 Jianman Lv1
Jianman Lv1 Wukang Liu
Wukang Liu Jun Lv
Jun Lv- 1College of Food Science and Technology, Huazhong Agricultural University, Wuhan, China
- 2Institute of Infection and Immunity, Taihe Hospital, Hubei University of Medicine, Shiyan, China
- 3Aquatic Preservation and Processing Technology, Guangxi Academy of Fishery Science, Nanning, China
Red vinasse acid has a distinct flavor and a vivid red color that are directly tied to the intricate metabolic activities of microorganisms that produce it. In this study, metagenomic technology was used to mine its functional genes and examine the microbial diversity of red vinasse acid. The findings revealed the identification of 2,609 species, 782 genera, and 63 phyla of microorganisms, and the dominant genus was Lactobacillus. Amino acid metabolism and carbohydrate metabolism were significant activities among the 16,093 and 49,652 genes that were annotated in the evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, respectively. In gluconeogenesis, red vinasse acid encodes 194 genes controlling the transporter protein systems of different sugars and has key enzyme genes that catalyze the conversion of intracellular sugars into glycolytic intermediates. In amino acid flavor formation, red vinasse acid contains 32 control genes for branched-chain aminotransferase (BCAT), 27 control genes for aromatic-amino-acid transaminase (ArAT), 60 control genes for keto acid invertase, 123 control genes for alcohol/aldehyde dehydrogenase, and 27 control genes for acetyl esterase, which have the basis for the formation of strong flavor substances from amino acids.
Introduction
Red vinasse acid (Bai et al., 2020) is a kind of unique flavor food in Guangxi, China. It is a well-known regional specialty that is mostly produced in Guangxi Wuxuan and Guangxi Guiping. Bright crimson, crisp., and delectable red vinasse acid is pickled with red rice as seeds. Red yeast rice was fermented at a specified temperature and duration under the combined influence of Monascus, Lactobacillus, Yeast, and other microorganisms to create red vinasse acid (Bai et al., 2020). Its diverse microbial community, which includes the dominant fungus Monascus and the dominant bacteria Lactobacillus, is strongly associated with the development of red vinasse acid’s distinctive flavor and functional features. Numerous secondary metabolites, such as Monascus pigments (MPs), monacolin K, γ-aminobutyric acid (GABA), and dimerumic acid, can be produced by Monascus (Aniya et al., 2000; Diana et al., 2014; Chen et al., 2015). In addition to effectively lowering blood pressure, lowering plasma cholesterol, preventing obesity, improving anti-inflammatory activity, preventing cancer, lowering blood sugar levels, and having anti-inflammatory and anti-tumor properties, MPs can replace some synthetic pigments in the food industry (Nam et al., 2014; Zhang et al., 2021). The most important effect of GABA is blood pressure regulation. Numerous studies have shown that GABA can reduce high blood pressure in animals and humans (Yoshimura et al., 2010; Yang et al., 2012; Pouliot-Mathieu et al., 2013; Tsai et al., 2013). Monacolin K can block cholesterol synthesis pathways, reduce cholesterol synthesis, and thus play a role in lowering blood lipids (Hsieh et al., 2013; Ajdari et al., 2014; Dikshit and Tallapragada, 2015, 2016). Lactobacillus has a variety of probiotic functions, such as promoting the absorption of nutrients, improving human immunity, and maintaining the balance of intestinal flora (Arnold et al., 2018; Riaz Rajoka et al., 2018; Sandes et al., 2018; Chen et al., 2020). Therefore, Red vinasse acid thus plays a significant role in healthcare. In recent years, it has been possible to estimate the microbiological diversity of red vinasse acid detected by 16srRNA technology only at the genus level and not with sufficient accuracy at the species level. The discovery of functional genes has some restrictions.
New insights into the roles played by natural microbial communities have been made possible by the introduction of metagenomic methods (Knight et al., 2018). As a result, the integration of the genetic, taxonomic, and functional diversity of the microbial community has drawn more and more attention. A study technique called metagenomics uses gene sequences to analyze the microbial data contained in environmental samples (Mori et al., 2008; Xie et al., 2020). It has developed into a crucial tool for researching microbial diversity and community traits because of its ability to swiftly and reliably gather a wealth of biological data and extensive microbiological information. A metagenomic technique, as opposed to 16srRNA high-throughput sequencing, can not only fully mine the microbial genome information but also study the function and metabolic pathways. Based on this information, it is more convenient to mine the biodiversity, community structure, functional characteristics, and relationships of microflora (Li et al., 2017; Belleggia et al., 2020). Importantly, when compared to conventional rRNA gene sequencing, metagenomic sequencing can identify novel species with greater resolution and accuracy (Chen et al., 2017). Our understanding of the microbial communities in fermented foods has significantly improved over the past several decades as a result of advancements in next-generation DNA sequencing and high-throughput computational methods (Arıkan et al., 2020). Studies on wines (Johansson et al., 2021), kimchi (Kim et al., 2021), fermented Chinese xiaoqu (Su et al., 2020), dajiang-meju (Xie et al., 2019), and Turkish kefir grains (Ilıkkan and Bağdat, 2021) are a few examples.
Studies on the microbiological diversity of red vinasse acid have been conducted recently. Further study is necessary since it is unclear what species of microorganisms exist and because functional genes are nearly at the blank stage. In order to provide a theoretical foundation for the development of the red vinasse acid industry and the development of Monascus resources, metagenomics, and bioinformatics analysis methods were used in this study to identify the core microbial groups at the ‘species’ level of taxonomic status, analyze the microbial diversity of red vinasse acid, and excavate functional genes in group microorganisms.
Materials and methods
Experimental materials
Composition and production process of red vinasse acid
Fresh edible fruits and vegetables (or edible fruit and vegetable salted billet), red vinasse as the main raw material, then add edible salt, add or not add sugar (sugar or rock sugar, etc.), rice-flavored white wine, garlic, chili pepper, star anise, and other auxiliary ingredients, through the raw and auxiliary materials pretreatment, cleaning, drying, mixing, vinasse fermentation, packaging, and other processes made ready to eat red vinasse acid (Bai et al., 2020).
Source and storage of experimental samples
The samples of red vinasse acid were collected from Guangxi in June, 2021, and then brought back to the laboratory and sent to Biomarker Technologies for metagenome sequencing immediately, and the remaining samples were stored in the laboratory refrigerator at 4°C.
Total DNA extraction
Take about 500 mg of red vinasse acid tissue sample into a domestic 2 ml centrifuge tube, add pre-cooled steel beads, place the taken tissue sample in liquid nitrogen, and grind it thoroughly at 1,500 rpm for DNA extraction.
Complete the nucleic acid extraction using the CTAB procedure, measure the concentration of the extracted nucleic acid using the Qbiut (Invitrogen, model Qubit3.0, reagent Qubit TM dsDNA HS Assay Kit), and assess the integrity using 1% agarose gel electrophoresis. Red vinasse acid sample processing by Biomarker Technologies included DNA extraction and metagenome sequencing.
Metagenome library construction and sequencing methods
Library construction methods
Genomic enzymatic disruption and end repair
1. Take 10 ng of genomic DNA (Qubit quantification) into a 96-well plate, make up to the corresponding volume with Unclease-free Water, finally add the corresponding digestion reagent (on ice), mix well by pipetting, centrifuge instantly, and perform the reaction immediately in a PCR instrument (Eppendorf).
2. Rapid Ligation Butter 3, ligase and index are added to the digestion product, blown, and mixed, centrifuged instantaneously, and placed in the PCR instrument for reaction.
Conjugate product purification
1. Add a certain volume of magnetic beads to the conjugate product, mix thoroughly, incubate at room temperature, and place on a magnetic stand, let stand, and carefully remove the supernatant.
2. Add freshly prepared 80% ethanol to the tube, let it stand on the magnetic rack for 30s, and remove the supernatant.
3. Repeat Step 2 once, after the second wash, centrifuge instantaneously and remove the supernatant with a 10 μl gun.
4. Place the reaction tube on the magnetic rack, open the cap and air dry until the surface of the magnetic beads is lusterless and has fine cracks.
5. Remove the reaction tube from the magnetic rack, add Unclease-free Water to elute DNA, blow and mix well, incubate at room temperature, and then rest on the magnetic rack, carefully aspirate the supernatant into a new PCR tube.
PCR amplification
1. Add various reagents required for PCR reaction, mix well, and centrifuge.
2. According to the PCR reaction conditions, put it into the PCR instrument for PCR amplification.
PCR product fragment screening
1. Instantaneously centrifuge the PCR reaction tube, add the corresponding volume (0.6×) of magnetic beads, and mix thoroughly.
2. Incubate at room temperature, then place on a magnetic stand, and carefully transfer the supernatant to a new PCR tube.
3. Aspirate the corresponding volume (0.12×) of beads into the supernatant of Step 2, gently blow and mix well, incubate at room temperature, then place on a magnetic stand, let stand, carefully remove the supernatant.
4. Add freshly prepared 80% ethanol to the reaction tube, let it stand on the magnetic rack for 30s, remove the supernatant, and repeat the Step 1 time.
5. Open the cap of the tube, air dry until the surface of the magnetic beads is lusterless with fine cracks, add Unclease-free Water to elute the library, blow and mix, let stand at room temperature, let stand on the magnetic stand, carefully aspirate the supernatant into a new 1.5 ml centrifuge tube, and store at −20°C for backup.
Library quality control
The library was quality-checked by Qsep-400 and the library concentration was quantified using Qubit 3.0, and meets the following indicators can be tested on the machine: Concentration ≥ 1 ng/μl, center value of fragment 430–530 bp, average value between 420 and 580 bp, normal distribution of peak pattern, single fragment without spurious peak.
Sequencing methods
The constructed libraries were sequenced using illumina novaseq6000 (San Diego, CA, United States). The kit NovaSeq 6,000 S4 Reagent Kit (San Diego, CA, United States) was used for sequencing.
Data analysis
Sequencing data quality control
The raw sequences (Raw reads) obtained from sequencing contain low quality sequences. To ensure the quality of information analysis, raw reads need to be filtered to obtain clean reads for subsequent information analysis. The main steps of data filtering are as follows.
1. Filtering raw tags to obtain high quality sequencing data (clean tags) using the software Trimmomatic (version v0.33 parameters PE LEADING:3 TRAILING:3 SLIDINGWINDOW:50:20 MINLEN:120).
2. Use bowtie2 (version v2.2.4 parameter—seed 123,456-I 200-X 1000—un-conc) to compare with host genome sequence to remove host contamination (if a host reference genome is provided).
Metagenome assembly
Metagenome assembly was performed using the software MEGAHIT (Li et al., 2015; version v1.1.2 default parameters), filtering contig sequences shorter than 300 bp. The assembly results were evaluated using QUAST (Gurevich et al., 2013; version v2.3 default parameters) software.
Metagenome component analysis
Gene prediction
MetaGeneMark (Zhu et al., 2010; version v.3.26 default parameters) software (http://exon.gatech.edu/meta_gmhmmp.cgi, Version 3.26) was used to identify coding regions in the genome using default parameters (parameters -A -D -f G).
Construction of non-redundant gene sets
Redundancy was removed using MMseq2 software (https://github.com/soedinglab/mmseqs2, Version 11-e1a1c) with the similarity threshold set to 95% and the coverage threshold set to 90%.
Functional annotation analysis
eggNOG functional annotation
Evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG; Powell et al., 2014; evolutionary genealogy of genes: Non-supervised Orthologous Groups) is a database of biological direct homologous gene clusters, which is a continuous update of the COG database. For specific annotation, the protein sequences of non-redundant genes are compared with the eggNOG database by BLAST (diamond v0.9.29 matching threshold E-value 1e−5) to find the most similar sequence in the eggNOG database, and the annotation and classification information corresponding to this sequence is the annotation and classification information of the corresponding sequenced genomic genes.
KEGG functional annotation
Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa et al., 2004) is a database for collecting genomes, biological pathways, diseases, drugs, and chemicals. For specific annotation, the protein sequences of non-redundant genes are compared with those included in the KEGG database by BLAST (diamond v0.9.29 matching threshold E-value 1e−5) to find the most similar sequence in the KEGG database, and the annotation information, corresponding KO number, and position in the KO corresponding pathway of the sequence is the annotation information of the corresponding gene in the sequenced genome. The annotation information, KO number and position in the biological process pathway of the corresponding gene in the KEGG database is the sequence with KO sequence number.
CAZy function annotation
Carbohydrate-active enzymes database (CAZy; Cantarel et al., 2009) is a database of carbohydrate-active enzymes, formed by the collection and classification of published literature and related proteins, and carefully maintained by experts. For the specific annotation, the hmmer (version 3.0) software was used to compare the protein sequences of the non-redundant genes separately with the Hidden Markov Model of each family of the CAZy database (matching parameters default, default screening threshold “if alignment >80aa, use E-value <1e−5, otherwise use E-value <1e−3; covered fraction of HMM > 0.3”) to find all families that satisfy the filtering threshold, so that the carbohydrate-active enzymes in the genome can be identified and analyzed for the presence of several conserved carbohydrate-related functional domains.
Species annotation and statistics
Based on the species information of the above non-redundant genes compared to the sequences in Nr, the species composition and relative abundance information of the samples were obtained.
Results
Microbial diversity analysis of red vinasse acid
The red vinasse acid sample’s genome had a length of 75, 320, and 253 bp, 39,965 sequences, and a G + C content of 50.46 percent. Red vinasse acid gene species abundance annotation revealed 63 phyla, 782 genera, and 2,609 microorganisms. There are three groups of advantage bacteria among them with relative contents above 1% at the phylum level, seven groups at the genus level, and nine groups at the species level (Figure 1).

Figure 1. Krona diagram of the microbial community of red vinasse acid.
At the phylum level, Firmicutes, Proteobacteria, and Ascomycota were the dominant phyla, with relative contents of 38.44, 16.90, and 16.78%, respectively, accounting for 72.12% of the total content. At the genus level, the dominant genera include Lactobacillus, Pediococcus, Aspergillus, Burkholderia, Klebsiella, Monascus, and Penicillium, with relative contents of 21.59, 16.1, 10.25, 10.02, 2.74, 1.33, and 1.06%, respectively, accounting for 63.13% of the total content. At the species level, there are nine dominant strains with relative content of more than 1%, including Pediococcus acidilactici, Burkholderia multivorans, Lactobacillus sp. BCRC 12945, Lactobacillus panis, Lactobacillus fermentum, Lactobacillus phage Bacchae, Klebsiella pneumoniae, Aspergillus wentii, and Monascus purpureus. The relative contents were 15.47, 6.84, 5.19, 4.11, 3.97, 2.08, 1.96, 1.36, and 1.12%, respectively.
Annotation analysis of common database
Annotation analysis of eggNOG database
According to the eggNOG database, 16,093 genes of red vinasse acid showed functional annotation, and these genes can be classified into 23 categories based on their functions. Remove the R (general function prediction only) and S (function unknown) parts with unknown function, in which the number of genes greater than 300 is transcription (1086), intracellular trafficking, secretion, and vesicular transport (589), carbohydrate transport and metabolism (582), replication, recombination, and repair (484), posttranslational modification, protein turnover, chaperones (474), signal transduction mechanisms (418), cell wall/membrane/envelope biogenesis (393), inorganic ion transport and metabolism (352), amino acid transport and metabolism (330), and translation, ribosomal structure, and biogenesis (301). In addition to the basic life activities of microorganisms, metabolic activities are mainly carbohydrate metabolism, inorganic ion transport and metabolism, and amino acid transport and metabolism (Figure 2).
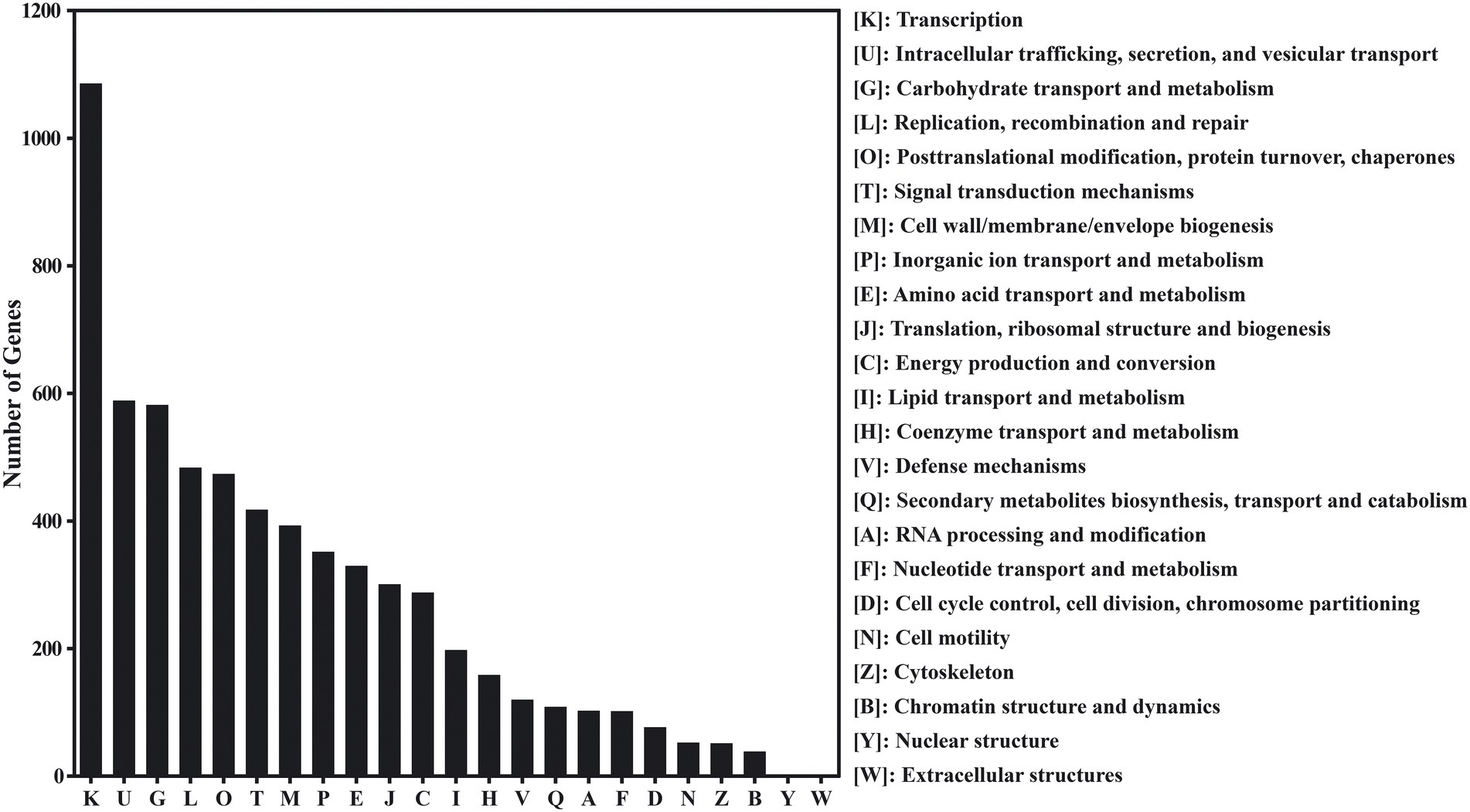
Figure 2. The evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) gene’s functional classification in red vinasse acid.
Annotation analysis of KEGG database
49,652 different genes in red vinasse acid were annotated, belonging to 21 KEGG pathways in four categories of the KEGG database (Figure 3). The four categories are: metabolism (35,782, 72.07%), genetic information processing (8,272, 16.66%), cellular processes (3,670, 7.39%), and environmental information processing (1,928, 3.88%). Carbohydrate metabolism (10,375), amino acid metabolism (7,057), nucleotide metabolism (4,005), replication and repair (3,516), metabolism of cofactors and vitamins (3,284), energy metabolism (2,849), lipid metabolism (2529), and translation (2,029) are among the eight KEGG pathways that enrich differentially expressed genes (2,029). The two most significant metabolic processes among them are glucose and amino acid metabolism, which are identical to the findings of eggNOG’s functional annotation.
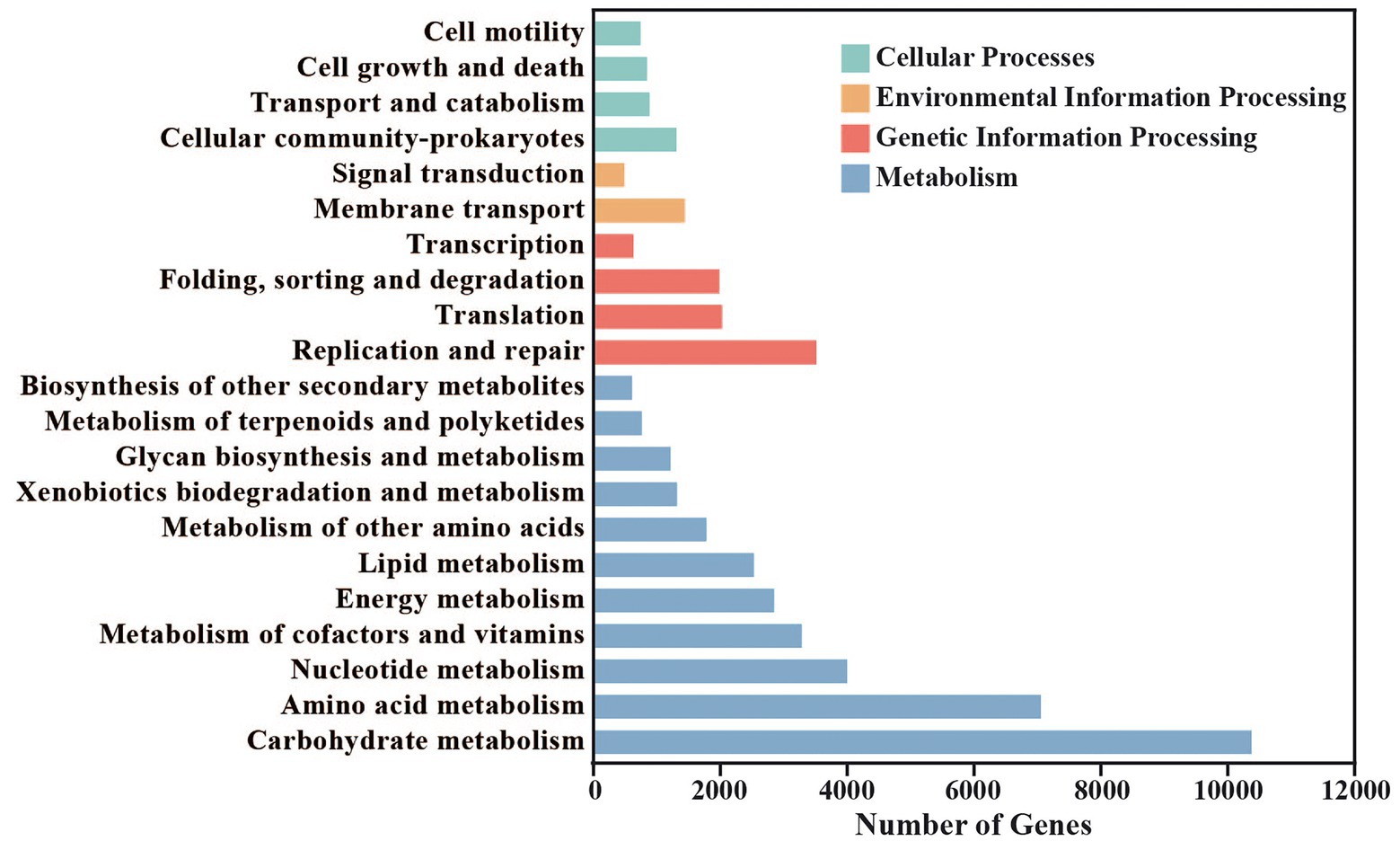
Figure 3. Statistics of Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathway in red vinasse acid.
Annotation analysis of CAZy database
The CAZy database can divide different types of carbohydrate-active enzymes into six protein families, including glycoside hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), and auxiliary activities (AAs). Compared with the CAZy database, a total of 3,328 carbohydrate-active enzymes were identified (Figure 4), of which glycosyl transferases (GTs, 31.3%) and glycoside hydrolases (GHs, 28.3%) were the most. Then, carbohydrate esterases (CEs, 21.4%), auxiliary activities (AAs, 11.5%), and carbohydrate-binding modules (CBMs, 6.7%). Polysaccharide lyases (PLs, 1.0%) were the least.
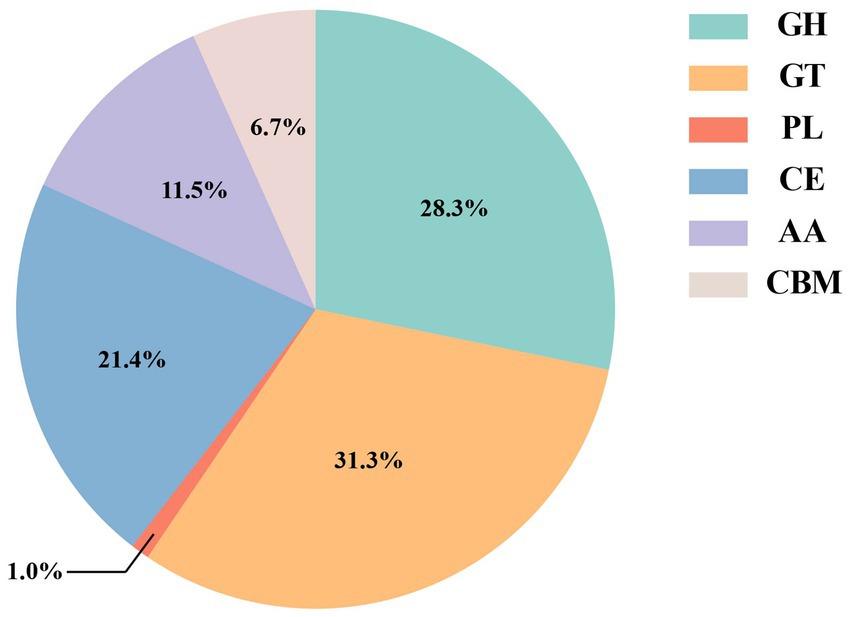
Figure 4. Carbohydrate-active enzymes database (CAZy) statistics in red vinasse acid.
Gene analysis of glucose metabolism
Sugar transport system
In the sugar transport system, mannitol, fructose, mannose, and sucrose can be transported by PTS (phosphotransferase system) simultaneously. A total of 27, 49, 34, and 17 control genes were found in red vinasse acid, respectively, indicating that the ability of red vinasse acid microorganisms to transport different sugars was different during fermentation, and the ability of microorganisms to transport fructose was strong during fermentation. Lactose and maltose can be transported through the PTS transport system, ABC transport protein, and permease. One PTS transport system for lactose and maltose was found in the red vinasse acid gene, but the ABC transport protein and permease gene for lactose and maltose were not found, indicating that the transport of lactose and maltose by microorganisms in red vinasse acid was mainly carried out through the PTS transport system. Galacitol can only be transported through the PTS transport system, and 16 related genes were found in red vinasse acid. Gluconate, fructose, and oligosaccharides can also be used to enter the strain cells by using relevant permeability proteins, and 10, 0, and 14 genes were retrieved respectively, indicating that fructose is mainly transported by microorganisms in red vinasse acid through the PTS transport system (Table 1).
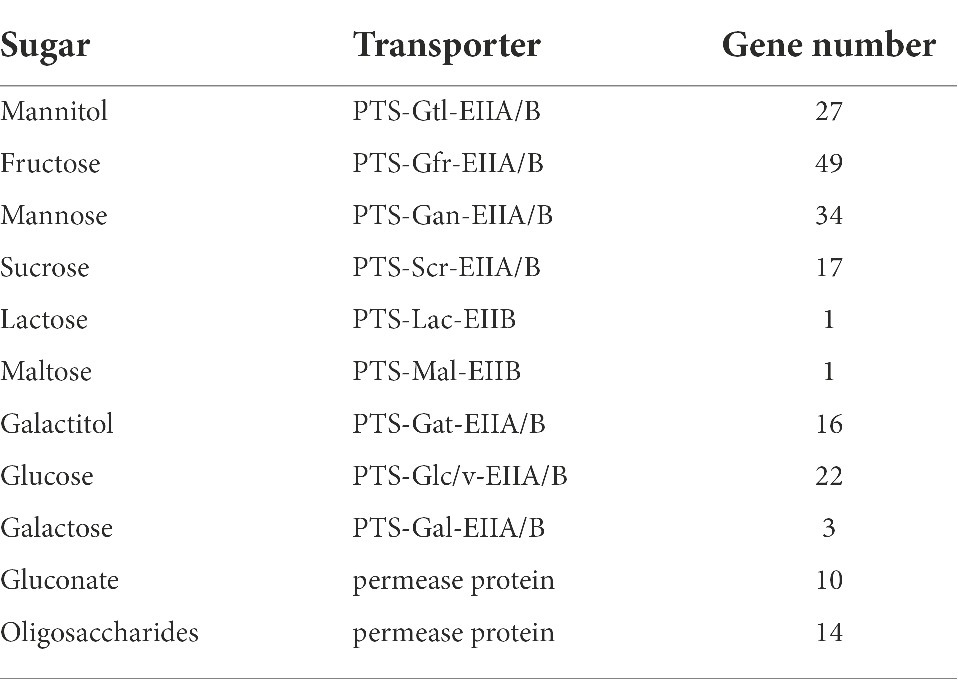
Table 1. Gene analysis of the sugar transport system in red vinasse acid.
Transformation of D-mannitol 1-phosphate
The PTS transport system phosphorylates sugars before they are delivered to cells. Mannitol was phosphorylated while being transported by the PTS system, and after reaching the cells, D-mannitol 1-phosphate was eventually produced. The latter is often transformed by mannitol-1-phosphate 5-dehydrogenase (EC: 1.1.1.17) into D-Fructose 6-phosphate, an intermediate glycolysis product. In conjunction with Tables 1, 2, red vinasse acid has 14 mannitol-1-phosphate 5-dehydrogenase genes and the mannitol PTS transport system, allowing mannitol to be transformed into the intermediate of the glycolysis pathway known as D-Fructose 6-phosphate. Table 2 demonstrates that there are seven species of bacteria and one species of fungi at the genus level, with Lactobacillus being the predominate genus and playing a significant role in the transfer of mannitol (Table 2; Chen et al., 2014).
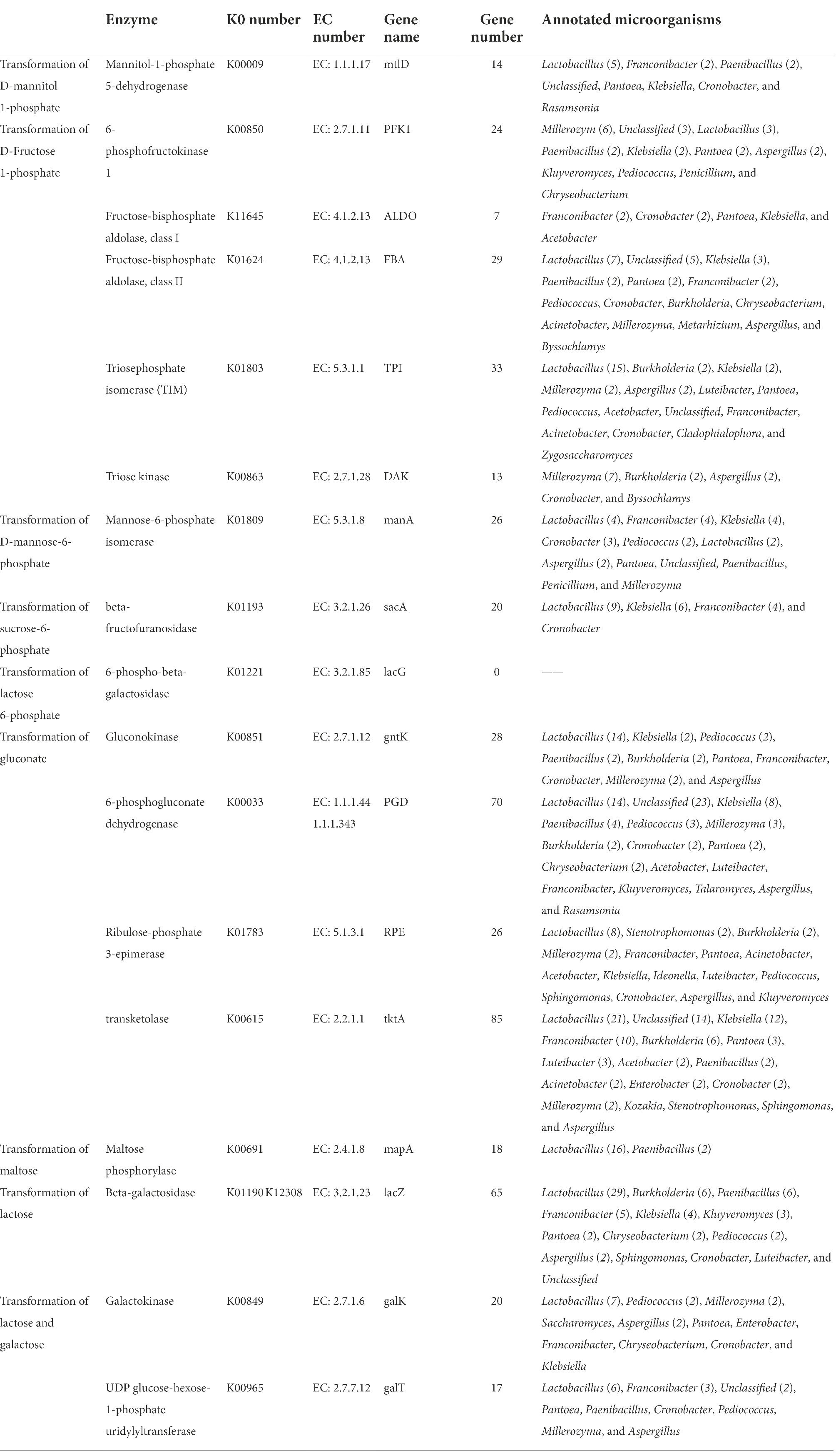
Table 2. Genes encoding enzymes involved in conversion sugar into intermediates of glycolysis pathway.
Transformation of D-fructose 1-phosphate
Fructose was finally transported into cells through the PTS system to form D-Fructose 1-phosphate, which was generally converted into the intermediate product of glycolysis D-glyceraldehyde 3-phosphate through three pathways. There are three combinations of key enzymes, namely, phosphofructokinase (EC: 2.7.1.11) and fructose-2-phosphate aldolase, fructose-bisphosphate aldolase (EC: 4.1.2.13) and triosephosphate isomerase (EC: 5.3.1.1), and fructose-2-phosphate aldolase and triose kinase (EC: 2.7.1.28). Combined with Tables 1, 2, red vinasse acid contains fructose PTS transport system, 36 fructose-bisphosphate aldolase and 33 triphosphate isomerase genes, which can convert intracellular D-Fructose 1-phosphate into D-glyceraldehyde 3-phosphate by the second pathway. Fructose-bisphosphate aldolase (EC: 4.1.2.13) comes in two varieties: fructose-bisphosphate aldolase class I and class II, as indicated in Table 2. Fructose-bisphosphate aldolase and class I annotated five kinds of microorganisms, and the dominant genera were Franconibacter and Cronobacter. Fructose-bisphosphate aldolase and class II annotated 15 species of microorganisms, and the dominant genus was Lactobacillus. Triosephosphate isomerase (EC:5.3.1.1) annotated 15 species of microorganisms, including 11 species of bacteria and four species of fungi. The dominant genera of bacteria were Lactobacillus and the dominant genera of fungi were Millerozyma and Aspergillus. Therefore, Lactobacillus plays a major role in the transport of fructose.
Transformation of D-mannose-6-phosphate
Mannatose was transported to cells through the PTS system to form D-mannose-6-phosphate, which was directly converted into the intermediate product β-D-Fructose 6 phosphate in the glycolysis pathway catalyzed by mannose-6-phosphate isomerase (EC: 5.3.1.8). Combining Tables 1, 2, it is known that red vinasse acid contains a mannose PTS transport system and 26 mannose-6-phosphate isomerase genes, so mannose can be converted into β-D-Fructose 6 phosphate, which is the intermediate of the glycolysis pathway. As shown in Table 2, nine species of bacteria and three species of fungi were annotated in the transport process. The dominant bacterial genera were Franconibacter, Klebsiella, and Lactobacillus, and the dominant genus of fungi was Aspergillus, indicating that the transport of mannose was mainly completed by the above microorganisms.
Transformation of sucrose-6-phosphate
Sucrose was transported to cells by the PTS system to form sucrose 6-phosphate, which was then converted to D-glucose 6-phosphate and β-D-fructose under the catalysis of β-fructofuranosidase (EC: 3.2.1.26). Combined with Tables 1, 2, red vinasse acid contains the PTS transport system of sucrose and encodes 20 genes of β-fructofuranosidase, so red vinasse acid can be transported to intracellular by extracellular sucrose to further produce D-glucose 6-phosphate and β-D-fructose. As shown in Table 2, four kinds of microorganisms are noted. All of them are bacteria, and the dominant genus is Lactobacillus.
Transformation of lactose 6-phosphate
The product of lactose transported to cells via the PTS system is 6-phosphate lactose, which is catalyzed by 6-phospho-β-galactosidase (EC: 3.2.1.85) to form β-D-glucose and D-galactose-6-phosphate. Combined with Tables 1, 2, the PTS transport system contained lactose, but the gene encoding 6-phospho-β-galactosidase was not found, so the transport of lactose could not be realized by red vinasse acid.
Transformation of gluconate
Gluconate transported to the cells can further form the intermediate substance β-D-fructose 6-phosphate in the glycolysis pathway, but this process needs to be catalyzed by four enzymes: glucokinase (EC: 2.7.1.12), 6-phosphogluconate dehydrogenase (EC:1.1.1.44 1.1.1.343), ribulose-phosphate 3-epimerase (EC: 5.1.3.1), and transketolase (EC: 2.2.1.1). The control genes of four enzymes were found in red vinasse acid, and the number of genes was 28, 70, 26, and 85, respectively, so red vinasse acid can use extracellular gluconate to further produce β-D-fructose-6-phosphate and participate in the pathway of glycolysis. As shown in Table 2, glucokinase annotated 10 species of microorganisms, including eight species of bacteria and two species of fungi, and the dominant genus was Lactobacillus. Twelve species of bacteria, with Unclassified and Lactobacillus as the most prevalent genera, and five species of fungi, with Millerozyma as the most prevalent genus, were found to contain 6-phosphate gluconate dehydrogenase. Ribulose-3-phosphate isomerase annotated three species of fungi and 13 species of bacteria. Millerozyma was the main fungus genus, whereas Lactobacillus was the leading genus of bacteria. The predominant genus among the 17 microorganism species for which transketolase was annotated was Lactobacillus. As a result, Lactobacillus is crucial to the transfer of gluconate.
Transformation of maltose
After entering the bacteria cells, maltose can be converted into D-glucose under the action of maltose phosphorylase (EC: 2.4.1.8) and enter the glycolysis pathway directly. 18 genes of maltose phosphorylase are encoded in red vinasse acid, so maltose can be converted into D-glucose, an intermediate product of the glycolysis pathway. As shown in Table 2, maltose phosphorylase was annotated to two microorganisms, and Lactobacillus was the dominant genus.
Transformation of lactose
Lactose enters the cell through the PTS transport system and produces glycolysis intermediates through two pathways (Table 2). The lactase transferred into the cell was hydrolyzed into α-D-glucose and D-glucose-1-phosphate by pathways I and II. In pathway I, there are 70 genes encoding β-galactosidase (EC: 3.2.1.23), and in pathway II, the number of genes encoding β-galactosidase (EC: 3.2.1.23)/galactokinase (EC: 2.7.1.6)/UDP- glucose-hexose-1-phosphate uridylyltransferase (EC: 2.7.7.12) is 70, 20, and 17, respectively. Therefore, the intracellular lactose in red vinasse acid can be hydrolyzed into α-D-glucose and D-glucose-1-phosphate by pathways I and II. As shown in Table 2, β-galactosidase, galactokinase, and UDP- glucose-hexose-1-phosphate uridine invertase were annotated to 14, 11, and 9 species of microorganisms, respectively, and all the dominant bacteria were Lactobacillus.
Transformation of galactose
Galactose could transform D-glucose 1-phosphate under the catalysis of galactokinase (EC: 2. 7. 1. 6) and UDP-glucose-hexose-1-phosphate uridylyltransferase (EC: 2.7.7.12), and 20 and 17 coding genes were found and annotated for 11 and nine species of microorganisms, respectively. All the dominant bacteria were Lactobacillus. And red vinasse acid contains the PTS transport system of galactose, so galactose can be converted into D-glucose 1-phosphate.
Amino acid flavor formation pathway
The pathway of amino acid flavor formation means that amino acids are transformed into corresponding ketoacids by a transaminase. Which are unstable, converted into corresponding aldehydes, further dehydrogenated to alcohols, and then esterified to corresponding esters. These metabolites are important flavor components in food.
Transaminase
The transamination of branched-chain amino acids, aromatic amino acids, and methionine can be catalyzed by different transaminases. Branched-chain amino acid aminotransferase (BCAT) has activity for branched-chain amino acids and methionine, while aromatic-amino-acid transaminase (ArAT) has catalytic activity for aromatic amino acids, leucine, and methionine. As shown in Table 3, 32 BCAT genes (ilvE) were identified, and 15 kinds of microorganisms were annotated with red vinasse acid genes, and the dominant strain was Aspergillus. And 27 ArAT genes (tyrB, ARO8, and ARO93) were found, which were, respectively, annotated to seven, six, and two kinds of microorganisms. The dominant bacterial genera were Burkholderia, Debaryomyces, and Millerozyma, respectively. According to the findings, the key players in the fermentation of red vinasse acid were Aspergillus, Burkholderia, Debaryomyces, and Millerozyma, which catalyzed the synthesis of branched-chain amino acids, aromatic amino acids, and methionine. Figures 5, 6 respectively depict the metabolic and synthesis processes for aromatic amino acids and branched-chain amino acids.
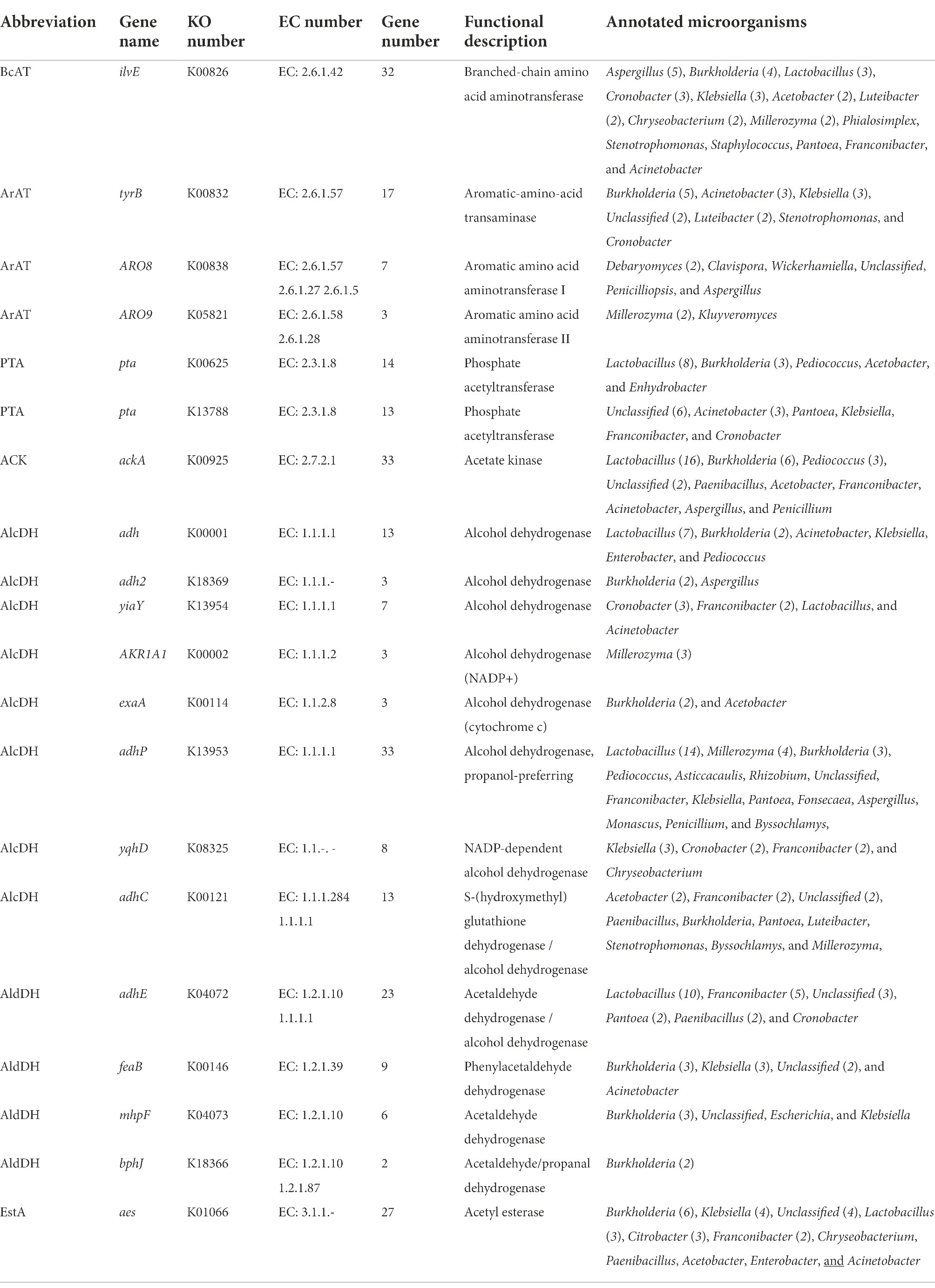
Table 3. Genes of encoding enzymes involved in transamination pathway in red vinasse acid.

Figure 5. Synthesis and metabolic pathway of branched-chain amino acids.
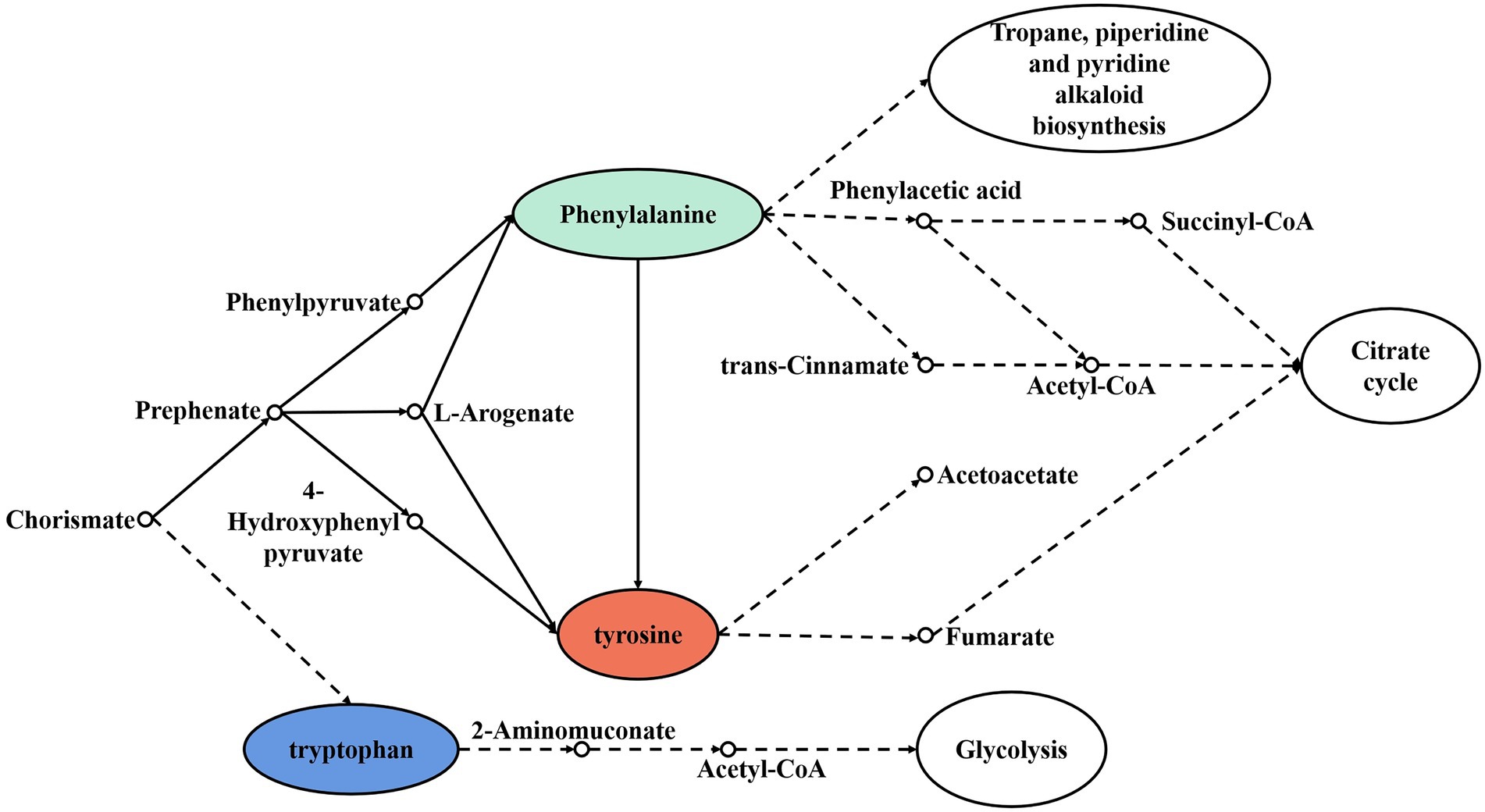
Figure 6. Synthesis and metabolic pathway of aromatic amino acids.
Ketoacid invertase
Keto acids can be converted in three ways. The first way is that keto acids can be directly converted into carboxylic acids by oxidative decarboxylation. In this pathway, keto acid dehydrogenase (KaDH), phosphate acetyltransferase (PTA), and acetate kinase (ACK) showed activity on the matrix of branched-chain amino acids. As shown in Table 3, there are genes encoding PTA (27) and ACK (33) in the genomic DNA of red vinasse acid, which were annotated to 11 and 7 microorganisms, respectively, indicating that keto acids of the above amino acids can be converted into corresponding carboxylic acids in the process of red vinasse acid fermentation, and the dominant genus was Lactobacillus.
The second pathway is that keto acids are decomposed into carboxylic acids by carboxylic acid dehydrogenase (HycDH). HycDH has two stereospecific enzymes, D-carboxylate dehydrogenase (D-HycDH), and L-carboxylate dehydrogenase (L-HycDH). At present, although it cannot be proved that this approach can directly lead to the formation of flavored substances, it can reduce the synthesis of flavored substances by shunting the precursors of flavor substances into non-flavored products. These two carboxylic acid dehydrogenases were not found in the genomic DNA of red vinasse acid, so the overall flavor will not be reduced because of the above pathways.
The third way is the conversion of α-ketoacid to corresponding aldehydes by α-ketoacid decarboxylase (KdcA). The gene encoding keto acid decarboxylase was not found in red vinasse acid, which indicated that the microorganisms in red vinasse acid could not carry out the latter two forms of transformation, and the first keto-acid transformation was mainly used in the fermentation process, and Lactobacillus was the main function.
Alcohol and aldehyde dehydrogenase
Aldehyde dehydrogenase and alcohol dehydrogenase can catalyze the conversion of aldehydes (alcohols) into corresponding alcohols and carboxylic acids, and the control genes are aldehyde dehydrogenase (AldDH) and alcohol dehydrogenase (AlcDH), respectively. In the red vinasse acid gene, 83 genes encoding alcohol dehydrogenase (AlcDH) and 40 genes encoding aldehyde dehydrogenase (AldDH) were found. Alcohol dehydrogenase (AlcDH) includes adh, adh2, yiaY, AKR1A1, exaA, adhP, yqhD, and adhC. It is noted that the dominant genus is Lactobacillus, and the fungus Millerozyma also plays an important role. Aldehyde dehydrogenase (AldDH) includes adhE, feaB, mhpF, and bphJ, and the dominant genus is Lactobacillus. Therefore, in the process of catalyzing aldehydes (alcohols) into corresponding alcohols and carboxylic acids, Lactobacillus was the main microorganism.
Discussion
Red vinasse acid has a rich aroma and unique flavor, which is closely related to its complex microbial metabolic activities. It has been reported that in fermented food, the different dynamic patterns of free sugars and amino acids imply different co-metabolisms, for example, free sugars might derive from polysaccharides through hydrolysis and be consumed by lactic acid bacteria, whereas amino acids produced through proteolysis of proteins are consumed by fungi (Mouritsen et al., 2017).
In this study, macrogenomic analysis revealed the microbial diversity and functional properties of the microbial community. We annotated the species abundance of the red vinasse acid gene, and the dominant genera included Lactobacillus, Pediococcus, Aspergillus, Burkholderia, Klebsiella, Monascus, and Penicillium (Figure 1). Lactobacillus can produce lactic acid and a variety of antibacterial substances. Lactobacillus intracellular carbohydrate active enzymes are specific (Swanson et al., 2020). The amount of lactic acid produced during fermentation has an impact on the quality of the fermentation. In addition to improving the nutritional value and quality of food, Pediococcus also has great resistance to acid and high temperatures. Additionally, Pediococcus displays biological properties that fight cancer and lower cholesterol (Zhang et al., 2019). Monascus is the main beneficial fungus of red vinasse acid fermentation, which gives red vinasse acid pigment and a unique flavor. Due to the lack of sterilization technology in the production of traditional red vinasse acid, the community information of microorganisms in red vinasse acid is complex and diverse, so in addition to beneficial microbes such as Lactobacillus and Monascus, there are still a small number of spoilage microbes and detrimental microbes in the red vinasse acid, such as Burkholderia and Klebsiella. The comparative analyses that can be performed are limited by the relatively few reports on the microbial diversity of red vinasse acid at the species level, and our macrogenomic analysis of red vinasse acid has enriched the information on the specific category information of microbial diversity of red vinasse acid. Compared with the eggNOG and KEGG databases, 16,093 and 49,652 genes were annotated, respectively. The main metabolic activities were amino acid metabolism and carbohydrate metabolism (Figures 2, 3). Glycosyl transferases and glycoside hydrolases were the most abundant in the CAZy database analysis (Figure 4). Glycoside hydrolases hydrolyze the glycosidic bonds of various sugar-containing compounds such as oligosaccharides and polysaccharides by internal or external digestion and generate monosaccharides, oligosaccharides, or sugar complexes, thus playing an important role in the synthesis of oligosaccharides and aromatic glycosides, and the glycosylation of amino acids and peptides. Glycosyl transferases catalyze the binding of activated sugars to non-receptor molecules, such as proteins, oligosaccharides, lipids, and small molecules (Chen et al., 2013; Cai et al., 2018; Huang et al., 2018). The abundant glycoside hydrolase and glycosyl transferase in red vinasse acid provide the basis for the formation, transfer, and further metabolism of monosaccharides and oligosaccharides.
In the pathway of glucose metabolism, red vinasse acid encodes the sugar transport system genes of mannitol, fructose, mannose, and sucrose, and retrieves the genes of gluconate and oligosaccharide related permeability proteins (Table 1). It also has the key enzyme genes that catalyze intracellular D-mannitol-1-phosphate, D-fructose-1-phosphate, D-mannose-6-phosphate, sucrose-6-phosphate, gluconate, maltose, lactose, and galactose, and has the basis for transforming them into glycolysis intermediates, and the main dominant bacterial genus of the glucose metabolism pathway is Lactobacillus (Table 2). Lactobacillus uses the relevant transport mechanism to transport various sugar substances from outside the cell to the cell, and then transformed into intermediate products of the glycolytic pathway through the catalysis of various related enzymes. These substances enter the glycolytic pathway and finally produce pyruvate, the precursor substance of lactic acid, through the catalysis of related enzymes. Pyruvate can be catalyzed by L-lactate dehydrogenase or D-lactate dehydrogenase to ultimately produce L-lactate or D- lactate (Kleerebezemab et al., 2000; Wang et al., 2010), resulting in a unique flavor.
In the process of amino acid flavor formation, the control genes of 32 branched-chain amino acid aminotransferase (BCAT), 27 aromatic-amino-acid transaminases (ArAT), 60 ketoacid invertase, 123 alcohol/aldehyde dehydrogenases, and 27 acetyl esterases were encoded by red vinasse acid (Table 3). Amino acids can be transformed into corresponding keto acids by transaminase, and then into corresponding aldehydes, further dehydrogenation to alcohols, and then esterification to corresponding esters, which have the basis for the formation of strong ester flavors, and the main dominant bacteria genus during amino acid flavor formation is Lactobacillus. Figure 5 shows the synthesis and metabolic pathway of branched-chain amino acids (Leucine, Valine, and Isoleucine), which is a crosstalk and a simplified process of the KEGG metabolic pathway ko00290 and ko00280. The solid line in the diagram represents the direct process and the dashed line represents the indirect process. The degradation of Valine and Isoleucine to Propanoyl-CoA is catalyzed by a series of enzymes, and Propanoyl-CoA then indirectly enters the following three metabolic pathways: Biosynthesis of 12-, 14-, and 16-membered macrolides, Propanoate metabolism, and Biosynthesis of type II polyketide backbone. Leucine is metabolized to Acetyl-CoA by a series of enzymes, and Acetyl-CoA enters indirectly into the Biosynthesis of type II polyketide backbone and the Citrate cycle (TCA cycle), which further functions in two metabolic pathways. Figure 6 shows the synthesis and metabolic pathway of aromatic amino acids (Phenylalanine, Tyrosine, and Tryptophan), which is a crosstalk and a simplified process of the KEGG metabolic pathway ko00400, ko00360, ko00350, and ko00380. Tryptophan is metabolically involved in the glycolytic process, Tyrosine in the tricarboxylic acid cycle, and Phenylalanine in the Tropane, piperidine, and pyridine alkaloid biosynthesis and Citrate cycle. We show the synthesis and metabolic pathways of branched-chain amino acids and aromatic amino acids in this study in the form of pictures, which are simplified to mainly describe the relationship between metabolic pathways and amino acids compared to the complete metabolic pathways in KEGG, in order to highlight the wide range and importance of amino acids in the process of biological activities, as well as to provide a targeted direction for flavor studies in amino acid metabolism.
The primary metabolic functions of red vinasse acid and the essential enzyme genes for sugar and amino acid metabolism were further discovered based on an examination of the structure and makeup of red vinasse acid bacteria. Analysis of dominant strain roles in the major metabolic pathways and further confirmation of the expression of important enzyme genes. It is anticipated that it will offer a theoretical foundation for the enhancement of red vinasse acid quality and the mining of microbial functional gene pools.
Data availability statement
The data presented in the study are deposited in the National Center for Biotechnology Information repository, accession numbers are SRR21707708, SRR21707707, SRR21707706, SRR21707705, SRR21707704, SRR21707703.
Author contributions
JiL, YY, and YZ contributed to conception and design of the study. JiL and YY organized the database. JiL wrote the first draft of the manuscript. YZ performed the statistical analysis. WL, MC, and HM wrote sections of the manuscript. AG and JuL contributed to funding acquisition, resources, supervision, and writing–review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from the Major Science and Technology Fund of Guangxi Province (AB21196020) and (AB20297006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ajdari, Z., Abd Ghani, M., Khan Ayob, M., Bayat, S., Mokhtar, M., Abbasiliasi, S., et al. (2014). Hypocholesterolemic activity of Monascus fermented product in the absence of Monacolins with partial purification for functional food applications. Sci. World J. 2014:252647. doi: 10.1155/2014/252647
Aniya, Y., Ohtani, I. I., Higa, T., Miyagi, C., Gibo, H., Shimabukuro, M., et al. (2000). Dimerumic acid as an antioxidant of the mold, Monascus Anka. Free Radical BioMed. 28, 999–1004. doi: 10.1016/S0891-5849(00)00188-X
Arıkan, M., Mitchell, A. L., Finn, R. D., and Gürel, F. (2020). Microbial composition of Kombucha determined using amplicon sequencing and shotgun metagenomics. J. Food Sci. 85, 455–464. doi: 10.1111/1750-3841.14992
Arnold, J. W., Simpson, J. B., Roach, J., Bruno-Barcena, J. M., and Azcarate-Peril, M. A. (2018). Prebiotics for lactose intolerance: variability in Galacto-oligosaccharide utilization by intestinal Lactobacillus rhamnosus. Nutrients 10:1517 doi: 10.3390/nu10101517
Bai, Y., Lan, W., and Lin, Y. (2020). Nutrition and safety evaluation of Guangxi red vinasse acid (in Chinese). Food Industry, 41, 340–344.
Belleggia, L., Aquilanti, L., Ferrocino, I., Milanović, V., Garofalo, C., Clementi, F., et al. (2020). Discovering microbiota and volatile compounds of surströmming, the traditional Swedish sour herring. Food Microbiol. 91:103503. doi: 10.1016/j.fm.2020.103503
Cai, H., Zhang, T., Zhang, Q., Luo, J., Cai, C., and Mao, J. (2018). Microbial diversity and chemical analysis of the starters used in traditional Chinese sweet rice wine. Food Microbiol. 73, 319–326. doi: 10.1016/j.fm.2018.02.002
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, 233–238. doi: 10.1093/nar/gkn663
Chen, G., Chen, C., and Lei, Z. (2017). Meta-omics insights in the microbial community profiling and functional characterization of fermented foods. Trends Food Sci. Technol. 65, 23–31. doi: 10.1016/j.tifs.2017.05.002
Chen, M., Guo, W., Li, Q., Xu, J., Cao, Y., Liu, B., et al. (2020). The protective mechanism of Lactobacillus plantarum FZU3013 against non-alcoholic fatty liver associated with hyperlipidemia in mice fed a high-fat diet. Food Funct. 11, 3316–3331. doi: 10.1039/c9fo03003d
Chen, W., He, Y., Zhou, Y., Shao, Y., Feng, Y., Li, M., et al. (2015). Edible filamentous fungi from the species Monascus: early traditional fermentations, modern molecular biology, and future genomics. Compr. Rev. Food Sci. F. 14, 555–567. doi: 10.1111/1541-4337.12145
Chen, B., Wu, Q., and Xu, Y. (2014). Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 179, 80–84. doi: 10.1016/j.ijfoodmicro.2014.03.011
Chen, S., Xu, Y., and Qian, M. C. (2013). Aroma characterization of Chinese Rice wine by gas chromatography–Olfactometry, chemical quantitative analysis, and aroma reconstitution. J. Agric. Food Chem. 61, 11295–11302. doi: 10.1021/jf4030536
Diana, M., Quílez, J., and Rafecas, M. (2014). Gamma-aminobutyric acid as a bioactive compound in foods: a review. J. Funct. Foods 10, 407–420. doi: 10.1016/j.jff.2014.07.004
Dikshit, R., and Tallapragada, P. (2015). Bio-synthesis and screening of nutrients for lovastatin by Monascus sp. under solid-state fermentation. J. Food Sci. Technol. 52, 6679–6686. doi: 10.1007/s13197-014-1678-y
Dikshit, R., and Tallapragada, P. (2016). Statistical optimization of lovastatin and confirmation of nonexistence of citrinin under solid-state fermentation by Monascus sanguineus. J. Food Drug Anal. 24, 433–440. doi: 10.1016/j.jfda.2015.11.008
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hsieh, C., Lu, Y., Lin, S., Lai, T., and Chiou, R. Y. Y. (2013). Stability of Monacolin K and Citrinin and biochemical characterization of red-Koji vinegar during fermentation. J. Agric. Food Chem. 61, 7276–7283. doi: 10.1021/jf401542q
Huang, Z., Hong, J., Xu, J., Li, L., Guo, W., Pan, Y., et al. (2018). Exploring core functional microbiota responsible for the production of volatile flavour during the traditional brewing of Wuyi Hong Qu glutinous rice wine. Food Microbiol. 76, 487–496. doi: 10.1016/j.fm.2018.07.014
Ilıkkan, Ö. K., and Bağdat, E. Ş. (2021). Comparison of bacterial and fungal biodiversity of Turkish kefir grains with high-throughput metagenomic analysis. LWT 152:112375. doi: 10.1016/j.lwt.2021.112375
Johansson, L., Nikulin, J., Juvonen, R., Krogerus, K., Magalhaes, F., Mikkelson, A., et al. (2021). Sourdough cultures as reservoirs of maltose-negative yeasts for low-alcohol beer brewing. Food Microbiol. 94:103629. doi: 10.1016/j.fm.2020.103629
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, 277–280. doi: 10.1093/nar/gkh063
Kim, E., Cho, E., Yang, S., Kim, M., and Kim, H. (2021). Novel approaches for the identification of microbial communities in kimchi: MALDI-TOF MS analysis and high-throughput sequencing. Food Microbiol. 94:103641. doi: 10.1016/j.fm.2020.103641
Kleerebezemab, M., Hols, P., and Hugenholtz, J. (2000). Lactic acid bacteria as a cell factory: rerouting of carbon metabolism in Lactococcus lactis by metabolic engineering. Enzym. Microb. Technol. 26, 840–848. doi: 10.1016/S0141-0229(00)00180-0
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. doi: 10.1038/s41579-018-0029-9
Li, W., Han, Y., Yuan, X., Wang, G., Wang, Z., Pan, Q., et al. (2017). Metagenomic analysis reveals the influences of milk containing antibiotics on the rumen microbes of calves. Arch. Microbiol. 199, 433–443. doi: 10.1007/s00203-016-1311-8
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Mori, T., Mizuta, S., Suenaga, H., and Miyazaki, K. (2008). Metagenomic screening for Bleomycin resistance genes. Appl. Environ. Microbiol. 74, 6803–6805. doi: 10.1128/AEM.00873-08
Mouritsen, O. G., Duelund, L., Calleja, G., and Frøst, M. B. (2017). Flavour of fermented fish, insect, game, and pea sauces: Garum revisited. Int. J. Gastron. Food Sci. 9, 16–28. doi: 10.1016/j.ijgfs.2017.05.002
Nam, K., Choe, D., and Shin, C. S. (2014). Antiobesity effect of a jelly food containing the L-tryptophan derivative of Monascus pigment in mice. J. Funct. Foods 9, 306–314. doi: 10.1016/j.jff.2014.05.001
Pouliot-Mathieu, K., Gardner-Fortier, C., Lemieux, S., St-Gelais, D., Champagne, C. P., and Vuillemard, J. C. (2013). Effect of cheese containing gamma-aminobutyric acid-producing acid lactic bacteria on blood pressure in men. Pharmanutrition 1, 1–8. doi: 10.1016/j.phanu.2013.06.003
Powell, S., Forslund, K., Szklarczyk, D., Trachana, K., Roth, A., Huerta-Cepas, J., et al. (2014). eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42, 231–239. doi: 10.1093/nar/gkt1253
Riaz Rajoka, M. S., Zhao, H., Lu, Y., Lian, Z., Li, N., Hussain, N., et al. (2018). Anticancer potential against cervix cancer (HeLa) cell line of probiotic lactobacillus casei and lactobacillus paracasei strains isolated from human breast milk. Food Funct. 9, 2705–2715. doi: 10.1039/C8FO00547H
Sandes, S., Acurcio, L., Nicoli, J., and Neumann, E. (2018). In vitro and in vivo evaluation of two potential probiotic lactobacilli isolated from cocoa fermentation (Theobroma cacao L.). J. Funct. Foods 47, 184–191. doi: 10.1016/j.jff.2018.05.055
Su, C., Zhang, K., Cao, X., and Yang, J. (2020). Effects of Saccharomycopsis fibuligera and Saccharomyces cerevisiae inoculation on small fermentation starters in Sichuan-style Xiaoqu liquor. Food Res. Int. 137:109425. doi: 10.1016/j.foodres.2020.109425
Swanson, K. S., Gibson, G. R., Hutkins, R., Reimer, R. A., Reid, G., Verbeke, K., et al. (2020). The international scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of synbiotics. Nat. Rev. Gastroenterol. Hepatol. 17, 687–701. doi: 10.1038/s41575-020-0344-2
Tsai, C., Chiu, T., Ho, C., Lin, P., and Wu, T. (2013). Effects of anti-hypertension and intestinal microflora of spontaneously hypertensive rats fed gamma aminobutyric acid-enriched Chingshey purple sweet potato fermented milk by lactic acid bacteria. Afr. J. Microbiol. Res. 7, 932–940.
Wang, L., Zhao, B., Liu, B., Yang, C., Yu, B., Li, Q., et al. (2010). Efficient production of l-lactic acid from cassava powder by lactobacillus rhamnosus. Bioresour. Technol. 101, 7895–7901. doi: 10.1016/j.biortech.2010.05.018
Xie, M., An, F., Zhao, Y., Wu, R., and Wu, J. (2020). Metagenomic analysis of bacterial community structure and functions during the fermentation of da-jiang, a Chinese traditional fermented food. LWT 129:109450. doi: 10.1016/j.lwt.2020.109450
Xie, M., Wu, J., An, F., Yue, X., Tao, D., Wu, R., et al. (2019). An integrated metagenomic/metaproteomic investigation of microbiota in dajiang-meju, a traditional fermented soybean product in Northeast China. Food Res. Int. 115, 414–424. doi: 10.1016/j.foodres.2018.10.076
Yang, N. C., Jhou, K. Y., and Tseng, C. Y. (2012). Antihypertensive effect of mulberry leaf aqueous extract containing γ-aminobutyric acid in spontaneously hypertensive rats. Food Chem. 132, 1796–1801. doi: 10.1016/j.foodchem.2011.11.143
Yoshimura, M., Toyoshi, T., Sano, A., Izumi, T., Fujii, T., Konishi, C., et al. (2010). Antihypertensive effect of a γ-aminobutyric acid rich tomato cultivar 'DG03-9′ in spontaneously hypertensive rats. J. Agric. Food Chem. 58, 615–619. doi: 10.1021/jf903008t
Zhang, L., Xu, D., Wang, F., and Zhang, Q. (2019). Antifungal activity of Burkholderia sp. HD05 against Saprolegnia sp. by 2-pyrrolidone-5-carboxylic acid. Aquaculture 511:634198. doi: 10.1016/j.aquaculture.2019.06.012
Zhang, R., Yu, J., Guo, X., Li, W., Xing, Y., and Wang, Y. (2021). Monascus pigment-mediated green synthesis of silver nanoparticles: catalytic, antioxidant, and antibacterial activity. Appl. Organomet. Chem. 35:e6120. doi: 10.1002/aoc.6120
Keywords: red vinasse acid, metagenome, microbial diversity, glucose metabolism, functional genes, amino acid
Citation: Lv J, Ye Y, Zhong Y, Liu W, Chen M, Guo A, Lv J and Ma H (2022) Microbial diversity and functional genes of red vinasse acid based on metagenome analysis. Front. Microbiol. 13:1025886. doi: 10.3389/fmicb.2022.1025886
Edited by:
Biao Suo, Henan Agricultural University, ChinaReviewed by:
Zhen Feng, Northeast Agricultural University, ChinaJun Dai, Hubei University of Technology, China
Copyright © 2022 Lv, Ye, Zhong, Liu, Chen, Guo, Lv and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Lv, dGFpaGVsdmp1bjEyM0AxNjMuY29t; Ailing Guo, YWlsaW5nZ3VvMjM0QDE2My5jb20=