doi: 10.1371/journal.pone.0075794
Shao, F., Golstein, C., Ade, J., Stoutemyer, M., Dixon, J. E., and Innes, R. W. (2003). Cleavage of Arabidopsis PBS1 by a bacterial type III effector. Science 301, 1230–1233. doi: 10.1126/science.1085671
Shao, F., Merritt, P. M., Bao, Z., Innes, R. W., and Dixon, J. E. (2002). A Yersinia effector and a Pseudomonas avirulence protein define a family of cysteine proteases functioning in bacterial pathogenesis. Cell 109, 575–588. doi: 10.1016/S0092-8674(02)00766-3
Silby, M. W., Cerdeño-Tárraga, A. M., Vernikos, G. S., Giddens, S. R., Jackson, R. W., Preston, G. M., et al. (2009). Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 10:R51. doi: 10.1186/gb-2009-10-5-r51
Spotts, R. A., Wallis, K. M., Serdani, M., and Azarenko, A. N. (2010). Bacterial canker of sweet cherry in Oregon—Infection of horticultural and natural wounds, and resistance of cultivar and rootstock combinations. Plant Dis. 94, 345–350. doi: 10.1094/pdis-94-3-0345
Templeton, M. D., Warren, B. A., Andersen, M. T., Rikkerink, E. H., and Fineran, P. C. (2015). Complete DNA sequence of Pseudomonas syringae pv. actinidiae, the causal agent of kiwifruit canker disease. Genome Announc. 3, e01054–e01115. doi: 10.1128/genomeA.01054-15
Wang, P. W., Morgan, R. L., Scortichini, M., and Guttman, D. S. (2007). Convergent evolution of phytopathogenic pseudomonads onto hazelnut. Microbiology 153, 2067–2073. doi: 10.1099/mic.0.2006/001545-0
Wei, W., Plovanich-Jones, A., Deng, W., Jin, Q., Collmer, A., Huang, H., et al. (2000). The gene coding for the Hrp pilus structural protein is required for type III secretion of Hrp and Avr proteins in Pseudomonas syringae pv. tomato. Proc. Natl. Acad. Sci. U. S. A. 97, 2247–2252. doi: 10.1073/pnas.040570097
Xiang, T., Zong, N., Zou, Y., Wu, Y., Zhang, J., Xing, W., et al. (2008). Pseudomonas syringae effector AvrPto blocks innate immunity by targeting receptor kinases. Curr. Biol. 18, 74–80. doi: 10.1016/j.cub.2007.12.020
Yang, L., Teixeira, P. J., Biswas, S., Finkel, O. M., He, Y., Salas-Gonzalez, I., et al. (2017). Pseudomonas syringae type III effector HopBB1 promotes host transcriptional repressor degradation to regulate phytohormone responses and virulence. Cell Host Microbe 21, 156–168. doi: 10.1016/j.chom.2017.01.003
Young, J. M. (1987). New plant disease record in New Zealand: Pseudomonas syringae pv. persicae from nectarine, peach, and Japanese plum. N. Z. J. Agric. Res. 30, 235–247. doi: 10.1080/00288233.1987.10430502
Young, J. M. (2010). Taxonomy of Pseudomonas syringae. J. Plant Pathol. 92, S1.5–S1.14. doi: 10.4454/jpp.v92i1sup.2501
Zembek, P., Danilecka, A., Hoser, R., Eschen-Lippold, L., Benicka, M., Grech-Baran, M., et al. (2018). Two strategies of Pseudomonas syringae to avoid recognition of the HopQ1 effector in Nicotiana species. Front. Plant Sci. 9:978. doi: 10.3389/fpls.2018.00978
Zhang, J., Zhou, M., Liu, W., Nie, J., and Huang, L. (2022). Pseudomonas syringae pv. actinidiae effector HopAU1 interacts with calcium-sensing receptor to activate plant immunity. Int. J. Mol. Sci. 23:508. doi: 10.3390/ijms23010508
Zhao, W., Jiang, H., Tian, Q., and Hu, J. (2015). Draft genome sequence of Pseudomonas syringae pv. persicae NCPPB 2254. Genome Announc. 3, e00555–e00565. doi: 10.1128/genomeA.00555-15
Zhou, L. H., Han, Y., Ji, G. H., Wang, Z. S., and Liu, F. (2012). First report of bacterial leaf spot disease caused by Pseudomonas syringae pv. syringae on Panax notoginseng. Plant Dis. 97:685. doi: 10.1094/PDIS-11-12-1047-PDN
Zhu, M., Shao, F., Innes, R. W., Dixon, J. E., and Xu, Z. (2004). The crystal structure of Pseudomonas avirulence protein AvrPphB: A papain-like fold with a distinct substrate-binding site. Proc. Natl. Acad. Sci. U. S. A. 101, 302–307. doi: 10.1073/pnas.2036536100
Keywords: Pseudomonas syringae species complex, comparative genomics, pathogenicity, co-evolution, host adaptation
Citation: Ruinelli M, Blom J, Smits THM and Pothier JF (2022) Comparative Genomics of Prunus-Associated Members of the Pseudomonas syringae Species Complex Reveals Traits Supporting Co-evolution and Host Adaptation. Front. Microbiol. 13:804681. doi: 10.3389/fmicb.2022.804681
Edited by:
Neha Potnis, Auburn University, United StatesReviewed by:
Mustafa Ojonuba Jibrin, Wenatchee Tree Fruit Research and Extension Center, United StatesVittoria Catara, University of Catania, Italy
Copyright © 2022 Ruinelli, Blom, Smits and Pothier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joël F. Pothier, am9lbC5wb3RoaWVyQHpoYXcuY2g=