
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol., 09 November 2022
Sec. Virology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1044316
 Cora M. Holicki†
Cora M. Holicki† Felicitas Bergmann†Franziska StoekAnsgar Schulz
Felicitas Bergmann†Franziska StoekAnsgar Schulz Martin H. GroschupUte Ziegler
Martin H. GroschupUte Ziegler Balal Sadeghi*
Balal Sadeghi*Usutu virus (USUV) is a mosquito-borne zoonotic virus and one of the causes of flavivirus encephalitis in birds and occasionally in humans. USUV rapidly disperses in a susceptible host and vector environment, as is the case in South and Central Europe. However, compared to other flaviviruses, USUV has received less research attention and there is therefore limited access to whole-genome sequences and also to in-depth phylogenetic and phylodynamic analyses. To ease future molecular studies, this study compares first- (partial sequencing via Sanger), second- (Illumina), and third-generation (MinION Nanopore) sequencing platforms for USUV. With emphasis on MinION Nanopore sequencing, cDNA-direct and target-enrichment (amplicon-based) sequencing approaches were validated in parallel. The study was based on four samples from succumbed birds commonly collected throughout Germany. The samples were isolated from various sample matrices, organs as well as blood cruor, and included three different USUV lineages. We concluded that depending on the focus of a research project, amplicon-based MinION Nanopore sequencing can be an ideal cost- and time-effective alternative to Illumina in producing optimal genome coverage. It can be implemented for an array of lab- or field-based objectives, including among others: phylodynamic studies and the analysis of viral quasispecies.
Usutu virus (USUV), a neglected Old World flavivirus, causes annually reoccurring epizootics in the avian fauna and sporadic human infections. The enzootic transmission cycle involves ornithophilic mosquitoes (Culex spp.) as vectors and birds as reservoir and amplifying hosts. Blackbirds (Turdus merula) are particularly susceptible but also other passerine species including song thrushes (Turdus philomelos), common kingfishers (Alcedo atthis), house sparrows (Passer domesticus), canaries (Serinus canaria forma domestica), common starlings (Sturnus vulgaris), and magpies (Pica pica) as well as birds of prey such as owls (Strigiformes) can become infected (Becker et al., 2012; Saiz and Blazquez, 2017). USUV is an enveloped virus with a diameter of approximately 40–60 nm. It has a positive sense single-stranded genome of 11,064 nucleotides (nt) harboring a type I cap structure but lacking a poly-A tail (Bakonyi et al., 2004). The genome comprises an open reading frame encoding a single polyprotein (3,434 amino acids) that is cleaved by viral and cellular proteases into three structural (capsid C, premembrane prM, and envelope E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Figure 1) (Calisher and Gould, 2003).
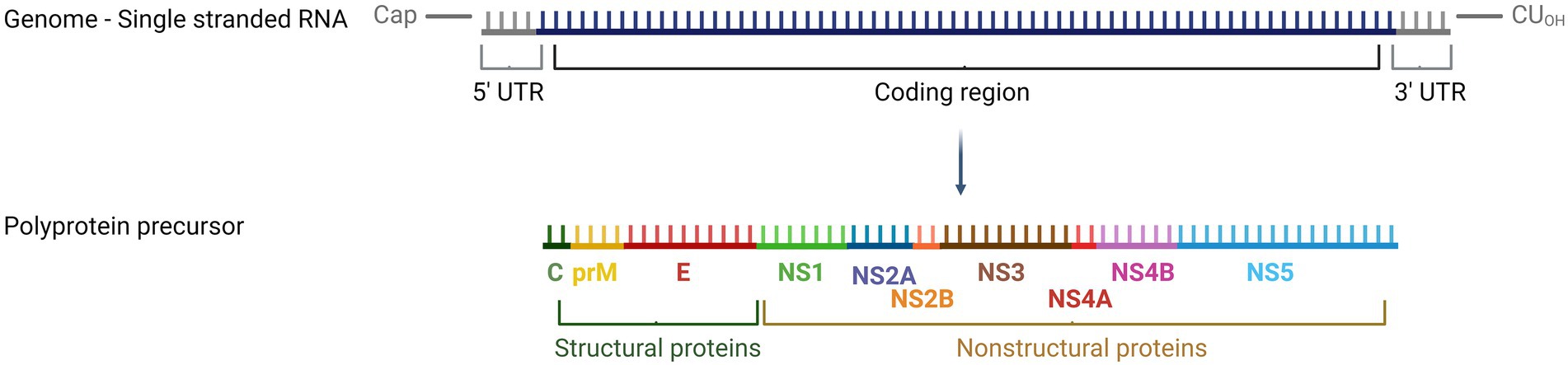
Figure 1. Schematic view of the USUV genome structure. Created with BioRender.com.
With its origin in South Africa (Williams et al., 1964), USUV reached Europe at the turn of the 21st century and was detected in Italy in 1996 retrospectively (Weissenböck et al., 2013) and in Austria in 2001 (Weissenböck et al., 2002). From there onwards, it rapidly spread to neighboring countries including Hungary, Switzerland, Croatia, Serbia, Slovakia, and the Czech Republic (Bakonyi et al., 2007; Steinmetz et al., 2011; Hubálek et al., 2014; Kemenesi et al., 2018; Čabanová et al., 2019). In 2010, USUV was detected for the first time in the southwest of Germany in Culex pipiens mosquitoes from Weinheim, Baden-Württemberg (Jöst et al., 2011). Mass mortality events of Eurasian blackbirds (Turdus merula) followed in 2011, 2016, and 2018 (Becker et al., 2012; Ziegler et al., 2016; Cadar et al., 2017; Lühken et al., 2017; Michel et al., 2019; Zecchin et al., 2021). Concurrently, the geographic range of USUV expanded from the southwest towards the east (Saxony and Saxony-Anhalt) and north (Lower Saxony, Bremen, and Schleswig-Holstein) until in 2018, all national federal states were afflicted (Sieg et al., 2017; Michel et al., 2018, 2019).
USUV isolates cluster into eight distinct lineages (Africa 1, 2, and 3 and Europe 1, 2, 3, 4, and 5), of which many cocirculate throughout Europe (Nikolay, 2015; Engel et al., 2016; Bakonyi et al., 2017; Cadar et al., 2017; Calzolari et al., 2017; Hönig et al., 2019; Zecchin et al., 2021). Until now, five of these lineages have been recorded in Germany: Africa 2, 3, Europe 2, 3, and 5 (Cadar et al., 2015, 2017; Ziegler et al., 2016; Michel et al., 2019). Three are still regularly isolated to date: USUV lineages Europe 3 and Africa 3 are spread throughout Germany (Ziegler et al., 2022), while lineage Europe 2 is only present in the east (Michel et al., 2019; Santos et al., 2021; Ziegler et al., 2022). By contrast, USUV Africa 2 was only found in Germany in the past: in Berlin (2015, 2017, 2018), in Leipzig (2017, 2018), and in Hannover (2018) (Ziegler et al., 2016; Michel et al., 2019; Santos et al., 2021; Störk et al., 2021). USUV Europe 5 was proposed as a novel putative lineage in Bonn in 2014 (Cadar et al., 2015) yet was never isolated again in subsequent years.
Even though partial USUV sequences are available from 2017 to 2020 (Michel et al., 2019; Ziegler et al., 2022), whole-genome sequences of USUV isolates from birds in Germany are limited (Becker et al., 2012; Ziegler et al., 2016; Störk et al., 2021). This makes it difficult to assess geographic and temporal dynamics of USUV in Germany. Whole-genome sequencing (WGS) can also be used to track cases or clusters in outbreak investigations. To ease the implementation of future phylogeographic and phylodynamic USUV studies, we aimed to compare possible sequencing platforms for USUV. In this study, we are comparing traditional low-throughput Sanger sequencing with innovative next-generation (second- and third-generation (SGS and TGS)) high-throughput sequencing technologies. Existing whole-genome sequences in the past were generated using SGS based on Illumina (Illumina, San Diego, CA, United States of America) (Ziegler et al., 2016) or Roche 454 Genome Sequencer FLX system instrument (Roche, Mannheim, Germany) (Becker et al., 2012). In this study, we therefore used TGS (long-read), amplicon and direct DNA sequencing via MinION by oxford Nanopore-technology (ONT, Oxford Science Park, the United Kingdom), in order to compare the previously used SGS (short-read), a reversible dye terminator technology via Illumina, to that of TGS.
Liver of a Eurasian blackbird (sample 1), blood coagulum of a great grey owl (Strix nebulosa, sample 2), kidney of another great grey owl (sample 3), and cell-culture supernatant (brain) of a steamer duck (Tachyeres sp., sample 4), which were found dead in Germany in 2019, were submitted to the national reference laboratory at the Friedrich-Loeffler-Institut (FLI) (Table 1). Viral RNA of samples 1–3 was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. RNA from cell-culture supernatant (sample 4) was isolated using the QIAamp Viral RNA Mini Kit (Qiagen) following the manufacturer’s instructions. We selected samples that covered the three most common lineages in Germany (Europe 2, Europe 3, and Africa 3), that were collected from different federal states, and where the RNA was of good quality with high genome copy numbers per μl. USUV infection was detected in all four birds using a published real-time quantitative polymerase chain reaction (RT-qPCR) assay (Jöst et al., 2011) with cycle threshold (Ct) values between 12.22 and 15.11, corresponding to genome copy numbers of 8.31 × 108 and 1.36 × 108 per μl, respectively.
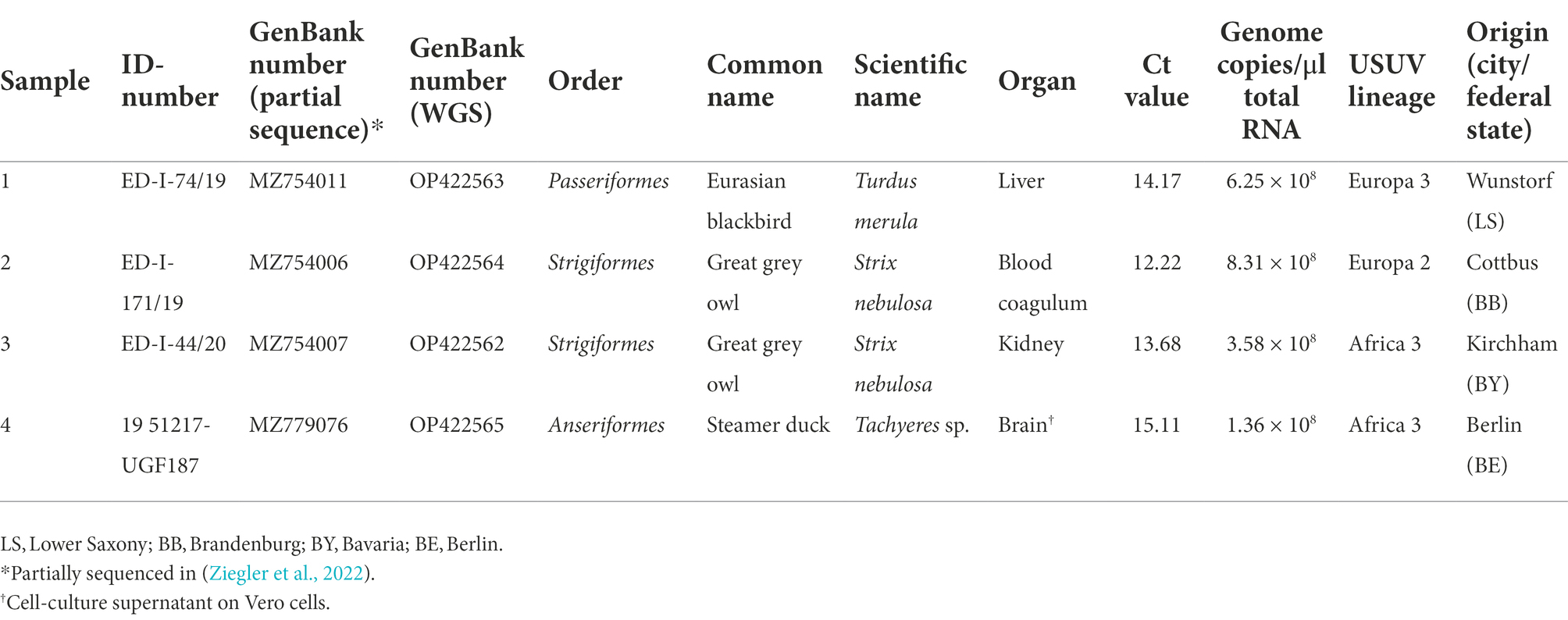
Table 1. Sample information.
For direct cDNA sequencing, synthesis of complementary DNA (cDNA) was performed via the multiplex PCR as previously described by Quick et al., 2017 using the SuperScript IV First-Strand Synthesis System (Cat. no. 18091050; Invitrogen by Thermo Fisher Scientific, Darmstadt, Germany) and random primers (Invitrogen) for reverse transcription (Table 2). After cDNA synthesis, the Rapid Barcoding Sequencing Kit (SQK-RBK004; ONT) and the Flow Cell Priming Kit (EXP-FLP002; ONT) were used according to the manufacturer’s instructions after a purification step with the Agencourt AMPure XP beads (Agencourt, Beckman-Coulter, United States) (Table 2).

Table 2. List of kits needed for direct DNA sequencing (MinION direct) and amplicon sequencing (MinION amplicon).
For amplicon sequencing, a conventional whole-genome PCR for USUV was performed as previously described (Oude Munnink et al., 2019). For this purpose, 32 primer pairs were used in two different reactions that generate overlapping amplicons with a length of 500 base pairs (bp) using the AccuPrime Taq DNA Polymerase High Fidelity (Cat. no. 12346-086; Invitrogen). For the quantification of the purified PCR products, NanoDrop (NanoDrop 2000c Spectrophotometer; Thermo Fisher Scientific) measurement was performed. The presence of amplicons of the correct length was determined by gel electrophoresis (1.5% agarose gel). To remove possible chemical contaminations, which could affect library preparation efficiency and sequencing quality, a purification step with Agencourt AMPure XP beads (Agencourt) was performed. To prepare the end-prep mix and barcoding/ligation master mixes, NEBNext Ultra II End Repair/dA-Tailing Module, NEBNext Ultra II Ligation Module, and NEBNext Quick Ligation Module (New England Biolabs, Ipswich, United States) were used in a separate hood for the preparation of master mixes to avoid cross-contaminations. Using the 1 D Native barcoding genomic DNA Kit (with EXP-NBD104 and SQK-LSK109; ONT) and the Flow Cell Priming Kit (EXP-FLP002; ONT) MinION sequencing was performed according to the manufacturer’s instructions. Spot-ON flow cells (R9.4.1; ONT) were used in a MinION MK1c instrument (ONT) for both approaches. Typically, 8 h of sequencing was sufficient to generate enough data for 6 samples multiplexed on one flow cell.
Viral nucleic acid was directly sent on dry ice to Eurofins Genomics Europe Sequencing GmbH, Ebersberg, Germany, and was sequenced via a special service for RNA-virus Sequencing (INVIEW virus sequencing, Eurofins Genomics Europe Sequencing GmbH). Sequencing was performed using proprietary methods of Eurofins Genomics Europe Sequencing GmbH and the Illumina NovaSeq 6,000 platform (2 × 150 Sequence mode). Library preparation was performed with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) and quality measurement of the mRNA was completed using the Fragment analyzer (ABI DNA analyzer; Applied Biosystems by Thermo Fischer Scientific).
For Sanger sequencing, USUV-specific oligonucleotide primers (3 primer pairs) were chosen to amplify a partial segment of the envelope-coding gene (length of 1,066 nt), according to Eiden et al. (2018) and Ziegler et al. (2022). For the amplification, a SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen) and the multi-block PCR thermal cycler Biometra TRIO (Analytik Jena GmbH, Jena, Germany) were used. A purification was performed using gel electrophoresis (1.5% agarose gel). For visualization with blue light, the gel was stained with the SYBR Safe DNA gel stain (Invitrogen). DNA bands were cut out and purified with the Wizard SV Gel and PCR Clean-Up System (Promega, Walldorf, Germany). Samples were sequenced via the TubeSeq service of Eurofins (Eurofins Genomics Europe Sequencing GmbH). The Sanger partial sequences were already published in Ziegler et al., 2022, yet for comparison purposes, various sequencing methods and results were included in this study.
Raw read quality was assessed using FastQC (Sena Brandine and Smith, 2019) and residual adapter sequences were trimmed using Cutadapt v4.1 (Martin, 2011). Trimmed reads were mapped against the USUV reference genome (GenBank accession number: NC_006551.1; Moureau et al., 2015) using Bowtie2 v2.4.5 (Langmead and Salzberg, 2012). The amplicon coverage was normalized to 50 using BBNorm (Bushnell, 2014) after which a de novo assembly was performed using SPAdes (Bankevich et al., 2012). Raw, quality-controlled reads were mapped back against the obtained consensus genome using Geneious Prime 2021.0.1 (Biomatters Ltd., Auckland, New Zealand).
Fast5 raw data reads were demultiplexed using Guppy v4.5.4 (Wick et al., 2019) and Porechop v0.2.4.1 Primers were trimmed and reads were quality controlled to a minimal length of 200 bp and a median Phred score of 7 using QUASR (Watson et al., 2013). First, a reference-based alignment against the selected USUV reference genomes v23 (Goodacre et al., 2018) was performed in KMA (k-mer alignment) (Clausen et al., 2018). The consensus genome was extracted and compared to the non-redundant database using Blastn (Altschul et al., 1990). Subsequently, the closest relative sequence was selected and used for a second reference-based alignment using the quality-controlled reads in Minimap2 (Li, 2018). Consensus sequences were visualized with Geneious Prime 2021.0.1 (Biomatters Ltd.).
The Muscle algorithm was used to align the sequences (Edgar, 2004). The best model of nucleotide substitutions (GTR + I + G4) was selected using jModeltest v.2 (Darriba et al., 2012), and maximum likelihood trees were reconstructed using PAUP* v.4 (Swofford, 2003). Reliability of the obtained tree topologies was performed by bootstrap testing (1,000 replicates), and finalized trees were reconstructed with FigTree v.1.4.3 (Rambaut, 2012).
In this study, we compared the performance of Nanopore and Illumina sequencing platforms, using three different methods: direct DNA sequencing and amplicon sequencing were performed with the MinION device and RNA sequencing was done with Illumina. For MinION direct (Nanopore) and massive parallel sequencing (Illumina), no amplicon-enrichment steps were used. For Illumina, the average and maximum USUV target sequence read lengths were as expected short with 148 and 246 nt, respectively. For MinION direct and amplicon, the USUV target sequence read lengths were, on average 282 nt and 578 nt, respectively, and the maximum lengths were 631 nt and 1,072 nt, respectively. Even though, the average read lengths of the USUV target sequences were therefore longer for MinION amplicon (578 bp vs. 282 bp), this was not the case for the non-target sequences, where MinION direct produced longer read lengths compared to MinION amplicon (≈1,750 bp vs. 500 bp). Total
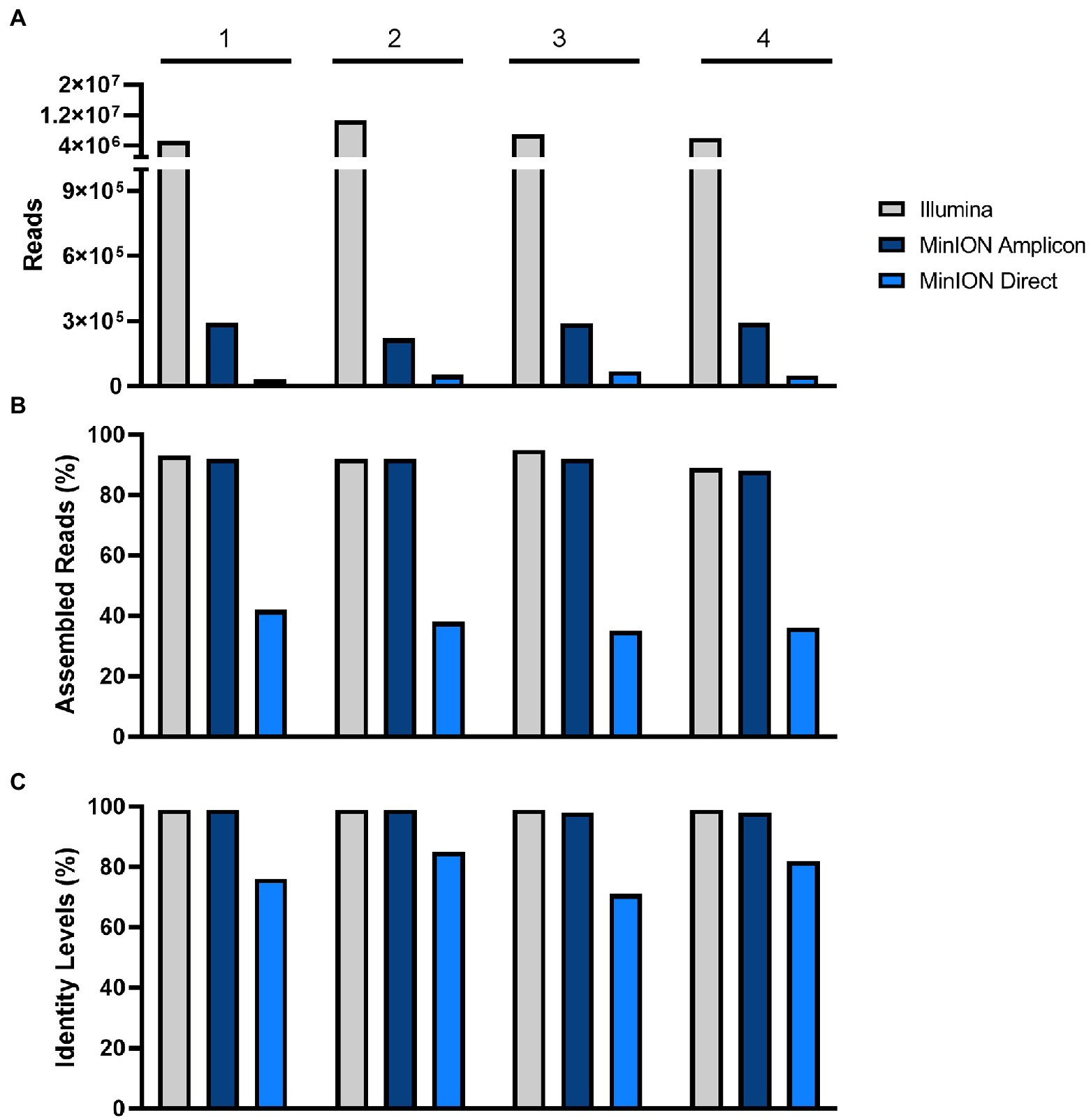
Figure 2. Comparison of (A) total read counts, (B) assembled reads (%), and (C) identity levels (%). in Illumina, MinION amplicon, and MinION direct in samples 1–4.
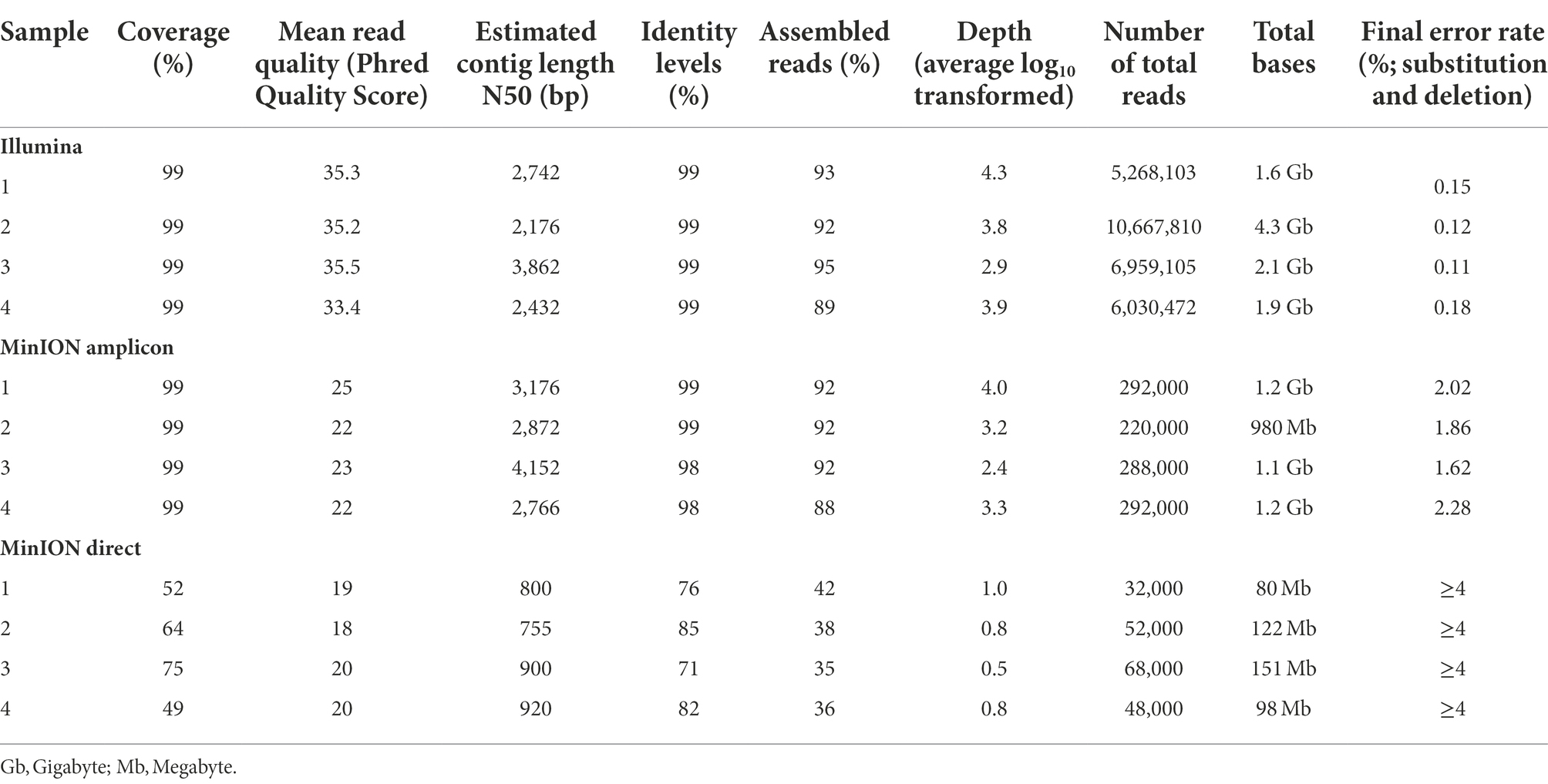
Table 3. Comparison of the most important whole-genome sequencing quality parameters in Illumina, MinION amplicon, and MinION direct.
MinION amplicon sequencing and RNA sequencing with Illumina had a higher efficiency in comparison to the results from the MinION direct DNA sequencing protocol. The data of the MinION amplicon and Illumina sequencing indicate that more specific reads can be obtained with these methods (Table 3; Figure 2). Nonetheless, a minimal depth (100.5–101 reads) and genome coverage (49–75%) were obtained using MinION direct DNA sequencing (Table 3; Figure 3). With the exception of direct DNA sequencing, genome assembly was successfully completed for all four samples using various sequencing platforms: MinION amplicon and Illumina (Table 3). Identities of more than 98% with consensus sequences based on the reference strain were achieved for all four genomes (Table 3; Figure 2). We measured the error rates of the various sequencing platforms by aligning the generated reads to the appropriate reference sequences. Subsequently, the nanopore assemblies were compared to the data generated by Illumina, which was used to assemble the USUV virus genomes. The error rate metrics from Illumina sequencing were better than those from MinION amplicon. The error rates were estimated at 0.15, 0.12, 0.11, and 0.18% for Illumina and 2.02, 1.86, 1.62, and 2.28% for MinION amplicon for samples 1–4, respectively.
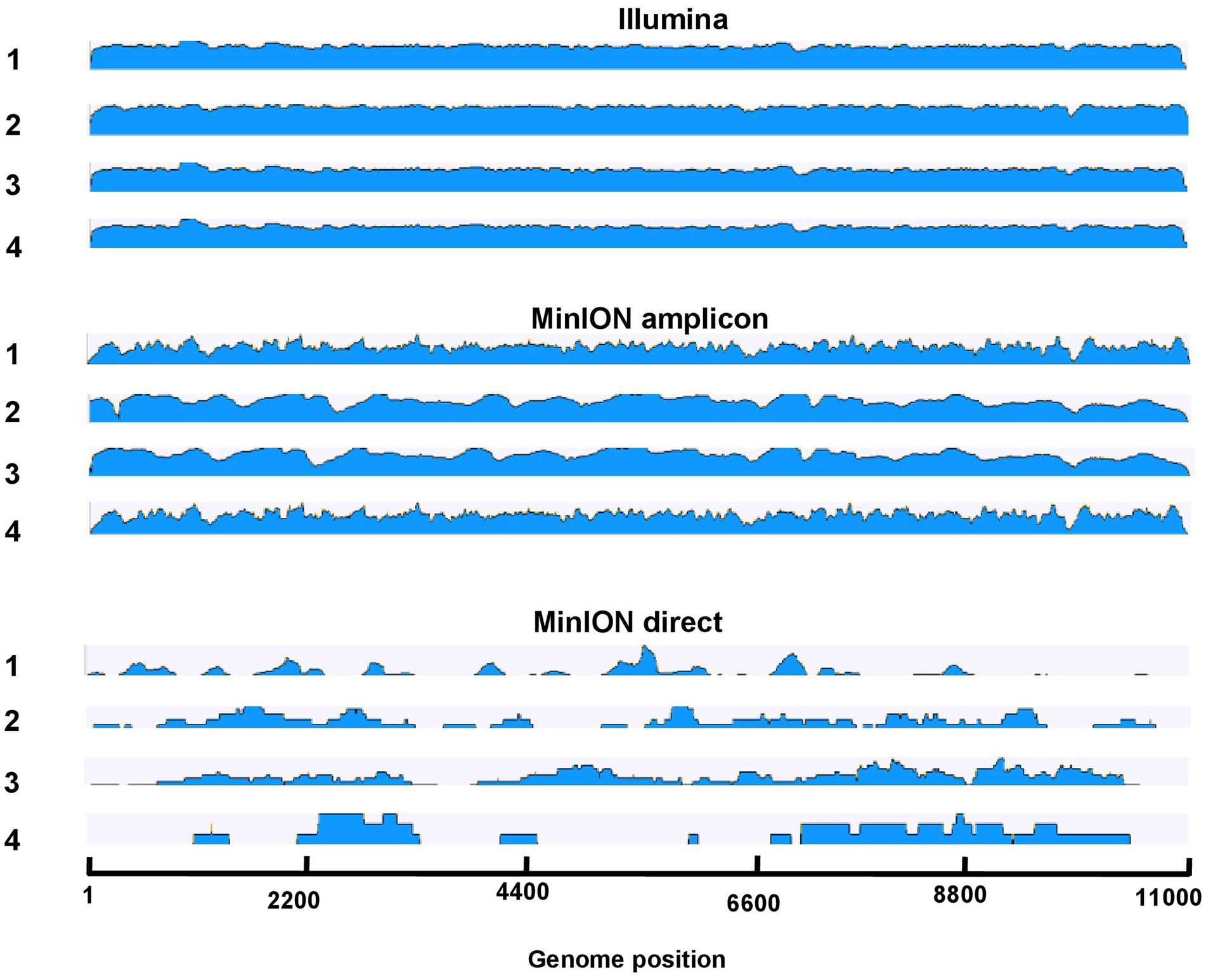
Figure 3. Overview of read coverage of sequenced genomes in Illumina, MinION amplicon, and MinION direct in samples 1–4. The scale represents the log transformed coverage depth (min: 101 and max: 106).
In comparison to the other two sequencing approaches tested, only an incomplete genome coverage including many gaps in the sequences was obtained from MinION direct DNA sequencing. The best quality and quantity of sequencing results were achieved by Illumina sequencing (Table 3). For each sample, Illumina produced on average more reads (total reads: ≈7.2 million reads; USUV reads: ≈6.2 million reads) with more depth (103.7 reads). The percentage of USUV target reads generated with the Illumina platform for the four samples (1–4) was 79, 91, 87, and 85%, respectively. On the other hand, MinION direct DNA sequencing turned out to be the cheapest alternative compared to other SGS and TGS protocols (Tables 2 and 4). As commonly implemented in virus diagnostics, partial sequencing in this study sufficed in identifying the correct USUV lineage of a sample (Ziegler et al., 2022). This can be derived from the three identical phylogenetic trees generated from the various sequencing techniques (one shown as exemplary in Figure 4).
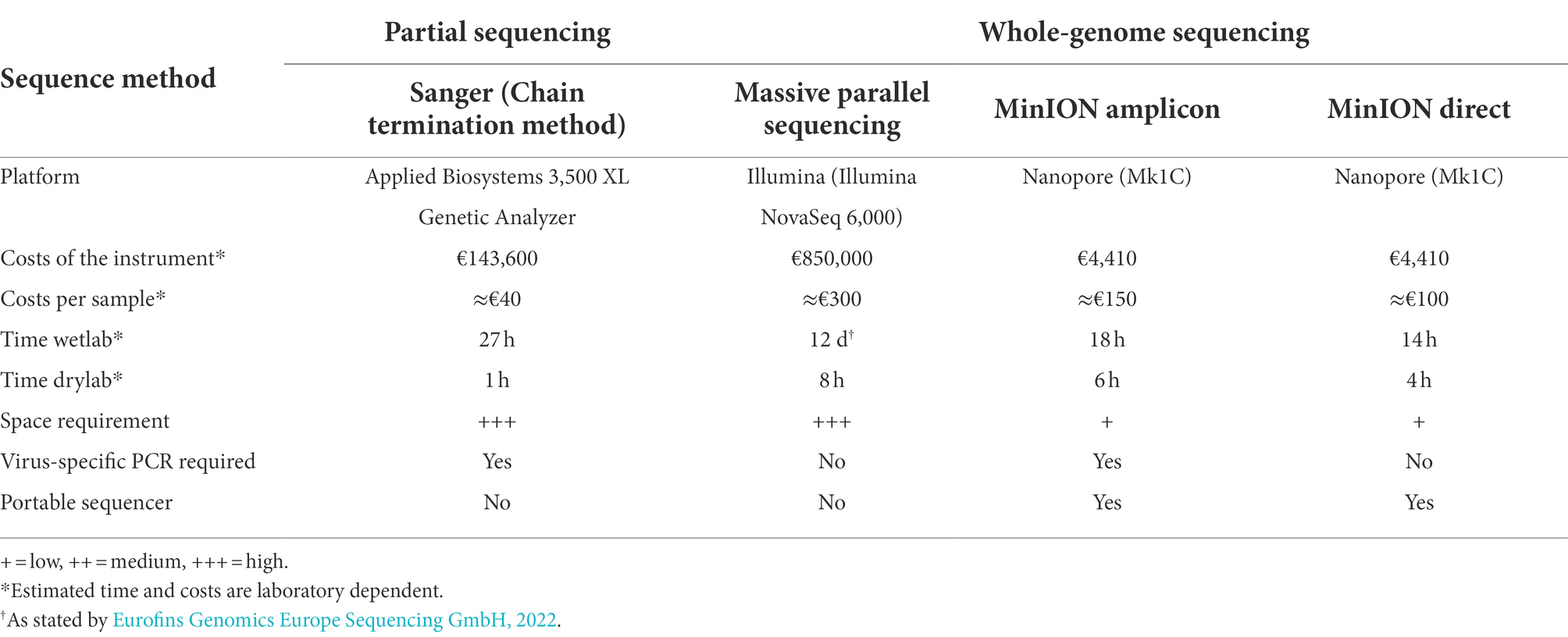
Table 4. Comparison of the most important genome sequencing variables in Sanger (partial genome sequencing), and Illumina, MinION amplicon, and MinION direct (whole-genome sequencing).
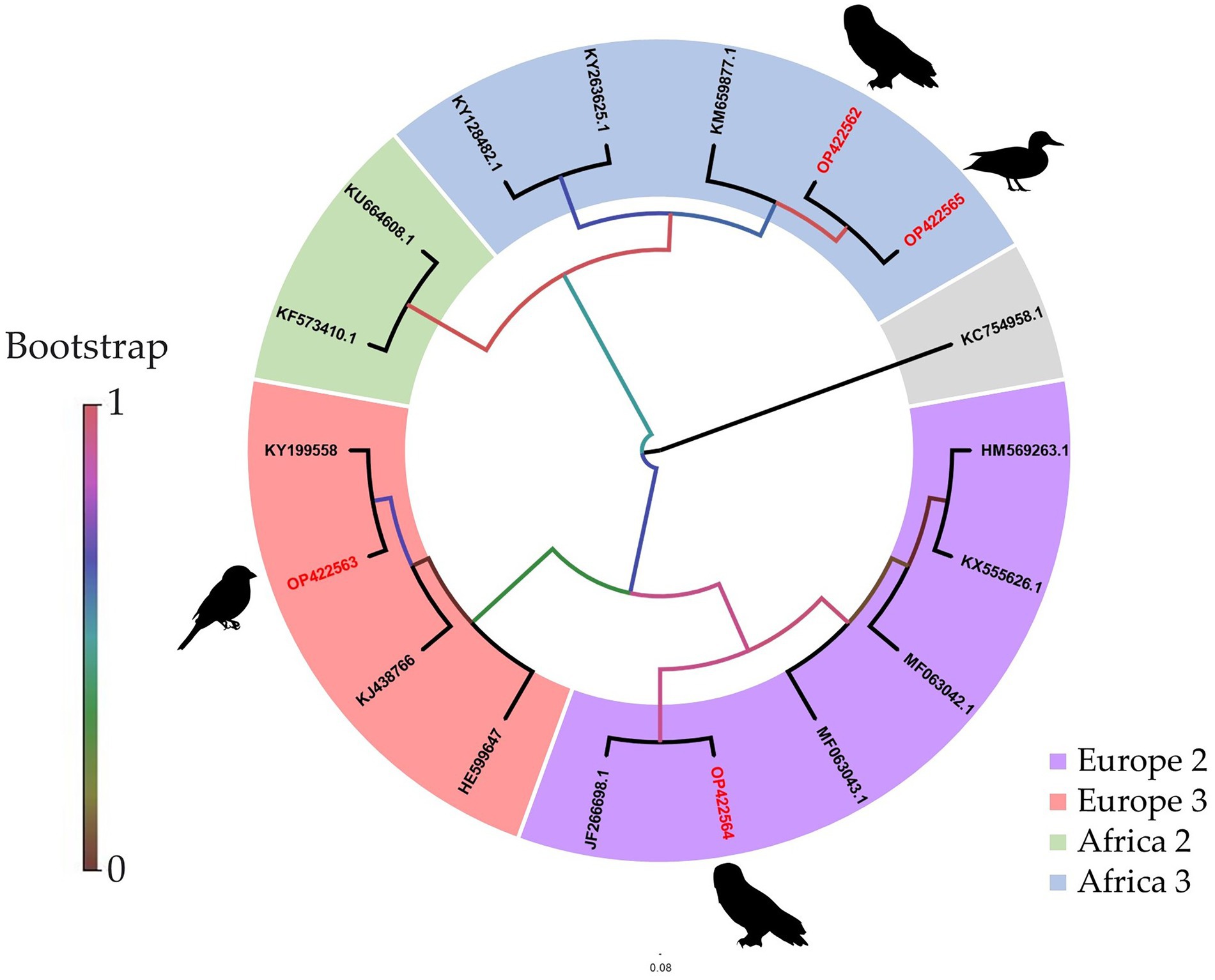
Figure 4. Phylogenetic analysis of USUV whole-genome strains detected in birds from Germany. The sequences from Germany generated in this study via MinION amplicon are highlighted in red. Sequences are labeled by codes containing the GenBank accession number. The phylogenetic trees were identical independent of the platform used (Sanger, Illumina, or MinION amplicon) and therefore only one is illustrated here.
In recent years, WGS has become a reliable alternative and complement to “traditional” sequencing techniques. As a result, we assessed the efficiency of WGS benchtop equipment as a tool for real-time genomics in rapid-response diagnostics. Different sequencing approaches were also evaluated based on the time to result, the costs of the sequencing instrument, the costs per sample sequenced, the specificity, and the suitability for sensitive WGS (Table 4). The MinION direct DNA sequencing results demonstrated that the entire genome could not be covered using this method. Therefore, comparison to Illumina and amplicon MinION sequencing is pointless. Illumina and amplicon MinION sequencing require the same benchtop laboratory tools for library preparation (Figure 5), although the MinION is more efficient and versatile in terms of run time (Table 4). It was possible to achieve a sequence consensus accuracy of 90% within a few hours of the sequencing run, as opposed to an incompressible run time of 20 h for the Illumina system. While the Illumina system is stand-alone, at least during the initial stages of bioinformatics, such as base calling and demultiplexing, MinION requires a strong laptop computer. Even though ONT has recently increased the sequencing accuracy of the MinION (currently, Nanoporetech has developed Bonito, A PyTorch Basecaller for Oxford Nanopore Reads with higher accuracy), raw read error rates remain higher than those of Illumina (around 0.2%), requiring a greater read depth to produce a reliable consensus sequence.
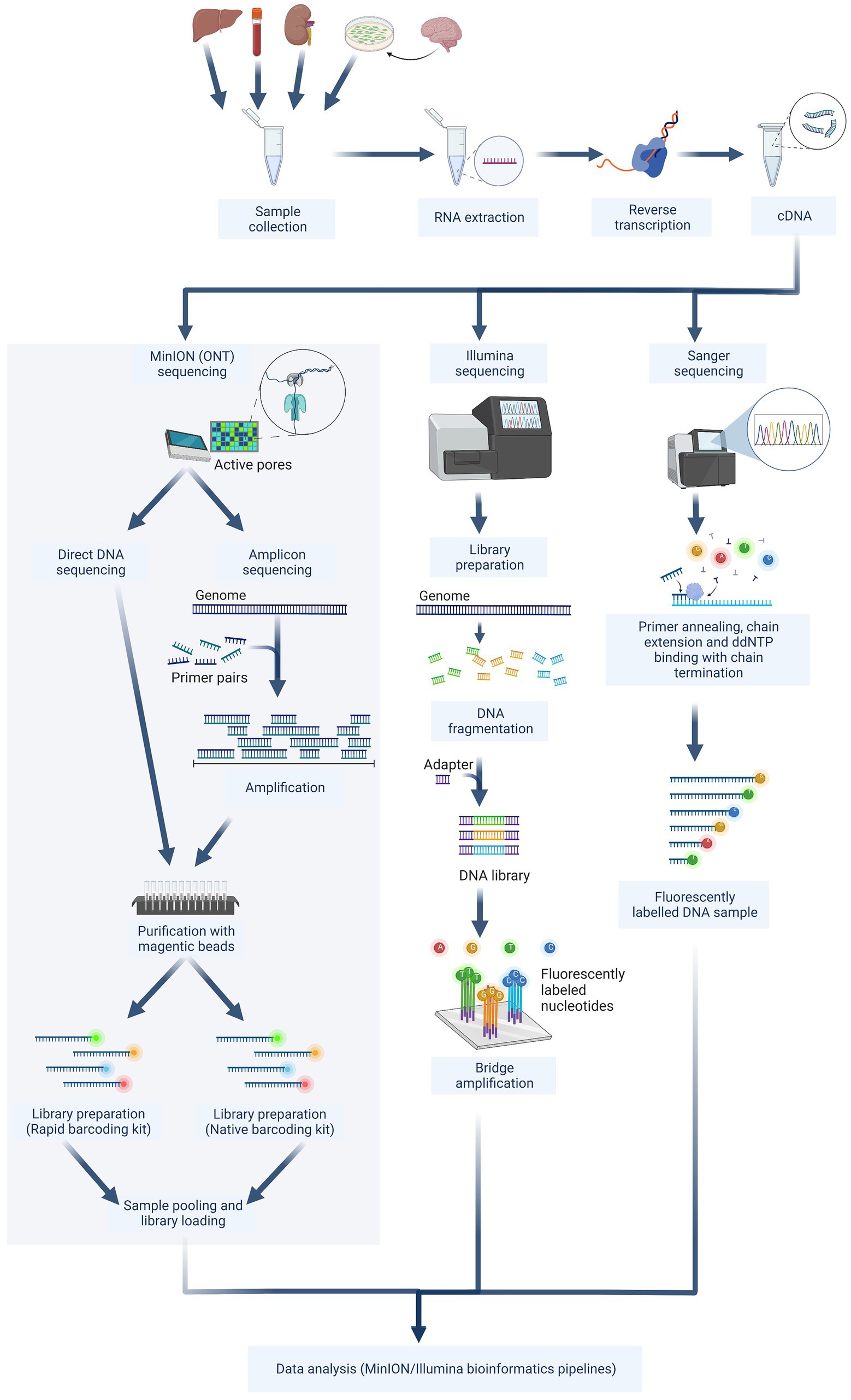
Figure 5. The next-generation sequencing workflow contains three sequencing approaches. The schematic figure was drawn with Biorender.com. ddNTP, Dideoxyadenosine triphosphate.
To date, USUV epidemiological surveillance programs have focused primarily on the molecular (RT-qPCR) and serological detection (virus neutralization tests and enzyme-linked immunosorbent assays) of USUV-specific genome and antibodies, respectively. Passive and active surveillance efforts of wild birds and captive zoological birds are regularly performed, among others in Austria (Bakonyi et al., 2007; Chvala et al., 2007; Meister et al., 2008; Rubel et al., 2008), Belgium (Rouffaer et al., 2018; Benzarti et al., 2020), France (Vittecoq et al., 2013; Roesch et al., 2019; Constant et al., 2020), Germany (Ziegler et al., 2015, 2022; Michel et al., 2018, 2019), Hungary (Bakonyi et al., 2007; Weidinger et al., 2020), Italy (Manarolla et al., 2010; Savini et al., 2011; Tamba et al., 2011; Giglia et al., 2021; Lauriano et al., 2021; Scaramozzino et al., 2021; Zecchin et al., 2021; Mancuso et al., 2022), the Netherlands (Lim et al., 2018), Spain (Jurado-Tarifa et al., 2016; Marzal et al., 2022), and the United Kingdom (Buckley et al., 2003; Horton et al., 2013; Folly et al., 2020). Nonetheless, a genomic surveillance of USUV is rare, with a limited number of available full-genome sequences. The majority of these derived from initial USUV detections in European countries and resultant surveillance efforts, including Italy (Savini et al., 2011; Calzolari et al., 2013; Gaibani et al., 2013; Zecchin et al., 2021), Germany (Jöst et al., 2011; Becker et al., 2012; Cadar et al., 2017; Santos et al., 2021), and the Netherlands (Cadar et al., 2017; Oude Munnink et al., 2020b). Individual sequences were isolated in Austria (Bakonyi et al., 2004, 2017; Chvala et al., 2007; Weidinger et al., 2020), Belgium (Garigliany et al., 2014, 2017; Benzarti et al., 2020), France (Cadar et al., 2017), Hungary (Bakonyi et al., 2007), and Spain (Vázquez et al., 2011; Höfle et al., 2013; Bakonyi et al., 2014). Outside of Europe, USUV has been fully sequenced from human patients, rodents, and vectors (Culex spp.) from the Central African Republic and Senegal, Africa (Bakonyi et al., 2004; Nikolay et al., 2013; Diagne et al., 2019). Similarly, reference strains are accessible from mosquitoes from Israel (Culex spp. and Aedes albopictus; Mannasse et al., 2017). The aim of this study was to test MinION Nanopore sequencing, promoted as a cost- and time-effective sequencing technique, for its user-friendly application in ongoing phylogenetic analyses and to compare the results to those obtained by first- and second-generation sequencing platforms.
NGS techniques, including MinION Nanopore and Illumina platforms, allow the simultaneous deep sequencing of millions of base pairs from multiple samples, while Sanger et al. (1977) can only sequence one DNA fragment at a time. Sanger is still the “gold-standard” in screening virus variants (i.e., sequencing single amplicon targets), especially when screening low sample numbers. As described in a previous study (Ziegler et al., 2022), Sanger sequencing consistently unraveled the USUV lineage affiliations of the tested samples. By contrast, NGS whole-genome sequencing platforms are ideal when a higher target sequence size is required. They can generate a more comprehensive sequencing dataset with greater depth and are, therefore, more sensitive in detecting low-frequency variants (<1%) as well as discovering novel gene variants in combination with a higher number of low-abundance samples (low input quantity and quality; Shendure et al., 2017; Young and Gillung, 2020). Through mass parallelization of sequencing reactions, NGS decreased the necessary costs and turnaround time per sample and revolutionized genomics (Margulies et al., 2005). It opened the door to an array of approaches including viral evolution, metagenomics, and transcriptomics in the study of quasispecies (Radford et al., 2012; Quer et al., 2022). These are genetically closely related viruses that can exist concurrently in a susceptible vector or host due to the high mutation rate of RNA viruses. This has been described for numerous flaviviruses, such as Zika virus (van Boheemen et al., 2017) and West Nile virus (Dridi et al., 2015). Understanding the generation and composition of these quasispecies can give insights into virus adaptation and immune escape mechanisms.
TGS stands for single-molecule sequencing as well as real-time sequencing with the possibility of omitting the requirement for DNA amplification (Heather and Chain, 2016). When directly comparing SGS, via Illumina or Ion Torrent (Thermo Fisher Scientific), with TGS, via for example SMRT (Single molecule real time; Pacific Biosciences, United States) or MinION (ONT), the main difference is the length of the reads. An advantage of SGS is the high throughput with fairly low error rates (≈1–2.4%) compared to TGS (≈10–15%). The disadvantage is linked to the shorter read lengths (75–600 nt/read) compared to TGS (60 K–2 M bp), which can limit the detection of mutations (deletions or insertions) as well as the characterization of repetitive genomic regions (Quer et al., 2022). In this study, however, both the SGS (Illumina) and the TGS techniques via amplicons (MinION; excluding direct sequencing) produced similar results with high identity levels of 98–99%. Not surprisingly, the only difference being that Illumina produced significantly more reads than MinION with an average of 7,231,373 compared to 273,000 reads, respectively. This difference in read counts and consequently also in depth could be relevant when evaluating mutation sites, primarily those in highly variable regions. It can be concluded that both platforms are equally suitable for genotype classification. However, it should not be disregarded that due to the novelty of Nanopore sequencing with its quick turnaround, there is a flood of continuously tailored flow cells, library preparation kits, protocols, and analytical software on the market, making a standardization of results as for Illumina more difficult (Stefan et al., 2022).
For the Nanopore platform via MinION an amplicon-based sequencing technique was used as well as a newly developed direct DNA sequencing method. Studies directly comparing these two strategies are still scarce, even though both of their applications were tested for example for Porcine Reproductive and Respiratory Syndrome Virus (PPRSV) (Tan et al., 2019) and the mycovirus, CHV-1 (Leigh et al., 2020). Amplicon-based sequencing using specific USUV primers has the advantage that lower viral titers can be successfully sequenced than in a non-selective approach including direct/SISPA (Sequence-independent, single-primer amplification) sequencing (Reyes and Kim, 1991). One of the limitations, however, is that the overlapping primer pairs need to be regularly revised to cover all available USUV sequences to try and reduce the possibility of missing out on certain genomic variations (Cotten et al., 2014; Cadar et al., 2017; Oude Munnink et al., 2020a,b). Aside from optimizing variant detection, primer concentrations need to be tailored to allow an even amplicon balance and a good coverage (Gohl et al., 2020). In this study, the primers used had already been published by Oude Munnink et al., 2019 and were mixed into two separated mixes to avoid overlapping fragments. After the analysis of USUV sequences via MinION amplicon (Figure 5), results indicated that the coverage of specific genome positions needs to be improved (for example positions between 1 and 1,000 bp, 1,800 and 2,500 bp, and 9,500 and 10,500 bp). Accordingly, the used primers should still be fine-tuned for future studies, as certain primer pairs produced more coverage than others (Santos et al., 2021).
An alternative to amplicon-based sequencing would be a random amplification approach via direct DNA sequencing kits or SISPA, as often used for metagenomic studies. These methods have an even shorter turnaround time, an easily deployable protocol, and lack systematic sequencing bias, therefore reducing the need for regular reappraisal of the protocol. Metagenomics allow the detection of new as well as known highly diverse viral agents without prior clinical knowledge (Kafetzopoulou et al., 2018). In this study, we tested direct sequencing of USUV cDNA but were not able to produce a sufficient dataset. It resulted in a smaller data set than amplicon-based sequencing, with lower read numbers (32,000 compared to 22,000 reads), quality of reads (18 compared to 25 Phred Quality Score), quantity of assembled reads (36% compared to 92%), and identity levels (71% compared to 92%). Due to the very short contig lengths (<1,000 bp) and the numerous gaps in the sequence, a poor coverage was observed and it was not possible to generate a consensus sequence based on the direct approach. Base calling errors observed as substitutions, insertions/deletions (indels), and ambiguous bases were identified in the generated data. These errors were more frequent in samples with lower total or target read counts. Allowing a longer sequencing time of the MinION device did not increase data yield and quality. It appears that the sequencing of cDNA without further target-amplification steps is less efficient at producing the required coverage of the region of interest, thereby requiring a higher DNA input. Consequently, in our study, a success was more guaranteed when enriching the input DNA with PCR amplification using target-specific primers. Skipping prior enrichment- and PCR-steps, this technique (cDNA + MinION rapid barcoding kit) is optimized for speed (>1 d) and simplicity and not for the achievement of high throughput. Such untargeted methods are not optimal for large-scale surveillance programs with varying sample quality as they have a lower detection limit requiring higher clinical titers (Faria et al., 2017; Quick et al., 2017). Therefore, in this study, direct sequencing appeared to be more error-prone, and for future phylogenetic studies with known pathogens like USUV, focus on targeted amplicon-based sequencing efforts is recommended.
Future improvement strategies aside from refining the primer pairs/mixes could include testing which multiplexing strategy produces the best data yield and data evenness and whether the incorporation of a washing step of the flow cell can enable the reuse of flow cells, to reduce the sequencing cost per sample. Furthermore, no difference in quality could be observed between the full-genome from cell-culture supernatant and that from organ samples, making an enrichment via culture isolation unnecessary in the presence of high viral loads (Houldcroft et al., 2017). Future studies can, therefore, use a broad panel of sample types (e.g., RNA from blood and tissue samples). Similarly, they can also include samples with higher Ct values (≥30) in the diagnostic RT-qPCR. As already described by other studies, it could also be tested whether the prior Agencourt AMPure XP bead (Agencourt) clean-up step of the unbarcoded amplicons could be omitted (Freed and Silander, 2021).
In this work, we compare four sequencing approaches using USUV and discuss their performance in terms of sensitivity and WGS comprehensiveness. In conclusion, Nanopore technologies including MinION amplicon have boosted WGS on a global scale. Due to the simplicity and portability of the sequencer, sequencing can now be deployed outside of core services, as in the field, as shown for Ebola (Hayden, 2015). Furthermore, data can be obtained at a much shorter turnaround (real-time sequencing) allowing a quick and robust analysis of samples during epidemics or ongoing surveillance efforts. Even though the yield quality is not as high as after employing SGS, it suffices to monitor the emergence and spread of viruses in the frame of phylogeographic studies. Keeping in mind that the platform best suited for a project not only depends on the research question to be answered but also on the scale of the project, the allocated time frame, the available budget, and the available amenities and know-how. Even a combination of approaches may be essential to acquire the most accurate genome assemblies of novel pathogens or highly rare variants (Neal-McKinney et al., 2021). This work provides a deep insight into the characteristics of each method and helps to ease decision-making.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, OP422563; https://www.ncbi.nlm.nih.gov/genbank/, OP422564; https://www.ncbi.nlm.nih.gov/genbank/, OP422562; https://www.ncbi.nlm.nih.gov/genbank/, OP422565.
CH, FB, BS, MG, and UZ: conceptualization. CH, FB, FS, AS, and BS: methodology. CH, FB, and BS: data curation, software, formal analysis, writing—original draft preparation, and visualization. CH, FB, BS, FS, AS, UZ, and MG: validation and writing—review and editing. CH and FB: investigation. UZ and MG: resources, project administration, and funding acquisition. BS, UZ, and MG: supervision. All authors contributed to the article and approved the submitted version.
This work was financially supported by the German Federal Ministry of Food and Agriculture (BMEL) through the Federal Office for Agriculture and Food (BLE grant number 2819113919), by the German Ministry of Education and Research (BMBF grant number 01KI2026D) as well as in the frame of the German Centre for Infection Research (BMBF-DZIF grant TTU EI 01.804_00).
We would like to thank Monika Rinder (Clinic for Birds, Small Mammals, Reptiles, and Ornamental Fish, Centre for Clinical Veterinary Medicine, Ludwig Maximilians University Munich), Sabine Bock (Berlin-Brandenburg State Laboratory, Frankfurt (Oder)), Karolin Schütte (Wildlife Rescue and Conservation Center, Sachsenhagen), and the staff of the wildlife park in Cottbus for providing the samples from the four USUV positive birds, which were collected in the nationwide wild bird surveillance network for zoonotic arthropod-borne viruses. We also thank Katja Wittig and Cornelia Steffen (both from FLI) for their excellent technical assistance. We are grateful to Anne Günther, Pauline Santos, and Jacqueline King (all from FLI) for their theoretical support concerning amplicon-based WGS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bakonyi, T., Busquets, N., and Nowotny, N. (2014). Comparison of complete genome sequences of Usutu virus strains detected in Spain, Central Europe, and Africa. Vector Borne Zoonotic Dis. 14, 324–329. doi: 10.1089/vbz.2013.1510
Bakonyi, T., Erdélyi, K., Brunthaler, R., Dán, Á., Weissenböck, H., and Nowotny, N. (2017). Usutu virus, Austria and Hungary, 2010-2016. Emerg. Microbes Infect. 6:e85. doi: 10.1038/emi.2017.72
Bakonyi, T., Erdélyi, K., Ursu, K., Ferenczi, E., Csörgo, T., Lussy, H., et al. (2007). Emergence of Usutu virus in Hungary. J. Clin. Microbiol. 45, 3870–3874. doi: 10.1128/JCM.01390-07
Bakonyi, T., Gould, E. A., Kolodziejek, J., Weissenböck, H., and Nowotny, N. (2004). Complete genome analysis and molecular characterization of Usutu virus that emerged in Austria in 2001: comparison with the south African strain SAAR-1776 and other flaviviruses. Virology 328, 301–310. doi: 10.1016/j.virol.2004.08.005
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Becker, N., Jöst, H., Ziegler, U., Eiden, M., Höper, D., Emmerich, P., et al. (2012). Epizootic emergence of Usutu virus in wild and captive birds in Germany. PLoS One 7:e32604. doi: 10.1371/journal.pone.0032604
Benzarti, E., Sarlet, M., Franssen, M., Cadar, D., Schmidt-Chanasit, J., Rivas, J. F., et al. (2020). Usutu virus epizootic in Belgium in 2017 and 2018: evidence of virus Endemization and ongoing introduction events. Vector Borne Zoonotic Dis. 20, 43–50. doi: 10.1089/vbz.2019.2469
Buckley, A., Dawson, A., Moss, S. R., Hinsley, S. A., Bellamy, P. E., and Gould, E. A. (2003). Serological evidence of West Nile virus, Usutu virus and Sindbis virus infection of birds in the UK. J. Gen. Virol. 84, 2807–2817. doi: 10.1099/vir.0.19341-0
Bushnell, B. (2014). BBnorm: Kmer-based error-correction and normalization tool (from the BBTools package). In SourceForge repository. Available at: https://sourceforge.net/projects/bbmap/ (Accessed July 4, 2022).
Čabanová, V., Šikutová, S., Straková, P., Šebesta, O., Vichová, B., Zubríková, D., et al. (2019). Co-circulation of West Nile and Usutu Flaviviruses in mosquitoes in Slovakia, 2018. Viruses 11:639. doi: 10.3390/v11070639
Cadar, D., Bosch, S., Jöst, H., Börstler, J., Garigliany, M.-M., Becker, N., et al. (2015). Putative lineage of novel African Usutu virus, Central Europe. Emerg. Infect. Dis. 21, 1647–1650. doi: 10.3201/eid2109.142026
Cadar, D., Lühken, R., van der Jeugd, H., Garigliany, M., Ziegler, U., Keller, M., et al. (2017). Widespread activity of multiple lineages of Usutu virus, western Europe, 2016. Euro. Surveill. 22:pii=30452. doi: 10.2807/1560-7917.ES.2017.22.4.30452
Calisher, C. H., and Gould, E. A. (2003). Taxonomy of the virus family Flaviviridae Advances in virus research Elsevier, 1–19.
Calzolari, M., Bonilauri, P., Bellini, R., Albieri, A., Defilippo, F., Tamba, M., et al. (2013). Usutu virus persistence and West Nile virus inactivity in the Emilia-Romagna region (Italy) in 2011. PLoS One 8:e63978. doi: 10.1371/journal.pone.0063978
Calzolari, M., Chiapponi, C., Bonilauri, P., Lelli, D., Baioni, L., Barbieri, I., et al. (2017). Co-circulation of two Usutu virus strains in northern Italy between 2009 and 2014. Infect. Genet. Evol. 51, 255–262. doi: 10.1016/j.meegid.2017.03.022
Chvala, S., Bakonyi, T., Bukovsky, C., Meister, T., Brugger, K., Rubel, F., et al. (2007). Monitoring of Usutu virus activity and spread by using dead bird surveillance in Austria, 2003-2005. Vet. Microbiol. 122, 237–245. doi: 10.1016/j.vetmic.2007.01.029
Clausen, P. T. L. C., Aarestrup, F. M., and Lund, O. (2018). Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 19:307. doi: 10.1186/s12859-018-2336-6
Constant, O., Bollore, K., Clé, M., Barthelemy, J., Foulongne, V., Chenet, B., et al. (2020). Evidence of exposure to USUV and WNV in zoo animals in France. Pathogens 9:1005. doi: 10.3390/pathogens9121005
Cotten, M., Petrova, V., Phan, M. V. T., Rabaa, M. A., Watson, S. J., Ong, S. H., et al. (2014). Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J. Virol. 88, 11056–11069. doi: 10.1128/JVI.01333-14
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Diagne, M. M., Ndione, M. H. D., Di Paola, N., Fall, G., Bedekelabou, A. P., Sembène, P. M., et al. (2019). Usutu virus isolated from rodents in Senegal. Viruses 11:181. doi: 10.3390/v11020181
Dridi, M., Rosseel, T., Orton, R., Johnson, P., Lecollinet, S., Muylkens, B., et al. (2015). Next-generation sequencing shows West Nile virus quasispecies diversification after a single passage in a carrion crow (Corvus corone) in vivo infection model. J. Gen. Virol. 96, 2999–3009. doi: 10.1099/jgv.0.000231
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eiden, M., Gil, P., Ziegler, U., Rakotoarivony, I., Marie, A., Frances, B., et al. (2018). Emergence of two Usutu virus lineages in Culex pipiens mosquitoes in the Camargue, France, 2015. Infect. Genet. Evol. 61, 151–154. doi: 10.1016/j.meegid.2018.03.020
Engel, D., Jöst, H., Wink, M., Börstler, J., Bosch, S., Garigliany, M.-M., et al. (2016). Reconstruction of the evolutionary history and dispersal of Usutu virus, a neglected emerging arbovirus in Europe and Africa. MBio 7, e01938–e01915. doi: 10.1128/mBio.01938-15
Eurofins Genomics Europe Sequencing GmbH (2022). INVIEW Virus Sequencing. Available at: https://eurofinsgenomics.eu/en/next-generation-sequencing/ngs-built-for-you/inview-virus/ [Accessed October 11, 2022].
Faria, N. R., Quick, J., Claro, I. M., Thézé, J., Jesus, J. G.De, Giovanetti, M., et al. (2017). Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546, 406–410. doi: 10.1038/nature22401
Folly, A. J., Lawson, B., Lean, F. Z., McCracken, F., Spiro, S., John, S. K., et al. (2020). Detection of Usutu virus infection in wild birds in the United Kingdom, 2020. Euro Surveill. 25:2001732. doi: 10.2807/1560-7917.ES.2020.25.41.2001732
Freed, N., and Silander, O. (2021). SARS-CoV2 genome sequencing protocol (1,200 bp amplicon “midnight” primer set, using Nanopore rapid kit) version 6: Protocols.Io. [Accessed August 10, 2022].
Gaibani, P., Cavrini, F., Gould, E. A., Rossini, G., Pierro, A., Landini, M. P., et al. (2013). Comparative genomic and phylogenetic analysis of the first Usutu virus isolate from a human patient presenting with neurological symptoms. PLoS One 8:e64761. doi: 10.1371/journal.pone.0064761
Garigliany, M.-M., Linden, A., Gilliau, G., Levy, E., Sarlet, M., Franssen, M., et al. (2017). Usutu virus, Belgium, 2016. Infect. Genet. Evol. 48, 116–119. doi: 10.1016/j.meegid.2016.12.023
Garigliany, M.-M., Marlier, D., Tenner-Racz, K., Eiden, M., Cassart, D., Gandar, F., et al. (2014). Detection of Usutu virus in a bullfinch (Pyrrhula pyrrhula) and a great spotted woodpecker (Dendrocopos major) in north-West Europe. Vet. J. 199, 191–193. doi: 10.1016/j.tvjl.2013.10.017
Giglia, G., Agliani, G., Munnink, B. B. O., Sikkema, R. S., Mandara, M. T., Lepri, E., et al. (2021). Pathology and pathogenesis of Eurasian blackbirds (Turdus merula) naturally infected with Usutu virus. Viruses 13:1481. doi: 10.3390/v13081481
Gohl, D. M., Garbe, J., Grady, P., Daniel, J., Watson, R. H. B., Auch, B., et al. (2020). A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genomics 21:863. doi: 10.1186/s12864-020-07283-6
Goodacre, N., Aljanahi, A., Nandakumar, S., Mikailov, M., and Khan, A. S. (2018). A reference viral database (RVDB) to enhance bioinformatics analysis of high-throughput sequencing for novel virus detection. mSphere 3:e00069-18. doi: 10.1128/mSphereDirect.00069-18
Hayden, E. C. (2015). Pint-sized DNA sequencer impresses first users. Nature 521, 15–16. doi: 10.1038/521015a
Heather, J. M., and Chain, B. (2016). The sequence of sequencers: the history of sequencing DNA. Genomics 107, 1–8. doi: 10.1016/j.ygeno.2015.11.003
Höfle, U., Gamino, V., Mera, I. G. F.De, Mangold, A. J., Ortíz, J.-A., and La Fuente, J.De. (2013). Usutu virus in migratory song thrushes, Spain. Emerg. Infect. Dis. 19, 1173–1175. doi: 10.3201/eid1907.130199
Hönig, V., Palus, M., Kaspar, T., Zemanova, M., Majerova, K., Hofmannova, L., et al. (2019). Multiple lineages of Usutu virus (Flaviviridae, Flavivirus) in blackbirds (Turdus merula) and mosquitoes (Culex pipiens, cx. Modestus) in the Czech Republic (2016-2019). Microorganisms 7:568. doi: 10.3390/microorganisms7110568
Horton, D. L., Lawson, B., Egbetade, A., Jeffries, C., Johnson, N., Cunningham, A. A., et al. (2013). Targeted surveillance for Usutu virus in British birds (2005−2011). Vet. Rec. 172:17. doi: 10.1136/vr.101275
Houldcroft, C. J., Beale, M. A., and Breuer, J. (2017). Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 15, 183–192. doi: 10.1038/nrmicro.2016.182
Hubálek, Z., Rudolf, I., Čapek, M., Bakonyi, T., Betášová, L., and Nowotny, N. (2014). Usutu virus in blackbirds (Turdus merula), Czech Republic, 2011-2012. Transbound. Emerg. Dis. 61, 273–276. doi: 10.1111/tbed.12025
Jöst, H., Bialonski, A., Maus, D., Sambri, V., Eiden, M., Groschup, M. H., et al. (2011). Isolation of usutu virus in Germany. Am. J. Trop. Med. Hyg. 85, 551–553. doi: 10.4269/ajtmh.2011.11-0248
Jurado-Tarifa, E., Napp, S., Lecollinet, S., Arenas, A., Beck, C., Cerdà-Cuéllar, M., et al. (2016). Monitoring of West Nile virus, Usutu virus and Meaban virus in waterfowl used as decoys and wild raptors in southern Spain. Comp. Immunol. Microbiol. Infect. Dis. 49, 58–64. doi: 10.1016/j.cimid.2016.10.001
Kafetzopoulou, L. E., Efthymiadis, K., Lewandowski, K., Crook, A., Carter, D., Osborne, J., et al. (2018). Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro Surveill. 23:1800228. doi: 10.2807/1560-7917.ES.2018.23.50.1800228
Kemenesi, G., Buzás, D., Zana, B., Kurucz, K., Krtinic, B., Kepner, A., et al. (2018). First genetic characterization of Usutu virus from Culex pipiens mosquitoes Serbia, 2014. Infect. Genet. Evol. 63, 58–61. doi: 10.1016/j.meegid.2018.05.012
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lauriano, A., Rossi, A., Galletti, G., Casadei, G., Santi, A., Rubini, S., et al. (2021). West Nile and Usutu viruses’ surveillance in birds of the province of Ferrara, Italy, from 2015 to 2019. Viruses 13:1367. doi: 10.3390/v13071367
Leigh, D. M., Schefer, C., and Cornejo, C. (2020). Determining the suitability of MinION’s direct RNA and DNA amplicon sequencing for viral subtype identification. Viruses 12:801. doi: 10.3390/v12080801
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Lim, S. M., Geervliet, M., Verhagen, J. H., Müskens, G. J. D. M., Majoor, F. A., Osterhaus, A. D. M. E., et al. (2018). Serologic evidence of West Nile virus and Usutu virus infections in Eurasian coots in the Netherlands. Zoonoses Public Health 65, 96–102. doi: 10.1111/zph.12375
Lühken, R., Jöst, H., Cadar, D., Thomas, S. M., Bosch, S., Tannich, E., et al. (2017). Distribution of Usutu virus in Germany and its effect on breeding bird populations. Emerg. Infect. Dis. 23, 1994–2001. doi: 10.3201/eid2312.171257
Manarolla, G., Bakonyi, T., Gallazzi, D., Crosta, L., Weissenböck, H., Dorrestein, G. M., et al. (2010). Usutu virus in wild birds in northern Italy. Vet. Microbiol. 141, 159–163. doi: 10.1016/j.vetmic.2009.07.036
Mancuso, E., Cecere, J. G., Iapaolo, F., Di Gennaro, A., Sacchi, M., Savini, G., et al. (2022). West Nile and Usutu virus introduction via migratory birds: a retrospective analysis in Italy. Viruses 14:416. doi: 10.3390/v14020416
Mannasse, B., Mendelson, E., Orshan, L., Mor, O., Shalom, U., Yeger, T., et al. (2017). Usutu Virus RNA in Mosquitoes, Israel, 2014-2015. Emerg. Infect. Dis. 23, 1699–1702. doi: 10.3201/eid2310.171017
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Marzal, A., Ferraguti, M., Muriel, J., Magallanes, S., Ortiz, J. A., García-Longoria, L., et al. (2022). Circulation of zoonotic flaviviruses in wild passerine birds in Western Spain. Vet. Microbiol. 268:109399. doi: 10.1016/j.vetmic.2022.109399
Meister, T., Lussy, H., Bakonyi, T., Sikutová, S., Rudolf, I., Vogl, W., et al. (2008). Serological evidence of continuing high Usutu virus (Flaviviridae) activity and establishment of herd immunity in wild birds in Austria. Vet. Microbiol. 127, 237–248. doi: 10.1016/j.vetmic.2007.08.023
Michel, F., Fischer, D., Eiden, M., Fast, C., Reuschel, M., Müller, K., et al. (2018). West Nile virus and Usutu virus monitoring of wild birds in Germany. Int. J. Environ. Res. Public Health 15:171. doi: 10.3390/ijerph15010171
Michel, F., Sieg, M., Fischer, D., Keller, M., Eiden, M., Reuschel, M., et al. (2019). Evidence for West Nile virus and Usutu virus infections in wild and resident birds in Germany, 2017 and 2018. Viruses 11. doi: 10.3390/v11070674
Moureau, G., Cook, S., Lemey, P., Nougairede, A., Forrester, N. L., Khasnatinov, M., et al. (2015). New insights into flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS One 10:e0117849. doi: 10.1371/journal.pone.0117849
Neal-McKinney, J. M., Liu, K. C., Lock, C. M., Wu, W.-H., and Hu, J. (2021). Comparison of MiSeq, MinION, and hybrid genome sequencing for analysis of campylobacter jejuni. Sci. Rep. 11:5676. doi: 10.1038/s41598-021-84956-6
Nikolay, B. (2015). A review of West Nile and Usutu virus co-circulation in Europe: how much do transmission cycles overlap? Trans. R. Soc. Trop. Med. Hyg. 109, 609–618. doi: 10.1093/trstmh/trv066
Nikolay, B., Dupressoir, A., Firth, C., Faye, O., Boye, C. S., Diallo, M., et al. (2013). Comparative full length genome sequence analysis of Usutu virus isolates from Africa. Virol. J. 10:217. doi: 10.1186/1743-422X-10-217
Oude Munnink, B. B., Kik, M., Bruijn, N. D.De, Kohl, R., van der Linden, A., Reusken, C. B. E. M., et al. (2019). Towards high quality real-time whole genome sequencing during outbreaks using Usutu virus as example. Infect. Genet. Evol. 73, 49–54. doi: 10.1016/j.meegid.2019.04.015
Oude Munnink, B. B., Münger, E., Nieuwenhuijse, D. F., Kohl, R., van der Linden, A., Schapendonk, C. M. E., et al. (2020a). Genomic monitoring to understand the emergence and spread of Usutu virus in the Netherlands, 2016-2018. Sci. Rep. 10:2798. doi: 10.1038/s41598-020-59692-y
Oude Munnink, B. B., Nieuwenhuijse, D. F., Sikkema, R. S., and Koopmans, M. (2020b). Validating whole genome Nanopore sequencing, using Usutu virus as an example. J. Vis. Exp. doi: 10.3791/60906
Quer, J., Colomer-Castell, S., Campos, C., Andrés, C., Piñana, M., Cortese, M. F., et al. (2022). Next-generation sequencing for confronting virus pandemics. Viruses 14:600. doi: 10.3390/v14030600
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi: 10.1038/nprot.2017.066
Radford, A. D., Chapman, D., Dixon, L., Chantrey, J., Darby, A. C., and Hall, N. (2012). Application of next-generation sequencing technologies in virology. J. Gen. Virol. 93, 1853–1868. doi: 10.1099/vir.0.043182-0
Rambaut, A. (2012). FigTree version 1.4.3. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (Accessed August 10, 2022).
Reyes, G. R., and Kim, J. P. (1991). Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol. Cell. Probes 5, 473–481. doi: 10.1016/s0890-8508(05)80020-9
Roesch, F., Fajardo, A., Moratorio, G., and Vignuzzi, M. (2019). Usutu virus: an arbovirus on the rise. Viruses 11:640. doi: 10.3390/v11070640
Rouffaer, L. O., Steensels, M., Verlinden, M., Vervaeke, M., Boonyarittichaikij, R., Martel,, et al. (2018). Usutu virus epizootic and plasmodium coinfection in Eurasian blackbirds (Turdus merula) in Flanders, Belgium. J. Wildl. Dis. 54, 859–862. doi: 10.7589/2017-07-163
Rubel, F., Brugger, K., Hantel, M., Chvala-Mannsberger, S., Bakonyi, T., Weissenböck, H., et al. (2008). Explaining Usutu virus dynamics in Austria: model development and calibration. Prev. Vet. Med. 85, 166–186. doi: 10.1016/j.prevetmed.2008.01.006
Saiz, J.-C., and Blazquez, A.-B. (2017). Usutu virus: current knowledge and future perspectives. VAAT 9, 27–40. doi: 10.2147/VAAT.S123619
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Santos, P. D., Michel, F., Wylezich, C., Höper, D., Keller, M., Holicki, C. M., et al. (2021). Co-infections: simultaneous detections of West Nile virus and Usutu virus in birds from Germany. Transbound. Emerg. Dis. 69, 776–792. doi: 10.1111/tbed.14050
Savini, G., Monaco, F., Terregino, C., Di Gennaro, A., Bano, L., Pinoni, C., et al. (2011). Usutu virus in Italy: an emergence or a silent infection? Vet. Microbiol. 151, 264–274. doi: 10.1016/j.vetmic.2011.03.036
Scaramozzino, P., Carvelli, A., Bruni, G., Cappiello, G., Censi, F., Magliano, A., et al. (2021). West Nile and Usutu viruses co-circulation in Central Italy: outcomes of the 2018 integrated surveillance. Parasit. Vectors 14:243. doi: 10.1186/s13071-021-04736-z
Shendure, J., Balasubramanian, S., Church, G. M., Gilbert, W., Rogers, J., Schloss, J. A., et al. (2017). DNA sequencing at 40: past, present and future. Nature 550, 345–353. doi: 10.1038/nature24286
Sieg, M., Schmidt, V., Ziegler, U., Keller, M., Höper, D., Heenemann, K., et al. (2017). Outbreak and Cocirculation of three different Usutu virus strains in eastern Germany. Vector Borne Zoonotic Dis. 17, 662–664. doi: 10.1089/vbz.2016.2096
Sena Brandine, G.de, and Smith, A. D. (2019). Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res. 8:1874. doi: 10.12688/f1000research.21142.1
Stefan, C. P., Hall, A. T., Graham, A. S., and Minogue, T. D. (2022). Comparison of Illumina and Oxford Nanopore sequencing Technologies for Pathogen Detection from clinical matrices using molecular inversion probes. J. Mol. Diagn. 24, 395–405. doi: 10.1016/j.jmoldx.2021.12.005
Steinmetz, H. W., Bakonyi, T., Weissenböck, H., Hatt, J.-M., Eulenberger, U., Robert, N., et al. (2011). Emergence and establishment of Usutu virus infection in wild and captive avian species in and around Zurich, Switzerland—genomic and pathologic comparison to other central European outbreaks. Vet. Microbiol. 148, 207–212. doi: 10.1016/j.vetmic.2010.09.018
Störk, T., Le Roi, M.De, Haverkamp, A.-K., Jesse, S. T., Peters, M., Fast, C., et al. (2021). Analysis of avian Usutu virus infections in Germany from 2011 to 2018 with focus on dsRNA detection to demonstrate viral infections. Sci. Rep. 11:24191. doi: 10.1038/s41598-021-03638-5
Swofford, D. L. (2003). PAUP*: Phylogenetic analysis using parsimony (* and other methods), v. 4.0. Sunderland, MA: Sinauer Associates.
Tamba, M., Bonilauri, P., Bellini, R., Calzolari, M., Albieri, A., Sambri, V., et al. (2011). Detection of Usutu virus within a West Nile virus surveillance program in northern Italy. Vector Borne Zoonotic Dis. 11, 551–557. doi: 10.1089/vbz.2010.0055
Tan, S., Dvorak, C. M. T., and Murtaugh, M. P. (2019). Rapid, unbiased PRRSV strain detection using MinION direct RNA sequencing and bioinformatics tools. Viruses 11:1132. doi: 10.3390/v11121132
van Boheemen, S., Tas, A., Anvar, S. Y., van Grootveld, R., Albulescu, I. C., Bauer, M. P., et al. (2017). Quasispecies composition and evolution of a typical Zika virus clinical isolate from Suriname. Sci. Rep. 7:2368. doi: 10.1038/s41598-017-02652-w
Vázquez, A., Jimenez-Clavero, M., Franco, L., Donoso-Mantke, O., Sambri, V., Niedrig, M., et al. (2011). Usutu virus: potential risk of human disease in Europe. Euro Surveill. 16. Review
Vittecoq, M., Lecollinet, S., Jourdain, E., Thomas, F., Blanchon, T., Arnal, A., et al. (2013). Recent circulation of West Nile virus and potentially other closely related flaviviruses in southern France. Vector Borne Zoonotic Dis. 13, 610–613. doi: 10.1089/vbz.2012.1166
Watson, S. J., Welkers, M. R. A., Depledge, D. P., Coulter, E., Breuer, J. M., De Jong, M. D., et al. (2013). Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 368:20120205. doi: 10.1098/rstb.2012.0205
Weidinger, P., Kolodziejek, J., Bakonyi, T., Brunthaler, R., Erdélyi, K., Weissenböck, H., et al. (2020). Different dynamics of Usutu virus infections in Austria and Hungary, 2017-2018. Transbound. Emerg. Dis. 67, 298–307. doi: 10.1111/tbed.13351
Weissenböck, H., Bakonyi, T., Rossi, G., Mani, P., and Nowotny, N. (2013). Usutu virus, Italy, 1996. Emerg. Infect. Dis. 19, 274–277. doi: 10.3201/eid1902.121191
Weissenböck, H., Kolodziejek, J., Url, A., Lussy, H., Rebel-Bauder, B., and Nowotny, N. (2002). Emergence of Usutu virus, an African mosquito-borne flavivirus of the Japanese encephalitis virus group, Central Europe. Emerg. Infect. Dis. 8, 652–656. doi: 10.3201/eid0807.020094
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20:129. doi: 10.1186/s13059-019-1727-y
Williams, M. C., Simpson, D. I., Haddow, A. J., and Knight, E. M. (1964). The isolation of West Nile virus from man and of Usutu virus from the bird-biting mosquito Mansonia Aurites (Theobald) in the Entebbe area of Uganda. Ann. Trop. Med. Parasitol. 58, 367–374. doi: 10.1080/00034983.1964.11686258
Young, A. D., and Gillung, J. P. (2020). Phylogenomics — principles, opportunities and pitfalls of big-data phylogenetics. Syst. Entomol. 45, 225–247. doi: 10.1111/syen.12406
Zecchin, B., Fusaro, A., Milani, A., Schivo, A., Ravagnan, S., Ormelli, S., et al. (2021). The central role of Italy in the spatial spread of USUTU virus in Europe. Virus Evol. 7:veab048. doi: 10.1093/ve/veab048
Ziegler, U., Bergmann, F., Fischer, D., Müller, K., Holicki, C. M., Sadeghi, B., et al. (2022). Spread of West Nile virus and Usutu virus in the German bird population, 2019-2020. Microorganisms 10, 60–66. doi: 10.3390/microorganisms10040807
Ziegler, U., Fast, C., Eiden, M., Bock, S., Schulze, C., Hoeper, D., et al. (2016). Evidence for an independent third Usutu virus introduction into Germany. Vet. Microbiol. 192, 60–66. doi: 10.1016/j.vetmic.2016.06.007
Keywords: USUV, Nanopore sequencing, MinION, long amplicon, Illumina
Citation: Holicki CM, Bergmann F, Stoek F, Schulz A, Groschup MH, Ziegler U and Sadeghi B (2022) Expedited retrieval of high-quality Usutu virus genomes via Nanopore sequencing with and without target enrichment. Front. Microbiol. 13:1044316. doi: 10.3389/fmicb.2022.1044316
Edited by:
Yongming Sang, Tennessee State University, United StatesReviewed by:
Alice Fusaro, Experimental Zooprophylactic Institute of the Venezie (IZSVe), ItalyCopyright © 2022 Holicki, Bergmann, Stoek, Schulz, Groschup, Ziegler and Sadeghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balal Sadeghi, QmFsYWwuc2FkZWdoaUBmbGkuZGU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.