
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 07 December 2022
Sec. Terrestrial Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1035247
 Sophie I. Holland1
Sophie I. Holland1 Xabier Vázquez-Campos2
Xabier Vázquez-Campos2 Haluk Ertan3,4
Haluk Ertan3,4 Richard J. Edwards2
Richard J. Edwards2 Michael J. Manefield1,3
Michael J. Manefield1,3 Matthew Lee1*
Matthew Lee1*Dichloromethane (DCM; CH2Cl2) is a widespread pollutant with anthropogenic and natural sources. Anaerobic DCM-dechlorinating bacteria use the Wood–Ljungdahl pathway, yet dechlorination reaction mechanisms remain unclear and the enzyme(s) responsible for carbon-chlorine bond cleavage have not been definitively identified. Of the three bacterial taxa known to carry out anaerobic dechlorination of DCM, ‘Candidatus Formimonas warabiya’ strain DCMF is the only organism that can also ferment non-chlorinated substrates, including quaternary amines (i.e., choline and glycine betaine) and methanol. Strain DCMF is present within enrichment culture DFE, which was derived from an organochlorine-contaminated aquifer. We utilized the metabolic versatility of strain DCMF to carry out comparative metaproteomics of cultures grown with DCM or glycine betaine. This revealed differential abundance of numerous proteins, including a methyltransferase gene cluster (the mec cassette) that was significantly more abundant during DCM degradation, as well as highly conserved amongst anaerobic DCM-degrading bacteria. This lends strong support to its involvement in DCM dechlorination. A putative glycine betaine methyltransferase was also discovered, adding to the limited knowledge about the fate of this widespread osmolyte in anoxic subsurface environments. Furthermore, the metagenome of enrichment culture DFE was assembled, resulting in five high quality and two low quality draft metagenome-assembled genomes. Metaproteogenomic analysis did not reveal any genes or proteins for utilization of DCM or glycine betaine in the cohabiting bacteria, supporting the previously held idea that they persist via necromass utilization.
Dichloromethane (DCM; CH2Cl2) is a widespread compound with a range of natural and anthropogenic sources. The latter accounts for 70% of the ~900 Gg DCM produced annually, while the former consists primarily of oceanic sources and biomass burning (Gribble, 2010). As a result of extensive industrial use, substantial DCM contamination in the environment continues to increase (Shestakova and Sillanpää, 2013; Hossaini et al., 2015; Leedham Elvidge et al., 2015), posing a threat to human and environmental health (Agency for Toxic Substances and Disease Registry, 2000; Hossaini et al., 2017). The compound is a priority pollutant in Europe (European Parliament Council of the European Union, 2013), the US (United States Environmental Protection Agency, 1977), and China (Ministry of Environmental Protection (MEP), 2017). Being denser than water, DCM that escapes into groundwater migrates downwards into anoxic zones. Despite this, microbial transformation of DCM in anoxic environments remains poorly understood, with only three organisms known to conserve energy via anaerobic DCM metabolism. Dehalobacterium formicoaceticum strain DMC, the only one of these in pure culture, ferments DCM to formate and acetate (Mägli et al., 1996). ‘Candidatus Dichloromethanomonas elyunquensis’ strain RM mineralizes DCM to H2 and CO2 (Kleindienst et al., 2017; Chen et al., 2020), while ‘Candidatus Formimonas warabiya’ strain DCMF ferments DCM to acetate (Holland et al., 2021). All three bacteria are members of the Peptococcaceae family and use the Wood–Ljungdahl pathway for DCM metabolism in varying ways (Mägli et al., 1998; Chen et al., 2017; Holland et al., 2019; Kleindienst et al., 2019; Holland et al., 2021).
Cell suspension assays with D. formicoaceticum suggested that DCM dechlorination was corrinoid-dependent (Mägli et al., 1996). Recent genomic and proteomic analysis of ‘Ca. Dichloromethanomonas elyunquensis’ and D. formicoaceticum highlighted a cluster of corrinoid-dependent methyltransferase genes thought to be involved in DCM catabolism – the mec cassette – and identified a homologous cassette in the strain DCMF genome (Kleindienst et al., 2019; Murdoch et al., 2022). Notably, ‘Ca. Dichloromethanomonas elyunquensis’ also encodes and expresses reductive dehalogenases in the presence of DCM (Kleindienst et al., 2019), which are absent from the other two organisms’ genomes (Chen et al., 2017; Holland et al., 2019). However, ‘Ca. Dichloromethanomonas elyunquensis’ and D. formicoaceticum are obligate DCM degraders, and demonstrably unable to metabolize any other chlorinated methanes (Mägli et al., 1996; Kleindienst et al., 2017). In light of this, as well as the difference in end products and previous dual carbon-chlorine isotope analyses (Chen et al., 2018), the three DCM-degrading bacteria are hypothesized to utilize differing dechlorination mechanisms, but these are not yet fully understood.
The strain DCMF genome encodes an abundance of predicted corrinoid-dependent methyltransferase genes (Holland et al., 2019), including 82 in the MttB superfamily, which contains methylamine and quaternary amine methyltransferases, as well as many of uncharacterized substrate specificity (Ferguson and Krzycki, 1997; Ticak et al., 2014). Corrinoid-dependent methyltransferase systems are typically found in methanogenic archaea and homoacetogenic bacteria and are comprised of three main components: a methyltransferase I (MTI), methyltransferase II (MTII), and cognate corrinoid protein (CoP). MTI transfers a methyl group from the substrate onto the CoP, from which MTII transfers it to the final receiving compound, typically coenzyme M in methanogens or tetrahydrofolate (THF) in acetogenic bacteria (reviewed in Ragsdale, 2008). A reductive activator of corrinoid-dependent enzymes (RACE) protein may also be required to reactivate the corrinoid cofactor (Ragsdale, 2008), although this is unlikely to be required every reaction cycle (Drummond et al., 1993; Menon and Ragsdale, 1999). In bacteria, these methyltransferases can demethylate chloromethane (Vannelli et al., 1999), methanol (van der Meijden et al., 1984), quaternary amines (Ticak et al., 2014; Picking et al., 2019; Kountz et al., 2020), and methoxylated aromatic compounds such as vanillate (Kaufmann et al., 1997).
Strain DCMF is a methylotrophic acetogen that ferments a number of these compounds – methanol, choline (N,N,N-trimethylethanolamine), and glycine betaine (N,N,N-trimethylglycine) – as well as DCM (Holland et al., 2021). Glycine betaine is a quaternary amine with substantial environmental roles, including widespread use as an osmoprotectant across all domains of life (Beers, 1967; Larher et al., 1982; Csonka, 1989). Strain DCMF is the dominant lineage in enrichment culture DFE, although its relative abundance varies from ~7% to 75% throughout a single substrate pulse with DCM or glycine betaine (Holland et al., 2021). Despite this, previous investigation of the strain DCMF genome, growth curve mass balances, and strain DCMF-free enrichment cultures have all suggested that it is highly unlikely the cohabitant bacteria are involved in primary substrate metabolism (Holland et al., 2021). However, it remains unclear how they persist in culture DFE. Based on physiological and genomic information, strain DCMF demethylates glycine betaine in a stepwise manner to N,N-dimethylglycine and then sarcosine (N-methylglycine), before being reductively cleaving sarcosine to monomethylamine and acetate. Methyltransferases likely catalyze methyl group transfer from glycine betaine and dimethylglycine onto THF and the resulting methyl-THF enters the Wood–Ljungdahl pathway resulting in acetate production (Holland et al., 2019, 2021). While glycine betaine methyltransferases have previously been characterized in Desulfitobacterium hafniense (Ticak et al., 2014) and Acetobacterium woodii (Lechtenfeld et al., 2018), there is only a single proteomic study (in Sporomusa ovata strain An4) investigating this enzyme (Visser et al., 2016).
Here, we aimed to determine the enzymes involved in DCM and glycine betaine metabolism in strain DCMF via comparative metaproteomics of enrichment culture DFE. This addressed knowledge gaps surrounding the proteins involved in DCM dechlorination and glycine betaine fermentation in this organism, and more broadly in anoxic subsurface environments. Methyltransferase systems were implicated in both metabolisms, providing gene targets for further study.
To assemble the culture DFE metagenome, a previously described set of PacBio-sequenced, non-redundant (NR) contigs that did not map to the strain DCMF genome (“NR contaminants”; Holland et al., 2019) were assembled using Metaflye v2.7.1 (Kolmogorov et al., 2020) with parameters --pacbio-raw --genome-size 20 m --meta --plasmids. The assembled contigs were frameshift-corrected using DIAMOND v0.9.31 (Buchfink et al., 2014) and MEGAN Community Edition v6.19.9 (Huson et al., 2016), as previously described (Arumugam et al., 2019).
“NR contaminants” reads, as well as those from previous Illumina sequencing efforts of culture DFE (SRA accession number SRR5179547; Holland et al., 2019), were mapped to the frameshift-corrected contigs using BBMap v38.51 and Bowtie2 v2.3.5.1, respectively. Taxonomy was assigned to contigs using Kaiju v1.7.2. This information was parsed into anvi’o (installed from the master Github repository on 1 October 2020; Eren et al., 2021) for manual binning. CheckM v1.1.3 (Parks et al., 2015) was used to assess completeness and contamination of each bin and taxonomy was assigned with GTDB-tk v2.0.0 (Chaumeil et al., 2019) against release R07-RS207. Gene calling was performed with Prokka v1.14.5 (Seemann, 2014). Predicted proteins were annotated with orthologous groups and KEGG database information using EggNOG-mapper v2.0 (database v5.0; Huerta-Cepas et al., 2019) and with subcellular localization using PSORTb v3.0.2 (Yu et al., 2010).
Metabolic pathways in the MAGs were determined via BlastKOALA (Kanehisa et al., 2016), dbCAN2 (Zhang et al., 2018) predicted Carbohydrate Active enZymes (CAZymes) and hydrogenase catalytic subunits were classified using HydDB (Søndergaard et al., 2016).
Culture DFE was grown in anaerobic, defined minimal mineral salts medium as previously described (Holland et al., 2019). The medium was buffered to pH 6.8–7.0 via addition of NaHCO3 (2.5 g L-1) and sparging with N2:CO2 (4,1). For metaproteomic analysis, 200 ml cultures were amended with either DCM (2 mM) or glycine betaine (5 mM; n = 6 for each). Cultures were incubated statically at 30°C in the dark. DCM was quantified by GC-FID and glycine betaine by LC–MS/MS (Holland et al., 2021).
Cells were harvested from cultures when ~80% of the substrate was depleted (Supplementary Figure S1). Biomass from two culture flasks (i.e., 400 ml total) were combined to produce triplicate samples for metaproteomic analysis of each substrate condition. Strain DCMF cells were enumerated in cultures via quantitative real-time PCR (qPCR) of the strain DCMF 16S rRNA gene with primers Dcm775F/Dcm930R, as previously described (Holland et al., 2019). Total bacterial 16S rRNA genes were quantified similarly, with primers Eub1048/Eub1194 (Holland et al., 2021). Strain DCMF 16S rRNA gene copy numbers were converted to cell numbers by dividing by four (the number of 16S rRNA gene copies in the strain DMCF genome).
Cultures were centrifuged at 8,000 × g at 4°C for 30 min and then resuspended in 120 μl protein extraction buffer (50 mM 3-(N-morpholino)propanesulfonic acid [pH 7], 4% sodium dodecylsulfate, 50 mM NaCl, 100 μM EDTA, 100 μM MgCl2). Mixtures were transferred to 2 ml tubes containing 0.06 g glass beads (150–212 μm, Sigma, North Ryde, Australia) and a ¼” ceramic sphere (MP Bio, Seven Hills, Australia) and bead-beat at 1,800 rpm for 5 min (PowerLyzer 24 Homogenizer, Qiagen, Chandstone Centre, Australia). Tubes were centrifuged at 16,000 × g for 10 min and the supernatants (i.e., crude protein extracts) transferred to fresh, 1.5 ml tubes to repeat the centrifugation.
Protein yield was quantified using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Scoresby, Australia) with crude protein extracts diluted 1:250 in MilliQ water. Bovine serum albumin was used to create a seven-point standard curve (0.5–40 μg ml-1).
Filter-aided sample preparation (FASP) was used to prepare the crude protein extracts for peptide identification (Wiśniewski, 2017). Samples were diluted to a concentration of ~1 μg μl-1 in 50 mM NH4HCO3. A total of 15.8 μg protein from each sample was transferred to a 1.5 ml microfuge tube with 5 mM dithiothreitol and incubated at 37°C for 30 min. Samples were loaded onto Amicon Ultra-0.5 30 kDa centrifugal filter units (Merck, Bayswater, Australia) with 200 μl UA solution (8 M urea in 100 mM Tris–HCl, pH 8.5). Filters were centrifuged at 14,000 × g for 15 min before another 200 μl UA was added to each and the centrifugation repeated. Proteins were alkylated by addition of 100 μl iodoacetamide solution (50 mM iodoacetamide in UA) and mixing at 600 rpm for 1 min prior to incubating statically in the dark for 20 min. Filters were centrifuged at 14,000 × g for 10 min. UA (100 μl) was added to each filter before centrifuging at 14,000 × g for 15 min, twice. Then, 50 mM NH4HCO3 (100 μl) was added to each filter before centrifuging at 14,000 × g for 10 min; repeated twice more. Proteolytic cleavage into peptides was performed by addition of trypsin (1:100 enzyme:protein ratio) in 40 μl NH4HCO3 and mixing at 600 rpm for 1 min. Filters were incubated in a 37°C wet chamber overnight, then transferred to fresh collection tubes and centrifuged at 14,000 × g for 10 min. A final 20 μl NH4HCO3 was added to each filter before centrifuging at 14,000 × g for 10 min; this was repeated once more. Eluent was stored at -80°C.
Peptide lysates were separated by nanoLC on an UltiMate™ 3,000 RSLCnano ultra performance liquid chromatograph and autosampler system (Dionex, Scoresby, Australia). Samples (2.5 μl) were concentrated and desalted onto a micro C18 precolumn (300 μm × 5 mm, Dionex) with water:acetonitrile (98:2, 0.2% TFA) at 15 μl min-1. After a 4 min wash the pre-column was switched (10 port UPLC valve, Valco, Houston, TX) into line with a fritless nano column (75 μm × 15 cm) containing C18AQ media (1.9 μ, 120 Å, Dr. Maisch). Peptide lysates were eluted using a linear gradient of water:acetonitrile (98:2, 0.1% formic acid) to water:acetonitrile (64:36, 0.1% formic acid) at 200 nl min-1 over 30 min. High voltage 2000 V was applied to low volume Titanium union (Valco) and the tip positioned ~0.5 cm from the heated capillary (T = 275°C) of an Orbitrap Fusion Lumos (Thermo Electron, Scoresby, Australia) mass spectrometer. Positive ions were generated by electrospray and the Fusion Lumos operated in data dependent acquisition mode. A survey scan m/z 350 – 1,750 was acquired (resolution = 120,000 at m/z 200, with an accumulation target value of 400,000 ions) and lockmass enabled (m/z 445.12003). Data-dependent tandem MS analysis was performed using a top-speed approach (cycle time of 2 s). MS/MS spectra were acquired by HCD (normalized collision energy = 30) fragmentation and the ion-trap was selected as the mass analyser. The intensity threshold for fragmentation was set to 25,000. A dynamic exclusion of 20 s was applied with a mass tolerance of 10 ppm.
Mass spectra files were searched against a custom database of all predicted proteins in the DFE metagenome using MaxQuant v1.6.17.0 (Cox et al., 2014). Enzyme specificity was trypsin/P with a maximum of two missed cleavages. Fixed (carbamidomethylation of cysteine) and variable (oxidation of methionine and N terminal acetylation) modifications were selected. Minimum peptide length was seven amino acids and maximum peptide mass 4,600 Da. The mass tolerance was set to 4.5 ppm for the MS and 0.5 Da for the MS/MS. ‘LFQ’ and ‘Match between runs’ were selected. The PSM and protein False Discovery Rate (FDR) were both 0.01. The mass spectrometry and proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD037334.
Statistical analysis of the MaxQuant output was performed in Perseus v1.6.13.0 (Tyanova et al., 2016). Proteins identified by site, reverse sequences, only one unique peptide, and potential contaminants were removed. Proteins were filtered to retain only those present in all three replicates of at least one substrate condition. Label free quantitative (LFQ) intensities were log2 transformed and missing values were imputed from a Gaussian distribution (down shift 1.8, width 0.3, relative to the standard deviation of each column). Triplicate-averaged values were Z-score transformed within each column to determine protein abundance relative to overall expression with each substrate. Only proteins in the ‘Majority Protein ID’ column were considered present, i.e., those where all proteins listed in a group had at least half the peptides that the leading protein had. Where >1 proteins were included in the ‘Majority Protein ID’ group, they were included for analysis and are listed separately, but marked as ‘Ambiguous’ and treated with appropriate caution in interpreting any results. Triplicate LFQ values were directly compared via multiple t-tests to create a volcano plot (S0 = 0.1, 250 randomizations, substrate grouping not preserved in randomizations). Proteins were considered differentially abundant if they had a FDR < 0.01.
To investigate the persistence of cohabiting bacteria in culture DFE and provide a taxa-specific platform for metaproteomic analysis, metagenomic assembly of a set of previously described (Holland et al., 2019) non-redundant, non-strain DCMF PacBio sequencing reads (“NR Contaminants”) was carried out. Sequencing details were previously reported (Holland et al., 2019) but assembly of this subset of reads was not previously attempted. A total 195,364 long reads (732,500,489 bp) assembled into 330 contigs (total size 13,752,672 bp; Supplementary Table S1). Reads were deposited in the NCBI SRA (SRX9412577).
Manual binning with anvi’o resulted in 10 bins (Supplementary Figure S2). Based on their completeness, contamination, and the presence of 16S rRNA genes, these comprise five high quality and two low quality draft metagenome-assembled genomes (MAGs; Supplementary Table S2), and three undetermined bins (“UNK-1/2/3”). Taxonomic assignment of the MAGs revealed unclassified lineages of Bacteroidales (henceforth referred to as “DFE-LEN”), Ignavibacteria (“DFE-IGN”), Cupidesulfovibrio (“DFE-NIT”; a recently proposed novel genus based on Desulfovibrio oxamicus and Desulfovibrio termitidis (Wan et al., 2021)), Synergistales (“DFE-SYN”), and Rectinema (“DFE-TRE1”; Table 1). Taxonomic assignment was not performed for bins below 50% completeness (“DFE-BAC” and “DFE-TRE2”). High-quality MAGs were deposited in NCBI (GenBank accessions in Supplementary Table S2).
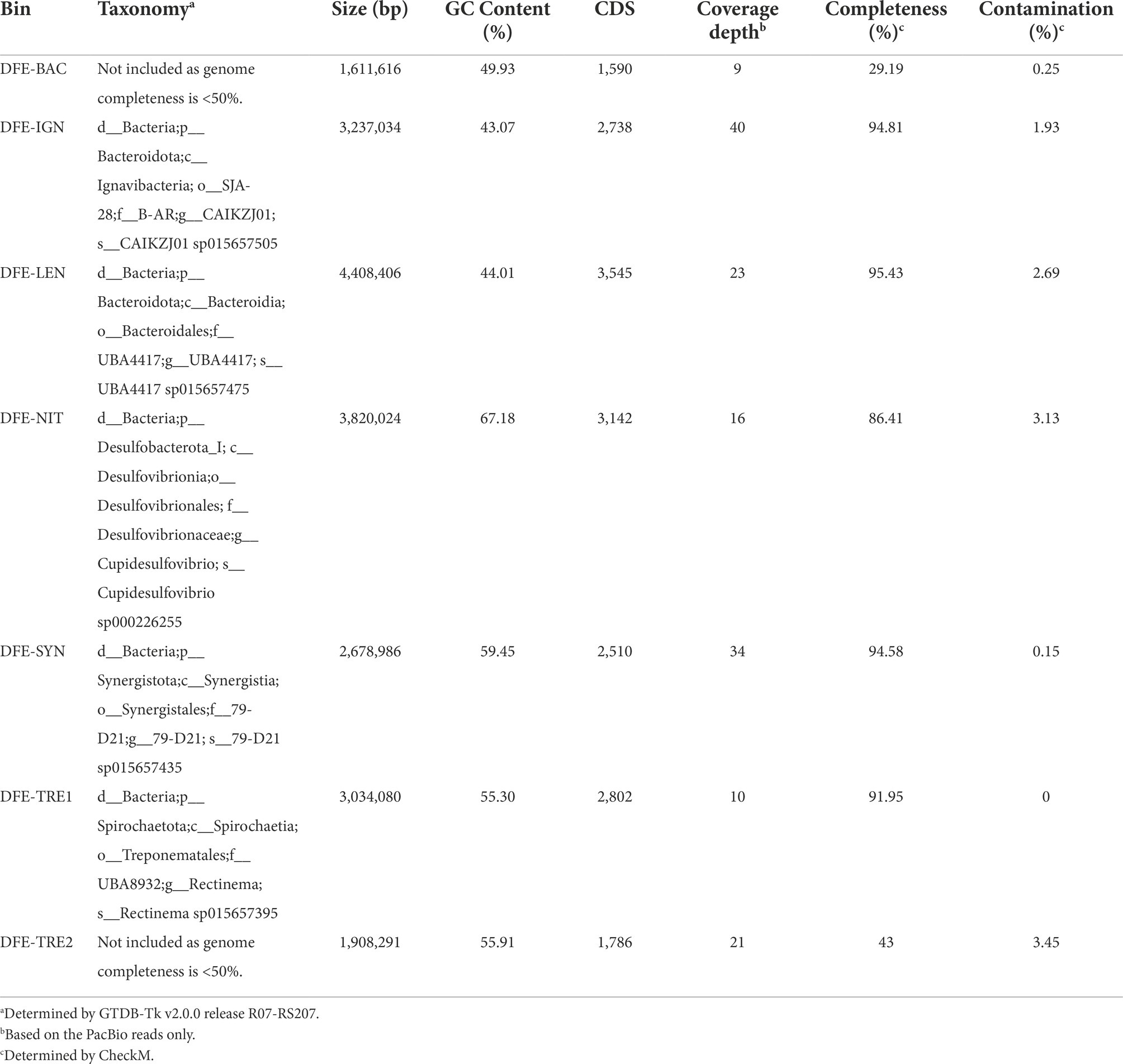
Table 1. Overview of the MAGs assembled from the DFE culture metagenome.
Annotation of the bins resulted in 1,590 coding sequences (CDS) in the smallest and least complete MAG (DFE-BAC) and 3,545 CDS in the largest (DFE-LEN; Table 1, full annotation available in Supplementary Table S3). DFE-IGN was omitted from further analysis as it was lost from the culture through subcultivation in the 3 years (~10 transfers) between PacBio sequencing and metaproteomic analysis.
Label-free quantitative metaproteomic analysis of DFE cultures grown with DCM or glycine betaine identified 1,713 proteins across the two substrate conditions (Supplementary Table S4). The IMG annotation of the strain DCMF genome was used throughout (JGI genome ID 2718217647). While only those proteins present in all three replicates of at least one substrate conditions (i.e., DCM or glycine betaine) were included, missing values were imputed for all reported analyses (see Methods). Most proteins (81%) were from strain DCMF (1,384), followed by DFE-SYN (134), DFE-LEN (60), DFE-NIT (43), DFE-TRE2 (42), DFE-TRE1 (41), DFE-BAC (3), and UNK-1/2/3 (5). There were 409 significantly differentially abundant strain DCMF proteins (FDR 0.01; Figure 1; Supplementary Table S5). While the relative abundance of bacterial community members may have some bearing on the direct comparison of protein abundance between DCM- and glycine betaine-amended cultures, it is unlikely to play a large role in the data presented here for two reasons. Firstly, there was no significant difference in strain DCMF abundance between DCM vs. glycine betaine-amended cultures (either before or after duplicate culture samples were combined), as quantified by qPCR of the DCMF 16S rRNA gene (p > 0.05, two-tailed unpaired homoscedastic t-test; Supplementary Table S6). Secondly, we previously found no significant difference in the Shannon Diversity Index between culture DFE microcosms amended with different substrates (Kruskal–Wallis value of p 0.0976; Holland et al., 2021). Strain DCMF represented at least 73% relative abundance in the community when the metaproteomic samples were harvested (Supplementary Table S6).
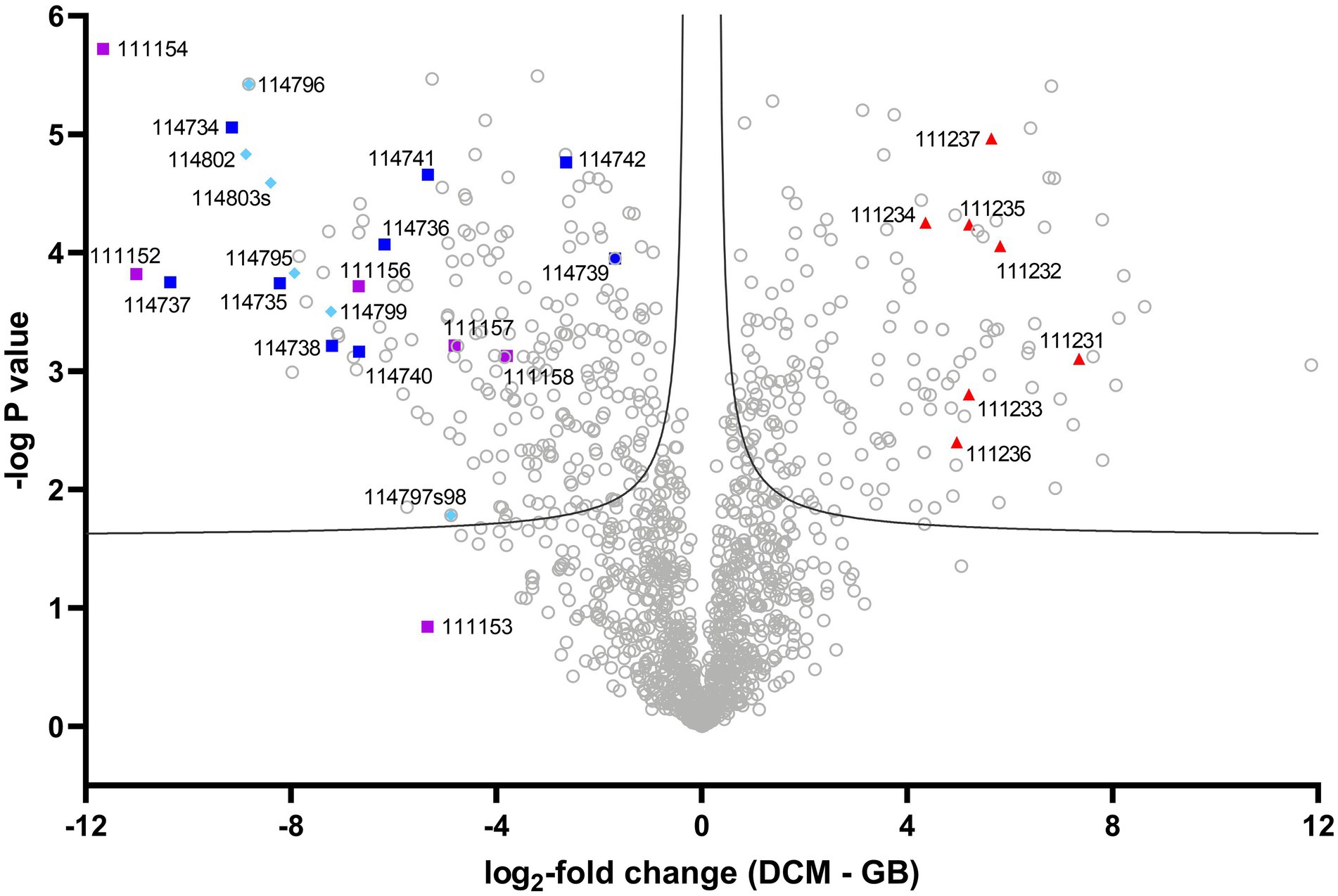
Figure 1. Volcano plot of strain DCMF protein expression with DCM and glycine betaine. Log2-fold change (LFC) in abundance is shown as the difference between DCM and glycine betaine label-free quantitative intensity. A false discovery rate of 0.01 was the significance boundary, indicated by two lines on the graph. Proteins in the DCM-associated mec cassette are labeled as red triangles, those from the complete glycine betaine methyltransferase gene cluster by dark blue squares, those in the incomplete glycine betaine and/or dimethylglycine methyltransferase gene cluster by purple squares, and those from the sarcosine reductase gene cluster by light blue diamonds. IMG gene loci (prefaced by ‘Ga018035_’) are indicated for these proteins of interest. A full list of all significantly differentially abundant proteins is included in Supplementary Table S5.
The unique ability of strain DCMF to grow on substrates other than DCM allowed, for the first time, a statistical comparison of protein abundance in cells fermenting DCM vs. cells fermenting an alternative substrate (glycine betaine), where previous studies only examined fed vs. starvation conditions (Kleindienst et al., 2019; Murdoch et al., 2022). DCM- and glycine betaine-amended cultures were compared to determine the log2-fold change (LFC) of each protein. Proteins of interest were identified as those with particularly high/low LFC, as well as searches for homologs to proteins previously reported to be involved in DCM and glycine betaine metabolism in the literature (i.e., the mec cassette, glycine betaine methyltransferases, and glycine/betaine/sarcosine reductases).
Proteins that were highly abundant under both substrate conditions were also analyzed. Within strain DCMF, this included proteins for the Wood–Ljungdahl pathway, conversion of acetyl-CoA to acetate, and six of eight subunits for an FOF1-type ATP synthase (Figure 2; Supplementary Table S4, labeled in the Pathway/Function column as “Wood–Ljungdahl Pathway” and “Energy conservation,” respectively). From the complete Rnf complex and NADH:ubiquinone reductase (complex I) encoded in the genome (Holland et al., 2019), three subunits from the former (RnfBCG, Ga0180325_113065, Ga0180325_113068, Ga0180325_113070) and 10 subunits from the latter (NuoBCDEFGHIJM, Ga0180325_115678-80, Ga0180325_11791-92, Ga0180325_11330, Ga0180325_115681-83, Ga0180325_115686) were found in the proteome (Supplementary Table S4, “Energy conservation”). Two proteins for a putative K+ or Na+-stimulated pyrophosphate-energized sodium pump (Ga0180325_113285, Ga0180325_113311) were also highly abundant with all substrates (Supplementary Table S4, “Energy conservation”). Cumulatively, this suggests that strain DCMF generates energy through a chemiosmotic mechanism as well as substrate level phosphorylation (i.e., fermentation). Furthermore, proteins for a flagellum and chemotaxis indicated that strain DCMF is motile and responds to environmental cues (Supplementary Table S4, “Flagellum” and “Chemotaxis”).
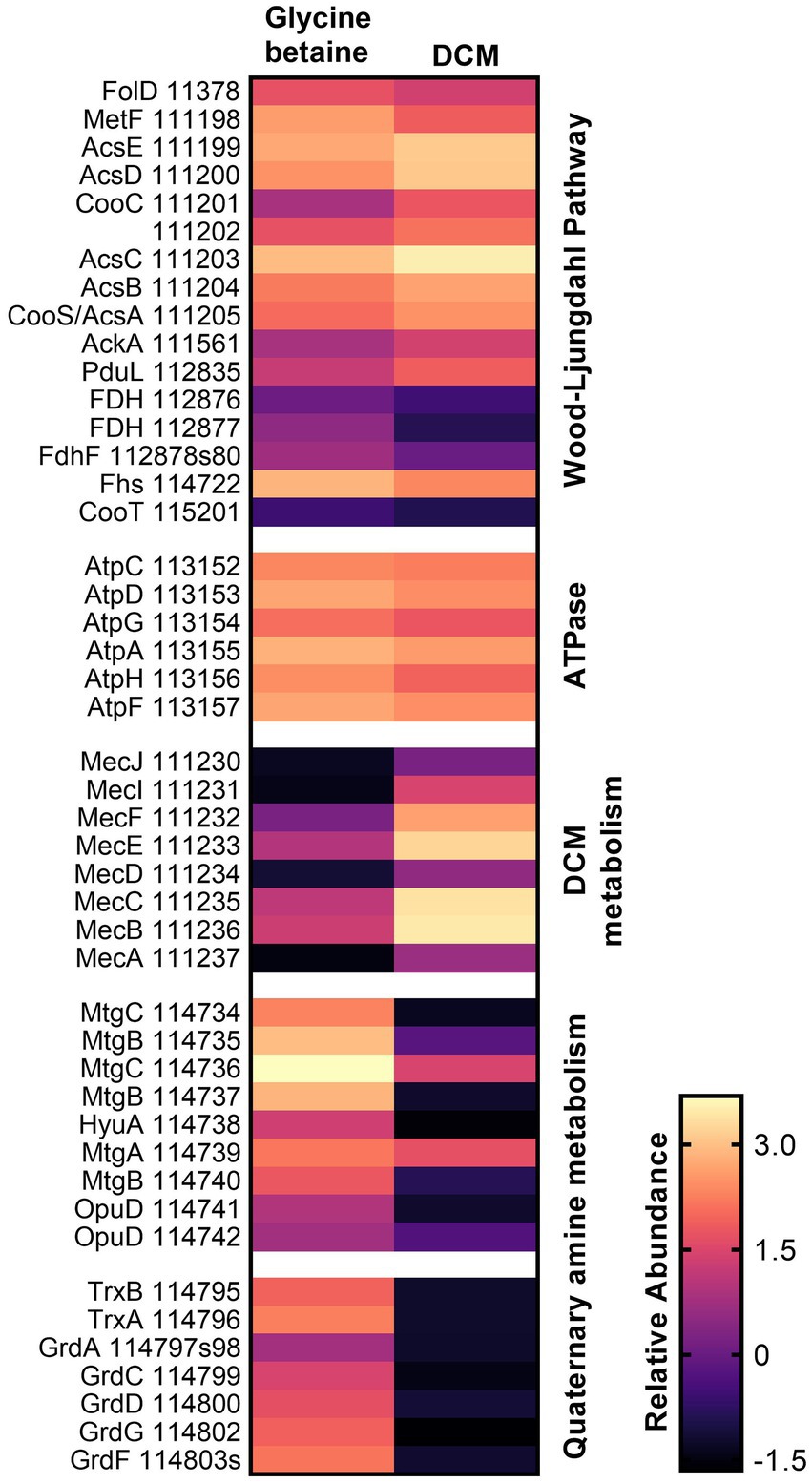
Figure 2. Heatmap of relative protein abundance for genes/pathways of interest in DCM and glycine betaine-amended strain DCMF cells. Proteins for the Wood–Ljungdahl pathway and a FOF1-type ATPase were highly abundant under both growth conditions, while proteins in the DCM-associated mec cassette were significantly more abundant in DCM-grown cells, and proteins in a putative glycine betaine methyltransferase gene cluster and a sarcosine reductase gene cluster were significantly more abundant in glycine betaine-grown cells. Rows are labeled with gene symbols and the strain DCMF IMG gene loci (each prefaced with “Ga0180325_”).
The strain DCMF genome encodes an abundance of corrinoid-dependent methyltransferase gene components, i.e., MTI, MTII and CoPs (Holland et al., 2019). Many of these were identified in the proteome and had significantly differential abundance between DCM- and glycine betaine-amended cultures. For example, in DCM-grown cells, a protein in the monomethylamine methyltransferase mtmB superfamily (i.e., a likely MTII) had the highest LFC (Ga0180325_111810, +11.87; Figure 1; Supplementary Table S5). The mec cassette, which was recently suggested to be involved in anaerobic DCM dechlorination (Murdoch et al., 2022), also stood out as significantly more abundant, with an average LFC of 5.34 (Figure 2; Table 2). This eight-gene cassette includes several corrinoid-dependent methyltransferase components, a two-component regulatory system and a cation exchange protein (Table 2). Importantly for the function of these methyltransferases (and despite the presence of 50 μg L-1 cyanocobalamin in the culture medium) strain DCMF encodes a complete corrinoid biosynthesis pathway (Holland et al., 2019) and 14 of these 25 proteins were detected in the proteome (Supplementary Table S4, “Corrinoid biosynthesis”).
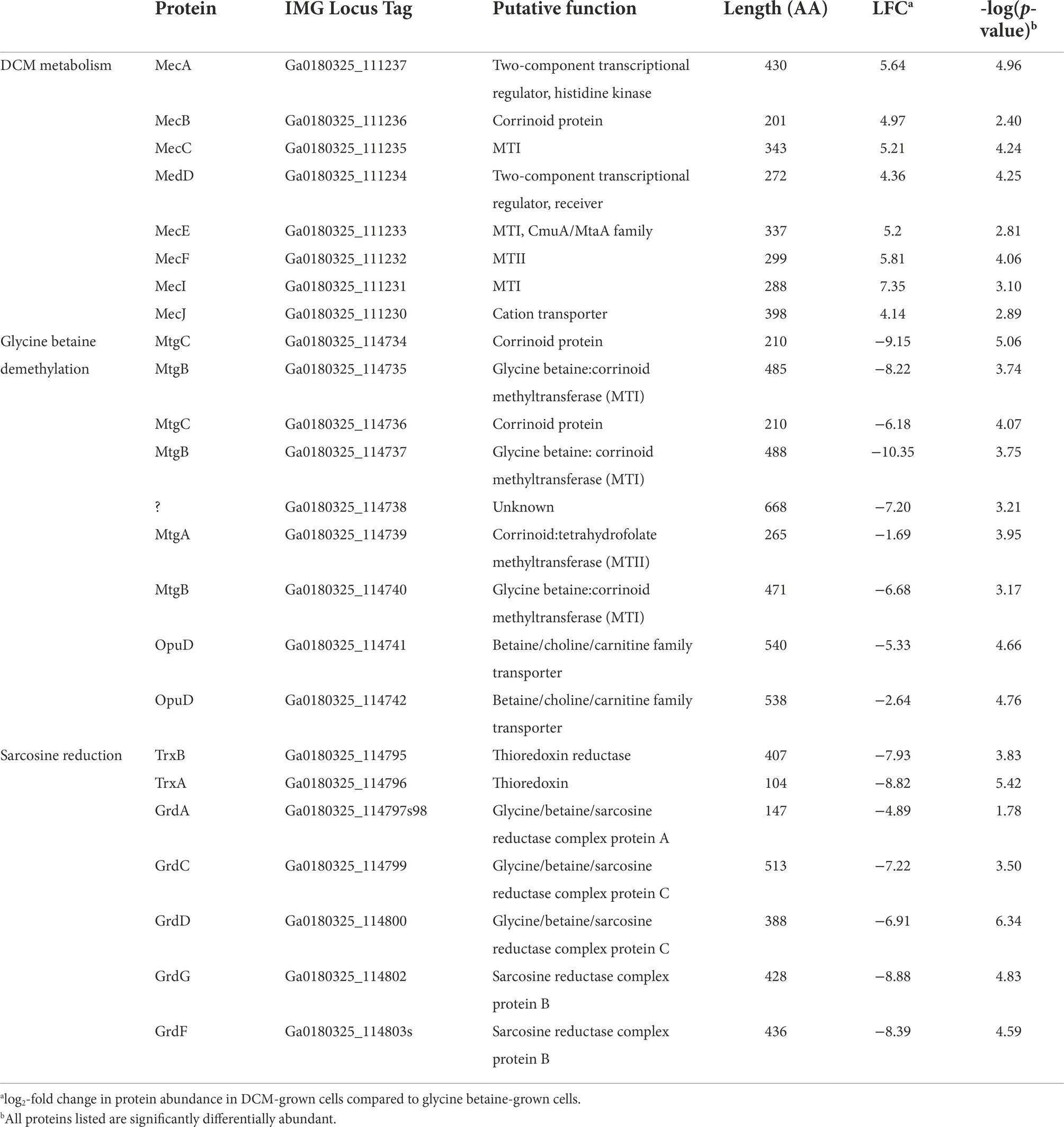
Table 2. List of proteins putatively involved in DCM and glycine betaine metabolism in strain DCMF.
The significant abundance of proteins in the mec cassette in DCM-amended strain DCMF cells provides experimental evidence supporting the hypothesis that these proteins are responsible for anaerobic dechlorination of DCM (Murdoch et al., 2022). Functional annotation of the proteins also supported previously suggested roles, that were based on expression of the mec cassette in D. formicoaceticum and ‘Ca. Dichloromethanomonas elyunquensis’ (Murdoch et al., 2022). Transcription of the cluster may be regulated directly in response to DCM, as the sensor histidine kinase (Ga0180325_111237, MecA) in the two-component transcriptional regulatory system harbors a PocR domain (Supplementary Table S7), which can bind DCM or other small hydrocarbons (Anantharaman and Aravind, 2005). Protein Ga0180325_111233 (MecE), classed as an MtaA/CmuA family methyltransferase (Supplementary Table S7), is the most likely candidate for the initial dechlorination of DCM. The MtaA/CmuA family contains methanol and chloromethane MTI proteins (van der Meijden et al., 1984; Vannelli et al., 1999). Chloromethane dechlorination is a methyltransfer reaction catalyzed by two subunits in aerobic methylotrophs: a fused MTI-CoP (CmuA) and an MTII (CmuB; Vannelli et al., 1998, 1999; Studer et al., 1999). A similar system is thought to operate in the anaerobe Acetobacterium dehalogenans (Meßmer et al., 1996; Wohlfarth and Diekert, 1997) and it is therefore feasible that MecE could act on DCM under anoxic conditions. The MtaA/CmuA protein family sits within the uroporphyrinogen decarboxylase (URO-D) superfamily, and two other proteins in the same gene cluster were also classified within this superfamily (Ga0180325_111231, MecI, and Ga0180325_111235, MecC; Supplementary Table S7). However, their role here remains unclear and as such, participation in DCM dechlorination cannot be excluded. We further hypothesize that protein Ga0180325_111232 (MecF) acts as an MTII, given that it can likely bind THF via a pterin-binding site (Supplementary Table S7) and may therefore catalyze the formation of 5,10-methylene-THF from DCM or a dechlorinated intermediate. Finally, protein Ga0180325_111236 (MecB) encodes a B12-binding site (Supplementary Table S7) and could thus act as the CoP for the proposed methyltransferase.
Full mec cassette homologs are encoded in DCM-fermenting bacteria D. formicoaceticum (CEQ75_RS03275–30) and ‘Ca. Dichloromethanomonas elyunquensis’ (AWM53_02086–85 and AWM53_01378-83), as well as the non-DCM-fermenting bacterium Dehalobacter restrictus strain UNSWDHB, which respires trichloromethane. Partial cassettes are also found in D. restrictus strains CF (which also respires trichloromethane) and DCA (which respires 1,2-dichloroethane; Murdoch et al., 2022). Within DCM-fermenting bacteria and D. restrictus UNSWDHB, the gene cassette is highly conserved in terms of genetic synteny and protein sequence (75–94% amino acid identity; Figure 3). Outside of the mec cassette homologs, the methyltransferase components share <45% amino acid identity to their closest characterized homologs (Supplementary Table S8).
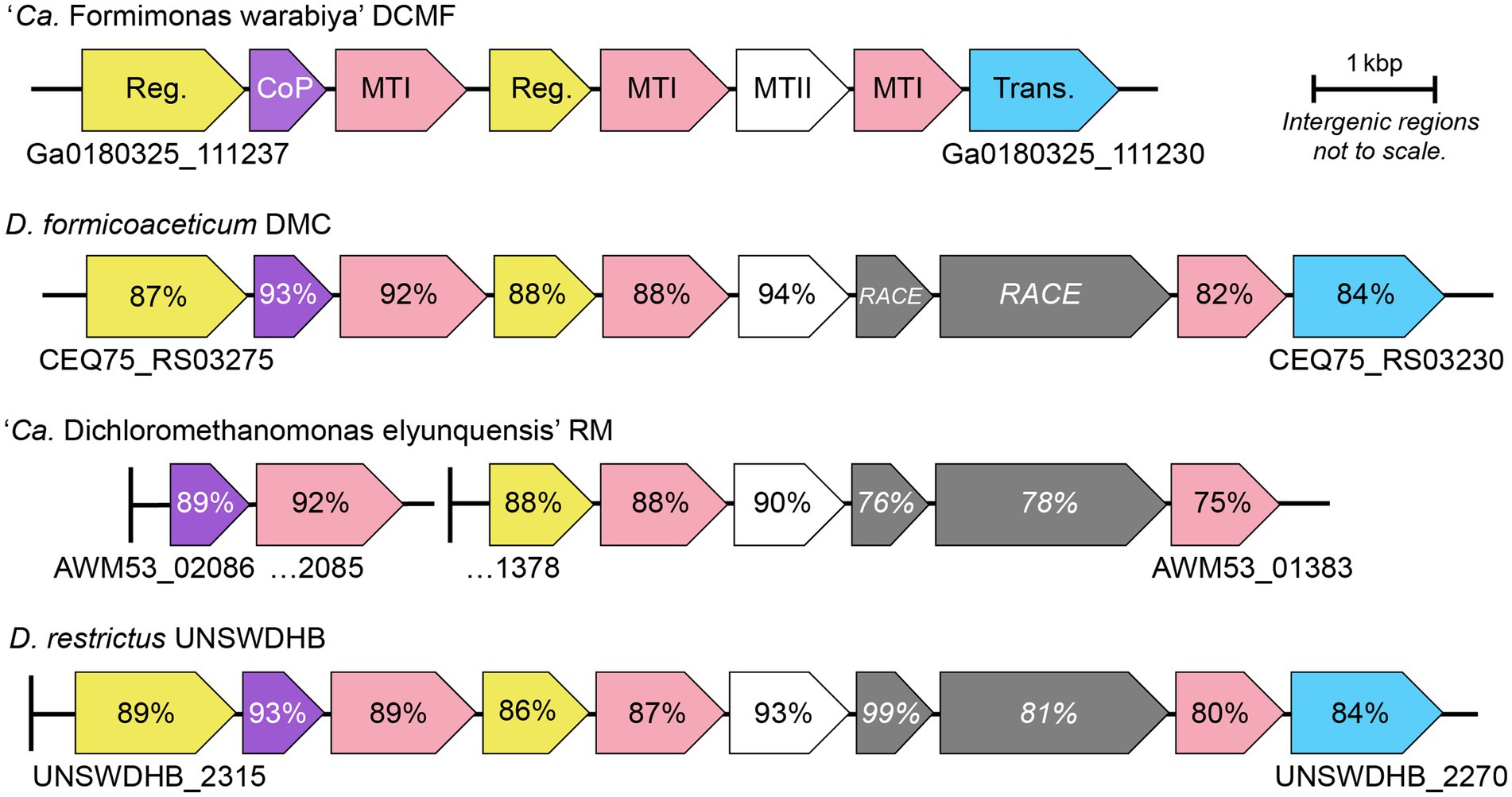
Figure 3. Genetic organization of the mec cassette in strain DCMF and other anaerobic chlorinated methane-degrading bacteria. Loci are shown below the first and last gene in each cluster. Percentage sequence identity to strain DCMF is shown for each homologous protein; percentage identity for the DUF proteins (italicized) is in relation to D. formicoaceticum. Vertical lines represent contig boundaries. Reg, regulator; CoP, corrinoid protein; MTI, methyltransferase I; MTII, methyltransferase II; Trans, transporter; RACE, reductive activator of corrinoid-dependent enzymes.
Strain DCMF lacks two homologous proteins that are present in all other mec cassettes - MecGH (Murdoch et al., 2022). These two proteins are likely involved in reactivation of the methyltransferase corrinoid cofactor (Ferguson et al., 2009; Schilhabel et al., 2009; Price et al., 2018; Murdoch et al., 2022). It is unusual that these genes are absent from the mec cassette in strain DCMF, as corrinoid reactivation is integral to the function of methyltransferase systems. However, homologs to mecG and mecH are present elsewhere in the strain DCMF genome (Ga0180325_115323 and Ga0180325_114747, respectively) and were expressed in the proteome. Although they were not significantly more abundant in DCM-amended cells, their expression was above average (i.e., the LFQ Z-score was >0; Supplementary Table S4).
Based on the functional annotation of the highly abundant proteins in the DCM-associated gene cluster, we propose a putative mechanism for DCM dechlorination in strain DCMF. Reduced Co(I) in the CoP (Ga0180325_111236, MecB), acting in concert with the MTI protein (Ga0180325_111233, MecE), cleaves one chlorine via nucleophilic attack, echoing the role of corrinoid cofactors in reductive dehalogenases (Holliger et al., 1999) and producing a CH2Cl-CoP intermediate (Figure 4A). Nitrogen heteroatoms (N5 and N10) in THF, bound by an MTII protein (Ga0180325_111232, MecF), can then attack the transient CH2Cl-CoP intermediate in a concerted fashion, eliminating the remaining chloride and cleaving the CoP to form 5,10-methylene-THF (Figure 4). This echoes the model recently proposed by Murdoch et al. (2022). Both schemas suggest an unusual departure from the typical biochemistry of methyltransferase reactions, which do not include chlorinated substituents (Kremp and Müller, 2021).
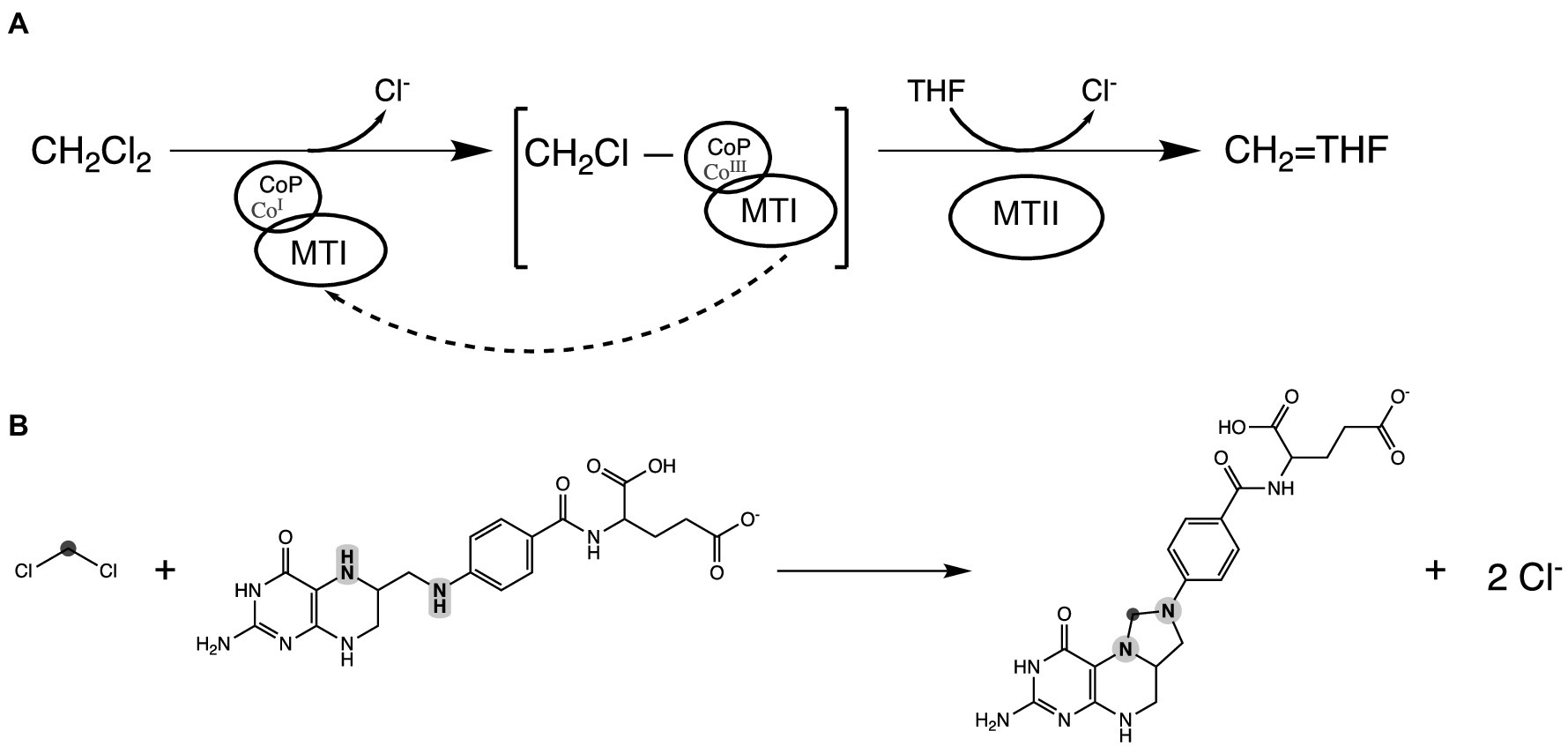
Figure 4. Proposed DCM dechlorination schema in strain DCMF. (A) Reduced Co(I) corrinoid protein (CoP), acting in concert with a methyltransferase I protein (MTI), cleaves the first chlorine from DCM, producing a transient CH2Cl-CoP intermediate. Tetrahydrofolate (THF), bound to a methyltransferase II protein (MTII), then attacks the remaining carbon-chlorine bond, ultimately producing 5,10-methylene-tetrahydrofolate. The Co (III) in the corrinoid protein is reduced back to Co (I) by putative reductive activators of corrinoid-dependent enzymes (RACE) proteins, although this may not be required every reaction cycle. (B) The overall reaction showing DCM and THF producing 5,10-methylene-tetrahydrofolate. The carbon in DCM is highlighted in dark gray; the N5 and N10 nitrogen heteroatoms in tetrahydrofolate are highlighted in light gray.
Proteomic experiments with ‘Ca. Dichloromethanomonas elyunquensis’ and D. formicoaceticum grown on DCM showed the presence of all genes in the cluster, with the methyltransferases among the most abundant (Kleindienst et al., 2019; Murdoch et al., 2022). However, it is notable that ‘Ca. Dichloromethanomonas elyunquensis’ also expressed reductive dehalogenases, which are expected to play a role in DCM dechlorination (Kleindienst et al., 2019). Although putative roles were initially discussed for the reductive dehalogenases (Kleindienst et al., 2019), it is not yet clear how they might act in concordance with the mec cassette proteins (Murdoch et al., 2022). If the mec cassette alone is responsible for DCM dechlorination, then it is unclear what role the reductive dehalogenases have in ‘Ca. Dichloromethanomonas elyunquensis’. Conversely, if reductive dehalogenases are required for DCM dechlorination in ‘Ca. Dichloromethanomonas elyunquensis’, then it is not clear what alternative enzymes or pathways for DCM dechlorination exist in strain DCMF and D. formicoaceticum, that might act in concert with the mec cassette. Previous dual C—Cl isotope analysis implied that dechlorination in D. formicoaceticum proceeds via a bimolecular nucleophilic substitution pathway (SN2), while ‘Ca. Dichloromethanomonas elyunquensis’ utilizes a unimolecular (SN1) pathway (Chen et al., 2018). Here, the mechanism proposed above for strain DCMF utilizes a SN1 reaction, as the nitrogen in folic acid cofactors is a weak nucleophile. However, dual C—Cl isotope analysis and further biochemical characterization of the proteins in the strain DCMF mec cassette are required to elucidate the exact dechlorination mechanism.
While data here supports the role of the mec cassette in DCM dechlorination, the role of other significantly differentially abundant methyltransferases is less clear. For example, numerous methyltransferase components (20 MTI, 28 MTII and 24 CoP) from strain DCMF were identified in the metaproteome (Supplementary Table S4, “Methyltransferase”). This included 26 of the 82 MttB superfamily methyltransferases (i.e., MTIIs) encoded in the genome (Holland et al., 2019). Seven of these MttB superfamily proteins contain the noncanonical amino acid pyrrolysine (Ga0180325_111271p72, Ga0180325_111278p79, Ga0180325_111485p86, Ga0180325_114321p22, Ga0180325_114324p25, Ga0180325_115207p08, Ga0180325_115773p74). Non-pyrrolysine members of the MttB superfamily are widespread in Bacteria and Archaea and have been shown to encode an increasingly diverse substrate range including glycine betaine (Ticak et al., 2014), proline betaine (Picking et al., 2019), carnitine (Kountz et al., 2020), and γ-butyrobetaine (Ellenbogen et al., 2021). However, pyrrolysine-encoding methyltransferases have thus far only been associated with methanogenesis from methylated amines in Archaea (Ferguson and Krzycki, 1997; Burke et al., 1998; Paul et al., 2000). A limited number of other bacterial genera also encode MttB superfamily genes with the pyrrolysine residue, but to our knowledge, proteomic expression has not previously been observed (Ticak et al., 2014). Further experimental work to elucidate the function of these proteins in strain DCMF could provide insight into a potentially novel role for Pyl-MttB proteins, mirroring the expanding role of their non-pyrrolysine counterparts.
In addition to this, the protein with the greatest LFC in DCM-amended cells sits within the monomethylamine methyltransferase MtmB superfamily (Ga0180325_111810, LFC +11.87). However, monomethylamine was not added to DCM-amended microcosms and would only have been present in glycine betaine-amended microcosms as a metabolic end product, as it cannot be used for growth (Holland et al., 2021). Ga0180325_111810 shares ~30% amino acid sequence identity to other proteins in the monomethylamine methyltransferase superfamily (Supplementary Table S8) but is also lacking the characteristic pyrrolysine residue as described in Methylosarcina barkeri MS (Hao et al., 2002). There are two homologs to this protein in D. formicoaceticum, albeit with low percentage identities (30.32% to WP_089610783.1, 28.26% to WP_089610108.1), and no homologs in ‘Ca. Dichloromethanomonas elyunquensis’. This suggests that perhaps this non-Pyl MtmB superfamily protein has a role outside of monomethylamine demethylation, mirroring the recently reported expanded substrate range of non-Pyl MttB superfamily methyltransferases outlined above.
Non-Pyl-MttB superfamily methyltransferases were implicated in glycine betaine metabolism in strain DCMF. To our knowledge, this is only the second shotgun proteomic study of a glycine betaine-fermenting bacterium (Visser et al., 2016). In glycine betaine grown strain DCMF cells, Ga0180325_114734 – Ga0180325_114742 were among the most abundant proteins (Figures 1, 2; Supplementary Table S4) and included putative glycine betaine methyltransferases (Table 2). This nine-gene cluster included three MTIs (all within the MttB superfamily), a putative MTII, two CoPs, two glycine betaine transporters, and an N-methylhydantoinase A/oxoprolinase/acetone carboxylase (Table 2). These proteins were all significantly more abundant in cells grown with glycine betaine than DCM (FDR 0.01), with an average LFC of -6.38 (Figure 1; Table 2).
A second cluster of methyltransferase genes was identified amongst the most differentially abundant proteins (Ga0180325_111152 – Ga0180325_111158; Supplementary Table S4). This gene cluster included the two proteins with the largest LFC difference in glycine betaine-amended cells: Ga0180325_111154 (LFC -11.66) and Ga0180325_111152 (LFC -11.01; Figure 1; Supplementary Table S5). These two proteins are both in the MttB superfamily, and their closest characterized homologs are in Desulfitobacterium hafniense DCB-2 (Supplementary Table S8). However, it remains unclear whether they act in concert with the other components in this gene cluster, or with those in the cluster mentioned above. Not all proteins in this gene cluster were significantly differentially abundant (the putative CoP Ga0180325_111153 was not), nor were all identified in the metaproteome (Ga0180325_111155, a MtaA/CmuA family protein, was absent). Furthermore, this gene cluster contains only putative MTI, MTII and CoP proteins, with no other genes related to glycine betaine metabolism (e.g., transcriptional regulators or transporters) in the genetic vicinity.
A putative sarcosine reductase gene cluster was also identified: Ga0180325_114795–Ga0180325_114803s encodes a thioredoxin reductase (trxB), thioredoxin I (trxA), glycine/betaine/sarcosine reductase complex selenoprotein A (grdA), protein C (grdCD), and a predicted sarcosine-specific protein B (grdGF; Figure 2). A hypothetical protein (Ga0180325_114801) in the middle of the cluster was not identified in the proteome. Excluding that, all proteins were significantly more abundant (FDR 0.01) in glycine betaine amended cells, with an average LFC of -7.58 compared to DCM-amended cells (Figure 1; Table 2; Supplementary Table S5).
Data here supports a previous model for quaternary amine metabolism in strain DCMF that was based on genomic and physiological information (Holland et al., 2021). Briefly, glycine betaine is demethylated to dimethylglycine and then sarcosine; the latter is then reductively cleaved to produce monomethylamine and acetate (Holland et al., 2021). It remains unclear whether any one glycine betaine MTI and CoP pair can catalyze methyl transfer from both glycine betaine and dimethylglycine, as was cautiously suggested for S. ovata An4 (Visser et al., 2016). It is possible that that the multiple MTI and CoP subunits in the more complete glycine betaine methyltransferase cluster (Ga0180325_114734–Ga0180325_114742) may be specific to the two substrates, while the singular MTII can transfer the methyl group from either of the CoPs to THF. Alternatively, perhaps the less complete methyltransferase cluster discussed above (Ga0180325_111152–Ga0180325_111158) is specific to dimethylglycine, which would explain why it does not contain a glycine betaine transporter gene.
Comparison of the complete glycine betaine methyltransferase gene cluster with homologs from other glycine betaine-fermenting bacteria revealed two broad gene cluster architectures, which has not been previously reported. Experimentally proven glycine betaine methyltransferase operons in A. woodii DSM 1030 (Lechtenfeld et al., 2018) and Desulfitobacterium hafniense Y51 (Ticak et al., 2014) demonstrate genomic synteny, along with a homologous gene cluster identified in A. dehalogenans DSM 11527 (Figure 5A). This region is echoed in the second half of the glycine betaine gene cluster identified in strain DCMF (Figure 5). In contrast, the glycine betaine fermenting bacteria S. ovata DSM 2662, S. ovata An4, and ‘Candidatus Frackibacter sp.’ T328-2 all encode syntenic regions to the first half of the strain DCMF cluster (Figure 5B). For the S. ovata strains, these genes differ from those suggested to be responsible for glycine betaine demethylation in previous works, offering new candidates for further study (Visser et al., 2016; Lechtenfeld et al., 2018). These genes suggested here were among the numerous abundant methyltransferases during glycine betaine metabolism by S. ovata An4 (Visser et al., 2016), while proteomic studies of A. woodii DSM 1030 and D. hafniense Y51 have not yet been reported. Strain DCMF similarly expressed a range of methyltransferases with both DCM and glycine betaine-amended cultures, and investigation into the apparent redundancy of these proteins may yield interesting results.
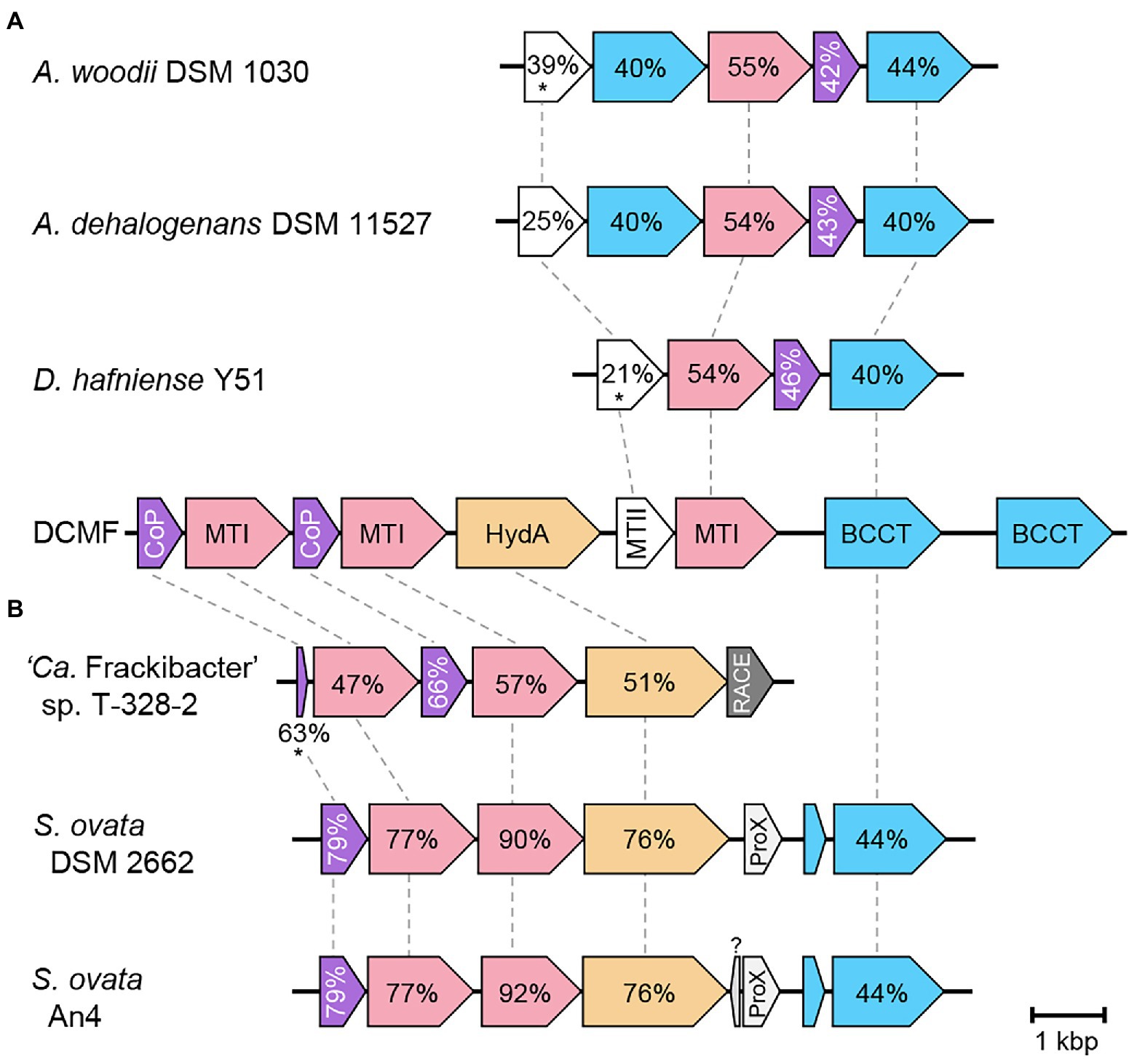
Figure 5. Genetic organization of the glycine betaine methyltransferase gene cluster in strain DCMF and homologs in other glycine betaine-fermenting bacteria. Homologous clusters fell into two distinct groups, with similarity to the second (A) or first (B) half of the strain DCMF gene cluster. Homologous proteins are linked by dotted lines and percentage amino acid sequence identity to strain DCMF written within. Values in the unlinked BCCT and CoP proteins in Panel A are to the strain DCMF homolog with the highest percentage amino acid sequence identity: Ga0180325_114741 or Ga0180325_114736, respectively, in all cases). Loci shown are: Acetobacterium woodii DSM 1030 Awo_c07520 – Awo_07560, Acetobacterium dehalogenans DSM 11527 A3KSDRAFT_02713 – A3KSDRAFT_02709, D. hafniense Y51 DSY3157 – DSY3154, strain DCMF Ga0180325_114734 – Ga0180325_114742, ‘Ca. Frackibacter sp.’ T328-2 AWU54_1980 – AWU54_1975, Sporomusa ovata DSM 2662 SOV_3c09370—SOV_3c09310, S. ovata An4 SpAn4DRAFT_2140 – SpAn4DRAFT_2133. CoP, corrinoid protein; MTI, methyltransferase I; HydA, hydantoinase; MTII, methyltransferase II; BCCT, betaine/carnitine/choline family transporter; RACE, reductive activator of corrinoid-dependent enzymes; ProX, extracellular glycine betaine ligand binding protein. Asterisks indicate proteins with <30% query coverage of the strain DCMF homolog.
It is worth noting that, while the role of DCM in the broader global carbon budget has only recently been reconsidered with increased significance (Hossaini et al., 2017; Murdoch et al., 2022), the links between glycine betaine and climate-active gasses have been reported for decades. In anoxic subsurface environments, it can be microbially transformed to methylamines (Naumann et al., 1983; King, 1984; Möller et al., 1986; Zindel et al., 1988; Heijthuijsen and Hansen, 1989; Mouné et al., 1999; Daly et al., 2016), which are then metabolized almost exclusively by methanogens, generating the potent greenhouse gas methane (Oremland et al., 1982; King, 1984, 1988; Oren, 1990; Ollivier et al., 1994; Daly et al., 2016). Additionally, some species of Methanococcoides can also utilize glycine betaine directly for methanogenesis (Watkins et al., 2014). The environmental relevance of strain DCMF therefore extends beyond DCM-contaminated sites, as the pathways involved in both DCM and glycine betaine degradation affect the flux of climate-active gasses from anoxic, subsurface environments.
None of the cohabitant MAGs contain a complete Wood–Ljungdahl pathway, which is consistent with a previous observation that culture DFE cannot grow autotrophically (Holland et al., 2021). Instead, the MAGs encode genes indicative of oxidative glycolysis for central carbon metabolism. Additionally, the cohabitant MAGs do not encode any corrinoid-dependent methyltransferases or corrinoid proteins, nor homologs to any of the genes in the mec cassette. However, DFE-SYN encoded multiple glycine/betaine/sarcosine reductase genes, including one cluster with all components of a reductase complex and thioredoxin (DFE_SYN_02509–DFE_SYN_02516). The reductase complex component B subunits gamma (DFE_SYN_02511) and alpha/beta (DFE_SYN_02514) were both identified in the metaproteome (Supplementary Table S4). While substrate specificity of this reductase could not be predicted based on sequence similarity to known enzymes (Supplementary Figure S3), there are no published reports of any Synergistetes utilizing glycine betaine or sarcosine, and both components were more highly expressed in DCM-amended cultures than those with glycine betaine (Supplementary Table S4). This suggests that they may be specific to glycine, rather than sarcosine or glycine betaine. Overall, metagenomic data supported previous experimental data suggesting that the cohabitant bacterial lineages are not able to consume any of the primary substrates added to culture DFE, but rather feed on expired cellular material, i.e., necromass (Holland et al., 2021).
In addition to this, we hypothesize that at least some of the cohabiting lineages provide a benefit to strain DCMF, given their persistence despite repeated attempts to isolate the bacterium (Holland et al., 2019, 2021). Cohabiting bacteria can provide various benefits to keystone species, including production of amino acids or essential cofactors for which the other is auxotrophic (e.g., Yan et al., 2012; Embree et al., 2015), or removing nitrogen-rich wastes that might otherwise accumulate to toxic levels (e.g., Christie-Oleza et al., 2017). However, in culture DFE, the exact nature of the syntrophic partnership between strain DCMF and the cohabiting bacteria remains unclear.
Nonetheless, lineages identified in culture DFE—Cupidesulfovibrio, Bacteroidales, Spirochaetes/Treponematales, Synergistetes—have previously been associated with hydrocarbon and organohalide-degrading cultures (Duhamel and Edwards, 2006; Kleinsteuber et al., 2008; Strąpoć et al., 2011; Taubert et al., 2012; Dong et al., 2018), where some reports have suggested that they persist via necromass recycling (Kleinsteuber et al., 2012; Lee et al., 2012; Taubert et al., 2012; Dong et al., 2018). Microbial necromass utilization is increasingly being considered as an important contributor to subsurface nutrient cycling (Simpson et al., 2007; Liang et al., 2011). At contaminated sites in particular, it may enhance bioremediation, as the production of hydrogen, acetate, and ethanol can both stimulate microbial blooms or serve as secondary substrates for co-metabolism (Horvath, 1972; Wrighton et al., 2014). Outside of necromass studies, similarly “self-feeding” mixed cultures have been described for dechlorination of trichloromethane and DCM (Wang et al., 2022), chlorobenzene (Liang et al., 2013), and 3-chlorobenzoate (Dolfing, 1986; Dolfing and Tiedje, 1987). Consumption of dead biomass can also remineralize or liberate important nutrients including nitrogen, phosphorous, trace elements, and cobalamins (Head et al., 2006; Christie-Oleza et al., 2017). Following in this theme are calls from other authors for the use of mixed consortia, rather than monocultures for biotechnological processes (Giri et al., 2020; Borchert et al., 2021). Diverse communities are more robust against environmental and ecological disturbances and have increased functionality due to synergistic interspecies interactions (Chiu et al., 2014; Pande and Kost, 2017; Giri et al., 2020; Pascual-García et al., 2020). Understanding microbial interactions in culture DFE, in which a chlorinated one-carbon compound has sustained a stable community for 10 years, can reveal new insights into syntrophic community dynamics and could aid the development of more robust mixed cultures for in situ bioremediation applications.
Proteomic study of the DCM-fermenting bacterium in culture DFE, ‘Ca. Formimonas warabiya’ strain DCMF, supports the role of a methyltransferase system encoded by the mec cassette in DCM dechlorination. The mec cassette encodes candidate genes for tracking anaerobic DCM metabolism in situ at contaminated sites, and this work lays the foundation for biochemical/structural characterization of DCM dechlorinating enzymes. Proteogenomic evidence for a putative glycine betaine methyltransferase in strain DCMF also added to our limited knowledge regarding the fate of this environmentally important compound in anoxic subsurface environments. Furthermore, analysis of metaproteogenomic data from the cohabiting linegaes in culture DFE supported the previous hypothesis that they are not involved in primary substrate metabolism, but rather persist via metabolism of necromass from spent cells.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
SH, MM, and ML conceived the study. SH performed the experiments, wrote the manuscript, and produced the figures. SH and HE analyzed the data. XV-C and RE advised and assisted with metagenomic assembly and analysis. RE and MM contributed the resources. All authors contributed to the article and approved the submitted version.
SH was supported by an Australian Government Research Training Program Scholarship. XV-C acknowledges support from the New South Wales State Government RAAP scheme and the National Collaborative Research Infrastructure Strategy. RE was funded by the Australian Research Council (ARC LP160100610).
Mass spectrometric analysis for this work was carried out at the Bioanalytical Mass Spectrometry Facility (BMSF), UNSW and was supported in part by infrastructure funding from the New South Wales Government as part of its co-investment in the National Collaborative Research Infrastructure Strategy. We are grateful to Ling Zhong at the BMSF for her assistance with LC–MS/MS. This research includes computations using the computational cluster Katana supported by Research Technology Services at UNSW Sydney. We acknowledge the Dharawal Language Program and thank the Eastern Zone Gujaga Aboriginal Corporation (Gujaga) for granting a non-exclusive license to use the placename, Warabiya, in the candidate species name for the bacterial lineage under investigation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1035247/full#supplementary-material
Agency for Toxic Substances and Disease Registry (2000). Toxicological profile for methylene chloride. Atlanta, GA.
Anantharaman, V., and Aravind, L. (2005). MEDS and PocR are novel domains with a predicted role in sensing simple hydrocarbon derivatives in prokaryotic signal transduction systems. Bioinformatics 21, 2805–2811. doi: 10.1093/bioinformatics/bti418
Arumugam, K., Bağcı, C., Bessarab, I., Beier, S., Buchfink, B., Górska, A., et al. (2019). Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data. Microbiome 7:61. doi: 10.1186/s40168-019-0665-y
Beers, J. R. (1967). The species distribution of some naturally occurring quaternary ammonium compounds. Comp. Biochem. Physiol. 21, 11–21. doi: 10.1016/0010-406X(67)90109-0
Borchert, E., Hammerschmidt, K., Hentschel, U., and Deines, P. (2021). Enhancing microbial pollutant degradation by integrating eco-evolutionary principles with environmental biotechnology. Trends Microbiol. 29, 908–918. doi: 10.1016/j.tim.2021.03.002
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Burke, S. A., Lo, S. L., and Krzycki, J. A. (1998). Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine. J. Bacteriol. 180, 3432–3440. doi: 10.1128/JB.180.13.3432-3440.1998
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics :btz848. doi: 10.1093/bioinformatics/btz848
Chen, G., Fisch, A. R., Gibson, C. M., Erin Mack, E., Seger, E. S., Campagna, S. R., et al. (2020). Mineralization versus fermentation: evidence for two distinct anaerobic bacterial degradation pathways for dichloromethane. ISME J. 14, 959–970. doi: 10.1038/s41396-019-0579-5
Chen, G., Murdoch, R. W., Mack, E. E., Seger, E. S., and Löffler, F. E. (2017). Complete genome sequence of Dehalobacterium formicoaceticum strain DMC, a strictly anaerobic dichloromethane-degrading bacterium. Genome Announc. 5, 18–19. doi: 10.1128/genomeA.00897-17
Chen, G., Shouakar-Stash, O., Phillips, E., Justicia-Leon, S. D., Gilevska, T., Sherwood Lollar, B., et al. (2018). Dual carbon–chlorine isotope analysis indicates distinct anaerobic dichloromethane degradation pathways in two members of Peptococcaceae. Environ. Sci. Technol. 52, 8607–8616. doi: 10.1021/acs.est.8b01583
Chiu, H.-C., Levy, R., and Borenstein, E. (2014). Emergent biosynthetic capacity in simple microbial communities. PLoS Comput. Biol. 10:e1003695. doi: 10.1371/journal.pcbi.1003695
Christie-Oleza, J. A., Sousoni, D., Lloyd, M., Armengaud, J., and Scanlan, D. J. (2017). Nutrient recycling facilitates long-term stability of marine microbial phototroph–heterotroph interactions. Nat. Microbiol. 2:17100. doi: 10.1038/nmicrobiol.2017.100
Cox, J., Hein, M. Y., Luber, C. A., Paron, I., Nagaraj, N., and Mann, M. (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. doi: 10.1074/mcp.M113.031591
Csonka, L. N. (1989). Physiological and genetic responses of bacteria to osmotic stress. Microbiol. Rev. 53, 121–147. doi: 10.1128/mr.53.1.121-147.1989
Daly, R. A., Borton, M. A., Wilkins, M. J., Hoyt, D. W., Kountz, D. J., Wolfe, R. A., et al. (2016). Microbial metabolisms in a 2.5-km-deep ecosystem created by hydraulic fracturing in shales. Nat. Microbiol. 1:16146. doi: 10.1038/nmicrobiol.2016.146
Dolfing, J. (1986). Hydrogen cycling in a three-tiered food web growing on the methanogenic conversion of 3-chlorobenzoate. FEMS Microbiol. Lett. 38, 293–298. doi: 10.1016/0378-1097(86)90005-4
Dolfing, J., and Tiedje, J. M. (1987). Growth yield increase linked to reductive dechlorination in a defined 3-chlorobenzoate degrading methanogenic coculture. Arch. Microbiol. 149, 102–105. doi: 10.1007/BF00425073
Dong, X., Greening, C., Brüls, T., Conrad, R., Guo, K., Blaskowski, S., et al. (2018). Fermentative Spirochaetes mediate necromass recycling in anoxic hydrocarbon-contaminated habitats. ISME J. 12, 2039–2050. doi: 10.1038/s41396-018-0148-3
Drummond, J. T., Huang, S., Matthews, R. G., and Blumenthal, R. M. (1993). Assignment of enzymatic function to specific protein regions of cobalamin-dependent methionine synthase from Escherichia coli. Biochemistry 32, 9290–9295. doi: 10.1021/bi00087a005
Duhamel, M., and Edwards, E. A. (2006). Microbial composition of chlorinated ethene-degrading cultures dominated by Dehalococcoides. FEMS Microbiol. Ecol. 58, 538–549. doi: 10.1111/j.1574-6941.2006.00191.x
Ellenbogen, J. B., Jiang, R., Kountz, D. J., Zhang, L., and Krzycki, J. A. (2021). The MttB superfamily member MtyB from the human gut symbiont Eubacterium limosum is a cobalamin-dependent γ-butyrobetaine methyltransferase. J. Biol. Chem. 297:101327. doi: 10.1016/j.jbc.2021.101327
Embree, M., Liu, J. K., Al-Bassam, M. M., and Zengler, K. (2015). Networks of energetic and metabolic interactions define dynamics in microbial communities. Proc. Natl. Acad. Sci. U. S. A. 112, 15450–15455. doi: 10.1073/pnas.1506034112
Eren, A. M., Kiefl, E., Shaiber, A., Veseli, I., Miller, S. E., Schechter, M. S., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6. doi: 10.1038/s41564-020-00834-3
European Parliament Council of the European Union (2013). Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy’ (2013) Official Journal L 226, p. 12. Available at https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32013L0039
Ferguson, D. J., and Krzycki, J. A. (1997). Reconstitution of trimethylamine-dependent coenzyme M methylation with the trimethylamine corrinoid protein and the isozymes of methyltransferase II from Methanosarcina barkeri. J. Bacteriol. 179, 846–852.
Ferguson, T., Soares, J. A., Lienard, T., Gottschalk, G., and Krzycki, J. A. (2009). RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic archaea. J. Biol. Chem. 284, 2285–2295. doi: 10.1074/jbc.M807392200
Giri, S., Shitut, S., and Kost, C. (2020). Harnessing ecological and evolutionary principles to guide the design of microbial production consortia. Curr. Opin. Biotechnol. 62, 228–238. doi: 10.1016/j.copbio.2019.12.012
Gribble, G. W. (2010). “Naturally occurring Organohalogen compounds - a comprehensive update” in Progress in the chemistry of organic natural products (Vienna: Springer Vienna), 12–13. doi: 10.1007/978-3-211-99323-1
Hao, B., Gong, W., Ferguson, T. K., James, C. M., Joseph, A., and Chan, M. K. (2002). A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 296, 1462–1466. doi: 10.1126/science.1069556
Head, I. M., Jones, D. M., and Röling, W. F. M. (2006). Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4, 173–182. doi: 10.1038/nrmicro1348
Heijthuijsen, J. H. F. G., and Hansen, T. A. (1989). Betaine fermentation and oxidation by marine Desulfuromonas strains. Appl. Environ. Microbiol. 55, 965–969.
Holland, S. I., Edwards, R. J., Ertan, H., Wong, Y. K., Russell, T. L., Deshpande, N. P., et al. (2019). Whole genome sequencing of a novel, dichloromethane-fermenting Peptococcaceae from an enrichment culture. PeerJ 7:e7775. doi: 10.7717/peerj.7775
Holland, S. I., Ertan, H., Montgomery, K., Manefield, M. J., and Lee, M. (2021). Novel dichloromethane-fermenting bacteria in the Peptococcaceae family. ISME J. 15, 1709–1721. doi: 10.1038/s41396-020-00881-y
Holliger, C., Wohlfarth, G., and Diekert, G. (1999). Reductive dechlorination in the energy metabolism of anaerobic bacteria. FEMS Microbiol. Rev. 22, 383–398. doi: 10.1111/j.1574-6976.1998.tb00377.x
Horvath, R. S. (1972). Microbial co-metabolism and the degradation of organic compounds in nature. Bacteriol. Rev. 36, 146–155. doi: 10.1128/br.36.2.146-155.1972
Hossaini, R., Chipperfield, M. P., Montzka, S. A., Leeson, A. A., Dhomse, S. S., and Pyle, J. A. (2017). The increasing threat to stratospheric ozone from dichloromethane. Nat. Commun. 8:15962. doi: 10.1038/ncomms15962
Hossaini, R., Chipperfield, M. P., Saiz-Lopez, A., Harrison, J. J., von Glasow, R., Sommariva, R., et al. (2015). Growth in stratospheric chlorine from short-lived chemicals not controlled by the Montreal protocol. Geophys. Res. Lett. 42, 4573–4580. doi: 10.1002/2015GL063783
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). EggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN Community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12, 1–12. doi: 10.1371/journal.pcbi.1004957
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kaufmann, F., Wohlfarth, G., and Diekert, G. (1997). Isolation of O-demethylase, an ether-cleaving enzyme system of the homoacetogenic strain MC. Arch. Microbiol. 168, 136–142. doi: 10.1007/s002030050479
King, G. M. (1984). Metabolism of trimethylamine, choline, and glycine betaine by sulfate-reducing and methanogenic bacteria in marine sediments. Appl. Environ. Microbiol. 48, 719–725. doi: 10.1080/01490458409377807
King, G. M. (1988). “Distribution and metabolism of quaternary amines in marine sediments” in Nitrogen cycling in coastal marine environments. eds. T. H. Blackburn and J. Soorensen (New York, NY: John Wiley & Sons), 143–173.
Kleindienst, S., Chourey, K., Chen, G., Murdoch, R. W., Higgins, S. A., Iyer, R., et al. (2019). Proteogenomics reveals novel reductive dehalogenases and methyltransferases expressed during anaerobic dichloromethane metabolism. Appl. Environ. Microbiol. 85, 1–16. doi: 10.1128/aem.02768-18
Kleindienst, S., Higgins, S. A., Tsementzi, D., Chen, G., Konstantinidis, K. T., Mack, E. E., et al. (2017). ‘Candidatus Dichloromethanomonas elyunquensis’ gen. Nov., sp. nov., a dichloromethane-degrading anaerobe of the Peptococcaceae family. Syst. Appl. Microbiol. 40, 150–159. doi: 10.1016/j.syapm.2016.12.001
Kleinsteuber, S., Schleinitz, K. M., Breitfeld, J., Harms, H., Richnow, H. H., and Vogt, C. (2008). Molecular characterization of bacterial communities mineralizing benzene under sulfate-reducing conditions. FEMS Microbiol. Ecol. 66, 143–157. doi: 10.1111/j.1574-6941.2008.00536.x
Kleinsteuber, S., Schleinitz, K. M., and Vogt, C. (2012). Key players and team play: anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl. Microbiol. Biotechnol. 94, 851–873. doi: 10.1007/s00253-012-4025-0
Kolmogorov, M., Bickhart, D. M., Behsaz, B., Gurevich, A., Rayko, M., Shin, S. B., et al. (2020). metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110. doi: 10.1038/s41592-020-00971-x
Kountz, D. J., Behrman, E. J., Zhang, L., and Krzycki, J. A. (2020). MtcB, a member of the MttB superfamily from the human gut acetogen Eubacterium limosum, is a cobalamin-dependent carnitine demethylase. J. Biol. Chem. 295, 11971–11981. doi: 10.1074/jbc.RA120.012934
Kremp, F., and Müller, V. (2021). Methanol and methyl group conversion in acetogenic bacteria: biochemistry, physiology and application. FEMS Microbiol. Rev. 45, 1–22. doi: 10.1093/femsre/fuaa040
Larher, F., Jolivet, Y., Briens, U., and Goas, U. (1982). Osmoregulation in higher plants halophytes: organic nitrogen accumulation in glycine, betaine, and proline during the growth of Aster tripolinum and Sueda macrocarpa under saline conditions. Plant Sci. Lett. 24, 201–210.
Lechtenfeld, M., Heine, J., Sameith, J., Kremp, F., and Müller, V. (2018). Glycine betaine metabolism in the acetogenic bacterium Acetobacterium woodii. Environ. Microbiol. 20, 4512–4525. doi: 10.1111/1462-2920.14389
Lee, M., Low, A., Zemb, O., Koenig, J., Michaelsen, A., and Manefield, M. (2012). Complete chloroform dechlorination by organochlorine respiration and fermentation. Environ. Microbiol. 14, 883–894. doi: 10.1111/j.1462-2920.2011.02656.x
Leedham Elvidge, E. C., Oram, D. E., Laube, J. C., Baker, A. K., Montzka, S. A., Humphrey, S., et al. (2015). Increasing concentrations of dichloromethane, CH2Cl2, inferred from CARIBIC air samples collected 1998-2012. Atmos. Chem. Phys. 15, 1939–1958. doi: 10.5194/acp-15-1939-2015
Liang, C., Cheng, G., Wixon, D. L., and Balser, T. C. (2011). An absorbing Markov chain approach to understanding the microbial role in soil carbon stabilization. Biogeochemistry 106, 303–309. doi: 10.1007/s10533-010-9525-3
Liang, X., Devine, C. E., Nelson, J., Sherwood Lollar, B., Zinder, S., and Edwards, E. A. (2013). Anaerobic conversion of chlorobenzene and benzene to CH4 and CO2 in bioaugmented microcosms. Environ. Sci. Technol. 47, 2378–2385. doi: 10.1021/es3043092
Mägli, A., Messmer, M., and Leisinger, T. (1998). Metabolism of dichloromethane by the strict anaerobe Dehalobacterium formicoaceticum. Appl. Environ. Microbiol. 64, 646–650.
Mägli, A., Wendt, M., and Leisinger, T. (1996). Isolation and characterization of Dehalobacterium formicoaceticum gen. Nov. sp. nov., a strictly anaerobic bacterium utilizing dichloromethane as source of carbon and energy. Arch. Microbiol. 166, 101–108. doi: 10.1007/s002030050362
Menon, S., and Ragsdale, S. W. (1999). The role of an iron-sulfur cluster in an enzymatic methylation reaction: methylation of CO dehydrogenase/acetyl-CoA synthase by the methylated corrinoid iron-sulfur protein. J. Biol. Chem. 274, 11513–11518. doi: 10.1074/jbc.274.17.11513
Meßmer, M., Reinhardt, S., Wohlfarth, G., and Diekert, G. (1996). Studies on methyl chloride dehalogenase and O-demethylase in cell extracts of the homoacetogen strain MC based on a newly developed coupled enzyme assay. Arch. Microbiol. 165, 18–25. doi: 10.1007/s002030050291
Ministry of Environmental Protection (MEP) (2017). “List of Chemicals Under Priority Control (First Batch). Action Plan for Prevention and Control of Water Pollution,” Order No.17 [2015] of the State Council, Ministry of Environmental Protection of China.
Möller, B., Hippe, H., and Gottschalk, G. (1986). Degradation of various amine compounds by mesophilic clostridia. Arch. Microbiol. 145, 85–90. doi: 10.1007/BF00413032
Mouné, S., Manac’h, N., Hirschler, A., Caumette, P., Willison, J. C., and Matheron, R. (1999). Haloanaerobacter salinarius sp. nov., a novel halophilic fermentative bacterium that reduces glycine-betaine to trimethylamine with hydrogen or serine as electron donors; emendation of the genus Haloanaerobacter. Int J Syst Biol 49, 103–112. doi: 10.1099/00207713-49-1-103
Murdoch, R. W., Chen, G., Kara Murdoch, F., Mack, E. E., Villalobos Solis, M. I., Hettich, R. L., et al. (2022). Identification and widespread environmental distribution of a gene cassette implicated in anaerobic dichloromethane degradation. Glob. Chang. Biol. 28, 2396–2412. doi: 10.1111/gcb.16068
Naumann, E., Hippe, H., and Gottschalk, G. (1983). Betaine: new oxidant in the Stickland reaction and methanogenesis from betaine and L-alanine by a clostridium sporogenes-Methanosarcina barkeri coculture. Appl. Environ. Microbiol. 45, 474–483. doi: 10.1128/aem.45.2.474-483.1983
Ollivier, B., Caumette, P., Garcia, J.-L., and Mah, R. A. (1994). Anaerobic bacteria from hypersaline environments. Microbiol. Rev. 58, 27–38. doi: 10.1128/mr.58.1.27-38.1994
Oremland, R. S., Marsh, L. M., and Polcin, S. (1982). Methane production and simultaneous sulphate reduction in anoxic, salt marsh sediments. Nature 296, 143–145. doi: 10.1038/296143a0
Oren, A. (1990). Formation and breakdown of glycine betaine and trimethylamine in hypersaline environments. Antonie Van Leeuwenhoek 58, 291–298. doi: 10.1007/BF00399342
Pande, S., and Kost, C. (2017). Bacterial unculturability and the formation of intercellular metabolic networks. Trends Microbiol. 25, 349–361. doi: 10.1016/j.tim.2017.02.015
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pascual-García, A., Bonhoeffer, S., and Bell, T. (2020). Metabolically cohesive microbial consortia and ecosystem functioning. Philos. Trans. R Soc. Lond. B Biol. Sci. 375:20190245. doi: 10.1098/rstb.2019.0245
Paul, L., Ferguson, D. J., and Krzycki, J. A. (2000). The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read- through amber codons. J. Bacteriol. 182, 2520–2529. doi: 10.1128/JB.182.9.2520-2529.2000
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Picking, J. W., Behrman, E. J., Zhang, L., and Krzycki, J. A. (2019). MtpB, a member of the MttB superfamily from the human intestinal acetogen Eubacterium limosum, catalyzes proline betaine demethylation. J. Biol. Chem. 294, 13697–13707. doi: 10.1074/jbc.RA119.009886
Price, M. N., Zane, G. M., Kuehl, J. V., Melnyk, R. A., Wall, J. D., Deutschbauer, A. M., et al. (2018). Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics. PLoS Genet. 14, 1–23. doi: 10.1371/journal.pgen.1007147
Ragsdale, S. W. (2008). “Catalysis of methyl group transfers involving tetrahydrofolate and B12” in Vitamins and Hormones (Elsevier), 293–324.
Schilhabel, A., Studenik, S., Vödisch, M., Kreher, S., Schlott, B., Pierik, A. Y., et al. (2009). The ether-cleaving methyltransferase system of the strict anaerobe Acetobacterium dehalogenans: analysis and expression of the encoding genes. J. Bacteriol. 191, 588–599. doi: 10.1128/JB.01104-08
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shestakova, M., and Sillanpää, M. (2013). Removal of dichloromethane from ground and wastewater: a review. Chemosphere 93, 1258–1267. doi: 10.1016/j.chemosphere.2013.07.022
Simpson, A. J., Simpson, M. J., Smith, E., and Kelleher, B. P. (2007). Microbially derived inputs to soil organic matter: are current estimates too low? Environ. Sci. Technol. 41, 8070–8076. doi: 10.1021/es8000932
Søndergaard, D., Pedersen, C. N. S., and Greening, C. (2016). HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6, 1–8. doi: 10.1038/srep34212
Strąpoć, D., Mastalerz, M., Dawson, K., Macalady, J., Callaghan, A. V., Wawrik, B., et al. (2011). Biogeochemistry of microbial coal-bed methane. Annu. Rev. Earth Planet. Sci. 39, 617–656. doi: 10.1146/annurev-earth-040610-133343
Studer, A., Vuilleumier, S., and Leisinger, T. (1999). Properties of the methylcobalamin:H4folate methyltransferase involved in chloromethane utilization by Methylobacterium sp. strain CM4. Eur. J. Biochem. 264, 242–249. doi: 10.1046/j.1432-1327.1999.00629.x
Taubert, M., Vogt, C., Wubet, T., Kleinsteuber, S., Tarkka, M. T., Harms, H., et al. (2012). Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J. 6, 2291–2301. doi: 10.1038/ismej.2012.68
Ticak, T., Kountz, D. J., Girosky, K. E., Krzycki, J. A., and Ferguson, D. J. (2014). A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 111, E4668–E4676. doi: 10.1073/pnas.1409642111
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
United States Environmental Protection Agency (1977). Priority Pollutant List. Washington, DC: US EPA.
van der Meijden, P., van der Drift, C., and Vogels, G. D. (1984). Methanol conversion in Eubacterium limosum. Arch. Microbiol. 138, 360–364. doi: 10.1007/BF00410904
Vannelli, T., Messmer, M., Studer, A., Vuilleumier, S., and Leisinger, T. (1999). A corrinoid-dependent catabolic pathway for growth of a Methylobacterium strain with chloromethane. Proc. Natl. Acad. Sci. U. S. A. 96, 4615–4620. doi: 10.1073/pnas.96.8.4615
Vannelli, T., Studer, A., Kertesz, M., and Leisinger, T. (1998). Chloromethane metabolism by Methylobacterium sp. strain CM4. Appl. Environ. Microbiol. 64, 1933–1936. doi: 10.1128/AEM.64.5.1933-1936.1998
Visser, M., Pieterse, M. M., Pinkse, M. W. H., Nijsse, B., Verhaert, P. D. E. M., de Vos, W. M., et al. (2016). Unravelling the one-carbon metabolism of the acetogen Sporomusa strain An4 by genome and proteome analysis. Environ. Microbiol. 18, 2843–2855. doi: 10.1111/1462-2920.12973
Wan, Y. Y., Luo, N., Liu, X.-L., Lai, Q.-L., and Goodfellow, M. (2021). Cupidesulfovibrio liaohensis gen. Nov., sp. nov., a novel sulphate-reducing bacterium isolated from an oil reservoir and reclassification of Desulfovibrio oxamicus and Desulfovibrio termitidis as Cupidesulfovibrio oxamicus comb. Int. J. Syst. Evol. Microbiol. 71, 1–9. doi: 10.1099/ijsem.0.004618
Wang, H., Yu, R., Webb, J., Dollar, P., and Freedman, D. L. (2022). Anaerobic biodegradation of chloroform and dichloromethane with a Dehalobacter enrichment culture. Appl. Environ. Microbiol. 88, e01970–e01921. doi: 10.1128/aem.01970-21
Watkins, A. J., Rousse, E. G., Parkes, R. J., and Sass, H. (2014). Glycine betaine as a direct substrate for methanogens (Methanococcoides spp.). Appl. Environ. Microbiol. 80, 289–293. doi: 10.1128/AEM.03076-13
Wiśniewski, J. R. (2017). “Filter-aided sample preparation: the versatile and efficient method for proteomic analysis” in Methods in Enzymology. Proteomics in Biology, Part A. ed. A. K. Shukla (Amsterdam, Netherlands: Elsevier BV), 15–27. doi: 10.1016/bs.mie.2016.09.013
Wohlfarth, G., and Diekert, G. (1997). Anaerobic dehalogenases. Curr. Opin. Biotechnol. 8, 290–295. doi: 10.1016/S0958-1669(97)80006-7
Wrighton, K. C., Castelle, C. J., Wilkins, M. J., Hug, L. A., Sharon, I., Thomas, B. C., et al. (2014). Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J. 8, 1452–1463. doi: 10.1038/ismej.2013.249
Yan, J., Ritalahti, K. M., Wagner, D. D., and Löffler, F. E. (2012). Unexpected specificity of interspecies cobamide transfer from Geobacter spp. to organohalide-respiring Dehalococcoides mccartyi strains. Appl. Environ. Microbiol. 78, 6630–6636. doi: 10.1128/AEM.01535-12
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). DbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zindel, U., Freudenberg, W., Rieth, M., Andreesen, J. R., Schnell, J., and Widdel, F. (1988). Eubacterium acidaminophilum sp. nov., a versatile amino acid-degrading anaerobe producing or utilizing H2 or formate - description and enzymatic studies. Arch. Microbiol. 150, 254–266. doi: 10.1007/BF00407789
Keywords: dichloromethane, anaerobic dechlorination, methyltransferase, glycine betaine, Wood–Ljungdahl pathway, metaproteomics, subsurface
Citation: Holland SI, Vázquez-Campos X, Ertan H, Edwards RJ, Manefield MJ and Lee M (2022) Metaproteomics reveals methyltransferases implicated in dichloromethane and glycine betaine fermentation by ‘Candidatus Formimonas warabiya’ strain DCMF. Front. Microbiol. 13:1035247. doi: 10.3389/fmicb.2022.1035247
Edited by:
Marcus A. Horn, Leibniz University Hannover, GermanyReviewed by:
Tobias Goris, German Institute of Human Nutrition Potsdam-Rehbruecke (DIfE), GermanyCopyright © 2022 Holland, Vázquez-Campos, Ertan, Edwards, Manefield and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew Lee, bWF0dGxlZUB1bnN3LmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.