 Muhammad Usama Marghoob
Muhammad Usama Marghoob Alejandro Rodriguez-Sanchez
Alejandro Rodriguez-Sanchez Asma Imran
Asma Imran Fathia Mubeen
Fathia Mubeen Lori Hoagland
Lori Hoagland
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 07 November 2022
Sec. Microbe and Virus Interactions with Plants
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1020175
This article is part of the Research TopicPlant Growth-Promoting Rhizobacteria (PGPR) and Plant Hormones: An Approach for Plant Abiotic Stress Management and Sustainable AgricultureView all 7 articles
Soil salinization and heavy metal (HM) contamination are major challenges facing agricultural systems worldwide. Determining how soil microbial communities respond to these stress factors and identifying individual phylotypes with potential to tolerate these conditions while promoting plant growth could help prevent negative impacts on crop productivity. This study used amplicon sequencing and several bioinformatic programs to characterize differences in the composition and potential functional capabilities of soil bacterial, fungal, and archaeal communities in five agricultural fields that varied in salinity and HM concentrations within the Indus basin region of Pakistan. The composition of bacteria with the potential to fix atmospheric nitrogen (N) and produce the enzyme 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase were also determined. Microbial communities were dominated by: Euryarchaeota (archaea), Actinobacteria, Proteobacteria, Planctomycetota, Firimicutes, Patescibacteria and Acidobacteria (bacteria), and Ascomycota (fungi), and all soils contained phylotypes capable of N-fixation and ACC-deaminase production. Salinity influenced bacterial, but not archaeal or fungal communities. Both salinity and HM altered the relative abundance of many phylotypes that could potentially promote or harm plant growth. These stress factors also appeared to influence the potential functional capabilities of the microbial communities, especially in their capacity to cycle phosphorous, produce siderophores, and act as symbiotrophs or pathotrophs. Results of this study confirm that farms in this region are at risk due to salinization and excessive levels of some toxic heavy metals, which could negatively impact crop and human health. Changes in soil microbial communities and their potential functional capabilities are also likely to affect several critical agroecosystem services related to nutrient cycling, pathogen suppression, and plant stress tolerance. Many potentially beneficial phylotypes were identified that appear to be salt and HM tolerant and could possibly be exploited to promote these services within this agroecosystem. Future efforts to isolate these phylotypes and determine whether they can indeed promote plant growth and/or carry out other important soil processes are recommended. At the same time, identifying ways to promote the abundance of these unique phylotypes either through modifying soil and crop management practices, or developing and applying them as inoculants, would be helpful for improving crop productivity in this region.
One of the biggest challenges facing agriculture in Pakistan and other arid agricultural regions worldwide is soil salinization, and models predict that this challenge is growing rapidly (Wang et al., 2020). When soils become saline, they contain excessive levels of various types of soluble salts that can stress crop plants. Adverse effects of soil salinity include reductions in soil fertility and inefficient plant metabolism, which can result in lower plant growth and reduced crop yields (Nawaz et al., 2020). The main causes of soil salinization in agricultural systems include water deficits, especially in areas with high rates of evapotranspiration, and the use of poor-quality irrigation water. One practice that is commonly used to alleviate adverse effects of soil salinity is to apply gypsum, however, this practice is not sustainable because of the high price of this amendment and negative effects that repeated applications can have on soil health over time (Nouri et al., 2017). Another way that has been suggested to try and overcome this challenge is to develop salt tolerant crops using transgenic approaches. However, this will be challenging since salt tolerance is likely controlled by quantitative traits, and it is currently difficult to correlate individual genetic elements with salt tolerance mechanisms (Ashraf and Foolad, 2013; Ben-Laouane et al., 2020). Consequently, alternative approaches to overcome the challenge of soil salinization in agricultural systems are needed (Machado and Serralheiro, 2017).
Another challenge facing agricultural systems in Pakistan and other areas of the world is the presence of elevated concentrations of heavy metals and metalloids in soil (HM). In particular, high concentrations of lead (Pb), chromium (Cr), zinc (Zn), cobalt (Co), arsenic (As), copper (Cu), nickel (Ni), cadmium (Cd), and mercury (Hg) are considered contaminants in soil (Batool et al., 2017; Rodríguez-Eugenio et al., 2018; Zea et al., 2022). High concentrations of these HM can be naturally present in some soils due to parent materials, and they can become elevated in others due to anthropogenic activities such as industrialization and mining (Shakir et al., 2016; Moryani et al., 2020; Marghoob et al., 2022). In addition to negatively impacting plant and human health, the presence of elevated concentrations of these HM can have devastating effects on critical agricultural and ecological processes in soil such as nutrient cycling and pathogen dynamics (Rodriguez-Sanchez et al., 2022). The presence of elevated concentrations of HM may be even more problematic in saline soils. For example, Cd bioavailability in soil and uptake into plants was increased in the presence of higher levels of soil salinity (Wang et al., 2014; Han J. et al., 2021), which would be highly problematic in agricultural systems growing edible crop plants.
One way to help overcome these challenges is to exploit beneficial soil microorganisms (Reeve et al., 2016). For example, some soil microbes can form intimate associations with plants, increasing their capacity to tolerate salinity and HM stress, resulting in greater crop productivity (Nawaz et al., 2021). One of the mechanisms responsible for these benefits is the capacity of some microbes to produce the enzyme, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase, which breaks down ACC, the main precursor for stress ethylene which reduces plant growth (Ben-Laouane et al., 2020). Other plant-associated microbes can promote the health and productivity of plants in high stress environments by fixing atmospheric nitrogen (N), solubilizing nutrients such as phosphorous (P), and acting as biocontrol agents against phytopathogens (Abdelrazek et al., 2020; Ben-Laouane et al., 2020; Mukhtar et al., 2020). As a result, many of these so-called “plant growth promoting” (PGP) microbes have been isolated and developed for use as inoculants to help address environmental and agricultural issues (Rilling et al., 2019).
Microbes with novel characteristics such as the capacity to promote plant growth under stress are often isolated from harsh environments. These microbes are known as extremophiles. Those with the potential to live in environments with high concentrations of soluble salts are called halophiles (Mukhtar et al., 2018). Learning more about ecological factors that influence the composition and potential functional role of halotolerant microbes with potential PGP activities could lead to the identification of practices that can help address critical agricultural challenges like salinity and HM stress. By leveraging new culture independent techniques and bioinformatics tools, it will be possible to overcome the challenge of identifying microbes that are difficult to culture and learn more about their potential functional roles in soil (Almeida and De Martinis, 2019). Therefore, the primary aim of this study was to characterize the diversity of microbial communities residing in the salt- and HM-affected agricultural lands of the Indus Basin in Pakistan. We also sought to determine how these stress factors can influence the composition of microbial communities with the potential to fix atmospheric N and produce ACC-deaminase, critical in agricultural systems. The Indus Basin is an ideal place to study these dynamics since this is an important agricultural region, it is suspected to contain soils with contrasting concentrations of soluble salts, and some fields have been reported to contain high concentrations of HM. To date, metagenomic surveys of soil microbial communities in salt- and HM-affected agricultural zones of Pakistan are scarce, and to the best of our knowledge, this will be the first report of soil microbial diversity and potential function in agricultural fields within the Mirpur Mathelo and Dhairk regions of the Indus Basin.
Soils were collected from five agricultural fields within the Indus Basin region and provinces of Punjab and Sindh, Pakistan expected to contain variable levels of soluble salts and possibly HM: Choa Saidan Shah (CSS), Chakwal, Punjab, 32.7485, 72.7574; Khewra Salt Mine Range (KSMR), Jhelum, Punjab; 32.6134, 73.0239, Pakka Anna (PA), Faisalabad, Punjab; 31.2435, 72.7998, Dhairki (D), Ghotki, Sindh; 27.945195, 69.671935, Mirpur Mathelo (MM), Ghotki, Sindh; 28.0211, 69.5490. The individual farm fields within each region were selected because they have similar crop rotations: specifically, wheat (Triticum aestivum) is cultivated in winter, and cotton (Gossypium hirsutum), maize (Zea mays), rice (Oryza sativa) and various pulse crops are cultivated in summer. Samples were collected at the land preparation stage for wheat cultivation during the months of October and November 2019. Soils were collected using a sterile soil sampling probe (2-cm width and 30.5-cm long) to a depth of 15-cm, placed in sterile containers, and transferred on ice to cold storage (4°C). At each location, three soil samples were collected, each consisting of multiple cores collected in a zig-zag pattern and pooled to account for heterogeneity at the site. The three fresh soil samples from each field were shipped on ice to the Microbial Physiology Lab, National Institute for Biotechnology and Genetic Engineering, Pakistan for chemical analysis, and to the Soil Microbial Ecology Lab, Purdue University, IN, United States, for DNA isolation and amplification in preparation for sequencing.
Soil texture and basic chemical parameters including pH, electrical conductivity (EC), total soil organic matter (SOM), exchangeable phosphorous (P), total N, and ionic concentrations of Cl− and water-soluble sodium (Na) and potassium (K) were determined using standard procedures previously described in the literature (Walkley and Black, 1934; Olsen, 1954; Richard, 1954; Imran et al., 2021; Khalid et al., 2021). Total concentrations of a wide range of elements including toxic heavy metals like Cr were quantified using a portable X-ray fluorescence analyzer (pXRF; Vanta M pXRF, with a Rh and W x-ray tube, Olympus, Waltham, Ma, United States). Briefly, 5 g of each soil were ground in a UDY grinder (3010SM/C, Seedburo, United States), packed into XRF sample cups and covered with 4.0 μm prolene film before being scanned with the pXRF. All analyses were conducted in triplicate.
Total genomic DNA was extracted from each soil sample (10 g each) using DNeasy PowerSoil Kits (QIAGEN, Germantown, MD, United States) per manufacturer’s instruction. The quality and quantity of the final product was checked using a NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, MA, United States), and stored at −20°C until use in PCR. The following primer pairs were used to amplify specific microbial taxa: 42F-376R targeting the V3-V4 region of the 16S rRNA gene for bacteria, ITS1F-ITS2 for fungi, 340F-806rB targeting the 16S rRNA gene for archaea, PolF-PolR for the nifH region of bacteria, and acdSF5-acdSR8 for the acdS gene of bacteria. PCR protocols (see Supplementary Table S1) were optimized based on previous studies conducted using the same primer sets (Op De Beeck et al., 2014; Bouffaud et al., 2018; Bahram et al., 2019; Wasimuddin et al., 2019; Yang et al., 2019). PCR products with desired band lengths were confirmed on 1% agarose gel using electrophoresis. After the first PCR, a 2nd PCR reaction was conducted to attach the CS1 and CS2 linkers in preparation for Illumina sequencing using the same conditions, but only 8 cycles. The final PCR products were stored at −20°C before shipping to the Core Genome Laboratory, Chicago, IL, United States1 where the products were diluted, tagged and subject to paired-end sequencing using Illumina NovaSeq.
A total of 20,932,024 raw sequences were received after sequencing for bacteria, 2,381,416 for archaea, 979,543 for fungi, 15,288,910 for nifH, and 25,929,149 for acdS. To ensure that only high-quality reads were used, the sequences were subject to the following screening methods: (1) contigs assembly, (2) elimination of homopolymers, chimeric sequences and ambiguous bases, and (3) additional screening, OTU clustering, and processing of the refined data.
Paired-end reads were merged into contigs using mothur v 1.44 (Schloss et al., 2009). Local alignment of paired-end reads was conducted using Needleman conditions, and differences in nucleotides in the same alignment position were resolved with an ambiguous base when the difference in Phred score was 6 or lower. For the acdS gene, only the overlap region was selected for further analysis (Bouffaud et al., 2018).
Contigs with ambiguous bases or those with homopolymers >8 bp were removed. VSEARCH (Rognes et al., 2015) de novo identification was used to remove contigs of chimeric nature, using 1.9 as the abundance skew, 0.3 score for chimera characterization, and 0.5 as the minimum divergence ratio. In addition, a minimum of three differences was allowed when comparing segments, 8 was used as a weight of no-votes, and 1.4 was used as a pseudo-count on no-votes.
Non-chimeric contigs were aligned against the SiLVA SEED v138 database using Needleman conditions in both directions as an additional tool for quality screening of bacterial and archaeal 16S rRNA gene regions. Contigs having different start and end positions from expected positions of the primers were eliminated for further processing. Next, hierarchical preclustering allowing 1 error/100 bp length was used to achieve denoised contigs and generate Amplicon Sequence Variants (ASV; Huse et al., 2010; Schloss, 2020). The SiLVA NR v138 database was used as a reference to classify ASVs against specified portions of the bacterial and archaeal 16S rRNA sequences. The K-nearest neighbor algorithm and a k-mer search of 8-bp length with a cutoff of 80% were used as quality checks. Only sequences that were successfully classified at phylum or lower levels were retained for further analysis.
For quality screening of the fungal ITS region and acdS and nifH genes, hierarchical preclustering approaches allowing 1 error/100 bp length was used to denoise non-chimeric contigs into ASVs. For taxonomic screening purposes, fungal ASVs were classified using the full public UNITE database (10/05/2021) with the k-nearest neighbor algorithm and a k-mer search of 8 bp length. Contigs that were unsuccessfully classified at the phylum or lower level were eliminated.
We obtained a total of 8,529,291 high quality reads for bacteria, 179,893 for archaea, 1,103,290 for fungi, and 36,638 and 4,408,719 for the nifH and acdS bacterial genes, respectively, after initial processing into AVSs. These high quality ASVs were subject to OTU clustering to better classify the sequences (Schloss, 2020) using the abundance-greedy clustering algorithm in VSEARCH (Westcott and Schloss, 2015). Matthew’s correlation coefficient was used to determine the accuracy of clusterization (Schloss et al., 2016). 97% similarity thresholds were used for the 16S rRNA and acdS genes of bacteria, 95% for the 16S rRNA gene of archaea and ITS gene of fungi, and 94% for the nifH bacterial gene. Elimination of singleton OTUs was performed and the most abundant sequence in each OTU was used as a representative sequence for that OTU.
Each representative sequence of bacterial and archaeal 16S rRNA genes was taxonomically classified using the SiLVA NR v138 database and the full public UNITE database (10/05/2021). For the fungal ITS region, the k-nearest neighbor algorithm and a k-mer search of 8 bp length conditions were applied. For the acdS and nifH genes, representative sequences of each OTU were classified against a selected group of sequences retrieved from the GenBank NT database. The collection of acdS sequences ranged from 160 to 1,500 bp in length, and for nifH, the collection ranged from 300 to 1,500 bp in length; both collections were retrieved from GenBank on 11/15/2021, and these databases were used to taxonomically classify each representative OTU using BLAST software (Altschul et al., 1991). A BLAST search was conducted using blastn default parameters and the best match retrieved for each sequence was considered an accurate taxonomic classification. After screening and assigning only good quality reads into OTUs on the basis of maximum similarity, there were a total of 71,737, 2,529, 1,693, 1,098 and 27,581 OTUs for bacteria, archaea, fungi and nifH, and acdS bacterial genes, respectively.
Hill-diversity indices of order 1 (Shannon index) and order 2 (Simpson index) were used to characterize α-diversity for the marker genes used in the study (Haegeman et al., 2013). Observations were made using mothur v1.44 (Schloss et al., 2009). Pairwise comparisons among soils from the five locations were conducted by computation of ANOVA-based Tukey’s post-hoc test over the mean values for the Shannon and Simpson indices using the software PAST (Rodriguez-Sanchez et al., 2022). Correlations between hill-diversity indices and soil chemical parameters were investigated by calculation of Pearson’s ρ correlation computed by PAST (Rodriguez-Sanchez et al., 2022).
For β-diversity analyses, PERmutational ANalysis Of Variance (PERMANOVA; Weiss et al., 2017), Principal Coordinates Analysis (PCA; Bian et al., 2017) and cluster analysis, were used as examples of quantitative and qualitative assessments for microbial community differences among the five soil samples. For all of these analyses, the ecological information was corrected to avoid zero values by the Bayesian Multiplicative replacement method using the package zCompositions implemented in R, and then centered-log ratio (CLR) transformed using the package robCompositions implemented in R. For PERMANOVA calculation, a Aitchison distance was used. For cluster analysis, the Unweighted Pair Group Method with Arithmetic mean (UPGMA) algorithm over the Aitchison distance were used for computation.
Distinct or unique phylotypes within each of the five soils were established based on their classification as an indicator species with significant differential abundance as compared to the other soils (Hartman et al., 2018). Phylotypes were classified as an indicator for one or more soils by computation of multilevel pattern analyses based on point biserial generalized correlation coefficient using 10,000 bootstrap permutations and the package indicspecies implemented in R (Hartman et al., 2018). Differential abundance of a phylotype was calculated using the CLR-transformed Kruskal-Wallis tests over the abundance of each phylotype using 128 Dirichlet-Monte Carlo simulations for correction of zero values and CLR transformation through the package ALDEx2 implemented in R.
Trophic modes and functional guilds within the fungal domain were classified using FUNGuild v1.1 with similar methodology reported before (Nguyen et al., 2016; Rodriguez-Sanchez et al., 2022). Briefly, taxonomic classification retrieved from the UNITE database (Köljalg et al., 2013) was used as input for FUNGuild v1.1. β-diversity analyses and determination of characteristic trophic modes were computed similar methods as described above.
Microbial phylotypes were considered dominant in a particular soil sample if their average relative abundance for all replicates was >0.5%. Dominant phylotypes were linked to soil chemical parameters through PCA. For this purpose, the fraction of the zero-corrected, centered log-ratio transformed OTU table was used. Computations were done for individual heavy metals concentrations and other chemical parameters separately. Soil chemical parameters were previously transformed through the LOG(X + 1) equation. Similar calculations were conducted for the fungal domain classified in trophic modes.
The representative sequence for each bacterial OTU generated above was used for metagenome prediction using the PICRUSt2 methodology (Douglas et al., 2020). Briefly, representative sequences were clustered de novo against a phylogenetic tree containing information of 20,000 unique bacterial genomes retrieved from the IMG database (11/08/2017). Sequences were clustered on this tree using HMMER2 software. Next, evaluation of clustering was assessed using EPA-ng software (Barbera et al., 2019), and then GAPPA software (Czech et al., 2020) was used to provide a new tree accounting for newly-added sequences. All parameters for calculation were default. For each representative sequence, both strands were used for calculation and the best result for each was kept for analyses.
Details of soil texture and standard soil chemical properties are provided in Table 1. Briefly, all soils were mildly (pH 7.4–7.8) to moderately (pH 8.5–9.0) alkaline in nature, ranging from pH 7.5 in the Khewra Salt Mine Range (KSMR) to pH 8.5 in Pakka Anna (PA). EC values confirmed that the soils varied in soluble salt concentrations, increasing from the upper toward the lower course of the river. KSMR and Choa Saidan Shah (CSS) fall under the category of moderately saline (EC 4–8), PA was on the low end of very strongly saline (EC > 16), and Dhairki (D) and Mirpur Mathelo (MM) were very strongly saline, according to Abrol et al. (1988). All soils had typical characteristics of saline soils (e.g.: they were low in OM and N, and had high ionic concentrations of K+, Na+, Cl−). Available P was below normal range for this region in CSS, while, PA and D met the recommended criteria, and KSMR and MM had higher than normal values.
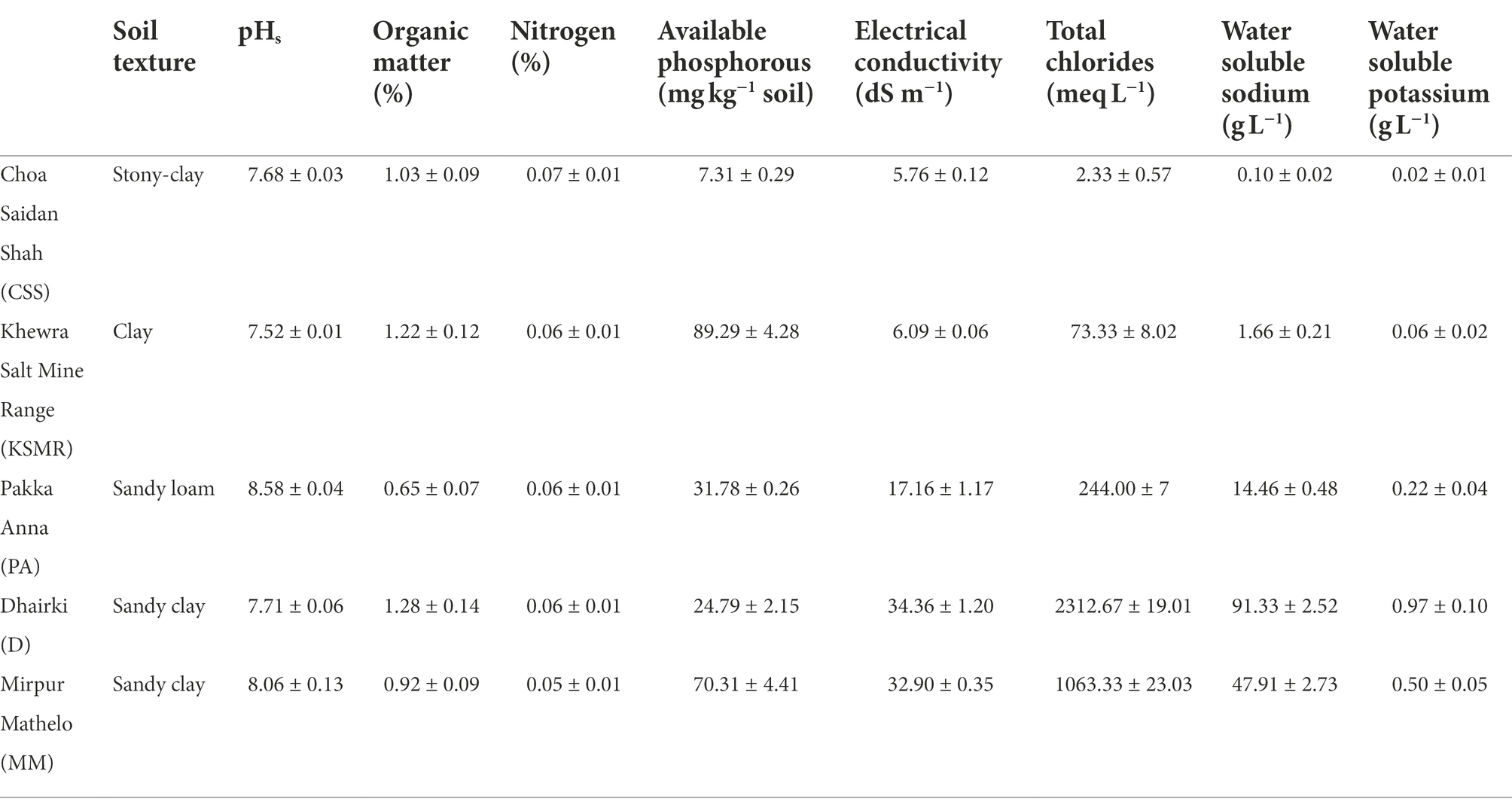
Table 1. Texture and chemical properties of soil samples collected from five farms in the Indus Basin Region, Pakistan.
Results of the pXRF analysis and FAO safety thresholds (Rodríguez-Eugenio et al., 2018) for a select set of heavy metal and metalloid concentrations are provided in Table 2. All soils had concentrations of Ni and Co that were above safety thresholds for agricultural soils. With the exception of KSMR, all soils also had concentrations of Cr that exceeded FAO safety thresholds. Only PA had As and Cu concentrations that were above the FAO safety threshold. All soils had concentrations of Pb, Cd and Zn below safety thresholds.
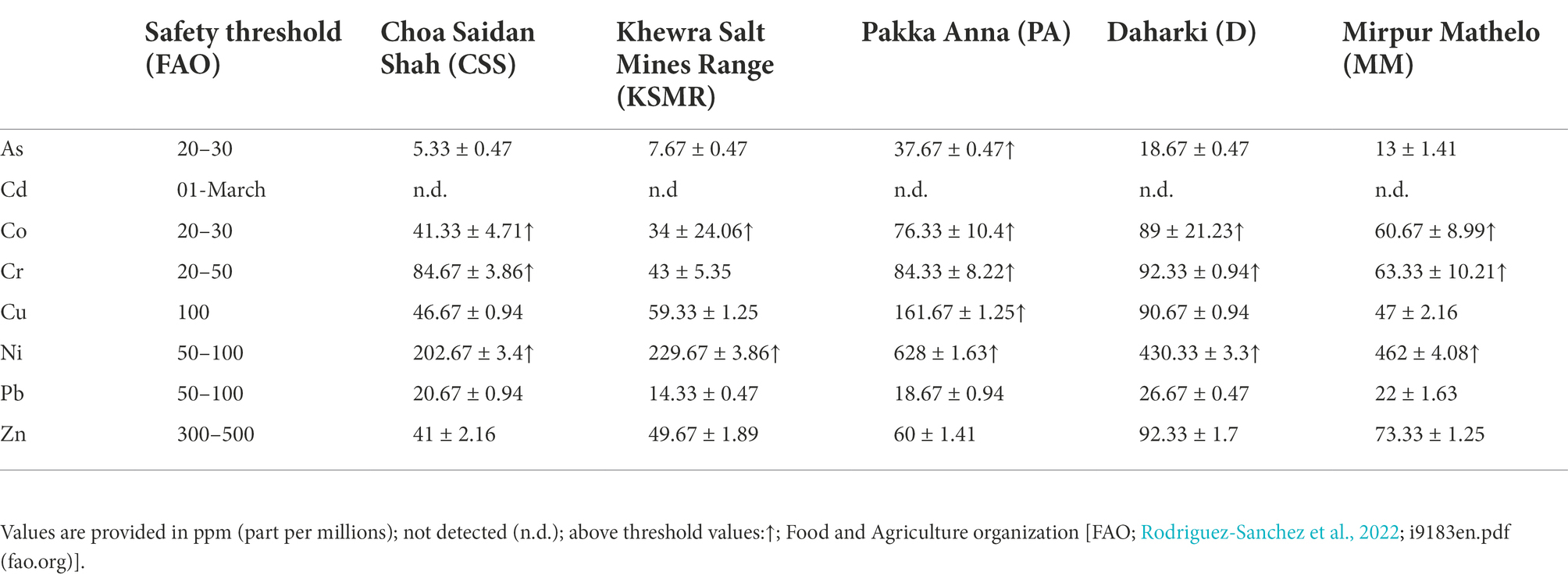
Table 2. Concentrations of heavy metals and metalloids in soil samples collected from five farms in the Indus Basin Region, Pakistan.
The relative abundance of archaeal, bacterial and fungal communities at the phylum levels are reported in Figure 1. The dominant archaeal phylum in all soils was Euryarchaeota (80.10%–95.28%) followed by Crenarchaeota (3.28%–19.23%). Other archaeal phyla such as Haloarchaeota Thermoplasmata and Hadaarchaeota represented <3% of all samples. The dominant bacterial phyla included Actinobacteria (9.22%–27.15%), Proteobacteria (9.83%–18.44%), Planctomycetota (4.78%–13.29%), Acidobacteria (4.96%–14.71%) and Patescibacteria (3.52%–12.12%). Other bacterial phyla such as Chloroflexi or Gemmatimonadota represented less than 10% of the total relative abundance in all of the soils. Fungal communities were dominated by the Ascomycota (54.26%–89.97%) and Chytridiomycota (1.14%–27.03%). The relative abundance of the fungal phylum Blastocladiomycota was present at a substantial level (16.88%) in the CSS soil only. Glomeromycota and Basidiomycota were present at low relative abundances in all soils (1.06%–8.10% and 0.77%–11.00%).
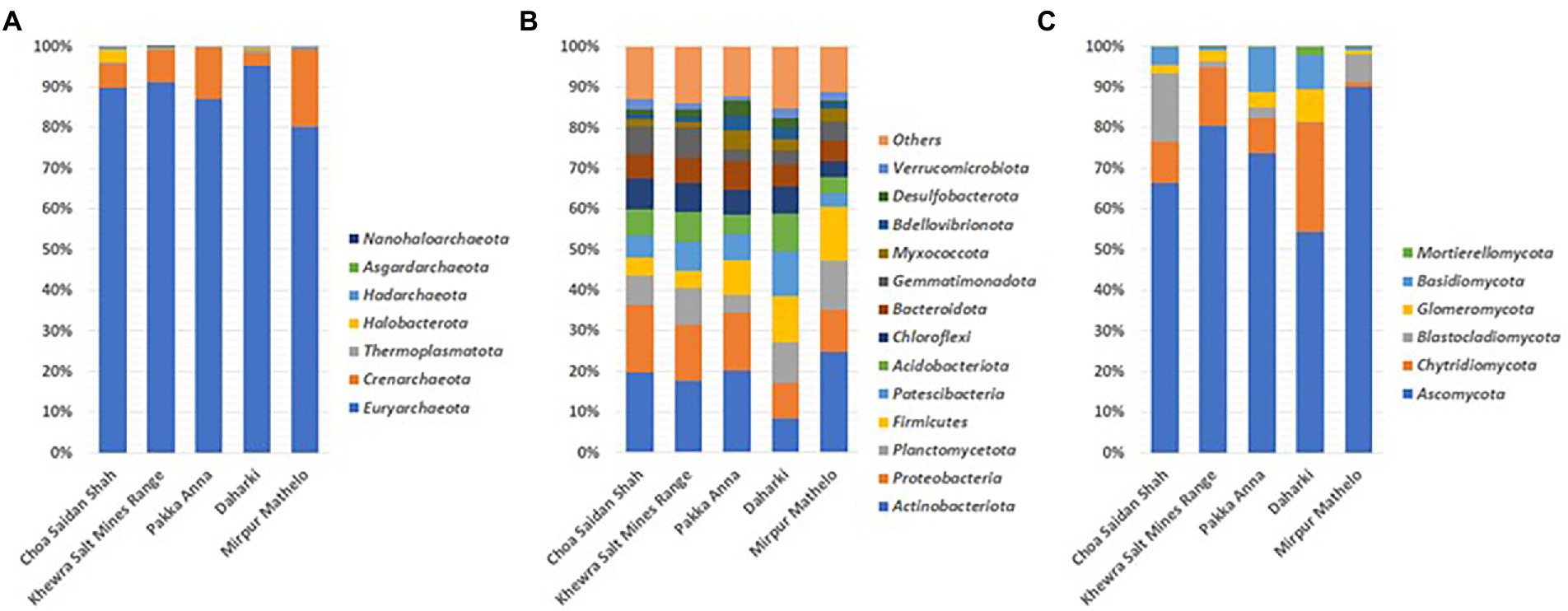
Figure 1. Relative abundance of dominant archaeal (A), bacterial (B), and fungal (C) phyla in five soil samples collected from agricultural fields in the Indus Basin Region, Pakistan.
Results of Shannon (diversity) and Simpson (evenness) α-diversity indices are shown in Figures 2 and Supplementary Figure S1, respectively. There were no significant differences in diversity or evenness between the archaeal and fungal domains (p < 0.05). In contrast, there were differences in diversity and evenness within the bacterial domain and the nifH gene, and in the diversity of the acdS functional gene. In general, CSS had higher diversity for all marker genes, followed by KSMR, PA, MM and D; differences in evenness followed the same order. Pearson’s ρ correlations indicated that lower bacterial, acdS, and nifH diversity were negatively correlated, and evenness was positively correlated, with salinity parameters (EC, Cl−, K+, Na+).
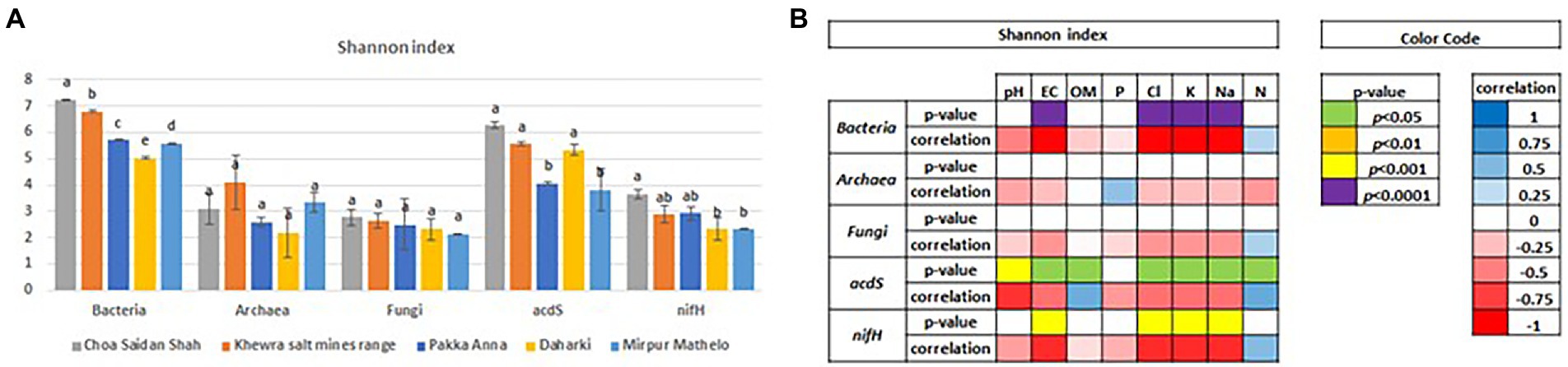
Figure 2. Differences in the alpha diversity of soil microbial communities in five soils collected from agricultural fields within the Indus River Basin, Pakistan, calculated using the Shannon Index (p < 0.05) (A), and Pearson’s ρ correlations between the Shannon index and select soil chemical properties (B). EC, Electric conductivity; OM, total organic matter; P, exchangeable phosphorous; CL, total concentrations of chloride; K, potassium; Na, sodium; N, nitrogen.
Results of the qualitative β-diversity analyses are shown in Figures 3 and Supplementary Figure S2, and results of the quantitative β-diversity analyses are shown in Supplementary Figure S3. CLR-transformed PCA analyses indicated that within the archaeal domain, MM and PA had distinct clusters, there was overlap between D and CSS, and no pattern for KSMR (Supplementary Figure S2). The bacterial domain clearly differentiated into three groups, with KSMR communities clustering apart from those in CSS, and communities from the other three locations clustered very closely together (Figure 3). For fungi, microbial communities within CSS were clearly separated from the rest of the samples, and fungal communities clustered with respect to trophic mode within MM clustered separately from the other soils analyzed (Supplementary Figure S2). Bacterial communities containing the acdS gene had a similar pattern of clustering to the bacterial domain, while those containing the nifH functional gene clustered in samples from CSS and MM, and communities in the other three soils overlapped (Figure 3). However, it is important to note that none of these were significantly different at the 95% confidence interval when evaluated using CLR-transformed PERMANOVA analyses (Supplementary Figure S3), so the results of the β-diversity analyses must be interpreted with caution. Cluster analyses showed similar patterns to those of PCA and PERMANOVA (Supplementary Figure S4).
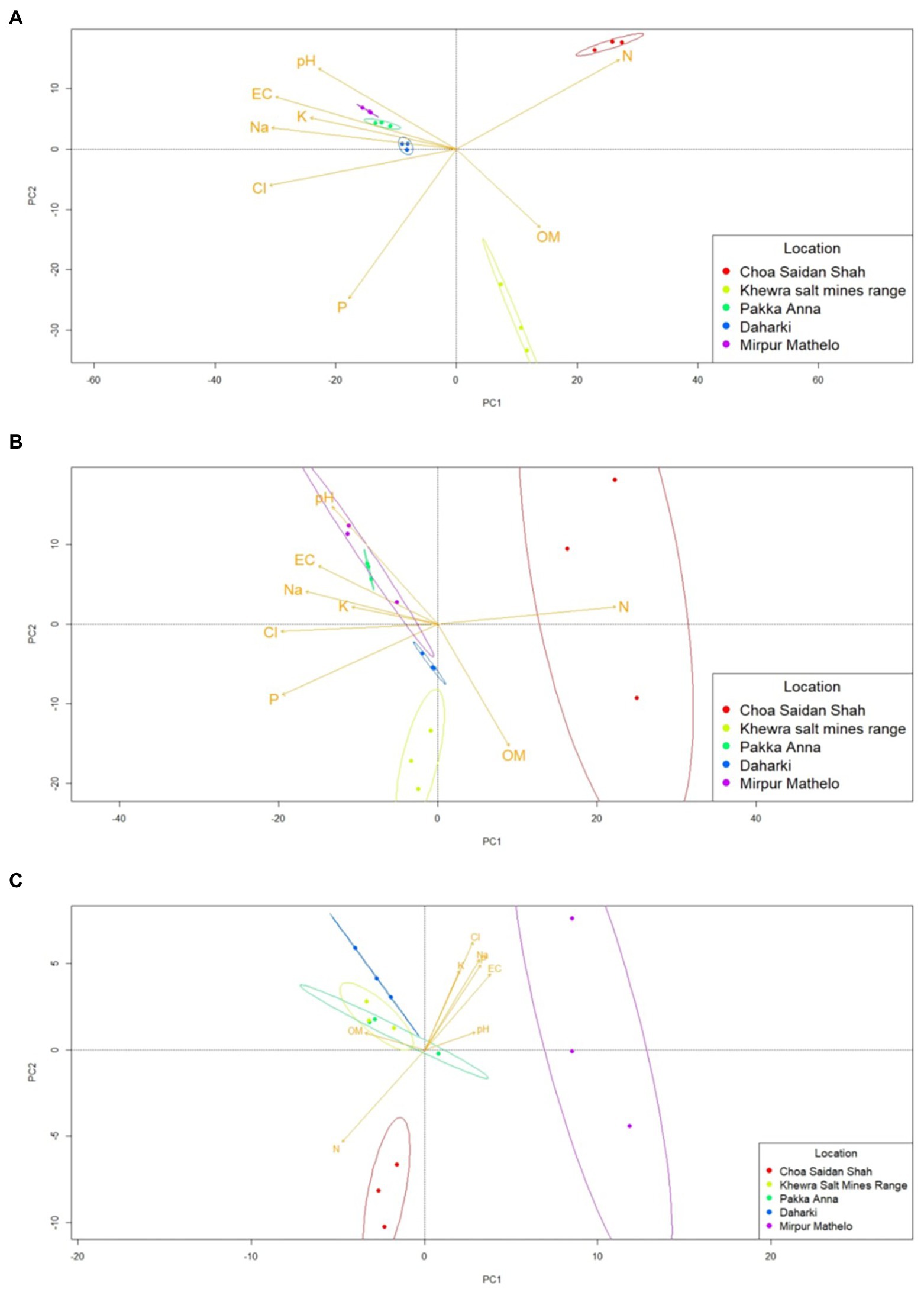
Figure 3. Differences in beta-diversity of soil microbial communities in five soils collected from agricultural fields within the Indus River Basin, Pakistan and correlations with chemical properties quantified using principal coordinates analyses (PCA). (A) bacteria; (B) nifH-containing bacteria; (C) acdS-containing bacteria; EC, electric conductivity; OM, organic matter; P, exchangeable phosphorous; CL, total concentrations of chloride; K, potassium; Na, sodium; N, nitrogen.
Within the bacterial domain, there were many dominant phylotypes that were distinct and therefore could be considered an indicator species within the five soils (Figure 4B; Supplementary Figure S4B). For example, within CSS and KSMR, Bryobacter and Gemmatimonas were distinct; within D, Fusibacter and members of Isospheraceae, Fibrobacteraceae and Acidobacteridota were distinct; within MM, Parcubacteria, Nocardiodaceae, Limnochordia and Polyangiales phylotypes were distinct; and finally, within PA, Saccharimonadales, Actinobacteria, Sphingobacterales and Acidimicrobiia members could be considered distinct or unique for the soils. There were fewer differences among the archaeal and fungal domains (Figures 4A,C; Supplementary Figures S4A,C). For example, within the archaea, individual Methanocaldococcus phylotypes were distinct among the soils, however, only CSS and KSMR had Crenarchaeota members as unique phylotypes. For fungi, the dominant phylotype Curvularia was unique to CSS and KSMR, one Aspergillus phylotype was unique for PA, and Ceratocystis adiposa, Microascus and a Xylariaceae member were unique in MM. No fungal trophic modes were characteristic of any of the sites analyzed (data not shown).
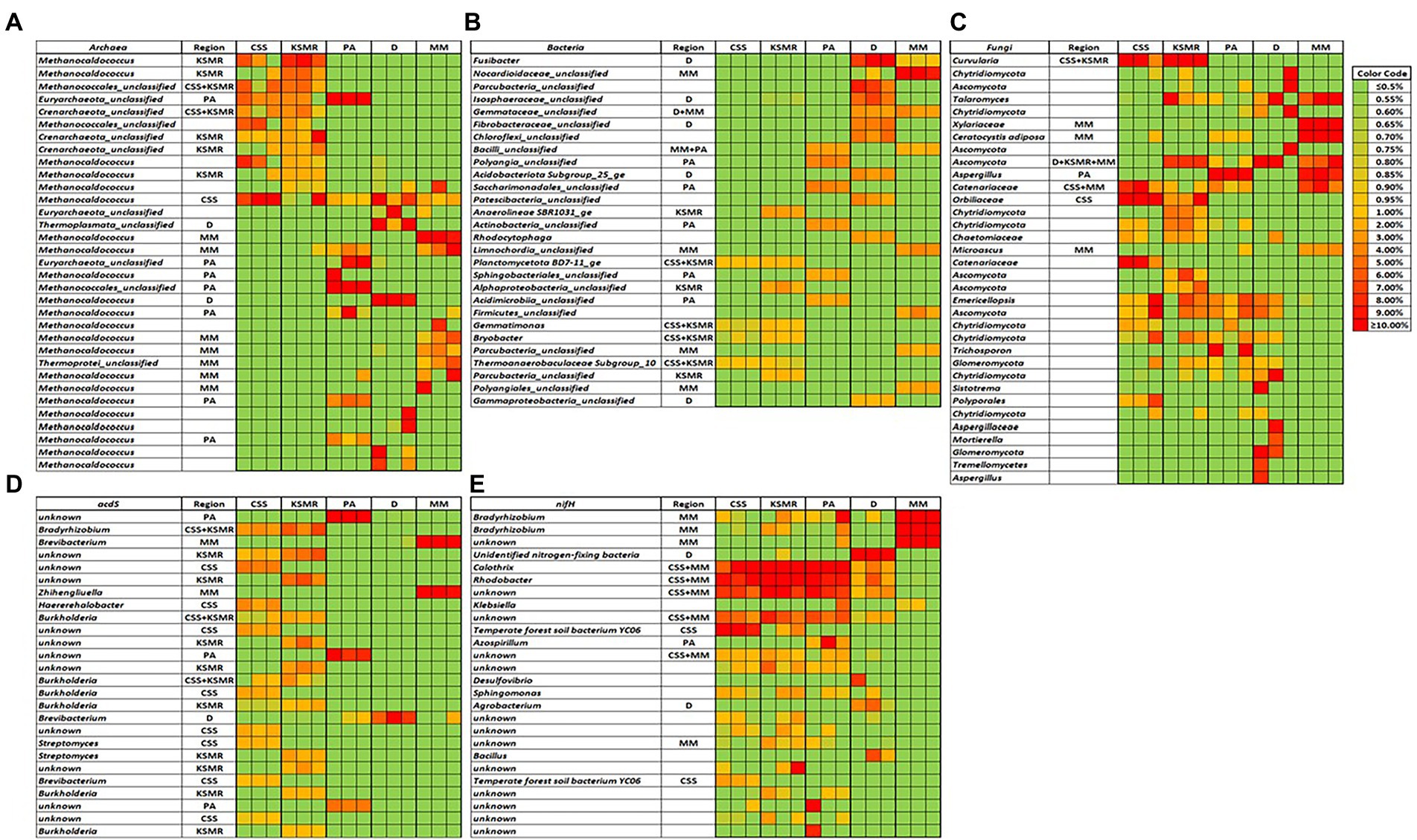
Figure 4. Heat maps showing the dominant soil archaeal (A), bacterial (B), and fungal (C), phylotypes, and bacteria with acdS (D), and nifH genes (E), in five soils collected from agricultural fields in the Indus River Basin, Pakistan, and whether they can be considered as indicator species for a particular region. CCS, Choa Saidan Shah; KSMR, Khewra salt mines range; PA, Pakka Anna; D, Daharki; MM, Mirpur Mathelo.
Interestingly, all of the five soils had acdS gene-containing microorganisms that were distinct (Figure 4D). In particular, KSMR hosted several unique Burkholderia Streptomyces, and Bradyrhizobium phylotypes; Bradyrhizobium, Haererehalobacter, Burkholderia, Streptomyces and Brevibacterium were unique in CSS; Brevibacterium and Zhihengliuella were unique in MM; Brevibacterium was unique in D; and, several unknown phylogenies were unique in PA.
For the nifH gene, surprisingly there were no dominant phylotypes that could be considered characteristic for any of the five soils, however, CSS, KSMR and PA had several unique indicator species among the dominant OTUs such as Calothrix or Rhodobacter (Figure 4E). Two phylotypes of Bradyrhizobium were unique in MM, and Azospirillum and Agrobacterium were unique in PA and D, respectively.
A PCA plot linking dominant bacterial phylotypes with soil chemical parameters indicated that there were three distinct clusters (Supplementary Figure S5B1). In one of them, representatives of Fusibacter, Parcubacteria, Patescibacteria, and Rhodocytophaga, among others, were positively correlated with salinity parameters (EC, K+, Na+, Cl−) and OM, and negatively correlated with pH. In contrast, in another cluster, representatives of Bacilli, Sacharimonadales, Parcubacteria, Firmicutes, and Polyangiales were negatively correlated with ion concentrations (K+, Na+,Cl−) and OM, and positively correlated with pH and N. Finally, in the third cluster, ion concentrations (K+, Na+, Cl−) and pH were positively correlated, and SOM and N were negatively correlated, with phylotypes belonging to Gemmatimonas, Bryobacter, Parcubacteria, Thermoanaerobaculaceae, and Anaerolineae, among others.
For the case of fungi, similar clustering of dominant phylotypes into three broad groups could be observed (Supplementary Figure S5C1). Those positively correlated with EC and SOM but negatively with pH were classified as Mortierella, Aspergillus, Trichosporon, Sistotrema, members of Chytridiomycota, Glomeromycota, and Tremllomycetes, among others. Those positively correlated with SOM and N and negatively correlated with EC were classified as Talaromyces, Ceratocystis, Aspergillus and Microascus. The cluster positively correlated with soil pH and EC and negatively correlated with SOM and N included members of Curvularia, Ascomycota, Chaetomiaceae, Orbiliaceae and Chytridiomycota, among others.
For trophic modes, two groups formed with respect to soil parameters (Supplementary Figure S5). One group was positively correlated with N and OM (trophic modes of pathotroph, symbiotroph, and pathotroph-symbiotroph), and the other were positively correlated with pH, P and concentrations of ionic concentrations of Cl, Na, and K (saprotrophs, saprotroph-symbiotroph, pathogen-saprotroph, and pathogen-saptrotroph-symbiotroph; Supplementary Figure S5F1). Pathotroph, symbiotroph, and pathotroph-symbiotroph were also positively correlated with Cu and As, whereas the other trophic modes were positively correlated with Cr, Co, Ni, Pb, and Zn (Supplementary Figure S5F2).
Within the archaea, dominant phylotypes related to Crenarchaeota were positively correlated with soil pH and EC, and negatively with SOM and N (Supplementary Figure S5A1).
For bacteria containing the acdS gene, Zhihengliuella was positively correlated with higher soil pH and EC, but negatively correlated with SOM and N, whereas Bradyrhizobium, Burkholderia, Haererehalobacter and Streptomyces were positively correlated with SOM and N, and negatively correlated with soil pH and EC (Supplementary Figure S5D1).
Finally, for bacteria containing the nifH gene, Bradyrhizobium, Azosporillum, Klebsiella and Desulfovibrio were positively correlated with SOM and N and negatively with salinity and pH, while Calothrix, Rhodobacter and Sphingomonas were positively correlated with salinity parameters and pH, and negatively correlated with SOM and N (Supplementary Figure S5E1).
Analyses to evaluate correlations with select heavy metal and metalloid concentrations indicated that the dominant bacterial communities also separated communities into three distinct clusters (Supplementary Figure S5B2). One cluster was positively correlated with Cr, Pb, Zn, and Co, and contained Fusibacter, a Parcubacteria member, Rhodocytophaga, and a Patescibacteria member, among others. Another group was positively correlated with As, Cu, and Ni, and included Gemmatimonas, Bryobacter, and a Parcubacteria member, among others. Finally, the third cluster was negatively correlated with all soil metal and metalloid concentrations, and included Polyangia and Parcubacteria members, among others.
Within the archaea, many Methanocaldococcus-affiliated and Crenarchaeota-affiliated phylotypes were positively correlated with concentrations of As, Ni and Cu (Supplementary Figure S5A1). Another cluster was negatively correlated with the concentrations of all metals and metalloids, and included Methanocaldococcus and Thermoprotei.
For fungi, several phylotypes were positively correlated with all of the heavy metals and metalloids such as Aspergillus, Sistotrema, and a few Ascomycota (Supplementary Figure S5C1). Another cluster contained fungal members that were negatively correlated with all heavy metals and metalloids, such as Emericellopsis or Trichosporon. Several phylotypes were positively correlated with Ni, As and Cu, such as Curvularia or Polyporales. Finally, another cluster contained fungal phylotypes that were positively correlated only with Pb, Zn, Cr, and Co, such as an Aspergillus and Ceratocystis adiposa.
Bacterial phylotypes containing the acdS gene clustered into three distinct groups. One cluster was positively correlated with all heavy metals and metalloids and included Zhihengliuella, Brevibacterium, Burkholderia and Streptomyces (Supplementary Figure S5D1). Other phylotypes classified as Burkholderia and Streptomyces were positively correlated only with Ni, As and Cu.
Finally, bacterial phylotypes containing the nifH gene clustered into two groups. One was positively correlated with Ni, As and Cu, and included Sphingomonas, Rhodobacter, Calothrix, among others (Supplementary Figure S5E2). The other group was positively correlated with concentrations of Pb, Zn, Cr and Co, and included Bradyrhizobium, Klebsiella, Azospirillum, Desulfovibrio, and Agrobacterium, among others.
Qualitative β-diversity metrics comparing the five agricultural soils indicated that the predicted metagenomes of the soil microbial communities clustered into 3 distinct groups: CSS and KSMR; PA and D; and MM (Figure 5). CSS and KSMR were positively correlated with OM and N, while PA and D were positively correlated with pH, P and salinity parameters. There were also significant differences in the relative abundance of some microbial genes with potential to help plants obtain nutrients and withstand abiotic stress among the five soils (Supplementary Table S2). For example, the relative abundance of microbes with genes for synthesis of ACC-deaminase (acdS) and nitrite reductase (nirK) were greater in CSS and D than the other soils. Differences in the potential for microbial communities to solubilize P were also apparent, with D having a greater relative abundance of microbes with acid phosphatase genes (appA and phoN), and CSS having a greater relative abundance of those with alkaline phosphatase genes (phoA, phoB). Microbes in D also had a greater potential relative abundance of genes for salicylate synthase (mbtI, irp9, ybtS), while PA had a greater relative abundance of the gene for isochorismate pyruvate lyase (pchB), and MM had a greater relative abundance of the gene for ferric uptake regulator (FUR). Finally, all soils except CSS contained microbes with a high relative abundance of the gene for indole-3-glycerol phosphate synthase (IGPS).
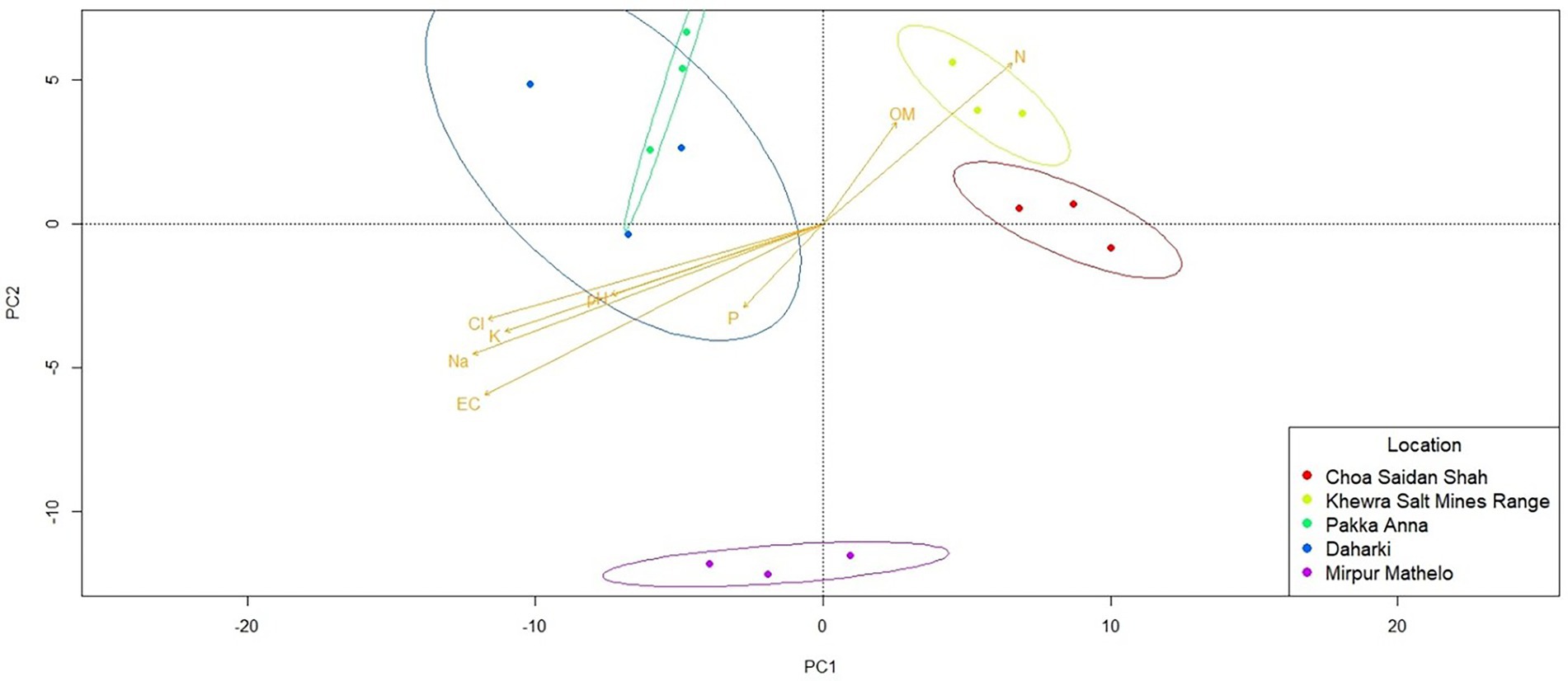
Figure 5. Differences in the predicted metagenome of soil microbial communities in five soils collected from agricultural fields within the Indus River Basin, Pakistan, and correlations with chemical properties quantified using principal coordinates analyses (PCA). EC, Electric conductivity; OM, total organic matter; P, exchangeable phosphorous; CL, total concentrations of chloride; K, potassium; Na, sodium; N, nitrogen.
Learning more about how soil salinity and elevated concentrations of HM can alter the composition and functional potential of soil microbial communities has potential to help aid in the development of new practices that can help farmers overcome these challenges. As predicted, agricultural soils in the Indus River Basin in Pakistan provided an ideal location to study these dynamics. Farm fields ranged from moderately saline upstream (CSS, KSMR), to very strongly saline downstream (PA, D, MM), but did not differ in other critical soil parameters such as pH and OM (Table 1), which are generally the strongest factors driving soil microbial community structure (Rodriguez-Sanchez et al., 2022). At the same time, concentrations of Ni, Co, and Cr were all greater at sites downstream relative to upstream (Table 2). Elevated concentrations of soluble salts in soils under irrigation in arable regions are common, and likely to accumulate at higher concentrations downstream due to deposition within irrigation networks (Syed et al., 2021). The exact source of HM in this region are unclear, though it could be due to soil factors such as soil parent materials, as well effluent from salt and coal mines, nearby chemical industries, and agricultural and municipal wastes (Ashraf et al., 2021). Regardless of the cause, the combined presence of high concentrations of soluble salts and HM is of concern given that both can harm crop plants, and salinity can increase bioavailability of HM like Cd and Pb (Acosta et al., 2011). Fortunately, others have reported opposite trends for HM like Cr, Ni, and As (Han J. et al., 2021). While we cannot predict how HM will behave in these soils, we can conclude that farms in the Indus Basin are at high risk for salinity and HM, so farmers should take steps to address these stress factors.
Identifying dominant microbial taxa that thrive within these agroecosystems can provide important clues about potential threats, as well as insights on how to better manage these systems. Microbial communities in all soils were dominated by taxa with high potential capacity to withstand abiotic stress (Figure 2), which is consistent with studies in similar ecosystems. For example, Rodriguez-Sanchez et al. (2022) also found Ascomycota to dominate soil fungal communities in an arid agricultural region subject to salt and HM stress in Peru. Dominance of the Ascomycota is these systems is likely due to the fact that these microbes generally contain a high number of genes and specific traits that make them more competitive in harsh environments (Egidi et al., 2019). Members of the Ascomycota play important roles in soil as saprotrophs, but others can cause plant diseases (Rodriguez-Sanchez et al., 2022). Acidobacteriota are commonly reported as one of the most abundant groups of bacteria found within saline soils, and thus, some have proposed that they can serve as bio-indicators for salt-tolerant communities (de Leon-Lorenzana et al., 2017; Yue et al., 2020; Cao et al., 2021). Microbes within the Acidobacteriota are difficult to cultivate and study under laboratory conditions, so their exact ecosystem functions are still unclear, though they are expected to play important roles in biogeochemical cycles and plant growth promotion (Kalam et al., 2020). Mukhtar et al. (2018) previously found Actinobacteria to be highly abundant in the rhizospheres of wheat plants collected in PA. Actinomycetes are well-known for their capacity to tolerate conditions of high soil pH, water (Pepper et al., 2015), and salinity stress, and some have been demonstrated to help plants withstand salt stress (Djebaili et al., 2020). Many genera within the Actinomycetes, such as Streptomycetes, are also noted for their capacity to both cause as well as prevent plant disease (Schrey and Tarkka, 2008). Finally, Euryarchaeota are widespread in terrestrial ecosystems worldwide, including many extreme environments (Hu et al., 2013). Many of the metabolic capabilities and potential functional roles of Euryarchaeota in soils remain unresolved, though they are expected to play critical roles in biogeochemical cycles (Hu et al., 2013). Consequently, future efforts to isolate microbes within these taxa and elucidate their specific functional roles are recommended as a means to promote crop growth and resilience against biotic and abiotic stress.
Determining how individual taxa respond to salinity and HM concentrations is also important for learning how to better manage these agroecosystems. Like other studies (Khan et al., 2020; Wang S. et al., 2021; Chen et al., 2022; Jiang et al., 2022) salinity had a strong influence on bacterial, but not fungal diversity (Figure 3; Supplementary Figure S1) in this study. This may be related to the fact that fungi are generally more resistant to salinity than bacteria (Rath et al., 2016, 2019). Some have theorized that this could be due, at least in part, to the capacity of fungi to retain Na+ within their hyphae, thereby preventing translocation to more sensitive tissues (Zhao et al., 2019). Unlike salinity, elevated concentrations of HM often influence both bacterial and fungal diversity, but in different directions. For example, Stefanowicz et al. (2008) observed a decrease in bacterial and increase in fungal diversity in response to HM along a pollution transect, indicating that HM may select for fungal taxa with unique capabilities to withstand HM stress. Rodriguez-Sanchez et al. (2022) noted that higher Co and Ni concentrations in particular, appeared to be important drivers of fungal community structure in Peru. In the present study, fungal diversity did not change in response to HM, but we did identify some phylotypes such as Aspergillus and Curvularia, that were positively correlated with increasing concentrations of Co and Ni (Supplementary Figure S5C2). Aspergillus and Curvularia have been noted for their strong capacity to withstand HM stress, which could be related to their capacity to counteract HM toxicity via efflux pumps, biosorption, and production of secondary metabolites (Torres-Cruz et al., 2018; Imran et al., 2020). Aspergillus play important roles in soil as saprophytes (Nayak et al., 2020). Curvularia have been noted to act as pathogens in some plant species, but promote plant growth in others via PGP activities such as P solubilization (Priyadharsini and Muthukumar, 2017). Finally, the absence of an effect of salinity or HM on α-diversity within the archaeal community was not surprising given the strong capacity of microbes within this to tolerate extreme environments, including high salinity (Matarredona et al., 2020). Nevertheless, the fact that there were potential differences in β-diversity indicate that some phylotypes, such as some of the Methanoncaldococcus, could be more sensitive to these stress factors.
A diverse set of potentially bacterial phylotypes were positively correlated with increasing concentrations of soluble salts and HM in the soils downstream (Supplementary Figure S5), and thus could be targets for future isolation and/or soil management efforts. Halotolerant Fusibacter spp. are noted for their capacity to tolerate highly saline (Serrano et al., 2017; Selivanova et al., 2018), and HM enriched environments (Serrano et al., 2017; Meng et al., 2021; Ghosh Roy et al., 2022). Fusibacter spp. are thought to contribute to many important agroecosystem services including decomposition of SOM (Qiu et al., 2021), heterotrophic denitrification (Han F. et al., 2021), and arsenate/sulfate reduction (Wang and Huang, 2021). Nocardioidaceae have previously been found to be among the most dominant phylotypes in wheat rhizospheres grown in Islamabad and Azad Jammu and Kashmir regions of Pakistan (Latif et al., 2020), and noted for their potential PGP capabilities (Yadav et al., 2017). Fibrobacteraceae have also been well-documented in many wheat production systems (Prudence et al., 2021), but while they are known to efficiently utilize wheat root exudates, their exact role in plant–soil systems is not yet clear. Limnochordia and Polyangiales have been linked with organic management practices commonly used to improve soil health in saline soils (Shang et al., 2020; Yu et al., 2020). Polyangiales are suspected to play an important role in cellulose degradation and carbon cycles (Braga et al., 2021). Finally, Isospheraceae have been isolated from extreme environments (Hewelke et al., 2020; Kaushik et al., 2020; Morrow et al., 2020), but their functional role in soils is still unclear.
A number of other bacterial and archaeal phylotypes such as Parcubacteria, Patescibacteria, and Crenarchaeota were also positively correlated with salinity and HM (Supplementary Figures S5A,B), regardless of their association with a particular soil, indicating that they could be broadly adapted to soils in this region. Parcubacteria and Patescibacteria are members of the novel candidate phyla radiation (CPR), which tend to have small-genomes, small cell size, and limited biosynthetic capabilities relative to other bacterial groups (Moreira et al., 2021). They are suspected to act as predatory bacteria that prey on other taxa, thereby helping to keep bacterial populations in check (Nelson and Stegen, 2015; Conte et al., 2018; Mora-Ruiz et al., 2018). They have also been shown to inhabit a diverse range of environments including plant rhizospheres, and have diverse functional capabilities for carbon (C) and N cycling (Danczak et al., 2017; Selivanova et al., 2018; Nicolas et al., 2021). While these taxa have not previously been noted to be adapted to saline environment, recent studies indicate that they are influenced by HM (Huang et al., 2021; Zhang et al., 2021). Crenarchaeota are widely recognized as important ammonium oxidizers in soil ecosystems (Treusch et al., 2005; Nicol and Schleper, 2006; Brochier-Armanet et al., 2008). Crenarchaeota have been reported in saline soils (Ren et al., 2018), and soils with high concentrations of HM (Li et al., 2020), though others have found them to be negatively correlated with salinity (Oueriaghli et al., 2013). This could help explain why some Crenarchaeota members were classified as characteristic phylotypes in CCS and KSMR.
Several fungal phylotypes including Mortierella, Sistotrema, Trichosporon, and Chaetomiaceae, were also positively correlated with salinity parameters, regardless of their connections with individual soils (Supplementary Figure S5B). The presence of Mortierella spp. well documented in saline soils and several studies have indicated that they have potential to promote plant growth in these systems (Bian et al., 2020; Ozimek and Hanaka, 2020; Sun et al., 2021). Similarly, Sistotrema spp. (Redou et al., 2015; Grum-Grzhimaylo et al., 2018), Trichosporon spp. (Liu et al., 2019; Sun et al., 2021), and Chaetomiaceae (Castelan-Sanchez et al., 2019), have all been reported to tolerate salinity stress and have potential PGP activities. Since Sistotrema and Trichosporon were also positively correlated with SOM in the present study, future studies should consider investigating whether methods that can increase SOM, such as growing cover crops or amending soils with composts (Hoagland et al., 2008; Rudisill et al., 2015; Reeve et al., 2016; Richardville et al., 2022), can enhance their populations in these soils.
Unfortunately, not all of the phylotypes with potential PGP activities identified in this study were positively correlated with salinity and/or HM. For example, Gemmatimonas and Curvularia, were more abundant in fields located upstream where salinity and HM levels were much lower (Figure 5). Yue et al. (2020) suggested that Gemmatimonas can play important roles in helping plants tolerate saline conditions. Curvularia has also been shown to help reduce salt stress in plants (Pan et al., 2018), possibly due to the capacity of these fungi to produce osmoprotectants (Bengyella et al., 2019). However, it is important to note that some Curvularia species have also been reported to cause diseases in crops like wheat (Sultana et al., 2019; Bozoğlu et al., 2022; Tan et al., 2022), consequently, more focused studies on the composition and role of individual species of Curvularia within these soils are recommended. At the same time, identifying ways to promote beneficial populations of Curvularia and Gemmatimonas also could be helpful. For example, Li et al. (2021) observed greater populations of Gemmatimonas in saline soils amended with organic fertilizers, and this was linked to greater abiotic stress resistance in crop plants.
All soils contained bacterial communities harboring nifH and acdS genes, and distribution patterns suggest that individual taxa may be better adapted to different salinity and HM concentrations. For example, some Calothrix phylotypes were positively correlated with both salinity and HM, as well as low N (Supplementary Figure S5B1). Jiang et al. (2019) also noted positive correlations between Calothrix and these soil properties, and suggested that these cyanobacteria likely play an important role in suppling N in habitats with low N fertility. Others have also noted that Sphingomonas spp. are highly tolerant of soluble salts and HM, and can promote plant growth under these stressful conditions (Asaf et al., 2020). Zhihengliuella have also been positively correlated with salinity (Chen et al., 2010; Aslam and Ali, 2018; Hajiabadi et al., 2021), and shown to promote plant growth via their capacity to fix atmospheric N, produce ACC deaminase and IAA, and mineralize N into plant available forms (Orhan and Demirci, 2020). Many Burkholderia spp. have been shown to have potential PGP activities (Zhao et al., 2014; Jung et al., 2018; Sarkar et al., 2018), and promote plant growth under saline conditions (Sarkar et al., 2018). In this study, Burkholderia were positively correlated with HM which is consistent with other studies (Li et al., 2020), but negatively correlated with salinity (Supplementary Figure S5B). Finally, two bacterial phylotypes, Klebsiella and Azospirillum, which are well known for their PGP activity, were negatively correlated with salinity in this study (Supplementary Figure S5E2). Others have noted that nifH activity in isolates from these genera can be negatively correlated with increasing salinity levels (Tripathi et al., 1998), though halotolerant Klebsiella strains linked with high N uptake in plants grown under saline conditions have also been isolated (Sharma et al., 2016). Similarly, a salt-resistant Klebsiella strain isolated from a wheat rhizosphere in the Bahawalpur district of Pakistan was shown to enhance the growth of this important crop under high salinity conditions via the production of exopolysaccharides (EPS) and indole acetic acid (IAA), and solubilization of Zn and P (Latif et al., 2022).
Predicting metabolic profiles in metagenomic studies using bioinformatic software such as PICRUSt2 (Douglas et al., 2020) is a robust and comparatively efficient technology in the field of microbial ecology, with strong potential to help infer implications of stress factors on soil microbial functions (Kori et al., 2019). When comparing soils in study, we found that functional capabilities within D and PA were positively correlated with salinity and HM (Figure 5), indicating that microbes within these soils may have developed unique functional capabilities to deal with these stress factors. In contrast, CSS and KSMR were positively correlated with SOM and N. The reason for these differences is unclear, though since salinity (Rath and Rousk, 2015) and HM (Aponte et al., 2020) can play significant roles in activities related to SOM decomposition and other elemental cycles, it is possible that increasing concentrations of salinity and HM are altering the capacity for microbes to influence these processes. For example, Wang et al. (2018) observed a negative correlation between salinity and denitrifying bacteria harboring the nirK gene (Supplementary Table S2). It is unclear why soil D also was enriched in nirK, though it is interesting to note that this soil also had greater abundance of microbes with the gene for pyrroloquinoline-quinone synthase (pqqC), and the highest levels of SOM (Table 1). The pqqC gene plays a critical role in SOM cycling, and not surprisingly, is often linked with greater SOC and total N levels (Shi et al., 2022).
Some of the soils in the present study were also linked with several genes associated with phosphorous (P) cycling, which is consistent with other studies demonstrating that many salt-tolerant bacteria with PGP capabilities harbor P solubilizing genes (Dey et al., 2021). CSS in particular, was linked with greater relative abundance of bacteria with alkaline phosphatases (phoA, phoB; Supplementary Table S2), which is likely related to the very low P level in this soil. In contrast, microbes with several acid-phosphate genes (phoN, aphA, appA) were enriched in soil D (Supplementary Table S2). It is unclear why these would be enriched in this soil given that it also had high pH (Supplementary Table S2), but these findings provide further evidence that this soil is unique and deserves additional attention in future studies. Unfortunately, increasing salinity levels are often associated with decreased activity of the enzymes associated with P solubilization (Cortes-Lorenzo et al., 2012), so future efforts to isolate salt and HM-tolerant phylotypes that are capable of carrying out this critical agroecosystem activity in the presence of these extreme conditions are needed.
Interestingly, several genes associated with siderophore production were distinct in some of the soils (Supplementary Table S2). For example, microbes with the ferric uptake regulator (FUR) gene were enriched in MM. The capacity for bacteria to leverage this approach to sequester iron under oxidative stress is an important adaptive trait in stressful environments (Duangurai et al., 2018; Wang H. et al., 2021). Microbes with genes associated with salicylate synthetase (mbtI, Irp9, ybtS) were enriched in D, and pchB, which is involved in salicylic acid synthesis, was enriched in PA (Supplementary Table S2). The capacity for microbes to synthesize secondary metabolites involved in salicylic acid production is linked with ferric-chelating siderophores, and can play a critical role in help microbes withstand biotic and abiotic stress (Gehring et al., 1998; Pelludat et al., 2003; Shelton and Lamb, 2018; Mishra and Baek, 2021). In addition, bacterial production of these compounds can help promote plant growth under iron-deficient environments (Mishra and Baek, 2021), and is thought to play a critical role in helping prevent plant disease (Köhl et al., 2019). Consequently, future efforts to isolate salt and HM-tolerant microbes with the capacity to produce these compounds could be very helpful in developing future inoculants to promote crop growth.
Finally, the capacity for microbes to thrive in stressful environments and promote plant growth will require isolation of microbes with high stress tolerance. Interestingly, all soils except CSS were enriched in bacteria with the gene for indole-3-glycerol phosphate synthase (IGPS), which is associated with tryptophan-independent indole acetic acid (IAA) production (Supplementary Table S2). Upregulation of IGPS under salinity stress has been observed in many studies (Ahmad et al., 2020; Rangseekaew et al., 2022), and is thought to be important in helping bacteria tolerate these extreme conditions. IAA is also an important plant hormone regulating root growth, and bacterial production of this compound has long been associated with potential PGP activity (Glick, 2012). Similarly, ACC-deaminase production, which is controlled by the gene acdS, is a critical PGP trait. D had a greater potential relative abundance of microbes with this gene (Supplementary Table S2), so again, this soil appears to be a particularly valuable soil for future efforts to isolate soil bacteria with PGP capabilities.
Unfortunately, using the FUNGuild tool (Nguyen et al., 2016), we noted a negative correlation between salinity and some HM on fungi with symbiotrophic activity, and positive correlation between pathotrophic fungi and N and some HM within the soils of this region (Supplementary Figure S5F1). Relationships between fungi with these trophic modes and soil chemical properties have been mixed in other studies (Lin et al., 2022; Zhu et al., 2022), indicating that other site-specific factors are likely to play a role in mediating these dynamics and further research is needed to elucidate the mechanisms responsible. Fortunately, fungi with saprotrophic capabilities were positively correlated with salinity and some metals, providing further evidence of the adaptability of this important group of fungi in these soils.
Agricultural fields within the Indus Basin are clearly at risk for salinity and HM stress, and farmers need new strategies to protect their crops and reduce human health risks. Results of this study demonstrate that several farm fields within this region contain indigenous microbial flora with unique capabilities to survive under these harsh conditions, and some may also be able to help promote plant growth. Overall, fields in location D appear to contain microbes with the most unique capabilities, and thus would provide a good starting point to begin to try and isolate unique taxa for future development as PGP inoculants. At the same time, we noted the dominance of microbes that could be a potential threat in these systems, as well as situations where microbes with well-known PGP capabilities were negatively impacted by salt and HM concentrations. Thus, future efforts to isolate and determine the exact functional roles of these microbes is recommended, along with strategies on how to better promote or prevent their abundance using different soil and crop management practices.
The data presented in the study are deposited in the GenBank SRA repository, accession number PRJNA869745.
MM: conceptualization, funding acquisition, data curation, formal analysis, software, investigation, writing—original draft, and review and editing. AR-S: conceptualization, data curation, formal analysis, software, validation, writing—original draft, and review and editing. AI: conceptualization, supervision, and review and editing. FM: conceptualization, supervision, validation, and review and editing. LH: conceptualization, funding acquisition, project administration, supervision, validation, and review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the International Research Support Initiative Program (IRSIP) fellowship to Ph.D. scholar MM, funded by Higher Education Commission (HEC), Pakistan IRSIP Fellowship No. (PIN) IRSIP 43, BMS 97.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1020175/full#supplementary-material
Abdelrazek, S., Simon, P., Colley, M., Mengiste, T., and Hoagland, L. (2020). Crop management system and carrot genotype affect endophyte composition and Alternaria dauci suppression. PLoS One 15:e0233783. doi: 10.1371/journal.pone.0233783
Abrol, I., Yadav, J. S. P., and Massoud, F. (1988). Salt-affected soils and their management. Rome: Food and Agriculture Organisation.
Acosta, J. A., Jansen, B., Kalbitz, K., Faz, A., and Martinez-Martinez, S. (2011). Salinity increases mobility of heavy metals in soils. Chemosphere 85, 1318–1324. doi: 10.1016/j.chemosphere.2011.07.046
Ahmad, E., Sharma, S. K., and Sharma, P. K. (2020). Deciphering operation of tryptophan-independent pathway in high indole-3-acetic acid (IAA) producing micrococcus aloeverae DCB-20. FEMS Microbiol. Lett. 367, 1–9. doi: 10.1093/femsle/fnaa190
Almeida, O. G. G., and De Martinis, E. C. P. (2019). Bioinformatics tools to assess metagenomic data for applied microbiology. Appl. Microbiol. Biotechnol. 103, 69–82. doi: 10.1007/s00253-018-9464-9
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. (1991). JANET. The GenBank BLAST server allows you to send a specially formatted mail message containing the nucleic acid or protein query sequence to the BLAST server at GenBank. A BLAST sequence similarity search is then performed against the specified database using the BLAST algorithm. Perspective 219, 555–565.
Aponte, H., Meli, P., Butler, B., Paolini, J., Matus, F., Merino, C., et al. (2020). Meta-analysis of heavy metal effects on soil enzyme activities. Sci. Total Environ. 737:139744. doi: 10.1016/j.scitotenv.2020.139744
Asaf, S., Numan, M., Khan, A. L., and Al-Harrasi, A. (2020). Sphingomonas: from diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 40, 138–152. doi: 10.1080/07388551.2019.1709793
Ashraf, A., Chen, X., and Ramamurthy, R. (2021). Modelling heavy metals contamination in groundwater of southern Punjab. Pakistan. Int. J. Environ. Sci. Technol. 18, 2221–2236. doi: 10.1007/s13762-020-02965-w
Ashraf, M., and Foolad, M. R. (2013). Crop breeding for salt tolerance in the era of molecular markers and marker-assisted selection. Plant Breed. 132, 10–20. doi: 10.1111/pbr.12000
Aslam, F., and Ali, B. (2018). Halotolerant bacterial diversity associated with Suaeda fruticosa (L.) forssk. Improved growth of maize under salinity stress. Agronomy 8:131. doi: 10.3390/agronomy8080131
Bahram, M., Anslan, S., Hildebrand, F., Bork, P., and Tedersoo, L. (2019). Newly designed 16S rRNA metabarcoding primers amplify diverse and novel archaeal taxa from the environment. Environ. Microbiol. Rep. 11, 487–494. doi: 10.1111/1758-2229.12684
Barbera, P., Kozlov, A. M., Czech, L., Morel, B., Darriba, D., Flouri, T., et al. (2019). EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68, 365–369. doi: 10.1093/sysbio/syy054
Batool, R., Marghoob, U., and Kalsoom, A. (2017). Estimation of exopolysaccharides (EPS) producing ability of Cr (VI) resistant bacterial strains from tannery effluent. J. Basic Appl. Sci. 13, 589–596. doi: 10.6000/1927-5129.2017.13.95
Bengyella, L., Iftikhar, S., Nawaz, K., Fonmboh, D. J., Yekwa, E. L., Jones, R. C., et al. (2019). Biotechnological application of endophytic filamentous Bipolaris and Curvularia: a review on bioeconomy impact. World J. Microbiol. Biotechnol. 35:69. doi: 10.1007/s11274-019-2644-7
Ben-Laouane, R., Baslam, M., Ait-El-Mokhtar, M., Anli, M., Boutasknit, A., Ait-Rahou, Y., et al. (2020). Potential of native arbuscular mycorrhizal fungi, rhizobia, and/or green compost as alfalfa (Medicago sativa) enhancers under salinity. Microorganisms 8:1695. doi: 10.3390/microorganisms8111695
Bian, G., Gloor, G. B., Gong, A., Jia, C., Zhang, W., Hu, J., et al. (2017). The gut microbiota of healthy aged Chinese is similar to that of the healthy young. Msphere 2:e00327-17. doi: 10.1128/mSphere.00327-17
Bian, X., Xiao, S., Zhao, Y., Xu, Y., Yang, H., and Zhang, L. (2020). Comparative analysis of rhizosphere soil physiochemical characteristics and microbial communities between rusty and healthy ginseng root. Sci. Rep. 10:15756. doi: 10.1038/s41598-020-71024-8
Bouffaud, M. L., Renoud, S., Dubost, A., Moenne-Loccoz, Y., and Muller, D. (2018). 1-Aminocyclopropane-1-carboxylate deaminase producers associated to maize and other Poaceae species. Microbiome 6:114. doi: 10.1186/s40168-018-0503-7
Bozoğlu, T., Derviş, S., Imren, M., Amer, M., Özdemir, F., Paulitz, T. C., et al. (2022). Fungal pathogens associated with crown and root rot of wheat in central, eastern, and southeastern Kazakhstan. J. Fungi 8:417. doi: 10.3390/jof8050417
Braga, L. P. P., Pereira, R. V., Martins, L. F., Moura, L. M. S., Sanchez, F. B., Patane, J. S. L., et al. (2021). Genome-resolved metagenome and metatranscriptome analyses of thermophilic composting reveal key bacterial players and their metabolic interactions. BMC Genomics 22:652. doi: 10.1186/s12864-021-07957-9
Brochier-Armanet, C., Boussau, B., Gribaldo, S., and Forterre, P. (2008). Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6, 245–252. doi: 10.1038/nrmicro1852
Cao, C., Tao, S., Cui, Z., and Zhang, Y. (2021). Response of soil properties and microbial communities to increasing salinization in the meadow grassland of Northeast China. Microb. Ecol. 82, 722–735. doi: 10.1007/s00248-021-01695-x
Castelan-Sanchez, H. G., Elorrieta, P., Romoacca, P., Linan-Torres, A., Sierra, J. L., Vera, I., et al. (2019). Intermediate-salinity systems at high altitudes in the peruvian Andes unveil a high diversity and abundance of bacteria and viruses. Genes 10:891. doi: 10.3390/genes10110891
Chen, H., Ma, K., Huang, Y., Fu, Q., Qiu, Y., and Yao, Z. (2022). Significant response of microbial community to increased salinity across wetland ecosystems. Geoderma 415:115778. doi: 10.1016/j.geoderma.2022.115778
Chen, Y. G., Tang, S. K., Zhang, Y. Q., Liu, Z. X., Chen, Q. H., He, J. W., et al. (2010). Zhihengliuella salsuginis sp. nov., a moderately halophilic actinobacterium from a subterranean brine. Extremophiles 14, 397–402. doi: 10.1007/s00792-010-0317-4
Conte, A., Papale, M., Amalfitano, S., Mikkonen, A., Rizzo, C., De Domenico, E., et al. (2018). Bacterial community structure along the subtidal sandy sediment belt of a high Arctic fjord (Kongsfjorden, Svalbard Islands). Sci. Total Environ. 619-620, 203–211. doi: 10.1016/j.scitotenv.2017.11.077
Cortes-Lorenzo, C., Rodriguez-Diaz, M., Lopez-Lopez, C., Sanchez-Peinado, M., Rodelas, B., and Gonzalez-Lopez, J. (2012). Effect of salinity on enzymatic activities in a submerged fixed bed biofilm reactor for municipal sewage treatment. Bioresour. Technol. 121, 312–319. doi: 10.1016/j.biortech.2012.06.083
Czech, L., Barbera, P., and Stamatakis, A. (2020). Genesis and Gappa: processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 36, 3263–3265. doi: 10.1093/bioinformatics/btaa070
Danczak, R. E., Johnston, M. D., Kenah, C., Slattery, M., Wrighton, K. C., and Wilkins, M. J. (2017). Members of the candidate phyla radiation are functionally differentiated by carbon- and nitrogen-cycling capabilities. Microbiome 5:112. doi: 10.1186/s40168-017-0331-1
De Leon-Lorenzana, A. S., Delgado-Balbuena, L., Dominguez-Mendoza, C., Navarro-Noya, Y. E., Luna-Guido, M., and Dendooven, L. (2017). Reducing salinity by flooding an extremely alkaline and saline soil changes the bacterial community but its effect on the archaeal community is limited. Front. Microbiol. 8:466. doi: 10.3389/fmicb.2017.00466
Dey, G., Banerjee, P., Sharma, R. K., Maity, J. P., Etesami, H., Shaw, A. K., et al. (2021). Management of phosphorus in salinity-stressed agriculture for sustainable crop production by salt-tolerant phosphate-solubilizing bacteria—a review. Agronomy 11:1552. doi: 10.3390/agronomy11081552
Djebaili, R., Pellegrini, M., Smati, M., Del Gallo, M., and Kitouni, M. (2020). Actinomycete strains isolated from saline soils: plant-growth-promoting traits and inoculation effects on Solanum lycopersicum. Sustainability 12:4617. doi: 10.3390/su12114617
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Duangurai, T., Indrawattana, N., and Pumirat, P. (2018). Burkholderia pseudomallei adaptation for survival in stressful conditions. Biomed. Res. Int. 2018, 1–11. doi: 10.1155/2018/3039106
Egidi, E., Delgado-Baquerizo, M., Plett, J. M., Wang, J., Eldridge, D. J., Bardgett, R. D., et al. (2019). A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 10:2369. doi: 10.1038/s41467-019-10373-z
Gehring, A. M., DeMoll, E., Fetherston, J. D., Mori, I., Mayhew, G. F., Blattner, F. R., et al. (1998). Iron acquisition in plague: modular logic in enzymatic biogenesis of yersiniabactin by Yersinia pestis. Chem. Biol. 5, 573–586. doi: 10.1016/S1074-5521(98)90115-6
Ghosh Roy, S., Wimpee, C. F., McGuire, S. A., and Ehlinger, T. J. (2022). Responses of bacterial taxonomical diversity indicators to pollutant loadings in experimental wetland microcosms. Water 14:251. doi: 10.3390/w14020251
Glick, B. R. (2012). Plant growth-promoting bacteria: mechanisms and applications. Scientifica 2012, 1–15. doi: 10.6064/2012/963401
Grum-Grzhimaylo, O. A., Debets, A. J. M., and Bilanenko, E. N. (2018). Mosaic structure of the fungal community in the Kislo-Sladkoe lake that is detaching from the White Sea. Polar Biol. 41, 2075–2089. doi: 10.1007/s00300-018-2347-9
Haegeman, B., Hamelin, J., Moriarty, J., Neal, P., Dushoff, J., and Weitz, J. S. (2013). Robust estimation of microbial diversity in theory and in practice. ISME J. 7, 1092–1101. doi: 10.1038/ismej.2013.10
Hajiabadi, A. A., Arani, A. M., Ghasemi, S., Rad, M. H., Etesami, H., Manshadi, S. S., et al. (2021). Mining the rhizosphere of halophytic rangeland plants for halotolerant bacteria to improve growth and yield of salinity-stressed wheat. Plant Physiol. Biochem. 163, 139–153. doi: 10.1016/j.plaphy.2021.03.059
Han, F., Li, X., Zhang, M., Liu, Z., Han, Y., Li, Q., et al. (2021). Solid-phase denitrification in high salinity and low-temperature wastewater treatment. Bioresour. Technol. 341:125801. doi: 10.1016/j.biortech.2021.125801
Han, J., Mammadov, Z., Kim, M., Mammadov, E., Lee, S., Park, J., et al. (2021). Spatial distribution of salinity and heavy metals in surface soils on the Mugan plain, the republic of Azerbaijan. Environ. Monit. Assess. 193:95. doi: 10.1007/s10661-021-08877-7
Hartman, K., van der Heijden, M. G. A., Wittwer, R. A., Banerjee, S., Walser, J. C., and Schlaeppi, K. (2018). Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 6:14. doi: 10.1186/s40168-017-0389-9
Hewelke, E., Górska, E. B., Gozdowski, D., Korc, M., Olejniczak, I., and Prędecka, A. (2020). Soil functional responses to natural ecosystem restoration of a pine forest peucedano-pinetum after a fire. Forests 11:286. doi: 10.3390/f11030286
Hoagland, L., Carpenter-Boggs, L., Granatstein, D., Mazzola, M., Smith, J., Peryea, F., et al. (2008). Orchard floor management effects on nitrogen fertility and soil biological activity in a newly established organic apple orchard. Biol. Fertil. Soils 45, 11–18. doi: 10.1007/s00374-008-0304-4
Hu, H. W., Zhang, L. M., Yuan, C. L., and He, J. Z. (2013). Contrasting Euryarchaeota communities between upland and paddy soils exhibited similar pH-impacted biogeographic patterns. Soil Biol. Biochem. 64, 18–27. doi: 10.1016/j.soilbio.2013.04.003
Huang, C. C., Liang, C. M., Yang, T. I., Chen, J. L., and Wang, W. K. (2021). Shift of bacterial communities in heavy metal-contaminated agricultural land during a remediation process. PLoS One 16:e0255137. doi: 10.1371/journal.pone.0255137
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Imran, M., Ahmad, I., Barasubiye, T., Abulreesh, H. H., and Samreen, S. (2020). Heavy metal tolerance among free-living fungi isolated from soil receiving long term application of wastewater. J. Pure Appl. Microbiol. 14, 157–170. doi: 10.22207/JPAM.14.1.17
Imran, M., Ashraf, M., and Awan, A. R. (2021). Growth, yield and arsenic accumulation by wheat grown in a pressmud amended salt-affected soil irrigated with arsenic contaminated water. Ecotoxicol. Environ. Saf. 224:112692. doi: 10.1016/j.ecoenv.2021.112692
Jiang, Z., Chen, J., Zhai, H., Zhou, F., Yan, X., Zhu, Y., et al. (2019). Kuroshio shape composition and distribution of filamentous diazotrophs in the East China Sea and southern yellow sea. J. Geophys. Res. Oceans 124, 7421–7436. doi: 10.1029/2019jc015413
Jiang, X., Van Horn, D. J., Okie, J. G., Buelow, H. N., Schwartz, E., Colman, D. R., et al. (2022). Limits to the three domains of life: lessons from community assembly along an Antarctic salinity gradient. Extremophiles 26:15. doi: 10.1007/s00792-022-01262-3
Jung, B. K., Hong, S.-J., Park, G.-S., Kim, M.-C., and Shin, J.-H. (2018). Isolation of Burkholderia cepacia JBK9 with plant growth-promoting activity while producing pyrrolnitrin antagonistic to plant fungal diseases. Appl. Biol. Chem. 61, 173–180. doi: 10.1007/s13765-018-0345-9
Kalam, S., Basu, A., Ahmad, I., Sayyed, R., El-Enshasy, H. A., Dailin, D. J., et al. (2020). Recent understanding of soil Acidobacteria and their ecological significance: a critical review. Front. Microbiol. 11:580024. doi: 10.3389/fmicb.2020.580024
Kaushik, R., Sharma, M., Gaurav, K., Jagadeeshwari, U., Shabbir, A., Sasikala, C., et al. (2020). Paludisphaera soli sp. nov., a new member of the family Isosphaeraceae isolated from high altitude soil in the Western Himalaya. Antonie Van Leeuwenhoek 113, 1663–1674. doi: 10.1007/s10482-020-01471-w
Khalid, M. U., Imran, M., Aslam, M., and Ashraf, M. (2021). The interactive effect of selenium and farmyard manure on soil microbial activities, yield and selenium accumulation by wheat (Triticum aestivum l.). Grains. J. Plant Growth Regul. 41, 2669–2677. doi: 10.1007/s00344-021-10465-5
Khan, S., Chen, N., Zhang, C., Wang, L., Han, C., Lu, K., et al. (2020). Soil fungal taxonomic diversity along an elevation gradient on the semi-arid Xinglong Mountain. Northwest China. Arch. Microbiol. 202, 2291–2302. doi: 10.1007/s00203-020-01948-2
Köhl, J., Kolnaar, R., and Ravensberg, W. J. (2019). Mode of action of microbial biological control agents against plant diseases: relevance beyond efficacy. Front. Plant Sci. 10:845. doi: 10.3389/fpls.2019.00845
Köljalg, U., Nilsson, R. H., Taylor, A. F. S., Bates, S. T., Thomas, D., Bengtsson-palme, J., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kori, J. A., Mahar, R. B., Vistro, M. R., Tariq, H., Khan, I. A., and Goel, R. (2019). Metagenomic analysis of drinking water samples collected from treatment plants of Hyderabad city and Mehran University employees cooperative housing society. Environ. Sci. Pollut. Res. Int. 26, 29052–29064. doi: 10.1007/s11356-019-05859-8
Latif, S., Bibi, S., Kouser, R., Fatimah, H., Farooq, S., Naseer, S., et al. (2020). Characterization of bacterial community structure in the rhizosphere of Triticum aestivum L. Genomics 112, 4760–4768. doi: 10.1016/j.ygeno.2020.07.031
Latif, M., Bukhari, S. A. H., Alrajhi, A. A., Alotaibi, F. S., Ahmad, M., Shahzad, A. N., et al. (2022). Inducing drought tolerance in wheat through exopolysaccharide-producing rhizobacteria. Agronomy 12:1140. doi: 10.3390/agronomy12051140
Li, H., Luo, N., Ji, C., Li, J., Zhang, L., Xiao, L., et al. (2021). Liquid organic fertilizer amendment alters rhizosphere microbial community structure and co-occurrence patterns and improves sunflower yield under salinity-alkalinity stress. Microb. Ecol. 84, 423–438. doi: 10.1007/s00248-021-01870-0
Li, S., Zhao, B., Jin, M., Hu, L., Zhong, H., and He, Z. (2020). A comprehensive survey on the horizontal and vertical distribution of heavy metals and microorganisms in soils of a Pb/Zn smelter. J. Hazard. Mater. 400:123255. doi: 10.1016/j.jhazmat.2020.123255
Lin, L., Jing, X., Lucas-Borja, M. E., Shen, C., Wang, Y., and Feng, W. (2022). Rare taxa drive the response of soil fungal guilds to soil salinization in the Taklamakan Desert. Front. Microbiol. 13:862245. doi: 10.3389/fmicb.2022.862245
Liu, H., Khan, M. Y., Carvalhais, L. C., Delgado-Baquerizo, M., Yan, L., Crawford, M., et al. (2019). Soil amendments with ethylene precursor alleviate negative impacts of salinity on oil microbial properties and productivity. Sci. Rep. 9:6892. doi: 10.1038/s41598-019-43305-4
Machado, R., and Serralheiro, R. (2017). Soil salinity: effect on vegetable crop growth. Management practices to prevent and mitigate soil salinization. Horticulturae 3:30. doi: 10.3390/horticulturae3020030
Marghoob, M. U., Noureen, A., Raza, A., Khan, W. S., Iftikhar, M., and Sher, F. (2022). Synthesis and toxicity assessment of environment friendly high yield ceria nanoparticles for biosafety. J. Environ. Chem. Eng. 10:107029. doi: 10.1016/j.jece.2021.107029
Matarredona, L., Camacho, M., Zafrilla, B., Bonete, M.-J., and Esclapez, J. (2020). The role of stress proteins in Haloarchaea and their adaptive response to environmental shifts. Biomol. Ther. 10:1390. doi: 10.3390/biom10101390
Meng, S., Peng, T., Pratush, A., Huang, T., and Hu, Z. (2021). Interactions between heavy metals and bacteria in mangroves. Mar. Pollut. Bull. 172:112846. doi: 10.1016/j.marpolbul.2021.112846
Mishra, A. K., and Baek, K. H. (2021). Salicylic acid biosynthesis and metabolism: a divergent pathway for plants and bacteria. Biomol. Ther. 11:705. doi: 10.3390/biom11050705
Mora-Ruiz, M. D. R., Cifuentes, A., Font-Verdera, F., Pérez-Fernández, C., Farias, M. E., González, B., et al. (2018). Biogeographical patterns of bacterial and archaeal communities from distant hypersaline environments. Syst. Appl. Microbiol. 41, 139–150. doi: 10.1016/j.syapm.2017.10.006
Moreira, D., Zivanovic, Y., López-Archilla, A. I., Iniesto, M., and López-García, P. (2021). Reductive evolution and unique predatory mode in the CPR bacterium Vampirococcus lugosii. Nat. Commun. 12, 2454–2411. doi: 10.1038/s41467-021-22762-4
Morrow, M. A., Pold, G., and DeAngelis, K. M. (2020). Draft genome sequence of a terrestrial planctomycete, Singulisphaera sp. strain gp187, isolated from forest soil. Microbiol. Resour. Announc. 9:00956–20. doi: 10.1128/MRA.00956-20
Moryani, H. T., Kong, S., Du, J., and Bao, J. (2020). Health risk assessment of heavy metals accumulated on PM2.5 fractioned road dust from two cities of Pakistan. Int. J. Environ. Res. Public Health 17:7124. doi: 10.3390/ijerph17197124
Mukhtar, S., Hirsch, A. M., Khan, N., Malik, K. A., Humm, E. A., Pellegrini, M., et al. (2020). Impact of soil salinity on the cowpea nodule microbiome and the isolation of halotolerant PGPR strains to promote plant growth under salinity stress. Phytobiomes J. 4, 364–374. doi: 10.1094/pbiomes-09-19-0057-r
Mukhtar, S., Mirza, B. S., Mehnaz, S., Mirza, M. S., McLean, J., and Malik, K. A. (2018). Impact of soil salinity on the microbial structure of halophyte rhizosphere microbiome. World J. Microbiol. Biotechnol. 34:136. doi: 10.1007/s11274-018-2509-5
Nawaz, A., Mubeen, F., Qamar, Z. U., Marghoob, M. U., Aziz, S., and Gross, H. (2021). Draft genome sequence of the halophilic strain Citrobacter braakii AN-PRR1, isolated from rhizospheric soil of rice (Oryza sativa L.) from Pakistan. Microbiol. Resour. Announc. 10, e00787–e00721. doi: 10.1128/MRA.00787-21
Nawaz, A., Shahbaz, M., Asadullah,, Imran, A., Marghoob, M. U., Imtiaz, M., et al. (2020). Potential of salt tolerant pgpr in growth and yield augmentation of wheat (Triticum aestivum L.) under saline conditions. Front. Microbiol. 2019:11. doi: 10.3389/fmicb.2020.02019
Nayak, S., Samanta, S., and Mukherjee, A. K. (2020). “Beneficial role of aspergillus sp. in agricultural soil and environment” in the Frontiers in soil and environmental microbiology. eds. S. K. Nayak and B. B. Mishra (Boca Raton, FL: CRC press), edn. 1. 7–36.
Nelson, W. C., and Stegen, J. C. (2015). The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 6:713. doi: 10.3389/fmicb.2015.00713
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nicol, G. W., and Schleper, C. (2006). Ammonia-oxidising Crenarchaeota: important players in the nitrogen cycle? Trends Microbiol. 14, 207–212. doi: 10.1016/j.tim.2006.03.004
Nicolas, A. M., Jaffe, A. L., Nuccio, E. E., Taga, M. E., Firestone, M. K., and Banfield, J. F. (2021). Soil candidate phyla radiation bacteria encode components of aerobic metabolism and co-occur with Nanoarchaea in the rare biosphere of rhizosphere grassland communities. Msystems 6, e01205–e01220. doi: 10.1128/mSystems.01205-20
Nouri, H., Chavoshi Borujeni, S., Nirola, R., Hassanli, A., Beecham, S., Alaghmand, S., et al. (2017). Application of green remediation on soil salinity treatment: a review on halophytoremediation. Process. Saf. Environ. Prot. 107, 94–107. doi: 10.1016/j.psep.2017.01.021
Olsen, S. R. (1954). Estimation of available phosphorus in soils by extraction with sodium bicarbonate. Washington DC: US Department of Agriculture.
Op De Beeck, M., Lievens, B., Busschaert, P., Declerck, S., Vangronsveld, J., and Colpaert, J. V. (2014). Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS One 9:e97629. doi: 10.1371/journal.pone.0097629
Orhan, F., and Demirci, A. (2020). Salt stress mitigating potential of halotolerant/halophilic plant growth promoting. Geomicrobiol J. 37, 663–669. doi: 10.1080/01490451.2020.1761911
Oueriaghli, N., Béjar, V., Quesada, E., and Martínez-Checa, F. (2013). Molecular ecology techniques reveal both spatial and temporal variations in the diversity of archaeal communities within the athalassohaline environment of Rambla Salada. Spain. Microb. Ecol. 66, 297–311. doi: 10.1007/s00248-013-0176-5
Ozimek, E., and Hanaka, A. (2020). Mortierella species as the plant growth-promoting fungi present in the agricultural soils. Agriculture 11:7. doi: 10.3390/agriculture11010007
Pan, X., Qin, Y., and Yuan, Z. (2018). Potential of a halophyte-associated endophytic fungus for sustaining Chinese white poplar growth under salinity. Symbiosis 76, 109–116. doi: 10.1007/s13199-018-0541-8
Pelludat, C., Brem, D., and Heesemann, J. (2003). Irp9, encoded by the high-pathogenicity island of Yersinia enterocolitica, is able to convert chorismate into salicylate, the precursor of the siderophore yersiniabactin. J. Bacteriol. 185, 5648–5653. doi: 10.1128/JB.185.18.5648-5653.2003
Pepper, I. L., Gerba, C. P., and Gentry, T. J. (2015). "Introduction to environmental microbiology," in the Environmental Microbiology. ed. 3, (Amsterdam: Elsevier), 3–8.
Priyadharsini, P., and Muthukumar, T. (2017). The root endophytic fungus Curvularia geniculata from Parthenium hysterophorus roots improves plant growth through phosphate solubilization and phytohormone production. Fungal Ecol. 27, 69–77. doi: 10.1016/j.funeco.2017.02.007
Prudence, S. M., Newitt, J. T., Worsley, S. F., Macey, M. C., Murrell, J. C., Lehtovirta-Morley, L. E., et al. (2021). Soil, senescence and exudate utilisation: characterisation of the paragon var. spring bread wheat root microbiome. Environ. Microbiome 16:12. doi: 10.1186/s40793-021-00381-2
Qiu, D., Zeng, X., Zeng, L., Li, G., and Shao, Z. (2021). Fusibacter ferrireducens sp. nov., an anaerobic, Fe()- and Sulphur-reducing bacterium isolated from mangrove sediment. Int. J. Syst. Evol. Microbiol. 71:004952. doi: 10.1099/ijsem.0.004952
Rangseekaew, P., Barros-Rodriguez, A., Pathom-Aree, W., and Manzanera, M. (2022). Plant beneficial deep-sea Actinobacterium, Dermacoccus abyssi MT1.1(T) promote growth of tomato (Solanum lycopersicum) under salinity stress. Biology 11:191. doi: 10.3390/biology11020191
Rath, K. M., Fierer, N., Murphy, D. V., and Rousk, J. (2019). Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 13, 836–846. doi: 10.1038/s41396-018-0313-8
Rath, K. M., Maheshwari, A., Bengtson, P., and Rousk, J. (2016). Comparative toxicities of salts on microbial processes in soil. Appl. Environ. Microbiol. 82, 2012–2020. doi: 10.1128/AEM.04052-15
Rath, K. M., and Rousk, J. (2015). Salt effects on the soil microbial decomposer community and their role in organic carbon cycling: a review. Soil Biol. Biochem. 81, 108–123. doi: 10.1016/j.soilbio.2014.11.001
Redou, V., Navarri, M., Meslet-Cladiere, L., Barbier, G., and Burgaud, G. (2015). Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl. Environ. Microbiol. 81, 3571–3583. doi: 10.1128/AEM.04064-14
Reeve, J. R., Hoagland, L. A., Villalba, J. J., Carr, P. M., Atucha, A., Cambardella, C., et al. (2016). Organic farming, soil health, and food quality: considering possible links. Adv. Agron. 137, 319–367. doi: 10.1016/bs.agron.2015.12.003
Ren, M., Zhang, Z., Wang, X., Zhou, Z., Chen, D., Zeng, H., et al. (2018). Diversity and contributions to nitrogen cycling and carbon fixation of soil salinity shaped microbial communities in Tarim Basin. Front. Microbiol. 9:431. doi: 10.3389/fmicb.2018.00431
Richard, L. (1954). “Diagnosis and improvement of saline and alkali soils” in Agricultural handbook (Washington DC: US Department of Agriculture) 60
Richardville, K., Egel, D., Flachs, A., Jaiswal, A., Perkins, D., Thompson, A., et al. (2022). Leaf mold compost reduces waste, improves soil and microbial properties, and increases tomato productivity. Urban Agri. Regional Food Syst. 7:e20022. doi: 10.1002/uar2.20022
Rilling, J., Acuña, J., Nannipieri, P., Cassan, F., Maruyama, F., and Jorquera, M. (2019). Current opinion and perspectives on the methods for tracking and monitoring plant growth–promoting bacteria. Soil Biol. Biochem. 130, 205–219. doi: 10.1016/j.soilbio.2018.12.012
Rodríguez-Eugenio, N., McLaughlin, M., and Pennock, D. (2018). Soil pollution: A hidden reality. Rome: FAO.
Rodriguez-Sanchez, A., Tomasek, A., McMillan, S., Yufra, S., Yupanqui, M., Rondon, R., et al. (2022). Composition and potential functional roles of soil fungal communities on arid farms in Arequipa (southern Peru) characterized using SMRT sequencing. Appl. Soil Ecol. 169:104228. doi: 10.1016/j.apsoil.2021.104228
Rognes, T., Mahé, F., Flouri, T., Quince, C., and Nichols, B. (2015). VSEARCH. A versatile open source tool for metagenomics. Peer J. 4:4. doi: 10.7717/peerj.2584
Rudisill, M. A., Bordelon, B. P., Turco, R. F., and Hoagland, L. A. (2015). Sustaining soil quality in intensively managed high tunnel vegetable production systems: a role for green manures and chicken litter. HortScience 50, 461–468. doi: 10.21273/HORTSCI.50.3.461
Sarkar, A., Pramanik, K., Mitra, S., Soren, T., and Maiti, T. K. (2018). Enhancement of growth and salt tolerance of rice seedlings by ACC deaminase producing Burkholderia sp. MTCC 12259. J. Plant Physiol. 231, 434–442. doi: 10.1016/j.jplph.2018.10.010
Schloss, P. D. (2020). Removal of rare amplicon sequence variants from 16S rRNA gene sequence surveys biases the interpretation of community structure data. bioRxiv [Preprint].
Schloss, P. D., Jenior, M. L., Koumpouras, C. C., Westcott, S. L., and Highlander, S. K. (2016). Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4:e1869. doi: 10.7717/peerj.1869
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schrey, S. D., and Tarkka, M. T. (2008). Friends and foes: Streptomycetes as modulators of plant disease and symbiosis. Antonie Van Leeuwenhoek 94, 11–19. doi: 10.1007/s10482-008-9241-3
Selivanova, E. A., Poshvina, D. V., Khlopko, Y. A., Gogoleva, N. E., and Plotnikov, A. O. (2018). Diversity of prokaryotes in planktonic communities of saline sol-iletsk lakes (Orenburg oblast, Russia). Microbiology 87, 569–582. doi: 10.1134/s0026261718040161
Serrano, A. E., Escudero, L. V., Tebes-Cayo, C., Acosta, M., Encalada, O., Fernandez-Moroso, S., et al. (2017). First draft genome sequence of a strain from the genus Fusibacter isolated from Salar de Ascotan in northern Chile. Stand Genomic Sci. 12:43. doi: 10.1186/s40793-017-0252-4
Shakir, S. K., Azizullah, A., Murad, W., Daud, M. K., Nabeela, F., Rahman, H., et al. (2016). “Toxic metal pollution in Pakistan and its possible risks to public health,” in Reviews of environmental contamination and toxicology, vol. 242. ed. W. P. Pim de Voogt (Berlin: Springer), 1–60.
Shang, L., Wan, L., Zhou, X., Li, S., and Li, X. (2020). Effects of organic fertilizer on soil nutrient status, enzyme activity, and bacterial community diversity in Leymus chinensis steppe in Inner Mongolia, China. PLoS One 15:e0240559. doi: 10.1371/journal.pone.0240559
Sharma, S., Kulkarni, J., and Jha, B. (2016). Halotolerant rhizobacteria promote growth and enhance salinity tolerance in peanut. Front. Microbiol. 7:1600. doi: 10.3389/fmicb.2016.01600
Shelton, C. L., and Lamb, A. L. (2018). Unraveling the structure and mechanism of the MST(ery) enzymes. Trends Biochem. Sci. 43, 342–357. doi: 10.1016/j.tibs.2018.02.011
Shi, W., Xing, Y., Zhu, Y., Gao, N., and Ying, Y. (2022). Diverse responses of pqqC- and phoD-harbouring bacterial communities to variation in soil properties of Moso bamboo forests. J. Microbial. Biotechnol. 15, 2097–2111. doi: 10.1111/1751-7915.14029
Stefanowicz, A. M., Niklińska, M., and Laskowski, R. (2008). Metals affect soil bacterial and fungal functional diversity differently. Environ. Toxicol. Chem. 27, 591–598. doi: 10.1897/07-288.1
Sultana, N., Khanzada, K. A., and Azeem, M. T. (2019). Black point of wheat in commercial varieties of Sindh. Pakistan. Int. J. Biol. Biotech. 16, 385–389.
Sun, L., Shao, S., and Bao, J. (2021). Microbial lipid fermentation of Trichosporon cutaneum in high saline water. Bioresour. Bioprocess. 8:71. doi: 10.1186/s40643-021-00424-z
Syed, A., Sarwar, G., Shah, S. H., and Muhammad, S. (2021). Soil salinity research in 21st century in Pakistan: its impact on availability of plant nutrients, growth and yield of crops. Commun. Soil Sci. Plant Anal. 52, 183–200. doi: 10.1080/00103624.2020.1854294
Tan, M., Fang, Y., Wu, Y., Reardon, R. C., and Qiang, S. (2022). First report of Curvularia intermedia Boedijn causing leaf blight disease on Microstegium vimineum in China. Plant Dis. 106:2526. doi: 10.1094/PDIS-10-21-2289-PDN
Torres-Cruz, T. J., Hesse, C., Kuske, C. R., and Porras-Alfaro, A. (2018). Presence and distribution of heavy metal tolerant fungi in surface soils of a temperate pine forest. Appl. Soil Ecol. 131, 66–74. doi: 10.1016/j.apsoil.2018.08.001
Treusch, A. H., Leininger, S., Kletzin, A., Schuster, S. C., Klenk, H. P., and Schleper, C. (2005). Novel genes for nitrite reductase and amo-related proteins indicate a role of uncultivated mesophilic Crenarchaeota in nitrogen cycling. Environ. Microbiol. 7, 1985–1995. doi: 10.1111/j.1462-2920.2005.00906.x
Tripathi, A., Mishra, B., and Tripathi, P. (1998). Salinity stress responses in the plant growth promoting rhizobacteria, Azospirillum spp. J. Biosci. 23, 463–471. doi: 10.1007/BF02936140
Walkley, A., and Black, I. A. (1934). An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 37, 29–38. doi: 10.1097/00010694-193401000-00003
Wang, H., Gilbert, J. A., Zhu, Y., and Yang, X. (2018). Salinity is a key factor driving the nitrogen cycling in the mangrove sediment. Sci. Total Environ. 631-632, 1342–1349. doi: 10.1016/j.scitotenv.2018.03.102
Wang, L., and Huang, D. (2021). Soil ammonia-oxidizing archaea in a paddy field with different irrigation and fertilization managements. Sci. Rep. 11:14563. doi: 10.1038/s41598-021-93898-y
Wang, F., Shi, Z., Biswas, A., Yang, S., and Ding, J. (2020). Multi-algorithm comparison for predicting soil salinity. Geoderma 365:114211. doi: 10.1016/j.geoderma.2020.114211
Wang, H. L., Tian, C. Y., Jiang, L., and Wang, L. (2014). Remediation of heavy metals contaminated saline soils: a halophyte choice? Environ. Sci. Technol. 48, 21–22. doi: 10.1021/es405052j
Wang, H., Zhang, Y., Bartlett, D. H., and Xiao, X. (2021). Transcriptomic analysis reveals common adaptation mechanisms under different stresses for moderately piezophilic bacteria. Microb. Ecol. 81, 617–629. doi: 10.1007/s00248-020-01609-3
Wang, S., Zuo, X., Awada, T., Medima-Roldán, E., Feng, K., Yue, P., et al. (2021). Changes of soil bacterial and fungal community structure along a natural aridity gradient in desert grassland ecosystems, Inner Mongolia. Catena 205:105470. doi: 10.1016/j.catena.2021.105470
Wasimuddin, W., Schläppi, K. B., Ronchi, F., Leib, S., Erb, M., and Ramette, A. N. (2019). Evaluation of primer pairs for microbiome profiling across a food chain from soils to humans within the one health framework. BioRxiv [Preprint].
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 1–18. doi: 10.1186/s40168-017-0237-y
Westcott, S. L., and Schloss, P. D. (2015). De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 3:e1487. doi: 10.7717/peerj.1487
Yadav, A. N., Verma, P., Kour, D., Rana, K. L., Kumar, V., Singh, B., et al. (2017). Plant microbiomes and its beneficial multifunctional plant growth promoting attributes. Int. J. Environ. Sci. Nat. Resour. 3, 1–8. doi: 10.19080/IJESNR.2017.03.555601
Yang, Q.-S., Dong, J.-D., Ahmad, M., Ling, J., Zhou, W.-G., Tan, Y.-H., et al. (2019). Analysis of nifH DNA and RNA reveals a disproportionate contribution to nitrogenase activities by rare plankton-associated diazotrophs. BMC Microbiol. 19, 188–112. doi: 10.1186/s12866-019-1565-9
Yu, J., Pavia, M. J., Deem, L. M., Crow, S. E., Deenik, J. L., and Penton, C. R. (2020). DNA-stable isotope probing shotgun metagenomics reveals the resilience of active microbial communities to biochar amendment in oxisol soil. Front. Microbiol. 11:587972. doi: 10.3389/fmicb.2020.587972
Yue, Y., Shao, T., Long, X., He, T., Gao, X., Zhou, Z., et al. (2020). Microbiome structure and function in rhizosphere of Jerusalem artichoke grown in saline land. Sci. Total Environ. 724:138259. doi: 10.1016/j.scitotenv.2020.138259
Zea, M., Souza, A., Yang, Y., Lee, L., Nemali, K., and Hoagland, L. (2022). Leveraging high-throughput hyperspectral imaing technology to detect cadmium stress in two leafy green crops and accelerate soil remediation efforts. Environ. Pollut. 292:118405. doi: 10.1016/j.envpol.2021.118405
Zhang, S., Yang, G., Hou, S., Zhang, T., Li, Z., and Du, W. (2021). Analysis of heavy metal-related indices in the Eboling permafrost on the Tibetan plateau. Catena 196:104907. doi: 10.1016/j.catena.2020.104907
Zhao, S., Liu, J. J., Banerjee, S., White, J. F., Zhou, N., Zhao, Z. Y., et al. (2019). Not by salinity alone: how environmental factors shape fungal communities in saline soils. Soil Sci. Soc. Am. J. 83, 1387–1398. doi: 10.2136/sssaj2019.03.0082
Zhao, K., Penttinen, P., Zhang, X., Ao, X., Liu, M., Yu, X., et al. (2014). Maize rhizosphere in Sichuan, China, hosts plant growth promoting Burkholderia cepacia with phosphate solubilizing and antifungal abilities. Microbiol. Res. 169, 76–82. doi: 10.1016/j.micres.2013.07.003
Keywords: salinization, metagenomics, PGP microbes, heavy metals, metabolite profiling, ACC-deaminase
Citation: Marghoob MU, Rodriguez-Sanchez A, Imran A, Mubeen F and Hoagland L (2022) Diversity and functional traits of indigenous soil microbial flora associated with salinity and heavy metal concentrations in agricultural fields within the Indus Basin region, Pakistan. Front. Microbiol. 13:1020175. doi: 10.3389/fmicb.2022.1020175
Edited by:
Kumar Pranaw, University of Warsaw, PolandReviewed by:
Rameshwar Tiwari, Ulsan National Institute of Science and Technology, South KoreaCopyright © 2022 Marghoob, Rodriguez-Sanchez, Imran, Mubeen and Hoagland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lori Hoagland, bGhvYWdsYW5AcHVyZHVlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.