 Chunming Tan
Chunming Tan Wang Zhao
Wang Zhao Weigeng Wen1,2,4
Weigeng Wen1,2,4 Zhenhua Ma
Zhenhua Ma- 1Tropical Aquaculture Research and Development Center of South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Sanya, China
- 2Key Laboratory of South China Sea Fishery Resources Exploitation and Utilization, Ministry of Agriculture and Rural Affairs, Guangzhou, China
- 3College of Food Science and Nutritional Engineering, China Agricultural University, Beijing, China
- 4Sanya Tropical Fisheries Research Institute, Sanya, China
The aim of this work was to reveal the changes in gut microbiota composition and immune responses of sea cucumber (Stichopus variegatus) after being affected by different doses of sulfamethoxazole. In this study, the bacterial 16S rRNA of gut microbiota were analyzed by high-throughput sequencing, and the activities of immune enzymes [lysozyme (LZM), phenoloxidase (PO), alkaline phosphatase (AKP), and acid phosphatase (ACP)] in the gut of S. variegatus were determined. The results showed that the gut microbiota presented a lower richness in the antibiotic treatment groups compared with the control group, and there were significant differences among the dominant bacteria of different concentration treatments. At the genus level, the abundance of Escherichia, Exiguobacterium, Acinetobacter, Pseudomonas, and Thalassotalea were significantly decreased in the 3 mg/L treatment group, while Vibrio was significantly increased. Furthermore, the 6 mg/L treatment group had less effect on these intestinal dominant bacteria, especially Vibrio. The changes in relative abundance of Vibrio at the species level indicated that lower concentrations of sulfamethoxazole could enhance the enrichment of Vibrio mediterranei and Vibrio fortis in S. variegatus more than higher concentrations of sulfamethoxazole. Meanwhile, the 3 mg/L treatment group significantly increased the activities of PO, AKP, and ACP, and decreased the activity of LZM. These results suggested that lower doses of sulfamethoxazole have a greater effect on the gut microbiota composition and immune responses in S. variegatus and may increase the risk of host infection.
Introduction
Stichopus variegatus is an important aquaculture species in China with high nutrition and economic value (Wang J. J. et al., 2015). In addition to being edible, S. variegatus also has many important medicinal components, such as active saponins, acid mucopolysaccharides, and sulfated polysaccharides (Thinh et al., 2018; Kokadir et al., 2021). These biologically active compounds are good functional food ingredients. Consequently, the aquaculture and consumption of S. varigatus is increasing. In recent years, with the expansion of intensive aquiculture of S. variegatus, the breeding industry began to frequently explode various bacterial diseases, and the dispensable and inexpensive method to deal with pathogenic infections is still to use a certain amount of antibiotics.
Sulfamethoxazole is a widespread antibiotic, because of their physicochemical properties and broad spectrum of against antibacterial activity, has been used in aquaculture to control the occurrence of aquatic animal diseases (Payán et al., 2011; Zhang et al., 2015). In China, there is a severe situation that sulfamethoxazole misuse or abuse is widespread, because of the weak public consciousness and lack of reasonable supervision, especially in the breeding of sea cucumbers. However, sulfamethoxazole has the characteristics of low bioavailability and high water solubility. The large amount of unabsorbed antibiotics not only poses a serious ecological threat to the environment, but also severely disturbs the intestinal homeostasis of organisms and leads to the emergence of antibiotic-resistant strains (Li et al., 2015; Zhao et al., 2019b). Studies showed that although antibiotics control the occurrence of diseases to some extent, it also affects the structure of animal intestinal microflora, studies on the changes of intestinal microflora in aquaculture animals caused by antibiotics also attracted attention (Stanton, 2013; Wang R. X. et al., 2015; Zhao et al., 2019b). Gut microbiota are important for host growth and survival, they can help the host facilitate digestion, maintain energy balance, prevent pathogen colonization, and improve immunity (Ming et al., 2020). It plays crucial roles in boosting the immune-related activities and degrading toxic compounds in the host (Tremaroli and Backhed, 2012; Purchiaroni et al., 2013; Zhao et al., 2016). Especially bacterial pathogens (e.g., Escherichia, Shigella, and Vibrio, etc.), of which Vibrio species are the main opportunistic bacterial pathogens responsible for diseases in marine animals (Kang et al., 2014). However, systematic analysis of the effects of antibiotics on the gut microbiota and immune responses in S. variegatus has not been performed.
The gut microbiota of aquatic animals is an integral part of intestinal tract, it is interdependent and mutually restrictive with the host and the aquatic environment in which it is resided (Wang et al., 2010). The homeostatic balance of the gut microbiota in sea cucumber is sensitive to these factors and causing diseases like rot skin syndrome, mycosis, and bacterial ulcer disease (Zhao et al., 2016; Zhang et al., 2018). Previously, the study on the gut microbiota of sea cucumber is mainly through the analysis of culturable microorganisms, however, 85–99% of bacteria in nature cannot be isolated and cultured (Zhang et al., 2011). Comprehensive information on animal gut microbiome failed to truly reflect, as only a small fraction of the microbes was isolated and characterized (Iacumin et al., 2009). Although the combination of 16S rRNA and genetic fingerprinting technologies can be used to identify the dominant microflora in the aquatic animal intestine (Shokralla et al., 2012), it is vulnerable to the effects of the gene copy number.
High-throughput sequencing was regarded as a new technology in microbiota study, which has been successfully used in many disciplines to analyze the complex bacterial ecosystem of the gut, such as phytoplankton, marine and intestinal environment, fermented food, and human health and disease (Qiao et al., 2019; Ming et al., 2020; Chen Y. et al., 2022). These new analytical approaches have significantly accelerated the characterization of animal bacteria in several phenotypes and diseases by metagenomic and 16S rRNA-based techniques (Zoetendal et al., 2008; Fre-Mont et al., 2013; Zhao et al., 2016). It provides a high number of reads at a relatively low cost, and thousands of bacteria can be identified in each sample by sequence analysis. Sequencing-based molecular techniques can provide a comprehensive dissect of the dynamics of gut microbiota and reveal the association between changes in microbiota composition and disease (Yu et al., 2013; Shang et al., 2018; Zhuang et al., 2019). In this study, a high-throughput sequencing approach was used to reveal the characteristics of S. variegatus gut microbiota after being affected by sulfamethoxazole. And the immune-related enzyme activities of S. variegatus were also analyzed. We aimed to investigate the advantages and disadvantages of sulfamethoxazole in aquaculture, and then provide a reference for the administration of sulfamethoxazole in the aquaculture industry.
Materials and methods
Sample collection
The S. variegatus were caught from an aquaculture base in West Island of Sanya in April 2019, bodyweight 152.17 ± 26.89 g. We randomly divided 90 sea cucumbers into three pools (500 L each, 30 sea cucumber per pool), and then allowed the sea cucumbers to acclimate for a week in the culture pool of the Tropical Aquaculture Research and Development Center. Feeding with artificial feed (composed of Sargassum powder, Sargassum thunbergii powder, sea mud, sand, and sea cucumber bait in equal proportion). After the acclimation period, the experiment started and continued for 12 days. During the experiment, sea cucumbers were fed artificial feed once daily at 16:00. The water temperature, salinity, and pH of the seawater during the period were (25 ± 0.5)°C, 32 ± 0.8, and 7.8 ± 0.2, respectively. The control group (HCG) was treated with no added sulfamethoxazole, the HATG treatment group were supplemented with sulfamethoxazole (3 mg/L water volume), while the HHC treatment group were supplemented with sulfamethoxazole (6 mg/L water volume). The complete intestine samples of S. variegatus were collected after the end of experimentation in a pool with sulfamethoxazole. Each group were performed in three replicates, and all samples were immediately stored at −80°C and processed within a month.
Microbial community structure analysis
Total genomic DNA of the samples was extracted using Stool DNA Kit (Refer to QC report for specific kit) according to manufacturer’s protocols. The V3 and V4 regions of 16S rRNA were amplified by specific primer combination (forward primers: 5′-CCTACGGRRBGCASCAGKVRVGAAT-3′ and reverse primers: 5′-GGACTACNVG GGTWTCTAATCC-3′). At the same time, indexed adapters were added to the ends of the 16S rRNA amplicons to generate indexed libraries ready for downstream NGS sequencing on Illumina Miseq. PCR reactions were carried out in 25 μl mixture containing 2.5 μl TransStart Buffer, 2 μl dNTPs, 1 μl each primer, and 20 ng template DNA. The PCR products were checked by 2% agarose gel electrophoresis and quantified the library to 10 nM using a Qubit fluorescence quantifier. The amplicon libraries were constructed using the TruSeq® DNA Library Prep Kit (Illumina Inc., San Diego, California, United States), and sequencing was performed on an Illumina MiSeq PE300 platform (Illumina, San Diego, CA, United States). The raw sequence datasets of samples obtained in this study have been uploaded to the NCBI Sequence Read Archive (SRA), BioProject PRJNA874578.
Quality filtering on merged sequences was performed by QIIME (version 9.1), and the sequences were compared with the reference database (RDP Gold database) using UCHIME algorithm to detect chimeric sequence. Effective sequences were clustered into operational taxonomic units (OTUs) using the clustering program VSEARCH (1.9.6) against the Silva 132 database pre-clustered at 97% sequence identity. The OTUs were taxonomically identified using the Ribosomal Database Program (RDP) classifier. The RDP classifier uses the Silva 132 database, which has taxonomic categories predicted to the genus level (Quast et al., 2013; Yilmaz et al., 2014). The OTU cluster information, rarefaction curves, and alpha diversity indexes (Shannon and Simpson index, Chao1 richness index, and Good’s coverage) were calculated by QIIME program (Kang et al., 2021). The beta diversity was carried out using UniFrac analysis (Hamady et al., 2010) and principal coordinates analysis (PCoA), the ANOSIM was used to perform a hypothesis test of the microbiome (Kim et al., 2021). The significant differences in community composition between groups based on community abundance data at genus level were analyzed by Metastats analysis. The Kruskal-Wallis test was used to detect taxa that exhibit differences in abundance between the two groups of microbial communities, and the multiple hypotheses testing of rare frequency data and false discovery rate (FDR) analysis were used to assess the significance of the observed differences.
Immune indexes
The activity of immune enzymes in the samples were tested with corresponding kits (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China), including lysozyme (LZM, No. A050-1), phenoloxidase (PO, No. H247), alkaline phosphatase (AKP, No. A059-2), and acid phosphatase (ACP, No. A060-2). The activity of LZM was determined according to the fact that LZM can lyse bacteria and enhance the light transmittance of turbid bacterial liquid, and the results were expressed as U per mg protein. The protein concentration of the samples was tested using a BCA kit (No. A045-3).
The content of PO was determined according to the kit instructions. It is based on the competition of enzymes and antibodies against solid phase antigens, followed by the formation of immune complexes. The results were expressed as mg per g protein.
Alkaline phosphatase and ACP can catalyze the formation of free phenol from disodium phenyl phosphate under acidic and basic conditions, respectively. The phenol reacts with 4-aminoantipyrine in alkaline solution and is oxidized by potassium ferricyanide to form red quinone derivatives, its enzymatic activity can be calculated based on the absorbance value at 510 nm (Wei et al., 2022). One unit of enzymatic activity is defined as the catalytic production of 1 μM phenol per milligram of protein per minute at 37°C, and the results were expressed as U per mg protein.
Statistical analysis
The data were processed using Excel 2016 and expressed as mean ± SD. The significant differences among samples were analyzed through SPSS v21.0 (SPSS Inc., Chicago, IL, United States). A one-way ANOVA and Duncan multiple comparison test were used to compare the effect of sulfamethoxazole on the differences for LZM, PO, AKP, and ACP. The p < 0.05 represented has a significant difference.
Results
Characteristics of the sequencing results and alpha diversity
A total of 664,189 high quality reads of 16S rRNA sequences were obtained from the nine samples with 73,799 sequence reads per sample, the average length of sequence reads was 459.75 bp, and the coverage reached 99.9% (Table 1). The rarefaction curves showed a similar pattern of reaching plateau and the final trend is saturated (Figure 1A). These results showed almost all the OTUs in the datasets have already been captured.
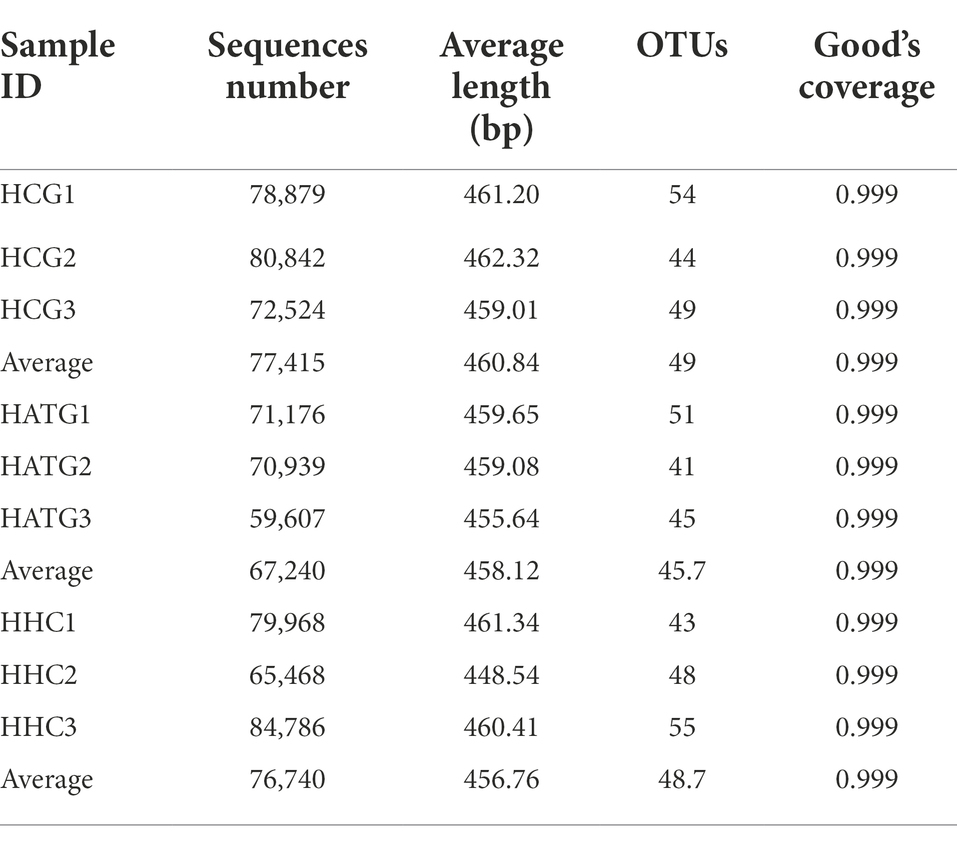
Table 1. Sample information and sequence statistics.
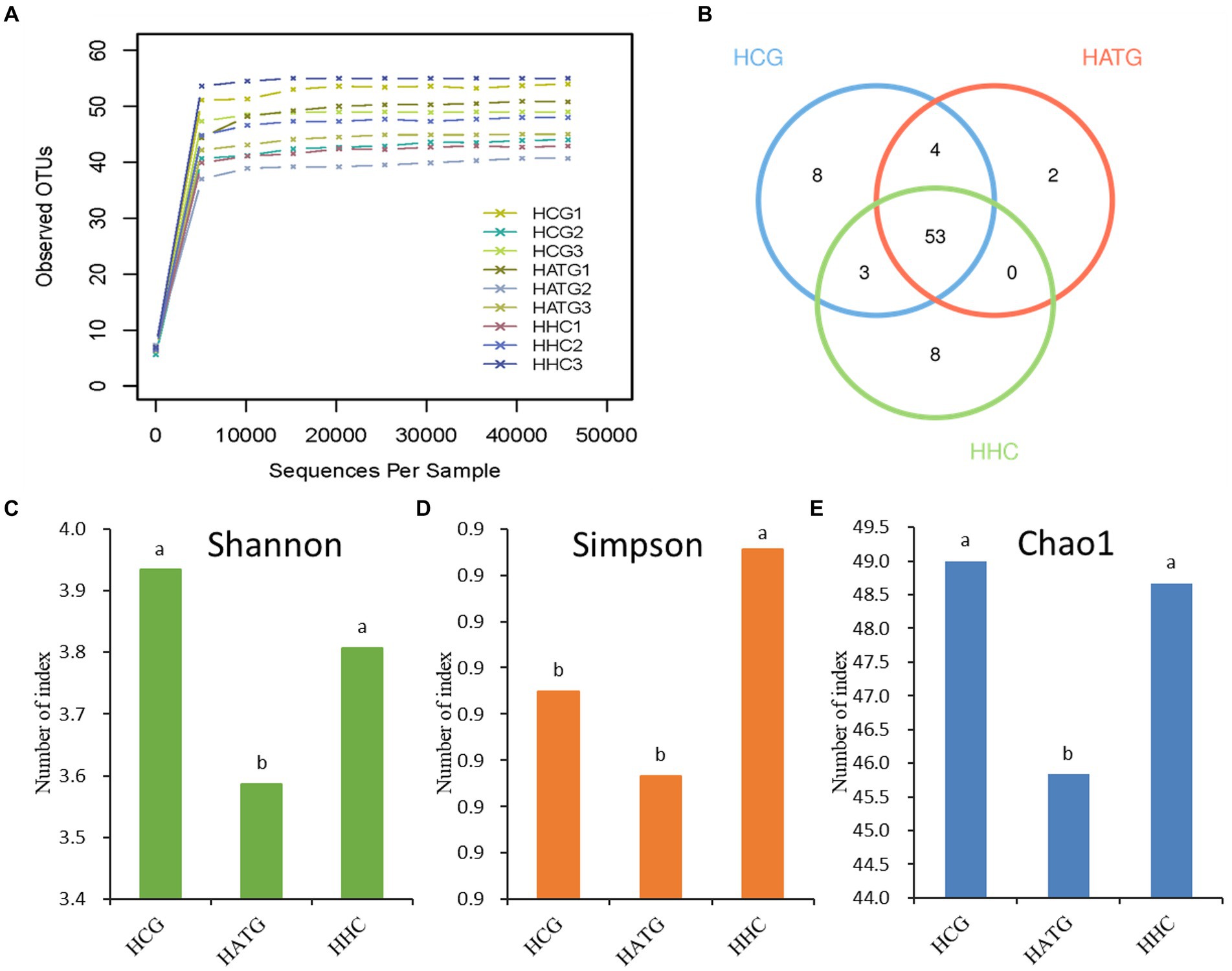
Figure 1. The Rarefaction Curve (A), Venn diagram (B), Shannon diversity index (C), Simpson diversity index (D), and Chao1 richness index (E) for various samples. The HCG, HATG, and HHC represent control group, 3 and 6 mg/L sulfamethoxazole treatment group, respectively. Different letters obtained by one-way ANOVA followed by Duncan’s test indicated significant differences at p < 0.05.
The Venn diagram could reflect the difference among HCG, HATG, and HHC groups. There were 68, 59, and 64 OTUs in HCG, HATG, and HHC, and the ratio of their common OTUs were 77.9, 89.8, and 82.8%, respectively (Figure 1B) It indicated that the sulfamethoxazole significantly changed the microbial composition in S. variegatus intestine.
The community diversity can generally be reflected by Shannon index and Simpson index, and the greater the Shannon value or the lower the Simpson value, the higher the species diversity. According to the results, two indices hinted that the control group (HCG) has the highest community diversity (Figures 1C,D). In addition, Chao proposed that the index of the number of OTUs in the sample estimated by the Chao1 algorithm can be well used for the estimation of species evaluation in ecology (Pitta et al., 2010). Chao1 index represent the community richness, the greater the Chao1 index, and the higher the community richness. As shown in Figure 1E, the highest Chao1 index is control group, while the lowest Chao1 index is HATG group, which suggest that the community richness of the gut microbiota of S. variegatus decreased after treated by sulfamethoxazole, and the low doses of sulfamethoxazole had a greater impact.
Analysis of beta diversity
The differences in microbiota between samples can be compared through calculating the distance between samples by using the richness and evolutionary information of sample sequences (Lozupone and Knight, 2012). In this study, the UniFrac analysis was used to calculate the distance matrix of samples, and the differences were reflected in a heat-map with color changes. The defined light and dark colors in the heat-map represent the degree of difference between the two samples. Compared with control group, the HATG treatment group had a higher difference coefficient in genus diversity (Figure 2A). This result indicated that the effect of HATG treatment group on the diversity of the gut microbiota of S. variegatus was greater than that of HHC group.
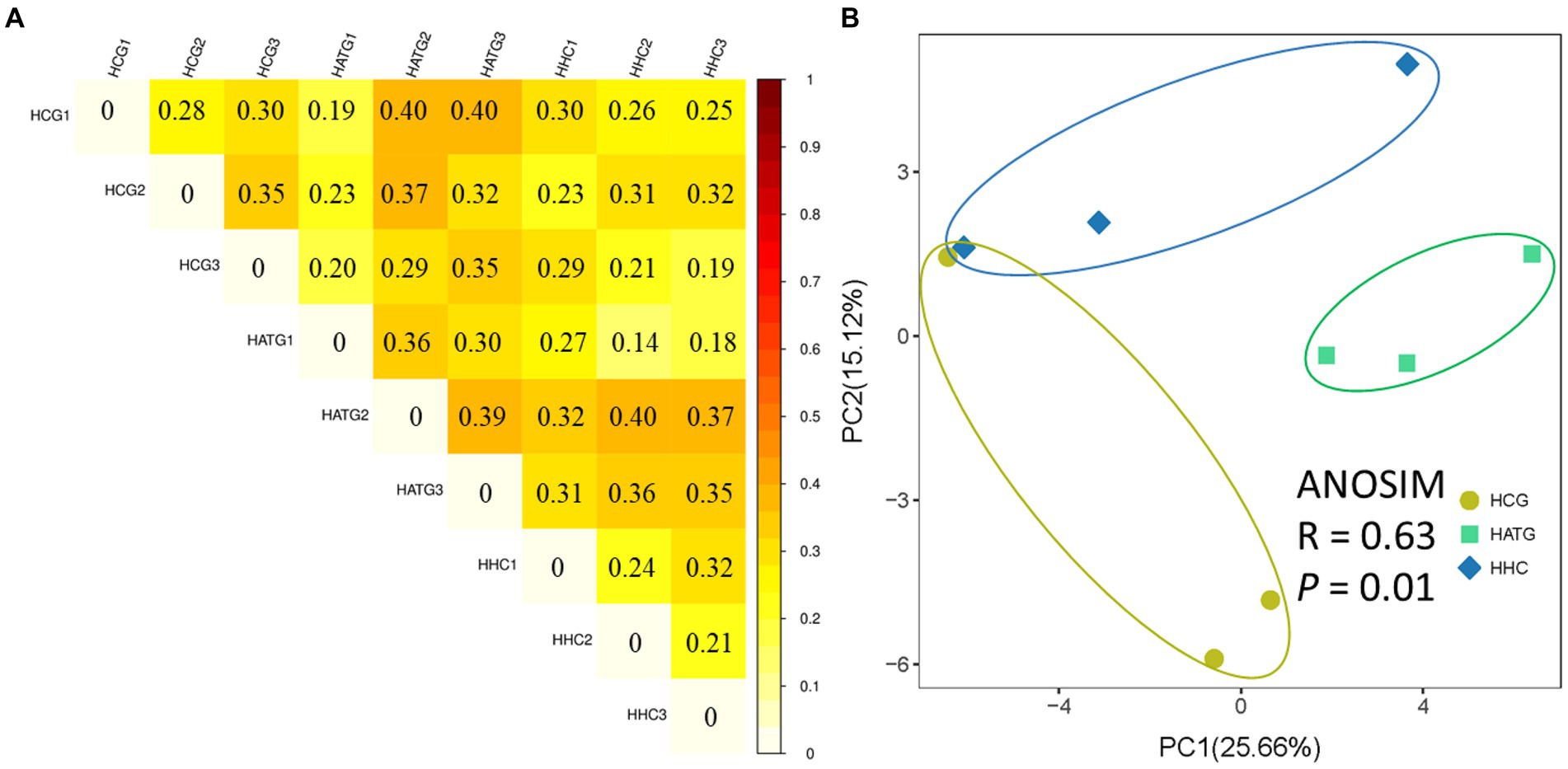
Figure 2. Unweighted UniFrac difference analysis of the diversity among samples (A) and principal co-ordinates analysis (PCoA) of bacterial community of Stichopus variegatus (B).
The PCoA analysis based on the relative abundance of OTUs showed that the differences in microbial community composition was significant (p < 0.05; Figure 2B). The HCG samples were clearly separated from the HATG and HHC samples, suggesting that the microbial community composition changed greatly and the effect of sulfamethoxazole on the microbial community composition was significant and greater at the low doses of sulfamethoxazole.
Changes in community abundance and composition
In this study, statistical analysis was used to observe the community structure of samples at different taxonomic levels. At the genus level, data for the top 30 microbial populations were analyzed. As shown in Figure 3, Vibrio, Escherichia-Shigella, Exiguobacterium, Acinetobacter, Helicobacter, and Pseudomonas constituted six common dominant genera in HCG, HATG, and HHC groups, which accounted for 56.74, 72.37, and 74.97% of the total sequencing number. However, Thalassotalea, Lactococcus, Anoxybacillus, and Pseudoalteromonas were also the dominant genera in the HCG group. Compared with control group, the abundant of Escherichia-Shigella, Exiguobacterium, Acinetobacter, Pseudomonas Thalassotalea, Lactococcus, Anoxybacillus, and Pseudoalteromonas were significantly decreased in the HATG groups, while Vibrio increased significantly. And the ratio of Vibrio in HATG group was much higher than that in the control group and HHC group, indicating that the Vibrio in gut microbiota of S. variegatus was more sensitive to the effects of lower concentrations of sulfamethoxazole (Figure 3).
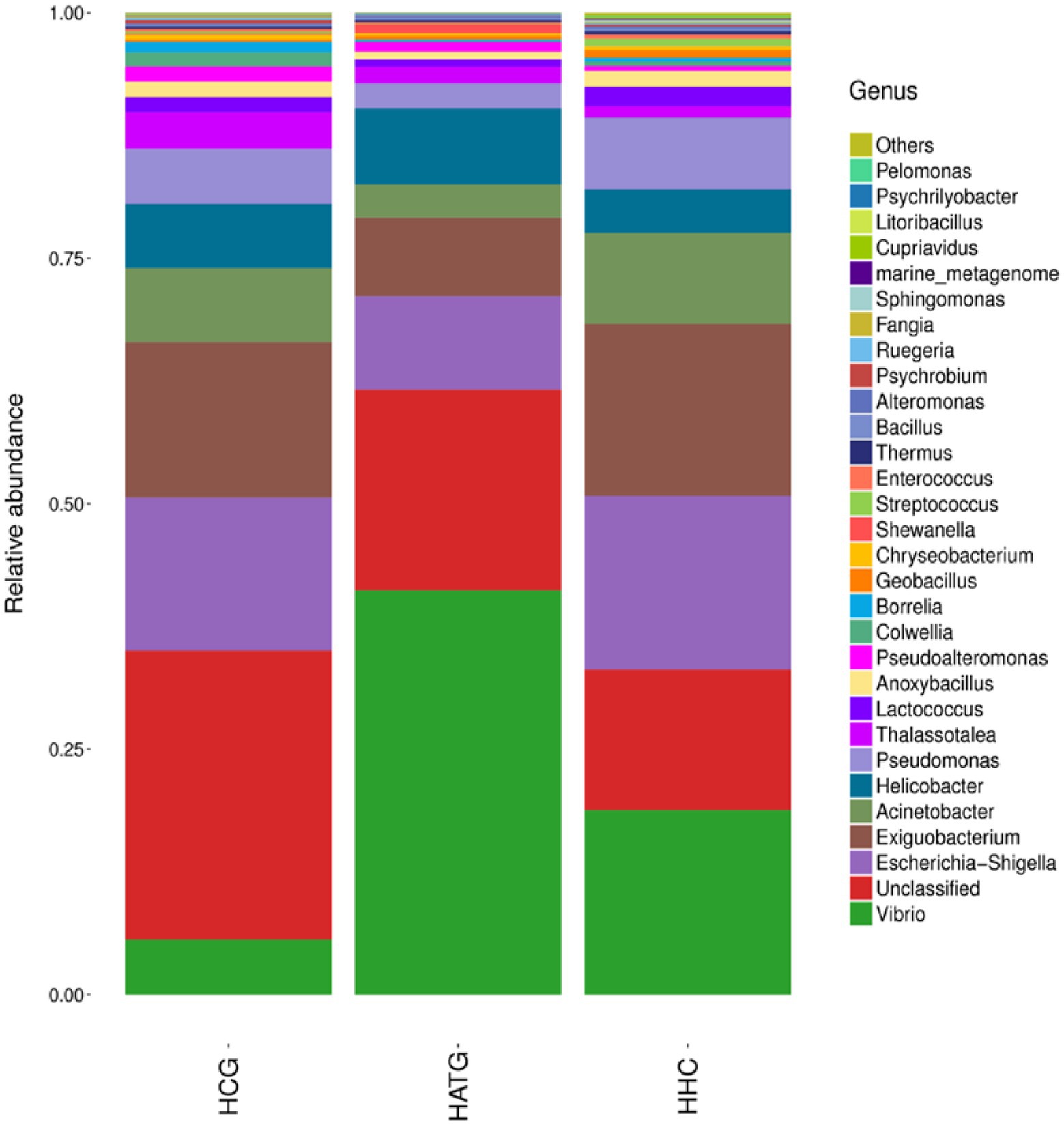
Figure 3. Composition and relative abundance of bacterial communities in genera of the different treatment groups.
Analysis of significant differences in community structure between groups
Based on community abundance of different groups, the five top genera significant differences between groups were compared. A Kruskal-Wallis test was used to detect the classification of abundance differences between two groups of microbial communities, and multiple hypothesis test and false detection rate (FDR) analysis of rare frequency data are carried out. As shown in Figure 4A, the Acinetobacter, Escherichia-Shigella, Exiguobacterium, Pseudomonas, and Vibrio were most significantly different (p < 0.05) between HCG and HATG. Among these five genera, Vibrio was significantly more abundant in HATG group compared with HCG group, while the opposite was observed for other genera. Compared with HATG group, the HHC showed a significantly higher bacterial community abundant in Acinetobacter, Anoxybacillus, Exiguobacterium, Lactococcus, and Pseudomonas (Figure 4B). The results indicated that lower concentration sulfamethoxazole was more effective in reducing the abundance of gut microbiota of S. variegatus, but greatly increased the abundance of Vibrio. This means that lower concentration sulfamethoxazole can play a better antibacterial effect, but increase the risk of Vibrio infection in S. variegatus.
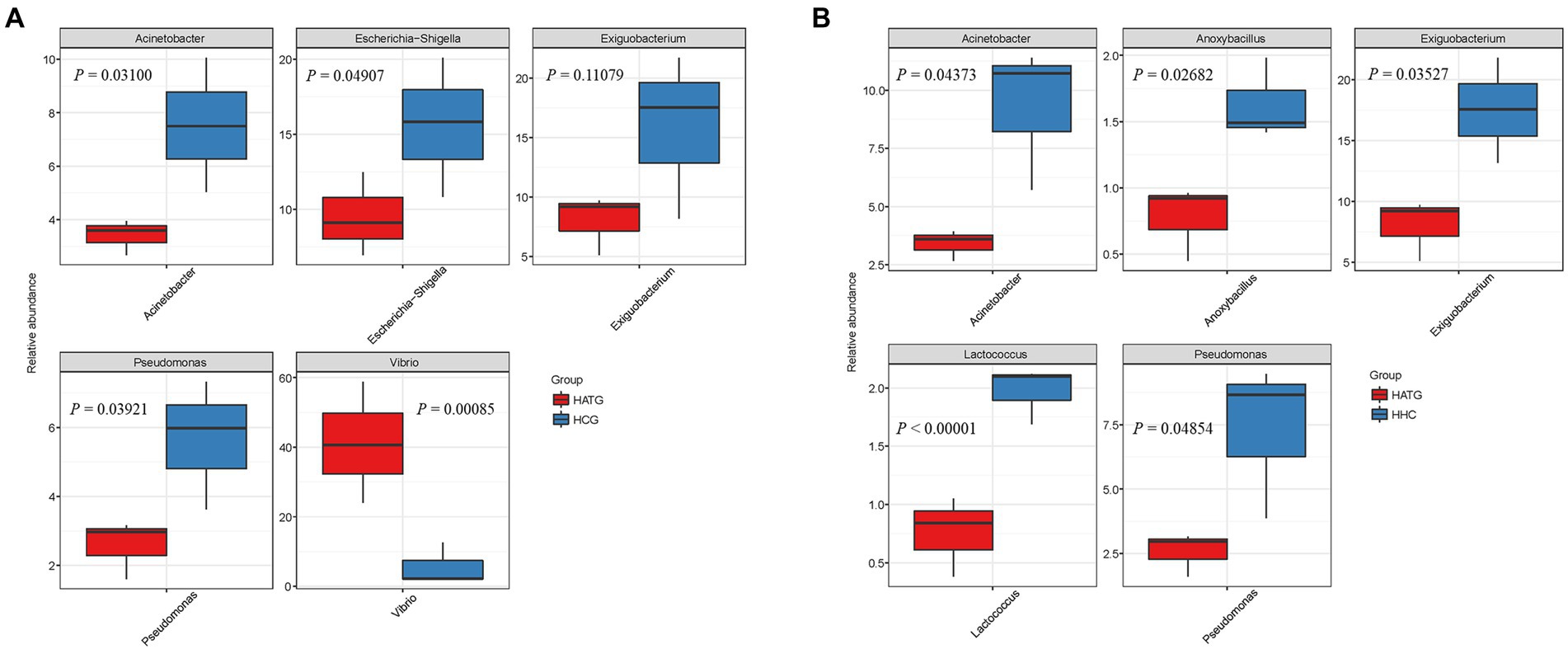
Figure 4. Comparison of relative abundance differences of the bacterial communities between groups, (A) group HCG and HATG, (B) group HATG and HHC. A Kruskal-Wallis test was used to detect the differences in abundance between the two groups of microbial communities.
Relative abundance changes of Vibrio species in Stichopus variegatus
Furthermore, the relative abundance of Vibrio at the species level was analyzed, as shown in Figure 5. In this study, two types of Vibrio species were identified, namely Vibrio mediterranei and Vibrio fortis. Compared to the group not treated with sulfamethoxazole (HCG), the abundance of V. mediterranei was significantly increased after sulfamethoxazole treatment at 3 mg/L (HATG). However, the 6 mg/L sulfamethoxazole treatment group (HHC) had a lower effect on V. mediterranei. And the effect of sulfamethoxazole on Vibrio fortis in the gut of S. variegatus was consistent with the trend of V. mediterranei. The results further indicated that lower concentrations of sulfamethoxazole could enhance the enrichment of Vibrio in S. variegatus more than higher concentrations of sulfamethoxazole.
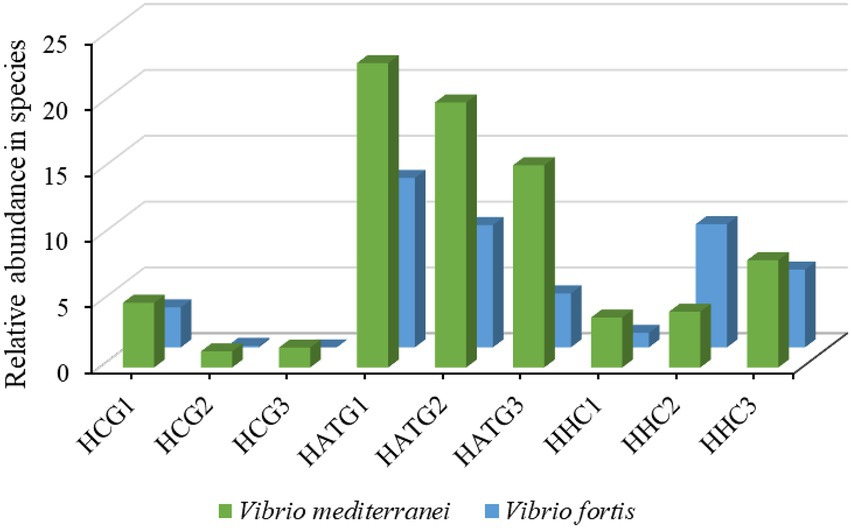
Figure 5. The relative abundance changes of Vibrio species of Intestinal microbiota in Stichopus variegatus.
Effects on the activities of immune-related enzymes in Stichopus variegatus
The LZM activity of S. variegatus was significantly changed after being affected by sulfamethoxazole (p < 0.05), as shown in Figure 6A. The activity of LZM decreased in both HATG and HHC treatment groups, and the decrease was more pronounced in the HATG treatment group. The changes in PO activity after the sulfamethoxazole exposure showed a trend of increasing with the concentration of sulfamethoxazole (Figure 6B). However, there was no significant difference between the HATG and HHC treatment groups (p > 0.05). The alkaline phosphatase and acid phosphatase activities in S. variegatus were also affected by sulfamethoxazole exposure, and they showed a similar trend. The lower concentration antibiotic treatment group significantly increased the activities of ACP and AKP, while there was no significant difference between the higher concentration antibiotic treatment group and the control group (Figures 6C,D).
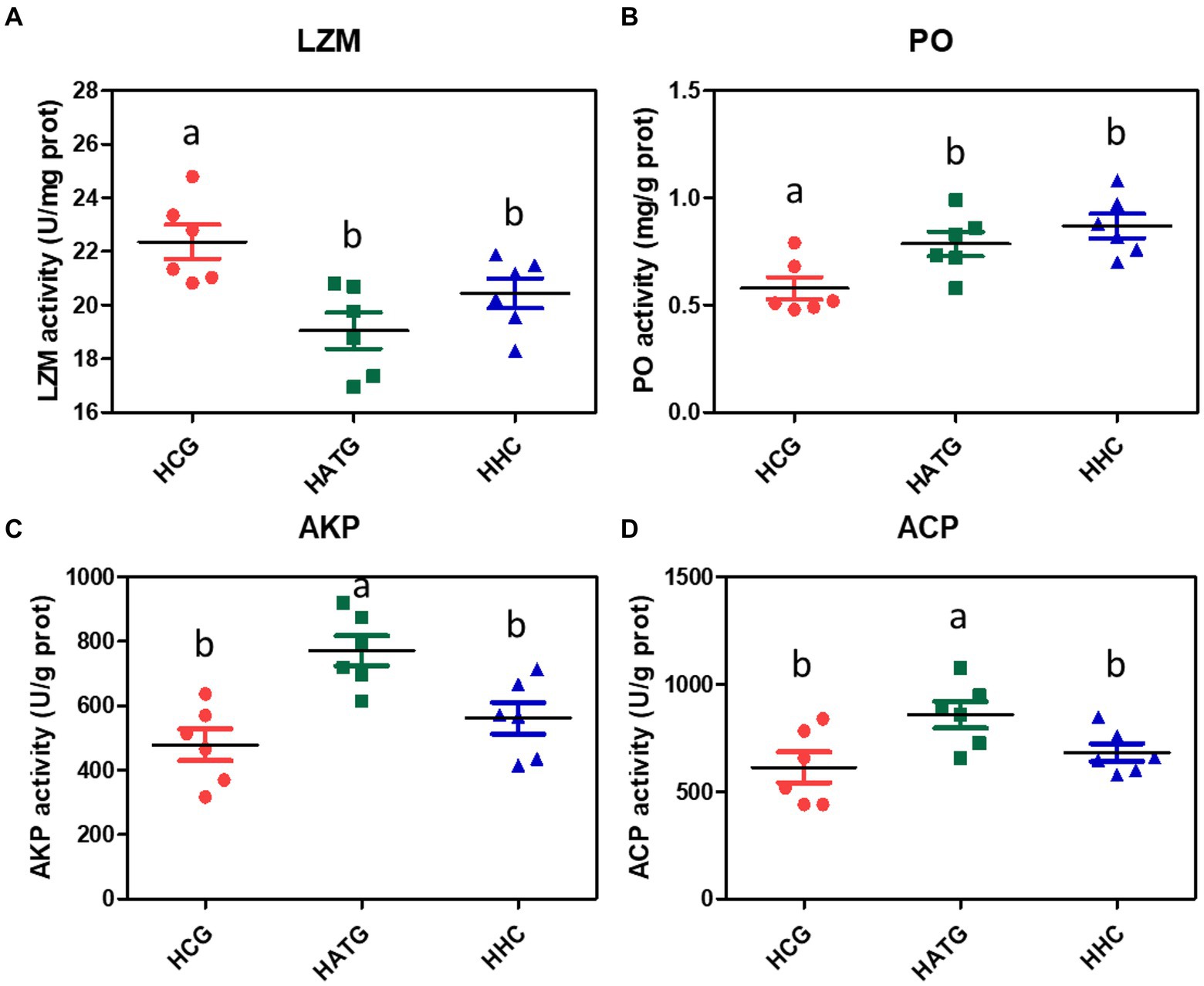
Figure 6. Changes in the activities of immune-related enzymes in Stichopus variegatus after treated by sulfamethoxazole. (A) lysozyme activity (LZM), (B) phenoloxidase activity (PO), (C) alkaline phosphatase activity (AKP), and (D) acid phosphatase (ACP). Data were expressed as mean ± SD (n = 6). The different superscript letters indicated significant differences from other groups (p < 0.05).
Correlation analysis between the bacterial abundance and immune enzymes
The correlation between microbiota and immune-related enzymes of S. variegatus was analyzed by Spearman correlation analysis (Figure 7). Dominant bacteria Vibrio, Anoxybacillus, Acinetobacter, Lactobacillus, and Enterococcus showed a significant correlation with the immune enzymes (p < 0.05). Among them, Vibrio had the highest abundance at the HATG group, which positively correlated with activities of ACP, AKP, and PO, and negatively correlated with LZM. While Lactobacillus showed a positive correlation with the activities of LZM, and negatively correlated with PO and AKP. Besides, Enterococcus, Acinetobacter, and Anoxybacillus showed a similar correlation with immune enzymes, which is positive correlation with the activities of LZM and PO, and negatively correlated with ACP and AKP.
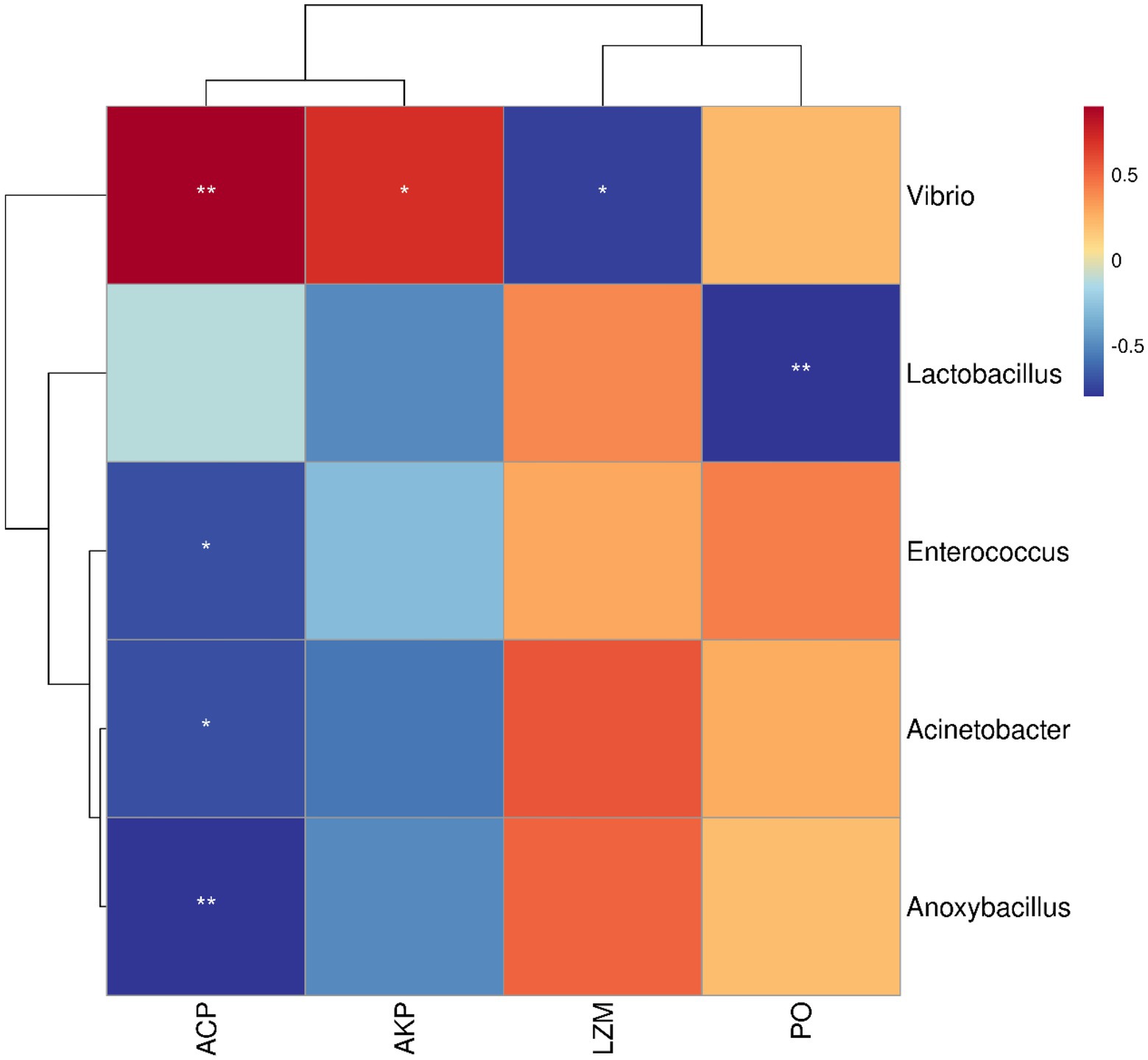
Figure 7. Analysis of the correlation between microbiota and immune enzymes. Microbiota was considered at the OTU level and statistically significant Spearman correlations were calculated among samples. * represented p < 0.05, ** represented p < 0.01.
Discussion
Accumulating scientific research-based evidence is driving our increased awareness of the significance of the antibiotics to the healthy and homeostatic operation of the aquatic animals (Wang J. J. et al., 2015; Manage, 2018). Antibiotics are gradually attracting global attention as a new type of pollutant. To investigate the effects of sulfamethoxazole on aquatic animals might provide a reference for the administration of sulfamethoxazole in aquaculture. In this work, high-throughput sequencing technique was applied to reveal the gut microbiome characteristics of S. variegatus after being affected by sulfamethoxazole, and a comprehensive analysis of the sequence abundance and diversity of the gut microbiota of the sulfamethoxazole treated and non-sulfamethoxazole treated groups were performed. We found that OTUs and some bacterial genera varied among different treatments, indicating the abundance and diversity of bacterial communities in samples were changed by different concentrations of sulfamethoxazole. Angelakis et al. (2013) reported that the effects of antibiotics on changes in gut microbiota and functional alterations in animals depend on the type of antibiotic, dose, and duration of exposure.
The community richness and diversity of the gut microbiota in sulfamethoxazole treatment groups decreased compared with control group. The decrease of diversity will break the dynamic balance of intestinal microecology and cause instability of community structure (Zhao et al., 2018). Liu et al. (2011) reported that there is a relative dynamic equilibrium between the bacteria in the intestine, or between the animals and the intestinal bacteria, this state is of great significance to the maintenance of the host’s normal physiological function. The imbalance of gut microbiota of the sea cucumber is easy to cause disease such as inflammatory bowel disease, and this change in the structure of the gut microbiota may also be one of the causes of frequent disease in spring and autumn (Zhang et al., 2006; Zhao et al., 2019a). In this study, the effect of 3 mg/L sulfamethoxazole treatment groups on gut microbiota is greater than in 6 mg/L sulfamethoxazole exposed group. This may be a result of varying degrees of suppression of the normal microflora in the intestine that are sensitive to the drugs, while insensitive strains multiply under the action of higher concentration of antibiotics and induce other bacteria to produce resistance. Previous studies have demonstrated that antibiotic resistance can be rapidly induced and spread among bacterial species, resulting in harmful effects on aquatic ecology and aquatic organisms (Wang and Schaffner, 2011). The report of Butts et al. (2019) on establishment of antibiotic-induced dysbacteriosis model also confirmed that low concentration antibiotics induce dysbacteriosis in a method that is superior to higher concentration of antibiotics. This might be low-dose antibiotics have a greater impact on their metabolic capacity and the spread of antibiotic-resistant bacterial strains (Duan et al., 2020). Besides, the enterotype, genotypes, and different antibiotic sensitivities of individuals may also lead to low-dose antibiotic treatment induces dysbiosis by decreasing the diversity of the microbiome.
Our results indicated that the composition of gut microbiota of S. variegatus in phylum level are consistent with previous studies that Proteobacteria and Firmicutes have high abundance (Pagán-Jiménez et al., 2019; Zhao et al., 2019a; Chen J. et al., 2022). At the genus level, the results of heat-map showed that the community richness of the gut microbiota of S. variegatus decreased after treated by sulfamethoxazole, and Vibrio, Escherichia, Exiguobacterium, Acinetobacter, Helicobacter, and Pseudomonas were the dominant bacteria have the highest richness at the genus level. As for Vibrio, Escherichia, and Acinetobacter that played important roles in aquaculture, which were noticeably abundant in the naturally occurring animals (Hakim et al., 2015). However, Vibrio and Escherichia were considered intestinal pathogens that can cause diseases in aquatic animals (Ming et al., 2020). The Vibrio in low-concentration sulfamethoxazole treatment group showed the greatest richness, and the proportion of Vibrio species after treated by sulfamethoxazole has increased greatly. This result is probably due to the fact that Vibrio species are the main opportunistic bacterial pathogens responsible for diseases in marine animals (Kang et al., 2014). However, the significantly decreased abundance of Escherichia and Acinetobacter may be due to their sensitivity to sulfamethoxazole. Fu et al. (2008) reported that Escherichia sensitive to sulfonamides, streptomycin, chloramphenicol, etc.
After analyzing differences in relative abundance, we observed that the genera between the two groups were significantly different, which suggested that sulfamethoxazole influenced the composition of gut microbiota, and the effect varies with the concentration. In the study, the effect of low concentration of sulfamethoxazole treatment on gut microbiota is much greater than higher concentration. The result was consistent with the characteristics of gastrointestinal microbiota in mice that induced by an antibiotic (Butts et al., 2019). The sulfamethoxazole significantly increased the ratio of Vibrio and decreased the proportion of Exiguobacterium, Acinetobacter, and Pseudomonas. Wang R. X. et al. (2015) showed that many Vibrio strains have high resistant rates to antibiotics. In China, marine Vibrio species are the predominant multidrug-resistant bacteria in the mariculture environments studied, which have long been recognized as important reservoirs and vehicles of antimicrobial resistance (Thompson et al., 2005; Dang et al., 2009). Thompson et al. (2004) showed that Vibrio species have the ability to readily develop and acquire antimicrobial resistance in response to selective pressure, as well as their ability to spread resistance through horizontal exchange of genetic material. And this suggests that Vibrio, Acinetobacter, Pseudomonas, and Exiguobacterium may play an important role in balancing gut microbiota of S. variegatus. However, this is the first report of an abundance of Pseudomonas in low concentration of sulfamethoxazole treatment groups and the Exiguobacterium in high concentration treatment groups, more research must be conducted.
According to Liu et al. (2020), the antibiotics exposure might evoke immune responses in healthy aquatic organisms. In this study, the LZM activity of S. variegatus was significantly inhibited after exposure to sulfamethoxazole environment. This might be sulfamethoxazole suppresses the ability of S. variegatus to resist pathogen. Strzepa and Szczepanik (2013) reported that the composition of the gut microbiota affects the immune response. Through correlation analysis between the bacterial abundance and immune enzymes we found that the dominant gut microbiota Vibrio, Anoxybacillus, Acinetobacter, Lactobacillus, and Enterococcus in S. variegatus showed a significant correlation with the immune enzymes (p < 0.05). Especially the Vibrio, which showed a strong correlation with the activities of ACP, AKP, and LZM. This is the reason why ACP, AKP, and LZM enzyme activities were significantly higher or lower in the HATG-treated groups. Therefore, the change of immune enzymes of S. variegatus related to the influence of sulfamethoxazole on the community structure of its gut microbiota. However, the treatment of sulfamethoxazole significantly increased the PO activity of S. variegatus, although there was no significant difference between HATG and HHC. This may be related to the increased proportion of Vibrio in the sea cucumber gut after sulfamethoxazole treatment, because phenoloxidase plays an important role in inhibiting or even killing pathogenic microorganisms (Liu et al., 2007). In addition, AKP and ACP activities showed a consistent pattern of changes, that is, treatment with lower concentrations of sulfamethoxazole significantly increased their activities. The results suggested that lower concentration sulfamethoxazole might make S. variegatus easier to against pathogens and foreign bodies. These results may be an important piece of evidence in the fact that S. variegatus is more susceptible to diseases in artificial aquaculture systems than in natural growth, but this needs to be confirmed further. In addition, it is necessary to conduct more research to characterize how antibiotic differences directly affect the bacterial populations present in intestinal samples.
Conclusion
The structure of the gut microbiota in S. variegatus changed dramatically after exposure to sulfamethoxazole. Sulfamethoxazole treatment resulted in a decrease in the community diversity, while increasing the abundance of pathogenic bacteria. The proportion of Vibrio species have increased greatly after treated by sulfamethoxazole, caused S. variegatus to be more susceptible to diseases, because the Vibrio species are the main opportunistic bacterial pathogens responsible for diseases in marine animals. Besides, the effect of lower concentration of sulfamethoxazole treatment on microbial diversity was greater than that of higher concentration treatment. Our findings provide preliminary guidance for the use of sulfamethoxazole in S. variegatus, and lay the foundation for the development of appropriate treatment regimens to address the impact on the gut microbiota. However, deeper research should be continued to reveal the effects of different sulfamethoxazole concentrations on particular types of microbes in the S. variegatus gut and thus evaluate their specific functional and molecular contributions to the gut microbiota.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA874578.
Author contributions
CT and GY conceived and designed the study. CT, WZ, and WW performed sampling and laboratory testing. CT, ZM, and XC analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Earmarked Fund for Hainan Natural Science Foundation for Youth (319QN341), Modern Agro-industry Technology Research System (CARS-49), and National Key R&D Program of China (2019YFD0900905).
Acknowledgments
The authors would like to thank the staffs of Sanya aquaculture base, who provided the assistance for samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Angelakis, E., Merhej, V., and Raoult, D. (2013). Related actions of probiotics and antibiotics on gut microbiota and weight modification. Lancet Infect. Dis. 13, 889–899. doi: 10.1016/S1473-3099(13)70179-8
Butts, C. A., Paturi, G., Stoklosinski, H., and Martell, S. (2019). Animal model of antibiotic induced gut microbiota dysbiosis. PRO 8, 11–16. doi: 10.3390/proceedings2019008011
Chen, Y., Li, P., He, W., Liao, L., Xia, B., Jiang, L., et al. (2022). Analysis of microbial community and the characterization of Aspergillus flavus in Liuyang Douchi during fermentation. LWT 154:112567. doi: 10.1016/j.lwt.2021.112567
Chen, J., Xu, Y., Liu, Y., Liu, K., Wu, Y., Zhang, Y., et al. (2022). Gut microbiota analysis and gene function prediction among young and adult Larus saundersi with habitat soil in the Yellow River Delta. Bioresour. Technol. Rep. 17:100960. doi: 10.1016/j.biteb.2022.100960
Dang, H. Y., Zhao, J. Y., Song, L. S., Chen, M., and Chang, Y. Q. (2009). Molecular characterizations of chloramphenicol-and oxytetracycline-resistant bacteria and resistance genes in mariculture waters of China. Mar. Pollut. Bull. 58, 987–994. doi: 10.1016/j.marpolbul.2009.02.016
Duan, H., Yu, L., Tian, F., Zhai, Q., Fan, L., and Chen, W. (2020). Antibiotic-induced gut dysbiosis and barrier disruption and the potential protective strategies. Crit. Rev. Food Sci. Nutr. 62, 1427–1452. doi: 10.1080/10408398.2020.1843396
Fre-Mont, M., Coomans, D., Massart, S., and De-Meirleir, K. (2013). High-throughput 16S rRNA gene sequencing reveals alterations of intestinal microbiota in myalgic encephalomyelitis/chronic fatigue syndrome patients. Anaerobe 22, 50–56. doi: 10.1016/j.anaerobe.2013.06.002
Fu, L., Ren, J., Li, J. Y., and Niu, S. P. (2008). Studies on drug sensitivity of salmonella and Escherichia colis from local pig farm in Xinjiang to 16 kinds of antibiotics. Xinjiang Agr. Sci. 36, 25–29. doi: 10.2967/jnmt.107.044081
Hakim, J. A., Koo, H., Dennis, L. N., Kumar, R., Ptacek, T., Morrow, C. D., et al. (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 6:1047. doi: 10.3389/fmicb.2015.01047
Hamady, M., Lozupone, C., and Knight, R. (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4, 17–27. doi: 10.1038/ismej.2009.97
Iacumin, L., Cecchini, F., Manzano, M., and Comi, G. (2009). Description of the microflora of sourdoughs by culture-dependent and culture-independent methods. Food Microbiol. 26, 128–135. doi: 10.1016/j.fm.2008.10.010
Kang, J., Hu, Y., Ding, Z., Ye, L., Li, H., Cheng, J., et al. (2021). Deciphering the shifts in microbial community diversity from material pretreatment to saccharification process of Fuyu-flavor Baijiu. Front. Microbiol. 12:705967. doi: 10.3389/fmicb.2021.705967
Kang, C. H., Kim, Y. G., Oh, S. G., Mok, J. S., Cho, M. H., and So, J. S. (2014). Antibiotic resistance of Vibrio harveyi isolated from seawater in Korea. Mar. Pollut. Bull. 86, 261–265. doi: 10.1016/j.marpolbul.2014.07.008
Kim, P. S., Shin, N. R., Lee, J. B., Kim, M. S., Whon, T. W., Hyun, D. W., et al. (2021). Host habitat is the major determinant of the gut microbiome of fish. Microbiome 9:166. doi: 10.1186/s40168-021-01113-x
Kokadir, S., Basilianus, E., Swandiny, G. F., and Abdillah, S. (2021). A review over sea cucumber (Stichopus variegatus): a potential candidate for nutraceuticals. J. Nutr. Food Sci. 6:42.
Li, W. H., Gao, L. H., Shi, Y. L., Liu, J. M., and Cai, Y. Q. (2015). Occurrence, distribution and risks of antibiotics in urban surface water in Beijing. Chin. Environ. Sci-Proc. Imp. 17, 1611–1619. doi: 10.1039/C5EM00216H
Liu, H., Jiravanichpaisal, P., Cerenius, L., Lee, B. L., Söderhäll, I., and Söderhäll, K. (2007). Phenoloxidase is an important component of the defense against Aeromonas hydrophila infection in a crustacean, Pacifastacus leniusculus. J. Biol. Chem. 282, 33593–33598. doi: 10.1074/jbc.M706113200
Liu, H. D., Wang, L., Liu, M., Wang, B. J., Jiang, K. Y., Ma, S. S., et al. (2011). The intestinal microbial diversity in Chinese shrimp (Fenneropenaeus chinensis) as determined by PCR–DGGE and clone library analyses. Aquaculture 317, 32–36. doi: 10.1016/j.aquaculture.2011.04.008
Liu, J., Wei, T., Wu, X., Zhong, H., Qiu, W., and Zheng, Y. (2020). Early exposure to environmental levels of sulfamethoxazole triggers immune and inflammatory response of healthy zebrafish larvae. Sci. Total Environ. 703:134724. doi: 10.1016/j.scitotenv.2019.134724
Lozupone, C., and Knight, R. (2012). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Manage, P. M. (2018). Heavy use of antibiotics in aquaculture: emerging human and animal health problems–a review. Sri Lanka J. Aqua. Sci. 23, 13–27. doi: 10.4038/sljas.v23i1.7543
Ming, J., Fu, Z., Ma, Z., Zhou, L., Zhang, Z., Song, C., et al. (2020). The effect of sulfamonomethoxine treatment on the gut microbiota of Nile tilapia (Oreochromis niloticus). MicrobiologyOpen 9:e1116. doi: 10.1002/mbo3.1116
Pagán-Jiménez, M., Ruiz-Calderón, J. F., Dominguez-Bello, M. G., and Garcıa-Arraras, J. E. (2019). Characterization of the intestinal microbiota of the sea cucumber Holothuria glaberrima. PLoS One 14:e0208011. doi: 10.1371/journal.pone.0208011
Payán, M. R., López, M. A. B., Fernández-Torres, R., Navarro, M. V., and Mochon, M. C. (2011). Hollow fiber-based liquid phase microextraction (HF-LPME) for a highly sensitive HPLC determination of sulfonamides and their main metabolites. J. Chromatogr. B 879, 197–204. doi: 10.1016/j.jchromb.2010.12.006
Pitta, D. W., Pinchak, W. E., Dowd, S. E., Osterstock, J., Gontcharova, V., Youn, E., et al. (2010). Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb. Ecol. 59, 511–522. doi: 10.1007/s00248-009-9609-6
Purchiaroni, F., Tortora, A., Gabrielli, M., Bertucci, F., Gigante, G., Ianiro, G., et al. (2013). The role of intestinal microbiota and the immune system. Eur. Rev. Med. Pharmacol. Sci. 38, 377–385. doi: 10.1038/npp.2012.191
Qiao, L., Chang, Z. Q., Li, J., Chen, Z., Yang, L. G., and Luo, Q. (2019). Phytoplankton community structure and diversity in the indoor industrial aquaculture system for Litopenaeus vannamei revealed by high–throughput sequencing and morphological identification. Aquac. Res. 50, 2563–2576. doi: 10.1111/are.14213
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Shang, B. J., Zuo, Z. H., Dou, C. M., Li, W. Y., and Sun, J. S. (2018). Analysis of the characteristics of Litopenaeus vannamei gut microbiota after being fed with two probiotics using high-throughput sequencing method. J. Fish. China 42, 113–122. doi: 10.11964/jfc.20180211184
Shokralla, S., Spall, J. L., Gibson, J. F., and Hajibabaei, M. (2012). Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 21, 1794–1805. doi: 10.1111/j.1365-294x.2012.05538.x
Stanton, T. B. (2013). A call for antibiotic alternatives research. Trends Microbiol. 21, 111–113. doi: 10.1016/j.tim.2012.11.002
Strzepa, A., and Szczepanik, M. (2013). Influence of natural gut flora on immune response. Postęp. Hig. Med. Dośw. 67, 908–920. doi: 10.5604/17322693.1064563
Thinh, P. D., Ly, B. M., Usoltseva, R. V., Shevchenko, N. M., Rasin, A. B., Anastyuk, S. D., et al. (2018). A novel sulfated fucan from Vietnamese sea cucumber Stichopus variegatus: isolation, structure and anticancer activity in vitro. Int. J. Biol. Macromol. 117, 1101–1109. doi: 10.1016/j.ijbiomac.2018.06.017
Thompson, F. L., Iida, T., and Swings, J. (2004). Biodiversity of vibrios. Microbiol. Mol. Biol. R. 68, 403–431. doi: 10.1128/MMBR.68.3.403-431.2004
Thompson, J. R., Pacocha, S., Pharino, C., Klepac-Ceraj, V., Hunt, D. E., Benoit, J., et al. (2005). Genotypic diversity within a natural coastal bacterioplankton population. Science 307, 1311–1313. doi: 10.1126/science.1106028
Tremaroli, V., and Backhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Wang, J. J., Li, D., Wang, Y. N., Wang, J. J., Liu, Y. P., and Chang, Y. Q. (2015). Bacterial flora in gut contents of sea cucumber Apostichopus japonicus at different culture periods. J. Dalian Ocean Univ. 30, 345–350. doi: 10.16535/j.cnki.dlhyxb.2015.04.001
Wang, H. H., and Schaffner, D. W. (2011). Antibiotic resistance: how much do we know and where to go from here? Appl. Environ. Microbiol. 77, 7093–7095. doi: 10.1128/AEM.06565-11
Wang, R. X., Wang, J. Y., Sun, Y. C., Yang, B. L., and Wang, A. L. (2015). Antibiotic resistance monitoring in vibrio spp. isolated from rearing environment and intestines of abalone Haliotis diversicolor. Mar. Pollut. Bull. 101, 701–706. doi: 10.1016/j.marpolbul.2015.10.027
Wang, Y. N., Zhu, S. W., and Chang, Y. Q. (2010). PCR-DGGE analysis of bacterial community composition in the intestine and aquaculture pond of Apostichopus japonicus. Prog. Fish. Sci. 31, 119–122. doi: 10.3969/j.issn.1000-7075.2010.03.017
Wei, H., Chen, M., Deng, Z., Zhao, W., Li, Y., Fang, W., et al. (2022). Immune and antioxidant responses of pearl oyster Pinctada maxima exposed to acute salinity stress. Aquac. Res. 53, 2439–2447. doi: 10.1111/are.15761
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Yu, L., Nicolaisen, M., Larsen, J., and Ravnskov, S. (2013). Organic fertilization alters the community composition of root associated fungi in Pisum sativum. Soil Biol. Biochem. 58, 36–41. doi: 10.1016/j.soilbio.2012.11.004
Zhang, W. J., Hou, H. M., Zhang, G. L., Li, Q. Y., and Du, C. M. (2011). Study on diversity of intestine cultivable microorganisms from Apostichopus japonicus. Sci. Technol. Food Ind. 36, 66–69. doi: 10.1097/RLU.0b013e3181f49ac7
Zhang, C. Y., Wang, Y. G., and Rong, X. J. (2006). Isolation and identification of causative pathogen for skin ulcerative syndrome in Apostichopus japonicus. J. Fish. China 44, 437–122. doi: 10.1360/aps050066
Zhang, Z., Xing, R., Lv, Z., Shao, Y., Zhang, W., Zhao, X., et al. (2018). Analysis of gut microbiota revealed Lactococcus garviaeae could be an indicative of skin ulceration syndrome in farmed sea cucumber Apostichopus japonicas. Fish Shellfish Immun. 80, 148–154. doi: 10.1016/j.fsi.2018.06.001
Zhang, Q. Q., Ying, G. G., Pan, C. G., Liu, Y. S., and Zhao, J. L. (2015). Comprehensive evaluation of antibiotics emission and fate in the river basins of China: source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 49, 6772–6782. doi: 10.1021/acs.est.5b00729
Zhao, Y. P., Li, B., Bai, D. Y., Huang, J. L., Shiraigo, W., Yang, L. H., et al. (2016). Comparison of fecal microbiota of mongolian and thoroughbred horses by high-throughput sequencing of the V4 region of the 16S rRNA gene. Asian Australas. J. Anim. Sci. 29, 1345–1352. doi: 10.5713/ajas.15.0587
Zhao, Y., Liu, H., Wang, Q., Li, B. J., Zhang, H. X., and Pi, Y. R. (2019a). The effects of benzo[a]pyrene on the composition of gut microbiota and the gut health of the juvenile sea cucumber Apostichopus japonicus Selenka. Fish Shellfish Immun. 93, 369–379. doi: 10.1016/j.fsi.2019.07.073
Zhao, Y., Wang, Q., Liu, H., Li, B., and Zhang, H. (2019b). High-throughput sequencing of 16S rRNA amplicons characterizes gut microbiota shift of juvenile sea cucumber Apostichopus japonicus feeding with three antibiotics. J. Oceanol. Limnol. 37, 1714–1725. doi: 10.1007/s00343-019-8308-5
Zhao, Y. C., Yuan, L., Wan, J. L., Sun, H. S., Wang, Y. Y., and Zhang, Q. (2018). Effects of a potential autochthonous probiotic Bacillus subtilis 2-1 on the growth and intestinal microbiota of juvenile sea cucumber, Apostichopus japonicus Selenka. J. Ocean Univ. China 17, 363–370. doi: 10.1007/s11802-018-3402-4
Zhuang, S., Li, Y., Jia, S., Hong, H., Liu, Y., and Luo, Y. (2019). Effects of pomegranate peel extract on quality and microbiota composition of bighead carp (Aristichthys nobilis) fillets during chilled storage. Food Microbiol. 82, 445–454. doi: 10.1016/j.fm.2019.03.019
Keywords: Stichopus variegatus, antibiotic, high-throughput sequencing, intestinal microbiota, immune enzymes
Citation: Tan C, Zhao W, Wen W, Chen X, Ma Z and Yu G (2022) Unraveling the effects of sulfamethoxazole on the composition of gut microbiota and immune responses in Stichopus variegatus. Front. Microbiol. 13:1032873. doi: 10.3389/fmicb.2022.1032873
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Junfeng Chen, Qufu Normal University, ChinaHenry Joseph Oduor Ogola, Jaramogi Oginga University of Science and Technology, Kenya
Copyright © 2022 Tan, Zhao, Wen, Chen, Ma and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Ma, zhenhua.ma@hotmail.com; Gang Yu, gyu0928@163.com