 Xiaoyan Wu
Xiaoyan Wu Ziyi Han1†
Ziyi Han1† Dongming Yu
Dongming Yu Jing Sun
Jing Sun Wenjie Tang
Wenjie Tang Shaojuan Liu
Shaojuan Liu- 1State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangdong Laboratory of Lingnan Modern Agriculture, National Engineering Research Center for Breeding Swine Industry, Guangdong Provincial Key Laboratory of Animal Nutrition Control, College of Animal Science, South China Agricultural University, Guangzhou, China
- 2Chongqing Academy of Animal Sciences, Chongqing, China
- 3Animal Breeding and Genetics Key Laboratory of Sichuan Province, Sichuan Animal Science Academy, Chengdu, China
- 4Livestock and Poultry Biological Products Key Laboratory of Sichuan Province, Sichuan Animtech Feed Co., Ltd., Chengdu, China
Methionine (Met) metabolism provides methyl groups for many important physiological processes and is implicated in multiple inflammatory diseases associated with the disrupted intestinal microbiota; nevertheless, whether intestinal microbiota determines Met metabolism in the host remains largely unknown. Here, we found that gut microbiota is responsible for host Met metabolism by using various animal models, including germ-free (GF) pigs and mice. Specifically, the Met levels are elevated in both GF pigs and GF mice that mainly metabolized to S-adenosine methionine (SAM) in the liver. Furthermore, antibiotic clearance experiments demonstrate that the loss of certain ampicillin- or neomycin-sensitive gut microbiota causes decreased Met in murine colon. Overall, our study suggests that gut microbiota mediates Met metabolism in the host and is a prospective target for the treatment of Met metabolism-related diseases.
Introduction
Methionine (Met) is one of the essential amino acids that could participate in the biosynthesis of various nitrogen-containing substances (Liu et al., 2021). Indeed, Met metabolism in the host mainly consists of four metabolic pathways surrounding the Met-homocysteine (hCy) cycle (Yin et al., 2018; Xu et al., 2020). The host Met-hCy cycle (hereinafter referred to as the Met cycle) is associated with transmethylation and the metabolism of amino acids such as serine (Ser), glycine (Gly), glutathione (GSH), and taurine (Tau; Xu et al., 2020). Specifically, Met reacts with ATP under the catalysis of methionine adenosyltransferase (MAT) to generate S-adenosine methionine (SAM), which then provides a methyl group to generate S-adenosine homocysteine (SAH; Clare et al., 2019). Homocysteine (hCy) generated by S-adenosylhomocysteine hydrolase (SAHH) is then remethylated by N5-methyltetrahydrofolate (CH3-THF) to generate Met, and vitamin B12 serves as a key coenzyme in this process (Lam et al., 2021). Of note, the Met cycle ensures Met level and the supply of methyl-donor SAM in liver; therefore, modulating Met cycle has therapeutic implications for many diseases, for example, alcoholic liver disease (ALD; Kharbanda, 2013). Moreover, the Met cycle in tumors has also received attention. For example, tumor initiation of lung cancer stem cells consumes large amounts of Met for histone methylation, resulting in Met depletion and exogenous Met dependence (Wang et al., 2019). Met cycle is also disrupted in liver tumors and is often associated with poor prognosis (Xu et al., 2020). Therefore, it is meaningful to fine-tune the Met metabolism in host. However, the factors affecting the Met metabolism are still unclear.
The gut has a complex microorganism community including bacteria, fungi, and viruses, which the composition is determined by natural colonization and subsequent environmental factors (Cani, 2018). Gut microbes play particularly important roles in regulating homeostasis by absorbing nutrients, producing metabolites, and cooperating or competing with other microorganisms in the intestinal tract (Osbelt et al., 2021). It is worth noting that the impact of the gut microbiota on host is not limited in gut, as supported by the findings of the gut-brain axis (Cryan et al., 2019), gut-lung axis (Dang and Marsland, 2019), and gut-liver axis (Tripathi et al., 2018). A study revealed that patients with quiescent inflammatory bowel disease (IBD) fatigue had reduced serum metabolites like Met, Ser and sarcosine (Sar), and decreased short-chain fatty acids (SCFAs)-producing gut microbiota such as Faecalibacterium prausnitzii (Borren et al., 2021). In a controlled study of Parkinson’s disease (PD), significant differences in the trajectories of Met and homoserine were found (Hertel et al., 2019). And the PD-associated gut microbiome analysis revealed increased abundance of Akkermansia muciniphila (A. muciniphila) and Bilophila wadsworthia involved in sulfur metabolism (Hertel et al., 2019). Additionally, A. muciniphila contributed more than 70% of Met secretion potential (Hertel et al., 2019), indicating the interaction between the gut microbiota and host Met cycle. Nevertheless, whether Met metabolism in host is related to the gut microbiota remains enigmatic.
Given the importance of Met metabolism in inflammation, cancer, and neurological diseases and the impact of microbial-host interactions on host physiology, biochemistry, and immune status, an in-depth analysis of gut microbial and host Met metabolism has profound implications. Microbiota-targeted interventions mainly include antibiotic treatment, probiotic addition, and fecal microbiota transplantation (Sun and Shen, 2018). Among them, antibiotic-induced intestinal dysbiosis is a common method that can lead to drastic changes in the intestinal microbiota (Andremont et al., 2021). Notably, germ-free (GF) animal models are currently important models for studying microbiome-host interactions. Unlike pharmacological interventions such as antibiotics, GF animals achieve true “sterility” and have distinct characteristics from specific pathogen-free (SPF) animals in gut development and/or immune system (Uzbay, 2019). Therefore, GF animals are valuable experimental models in cancer and metabolism-related diseases. Moreover, studies of Met metabolism have typically used in vitro cell or tissue cultures and validated in rodents (Wang et al., 2019; Xu et al., 2020). Genetically inbred mice in rodents are common experimental models that help scientists accomplish many studies, but they are not ideal models of the human immune system because of the differences in immune systems of humans and mice caused by evolutionary branches (Pulendran and Davis, 2020). Therefore, the GF pig model adopted in this paper may help to provide a more valuable reference.
As experimental subjects, pigs are ideal large animal models for studying nutrition or disease in pigs and humans due to their high similarity to humans in terms of anatomy, physiology, and immune system (Miller and Ullrey, 1987; Itoh et al., 2019). The GF pig model is therefore ideal for studying gut microbe-host Met metabolism. Importantly, dietary Met plays an important role in animal nutrition and is the second/third limiting amino acid in pig and piglet diets (Yang et al., 2021). Since Met is beneficial for the growth and development of pigs, most relevant studies have focused on the effects of dietary Met or Met-restricted diets on swine meat quality, growth performance, and intestinal function (Zeitz et al., 2019; Gondret et al., 2021; Yang et al., 2021). However, there are few studies concerning Met metabolism in pigs. Currently, only limited studies on plasma Met metabolism profiles and mammalian lifespan have been retrieved focusing on the relationship between porcine plasma Met metabolism and lifespan (Mota-Martorell et al., 2021).
Therefore, we used GF pig as an experimental model to explore the impact of gut microbiota on host Met metabolism that contributes to map the Met metabolism profile of the pig gut microbiome. We then explored the link between gut microbiota and host Met metabolism in GF mice and mice treated with different antibiotics, further expanding the generalizability of the experimental results and potentially providing new insights into Met metabolism-related diseases.
Materials and methods
Animal acquisition and breeding
GF and SPF pigs
GF pigs and SPF pigs were harvested by uterine dissection and then transferred to sterile feeding isolators (DOSSY Experimental Animals Co., Ltd., Chengdu, China) for decontamination. Next, the piglets were taken to rearing isolators (Class Biologically Clean Ltd., Madison, Wisconsin, United States; Qi et al., 2021; Zhou et al., 2021). The isolator operates in strict accordance with operating procedures within the barrier facilities.
The feeding conditions of the two groups of experimental animals are the same, and the specific steps refer to our previous experiments (Liu et al., 2022). For the first 21 days, they were fed Co60 γ-irradiated 4.8%-fat sterile milk powder (Jiangsu Anyou Group China) diluted with sterile water (Sun et al., 2018; Liu et al., 2022). The basal diet for pigs from day 22 was provided by the Institute of Animal Nutrition, Sichuan Agricultural University (Chengdu, China). In this study, pigs were obtained from the Technical Engineering Center for the Development and Utilization of Medical Animal Resources (Chongqing, China). All experimental protocols were approved and carried out under the guidance of the Laboratory Animal Ethics Committee of South China Agricultural University [Guangzhou, China; permit number SYXK (Guangdong) 2019–0136].
Eleven GF pigs were randomly selected, five of which were kept under GF conditions (GF pigs) and their anal swabs were collected weekly for microbial testing. Another six pigs served as controls (SPF pigs) by transplanting maternal feces. Control pigs were continuously orally administered fecal bacteria suspension (1 ml/d) for 3 days at 7 days of age and kept in sterile isolators (Liu et al., 2022).
For sample collection, on the last day of the experimental period, two groups of pigs were subjected to respiratory anesthesia with isoflurane after urine collection, and other samples were collected on a clean bench after blood collection to ensure that there was no bacterial contamination throughout the entire sampling process. Liver, bile, spleen, kidney, heart, and different intestinal segments and their contents were collected, including jejunum, ileum, cecum, colon, jejunum contents, cecal contents, colonic contents, rectal contents, and feces (Figure 1A). Samples were quickly frozen in liquid nitrogen and stored in an ultra-low temperature freezer at −80°C.
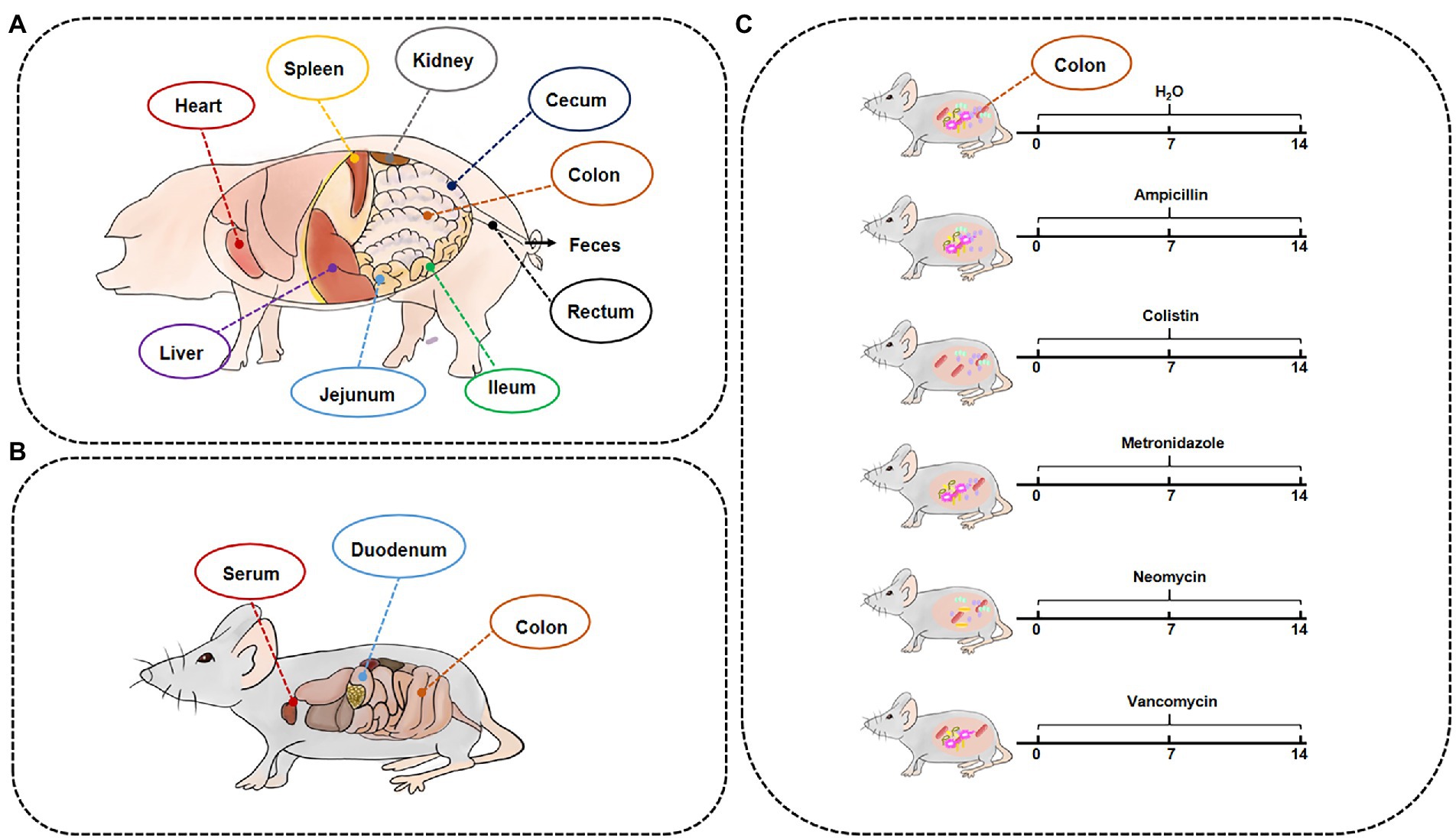
Figure 1. Timeline of experimental processing and sample collection information. The samples of GF/SPF pigs (A) and mice (B). (C) SPF mice were treated by different antibiotics for 2 weeks.
GF/SPF and antibiotic-treated mice
GF and SPF Kunming mice were bred at the Third Military Medical University (Chongqing, China). ICR mice were purchased from Liaoning Changsheng Biotechnology Co., Ltd. (Liaoning, China). GF mice were housed in sterile isolators and SPF Kunming and ICR mice were housed in sterile individual animal colonies (temperature, 25 ± 5°C; relative humidity, 55 ± 5%; dark light alternated every 12 h) with free access to drinking water and standard rodent chow.
For the Met cycle metabolite analysis experiments, GF (GF group, 4 weeks old, n = 10 males and 10 females) and SPF (SPF group, 4 weeks old, n = 10 males and 10 females) Kunming mice were treated with the same diet for 4 weeks, then were euthanized by CO2 asphyxiation, and blood, duodenum, and colon were collected (Figure 1B).
To establish an antibiotic-treated mouse model, 6-week-old female ICR mice (n = 12) were supplemented with ampicillin (MB1407, 1 mg/ml, meilunbio), colistin (MB1064, 1 mg/ml, meilunbio), metronidazole (B300250, 1 mg/ml, Aladdin), neomycin (MB1716, 1 mg/ml, meilunbio), and vancomycin (V301569, 0.5 mg/ml, Aladdin) in drinking water for 2 weeks. Mice were then euthanized by CO2 asphyxiation and colon samples were collected (Figure 1C).
Standard and sample preparation
The key metabolites in the Met cycle involved in this study are Met, SAM, SAH, and hCY. Accurately weighed 1.0 mg of these metabolites and dissolved each standard in 10 ml methanol–water (50:50, V/V) to obtain a 100 μg/ml stock solution. The standard solution was then serially diluted to 1,000, 500, 100, 10, 1, 0.5, 0.1, 0.01, 0.001 ng/ml. All metabolites were quantified by external standard method, and all standard solutions were stored at −20°C. The processing of tissue samples and fluid samples such as serum, urine, and bile was carried out according to our previous experiments (Liu et al., 2022), and stored at −20°C prior to UPLC-Orbitrap-MS/MS analysis.
UPLC-Orbitrap-MS/MS conditions
Met cycle-related metabolites were separated by Thermo Fisher Scientific UPLC system (Dionex UltiMate 3,000) with a C1s Hypersil Gold column (1.9 μm, 100 mm × 2.1 mm; Thermo Scientific). The main parameters and gradient elution procedure were based on our previous experiments (Liu et al., 2022).
Identification of metabolites
Metabolite identification refers to our previous experiments (Liu et al., 2022). Briefly, information such as retention time, mass (m/z), peak and fragment ion MS or MS/MS intensity could be obtained when the RAW file is opened with Xcalibur. The mass list of each MS scan was generated by the peak detection and chromatographic builder in the original data file, and then the chromatogram was built.
Data analysis and statistics
Xcalibur converted data in an instrument-specific format (*.raw) to a common data format (.XLS) that displays information on metabolites, including calculated amounts, retention times, and peak areas. The raw data can be accessed by the Metabolights repository,1 with the identifier MTBLS6154 (Haug et al., 2020).
All data are shown as mean ± SEM. Data between two groups were analyzed by (unpaired t-test, Prism 6.0) if the data exhibited Gaussian distribution and equal variance, and by (Welch-corrected unpaired t-test, Prism 6.0) if the data exhibited (Gaussian distribution but unequal variances). If the data were not normally distributed, a nonparametric test (Mann–Whitney U test, Prism 6.0) was performed. The Gaussian distribution of the data was analyzed by (D’Agostino-Pearson comprehensive normality test, Prism 6.0) and/or (Kolmogorov–Smirnov test, Prism 6.0). Data were analyzed for variance by (Brown-Forsythe test, Prism 6.0). Significant differences among 3 or more groups were passed (Bonferroni’s multiple comparison test, one-way ANOVA assessment, and multiple t-test, Prism 6.0). p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****) were considered statistically.
Results
Gut microbiota alters the met cycle of pig gut
In this study, we collected different intestinal contents of GF pigs and analyzed by UPLC (Figure 1A). Four key metabolites in the Met cycle were detected, including Met, SAM, SAH, and hCY (Figure 2A). Of note, SAM, generated by Met metabolism, is an important methyl donor that cooperates with the methylation in vivo, such as histone methylation (Yu et al., 2019). HCY from SAH deadenosine may regulate the one-carbon metabolism of Ser and Gly, the folate cycle of THF, or the generation of cysteine (Cys) to generate GSH or Tau (Selhub, 1999; Mentch et al., 2015; Jakubowski, 2019). As a result, Met level was significantly increased by almost 10-fold in the gut contents (including cecum, colon, rectal contents, and feces) of GF pigs compared to the control group (SPF), with the exception of the jejunum contents (Figure 2B). SAM, SAH, and hCY were not significantly altered in the jejunum contents of GF pigs (Figure 2C) that may be present at very low levels even below the detection limit in other intestinal contents. The experimental results showed that the absence of the gut microbiota led to a significant increase of Met levels in intestinal contents, suggesting that gut microbiota was involved in the regulation of the Met cycle in pig intestines.
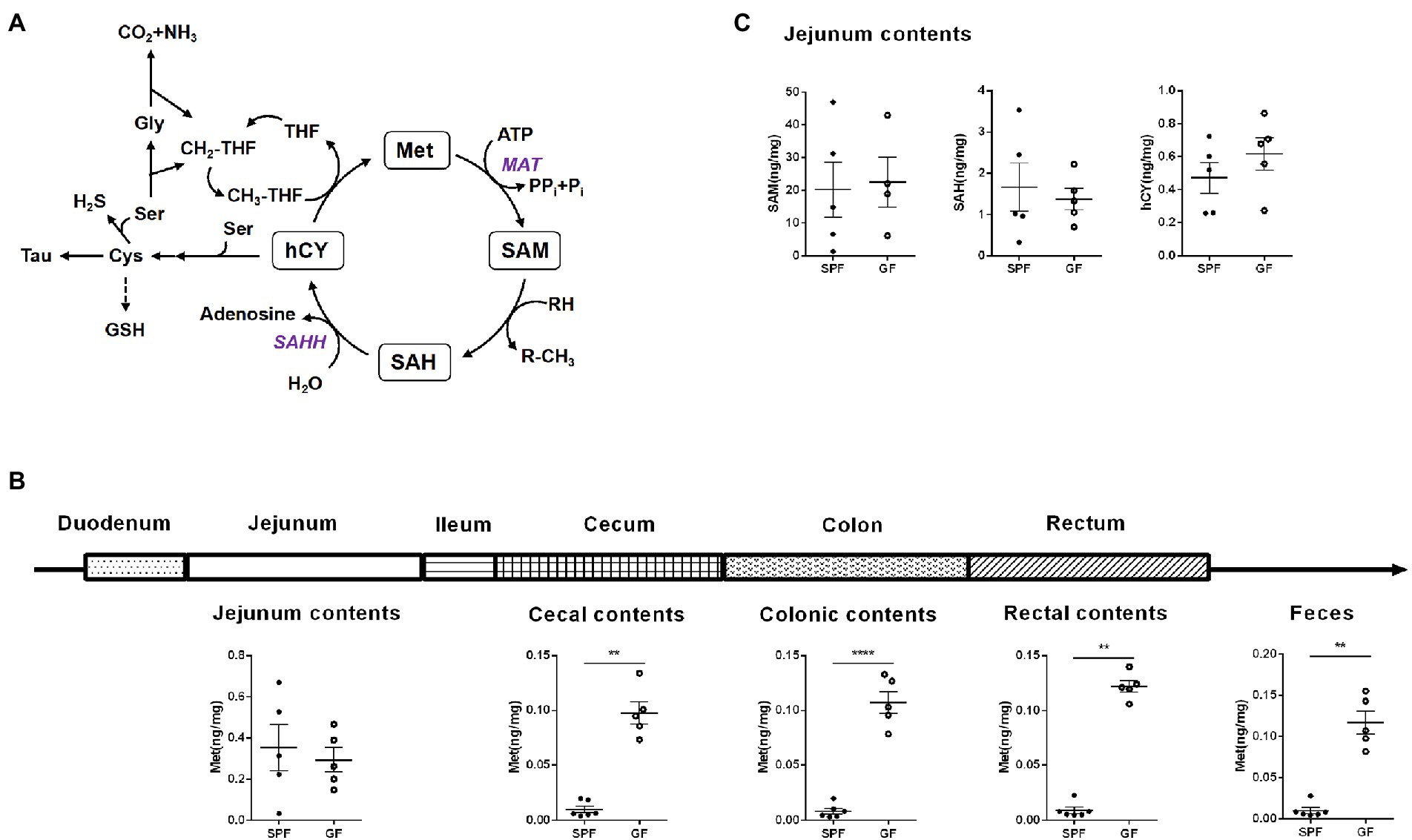
Figure 2. Gut microbiota alters the Met metabolism of pig gut. (A) Met cycle. Met consumes ATP under the catalysis of MAT to generate SAM, SAM provides methyl group to generate SAH, SAH generates hCY under the action of SAHH, and hCY obtains methyl group through CH3-THF in the folic acid cycle to generate Met. hCY participates in the generation of Cys, which, in turn, generates GSH or Tau. (B) Met content in different intestinal contents of GF pigs (n = 5) and SPF pigs (n = 6). (C) Contents of SAM, SAH, and hCY in the jejunum contents of GF pigs (n = 5) and SPF pigs (n = 6). Data were analyzed by unpaired t-test and represented as means ± SEM. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine; hCY: homocysteine; Ser: Serine; Cys: Cysteine; Tau: Taurine; GSH: Glutathione; Tau: Taurine; Gly: Glycine; THF: Tetrahydrofolate; MAT: Methionine adenosyltransferase; SAHH: S-adenosylhomocysteine hydrolase.
Gut microbiota regulates the met cycle in different intestinal segments of pigs
About 25% of the body’s trans-methylation and trans-sulfurization reactions occurred in the gastrointestinal tract, and 65% of the visceral first-pass effect of Cys occurred in the intestine (Bauchart-Thevret et al., 2009), indicating that intestinal absorption was crucial to Met cycle. Therefore, we examined Met cycle-associated metabolites in different intestinal segments. There was no significance in the four key metabolites in the jejunum (Figure 3A). Compared with SPF group, Met in the ileum of GF group was significantly decreased, and SAH showed a downward trend (Figure 3B). In the cecum and colon, Met was increased in GF group compared to SPF group, but SAM, SAH, and hCY were not significantly different (Figures 3C,D). Together, the increase of Met in GF pigs was mainly occurred in the large intestinal segment.
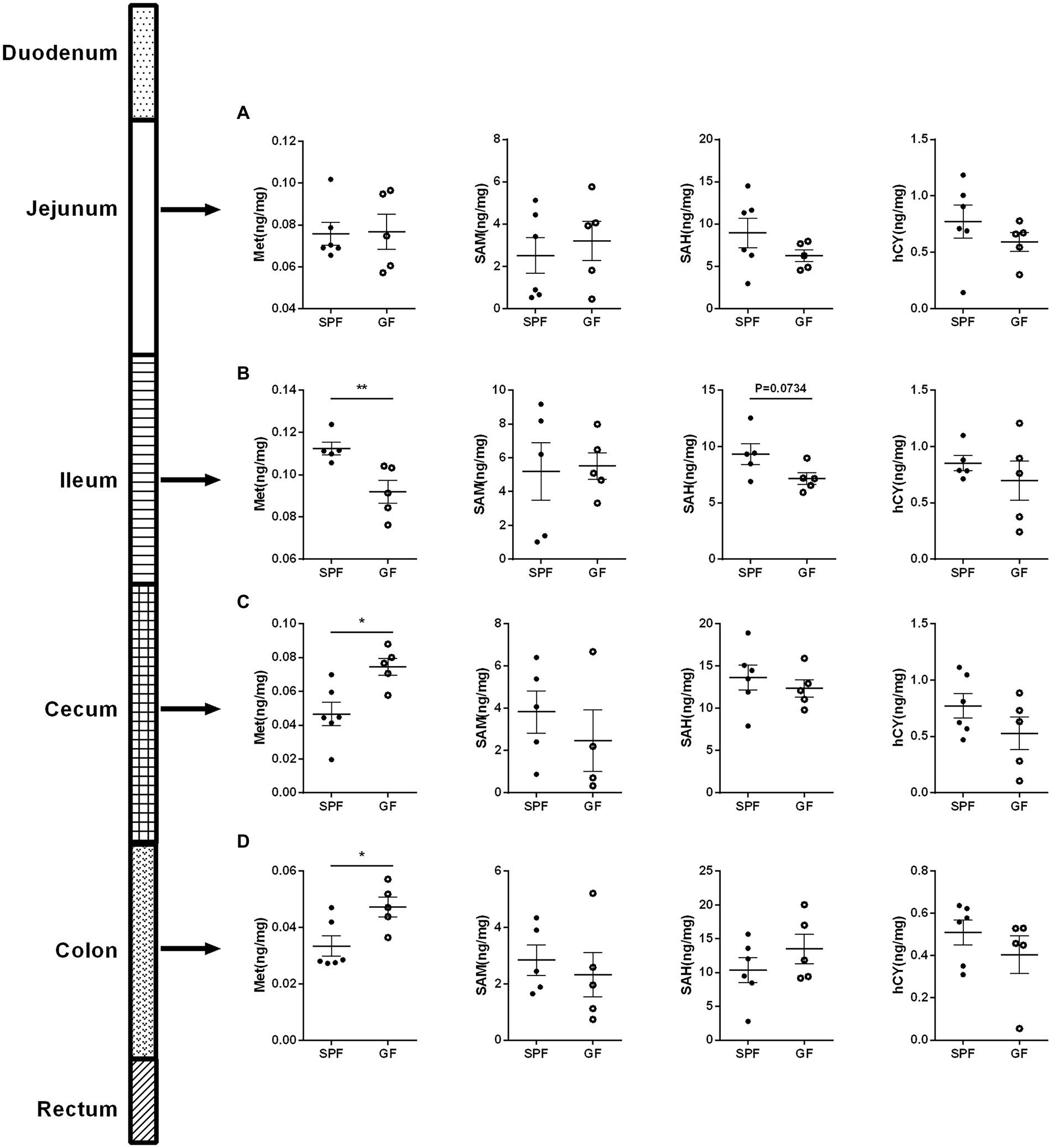
Figure 3. Gut microbiota regulates the Met metabolism in different intestinal segments of pigs. Contents of Met, SAM, SAH, hCY in (A) jejunum, (B) ileum, (C) cecum, and (D) colon of GF pigs (n = 5) and SPF pigs (n = 6). Data were analyzed by unpaired t-test and represented as means ± SEM. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine; hCY: homocysteine.
Gut microbiota remodels the met cycle in extraintestinal tissue and circulation system of pigs
The Met metabolites are absorbed by the intestinal epithelium and penetrate the basement membrane and then conducted on the extraintestinal tissues to further exert their effects through blood circulation and other means (Kharbanda, 2013; Xu et al., 2020). In addition, the liver is an important organ for Met metabolism that converts Met to SAM by MAT1A (Methionine adenosyltransferase 1A; Lu and Mato, 2012). Met-related metabolites are perturbed in both cardiovascular disease (CVD) and chronic kidney disease (CKD; Xiao et al., 2015; Yang et al., 2016), and the kidneys are involved in SAH metabolism (Garibotto et al., 2009).
We collected extraintestinal tissue samples from GF pigs and SPF pigs, including liver, spleen, kidney, and heart, and detected and analyzed the key metabolites of the Met cycle, respectively, by UPLC-Orbitrap-MS/MS. The results showed that Met in the liver of GF pigs decreased and SAM tended to increase (Figure 4A). There were no significant differences in Met, SAM, SAH, and hCY in spleen (Figure 4B), kidney (Figure 4C), and heart (Figure 4D) between GF and SPF groups. Analogously, blood, bile, and urine in the circulation system of both groups (GF and SPF) were detected by UPLC-Orbitrap-MS/MS. The results illustrated that Met was elevated in bile (Figure 5A) and serum (Figure 5B) but was no significant difference in urine (Figure 5C) in GF group. This result suggested that the lack of gut microbiota led to the increase of Met in the circulatory system, which was converted to SAM in the liver (Figure 6).
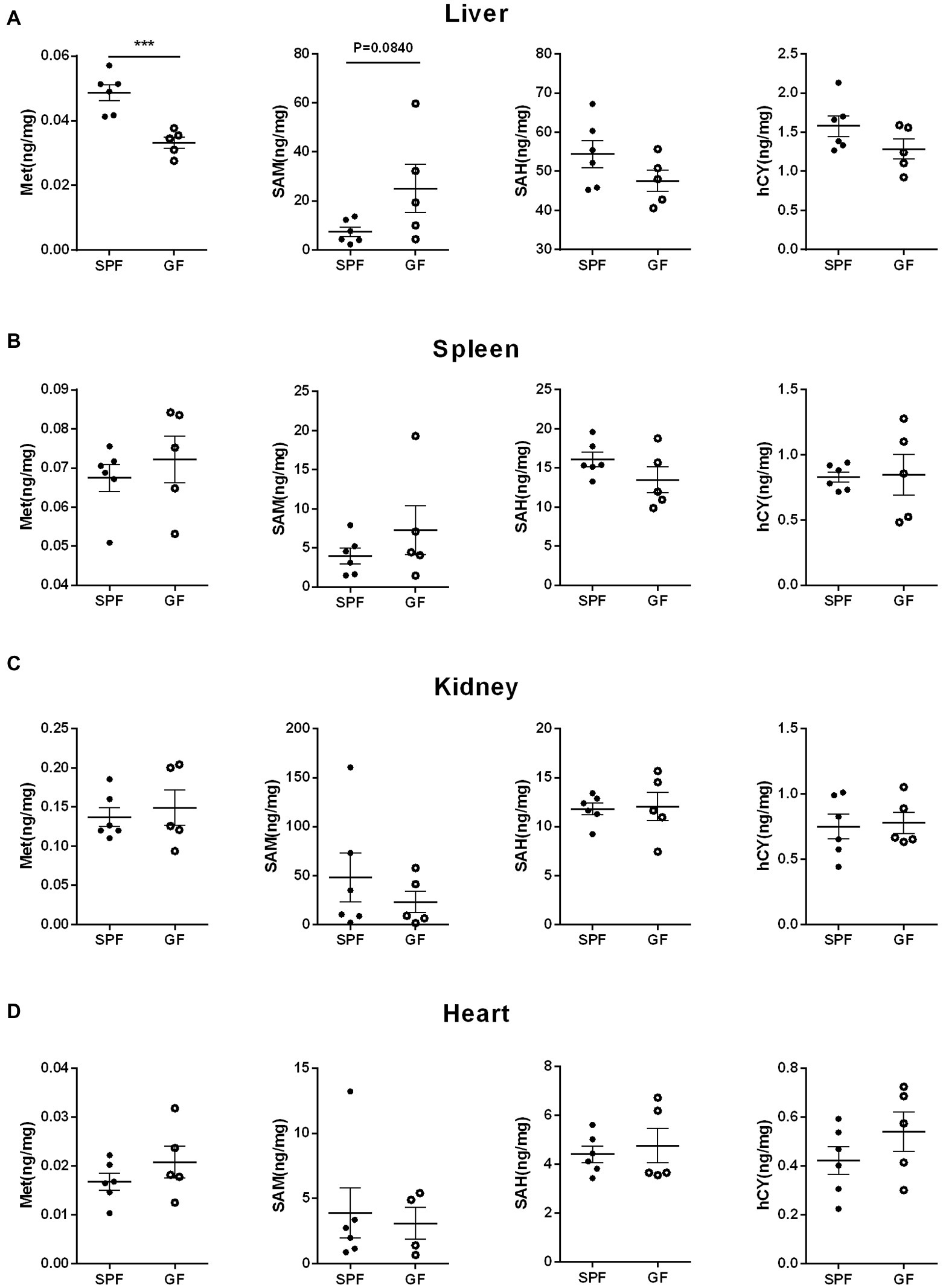
Figure 4. Gut microbiota remodels the Met metabolism in porcine extraintestinal tissue by gut microbiota. Contents of Met, SAM, SAH, hCY in (A) liver, (B) spleen, (C) kidney, (D) heart of GF pigs (n = 5) and SPF pigs (n = 6). Data were analyzed by unpaired t-test and represented as means ± SEM. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine; hCY: homocysteine.
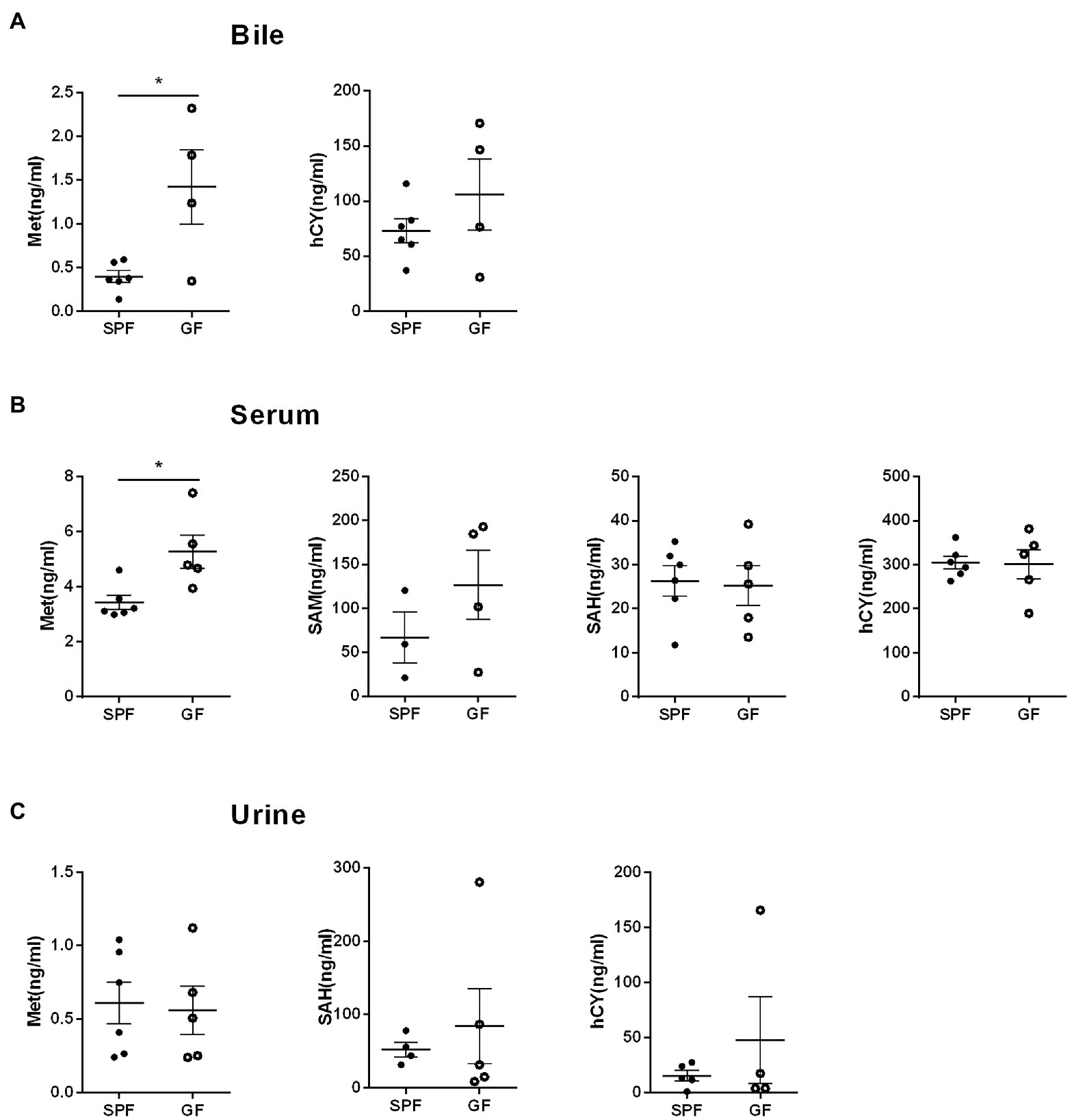
Figure 5. Gut microbiota modulates Met metabolism in porcine circulation system. (A) Met and hCY levels in bile. (B) Met, SAM, SAH, hCY levels in serum. (C) Met, SAH, hCY levels in urine of GF pigs (n = 5) and SPF pigs (n = 6). Data were analyzed by unpaired t-test and represented as means ± SEM. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine; hCY: homocysteine.
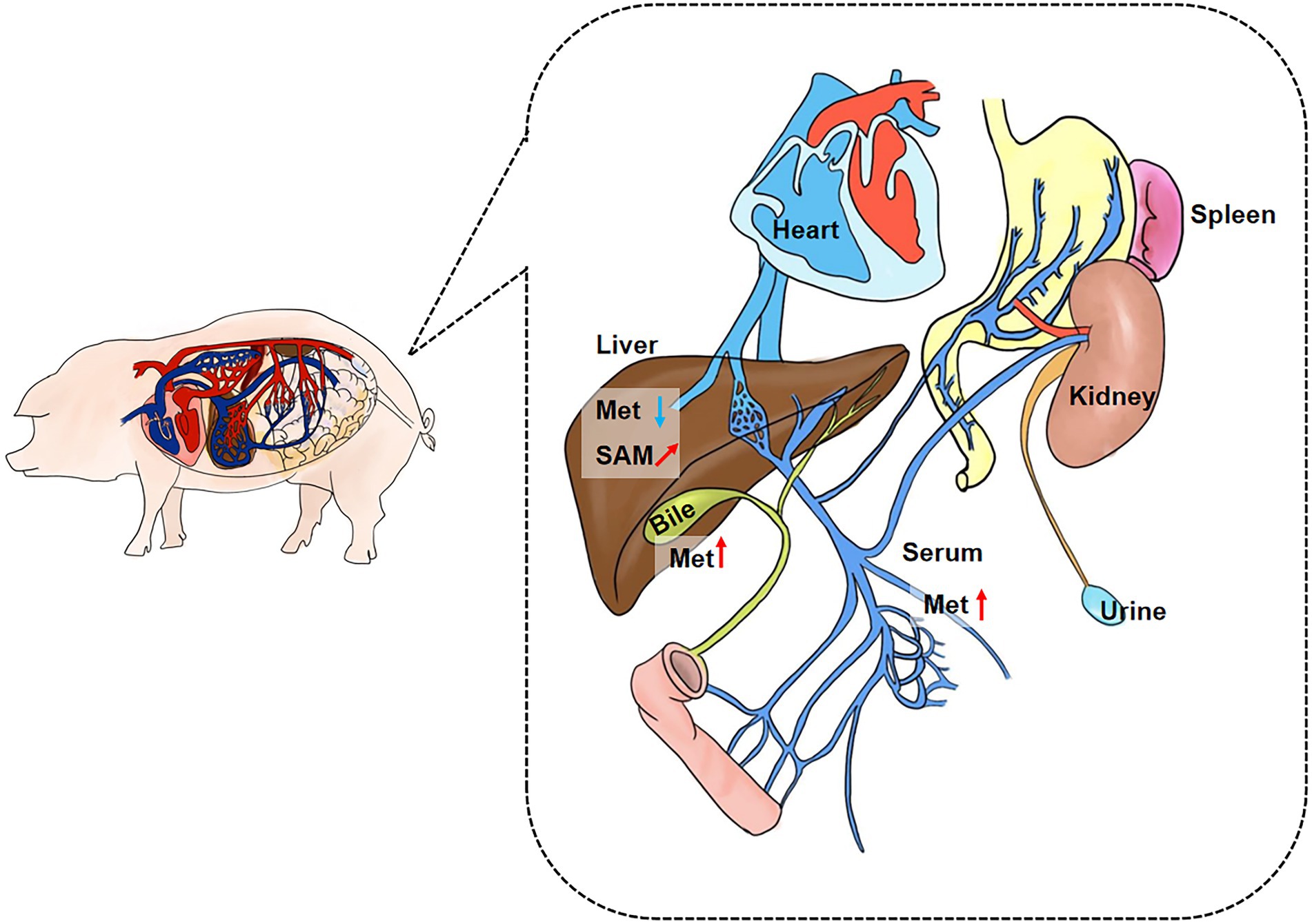
Figure 6. Summary plot of Met metabolic changes in GF porcine parenteral tissue and parenteral circulation. Met was increased in the overall circulation in GF pigs, and the elevated Met was mainly utilized in liver. Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine. *(“↗”arrows represent increasing trend, “⬆” arrows represent increases, and “⬇” arrows represent decreases.)
Gut microbiota modulates the met cycle of mice
By collecting samples from GF pigs and SPF pigs, we detected Met cycle-metabolites in the intestinal contents, different intestinal segments, extraintestinal tissues, and the circulation system. It was found that the absence of gut microbiota resulted in an overall increase in circulating Met and the elevated Met might be primarily utilized in liver. Next, we sought to translate the involvement of gut microbiota on Met metabolism to rodents. We collected duodenum, colon, and serum samples from GF and SPF mice and examined key Met cycle metabolites. Met was significantly increased and hCY was significantly decreased in the duodenum and colon of GF mice (Figures 7A,B). SAM in the colon of GF mice was significantly decreased (Figure 7B), which was consistent with the trend in pigs. There was no significant difference on Met in the serum of GF mice, but hCY was significantly reduced (Figure 7C). Thus, the findings further confirmed that the gut microbiota can modulate the Met metabolism and the absence of gut microbiota resulted in elevated Met and lower downstream SAM and/or hCY.
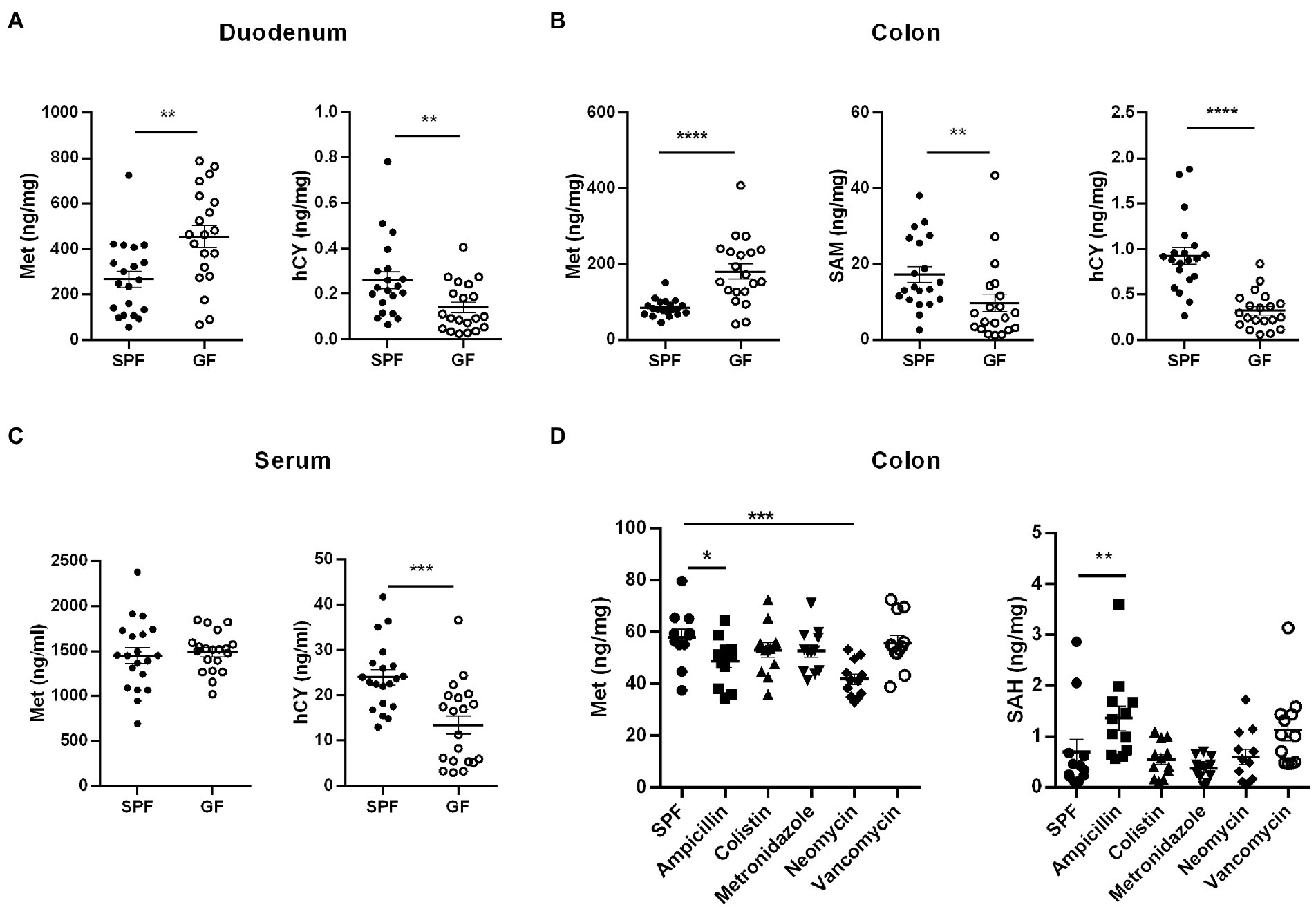
Figure 7. Influence of gut microbiota on Met metabolism in mice. (A) Met and hCY contents in duodenum of GF mice and SPF mice (n = 20). (B) Met, SAM, and hCY contents in colons of GF mice and SPF mice (n = 20). (C) Serum Met and hCY levels in GF mice and SPF mice (n = 20). (D) Met and SAH contents in colon of SPF mice (n = 12) after different antibiotic treatments. Data were analyzed by unpaired t-test (two groups) and one-way ANOVA assessment (>two groups), and represented as means ± SEM. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****). Met: Methionine; SAH: S-adenosine homocysteine; SAM: S-adenosine methionine; hCY: homocysteine.
In addition to host metabolism of Met, it has been demonstrated the existence of microbial metabolism of Met and Cys (Das et al., 2021). For example, gut microbes use cysteine desulfurase (CDS) to catalyze the synthesis of H2S from Cys (Wang et al., 2019). In the intestinal flora, Gram-positive Streptococcus, Clostridium, Staphylococcus aureus, Mycobacterium tuberculosis, Gram-negative Klebsiella, Enterobacter, Salmonella, Akkermansia spp., and Desulfovibrio are involved in the degradation of Cys (Carbonero et al., 2012). In the lower gastrointestinal tract, taurine, and isethionate are substrates for microbial metabolism, and microorganisms metabolize these organic sulfonates with the assistance of electron carriers to produce sulfide, ammonia, and other products (Laue et al., 1997). Therefore, to determine the types of microbes played pivotal roles in the regulation of host’s Met metabolism, we constructed a model of microbe deletion by treating mice with different antibiotics in drinking water for 2 weeks. The results showed decreased Met and increased SAH in colon of ampicillin-treated mice (Figure 7D). Neomycin treatment caused decreased colonic Met levels (Figure 7D). However, colonic Met and SAH were not significantly different between colistin, metronidazole, and vancomycin-treated mice (Figure 7D). Therefore, we speculated that part of the ampicillin- and/or neomycin-sensitive gut microbiota has taken a major role in regulating the Met cycle in host.
Discussion
Met metabolism is mainly composed of Met-cycle and produces the important methyl donor SAM, which participates in the biosynthesis of various proteins and DNA (Clare et al., 2019). SAM is demethylated to generate SAH and then SAH is deadenosine to generate hCY which may participate in the folate cycle and generate Cys (Lam et al., 2021). Cys produces metabolites, such as Tau or GSH, to regulate metabolic homeostasis (Selhub, 1999; Mentch et al., 2015; Jakubowski, 2019). The intestinal tract plays a key role in Met metabolism (Bauchart-Thevret et al., 2009). In addition to host intestinal transport or metabolism of Met (Zhang et al., 2014; Li et al., 2016; Romanet et al., 2021; To V et al., 2021), the gut microbiota also plays an important role in Met metabolism in the large intestine (Carbonero et al., 2012; Wang et al., 2019). Therefore, we used GF pigs as a model to explore the metabolites of the Met cycle and found that the gut microbiota was involved in regulating Met metabolism in host and the lack of gut microbiota led to the increase of Met in the gut and circulation system (Figure 8). Furthermore, we speculated that the liver is the main site of Met metabolism and utilization in GF pigs.
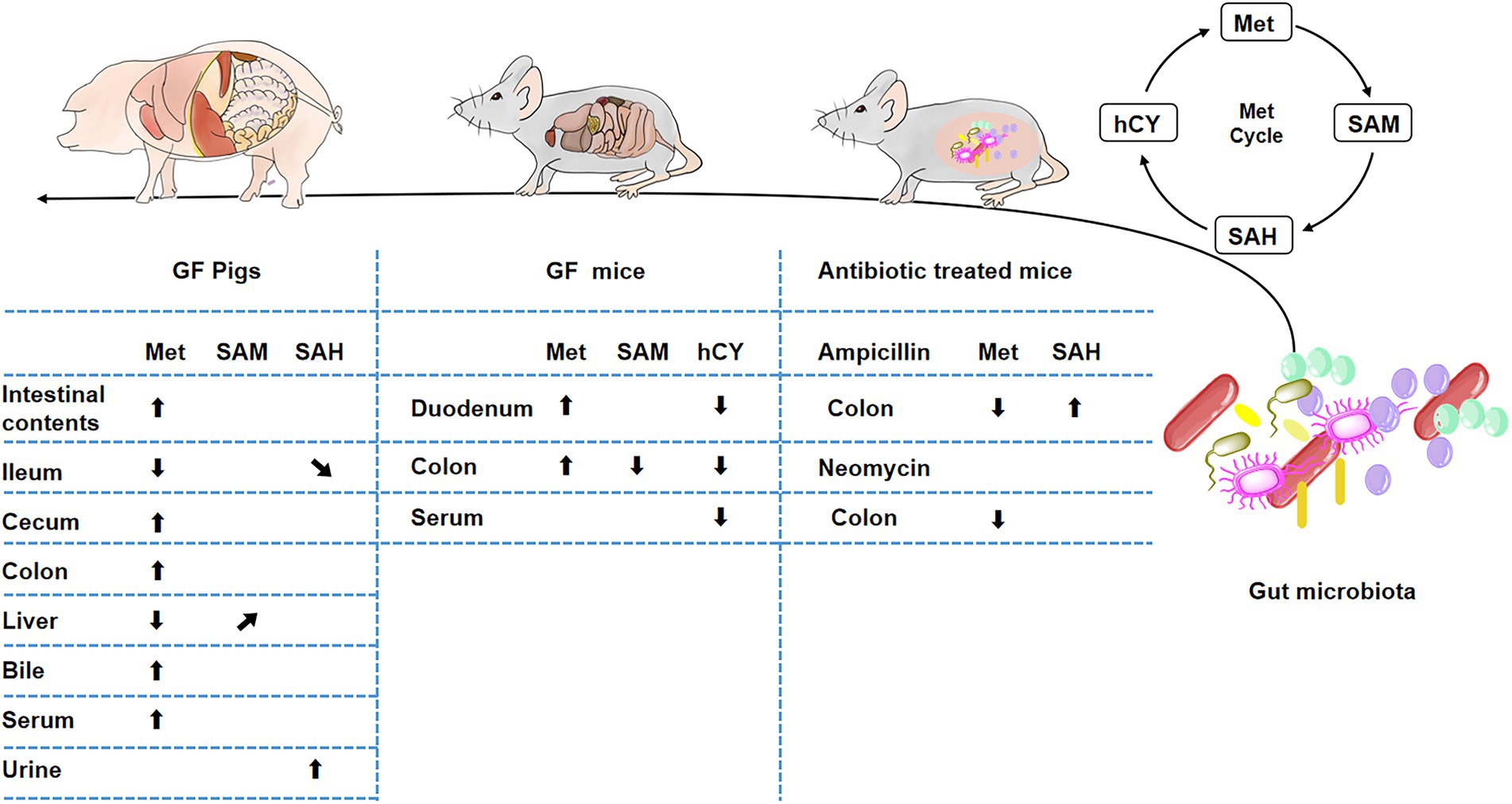
Figure 8. Influence of gut microbiota on Met metabolism. *(“⬆” arrows represent increases, “⬇” arrows represent decreases, “↗” arrows represent increasing trend, “↘” arrows represent decreasing trend).
The increase of Met in the gut is mainly concentrated in large intestine, while the Met in small intestine shows a decreasing trend. We speculate that it might be due to the changes in intestinal Met transporters. Amino acid absorption in the gut relies on transport across the apical and basolateral membranes of epithelial cells. As a neutral amino acid (NAA), Met is taken up across the apical membrane of intestinal epithelial cells by various Na+-dependent/ independent transporters, such as ASCT2 (SLC1A5), B0AT1 (SLC6A19), rBAT/b0,+AT (SLC3A1/SLC7A9), 4F2hc/y+LAT1 (SLC3A2/SLC7A7), and LAT4 (SLC43A2) primarily transports Met across the basolateral membrane (To V et al., 2021). Previous studies have demonstrated that the presence of ASCT2 and y+LAT1 in pig jejunum (Zhang et al., 2014; Li et al., 2016), B0AT1 in pig jejunum and ileum (Yi et al., 2019; Hu et al., 2021), and rBAT/b0,+AT in pig ileum (Yi et al., 2019). LAT4 is expressed in the human small intestine, especially the duodenum (Maric et al., 2021). A recent study has performed a segmented quantification of Met transporters in the porcine gastrointestinal tract and provided us with a more detailed description (Table 1). In addition to our previously mentioned Met transporters, LAT2, IMINO, SNAT2, and ATB0,+ are expressed in the porcine gastrointestinal tract (Romanet et al., 2021). Moreover, the research found that in Chd8+/− mice model of autism spectrum disorder (ASD), increased expression of neutral amino acid transporters Slc6a19, Slc7a8, and Slc7a15 resulted in increased serum glutamine and tryptophan levels (Yu et al., 2022). In Chd8+/− mice, the abundance of Bacteroides decreased, especially Bacteroides uniformis, and Bacteroides supplementation improved ASD-like behavior (Yu et al., 2022). In a piglet experiment, lysine restriction caused a decrease in SLC7A1 and SLC7A2, limiting lysine transport (Yin et al., 2017). Sequencing found that lysine restriction increased the abundance of Actinobacteria and Saccharibacteria in the gut, and functional analysis of the microbial community revealed that the differences were concentrated in amino acid metabolism and membrane transport (Yin et al., 2017). Thus, gut microbiota has the ability to modulate different amino acid transporters. Therefore, we hypothesize that the gut microbiota might have varying degrees of influences on the Met transporters in the gut. Furthermore, based on the changes of Met-related metabolites in the gut and the expression locations of Met transporters, we suppose that B0AT1, rBAT, and LAT4 might be partially inhibited, and/or ATB0+, ASCT2, and SNAT2 might be promoted. However, this conjecture still needs to be verified by detecting the expression of related transporters. In addition, the gut microbiota has been shown to be involved in regulating intestinal mucosal integrity (Wang et al., 2019; Paone and Cani, 2020), angiogenesis (Franks, 2013; Andriessen et al., 2016), both of which are associated with nutrient absorption. Furthermore, gut microbes can utilize substances in the Met cycle (Carbonero et al., 2012; Wang et al., 2019; Das et al., 2021), or metabolize to produce Met-related downstream substances (Rosario et al., 2021), or interact with substances in Met cycle (Roth and Mohamadzadeh, 2021). Whether these functions of gut microbes are related to the regulation of host Met metabolism in this paper requires further experimental verification.
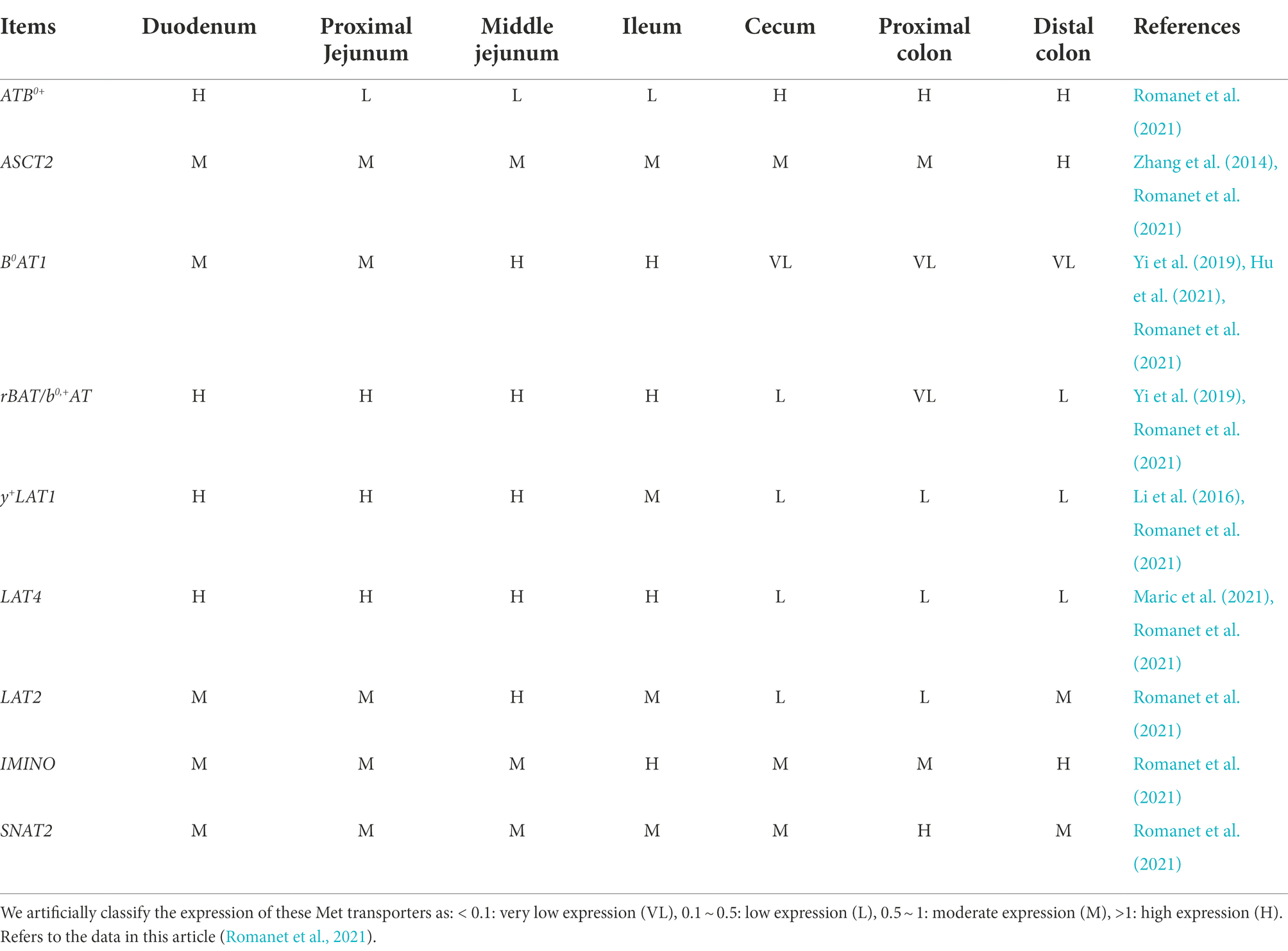
Table 1. mRNA Expression of Met transporters in different intestinal tracts of pigs.
According to the experimental results, we found that gut microbiota could regulate host Met metabolism, but the increase in Met caused by lack of gut microbes was not detected in the jejunum. The possible reasons are: (1) Jejunal microbial metabolism: Jejunal microbiota is mainly involved in the degradation of lipids and sugars (Lema et al., 2020), so amino acid changes are not obvious; (2) Jejunal microbial types: the abundance and diversity of microbiota in the jejunum are very low (about 104 ~ 107 CFU/ml), and are mainly facultative anaerobic bacteria, such as Streptococcus and Lactococcus, which are different from the large intestine (Martinez-Guryn et al., 2019; Seekatz et al., 2019); (3) Features of the jejunum: in order to facilitate the nutrient absorption, the jejunum is covered with a thin layer of loose mucus, and the intervening Paneth cells secrete antimicrobial peptides (AMPs; Guo et al., 2017; Lema et al., 2020). And the transit time is faster in the small intestine compared to the large intestine (Martinez-Guryn et al., 2019), so the environment of the jejunum is not conducive to the long-term colonization of most microorganisms.
In addition, we examined the Met cycle-metabolites in GF mice, further confirmed that gut microbiota depletion caused the increase of Met, which promoted its downstream metabolic responses to a decrease in hCY and SAM (Figure 8). A variety of gram-positive or negative bacteria are involved in Met metabolism (Carbonero et al., 2012), thus, we treated mice with five different antibiotics, including the beta-lactam antibiotic ampicillin, polymyxin antibiotic colistin, nitroimidazole derivative metronidazole, aminoglycoside antibiotic neomycin, and glycopeptide antibiotic vancomycin. However, we could not achieve a Met increase similar to GF-treated effects by treating mice with antibiotics to mimic microbe deficiency. In contrast, we found a decrease in intestinal Met in the ampicillin and neomycin-treated groups (Figure 8).
Ampicillin can prevent the synthesis of bacterial cell walls with the bacteriostatic and bactericidal effects, and its effect on Gram-positive bacteria is similar to that of penicillin. Gram-negative bacteria such as Escherichia coli are sensitive to ampicillin (Jõers et al., 2010), while Pneumococcus pneumoniae and Pseudomonas aeruginosa are not (Li et al., 2021; Brazel et al., 2022). Neomycin has a good bactericidal effect on Gram-negative bacteria, Gram-positive bacteria, and Mycobacterium tuberculosis, of which Escherichia coli is the most sensitive (Kadison et al., 1951; Bera et al., 2008). Combined with the experimental results, we could only conclude that the lack of certain ampicillin- or neomycin-susceptible microbes resulted in lower intestinal Met levels. Moreover, studies have shown that there are objective function differences between GF animals and SPF animals such as metabolism and immunity (Uzbay, 2019), and the elimination of a single type of microbe may cause the increase in others, thereby probably leading to more utilization of Met. Determining the key microbes in host Met metabolic remodeling requires specific inhibition of possible microbial species and detection of Met metabolites after recolonization in GF animals. In addition to the changes of Met metabolism, studies have shown that growth performance, nutrient digestibility, and skeletal muscle growth and development are affected in GF piglets (Qi et al., 2021; Zhou et al., 2021). Therefore, it may be that the effect of gut microbiota on Met metabolism in GF animals was caused by the changes of global growth characteristics to drive the regulation of nutrient metabolism. In contrast to antibiotic-treated mice, GF animals was sterile in terms of other organs. In view of recent findings, the diversity of lung microbiota has a certain correlation with the risk of bronchopulmonary dysplasia (BPD) in neonates, and the lung microbiota metagenome of BPD probably changed the host metabolome (Lohmann et al., 2014; Lal et al., 2018). Therefore, it is also possible that microbiota in lung and other organs are involved in metabolic regulation in host, resulting the change trend of intestinal Met in antibiotic-treated mice is opposite to that in GF mice in this study. In order to study the influence of other organ microorganisms on Met metabolism, it may be a feasible experimental idea to redetect the changes of Met metabolism by supplementing intestinal microbiota of GF animals with whole intestinal microbiota transplantation (WIMT; Li et al., 2020).
In conclusion, the gut microbiota was involved in host Met remodeling, and Met was increased in complete GF animals (GF pigs and GF mice). However, when the microbes were cleared with antibiotics, ampicillin and neomycin treatment instead resulted in a reduction in intestinal Met. Since Met metabolism is closely related to various inflammatory diseases (Borren et al., 2021), ALD, liver tumors (Kharbanda, 2013; Xu et al., 2020), CVD, CKD (Yang et al., 2016), and other diseases, precise regulation of Met metabolism would become an effective means to treat these diseases. Therefore, our study provided new insights into the prevention and/or treatment of the associated diseases by modulating gut microbes in animals and humans.
Data availability statement
The data presented in the study are deposited in the Metabolights repository, and the accession number is MTBLS6154.
Ethics statement
The animal study was reviewed and approved by the Laboratory Animal Ethical Commission of the South China Agricultural University [permit number SYXK (Guangdong) 2019–0136].
Author contributions
WT and SL designed the research and analyzed data. DY, JS, and LG assisted in the establishment of GF pigs and GF mice. XW, ZH, and BL participated in antibiotic-animal experiments. XW and BL prepared the figures and draft the manuscript. XW, ZH, WT, and SL revised and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Key R&D Program of China (2021YFD1301100), and Sichuan Science and Technology Programs (2021JDYZ0001 and 2021ZDZX0009).
Acknowledgments
All pigs were provided by the Technical Engineering Center for the Development and Utilization of Medical Animal Resources (Chongqing, China).
Conflict of interest
WT is employed by Sichuan Animtech Feed Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
A. muciniphila, Akkermansia muciniphila, ALD, Alcoholic liver disease, CH3-THF, N5-methyltetrahydrofolate, CKD: Chronic kidney disease, CVD, Cardiovascular disease, Cys, Cysteine, ESI, Electrospray ionization, GF, Germ-free, Gly, Glycine, GSH, Glutathione, HCD, High collision-induced dissociation, hCY, homocysteine, HHcy, hyperhomocysteinemia, IBD, Inflammatory bowel disease, MAT, Methionine adenosyltransferase, MAT1A, Methionine adenosyltransferase 1A, Met, Methionine, NAA, Neutral amino acid, PD, Parkinson’s disease, SAH, S-adenosine homocysteine, SAHH, S-adenosylhomocysteine hydrolase, SAM, S-adenosine methionine, Sar, Sarcosine, SCFAs, Short-chain fatty acids, Ser, Serine, SPF, Specific pathogen-free, Tau, Taurine,
Footnotes
References
Andremont, A., Cervesi, J., Bandinelli, P. A., Vitry, F., and de Gunzburg, J. (2021). Spare and repair the gut microbiota from antibiotic-induced Dysbiosis: state-of-the-art. Drug Discov. Today 26, 2159–2163. doi: 10.1016/j.drudis.2021.02.022
Andriessen, E. M., Wilson, A. M., Mawambo, G., Dejda, A., Miloudi, K., Sennlaub, F., et al. (2016). Gut microbiota influences pathological angiogenesis in obesity-driven Choroidal neovascularization. EMBO Mol. Med. 8, 1366–1379. doi: 10.15252/emmm.201606531
Bauchart-Thevret, C., Stoll, B., and Burrin, D. G. (2009). Intestinal metabolism of sulfur amino acids. Nutr. Res. Rev. 22, 175–187. doi: 10.1017/s0954422409990138
Bera, S., Zhanel, G. G., and Schweizer, F. (2008). Design, synthesis, and antibacterial activities of neomycin-lipid conjugates: Polycationic lipids with potent gram-positive activity. J. Med. Chem. 51, 6160–6164. doi: 10.1021/jm800345u
Borren, N. Z., Plichta, D., Joshi, A. D., Bonilla, G., Peng, V., Colizzo, F. P., et al. (2021). Alterations in fecal microbiomes and serum Metabolomes of fatigued patients with quiescent inflammatory bowel diseases. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association 19, 519–527. doi: 10.1016/j.cgh.2020.03.013
Brazel, E. B., Tan, A., Neville, S. L., Iverson, A. R., Udagedara, S. R., Cunningham, B. A., et al. (2022). Dysregulation of streptococcus Pneumoniae zinc homeostasis breaks ampicillin resistance in a pneumonia infection model. Cell Rep. 38:110202. doi: 10.1016/j.celrep.2021.110202
Cani, P. D. (2018). Human gut microbiome: hopes. Threats and Promises. Gut 67, 1716–1725. doi: 10.1136/gutjnl-2018-316723
Carbonero, F., Benefiel, A. C., Alizadeh-Ghamsari, A. H., and Gaskins, H. R. (2012). Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 3:448. doi: 10.3389/fphys.2012.00448
Clare, C. E., Brassington, A. H., Kwong, W. Y., and Sinclair, K. D. (2019). One-carbon metabolism: linking nutritional biochemistry to epigenetic programming of Long-term development. Annual rev. animal biosciences 7, 263–287. doi: 10.1146/annurev-animal-020518-115206
Cryan, J. F., O'Riordan, K. J., Cowan, C. S. M., Sandhu, K. V., Bastiaanssen, T. F. S., Boehme, M., et al. (2019). The microbiota-gut-brain Axis. Physiol. Rev. 99, 1877–2013. doi: 10.1152/physrev.00018.2018
Dang, A. T., and Marsland, B. J. (2019). Microbes, metabolites, and the gut-lung Axis. Mucosal Immunol. 12, 843–850. doi: 10.1038/s41385-019-0160-6
Das, M., Dewan, A., Shee, S., and Singh, A. (2021). The multifaceted bacterial cysteine Desulfurases: from metabolism to pathogenesis. Antioxidants (Basel, Switzerland) 10:997. doi: 10.3390/antiox10070997
Franks, I. (2013). Microbiota: gut microbes might promote intestinal angiogenesis. Nat. Rev. Gastroenterol. Hepatol. 10:3. doi: 10.1038/nrgastro.2012.227
Garibotto, G., Valli, A., Anderstam, B., Eriksson, M., Suliman, M. E., Balbi, M., et al. (2009). The kidney is the major site of S-Adenosylhomocysteine disposal in humans. Kidney Int. 76, 293–296. doi: 10.1038/ki.2009.117
Gondret, F., Le Floc'h, N., Batonon-Alavo, D. I., Perruchot, M. H., Mercier, Y., and Lebret, B. (2021). Flash dietary methionine supply over growth requirements in pigs: multi-facetted effects on skeletal muscle metabolism. Animal: an international journal of animal bioscience 15:100268. doi: 10.1016/j.animal.2021.100268
Guo, X., Li, J., Tang, R., Zhang, G., Zeng, H., Wood, R. J., et al. (2017). High fat diet alters gut microbiota and the expression of Paneth cell-antimicrobial peptides preceding changes of circulating inflammatory cytokines. Mediat. Inflamm. 2017, 9474896–9474899. doi: 10.1155/2017/9474896
Haug, K., Cochrane, K., Nainala, V. C., Williams, M., Chang, J., Jayaseelan, K. V., et al. (2020). Metabolights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444. doi: 10.1093/nar/gkz1019
Hertel, J., Harms, A. C., Heinken, A., Baldini, F., Thinnes, C. C., Glaab, E., et al. (2019). Integrated analyses of microbiome and longitudinal Metabolome data reveal microbial-host interactions on sulfur metabolism in Parkinson's disease. Cell Rep. 29, 1767–1777. doi: 10.1016/j.celrep.2019.10.035
Hu, P., Zhao, F., Wang, J., and Zhu, W. (2021). Metabolomic profiling reveals the effects of early-life Lactoferrin intervention on protein synthesis, energy production and Antioxidative capacity in the liver of suckling piglets. Food Funct. 12, 3405–3419. doi: 10.1039/d0fo01747g
Itoh, M., Mukae, Y., Kitsuka, T., Arai, K., Nakamura, A., Uchihashi, K., et al. (2019). Development of an Immunodeficient pig model allowing Long-term accommodation of artificial human vascular tubes. Nat. Commun. 10:2244. doi: 10.1038/s41467-019-10107-1
Jakubowski, H. (2019). Homocysteine modification in protein structure/function and human disease. Physiol. Rev. 99, 555–604. doi: 10.1152/physrev.00003.2018
Jõers, A., Kaldalu, N., and Tenson, T. (2010). The frequency of Persisters in Escherichia Coli reflects the kinetics of awakening from dormancy. J. Bacteriol. 192, 3379–3384. doi: 10.1128/jb.00056-10
Kadison, E. R., Volini, I. F., Hoffman, S. J., and Felsenfeld, O. (1951). Neomycin therapy; its use in virus pneumonia, tuberculosis and diseases caused by gram-negative bacteria. J. Am. Med. Assoc. 145, 1307–1312. doi: 10.1001/jama.1951.02920350001001
Kharbanda, K. K. (2013). Methionine metabolic pathway in alcoholic liver injury. Curr. Opin. Clin. Nutr. Metab. Care 16, 89–95. doi: 10.1097/MCO.0b013e32835a892a
Lal, C. V., Kandasamy, J., Dolma, K., Ramani, M., Kumar, R., Wilson, L., et al. (2018). Early airway microbial metagenomic and Metabolomic signatures are associated with development of severe Bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 315, L810–L815. doi: 10.1152/ajplung.00085.2018
Lam, A. B., Kervin, K., and Tanis, J. E. (2021). Vitamin B (12) impacts amyloid Beta-induced Proteotoxicity by regulating the methionine/S-Adenosylmethionine cycle. Cell Rep. 36:109753. doi: 10.1016/j.celrep.2021.109753
Laue, H., Denger, K., and Cook, A. M. (1997). Taurine reduction in anaerobic respiration of Bilophila Wadsworthia Rzatau. Appl. Environ. Microbiol. 63, 2016–2021. doi: 10.1128/aem.63.5.2016-2021.1997
Lema, I., Araújo, J. R., Rolhion, N., and Demignot, S. (2020). Jejunum: the understudied meeting place of dietary lipids and the microbiota. Biochimie 178, 124–136. doi: 10.1016/j.biochi.2020.09.007
Li, Y., Xia, L., Chen, J., Lian, Y., Dandekar, A. A., Xu, F., et al. (2021). Resistance elicited by sub-lethal concentrations of ampicillin is partially mediated by quorum sensing in pseudomonas Aeruginosa. Environ. Int. 156:106619. doi: 10.1016/j.envint.2021.106619
Li, X. G., Xu, G. F., Zhai, Z. Y., Gao, C. Q., Yan, H. C., Xi, Q. Y., et al. (2016). Cdx 2 increases Slc 7a7 expression and proliferation of pig intestinal epithelial cells. Oncotarget 7, 30597–30609. doi: 10.18632/oncotarget.8894
Li, N., Zuo, B., Huang, S., Zeng, B., Han, D., Li, T., et al. (2020). Spatial heterogeneity of bacterial colonization across different gut segments following inter-species microbiota transplantation. Microbiome 8:161. doi: 10.1186/s40168-020-00917-7
Liu, J., Huang, J., Xin, P., Liu, G., and Wu, J. (2021). Biomedical applications of methionine-based systems. Biomater. Sci. 9, 1961–1973. doi: 10.1039/d0bm02180f
Liu, B., Yu, D., Sun, J., Wu, X., Xin, Z., Deng, B., et al. (2022). Characterizing the influence of gut microbiota on host tryptophan metabolism with germ-free pigs. Animal Nutrition 11, 190–200. doi: 10.1016/j.aninu.2022.07.005
Lohmann, P., Luna, R. A., Hollister, E. B., Devaraj, S., Mistretta, T. A., Welty, S. E., et al. (2014). The airway microbiome of intubated premature infants: characteristics and changes that predict the development of Bronchopulmonary dysplasia. Pediatr. Res. 76, 294–301. doi: 10.1038/pr.2014.85
Lu, S. C., and Mato, J. M. (2012). S-Adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 92, 1515–1542. doi: 10.1152/physrev.00047.2011
Maric, S., Flüchter, P., Guglielmetti, L. C., Staerkle, R. F., Sasse, T., Restin, T., et al. (2021). Plasma Citrulline correlates with Basolateral amino acid transporter Lat 4 expression in human small intestine. Clinical nutrition (Edinburgh, Scotland) 40:2244. doi: 10.1016/j.clnu.2020.10.003
Martinez-Guryn, K., Leone, V., and Chang, E. B. (2019). Regional diversity of the gastrointestinal microbiome. Cell Host Microbe 26, 314–324. doi: 10.1016/j.chom.2019.08.011
Mentch, S. J., Mehrmohamadi, M., Huang, L., Liu, X., Gupta, D., Mattocks, D., et al. (2015). Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861–873. doi: 10.1016/j.cmet.2015.08.024
Miller, E. R., and Ullrey, D. E. (1987). The pig as a model for human nutrition. Annu. Rev. Nutr. 7, 361–382. doi: 10.1146/annurev.nu.07.070187.002045
Mota-Martorell, N., Jové, M., Berdún, R., and Pamplona, R. (2021). Plasma methionine metabolic profile is associated with longevity in mammals. Communications biology 4:725. doi: 10.1038/s42003-021-02254-3
Osbelt, L., Wende, M., Almási, É., Derksen, E., Muthukumarasamy, U., Lesker, T. R., et al. (2021). Klebsiella Oxytoca causes colonization resistance against multidrug-Resistant K. pneumoniae in the gut via cooperative carbohydrate competition. Cell Host Microbe 29, 1663–1679. doi: 10.1016/j.chom.2021.09.003
Paone, P., and Cani, P. D. (2020). Mucus barrier, Mucins and gut microbiota: the expected slimy partners? Gut 69, 2232–2243. doi: 10.1136/gutjnl-2020-322260
Pulendran, B., and Davis, M. M. (2020). The science and medicine of human immunology. Science 369:eaay4014. doi: 10.1126/science.aay4014
Qi, R., Sun, J., Qiu, X., Zhang, Y., Wang, J., Wang, Q., et al. (2021). The intestinal microbiota contributes to the growth and physiological state of muscle tissue in piglets. Sci. Rep. 11:11237. doi: 10.1038/s41598-021-90881-5
Romanet, S., Aschenbach, J. R., Pieper, R., Zentek, J., Htoo, J. K., Whelan, R. A., et al. (2021). Expression of proposed methionine transporters along the gastrointestinal tract of pigs and their regulation by dietary methionine sources. Genes Nutr. 16:14. doi: 10.1186/s12263-021-00694-4
Rosario, D., Bidkhori, G., Lee, S., Bedarf, J., Hildebrand, F., Le Chatelier, E., et al. (2021). Systematic analysis of gut microbiome reveals the role of bacterial Folate and Homocysteine metabolism in Parkinson's disease. Cell Rep. 34:108807. doi: 10.1016/j.celrep.2021.108807
Roth, W., and Mohamadzadeh, M. (2021). Vitamin B12 and gut-brain homeostasis in the pathophysiology of ischemic stroke. EBioMedicine 73:103676. doi: 10.1016/j.ebiom.2021.103676
Seekatz, A. M., Schnizlein, M. K., Koenigsknecht, M. J., Baker, J. R., Hasler, W. L., Bleske, B. E., et al. (2019). Spatial and temporal analysis of the stomach and small-intestinal microbiota in fasted healthy humans. mSphere 4:e00126-19. doi: 10.1128/mSphere.00126-19
Selhub, J. (1999). Homocysteine metabolism. Annu. Rev. Nutr. 19, 217–246. doi: 10.1146/annurev.nutr.19.1.217
Sun, M. F., and Shen, Y. Q. (2018). Dysbiosis of gut microbiota and microbial metabolites in Parkinson's disease. Ageing Res. Rev. 45, 53–61. doi: 10.1016/j.arr.2018.04.004
Sun, J., Zhong, H., Du, L., Li, X., Ding, Y., Cao, H., et al. (2018). Gene expression profiles of germ-free and conventional piglets from the same litter. Sci. Rep. 8:10745. doi: 10.1038/s41598-018-29093-3
To VMasagounder, K., and Loewen, M. E. (2021). Critical transporters of methionine and methionine hydroxyl analogue supplements across the intestine: what we know so far and what can be learned to advance animal nutrition. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 255:110908. doi: 10.1016/j.cbpa.2021.110908
Tripathi, A., Debelius, J., Brenner, D. A., Karin, M., Loomba, R., Schnabl, B., et al. (2018). The gut-liver Axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411. doi: 10.1038/s41575-018-0011-z
Uzbay, T. (2019). Germ-free animal experiments in the gut microbiota studies. Curr. Opin. Pharmacol. 49, 6–10. doi: 10.1016/j.coph.2019.03.016
Wang, J., Guo, X., Li, H., Qi, H., Qian, J., Yan, S., et al. (2019). Hydrogen Sulfide from Cysteine Desulfurase, Not 3-Mercaptopyruvate Sulfurtransferase, Contributes to Sustaining Cell Growth and Bioenergetics in E. Coli under Anaerobic Conditions. Front Microbiol 10:2357. doi: 10.3389/fmicb.2019.02357
Wang, Z., Yip, L. Y., Lee, J. H. J., Wu, Z., Chew, H. Y., Chong, P. K. W., et al. (2019). Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 25, 825–837. doi: 10.1038/s41591-019-0423-5
Wang, L., Zhu, L., and Qin, S. (2019). Gut microbiota modulation on intestinal mucosal adaptive immunity. J Immunol Res 2019, 4735040–4735010. doi: 10.1155/2019/4735040
Xiao, Y., Su, X., Huang, W., Zhang, J., Peng, C., Huang, H., et al. (2015). Role of S-Adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int. J. Biochem. Cell Biol. 67, 158–166. doi: 10.1016/j.biocel.2015.06.015
Xu, Q., Li, Y., Gao, X., Kang, K., Williams, J. G., Tong, L., et al. (2020). Hnf4α regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat. Commun. 11:3978. doi: 10.1038/s41467-020-17818-w
Yang, J., Fang, P., Yu, D., Zhang, L., Zhang, D., Jiang, X., et al. (2016). Chronic kidney disease induces inflammatory Cd40+ monocyte differentiation via Homocysteine elevation and DNA Hypomethylation. Circ. Res. 119, 1226–1241. doi: 10.1161/circresaha.116.308750
Yang, Z., Hasan, M. S., Humphrey, R. M., Htoo, J. K., and Liao, S. F. (2021). Changes in growth performance, plasma metabolite concentrations, and myogenic gene expression in growing pigs fed a methionine-restricted diet. Frontiers in bioscience (Landmark edition) 26, 413–422. doi: 10.52586/4956
Yi, H., Yang, G., Xiong, Y., Wu, Q., Xiao, H., Wen, X., et al. (2019). Integrated Metabolomic and proteomics profiling reveals the promotion of lactobacillus Reuteri Lr1 on amino acid metabolism in the gut-liver Axis of weaned pigs. Food Funct. 10, 7387–7396. doi: 10.1039/c9fo01781j
Yin, J., Han, H., Li, Y., Liu, Z., Zhao, Y., Fang, R., et al. (2017). Lysine restriction affects feed intake and amino acid metabolism via gut microbiome in piglets. Cell. Physiol. Biochem. 44, 1749–1761. doi: 10.1159/000485782
Yin, J., Ren, W., Chen, S., Li, Y., Han, H., Gao, J., et al. (2018). Metabolic regulation of methionine restriction in diabetes. Mol. Nutr. Food Res. 62:e1700951. doi: 10.1002/mnfr.201700951
Yu, W., Wang, Z., Zhang, K., Chi, Z., Xu, T., Jiang, D., et al. (2019). One-carbon metabolism supports S-Adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell 75:1147. doi: 10.1016/j.molcel.2019.06.039
Yu, Y., Zhang, B., Ji, P., Zuo, Z., Huang, Y., Wang, N., et al. (2022). Changes to gut amino acid transporters and microbiome associated with increased E/I ratio in Chd 8(+/−) mouse model of Asd-like behavior. Nat. Commun. 13:1151. doi: 10.1038/s41467-022-28746-2
Zeitz, J. O., Kaltenböck, S., Most, E., and Eder, K. (2019). Effects of L-methionine on performance, gut morphology and antioxidant status in gut and liver of piglets in relation to dl-methionine. J. Anim. Physiol. Anim. Nutr. 103, 242–250. doi: 10.1111/jpn.13000
Zhang, S., Ren, M., Zeng, X., He, P., Ma, X., and Qiao, S. (2014). Leucine stimulates Asct 2 amino acid transporter expression in porcine Jejunal epithelial cell line (Ipec-J2) through Pi3k/Akt/Mtor and Erk signaling pathways. Amino Acids 46, 2633–2642. doi: 10.1007/s00726-014-1809-9
Keywords: gut microbiota, methionine, SAM, hCY, germ-free pigs
Citation: Wu X, Han Z, Liu B, Yu D, Sun J, Ge L, Tang W and Liu S (2022) Gut microbiota contributes to the methionine metabolism in host. Front. Microbiol. 13:1065668. doi: 10.3389/fmicb.2022.1065668
Edited by:
Zhixiong He, Institute of Subtropical Agriculture (CAS), ChinaReviewed by:
Xin Fang, The First Affiliated Hospital of Nanchang University, ChinaZhiqian Xu, Henan University of Science and Technology, China
Copyright © 2022 Wu, Han, Liu, Yu, Sun, Ge, Tang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenjie Tang, d2VuaGFuMjhAMTI2LmNvbQ==; Shaojuan Liu, MjAxODEwMjUwMDNAc3R1LnNjYXUuZWR1LmNu
†These authors have contributed equally to this work