 Hans-Christian Slotved
Hans-Christian Slotved Thor Bech Johannesen
Thor Bech Johannesen Marc Stegger
Marc Stegger Kurt Fuursted
Kurt Fuursted- Department of Bacteria, Parasites and Fungi, Statens Serum Institut, Copenhagen, Denmark
Haemophilus influenzae is a gram-negative coccobacillus known to cause respiratory and invasive infections. It can possess a polysaccharide capsule that can be categorized into six different serotypes (i.e., Hia, Hib, Hic, Hid, Hie, and Hif) and non-encapsulated strains that are defined as non-typeable. Furthermore, H. influenzae can be characterized into eight biotypes (I–VIII). Traditionally, isolates have been serotyped and biotyped using phenotypic methods; however, these methods are not always reliable. In this study, we evaluate the use of whole-genome sequencing (WGS) for national surveillance and characterization of clinical Danish H. influenzae isolates. In Denmark, all clinical invasive isolates between 2014 and 2021 have been serotyped using a traditional phenotypic latex agglutination test as well as in silico serotyped using the in silico programs “hinfluenzae_capsule_characterization” and “hicap” to compare the subsequent serotypes. Moreover, isolates were also biotyped using a phenotypic enzyme test and the genomic data for the detection of the genes encoding ornithine, tryptophan, and urease. The results showed a 99–100% concordance between the two genotypic approaches and the phenotypic serotyping, respectively. The biotyping showed a 95% concordance between genotyping and phenotyping. In conclusion, our results show that in a clinical surveillance setting, in silico serotyping and WGS-based biotyping are a robust and reliable approach for typing clinical H. influenzae isolates.
Introduction
Haemophilus influenzae is a gram-negative coccobacillus that can cause respiratory infections as well as invasive infections such as septicemia and meningitis (Nørskov-Lauritsen et al., 2021). It is categorized into six serotypes (i.e., Hia, Hib, Hic, Hid, Hie, and Hif) according to the polysaccharide capsule, as well as non-encapsulated strains that are referred to as non-typeable H. influenzae (non-cap) (Potts et al., 2019). In the pre-vaccine era, H. influenzae serotype b (Hib) was a frequent cause of bacterial meningitis among young children (Bijlmer, 1991; Nørskov-Lauritsen, 2014; Jalalvand and Riesbeck, 2018), but after the introduction of Hib vaccines in the 1980s, the numbers diminished dramatically worldwide (Morris et al., 2008; Nørskov-Lauritsen, 2014). The Hib vaccine was introduced into the Danish immunization program in 1993 as a part of the diphtheria-tetanus-pertussis-polio combination vaccine recommended at 3, 5, and 12 months of age, with high vaccination coverage of 96% (12-month vaccination; 1 accessed 17 December 2021), and it has been estimated to have an effectiveness of more than 97% for the prevention of Hib-related meningitis (2 accessed 29 November 2021).
Haemophilus influenzae has traditionally been serotyped by phenotypic methods and PCR (Potts et al., 2019; Watts and Holt, 2019). Recently, two in silico approaches using whole-genome sequencing (WGS), “hinfluenzae_capsule_characterization” (Potts et al., 2019) and “hicap” (Watts and Holt, 2019), have been described that allow easier serotyping.
Haemophilus influenzae can furthermore be characterized based on eight biotypes defined by the presence and absence of the three enzymes, namely, tryptophanase (indole production), urease, and ornithine decarboxylase (Nørskov-Lauritsen, 2014).
In this study, we evaluated the use of two different in silico serotyping programs based on WGS for national surveillance of invasive clinical H. influenzae isolates from Denmark received between 2014 and 2021. Additionally, we investigated the clonal relatedness to serotype and biotype.
Materials and methods
Clinical isolates
Since October 2007, surveillance of invasive Hib infections in Denmark has included mandatory submission of isolates to the Neisseria and Streptococcus Reference Laboratory (NSR) at Statens Serum Institut (SSI) following an executive order (BEK nr 1102 of 20/09/2007), and although the surveillance specifies only Hib, the reference laboratory receives the majority of invasive H. influenzae isolates from all Danish regional laboratories of clinical microbiology.
All isolates were from invasive cases from mainland Denmark. An invasive case was defined as the presence of invasive H. influenzae in a patient who had a positive culture result for H. influenzae from cerebrospinal fluid (CSF), blood, or other normally sterile sites.
Data on serotypes and sample sites on all clinical H. influenzae isolates between 2014 and 2021 were retrieved from the Danish laboratory surveillance system at the Neisseria and Streptococcus Reference Laboratory (Figure 1). The WGS and phenotyping were performed on isolates consecutively when received during the study period. All isolates were grown on chocolate plates for both DNA preparation and phenotyping.
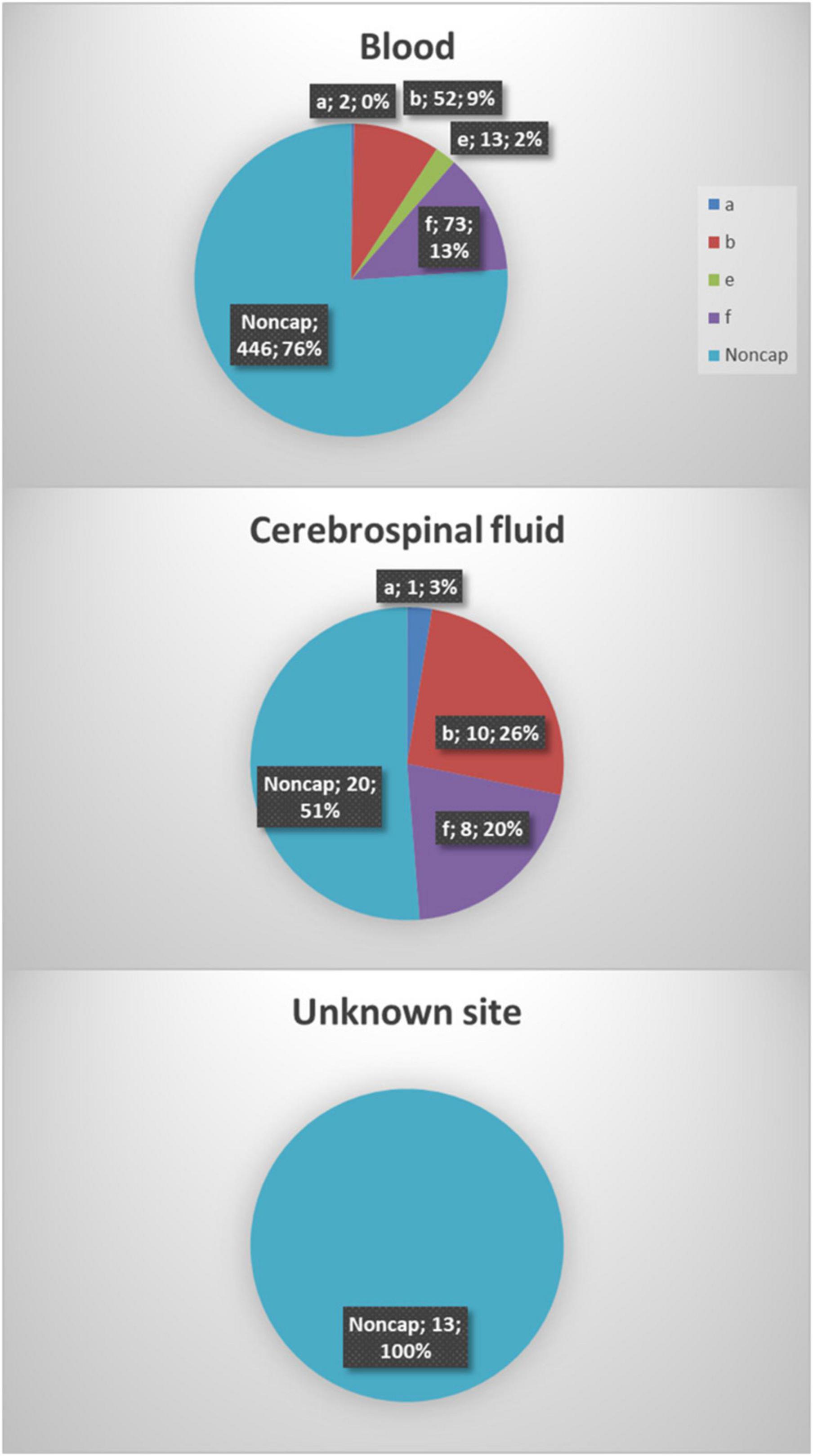
Figure 1. A total of six hundred thirty-eight Haemophilus influenzae isolates were included in the study. The majority of the isolates were from blood samples (91.8%, 586), 6.1% (39) were from cerebrospinal fluid, and information on the origin of 2% (13) of isolates was not available.
Species identification of Haemophilus influenzae isolates
The identification of H. influenzae was performed as previously described (Kilian, 1976; Fuursted et al., 2016). Briefly, species identification was performed on strains transferred directly from bacterial colonies using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Bruker Daltonics; Compass 1.4, Version 3.4, Build 3.4.76.0). Species identification was based on the standard MALDI-TOF score value [Biotyper database version MBT 6903 MSP Library (#1829023)] and confirmed with WGS data using KmerFinder.3
Phenotypic biotyping
From 2014 to 2019, the H. influenzae isolates were typed phenotypically into eight biotypes based on their variable production of tryptophanase (indole production), urease, and ornithine decarboxylase (ODC), as described in other studies (Kilian et al., 1979; Nørskov-Lauritsen, 2014). Phenotypic biotyping was not performed on the 2020–2021 isolates.
Serotyping
The isolates were serotyped using latex agglutination test (SSI Diagnostica, Denmark) specific for H. influenzae covering all six known serotypes (i.e., Hia, Hib, Hic, Hid, Hie, and Hif).
Whole-genome sequencing and assembly
Whole-genome sequencing was performed as previously described (Fuursted et al., 2016; Slotved et al., 2021). Briefly, genomic DNA was extracted using a DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany), and fragment libraries were constructed using a Nextera XT Kit (Illumina, San Diego, CA, USA) followed by either 150 or 250 bp paired-end sequencing on either the MiSeq or NextSeq 550 platform (Illumina, San Diego, CA, USA), respectively, according to the manufacturer’s instructions. The paired-end data were de novo assembled using the SKESA assembler (SKESA version 2.2) (Souvorov et al., 2018).
The genomic sequence data for the 638 clinical isolates have been deposited at the European Nucleotide Archive (ENA) under project no. PRJEB56415.
Genotyping
Genotyping was performed on all isolates using two in silico serotyping programs (Potts et al., 2019; Watts and Holt, 2019) with default parameters. The version used for the in silico program “hinfluenzae_capsule_characterization” was from the GitHub site: https://github.com/Vikash84/hinfluenzae_capsule_characterization, accessed 07-10-2022. The version used for the in silico program “hicap” was the version hicap 1.0.3: https://anaconda.org/bioconda/hicap, accessed 07-10-2022.
For isolates with deviations in phenotype/genotype, the genotype was additionally verified using BLASTN (BLAST 2.9.0 + version) using the sequences as described by Maaroufi et al. (2007) against the assembled genomes.
For the molecular detection of the biotype genes, partial sequences were used from the H. influenzae reference genome Rd KW20 (GenBank accession ID L42023); gene HI_0590 (1308 bp), gene HI_1389.1 (1434 bp), and gene HI_0535 (786 bp).
Multilocus sequence typing and phylogenetic analysis
Multilocus sequence typing was performed by uploading the assembled genomes to the PubMLST database (see text footnote 3, accessed 08-02-2022) to identify sequence types (ST) and corresponding clonal complexes (CC) for all isolates. STs sharing at least six of seven allelic variants were grouped into CCs (Shabayek and Spellerberg, 2018).
A phylogenetic tree was created based on single-nucleotide polymorphisms (SNPs) detected in the core genome of the isolate collection. SNPs were identified using NASP (Sahl et al., 2016) with BWAmem for mapping against the chromosome of H. influenzae isolate Rd KW20 (GenBank accession ID L42023), and GATK was set to remove positions with less than 10-fold depth and 90% unambiguous variant calls after removal of duplicated regions in the reference using NUCmer. The resulting SNP matrix was purged for recombination using Gubbins (Croucher et al., 2015). The phylogenetic tree was generated using a maximum-likelihood approach with IQ-TREE (Nguyen et al., 2015)4 and ModelFinder as implemented with 100 bootstraps before visualization using iTOL version 6 (Letunic and Bork, 2021).
Ethical considerations
The data and samples from patients were collected routinely for national surveillance purposes; therefore, no ethical approval or informed consent from patients or guardians was required. The study was approved by the Danish Data Protection Agency (record number 2007-41-0229). For further details on SSI’s permission to present and publish epidemiological data, see5 accessed 30-06-2022,6 accessed 30–06–2022). All presented data were anonymized.
Results
Characterization of the clinical isolates
Between 2014 and 2021, eight hundred seventy-five invasive H. influenzae isolates were received, and 638 (72.9%) of those isolates had both phenotypic and genotypic available data and were included in the study. All serotypes were represented except for serotypes c and d. One reference strain for Hic and two for Hid were included in the testing of capsular genes but not included in the clinical data (Table 1). The majority of the isolates were from blood cultures (91.8%), whereas 39 (6.1%) were from cerebrospinal fluid samples. No clinical information was available for the remaining 13 (2.0%) isolates (Figure 1).
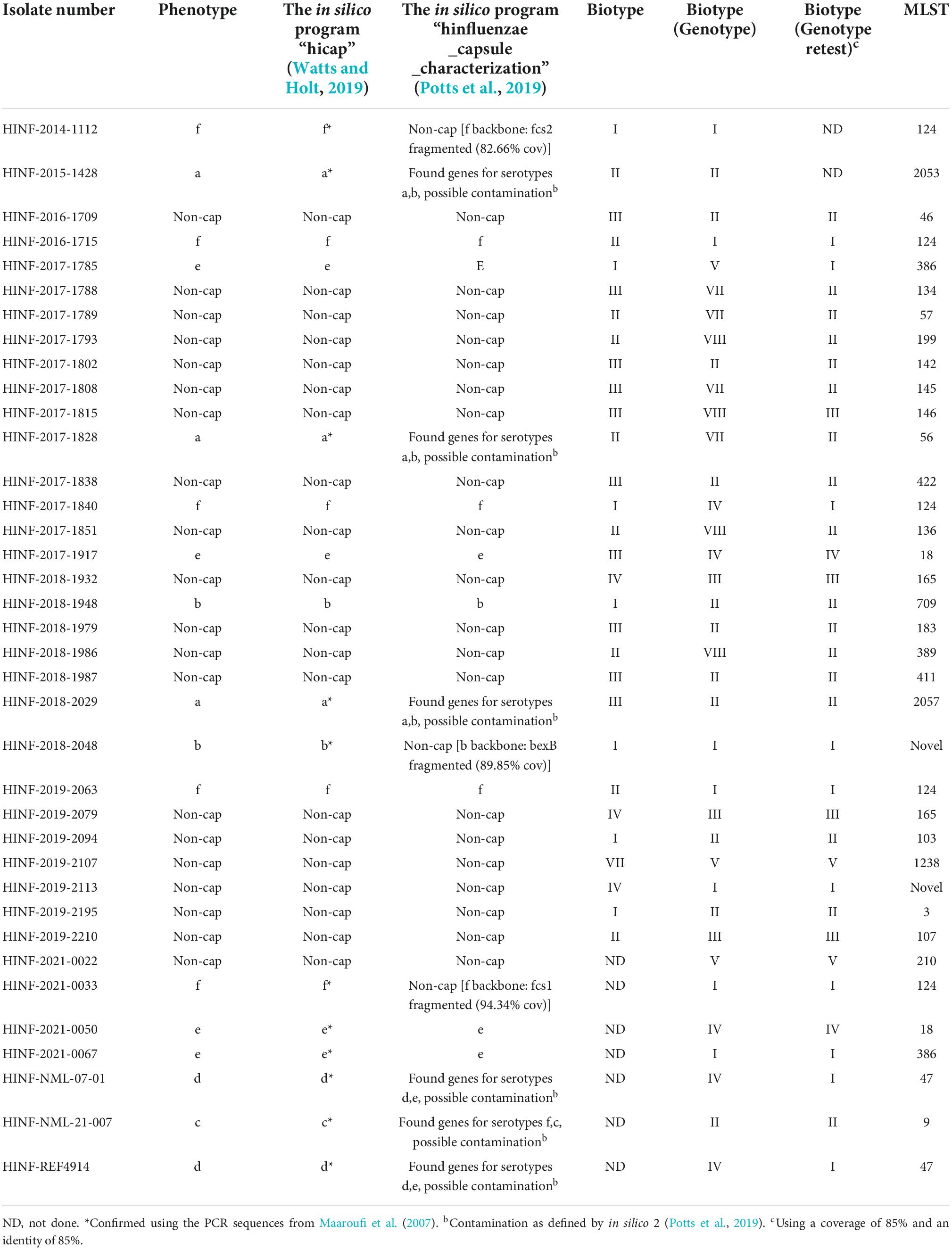
Table 1. List of isolates with divergent phenotype/genotype for serotype or biotype and missing values.
The overall material included 159 (24.9%) typeable and 479 (75.1%) non-typeable isolates (refer to Tables 2, 3).

Table 2. A total of eight hundred seventy-five invasive cases of Haemophilus influenzae were reported in Denmark from 2014 to 2021.
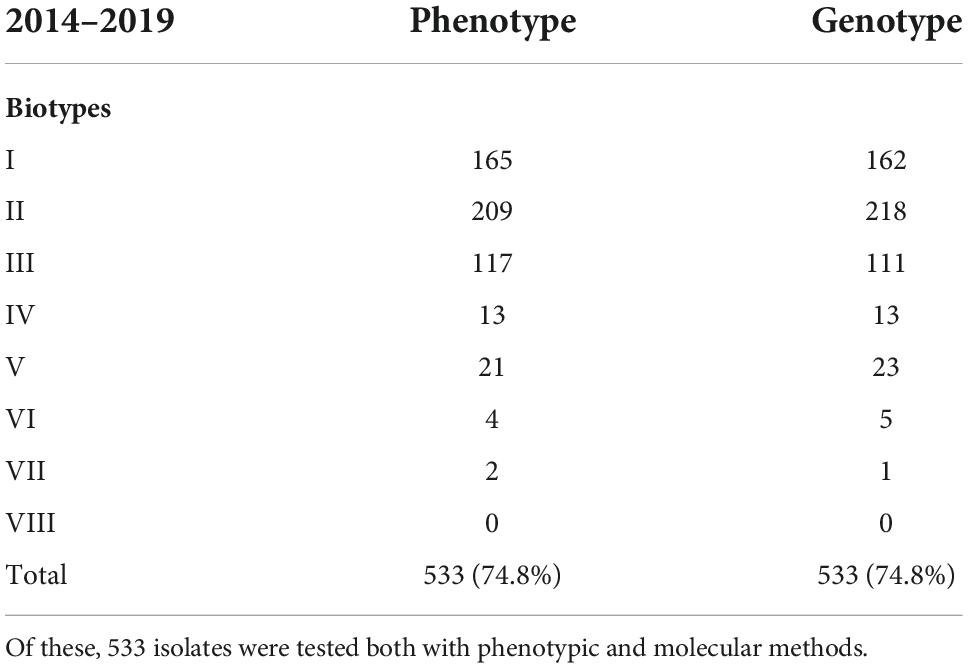
Table 3. A total of seven hundred and thirteen invasive cases of Haemophilus influenzae were reported in Denmark from 2014 to 2019.
Serotyping of the isolates
Across the collection, 638 (100%) and 632 (99.1%) isolates showed concordance between the phenotype and the in silico approaches developed by Watts and Holt (2019) and Potts et al., 2019; Tables 1, 2. One serotype b and two serotype f isolates were typed as non-cap isolates, while three isolates were labeled as contaminated due to matches against capsular genes for several H. influenzae serotypes (refer to Table 1). The Hic and Hid reference isolates were correctly identified with the Watts and Holt in silico method, while the Potts et al. in silico method described them as contaminated although detecting possible capsular genes of serotype c and serotype d, respectively (Table 1).
In our reference laboratory, the Watts and Holt in silico method was chosen as the reference method. Therefore, we did not perform a second WGS test of all the isolates (six isolates in total) showing contaminating results with the Potts et al. in silico method (Potts et al., 2019) (Table 1). This might have improved the typing results of the six isolates when using the Potts et al. (2019) in silico method.
Biotyping
The most common biotypes were biotype II (39%), I (31%), and III (22%), but all biotypes except for biotype VIII were observed. Of the 533 isolates (2014–2019) tested for both phenotype and genotype, 506 (94.9%) isolates showed concordance, while 27 (5.1%) isolates showed divergent biotypes (Tables 1, 3).
By lowering the coverage and identity to 85 and 85% when blasting for biotype sequences, it was possible to obtain complete concordance for both phenotype and genotype for eight of the 17 isolates with divergent biotypes (Table 1). Unfortunately, we were not able to retest the phenotype of the remaining 19 isolates, and we could therefore not rule out the possibility of diverting the biotype due to laboratory mistakes. The biotypes were not linked to a specific serotype, and each biotype was found to cover both capsular and non-capsular isolates (Figure 2).
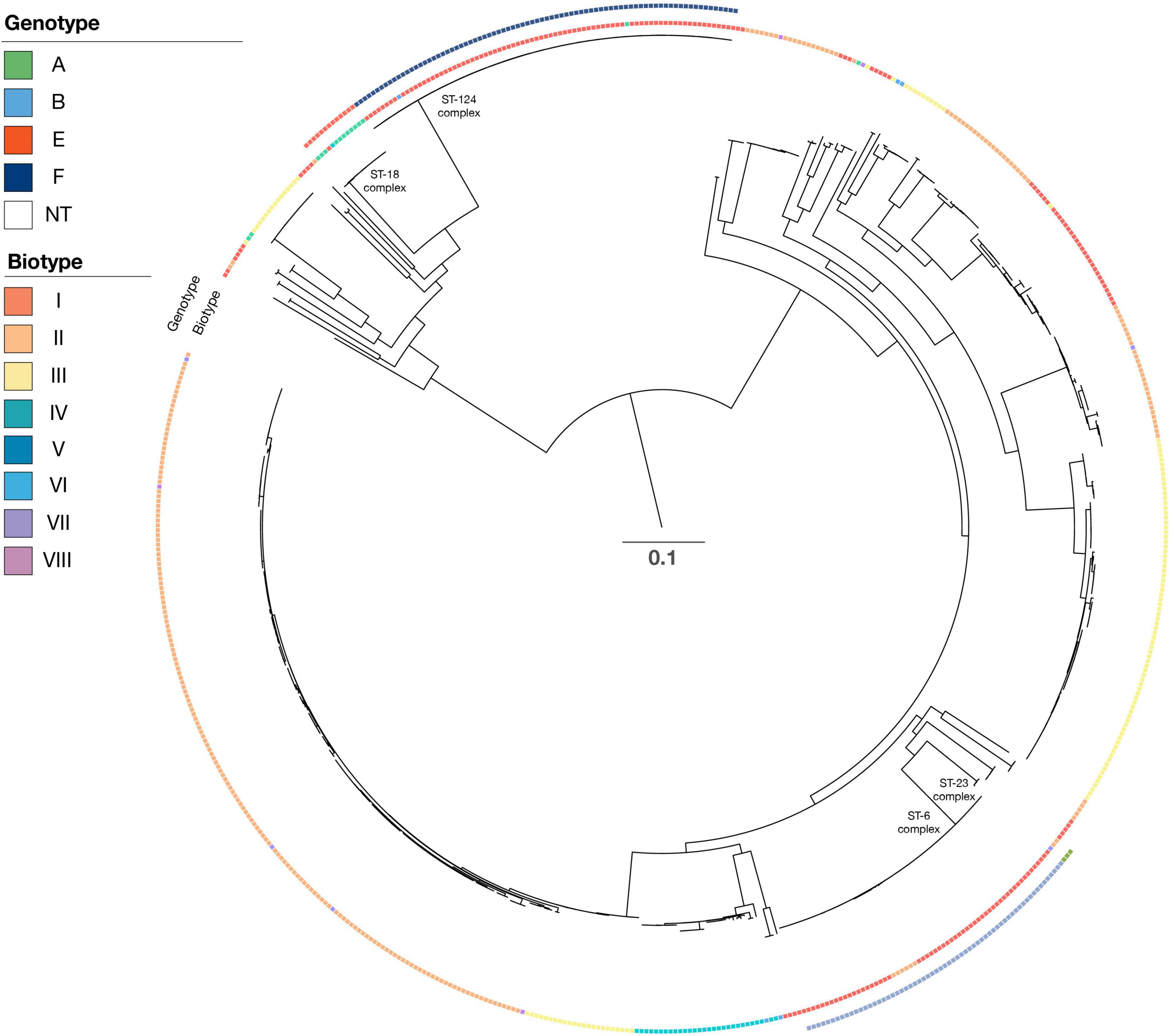
Figure 2. Phylogenetic tree of all isolates. For each isolate, data are presented for the serotype (genotype with color), biotype (biotype with color), and clonal complex. Isolate Hinf Rd KW20 (GenBank nb. L42023) was used as a reference strain in the SNP alignment. Scale bar indicate substitutions per side.
Multilocus sequence typing types and phylogenetic tree
The capsular H. influenzae (a–f) were all distributed among six different clonal complexes (Table 4) as follows: ST23 complex (ST56, ST2053, and ST2057), ST6 complex (ST6, ST95, ST190, ST206, ST709, and ST1448), ST18 complex (ST18, ST122, and ST386), and ST124 complex (ST124, ST598, and ST1739).

Table 4. Sequence type and clonal complex found among the Haemophilus influenzae isolates with a capsule.
The dominant ST types were ST6 (21 isolates), ST190 (28 isolates), and ST709 (six isolates), all of serotype b. ST18 consisted of 13 serotype e isolates, whereas ST124 consisted of 76 serotype f isolates. Finally, the four ST1739 were all serotype f. The reference strain serotype c belonged to the ST7 complex (ST9), and the two serotype d isolates belonged to the ST10 complex (ST47).
The H. influenzae non-cap isolates had 131 different STs, including 40 novel STs. The dominant STs for non-cap were ST103 (n = 29), ST12 (n = 15), and ST159 (n = 13), whereas novel STs were observed for 40 non-cap isolates (Table 5). None of the non-cap isolates were found to be clonal nor related phylogenetically to any of the capsular H. influenzae isolates.
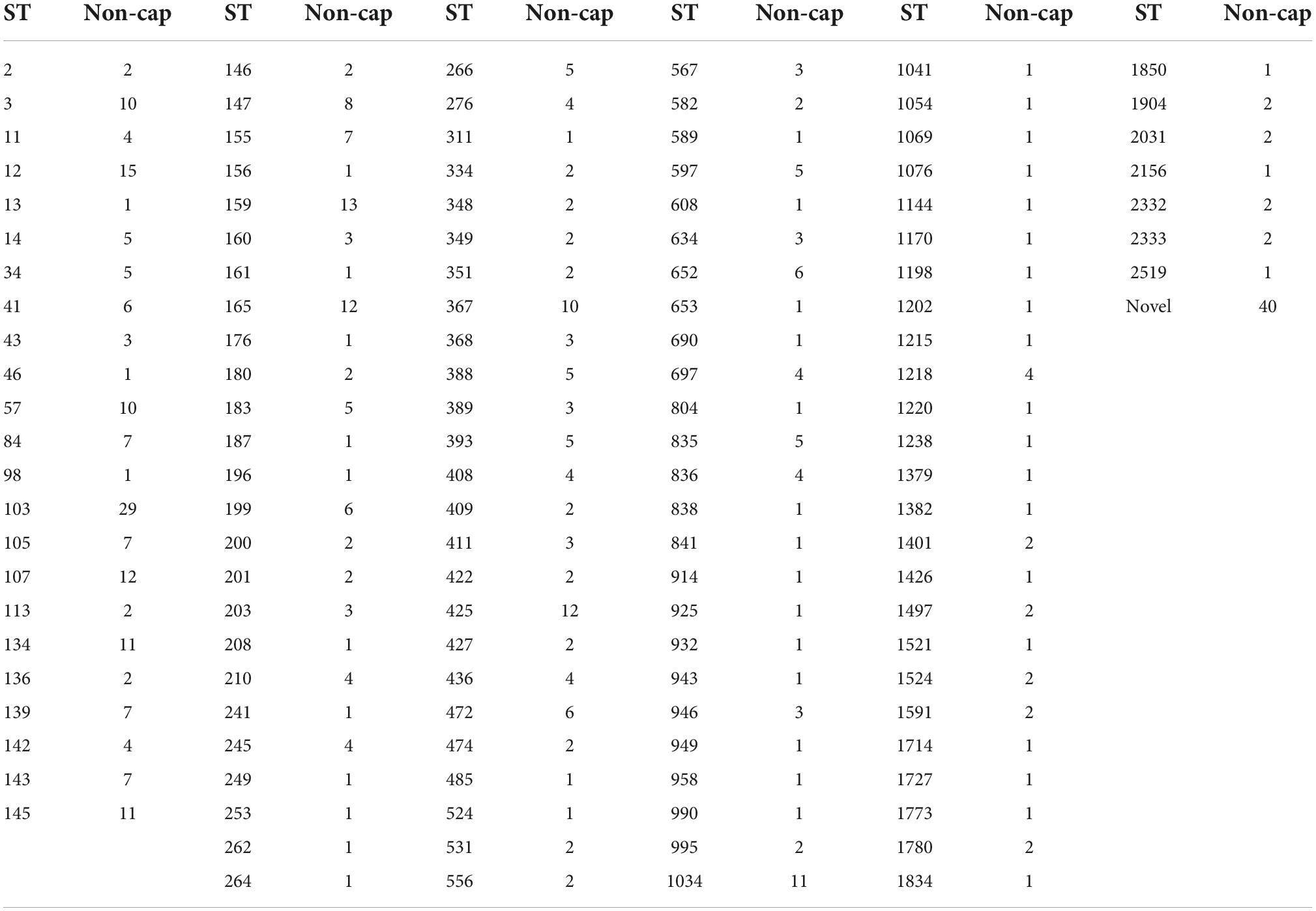
Table 5. Sequence types for Haemophilus influenzae non-cap isolates.
Phylogenetic analysis of all isolates (Figure 1) shows that all clinical isolates with a capsule clustered both according to serotype and clonal complex. The biotype did not show any type-specific clustering.
Discussion
The characterization of H. influenzae isolates has traditionally been based on phenotypic methods (Kilian, 1976; Kilian et al., 1979; Nørskov-Lauritsen, 2014). With the introduction of the Hib vaccine four decades ago, the importance of H. influenzae capsular serotype distribution has increased due to continued monitoring of vaccine efficacy (Satola et al., 2007).
Serotyping of H. influenzae isolates has for many years mainly been based on slide agglutination serotyping that detects the expressed capsule (Satola et al., 2007; Potts et al., 2019); however, the slide agglutination serotyping is not always considered reliable, and false-positive Hib designations have been reported (Maaroufi et al., 2007; Satola et al., 2007; Nørskov-Lauritsen, 2014; Potts et al., 2019). With the description of the capsular gene sequences and the development of molecular typing methods, new PCR and WGS-based procedures for H. influenzae serotyping have been presented (Maaroufi et al., 2007; Satola et al., 2007; Nørskov-Lauritsen, 2014; Potts et al., 2019; Watts and Holt, 2019). In the study by Potts et al. (2019), 675 invasive isolates were collected from surveillance programs from 27 states in the United States, and 13 isolates of other origins were compared between the in silico program “hinfluenzae_capsule_characterization” described serotypes and the serotype results obtained by both PCR and slide agglutination tests. They found that the WGS serotyping method was 99.9% concordant with the slide agglutination tests and completely concordant with RT-PCR. Watts and Holt (2019) tested the in silico program “hicap” using 41 publicly available isolate WGS sequences. In 40 of the 41 isolates (98%), they obtained the correct serotype. The difference between the two in silico approaches is that the in silico program “hicap” uses Prodigal (Hyatt et al., 2010) to identify open reading frames (ORFs) in genomic assemblies and BLAST (Camacho et al., 2009) to identify capsule genes based on a custom reference database. The serotype is then predicted based on the detection of capsule genes. The approach by Potts et al. (2019) performs BLAST directly on the genome assemblies and then parses the identified cap genes for truncations and internal stop codons before predicting a serotype, rather than using external software for gene prediction.
This study, representing 73% of all clinical isolates from 2014 to 2021 from Denmark, found 100% concordance between the capsular genotype and the phenotype (slide agglutination test). This is comparable to the observations by Potts et al. (2019) and Watts and Holt (2019). This is the first study to show that in silico serotyping programs are feasible in a national H. influenzae surveillance program with the notion that the Hib vaccine efficacy depends on the phenotypic expression of the Hib polysaccharide and not the presence of the capsular sequences.
As a complementary approach to capsular serotyping, a biotyping setup has been described by Kilian in 1976 (Kilian, 1976; Kilian et al., 1979; Nørskov-Lauritsen, 2014). The system is based on eight biotypes defined by variations in the production of tryptophanase (indole production), urease, and ornithine decarboxylase (ODC) (Nørskov-Lauritsen, 2014). However, because the biotype is not related to the capsular serotype and therefore not considered an important part of the efficacy monitoring of the Hib vaccine in Denmark, phenotypic biotyping has not been performed since 2019. However, comparing the phenotypic biotype with the molecular biotype obtained from isolates sampled from 2014 to 2019 did show a high concordance between phenotypic and molecular biotyping. In total, 27 isolates (5.1%) showed diverging biotypes. Adjusting the percentage of coverage and identity could improve the concordance between phenotypic and molecular biotyping (Table 1). However, there was a mixture of capsular H. influenzae isolates and non-cap H. influenzae isolates among each of the specifically detected biotypes (Figure 2).
While the isolates expressing a capsule could be linked to a specific CC, this was not the case for the non-cap H. influenzae isolates, which exhibited a broad diversity of ST types (Tables 4, 5, and Figure 2). Although the ST types in this study could be used to differentiate the capsular H. influenzae isolates from the non-cap isolates, it has previously been shown that MLST is not optimal for the identification of capsular H. influenzae isolates (Potts et al., 2019). However, we found that the MLST type can indicate the correct identification of the genotype because the capsular isolates appear to be part of only six clonal complexes (Table 4). Therefore, this can, in a national routine H. influenzae surveillance program for monitoring the effect of the Hib vaccine, be used as an additional confirmation of correct capsule identification, in that non-cap ST types belonging within these six CC or capsular isolates not belonging to these six CC, will need further evaluation before final capsule reporting.
Based on the results of this study, we recommend a workflow using genotyping either based on PCR (Maaroufi et al., 2007; Satola et al., 2007) or WGS (Potts et al., 2019; Watts and Holt, 2019). Because of the use of the Hib vaccine, we additionally suggest confirming the expression of the Hib capsule using a latex agglutination test for vaccine surveillance. As an additional verification of the serotype, we propose MLST typing as clinical capsular isolates appear to cluster in specific clonal complexes (Tables 4, 5), although the MLST itself cannot be used for serotype identification (Potts et al., 2019).
The weakness of this study is that by only looking at clinical isolates, the tested isolates representing all six serotypes are not equally distributed but dominated by serotypes f, b, and e (Table 2), while only a limited number of isolates with serotype a were detected, and none with serotype c and serotype d. The strength of the study is that the evaluation is based on clinical isolates received from Danish regional laboratories of clinical microbiology, and all the tested isolates showed a strong correlation between the results obtained with genotyping vs. phenotyping, in line with observations in other studies (Potts et al., 2019; Watts and Holt, 2019).
In conclusion, our study showed that there is a complete concordance between phenotypic serotyping and molecular-based methods for Danish clinical H. influenzae isolates. The concordance is 100% for both the described in silico serotyping methods, replacing laborious phenotypic methods with WGS-based in silico approaches in a clinical setting. In addition, biotyping shows a high concordance between molecular and phenotypic methods. Furthermore, in our Danish routine laboratory where epidemiological surveillance of H. influenzae for monitoring the effect of the Hib vaccine is an important routine task, the MLST type can be used as an indication of whether an isolated expresses a capsule or not. In that, all capsular-defined isolates in this study belonged to one of six clonal complexes depending on their capsule.
Data availability statement
The datasets presented in this study can be found in the online repository at the European Nucleotide Archive (ENA) under project no. PRJEB56415.
Author contributions
H-CS and KF designed the study, analyzed the data, and drafted the manuscript. All authors performed the genomic analyses and reviewed the data, contributed to the manuscript, and critically revised the manuscript, and have approved the final manuscript.
Acknowledgments
Mikkel Benjamin Nielsen and Nadia Xenia Olsen are acknowledged for their skillful laboratory work and input to this study. We acknowledge the Danish departments of clinical microbiology for submitting Haemophilus influenzae isolates for national surveillance throughout the study period. This study is a part of the national surveillance program for severe H. influenzae infections.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ https://statistik.ssi.dk//sygdomsdata#!/?vaccination=3&sex=3&landsdel=100&xaxis=Cohort&show=Graph&datatype=Vaccination
- ^ https://www.ssi.dk/vaccinationer/boernevaccination/sygdomsforekomst-foer-og-efter-vaccination
- ^ https://cge.food.dtu.dk/services/KmerFinder/
- ^ http://www.iqtree.org/
- ^ https://en.ssi.dk/research
- ^ https://en.ssi.dk/about-us
References
Bijlmer, H. A. (1991). World-wide epidemiology of Haemophilus influenzae meningitis; industrialized versus non-industrialized countries. Vaccine 9(Suppl. 1), S5–S9; discussion S25. doi: 10.1016/0264-410x(91)90172-3
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Fuursted, K., Hartmeyer, G. N., Stegger, M., Andersen, P. S., and Justesen, U. S. (2016). Molecular characterisation of the clonal emergence of high-level ciprofloxacin-monoresistant Haemophilus influenzae in the Region of Southern Denmark. J. Glob. Antimicrob. Resist. 5, 67–70. doi: 10.1016/j.jgar.2015.12.004
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jalalvand, F., and Riesbeck, K. (2018). Update on non-typeable Haemophilus influenzae-mediated disease and vaccine development. Expert Rev. Vaccines 17, 503–512. doi: 10.1080/14760584.2018.1484286
Kilian, M. (1976). A taxonomic study of the genus Haemophilus, with the proposal of a new species. J. Gen. Microbiol. 93, 9–62. doi: 10.1099/00221287-93-1-9
Kilian, M., Sørensen, I., and Frederiksen, W. (1979). Biochemical characteristics of 130 recent isolates from Haemophilus influenzae meningitis. J. Clin. Microbiol. 9, 409–412. doi: 10.1128/jcm.9.3.409-412.1979
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Maaroufi, Y., De Bruyne, J.-M., Heymans, C., and Crokaert, F. (2007). Real-time PCR for determining capsular serotypes of Haemophilus influenzae. J. Clin. Microbiol. 45, 2305–2308. doi: 10.1128/JCM.00102-07
Morris, S. K., Moss, W. J., and Halsey, N. (2008). Haemophilus influenzae type b conjugate vaccine use and effectiveness. Lancet Infect. Dis. 8, 435–443. doi: 10.1016/S1473-3099(08)70152-X
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nørskov-Lauritsen, N. (2014). Classification, identification, and clinical significance of Haemophilus and Aggregatibacter species with host specificity for humans. Clin. Microbiol. Rev. 27, 214–240. doi: 10.1128/CMR.00103-13
Nørskov-Lauritsen, N., Pedersen, N., Lam, J. U. H., Nielsen, H. L., Kobel, C. M., Hansen, D. S., et al. (2021). Haemophilus influenzae one day in Denmark: Prevalence, circulating clones, and dismal resistance to aminopenicillins. Eur. J. Clin. Microbiol. Infect. Dis. 40, 2077–2085. doi: 10.1007/s10096-021-04247-w
Potts, C. C., Topaz, N., Rodriguez-Rivera, L. D., Hu, F., Chang, H.-Y., Whaley, M. J., et al. (2019). Genomic characterization of Haemophilus influenzae: A focus on the capsule locus. BMC Genomics 20:733. doi: 10.1186/s12864-019-6145-8
Sahl, J. W., Lemmer, D., Travis, J., Schupp, J. M., Gillece, J. D., Aziz, M., et al. (2016). NASP: An accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2:e000074. doi: 10.1099/mgen.0.000074
Satola, S. W., Collins, J. T., Napier, R., and Farley, M. M. (2007). Capsule gene analysis of invasive Haemophilus influenzae: Accuracy of serotyping and prevalence of IS1016 among nontypeable isolates. J. Clin. Microbiol. 45, 3230–3238. doi: 10.1128/JCM.00794-07
Shabayek, S., and Spellerberg, B. (2018). Group B streptococcal colonization. Molecular Characteristics, and Epidemiology. Front. Microbiol. 9:437. doi: 10.3389/fmicb.2018.00437
Slotved, H. C., Fuursted, K., Kavalari, I. D., and Hoffmann, S. (2021). Molecular identification of invasive non-typeable group B Streptococcus isolates from Denmark (2015 to 2017). Front. Cell. Infect. Microbiol. 11:571901. doi: 10.3389/fcimb.2021.571901
Souvorov, A., Agarwala, R., and Lipman, D. J. (2018). SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 19:153. doi: 10.1186/s13059-018-1540-z
Keywords: Denmark, capsular genes, genotyping, serotyping, Haemophilus influenzae
Citation: Slotved H-C, Johannesen TB, Stegger M and Fuursted K (2022) Evaluation of molecular typing for national surveillance of invasive clinical Haemophilus influenzae isolates from Denmark. Front. Microbiol. 13:1030242. doi: 10.3389/fmicb.2022.1030242
Received: 28 August 2022; Accepted: 24 October 2022;
Published: 17 November 2022.
Edited by:
Bing Gu, Guangdong Provincial People’s Hospital, ChinaReviewed by:
Litian Ma, Fourth Military Medical University, ChinaHillary Crandall, The University of Utah, United States
Mary Slack, Griffith University, Australia
Copyright © 2022 Slotved, Johannesen, Stegger and Fuursted. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Christian Slotved, aGNzQHNzaS5kaw==