 Sinil Kim
Sinil Kim Hyerang Eom
Hyerang Eom Rutuja Nandre
Rutuja Nandre Yeon Jae Choi
Yeon Jae Choi Hojin Ryu
Hojin Ryu Hyeon-Su Ro
Hyeon-Su Ro
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 November 2022
Sec. Evolutionary and Genomic Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1034387
This article is part of the Research Topic Basidiomycete Fungi: From Biosystematics and Biodiversity to Biotechnology View all 21 articles
The evolution of mitochondria through variations in mitochondrial DNA (mtDNA) is one of the intriguing questions in eukaryotic cells. In order to assess the causes of the variations in mitochondria, the mtDNAs of the 21 strains of Lentinula edodes were assembled for this study, and analyzed together with four published mtDNA sequences. The mtDNAs were within the sizes of 117 kb ~ 122 kb. The gene number was observed consistent except for two mtDNAs, which carry a duplicated trnG1-trnG2 unit or a putative gene deletion. The size variation was largely attributed to the number of introns, repeated sequences, transposable elements (TEs), and plasmid-related sequences. Intron loss and gain were found from cox1, rnl, and rns of three mtDNAs. Loss of two introns in cox1 of KY217797.1 reduced its size by 2.7 kb, making it the smallest cox1 gene (8.4 kb) among the cox1s of the 25 mtDNAs, whereas gain of a Group II intron (2.65 kb) and loss of a Group I intron (1.7 kb) in cox1 of MF774813.1 resulted in the longest cox1 (12 kb). In rnl of L. edodes, we discovered four intron insertion consensus sequences which were unique to basidiomycetes but not ascomycetes. Differential incorporation of introns was the primary cause of the rnl size polymorphism. Homing endonucleases (HEGs) were suggestively involved in the mobilization of the introns because all of the introns have HEG genes of the LAGRIDADG or GIY-YIG families with the conserved HEG cleavage sites. TEs contributed to 11.04% of the mtDNA size in average, of which 7.08% was LTR-retrotransposon and 3.96% was DNA transposon, whereas the repeated sequences covered 4.6% of the mtDNA. The repeat numbers were variable in a strain-dependent manner. Both the TEs and repeated sequences were mostly found in the intronic and intergenic regions. Lastly, two major deletions were found in the plasmid-related sequence regions (pol2-pol3 and pol1-atp8) in the five mtDNAs. Particularly, the 6.8 kb-long deletion at pol2-pol3 region made MF774813.1 the shortest mtDNA of all. Our results demonstrate that mtDNA is a dynamic molecule that persistently evolves over a short period of time by insertion/deletion and repetition of DNA segments at the strain level.
Mitochondrion, the membrane-bound cellular organelle, is the powerhouse of eukaryotic cells that produces ATP through oxidative phosphorylation. It also involves in the cellular events like cell metabolism, calcium signaling, and apoptosis (Chan, 2006). Mitochondrion carries its own DNA (mtDNA), which is inherited independently of the nuclear DNA. The mitochondrial genome consists of genes for ATP generation, tRNAs, rRNAs, etc. The mtDNA size is highly dependent on organisms of which animals have relatively constant 16.4 kb mtDNAs whereas land plants have variable sizes with the average length of 412 kb (NCBI Genome1). The mtDNAs of the higher fungi, which include ascomycetes and basidiomycetes, have an average length of 65.2 kb and varies in size from 18.8 kb in Hanseniaspora uvarum (Pramateftaki et al., 2006) to 332 kb in Golovinomyces cichoracearum (Zaccaron and Stergiopoulos, 2021). Despite the size difference, the number of essential genes are relatively constant. The size diversity is mainly due to intron insertion, plasmid DNA insertion, partial duplication of mtDNA sequences, and changes in repetitive sequences (Hausner, 2003; Liu et al., 2020). The sequence variations make the mtDNA an active evolutionary genetic marker (Shen et al., 2015; Jelen et al., 2016; Robicheau et al., 2017; Zhang et al., 2017).
The mtDNA is acquired from one of the parent cells during the sexual reproduction(Sato and Sato, 2012). Cryptococcus neoformans, a basidiomycetous yeast, was observed to inherit the mtDNA in a uniparental way possibly through selective destruction of a mitochondrial type or through the emergence of bud where one parent is concentrated (Xu et al., 2000). Ustilago maydis also shows uniparental inheritance associated with a defined mating type, where the result is strongly affected by mating-type locus (Xu et al., 2000; Yan and Xu, 2003; Fedler et al., 2009). In the mating of filamentous basidiomyceteous fungi, two types of dikaryotic cells, which carry one of the two mtDNAs originated from the two monokaryotic cells, are generated through reciprocal entrance of nuclei to the cytoplasms of monokaryotic cells after hyphal fusion between monokaryotic hyphae with compatible mating types (Kim et al., 2019).
Lentinula edodes, shiitake mushroom, is a basidiomycete fungus that is a popular commercially cultivated mushroom. It is mainly found in East Asian countries, including China, Japan, and Korea (Menolli et al., 2022). Genomic DNA sequencing revealed variations in the genome size and gene number as the L. edodes B17 strain (Korea) consists of chromosomal DNA of 46.1 Mb with 13,028 predicted genes (Shim et al., 2016), while the L. edodes W1-26 strain (China) carries chromosomal DNA of 41.8 Mb with 14,889 predicted genes (Chen et al., 2016). The genetic diversity analysis using the mating type genes of 127 strains of L. edodes showed a high degree of diversity particularly in the A mating type locus of the wild strains (Ha et al., 2018). The diverse nature of the A mating type genes enabled the discrimination of nuclei in dikaryons as well as monokaryons of different origins (Kim et al., 2019). The mtDNA is another source of genetic information. Four mtDNAs of L. edodes have been reported thus far; they are AB697988.1, KY217797.1, MF774812.1, and MF774813.1 with the lengths of 121,394, 116,897, 119,134, and 115,116 bp, respectively. Gene annotation revealed the presence of 15 protein-coding genes (cob, atp6, 8, 9, nad1-6, 4 l, cox1-3, and rps3), two rRNAs (rns and rnl), 26 tRNAs, multiple intronic ORFs, and putative protein-coding genes (Song et al., 2019).
In recent years, we have analyzed the mtDNAs of different strains of L. edodes and found length polymorphism and the sequence variations in the mtDNAs of various origins, one of which was the presence of more than 25 variable-length tandem repeats in the intergenic and intronic regions, contributing the size polymorphism (Kim et al., 2019). For further understanding of the mtDNA diversification, we determined the mtDNA sequences in 20 selected strains from the 127 strains that were used for the A mating type analysis. This paper reports the detailed gene arrangement, variations in gene structure, and length polymorphism of the mtDNAs of L. edodes.
The strains of L. edodes used for the mtDNA analysis were chosen on the basis of the A mating type diversity as verified in our previous paper (Table 1; Ha et al., 2018). The strains were maintained on a potato-dextrose agar (PDA). For the preparation of total DNA, the mycelia were cultured in potato-dextrose broth (PDB) for 10 days at 25°C. The mycelia were harvested by centrifugation for 10 min at 3,000 × g. The harvested mycelia were washed with a PBS buffer and then dried with a kitchen towel. Approximately 100 mg of dried mycelium was flash-frozen in liquid nitrogen, and then genomic DNA was extracted using the GenExTM Plant kit (GeneAll, Seoul, Korea). The extracted DNA was measured using a Micro-spectrophotometer K5600 (BioFuture Inc., China).
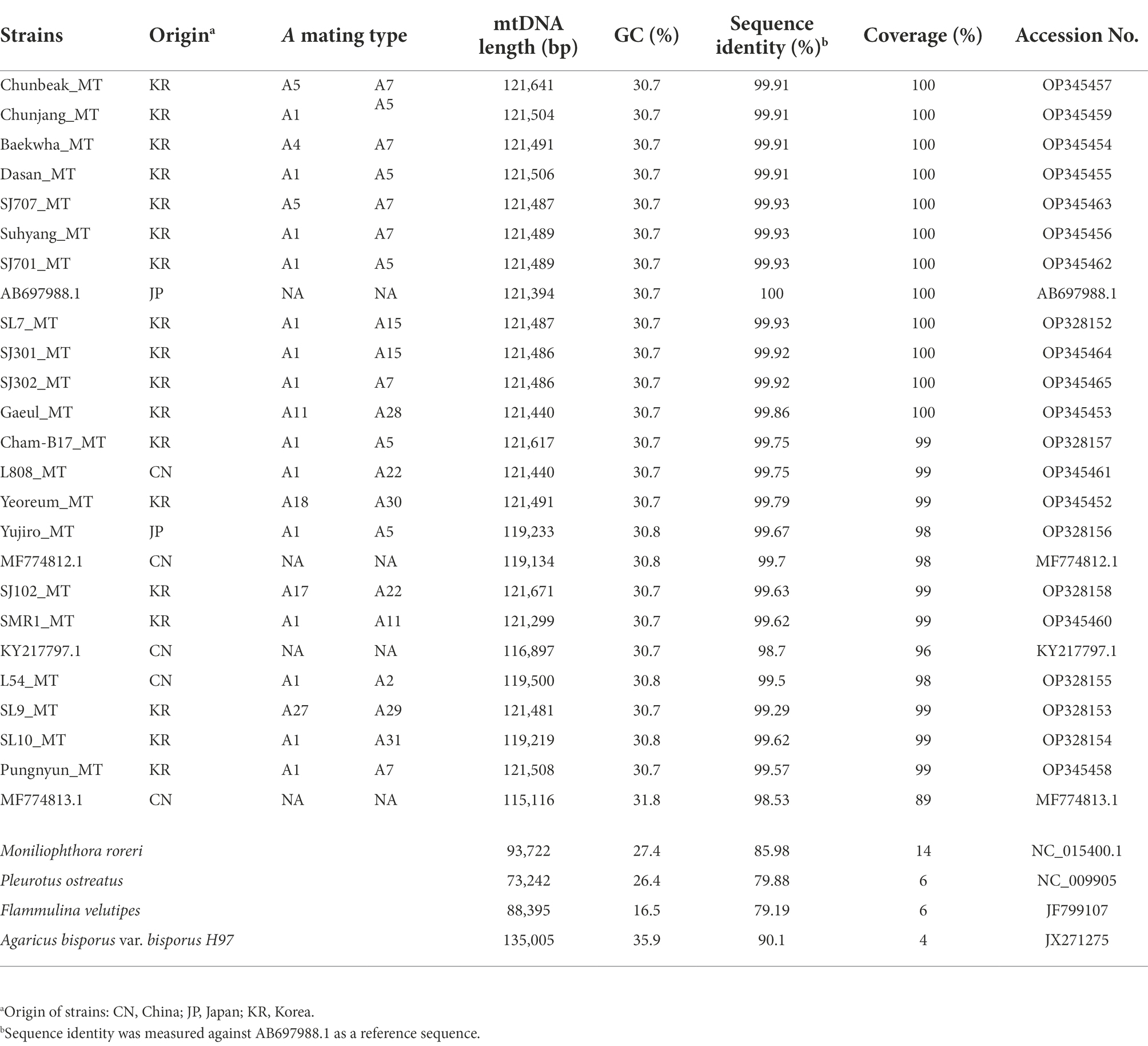
Table 1. Characteristics of mtDNAs of Lentinula edodes and some selected mushrooms.
The mtDNA sequences of the 21 selected L. edodes strains were assembled using the genomic DNA reads generated from the previous NGS sequencing (Shim et al., 2016; Lee et al., 2017). Briefly, the genomic DNA reads were mapped against AB697988.1 as a reference mtDNA by BWA (v.0.7.17; Li and Durbin, 2009). The mapped reads were assembled using SPAdes (v.3.12.0; Nurk et al., 2013), and the error-correction was performed using Pilon (v.1.22; Walker et al., 2014). The assembled mtDNA sequences were analyzed together with the four published L. edodes mtDNA sequences, including AB697988.1, KY217797.1, MF774812.1, and MF774813.1. MAFFT (v.7) online version2 was employed to compare the mtDNA sequences through multiple sequence alignment (Katoh et al., 2019). Phylogenetic tree was constructed using complete mtDNA sequences by the Neighbor-Joining method (Tajima-Nei model) with 1,000 times of Bootstrap resampling (Saitou and Nei, 1987).
The protein-coding genes, tRNAs, and introns in the mtDNA were annotated by MFannot and RNAweasel3 with manual corrections. The mtDNA sequences were deposited to GenBank with the accession numbers summarized in Table 1.
The polypeptide sequence of homing endonucleases (HEGs) in the introns of mitochondrial genes were obtained through direct translation by Translate program4 using the genetic code of ‘Mold, protozoan and coelenterate mitochondrial, mycoplasma/spiroplasma’. Multiple sequence analysis on the HEGs was performed by Clustal Omega5 using default parameters. Unrooted tree was constructed by Neighbor-Joining method. InterPro at EBI6 was used for protein domain analysis. The transmembrane domain in a putative protein between pol1 and atp8 was predicted using Phobius program (Käll et al., 2007).
Transposons in the mtDNA of L. eoddes were analyzed using CENSOR server (Kohany et al., 2006) and GIRI database7. Tandem repeats were identified using Tandem Repeat Finder8 (Benson, 1999).
The mtDNAs of 21 strains of L. edodes were assembled using genomic DNA reads. The sizes of the assembled mtDNAs were ranging from 119,219 bp to 121,671 bp (Table 1), which were similar to the previously published L. edodes mtDNA, such as AB697988.1 (121,394 bp), KY217797.1 (116,897 bp), MF774812.1 (119,134 bp), and MF774813.1 (115,116 bp; Yang et al., 2017; Song et al., 2019). The mtDNA of L. edodes was bigger than that of Pleurotus ostreatus (73,242 bp; Wang et al., 2008) and Flammulina velutipes (88,508 bp; Yoon et al., 2012), but smaller than that of Agaricus bisporus (135,005 bp; Férandon et al., 2013). SJ102_MT, which was the mtDNA from the SJ102 strain, containing rare composition of the A mating types (A17 and A22), was the largest with the length of 121,671 bp. GC contents were mostly 30.7% while the mtDNAs of SL10, Yujiro, L54, and MF774812.1 showed slightly higher GC content (30.8%). Interestingly, MF774813.1, the smallest mtDNA in this study, had the highest GC content (31.8%). The mtDNAs of some other mushrooms, such as A. bisporus, P. ostreatus, F. velutipes, and Moniliophthora roreri, had GC contents of 35.9, 26.4, 16.5, and 27.6%, respectively.
Phylogenetic analysis of the mtDNA sequences revealed the diversification of L. edodes mtDNA (Figure 1). Nonetheless, all the sequences were highly homologous, showing near 99% identity each other (Table 1). MF774813.1 was the least related sequence, showing 98.53% sequence homology with AB697988.1. The mtDNAs of 10 cultivated strains, including SJ701, SJ707, SJ301, SJ302, Suhyang, Baekhwa, Chunbaek, Chunjang, Dasan, and SL7, were highly homologous together with AB697988.1. Sequence identity among these sequences is more than 99.9%. The strains with these mtDNAs carry nuclei with the A mating type pairs mainly consisted of A1, A5, and A7, which are the major A mating types found in the cultivated strains (Kim et al., 2019). This indicates that the cultivated strains have been bred within very narrow genetic pool. The mtDNAs found from the strains with rare A mating types, such as Gaeul (A11, A28), Yeoreum (A18, A30), SJ102 (A17, A22), SL9 (A27, A29), and SL10 (A1, A31), had more mtDNA sequence variations, showing 99.86%, 99.79%, 99.63%, 99.26%, and 99.62% sequence identity, respectively, with AB697988.1 (Table 1). There was a tendency that the strains with rare A mating types have the mtDNAs of higher sequence variations.
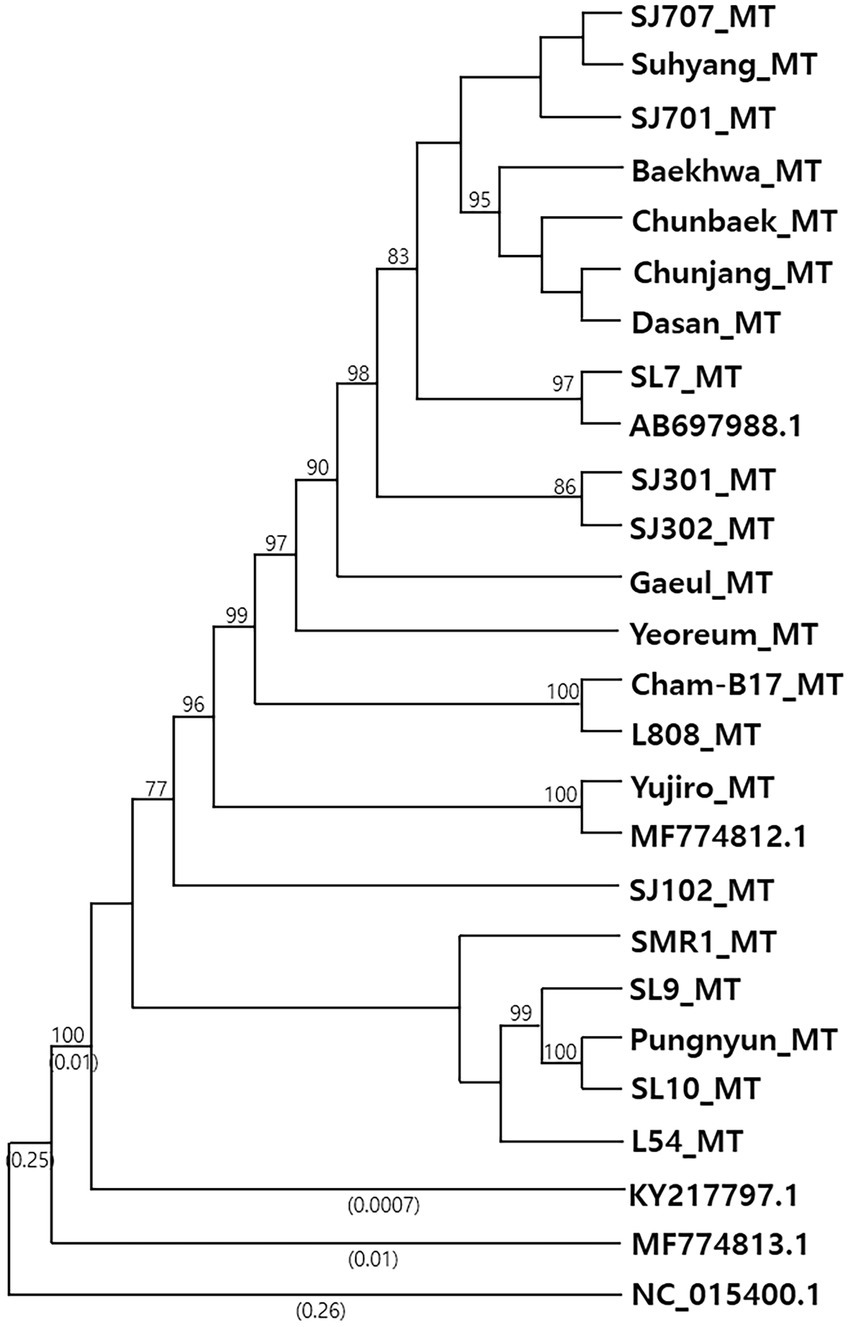
Figure 1. Phylogenetic analysis of Lentinula edodes mtDNAs. Phylogenetic tree was constructed using complete mtDNA sequences by the Neighbor-Joining method with 1,000 times of Bootstrap resampling. Bootstrap values greater than 70% are shown at branching points. Numbers enclosed in parentheses represent the branch lengths, and those shorter than 0.0005 are omitted. The mtDNA of Moniliophthora roreri (NC_015400.1) was used as an outgroup. Detailed information of the mtDNAs are summarized in Table 1.
The L. edodes mtDNAs consisted of 15 protein-coding genes (cob, atp6, 8, 9, nad1-6, 4 l, cox1-3, and rps3), two rRNAs (rns and rnl), 26 tRNAs, and three putative DNA polymerase genes (Figure 2), showing the same result with previously published paper (Song et al., 2019). The only difference in the gene number of all the assembled mtDNAs was the presence two additional trnGs, tRNA genes having anticodon for glycine, in the mtDNA of Cham-B17 (Supplementary Figure S1). The four identical copies of trnGs with three single nucleotide polymorphisms (SNPs) were tandemly arranged and separated by the ATTTAA motif. The SNP analysis revealed that the trnG1-trnG2 unit was duplicated in Cham-B17_MT (Supplementary Figure S1). The spatial arrangement of genes was essentially identical among the 25 mtDNAs but was highly different from that of M. roreri (NC_015400.1), a mushroom species belonged to Marasmiaceae family with the genus Lentinula (Supplementary Figure S2). Comparative analysis on the mtDNA structure revealed some major variations in the six mtDNA sequences (SL10_MT, L54_MT, Yujiro_MT, MF774812.1, MF774813.1, and KY217797.1; Figure 2, indicated by red arrows). The mtDNA of SL10 was shortened in rnl and rns by 954 bp and 1,353 bp, respectively. Deletion at the intergenic region (~2 kb) was observed in between pol1 and atp8 in the mtDNAs of L54, Yujiro, and MF774812.1. KY217797.1 was found to have two deleted regions, at the cox1 gene and the intergenic region described above. MF774813.1 contained multiple variations. It has the same deletion at rns with SL10_MT. The cox1 gene had an additional insertion which made it 1-kb larger than ordinary cox1 found in other mtDNAs (See below). Moreover, MF774813.1 was found to have a large deletion (6.8 kb) in between nad4 and pol3 in which pol2 was included. The pol2 gene was assumed to be non-functional because its ORF was broken into two pieces.
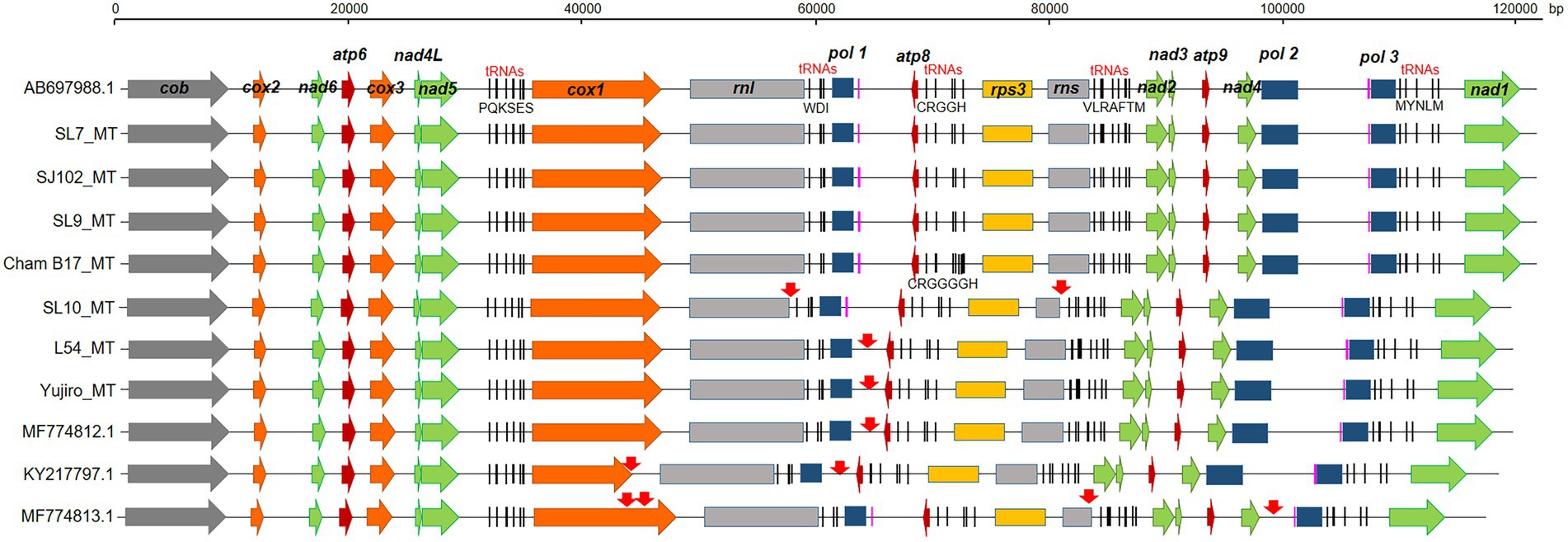
Figure 2. Gene arrangement of selected mtDNAs in different strains of Lentinula edodes. The mtDNA genes were annotated by MFannot. Regions of sequence variations are indicated by red arrows. Capital letters under the five tRNA regions indicate tRNAs having anticodons for corresponding amino acids. Two predicted tRNAs with unknown isotypes are colored in pink.
The major variations among the protein-coding genes were found in cox1 of KY217797.1 and MF774813.1. The cox1 gene of L. edodes mtDNA consisted of 8 exons separated by 7 introns (Figure 3A). All of the introns were group I introns, except for intron4 (group II; Supplementary Data S2) and have intronic ORFs encoding HEG of LAGRIDADG or GIY-YIG family. However, KY217797.1 lacked intron2 and intron3 which resulted in direct connection of exons 2–4 to a single exon. MF774813.1 was also devoid of intron2 but not intron3. Interestingly, MF774813.1 had a new intron insertion (2,653 bp) in exon5, dividing the exon5 (131 bp) into two exons of 34 bp and 100 bp. The inserted intron was a group II intron containing intronic ORFs homologous to reverse transcriptase/maturase found in Amanita thiersii (Li et al., 2020).
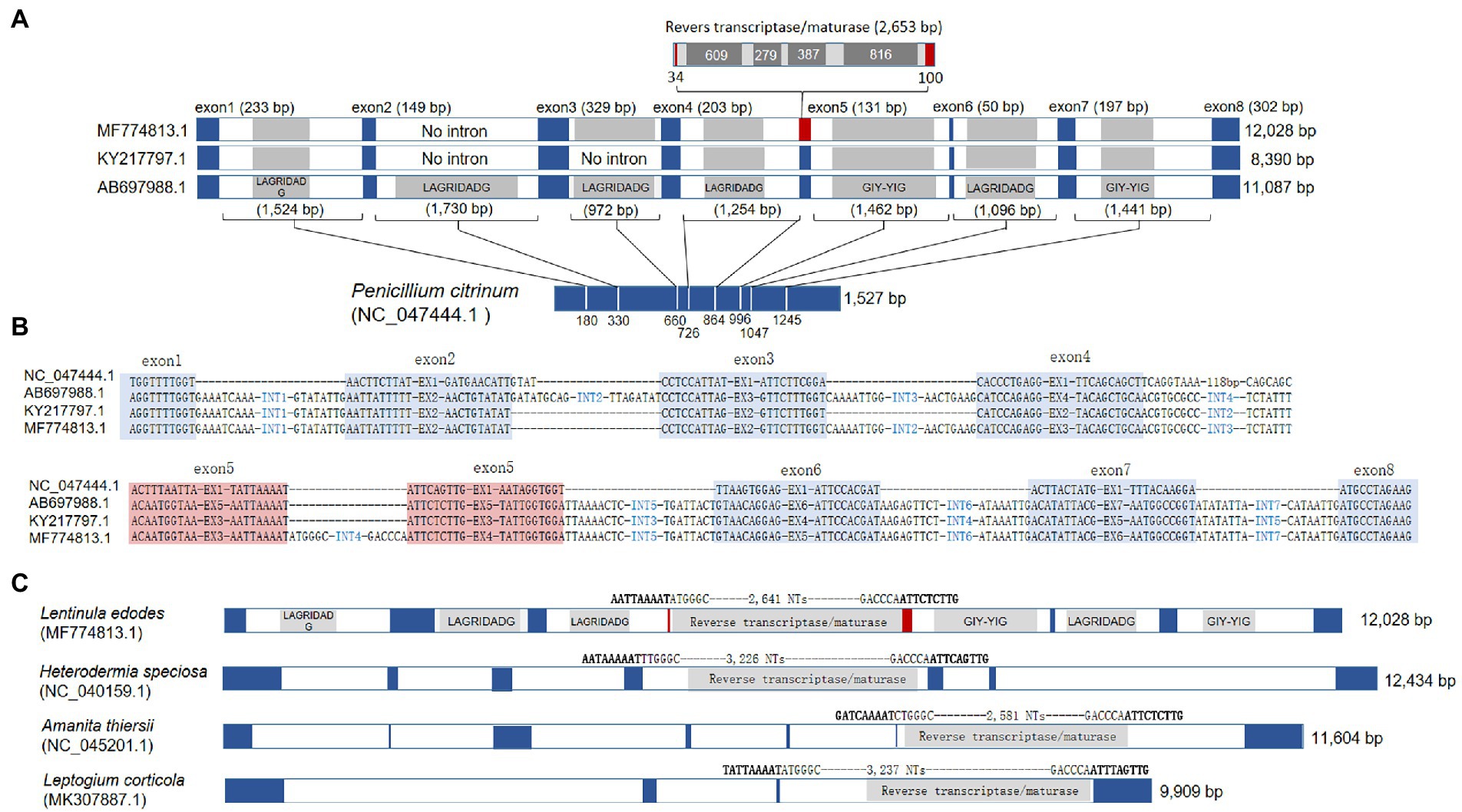
Figure 3. Gene structure variations of cox1. (A) Comparison of Lentinula edodes cox1 (Le_cox1) with archetypal cox1 in the mtDNA of Penicillium citrinum (Pc_cox1). Intronic homing nuclease genes are shaded inside introns. Intron insertion sites at Pc_cox1 are depicted by white lines with their positions from the 5′-end. The split exon by a group II intron in MF774813.1 is red-boxed. (B) Intron insertion sequences in cox1. Exonic regions are blue-shaded. The spilt exon in MF774813.1 are red-shaded. (C) The presence of homologous group II intron in different cox1 of fungal mtDNAs. Exons are colored in blue. The intron insertion sequence and the length of the group II inton are provided at the top of the introns.
The intron incorporation in the cox1 gene appears to occur independently. BLAST search with the exon sequence of L. edodes cox1 found intronless cox1s from various fungal mtDNAs, such as cox1 in the mtDNA of Penicillium citrinum (NC_047444.1) and P. chrysogenum (AM920464.1). The lengths of cox1 in these organisms are 1,527 bp (P. citrinum) and 1,608 bp (P. chrysogenum) which are close to the length of cox1 mRNA (1,602 bp) after removal of introns from 11 kb-long cox1 gene in L. edodes mtDNA. Comparative sequence analysis of the cox1 genes in L. edodes mtDNAs with that in P. citrinum revealed that each intron was inserted into a specific site possibly recognized by an HEG encoded by the intron (Figure 3A). For example, the first intron in cox1 of L. edodes was inserted into an exonic consensus sequence composed of AGGTTTTGGT and AATTATTTTT, which was conserved in P. citrinum as TGGTTTTGGT and AACTTCTTAT (Figure 3B). Similar insertion to a specific consensus sequence was also observed in the introns 2–7 (Figure 3B). Loss of introns in KY217797.1 and MF774813.1 resulted in the connection of the consensus sequences thereby connecting exons. These consensus sequences may serve for recognition and cleavage sites for intronic HEGs (summarized in Supplementary Table S1; Megarioti and Kouvelis, 2020). Exon4 in MF774813.1 (corresponding to exon5 in AB697988.1) was particularly of interest since it was divided by a unique intron, not observed in other mtDNAs of L. edodes. BLAST analysis using the intron sequence found that the homologous sequences were present in the mtDNA cox1 gene of A. thiersii, Heterodermia speciosa, and Leptogium corticola. All share a consensus sequence of AAAAT-ATT(C/T)(T/A) and encode a reverse transcriptase/maturase (Figure 3C).
Lentinula edodes mtDNA had a large rnl which encodes rRNA for ribosomal large subunit (LSU). rnl of L. edodes, 9,870 bp, was exceptionally large among LSU rRNAs of fungal mtDNAs (Figure 4). Fungi have rnl with the sizes ranging from 2.8 Kb to 5.3 Kb: 2,822 bp for Schizosaccharomyces pombe (NC_001326.1), 3,303 bp for Saccharomyces cerevisiae (NC_027264.1), 2,878 bp for Rhizopus oryzae (AY863212.1), 4,204 bp for Moniliophthora roreri (HQ259115.1), 5,070 bp for Flammulina velutipes (NC_021373.1), and 5,277 bp for Pleurotus ostreatus (NC_009905.1). Sequence analysis revealed that rnl of L. edodes contained three genes for intronic HEGs with the sizes of 1,498 bp, 947 bp, and 2,078 bp (Figure 4B). Each HEG gene encodes homing nuclease of GIY-YIG or LAGRIDADG. rnl of SL10_MT was the only mtDNA lacking the intronic region (934 bp) which contains the second LAGRIDADG HEG gene.
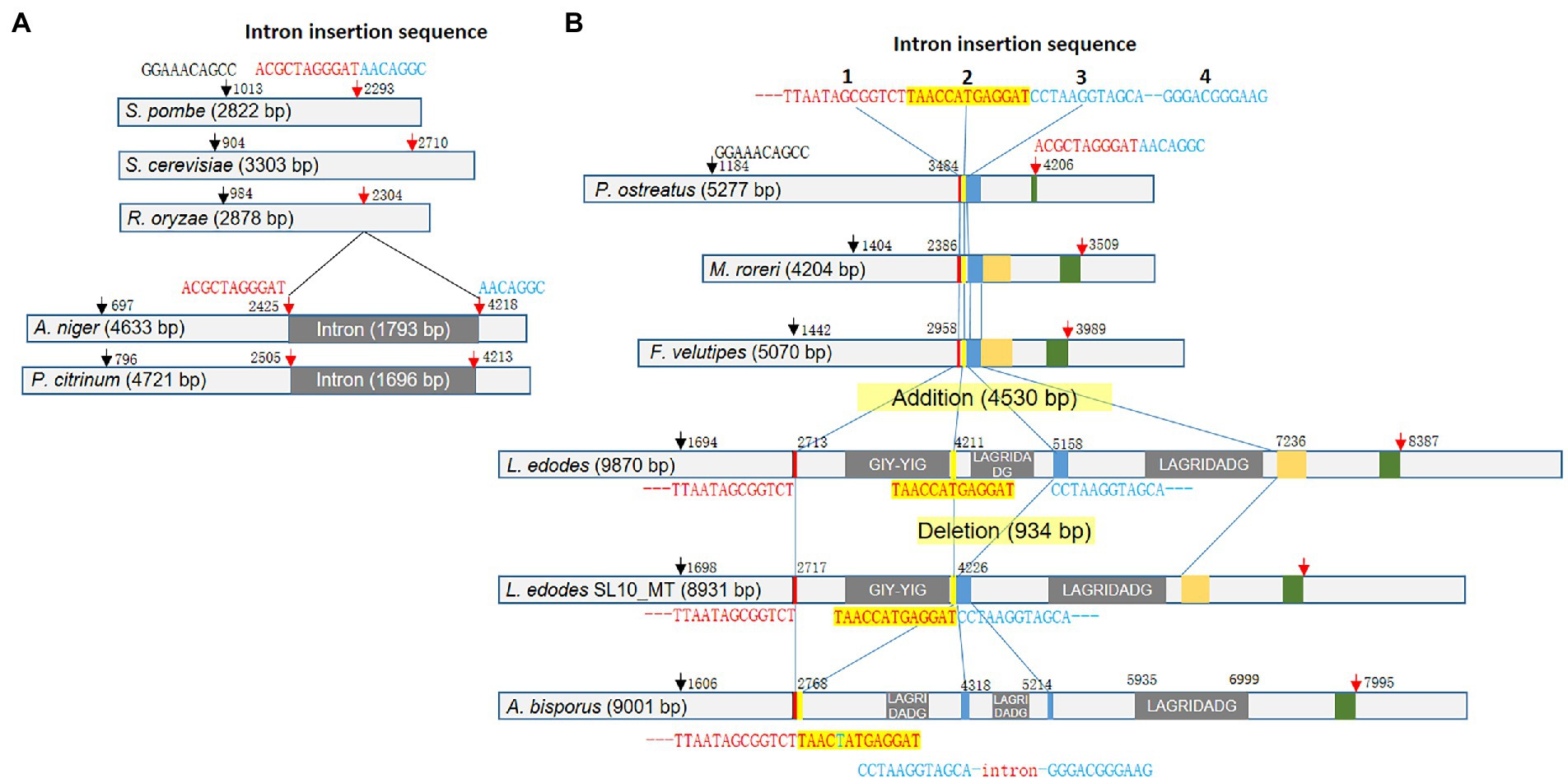
Figure 4. Variations of rnl in fungal mtDNAs. (A) Diversification of rnl in ascomycetes. The consensus sequences and the positions are depicted at the top of the gene scheme with arrows. Intron insertion splits the second consensus sequence into two pieces. (B) Diversification of rnl in basidiomycetes by intron insertion. The conserved sequences regions are color-boxed. The consensus sequences found in ascomycetes are also present in basidiomycetes (arrows). rnl in the mtDNA of L. edodes generally contains three introns with intronic HEG gene. However, SL10_MT lacked the second intron, resulting in the direct connection of the second and the third intron insertion sequences. Rnl of A. bisporus mtDNA has a new intron in between the third and the fourth intron insertion sequence.
For better understanding of the rnl size polymorphism, we performed detailed sequence analysis using rnl sequences in mtDNAs of dikaryotic fungi. Firstly, we found that the fungi have common signature sequences which include GGAAACAGCC at the 5′-region and ACGCTAGGGAT-AACAGGC at the 3′-region (Figure 4A). In ascomycetes, the latter sequence serves as the site of intron incorporation (ACGCTAGGGAT-intron-AACAGGC), resulting in increase of the size of rnl by the intron insertion (~1.7 kb) as shown in rnl of Aspergillus niger (NC_007445.1, 4,633 bp) and Penicillium citrinum (NC_047444.1, 5,277 bp; Figure 4A). However, basidiomycetes were found to use other signature sequences for the intron incorporation although the ACGCTAGGGAT-AACAGGC signature was highly conserved across fungal groups. Instead, basidiomycetes have a characteristic sequence less than 1 kb before ACGCTAGGGAT-AACAGGC that is a sequential combination of TTAATAGCGGTCT, TAACCATGAGGAT, and CCTAAGGTAGCA-115-NTs-GGGACGGGAAG (Figure 4B). These sequences were found as a connected sequence in basidiomycetes of intronless rnls such as P. ostreatus (5,277 bp), Moniliophthora rorei (4,204 bp), and F. velutipes (5,070 bp). However, these sequences became the signatures of intron insertion in the intron-containing rnl of mtDNAs. In L. edodes, the three sequences were separated by three units of introns, each of which encodes its own homing nuclease (Figure 4B). All the rnls in mtDNAs of L. edodes in this study, except for SL10_MT, had the triple incorporation of introns with the total size of 4,530 bp, resulting in increase of rnl to the size of 9,870 bp. This mobilization of intron in rnl through the signature sequences was further supported by the deletion of the second intron in SL10_MT, leaving TAACCATGAGGAT and CCTAAGGTAGCA-115-NTs-GGGACGGGAAG as a connected sequence. It was also supported by the mode of intron insertion in rnl of mtDNA in A. bisporus. The rnl in A. bisporus lacked the first intron encoding GIY-YIG homing nuclease, leaving TTAATAGCGGTCT and TAACTATGAGGAT as a connected sequence, while maintaining the second and the third introns in between TAACTATGAGGAT and CCTAAGGTAGCA and after GGGACGGGAAG (Figure 4B). One interesting finding here was the presence of an additional intron found inside the CCTAAGGTAGCA-115-NTs-GGGACGGGAAG signature in which the intron replaced the 115-bp internal sequence. It contains a truncated gene encoding the C-terminus of LAGRIDADG homing endonuclease. Therefore, it was suggested that basidiomycetes use the three consensus sequences as the sites of intron incorporation for the diversification of rnl in the mtDNA while ascomycetes use a single consensus sequence, also conserved in but not used by basidiomycetes.
The rns gene of L. edodes mtDNA, encoding rRNA for small subunit of ribosome (SSU), was also larger than that of other fungi, due to the intron incorporation dividing rns into two fragments (1,330 and 833 bp; Figure 5A). The 1,353 bp-long intron encoded a LAGRIDADG HEG. SL10_MT and MF774813.1 were the two mtDNAs lacking this intron among the 25 l. edodes mtDNAs. The consensus sequence at the intron insertion site was GAAATCCCTG-TTA(G)TATATTT (Supplementary Table S1).

Figure 5. Sequence variations in rns and other deletions in the mtDNA. (A) The occurrence of intronless rns in MF774813.1 and SL10_MT. (B) Deletion of sequence region found in four mtDNAs. The deleted region contains a putative protein-coding gene. Domain analysis failed to find any conserved domain but the putative protein was predicted to be a membrane protein with 5 transmembrane domains. (C) A large deletion found from MF774813.1.
Some mtDNAs, including MF774812.1, L54_MT, Yujiro_MT, and KY217797.1, lacked 1.9 kb DNA region located in between pol1 and atp8 (Figure 5B). The lacking region contained a gene (1,053 bp) which encodes a putative protein (350 aa) with 5 transmembrane domains. Lastly, there was a large deletion (6.8 kb) in between nad4 and pol3 in MF774813.1 (Figure 5C). Because of this deletion, MF774813.1 becomes the smallest among all L. edodes mtDNAs in spite of the large insertion at cox1 (Figure 3). pol2 was the only gene included in this region. BLAST analysis of the deleted pol2-encoding protein revealed that it was homologous to RNA polymerase of P. ostreatus linear mitochondrial plasmid mlp2 while pol3 was similar to DNA pol of M. roreri mitochondrial plasmid pMR3. Further phylogenetic analysis of pol1-pol3 showed that they were related to the mitochondrial plasmid DNAs of fungi, such as pPE1A of Pichia etchellsii and pHC2 of Hebeloma circinans (Supplementary Figure S3).
Thirteen homing endonuclease genes (HEGs) were discovered from the introns within cox1 (7), cox3 (1), nad1 (1), LSU rRNA (3), and SSU rRNA (1). Ten of them were LAGRIDADG domain containing HEGs while the remaining three, including intronic ORFs found in intron5 of cox1, intron1 of LSU, and intron7 of cox1, were GIY-YIG HEGs. Comparative sequence analysis with six S. cerevisiae HEGs, including I-SceI-IV, AI1 and AI2, and seven M. roreri HEGs, including MR_cox1 intron1-5, and MR_cox3 intron1 and intron2, showed that the HEGs were distributed in seven subgroups (Figure 6). The two LAGRIDADG domain-containing HEGs from intron 4 of cox1, intron 1 of cox3, which share significant homology with yeast mitochondrial I-SceI, forms a distinct group. The two LAGRIDADG domains were separated by 101 amino acid (aa) residues for both HEGs while 92 aa residues for I-SceI (Table 2). The yeast I-SceII (556 aa) and I-SceIV (630 aa) are long mtDNA intronic HEGs, which share highly homologous N-termini while the LAGRIDADG-containing C-termini are heterogeneous (Supplementary Data S1). The C-term of I-SceII was rather similar to HEG from intron3 of cox1. Both have two LAGRIDADG domains with the sequences of LAGLIDGDG and FS(V)GFFDADG separated by 97 aa residues. HEGs encoded by intron1 of nad1, intron of SSU, and intron1 of MR_cox3 formed a distinct group (Figure 6). HEGs in this group contained heterogeneous LAGRIDADG domains: VTGFFDAEA and ISGFVSGEG for SSU intron, WTGLIDGEG and IAGFVTGEG for MR_cox3 intron1, and VIGFSYAEV for nad1 intron1 (Table 2). Interestingly, HEG of nad1 lacked the second LAGRIDADG domain that makes this HEG as one of the two single LAGRIDADG domain-containing HEG among all L. edodes mitochondrial LAGRIDADG HEGs. HEG encoded by intron1 of cox1 shared high homology with I-SceIII, MR_cox1_intron2, and MR_cox3_intron2 (Supplementary Data S1). HEGs in intron2 and intron3 of LSU showed significant homology around the LAGRIDADG-domain, however, the former was a short HEG (196 aa) with a single LAGRIDADG whereas the latter was a long HEG (365 aa) with two LAGRIDADG-domains (Supplementary Data S1). HEG in intron2 of cox1 was highly homologous to HEG in intron1 of MR_cox1. There was three GIY-YIG HEGs in the mtDNA of L. edodes, which were found from intron5 and intron7 of cox1, and intron1 of LSU. Homologous protein was also found from intron5 of MR_cox1, which showed better homology with HEG from intron7 of cox1 than that from intron 5 of cox1 (Supplementary Data S1).
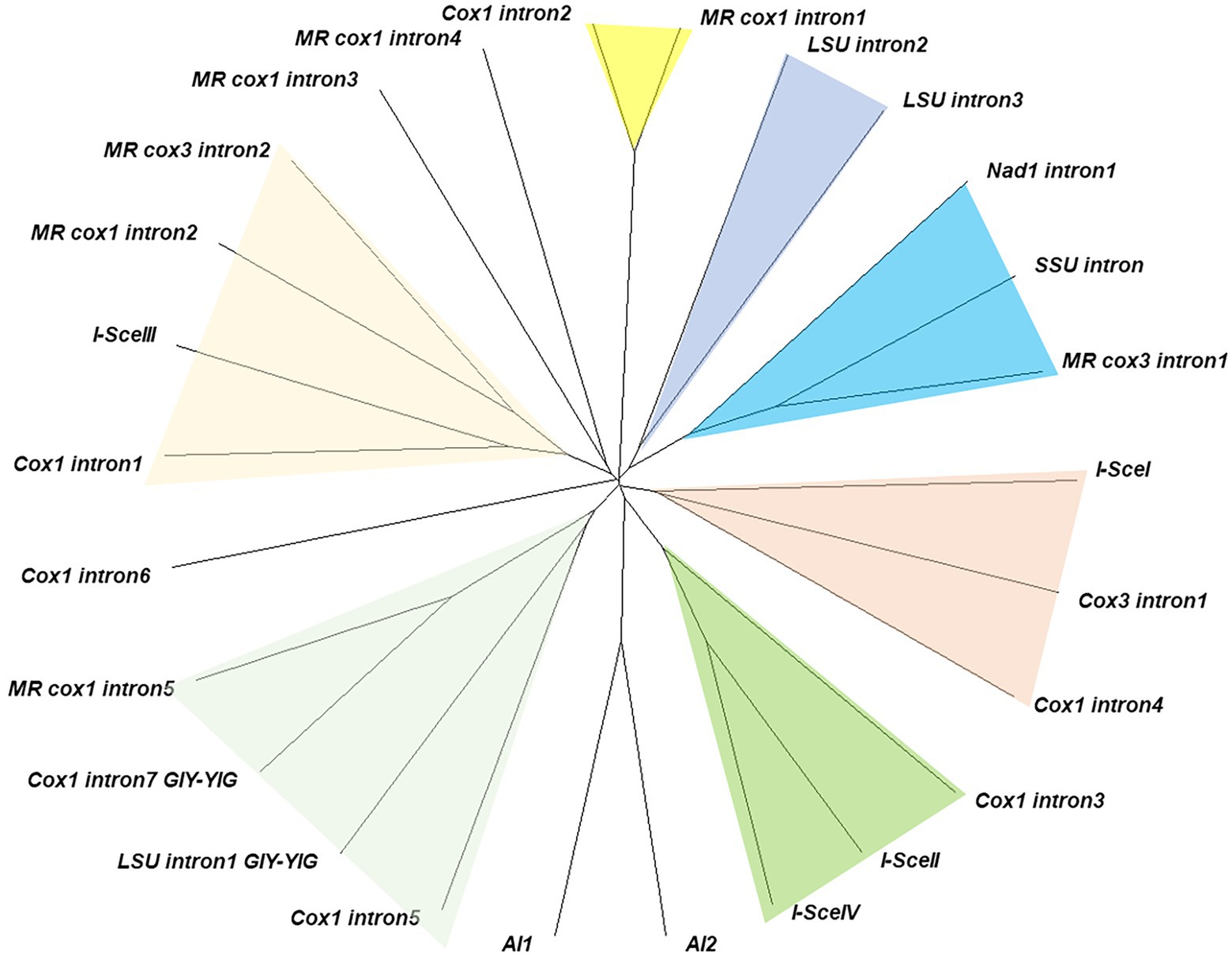
Figure 6. Grouping of intronic HEGs. Similar HEG groups are color-shaded. HEGs from introns in the genes of M. roreri are described as ‘MR gene name intron’ while HEGs of Saccharomyces cerevisiae are described following HEG naming rule. The unrooted tree was constructed using translated protein sequences by Neighbor-Joining method.
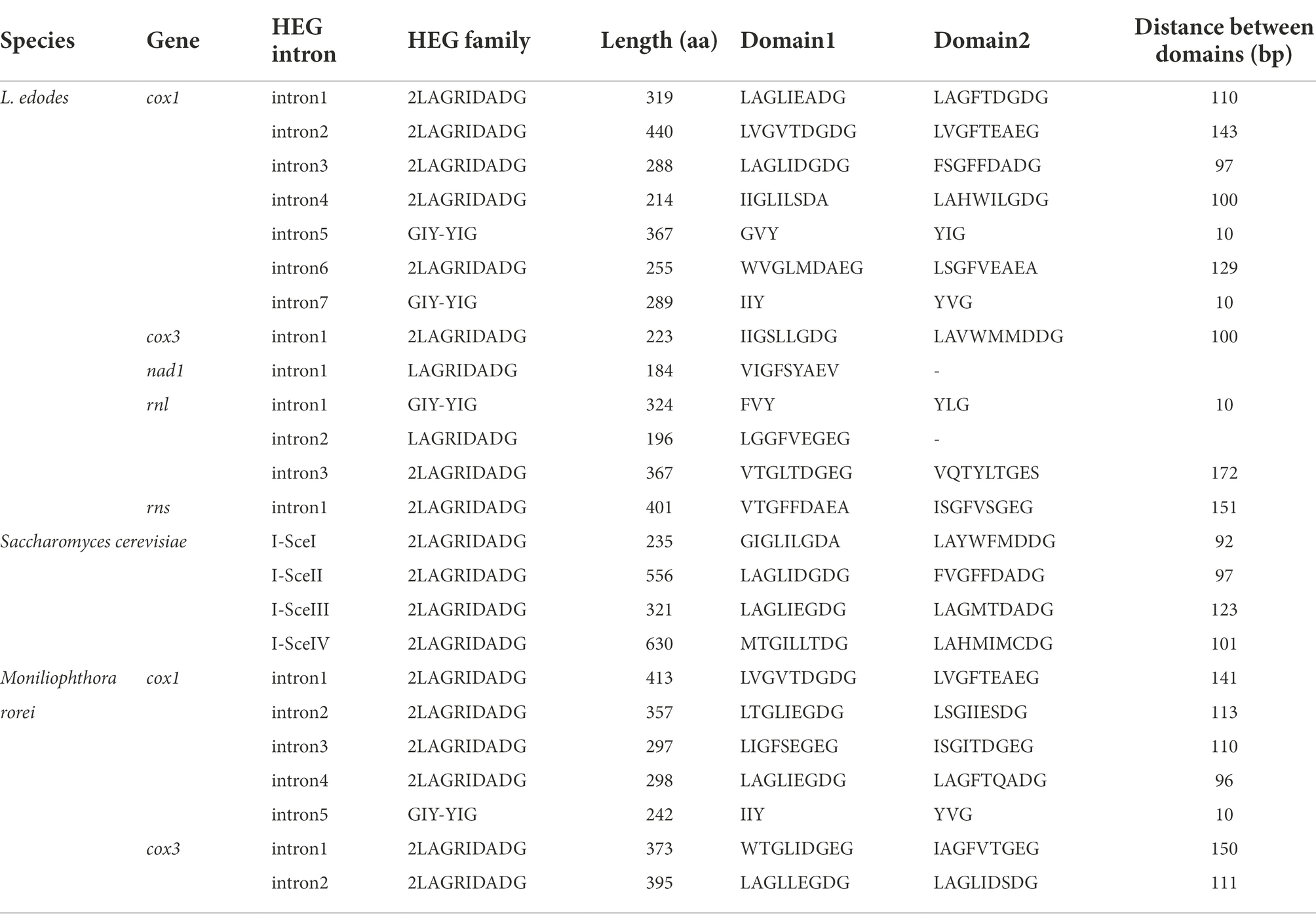
Table 2. Homing endonucleases in L. edodes mtDNAs.
Transposable elements (TEs) are some of the major factors in the genome modification. Computational analysis predicted that the L. edodes mtDNA contained 106.3 TE-related sequences in average (Figure 7A). However, there was no intact form remained. All of them were fragments of TEs with the sizes ranging from 50 bp to 250 bp. Sum of the total TE fragments was 13,322 bp per 120,700 bp mtDNA which covered 11.04% of the mitogenome (Figure 7A; Supplementary Table S2). LTR retrotransposon and DNA transposon were the major TEs with the composition of 7.08% (8,083 bp) and 3.96% (4,778 bp), respectively, while NonLTR retrotransposon (460 bp) was a minor TE family (Figure 7B). In LTR retrotransposon, LTR/gypsy and LTR/copia were most of the retrotransposons, covering 6,242 bp (53 count) and 1,777 bp (14.2 count), respectively, of the total retrotransposon content (8,543 bp; Figure 7A; Supplementary Table S3). Various DNA transposons, such as hAT, ExSpm/CACTA, and Mariner, were discovered with hAT as the sole major DNA transposon. 22.4 copies of hATs with the total length of 3,691 bp were scattered in different locations in the mtDNA (Figure 7A; Supplementary Table S3). TEs were largely located intronic and intergenic regions (Figure 8A, blue color). Ten of the 106.3 REs were found in the exonic regions of the protein-coding genes, including cox2 (2), nad6 (1), nad5 (2), cox1 (1), nad2 (2), and nad4 (2) (Figure 8A, black arrows). Of the protein-coding genes, cob, cox3, cox1, and nad1were found to carry TEs only in the intronic regions. Rnl had 7 TEs whereas none was included in rns.
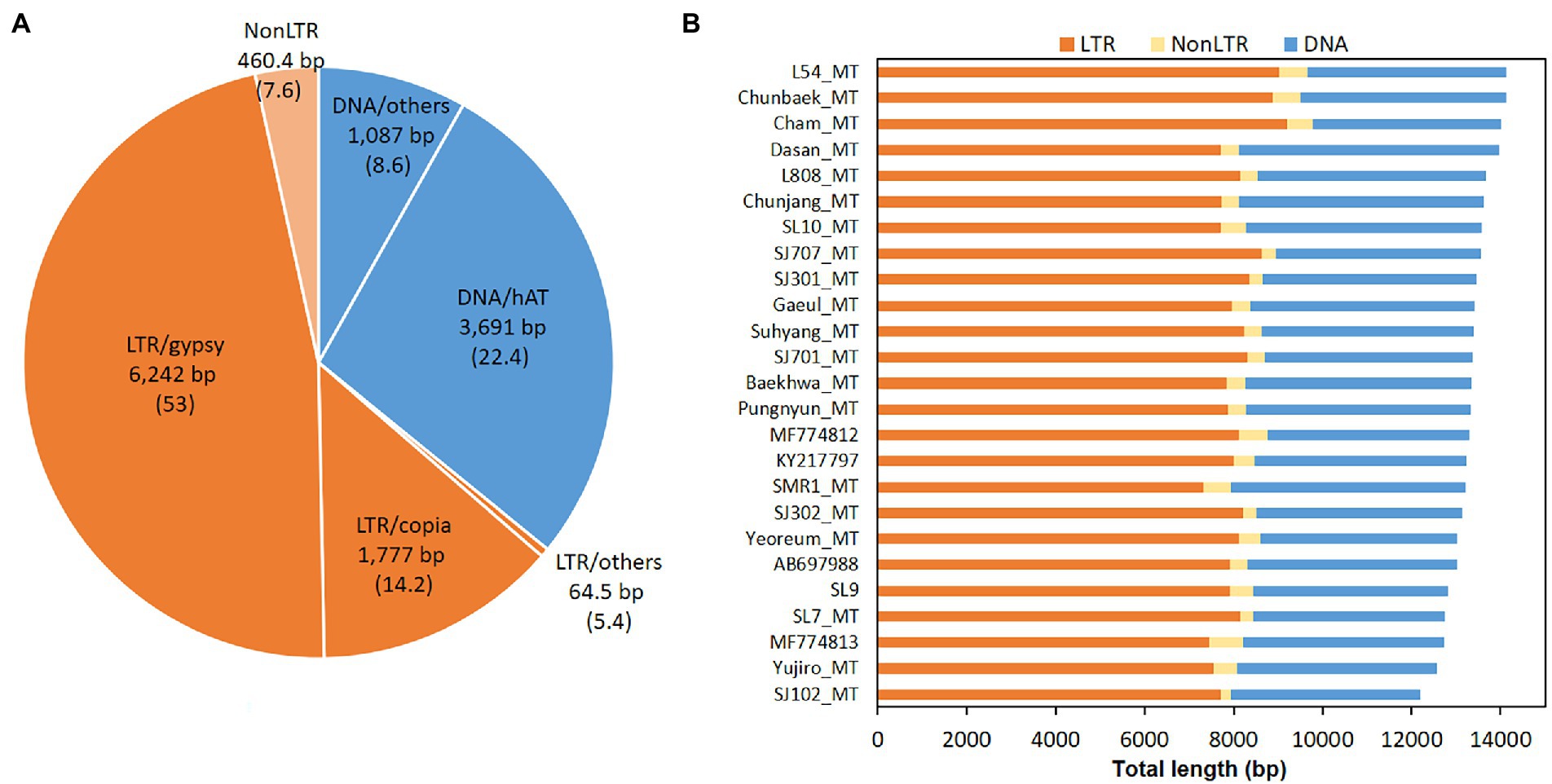
Figure 7. Transposable elements (TEs) in the mtDNA of L. edodes. TEs in 25 mtDNAs were predicted by Censor program. (A) Average contents of TEs. Retrotransposons are orange-colored while DNA transposons are in blue. Average number of each TE is described in parenthesis. (B) Total length of TEs in different mtDNAs.
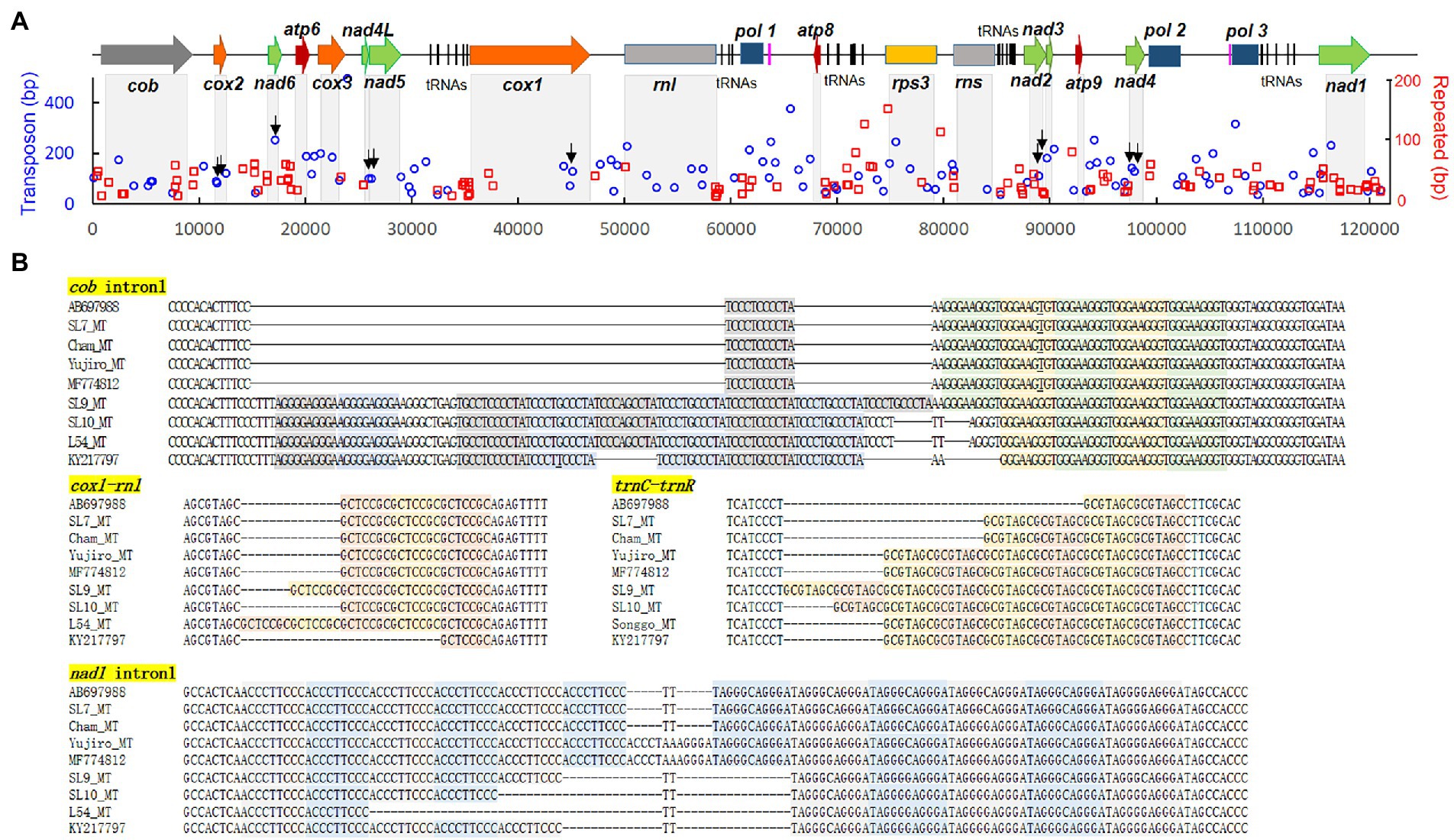
Figure 8. Repeated sequences in the mtDNA of L. edodes. (A) Locations of the repeated sequences (red rectangle) together with TEs (blue circle) along with the mtDNA sequence. The axis depicts the length of each TE or repeated sequence. Arrows indicate the positions of TEs in exons. (B) Sequence structure of repeated sequences at various locations in the mtDNA of different strains. The repeating units are colored differently.
Repeated sequences are frequently occurring DNA sequences in the genomes of living organisms. In our RepeatFinder analysis using AB697988.1, L. edodes mtDNA was found to contain 115 repeat sequences with the total length of 5,641 bp (4.6% of total mitogenome; Supplementary Data S2). Average length and copy number of the repeating unit were 12.7 bp and 5.2 copies. All of the repeats were found either inside the intron or in the intergenic region. In the intronic regions of protein-coding genes, 7 repeated sequences were found from nad1and cob, and 2 from cox1 and cox3 (Figure 8A; Supplementary Data S2). Some of the most frequent repeated sequences were ACCCTTCCC, TAGGGCAGGGA, GCTCCGC, TCCCTCCCCTA, and GGGAAGGGT as we previously reported (Kim et al., 2019). There were two types of repeats: one is the symmetrical repeats paring ACCCTTCCC with TAGGGCAGGGA occurred in intron1 of nad1 and intron2 of cob and TCCCTCCCCTA with GGGAAGGGT in intron1 of cob and intergenic region between trnC and trnR, another is direct repeats of GCTCCGC (nad1 intron1, nad6-atp6, and trnY-trnN), GCGTAGC (trnC-trnR), TATAA (atp9-nad4), etc. (Figure 8B). The repeating units occurred in different copy numbers in different mtDNAs. For example, a single copy of TCCCTCCCCTA was present in AB697988, SL7_MT, Cham-B17_MT, Yujiro_MT, and MF774812 while SL10_MT, L54_MT, and KY217797 had 5 copies and SL9_MT had 6 copies. GGGAAGGGT units occurred in 4–5 copies in opposite sides (Figure 8B).
In most eukaryotic species, the mitochondria are crucial cellular organelles in charge of energy metabolism. They have mtDNAs with a variety of sizes that contain their own genetic information, depending on the type of organism. The mtDNA size in higher fungi, which include ascomycetes and basidiomycetes, varies from 18.8 kb in the yeast H. uvarum (Pramateftaki et al., 2006) to 332 kb in the plant pathogen G. cichoracearum (Zaccaron and Stergiopoulos, 2021). The huge mtDNA of G. cichoracearum comprises extraordinarily large quantities of intron (207 kb, 62% of total mtDNA) and intergenic regions (104 kb, 31%), whereas the mtDNA of H. uvarum has primarily intronless genes and short intergenic sequences, suggesting that lateral gene transfer plays a great role in the evolution of mitochondria in fungal species.
In this study, we assessed the strain-level mtDNA variations using the mtDNA sequences of 25 different strains of L. edodes whose sizes range from 115,116 bp to 121,671 bp. The mtDNAs are homologous to each other with high sequence identity (Table 1). Particularly, the mtDNAs from the strains having frequently found A mating type pairs, consisted of A1, A5, and A7 (Kim et al., 2019), are essentially identical by sharing more than 99.9% sequence identity. The mtDNAs from the strains with the uncommon A mating types, however, show larger sequence variations (Table 1). The tendency found here shows that very few nuclear and mitochondrial genetic pools have been used to breed the cultivated strains.
The total number of mitochondrial genes within the 25 mtDNAs of L. edodes was constant, except for two mtDNAs. Cham-B17_MT carries two extra copies of homologous trnG separated by ATTTAA motifs, totaling four tandem trnGs, through duplication of trnG1-trnG2 unit (Figure 2; Supplementary Figure S1). The duplication found here could arise from replication slippage as discussed by Moritz and Brown (1987) using the ATTTAA motif as the repeating unit. Similar duplication of two tRNA unit has been found from the mtDNAs of Tagiades vajuna (Liu et al., 2017) and Odontoptilum angulatum (Liu et al., 2021). Another gene number variation was found from MF774813.1 in which the pol2 gene was deleted together with a large non-coding region (Figure 5C). The pol2 gene product is homologous to DNA polymerase type B (YP_009710628) of Amanita brunnescens, and is thought to be non-functional since it is broken. Since mtDNA replication depends on the activity of a nucleus-encoded DNA polymerase γ (MIP1; Foury, 1989), the true physiological function of the pol genes (pol1-pol3) is unknown. They can be remnants of mitochondrial plasmid DNAs which had been integrated into mtDNA and modified during further evolution (Weber et al., 1995; Wang et al., 2008; Férandon et al., 2013). Given the fact that the deletion only occurred in MF774813.1, the effect of the pol2 loss there deserves further investigation. Additionally, there was deletion of a putative gene in the intergenic region between pol1 and atp8 which was predicted to encode a 350 aa-membrane protein (Figure 5B). It appears to be nonessential since several mtDNAs are devoid of this sequence.
Some of the mitochondrial genes, including cox1, rnl, and rns, showed size variation that was caused by intron mobilization. For example, we found that L. edodes cox1 (Le_cox1) has been expanded to 11,087 bp by seven independent events of Group I intron insertion by the sequence comparison with an archetypal intronless cox1 from P. citrinum (1, 527 bp; Figure 3). Each intron insertion site retains its own consensus sequence, presumably recognized by an intronic HEG. In fact, most of the consensus sequences discovered here are homologous to the insertion sequences of HEGs in fungal mtDNAs described by Megarioti and Kouvelis (2020). The size variations in Le_cox1 were found from KY217797.1 and MF774813.1; the former has a variant Le_cox1 with a size of 8,390 bp, and the latter with a size of 12,028 bp. The size reduction in Le_cox1 of KY217797.1 is attributed to the absence of intron2 (1,720 bp) and intron3 (972 bp), resulting in the smallest Le_cox1. Providing the fact that KY217797.1 is more ancient mtDNA as shown by our phylogenetic analysis (Figure 1), Le_cox1 in KY217797.1 is conceivably an earlier version in which the intron insertion has not yet taken place. This is further corroborated by the finding of Le_cox1 in MF774813.1, which has intron3 but not intron2. Sequential insertion of intron3 and intron2 may occur to generate Le_cox1 of full size. Le_cox1 of MF774813.1 has another intriguing characteristic in that it evolves into a new form of cox1 by incorporating a new group II intron (2,653 bp) into exon4. The new intron is the only Group II intron discovered from the mtDNA of L. edodes, and because of its rarity, it is thought to have been recently acquired. Homologs have been discovered from cox1 of A. thiersii, Heterodermia speciosa, and Leptogium corticola (Figure 3C) with a distinct intron insertion sequence homologous to the exon binding sequence in cox1 of S. cerevisiae (Eskes et al., 2000; Lambowitz and Zimmerly, 2004), indicating lateral gene transfer may have mobilized this retroelement beyond the boundaries of species. The mtDNA of L. edodes has relatively large rnl (9,870 bp) for mushrooms when compared with rnls in P. ostreatus (5,277 bp), M. roreri (4,204 bp), and F. velutipes (5,070 bp; Figure 4). The size expansion here is attributed to the presence of three Group I introns, totaling 4,530 bp. Removal of them can result in rnl of the size close to above mentioned mushrooms. Investigation of the intron insertion sites reveals a serial connection of consensus sequence units composed of TTAATAGCGGTCT, TAACCATGAGGAT, CCTAAGGTAGCA, and GGGACGGGAAG through which mushrooms can incorporate at least three introns (Chen et al., 2021). In contrast to mushrooms, rnls of ascomycetes in this study have different consensus sequences which are composed of ACGCTAGGGAT and AACAGGC. A single intron with the size of 1.7 kb has been inserted in between these sequences. One noteworthy feature of these sequences is that they are also present in basidiomycetes but are never used for intron insertion.
Together with gene number variation and intron mobilization, transposable elements (TEs) and repeated sequences (RSs) also contributed to the mtDNA size variations in L. edodes. TEs and RSs cover 11.04 and 4.6% of the mtDNA, respectively, mostly locating intronic and intergenic regions. TEs in this study are predicted to be inactive since all of them are fragments of TEs. They may have originated from the nuclear genome (Farrelly and Butow, 1983; Alverson et al., 2010), but more research is need to verify their exact origin and significance in the evolution of mitochondria. RSs appear to occur in a random manner since different numbers of RSs have been detected even in the closely related mtDNAs (Figure 8B). They can occur during DNA replication through polymerase slippage and possibly in the DNA repair process (Bzymek and Lovett, 2001; Phadnis et al., 2005). Lastly, plasmid insertion is one of the factors causing mtDNA size expansion (Sederoff, 1984; Férandon et al., 2013; Mardanov et al., 2014). The mtDNA of Agaricus bisporus was reported to contain plasmid-related sequences in two separated regions, covering 5% of mtDNA size (Férandon et al., 2013). Genes found in these regions were DNA polymerase and RNA polymerase genes, which originated from pEM plasmid. Similarly, the mtDNA of Sclerotinia borealis has plasmid-related DNA pol and RNA pol genes with additional related remnant sequences (Mardanov et al., 2014). The mtDNA of L. edodes in this study also carries two complete pol genes and an incomplete one in two separated regions. Considering the region between pol2-pol3 and pol1-atp8 as plasmid-related sequences, they can contribute more than 10% of the total mtDNA in L. edodes.
In conclusion, our research demonstrates that replication errors, such as gene duplication and RS generation, and the mobilization of DNA fragments, including introns, TEs, and plasmids, are the primary factors that allow persistent evolution of mtDNA even within a species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, OP328153; https://www.ncbi.nlm.nih.gov/genbank/, OP328154; https://www.ncbi.nlm.nih.gov/genbank/, OP328155; https://www.ncbi.nlm.nih.gov/genbank/, OP345458; https://www.ncbi.nlm.nih.gov/genbank/, OP345460; https://www.ncbi.nlm.nih.gov/genbank/, OP328158; https://www.ncbi.nlm.nih.gov/genbank/, OP328156; https://www.ncbi.nlm.nih.gov/genbank/, OP345452; https://www.ncbi.nlm.nih.gov/genbank/, OP345461; https://www.ncbi.nlm.nih.gov/genbank/, OP328157; https://www.ncbi.nlm.nih.gov/genbank/, OP345453; https://www.ncbi.nlm.nih.gov/genbank/, OP345465; https://www.ncbi.nlm.nih.gov/genbank/, OP345464; https://www.ncbi.nlm.nih.gov/genbank/, OP328152; https://www.ncbi.nlm.nih.gov/genbank/, OP345462; https://www.ncbi.nlm.nih.gov/genbank/, OP345456; https://www.ncbi.nlm.nih.gov/genbank/, OP345463; https://www.ncbi.nlm.nih.gov/genbank/, OP345455; https://www.ncbi.nlm.nih.gov/genbank/, OP345454; https://www.ncbi.nlm.nih.gov/genbank/, OP345459; https://www.ncbi.nlm.nih.gov/genbank/, OP345457.
SK and HE analyzed the mtDNA gene structure and intron sequences. RN did the mtDNA annotation and contributed to the manuscript writing. YC did the mtDNA annotation. HL and HR generated the NGS sequences and provided the assembled mtDNA sequences. H-SR leaded the whole project from conceptualization to manuscript writing. All authors contributed to the article and approved the submitted version.
This work was supported by Golden Seed Project (Grant No: 213007–05-4-WTH21) from the Ministry of Agriculture, Food and Rural Affairs, Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1034387/full#supplementary-material
1. ^https://www.ncbi.nlm.nih.gov/genome/organelle/
2. ^https://mafft.cbrc.jp/alignment/server/
3. ^https://megasun.bch.umontreal.ca/RNAweasel/
4. ^https://web.expasy.org/translate/
5. ^https://www.ebi.ac.uk/Tools/msa/clustalo/
6. ^https://www.ebi.ac.uk/interpro/
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Jeffrey, K. B., and Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bzymek, M., and Lovett, S. T. (2001). Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. U. S. A. 98, 8319–8325. doi: 10.1073/pnas.111008398
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cells 125, 1241–1252. doi: 10.1016/J.CELL.2006.06.010
Chen, L., Gong, Y., Cai, Y., Liu, W., Zhou, Y., Xiao, Y., et al. (2016). Genome sequence of the edible cultivated mushroom Lentinula edodes (shiitake) reveals insights into lignocellulose degradation. PLoS One 11:e0160336. doi: 10.1371/journal.pone.0160336
Chen, C., Li, Q., Fu, R., Wang, J., Deng, G., Chen, X., et al. (2021). Comparative mitochondrial genome analysis reveals intron dynamics and gene rearrangements in two Trametes species. Sci. Rep. 11:2569. doi: 10.1038/s41598-021-82040-7
Eskes, R., Liu, L., Ma, H., Chao, M. Y., Dickson, L., Lambowitz, A. M., et al. (2000). Multiple homing pathways used by yeast mitochondrial group II introns. Mol. Cell. Biol. 20, 8432–8446. doi: 10.1128/MCB.20.22.8432-8446.2000
Farrelly, F., and Butow, R. (1983). Rearranged mitochondrial genes in the yeast nuclear genome. Nature 301, 296–301. doi: 10.1038/301296a0
Fedler, M., Luh, K.-S., Stelter, K., Nieto-Jacobo, F., and Basse, C. W. (2009). The a2 mating-type locus genes lga2 and rga2 direct uniparental mitochondrial DNA (mtDNA) inheritance and constrain mtDNA recombination during sexual development of Ustilago maydis. Genetics 181, 847–860. doi: 10.1534/genetics.108.096859
Férandon, C., Xu, J., and Barroso, G. (2013). The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 55, 85–91. doi: 10.1016/J.FGB.2013.01.009
Foury, F. (1989). Cloning and sequencing of the nuclear gene MIP1 encoding the catalytic subunit of the yeast mitochondrial DNA polymerase. J. Biol. Chem. 264, 20552–20560. doi: 10.1016/S0021-9258(19)47098-1
Ha, B., Kim, S., Kim, M., Moon, Y. J., Song, Y., Ryu, J. S., et al. (2018). Diversity of a mating type in Lentinula edodes and mating type preference in the cultivated strains. J. Microbiol. 56, 416–425. doi: 10.1007/s12275-018-8030-6
Hausner, G. (2003). “Fungal mitochondrial genomes, plasmids and introns,” in Fungal Genomics. eds. D. K. Arora and G. G. Khachatourians, vol. III (Amsterdam: Elsevier Science).
Jelen, V., de Jonge, R., Van de Peer, Y., Javornik, B., and Jakše, J. (2016). Complete mitochondrial genome of the Verticillium-wilt causing plant pathogen Verticillium nonalfalfae. PLoS One 11:e0148525. doi: 10.1371/journal.pone.0148525
Käll, L., Krogh, A., and Sonnhammer, E. L. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 35, W429–W432. doi: 10.1093/nar/gkm256
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kim, S., Song, Y., Ha, B., Moon, Y. J., Kim, M., Ryu, H., et al. (2019). Variable number tandem repeats in the mitochondrial DNA of Lentinula edodes. Genes (Basel) 10:542. doi: 10.3390/genes10070542
Kohany, O., Gentles, A. J., Hankus, L., and Jurka, J. (2006). Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7:474. doi: 10.1186/1471-2105-7-474
Lambowitz, A. M., and Zimmerly, S. (2004). Mobile group II introns. Ann. Rev. Genet. 38, 1–35. doi: 10.1146/annurev.genet.38.072902.091600
Lee, H.-Y., Moon, S., Shim, D., Hong, C. P., Lee, Y., Koo, C.-D., et al. (2017). Development of 44 novel polymorphic SSR markers for determination of shiitake mushroom (Lentinula edodes) cultivars. Genes 8:109. doi: 10.3390/genes8040109
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Q., He, X., Ren, Y., Xiong, C., Jin, X., Peng, L., et al. (2020). Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and asymbiotic amanita species. Front. Microbiol. 11:01382. doi: 10.3389/fmicb.2020.01382
Liu, W., Cai, Y., Zhang, Q., Shu, F., Chen, L., Ma, X., et al. (2020). Subchromosome-scale nuclear and complete mitochondrial genome characteristics of Morchella crassipes. Int. J. Mol. Sci. 21:483. doi: 10.3390/ijms21020483
Liu, F.-F., Li, Y.-P., Jakovlic, I., and Yuan, X.-Q. (2017). Tandem duplication of two tRNA genes in the mitochondrial genome of Tagiades vajuna (Lepidoptera: Hesperiidae). Eur. J. Entomol. 114, 407–415. doi: 10.14411/eje.2017.052
Liu, J., Xiao, J., Hao, X., and Yuan, X. (2021). Unique duplication of trnN in Odontoptilum angulatum (Lepidoptera: Pyrginae) and phylogeny within Hesperiidae. Insects 12:348. doi: 10.3390/insects12040348
Mardanov, A. V., Beletsky, A. V., Kadnikov, V. V., Ignatov, A. N., and Ravin, N. V. (2014). The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS One 9, 1–11. doi: 10.1371/journal.pone.0107536
Megarioti, A. H., and Kouvelis, V. N. (2020). The coevolution of fungal mitochondrial introns and their homing endonucleases (GIY-YIG and LAGLIDADG). Genome Biol. Evol. 12, 1337–1354. doi: 10.1093/gbe/evaa126
Menolli, N., Sanchez-Ramirez, S., Sanchez-Garcia, M., Wang, C., Patev, S., Ishikawa, N. K., et al. (2022). Global phylogeny of the shiitake mushroom and related Lentinula species uncovers novel diversity and suggests an origin in the Neotropics. Mol. Phylogenet. Evol. 173:107494. doi: 10.1016/j.ympev.2022.107494
Moritz, C., and Brown, W. M. (1987). Tandem duplications in animal mitochondrial DNAs: variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. U. S. A. 84, 7183–7187. doi: 10.1073/pnas.84.20.7183
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Research in Computational Molecular Biology. RECOMB 2013. eds. M. Deng, R. Jiang, F. Sun, and X. Zhang, Lecture Notes in Computer Science, vol. 7821 (Berlin, Heidelberg: Springer)
Phadnis, N., Sia, R. A., and Sia, E. A. (2005). Analysis of repeat-mediated deletions in the mitochondrial genome of Saccharomyces cerevisiae. Genetics 171, 1549–1559. doi: 10.1534/genetics.105.047092
Pramateftaki, P. V., Kouvelis, V. N., Lanaridis, P., and Typas, M. A. (2006). The mitochondrial genome of the wine yeast Hanseniaspora uvarum: a unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 6, 77–90. doi: 10.1111/j.1567-1364.2005.00018.x
Robicheau, B. M., Young, A. P., LaButti, K., Grigoriev, I. V., and Walker, A. K. (2017). The complete mitochondrial genome of the conifer needle endophyte, Phialocephala scopiformis DAOMC 229536 confirms evolutionary division within the fungal Phialocephala fortinii s.l. – Acephala appalanata species complex. Fungal Biol. 121, 212–221. doi: 10.1016/J.FUNBIO.2016.11.007
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sato, M., and Sato, K. (2012). Maternal inheritance of mitochondrial DNA. Autophagy 8, 424–425. doi: 10.4161/auto.19243
Sederoff, R. R. (1984). Structural variation in mitochondrial DNA. Adv. Genet. 22, 1–108. doi: 10.1016/S0065-2660(08)60038-3
Shen, X.-Y., Li, T., Chen, S., Fan, L., Gao, J., and Hou, C.-L. (2015). Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of Pleosporales. PLoS One 10:e0116466. doi: 10.1371/journal.pone.0116466
Shim, D., Park, S. G., Kim, K., Bae, W., Lee, G. W., Ha, B. S., et al. (2016). Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 223, 24–25. doi: 10.1016/j.jbiotec.2016.02.032
Song, X., Zhao, Y., Song, C., Chen, M., Huang, J., Bao, D., et al. (2019). Mitogenome types of two Lentinula edodes sensu lato populations in China. Sci. Rep. 9:9421. doi: 10.1038/s41598-019-45922-5
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, Y., Zeng, F., Hon, C. C., Zhang, Y., and Leung, F. C. C. (2008). The mitochondrial genome of the Basidiomycete fungus Pleurotus ostreatus (oyster mushroom). FEMS Microbiol. Lett. 280, 34–41. doi: 10.1111/j.1574-6968.2007.01048.x
Weber, B., Börner, T., and Weihe, A. (1995). Remnants of a DNA polymerase gene in the mitochondrial DNA of Marchantia polymorpha. Curr. Genet. 27, 488–490. doi: 10.1007/BF00311221
Xu, J., Ali, R. Y., Gregory, D. A., Amick, D., Lambert, S. E., Yoell, H. J., et al. (2000). Uniparental mitochondrial transmission in sexual crosses in Cryptococcus neoformans. Curr. Microbiol. 40, 269–273. doi: 10.1007/s002849910053
Yan, Z., and Xu, J. (2003). Mitochondria are inherited from the MATa parent in crosses of the basidiomycete fungus Cryptococcus neoformans. Genetics 163, 1315–1325. doi: 10.1093/genetics/163.4.1315
Yang, R., Li, Y., Song, X., Tang, L., Li, C., Tan, Q., et al. (2017). The complete mitochondrial genome of the widely cultivated edible fungus Lentinula edodes. Mitochondrial DNA Part B. 2, 13–14. doi: 10.1080/23802359.2016.1275839
Yoon, H., You, Y.-H., Woo, J.-R., Park, Y.-J., Kong, W.-S., Lee, B.-M., et al. (2012). The mitochondrial genome of the white-rot fungus Flammulina velutipes. J. General Appl. Microbiol. 58, 331–337. doi: 10.2323/jgam.58.331
Zaccaron, A. Z., and Stergiopoulos, I. (2021). Characterization of the mitochondrial genomes of three powdery mildew pathogens reveals remarkable variation in size and nucleotide composition. Microb. Genom. 7:000720. doi: 10.1099/mgen.0.000720
Keywords: Lentinula edodes, mitochondrial DNA, evolution, intron, repeats
Citation: Kim S, Eom H, Nandre R, Choi YJ, Lee H, Ryu H and Ro H-S (2022) Comparative structural analysis on the mitochondrial DNAs from various strains of Lentinula edodes. Front. Microbiol. 13:1034387. doi: 10.3389/fmicb.2022.1034387
Edited by:
Masoomeh Ghobad-Nejhad, Iranian Research Organization for Science and Technology, IranReviewed by:
Alexander N. Ignatov, Peoples' Friendship University of Russia, RussiaCopyright © 2022 Kim, Eom, Nandre, Choi, Lee, Ryu and Ro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyeon-Su Ro, cm9oeWVvbkBnbnUuYWMua3I=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.