 Hendor N. R. Jesus1
Hendor N. R. Jesus1 Danilo J. P. G. Rocha2
Danilo J. P. G. Rocha2 Rommel T. J. Ramos3Artur Silva3
Rommel T. J. Ramos3Artur Silva3 Bertram Brenig4
Bertram Brenig4 Aristóteles Góes-Neto5
Aristóteles Góes-Neto5 Mateus M. Costa6
Mateus M. Costa6 Siomar C. Soares7
Siomar C. Soares7 Vasco Azevedo5
Vasco Azevedo5 Eric R. G. R. Aguiar8
Eric R. G. R. Aguiar8 Luiz Martínez-Martínez9,10,11Alain Ocampo12,13
Luiz Martínez-Martínez9,10,11Alain Ocampo12,13 Sana Alibi14Alexis Dorta13,15
Sana Alibi14Alexis Dorta13,15 Luis G. C. Pacheco1,2*
Luis G. C. Pacheco1,2* Jesus Navas13,15*
Jesus Navas13,15*- 1Multicenter Post-Graduate Program in Biochemistry and Molecular Biology (PMBqBM), Institute of Health Sciences, Federal University of Bahia, Salvador, BA, Brazil
- 2Post-Graduate Program in Biotechnology, Institute of Health Sciences, Federal University of Bahia, Salvador, BA, Brazil
- 3Institute of Biological Sciences, Federal University of Para, Belém, PA, Brazil
- 4Institute of Veterinary Medicine, University of Göttingen, Göttingen, Germany
- 5Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil
- 6Laboratório de Microbiologia e Imunologia Animal (LAMIA), Universidade Federal Do Vale Do São Francisco, Petrolina, Pernambuco, Brazil
- 7Department of Immunology, Microbiology and Parasitology, Institute of Biological and Natural Sciences, Federal University of Triângulo Mineiro (UFTM), Uberaba, Minas Gerais, Brazil
- 8Department of Biological Sciences, State University of Santa Cruz, Ilhéus, BA, Brazil
- 9Unidad de Gestión Clínica, Hospital Universitario Reina Sofía, Córdoba, Spain
- 10Departamento de Microbiología, Universidad de Córdoba, Córdoba, Spain
- 11Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC), Córdoba, Spain
- 12Microbiology Service, University Hospital Marqués de Valdecilla, Santander, Spain
- 13Instituto de Investigación Valdecilla (IDIVAL), Santander, Spain
- 14Research Unit Analysis and Process Applied to the Environment, Rejiche, Tunisia
- 15BIOMEDAGE Group, Faculty of Medicine, Cantabria University, Santander, Spain
Corynebacterium amycolatum is a nonlipophilic coryneform which is increasingly being recognized as a relevant human and animal pathogen showing multidrug resistance to commonly used antibiotics. However, little is known about the molecular mechanisms involved in transition from colonization to the MDR invasive phenotype in clinical isolates. In this study, we performed a comprehensive pan-genomic analysis of C. amycolatum, including 26 isolates from different countries. We obtained the novel genome sequences of 8 of them, which are multidrug resistant clinical isolates from Spain and Tunisia. They were analyzed together with other 18 complete or draft C. amycolatum genomes retrieved from GenBank. The species C. amycolatum presented an open pan-genome (α = 0.854905), with 3,280 gene families, being 1,690 (51.52%) in the core genome, 1,121 related to accessory genes (34.17%), and 469 related to unique genes (14.29%). Although some classic corynebacterial virulence factors are absent in the species C. amycolatum, we did identify genes associated with immune evasion, toxin, and antiphagocytosis among the predicted putative virulence factors. Additionally, we found genomic evidence for extensive acquisition of antimicrobial resistance genes through genomic islands.
Introduction
Although Corynebacterium amycolatum (Collins et al., 1988) is commonly found in the normal microbiome of the human skin and mucosal membranes (Gladysheva et al., 2022), this microorganism is now regarded as a potential multidrug-resistant opportunistic pathogen, especially in nosocomial environments and particularly when it comes to immunocompromised patients (Konstantinidis and Tiedje, 2005; Carvalho et al., 2018; Borde et al., 2020). It has already been described as the causative agent of serious infections in both humans and animals. Focusing on human infections, C. amycolatum has been described as the underlaying agent of endocarditis (Konstantinidis and Tiedje, 2005), mastitis (Borde et al., 2020), ear infections (Sengupta et al., 2015), and neonatal sepsis (Berner et al., 1997).
Several studies have shown that C. amycolatum infections are often misidentified by culturing and subsequent phenotypic analysis of the isolates, making it difficult to implement appropriate therapeutic interventions (Funke et al., 1996; Zinkernagel et al., 1996; Wauters et al., 1998; Soltan Mohammadi et al., 2013). In this sense, it is essential to define better phenotypic and genetic markers that could improve the identification of pathogenic nonlipophilic members of the genus Corynebacterium, including C. amycolatum (Santos et al., 2017, 2018). C. amycolatum can be clearly distinguished from C. xerosis and C. imitans by means of MALDI-TOF mass spectrometry using the MALDI Biotyper system (Alibi et al., 2017). However, this technology is not always accessible to all clinical microbiology laboratories, in particular in developing countries. Besides, monitoring the phenotypic profiles of antimicrobial susceptibility is of fundamental importance, as several isolates have demonstrated multiple resistance to antibiotics, in particular to penicillins, clindamycin, aminoglycosides, and fluoroquinolones (Sánchez Hernández et al., 2003; Carvalho et al., 2018; Borde et al., 2020; Dragomirescu et al., 2020).
Previous studies by our group have already demonstrated the potential of comparative genomics to aid the understanding of variability in biochemical reactions commonly used to identify non-diphtherial Corynebacterium spp. which are difficult-to-differentiate from C. amycolatum in phenotypic tests, particularly C. xerosis (Santos et al., 2018). Besides, through comparative genomics we were able to identify specific target genes that can render reliable identification of C. striatum, C. amycolatum and C. xerosis clinical isolates, by multiplex PCR (Santos et al., 2017). More recently, different studies have been demonstrating the added value of whole-genome analyses to improve species circumscription in the genus Corynebacterium, including the study by Dover and collaborators (Dover et al., 2021) which proposes a new phylogenomic-based classification of the genus Corynebacterium, based on previous studies (Huson and Bryant, 2006), encompassing 19 phylogenetic groups; C. amycolatum belongs to the newly proposed group M, that also includes isolates of C. xerosis and C. freneyi (Dover et al., 2021). Noteworthy, all these previous studies were based on a limited number of isolates of the species C. amycolatum. Therefore, an extended pan-genomic analysis of the species can contribute to a better knowledge of the repertoire of gene families, and can aid the understanding of the taxonomy, pathogenicity, lifestyle, and resistome (Moradigaravand et al., 2018; Caputo et al., 2019; Kim et al., 2020).
In this study, we performed a pan-genomic analysis of the species C. amycolatum, including genome sequences of 26 isolates from different countries. Eight of these genomic sequences were newly generated in this work and were derived from clinical isolates of C. amycolatum from Spain and Tunisia, which presented multiple resistance to antimicrobial agents (Supplementary Tables S1, S2). Therefore, we can infer that the species C. amycolatum has an open pan-genome, with major horizontal acquisition of antimicrobial resistance genes through genomic islands and many virulence factors.
Materials and methods
Whole-genome sequencing of new clinical isolates and retrieval of Corynebacterium amycolatum genomic sequences from public databases
Next-generation sequencing was performed for eight new clinical isolates, which were identified as C. amycolatum / xerosis by the API Coryne biochemical battery and by MALDI-ToF mass spectrometry, according to standard protocols: strains FA111 and FA86 isolated at Farhat Hached Hospital (Sousse, Tunisia); strains VH1773, VH2077, VH2225, VH4147_1, VH4147_3, and VH6958 isolated at University Hospital Marqués de Valdecilla (Santander, Spain; please, see Supplementary Tables S1, S2 for clinical information and antimicrobial susceptibility profiles of the isolates). The isolates were cultured on blood agar plates for 48 h at 37°C, and the genomic DNA was extracted using the NucleoSpin Microbial DNA Kit (Macherey-Nagel). For next-generation sequencing using the Illumina HiSeq 2,500 platform (Illumina Inc.), sequencing libraries were prepared by the NEBNext® Fast DNA Fragmentation and Library Preparation Kit for Illumina® (New England Biolabs Inc.), as previously described (Rocha et al., 2020). Genome sequences were obtained for paired-end libraries with a minimum coverage of 1,000x. Genomic assemblies were obtained through the automated pipeline available at the PATRIC platform (Wattam et al., 2017) using SPAdes (Bankevich et al., 2012).
Eighteen additional genomic sequences for the species C. amycolatum (complete or draft) were retrieved from the National Center for Biotechnology Information (NCBI)’s GenBank (Tatusova et al., 2016).
Average nucleotide identity (ANIb) and TETRA
To certify that the genomic sequences are circumscribed within the C. amycolatum species, we performed average nucleotide identity by BLAST (ANIb) and tetranucleotide signature (TETRA) analyses through the JSpeciesWS platform (Richter et al., 2016).
Pan-genomic analysis
For standardization, all assembled genomic sequences were annotated using NCBI’s Prokaryotic Genome Annotation Pipeline (PGAP; Tatusova et al., 2016). Pan-genomic analysis was performed with the Bacterial Pan Genome Analysis (BPGA 1.3) tool (Chaudhari et al., 2016), using a 50% identity cut-off and the USEARCH pipeline for gene grouping (Edgar, 2010). BPGA uses the Power Law regression model (n = k. Nα) to determine whether the pan-genome is open (α ≤ 1) or closed (α > 1; Tettelin et al., 2005, 2008).
Functional annotations
The subgroups of the pan-genome were submitted for annotation of the Cluster of Orthologous Groups (COG) functional categories using the eggNOG-Mapper (Huerta-Cepas et al., 2017). The prediction of antibiotic resistance genes was performed in the Pathosystems Resource Integration Center (PATRIC) platform (Wattam et al., 2017) using the Comprehensive Antibiotic Resistance Database (CARD; Jia et al., 2017) and Database of Antibiotic-Resistant Organisms (NDARO).1 Virulence factors were evaluated through VFanalyzer and the Virulence Factor Database (VFDB; Liu et al., 2019). The key genes involved in the mycolic acid biosynthetic pathway were searched in the C. amycolatum genomes using the sequences and the method described by Dover and collaborators (Dover et al., 2021).
Predictions of genomic Islands, phages, and plasmid-derived sequences
IslandViewer 4 (Bertelli et al., 2017) was used for genomic islands prediction by integrating IslandPath-DIMOB (Hsiao et al., 2003), IslandPick (Langille et al., 2008), SIGI-HMM (Waack et al., 2006), and Islander (Hudson et al., 2015). Circular plots of the genomic sequences were plotted using BLAST Ring Image Generator (BRIG), including reference positions for antimicrobial resistance genes (AMR), virulence factors (VF), and genomic islands (GI; Alikhan et al., 2011).
Phage sequences were predicted with the Phage Search Tool Enhanced Release (PHASTER) platform (Arndt et al., 2016), which has approximately 187,000 phage sequences in the database. Plasmid searches were performed with the PlasmidFinder platform (Carattoli et al., 2014), which searches for plasmid replicons, and with the RFPlasmid platform (van der Graaf-van Bloois et al., 2021), which identifies plasmid sequences in contigs generated from short-read sequencing, by searching for specific proteins and plasmid replicons.
Deposit of genomic sequences in public databases
The genomic sequences generated in this study are publicly accessible through NCBI’s GenBank, with the respective accession numbers: JAFJMB000000000, JAFJMC000000000, JAFJMD000000000, JAFJME000000000, JAFJMF000000000, JAFJMG000000000, JAFJMH000000000, and JAFJMI000000000. A detailed description of the genomes can be found in Supplementary Table S1.
Results and discussion
General features of the Corynebacterium amycolatum genomes
Among the 26 C. amycolatum studied genomes, five were marked as complete genomes, and the remaining are in draft versions (see Supplementary Table S1). The estimated genome sizes range between 2.42 and 2.82 Mbp, with the G + C% content varying less than 1% between the isolates (58.6–59.0%). The numbers of annotated coding sequences (CDS) ranges from 2,038 (for isolate UMB1182) to 2,371 (for isolate FDAARGOS_991; see Supplementary Table S1).
The species assignment of the genomic sequences through ANIb (Figure 1) showed that the SK46 isolate was below the generally regarded cutoff value for species delineation (95.0%) when compared with other C. amycolatum isolates: identity between the SK46 and the NCTC7243 strain was 94.11%. Nevertheless, it has been described that the values above 94.0% are equivalent to 70% of DNA–DNA hybridization (DDH), a method considered as the gold standard for species identification (Richter and Rosselló-Móra, 2009). The results of the TETRA analysis were approximately 0.999, and the variation of the percentage of GC was ≤1%, reinforcing that the SK46 lineage is circumscribed within the species C. amycolatum (Figure 1).
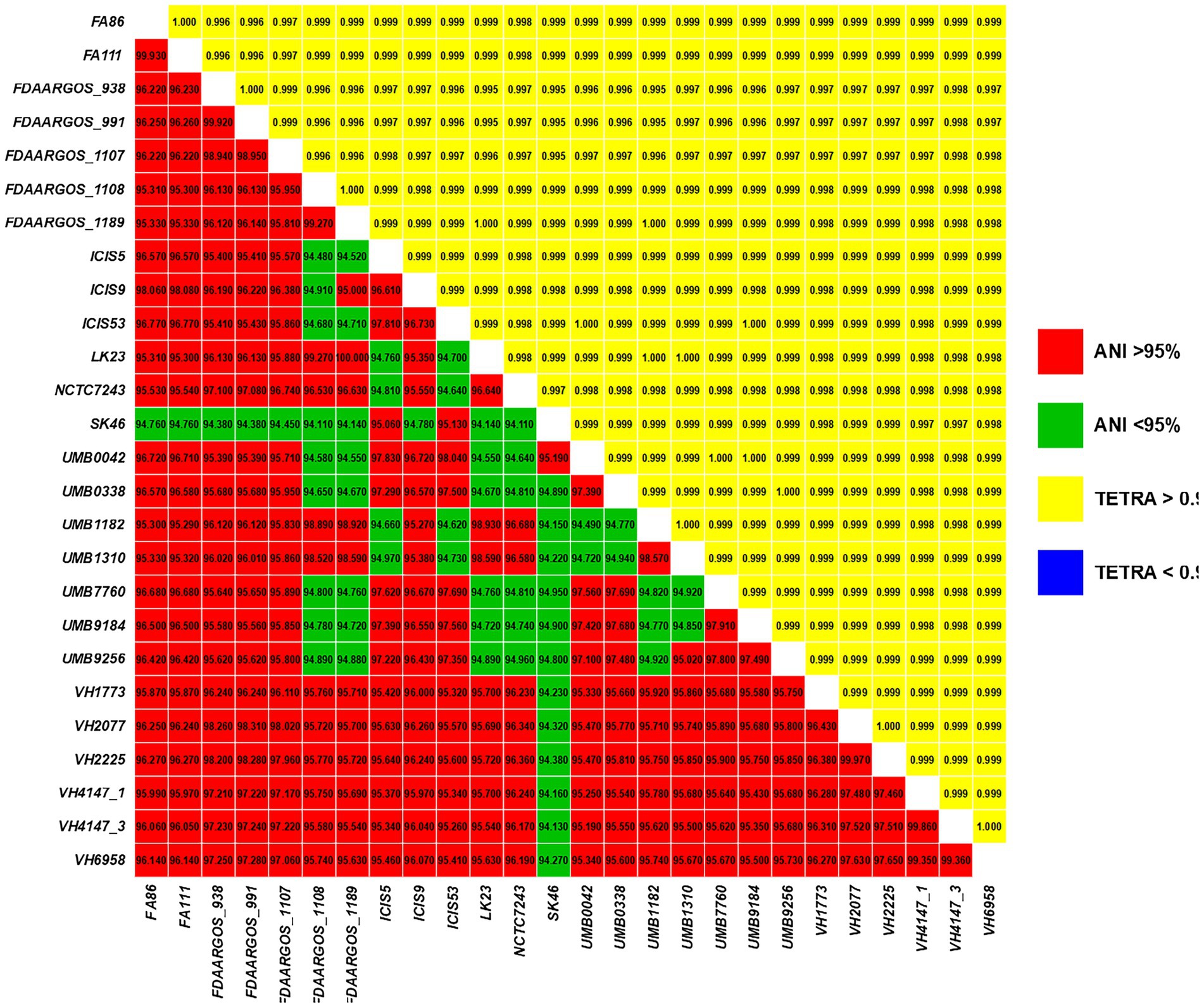
Figure 1. Pairwise comparisons of ANIb values (bottom triangle) and TETRA values (upper triangle) between all studied C. amycolatum genomic sequences.
Pan-genomic analysis and functional annotations by COGs
The C. amycolatum pan-genome has 3,280 predicted gene families (Figure 2), being 1,690 in the core genome (51.52%), 1,121 related to accessory genes (34.17%), and 469 related to unique genes (14.29%; Figure 3). The estimated α value of 0.854905 indicates an open pan-genome, and the predicted core genome stabilizes with approx. 1,641 gene families.
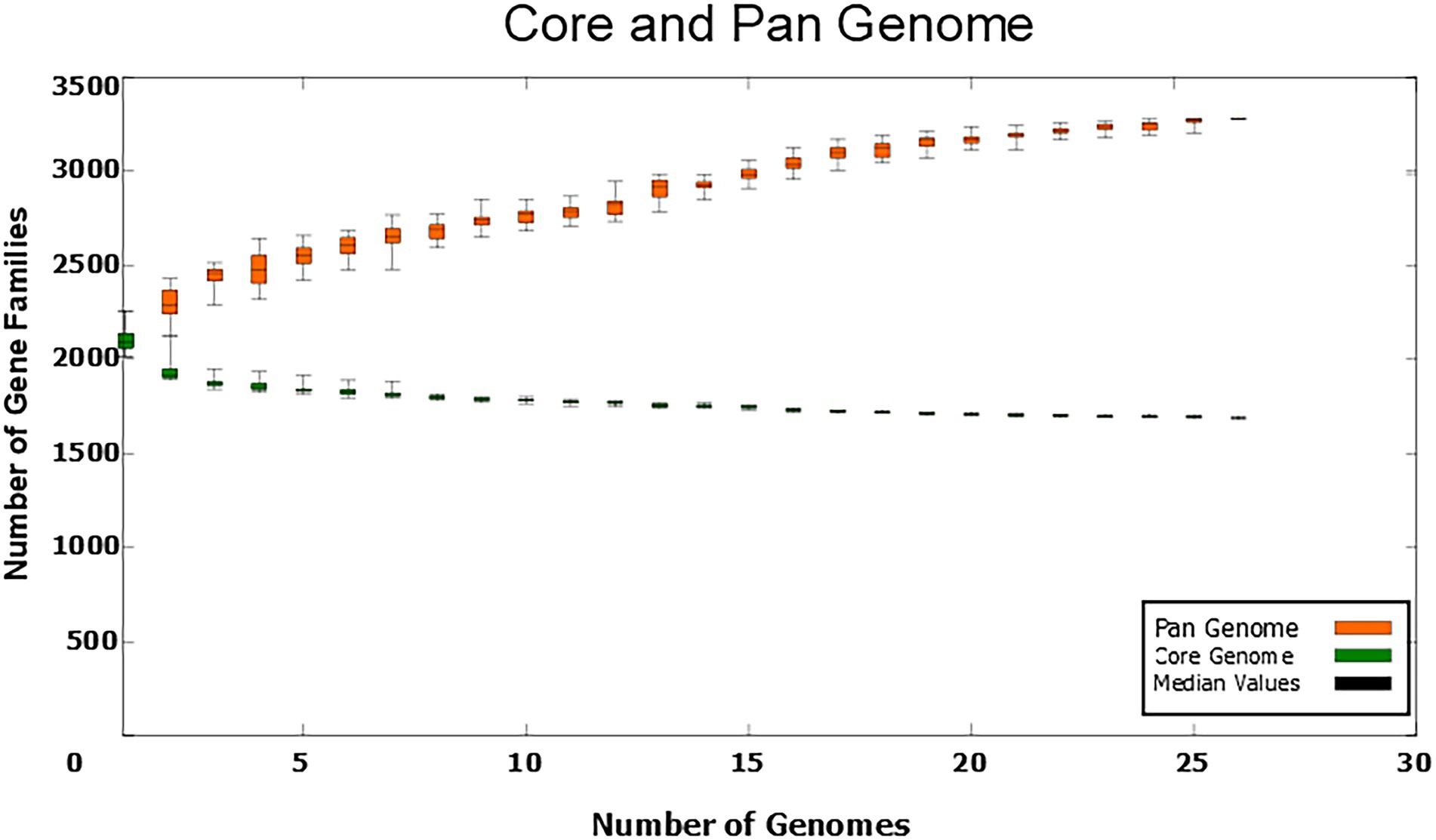
Figure 2. Numbers of gene families in the C. amycolatum pan-genome vs. numbers of new genomes added to the analysis.
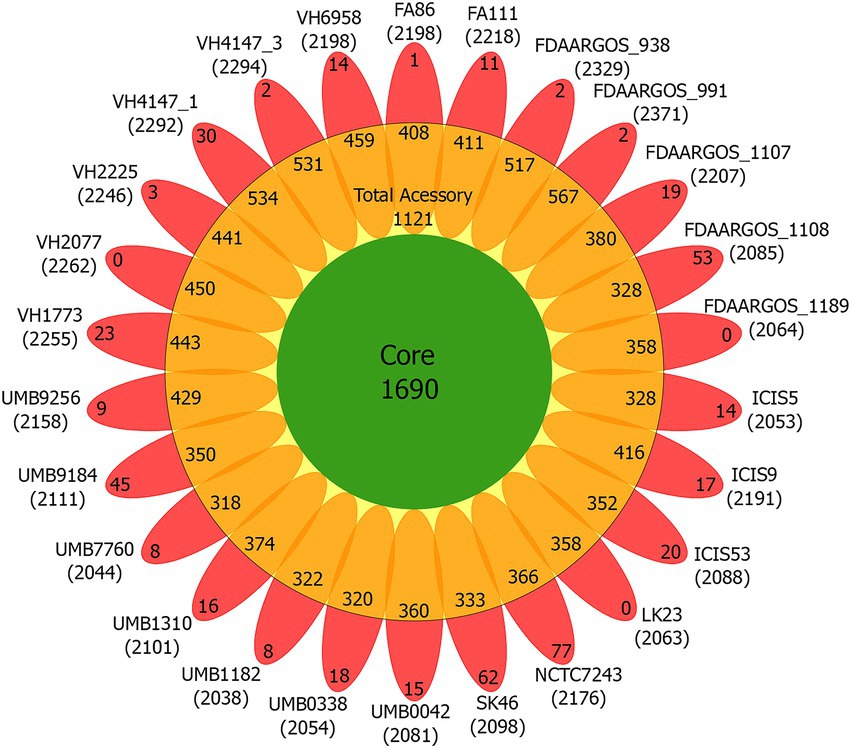
Figure 3. Flower plot diagram of the C. amycolatum pan-genome, showing core gene families (green), accessory genes present in each isolate (orange), and unique genes per genome (red). Within parentheses are the numbers of predicted coding sequences for each isolate.
The functional annotations of the pan-genomic subsets revealed that the core genome and accessory gene families are primarily classified in the ‘Metabolism’ category, with 582 and 216 annotated gene families (34.0 and 19.0%), respectively. Unique genes were mainly ranked in the ‘Information storage and processing’ category, with 72 genes in this class (15.0%). The main COG subcategories in the core genome were: translation, ribosomal structure and biogenesis (8.5%); amino acid transport and metabolism (6.7%); coenzyme transport and metabolism (5.8%); transcription (5.8%); and inorganic ion transport and metabolism (5.1%; Figure 4). Accessory genes were mainly involved in functions of replication, recombination, and repair (8.4%); inorganic ion transport and metabolism (6.1%); defense mechanisms (4.7%); transcription (4.1%); and amino acid transport and metabolism (3.7%). Unique genes were mostly related to biological functions of replication, recombination, and repair (10.4%); defense mechanisms (6.4%); transcription (4.3%); inorganic ion transport and metabolism (3.2%); and lipid transport and metabolism (2.1%). A total of 1,337 genes were labeled as ‘unknown function genes’, comprising 453 genes (26.8%) in the core genome, 589 (52.54%) in accessory genes, and 295 (62.89%) in the unique genes group (Figure 4).
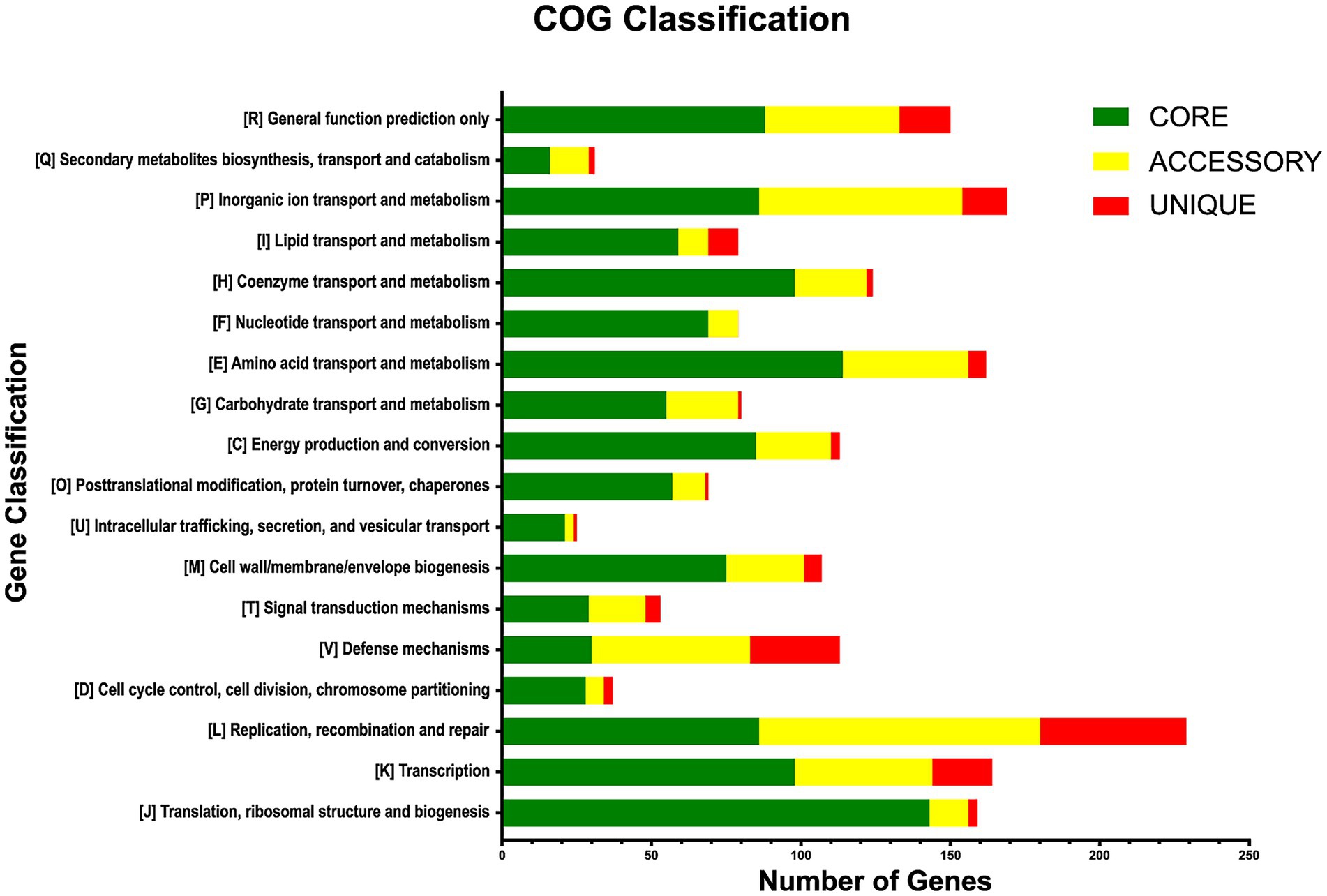
Figure 4. Functional annotations of the pan-genome subsets according to COG categories.
Prediction of antimicrobial resistance genes and genomic islands
The PATRIC platform identified nine antimicrobial resistance genes (AMRs) by automatic annotation of the C. amycolatum resistome (Figure 5). Only the rpsL gene was identified in all studied strains, containing mutations similar to those detected in streptomycin-resistant Mycobacterium tuberculosis isolates (Sreevatsan et al., 1996). Seven AMRs were placed in the accessory genome of C. amycolatum, which confer resistance to aminoglycosides, chloramphenicol, streptogramins, macrolides, lincosamides, and tetracycline: aac(3)-XI (aminoglycoside 3-N-acetyltransferase), identified in 15.0% of genomes; aph(3′)-Ia, aph(3″)-Ib, aph(6)-Id (aminoglycoside phosphotransferases), in 54% of isolates; cmx (efflux pump major facilitator superfamily, MFS), in 54% of isolates; ermX (Erm 23S ribosomal RNA methyltransferase), in 62% of isolates; and tetO (tetracycline resistance) in only 2 isolates. The tetW gene, encoding a ribosomal protection protein, was the single AMR gene found as unique in the pan-resistome of C. amycolatum, being detected only in the genomic sequence of isolate UMB9184 (Figure 5).
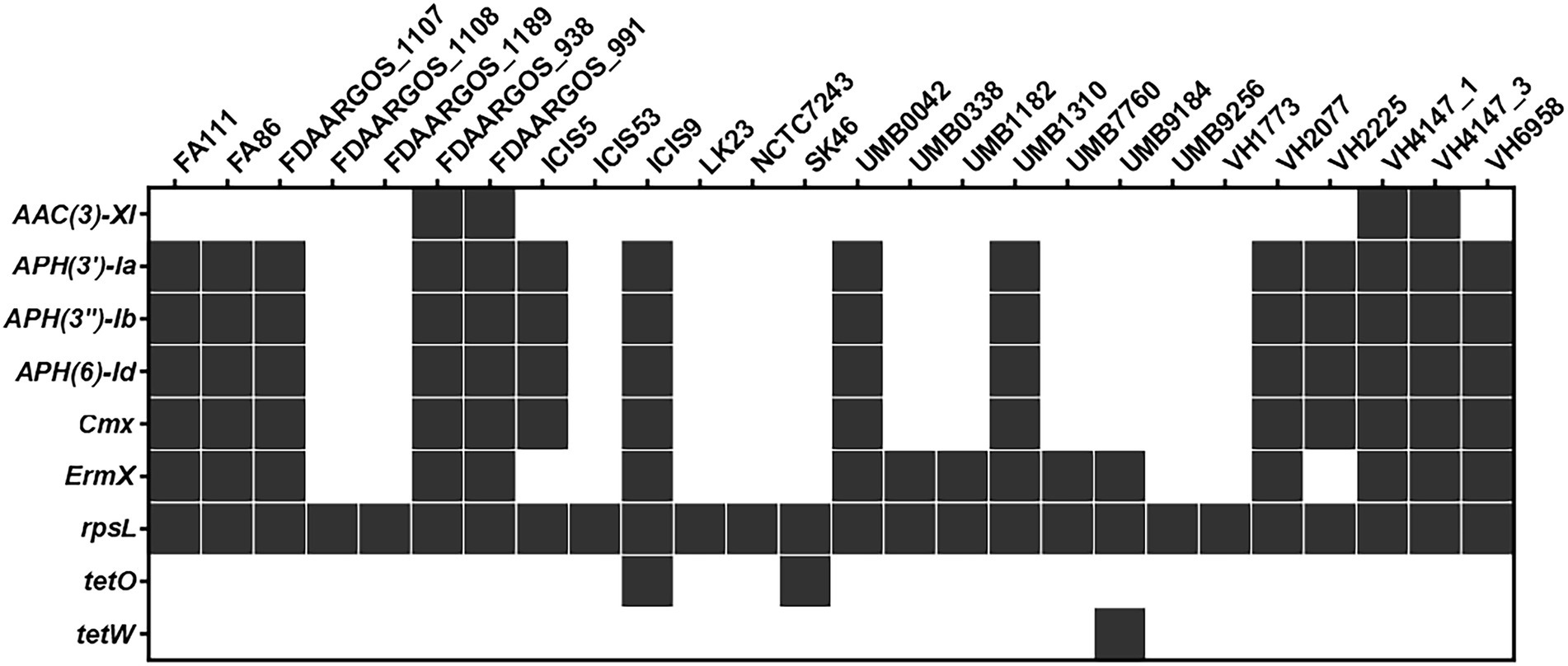
Figure 5. The C. amycolatum resistome predicted through automated annotation in the PATRIC platform.
Apart from the rpsL gene, all other predicted AMRs co-localize with predicted genomic islands in the studied genomes (Figure 6). The genes cmx, aph(3′)-Ia, aph(3″)-Ib, aph(6)-Id were consistently found within the exact genomic location (Figure 6), indicating a common mechanism of horizontal acquisition of AMR genes.
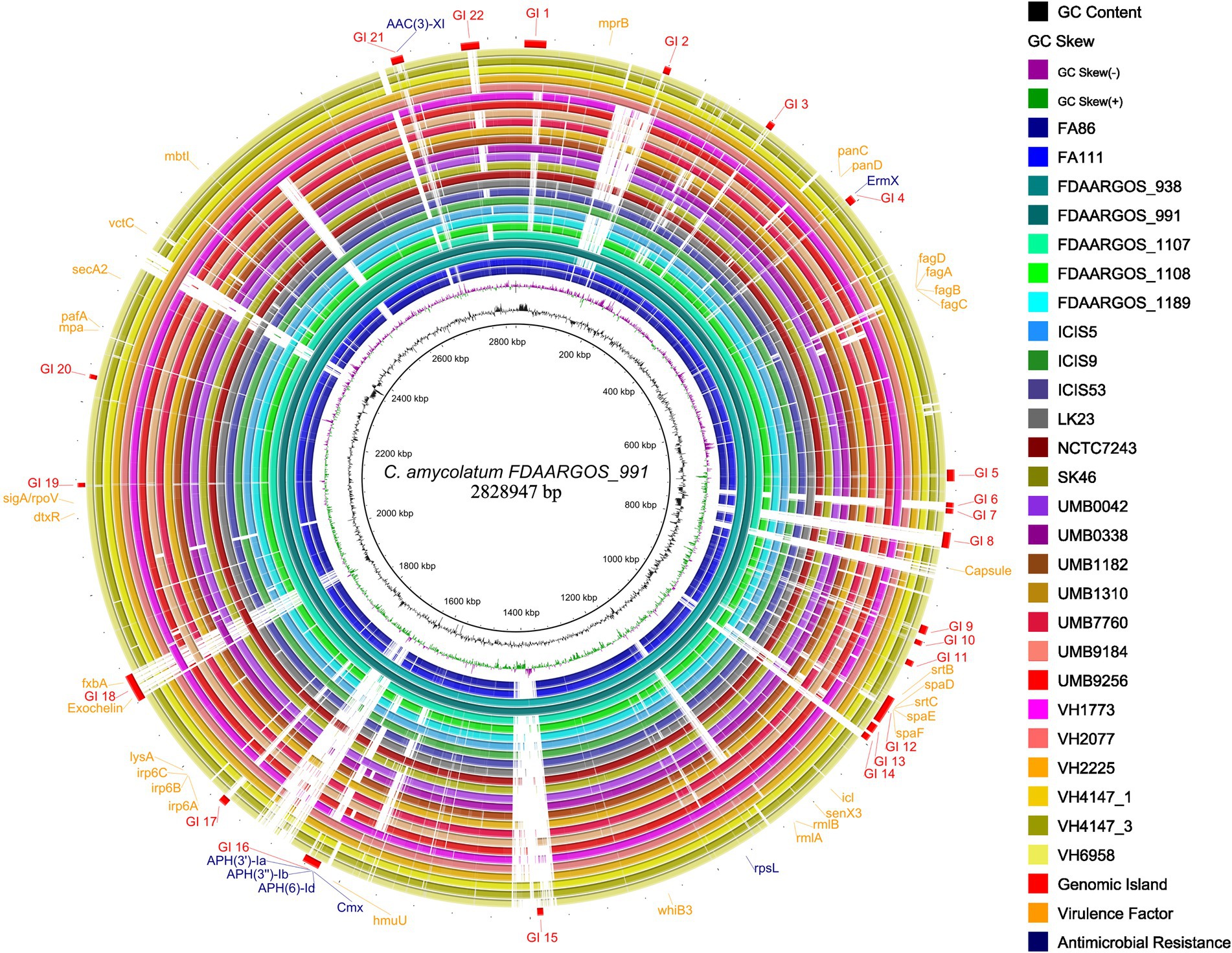
Figure 6. Circular genome plot showing the distribution of predicted genomic islands (GIs) in the studied C. amycolatum genomes. Most outer circle presents the positions of GIs and respective AMR genes. The genome sequence of the isolate FDAARGOS_911 was used as a reference.
Phages and plasmid-associated sequences in Corynebacterium amycolatum
The phage prediction detected 38 sequences (Supplementary Figure S1), the most frequent was the Corynebacterium Juicebox phage, present in 15 strains (Supplementary Figure S2), followed by the Corynebacterium phage SamW, identified in 6 strains. In total, 13 different phages were found. We identified the ermX gene within the Gordon phage Daredevil sequence in the C. amycolatum lineage UMB9184. The results generated by the RFPlasmid tool identified 36 sequences containing plasmid signatures among the studied strains, in which 27 AMR genes were present (Supplementary Table S2). This represents approximately 26% of the total predicted AMR genes. The ICIS5, ICIS9, VH2225, VH4147_1, and VH4147_3 strains presented sequences containing similar context with the AMR genes cmX, aph (6)-Id, aph(3″)-Ib, and aph(3′)-Ia. Analyzes performed with PlasmidFinder, however, did not detect any plasmid-related sequences, when searching for plasmid replicons.
Potential virulence factors
Forty-seven genes were found in the C. amycolatum pan-genome, which can be potentially associated with virulence functions (Figure 7). The majority of these virulence genes are present in the accessory genome (29 genes), while 12 genes are shared by all strains (core genome), and only 6 genes appear as unique to single isolates (Figure 7). Genes involved in iron acquisition are particularly enriched in this category of potential virulence genes, with 17 of those genes found in the species C. amycolatum. The operon ciuABDE, coding for an ABC-type siderophore transporter system (Kunkle and Schmitt, 2005), was found only in the strain NCTC7243. The fagABCD operon, coding for iron-siderophore transport through the membrane (Billington et al., 2002), was located entirely in 15 C. amycolatum genomes and partially found in additional 5 genomes. Twenty-four genomes also presented genes coding for the complete heterodimeric transporter Irp6ABC (Qian et al., 2002), while 2 genomes showed an incomplete coding potential. Additionally, the gene hmuU involved in the heme-transporter system hmuTUV of C. diphtheriae and C. ulcerans (Drazek et al., 2000) was found in all C. amycolatum genomes. An ortholog of the vctC gene that is part of the vctPDGC heme-transportation system in Vibrio cholerae (Wyckoff and Payne, 2011) was also found in 24 C. amycolatum genomes. Regarding siderophore biosynthesis pathways, we found orthologs in all studied genomes for the genes mbtI from Mycobacterium tuberculosis (Mori et al., 2020) and fxbA from Mycobacterium smegmatis. The latter is part of the biosynthetic pathway of mycobacterial exochelin (lipid- and water-soluble siderophore; Ojha and Hatfull, 2007) and was only found in two C. amycolatum isolates (FDAARGOS_938 and FDAARGOS_991).
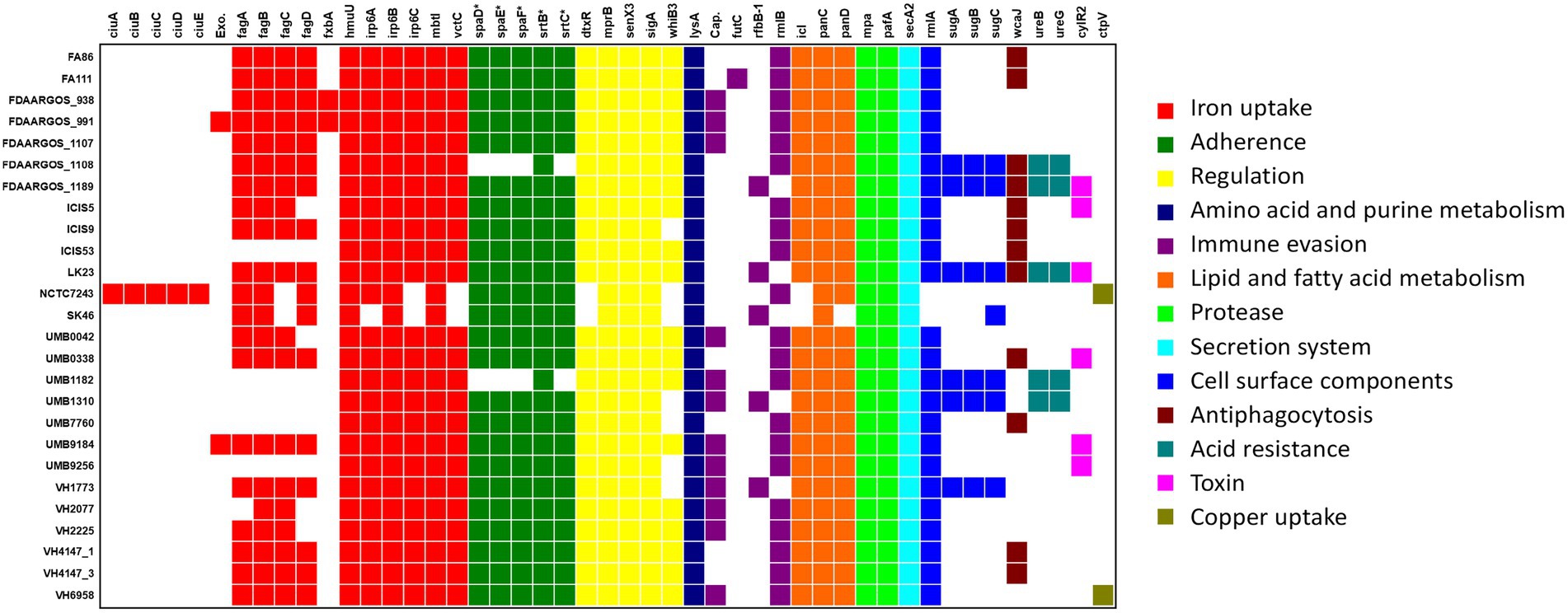
Figure 7. Distribution of virulence factors throughout the C. amycolatum genomic sequences. According to the legend, colors indicate the functional classes of the identified genes. Exo = exochelin coding sequence (ortholog to M. smegmatis gene); Cap = capsule related gene (ortholog to Acinetobacter spp). * Denotes gene sequences annotated by manual curation after automatic prediction.
Gene sequences coding for SpaD-like pili were predicted in most C. amycolatum isolates (Figure 7). In this adherence machinery, the proteins SpaD, SpaE, and SpaF form a filamentous structure that remains anchored to the bacterial surface and needs the sortases SrtB and SrtC for the anchoring step (Gaspar and Ton-That, 2006). These adherence structures are involved in essential pathogenicity functions that include host tissue colonization (Swaminathan et al., 2007), adherence under mechanical stress conditions (Echelman et al., 2016), and biofilm biogenesis (Swaminathan et al., 2007).
All C. amycolatum genomes presented genes coding for a functional ATP-dependent proteasome system, namely mpA (Mycobacterium proteasome ATPase) and pafA (proteasome accessory factor A; Pearce et al., 2008). Interestingly, six C. amycolatum genomes presented the cylR2 gene, whose product acts as a repressor of the cytolysin operon in Enterococcus faecalis (Haas et al., 2002).
The virulence genes fxbA, exc (exochelin), and the operons ciuABDE, sugABC, spaDEF plus the sortase genes srtB and srtC were mainly predicted within the context of genomic islands, showing their role in horizontal acquisition of variable genes.
These results were obtained from the VFDB database, which gathers information about virulence factors from studies that evaluated the ability of mutants to develop disease in the host (Liu et al., 2022). In this sense, our results reinforce the relevance of genes coding for pili (Broadway et al., 2013; Oliveira et al., 2017) and siderophores (Kunkle and Schmitt, 2003; Ibraim et al., 2019) in the Corynebacterium genus. Importantly, some studies have already discussed the important roles these genes play not only in virulence, but also in adaptation to distinct niches (Swierczynski and Ton-That, 2006; Tauch and Burkovski, 2015). Although we did not identify classic corynebacterial virulence factors in C. amycolatum, which are commonly associated with known pathogens of the Corynebacterium genus, such as diphtheria toxin, phospholipase D, and hemolysins (Dorella et al., 2006; Parveen et al., 2019), we were able to detect genes associated with immune evasion, antiphagocytosis, and toxins, that may be relevant to the pathogenicity of this species.
Mycolic acids are essential components of the cell wall of most Actinobacteria (Collins et al., 1982; Ioneda, 1993). They play a crucial role in the interaction of M. tuberculosis with host cells (Korf et al., 2005). However, C. amycolatum lacks corynomycolic acids in its cell wall (Barreau et al., 1993). The search for key genes of the mycolic acid biosynthesis pathway in C. amycolatum showed the absence of the essential genes (Supplementary Figure S3), especially the fadD32-pks13-accD4 operon (Portevin et al., 2005; Gavalda et al., 2009) and the cmrA gene (Lea-Smith et al., 2007) involved in mycolic acid condensation, then confirming that the species C. amycolatum does not have the genetic potential to synthesize mycolic acids; these findings corroborate the results from previous genomic studies of C. amycolatum (Daffé and Draper, 1997; Daffé, 2005; Baek et al., 2018; Dover et al., 2021).
Conclusion
The C. amycolatum pan-genome demonstrated an open status, which corroborates the high number of predicted genomic islands containing antimicrobial resistance genes (AMRs) and sequences coding for potential virulence factors. These biological functions are mainly acquired through horizontal gene transfer in the species. Notably, the high number of horizontally-acquired virulence genes that code for functions related to adaptation to the host organism, such as iron acquisition and adherence, may aid in the understanding of the pathogenic potential of this generally-regarded as commensal microorganism. In addition, the fact that we identified a genomic island containing genes that confer resistance to aminoglycosides and chloramphenicol in more than 50% of the studied isolates demonstrates the importance of unambiguous identification of this potentially pathogenic microorganism by clinical microbiology laboratories.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
HJ, DR, EA, and LP: investigation, formal analysis, methodology, software, data curation, visualization, and writing – original draft. RR, AS, BB, AG-N, MC, SS, VA, LM-M, AO, and SA: resources, project administration, and writing – review and editing. LP and JN: conceptualization, funding, project administration, supervision, and writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was partially supported by grants from FAPESB, CNPq, CAPES, FINEP, and RECOM Network, in Brazil. HJ was recipient of a PhD scholarship from FAPESB. LP was recipient of a research fellowship from CNPq.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1011578/full#supplementary-material
Footnotes
References
Alibi, S., Ferjani, A., Boukadida, J., Ruiz de Alegría, C., Martínez-Martínez, L., and Navas, J. (2017). Evaluation of the VITEK-MS matrix-assisted laser desorption/ionization time-of-flight mass spectrometry system for the identification of clinical Corynebacterium species. Rev. Esp. Quimioter. 30, 57–58.
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Baek, I., Kim, M., Lee, I., Na, S.-I., Goodfellow, M., and Chun, J. (2018). Phylogeny trumps chemotaxonomy: a case study involving Turicella otitidis. Front. Microbiol. 9:834. doi: 10.3389/fmicb.2018.00834
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barreau, C., Bimet, F., Kiredjian, M., Rouillon, N., and Bizet, C. (1993). Comparative chemotaxonomic studies of mycolic acid-free coryneform bacteria of human origin. J. Clin. Microbiol. 31, 2085–2090. doi: 10.1128/jcm.31.8.2085-2090.1993
Berner, R., Pelz, K., Wilhelm, C., Funke, A., Leititis, J. U., and Brandis, M. (1997). Fatal sepsis caused by Corynebacterium amycolatum in a premature infant. J. Clin. Microbiol. 35, 1011–1012. doi: 10.1128/jcm.35.4.1011-1012.1997
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Billington, S. J., Esmay, P. A., Songer, J. G., and Jost, B. H. (2002). Identification and role in virulence of putative iron acquisition genes from Corynebacterium pseudotuberculosis. FEMS Microbiol. Lett. 208, 41–45. doi: 10.1111/j.1574-6968.2002.tb11058.x
Borde, K., Rao, V., Shah, M., Pavani, N., and Anand, M. (2020). Not always a commensal: a case of mastitis by Corynebacterium amycolatum. IDCases 20:e00728. doi: 10.1016/j.idcr.2020.e00728
Broadway, M. M., Rogers, E. A., Chang, C., Huang, I.-H., Dwivedi, P., Yildirim, S., et al. (2013). Pilus Gene Pool variation and the virulence of Corynebacterium diphtheriae clinical isolates during infection of a nematode. J. Bacteriol. 195, 3774–3783. doi: 10.1128/JB.00500-13
Caputo, A., Fournier, P. E., and Raoult, D. (2019). Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct 14:5. doi: 10.1186/s13062-019-0234-0
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carvalho, R. V., Lima, F., Santos, C., Souza, M. C., Silva, R., and Mattos-Guaraldi, A. L. (2018). Central venous catheter-related infections caused by Corynebacterium amycolatum and other multiresistant non-diphtherial corynebacteria in paediatric oncology patients. Braz. J. Infect. Dis. 22, 347–351. doi: 10.1016/j.bjid.2018.07.004
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA – an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Collins, M. D., Burton, R. A., and Jones, D. (1988). Corynebacterium amycolatum sp. nov. a new mycolic acid-less Corynebacterium species from human skin. FEMS Microbiol. Lett. 49, 349–352. doi: 10.1111/j.1574-6968.1988.tb02755.x
Collins, M. D., Goodfellow, M., and Minnikin, D. E. (1982). Fatty acid composition of some mycolic acid-containing coryneform bacteria. J. Gen. Microbiol. 128, 2503–2509. doi: 10.1099/00221287-128-11-2503
Daffé, M. (2005). “The cell envelope of corynebacteria,” in Corynebacterium glutamicum, eds. L. Eggeling and M. Bott. (Boca Raton, FL: CRC Press), 121–148.
Daffé, M., and Draper, P. (1997). The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39, 131–203. doi: 10.1016/S0065-2911(08)60016-8
Dorella, F. A., Pacheco, L. G. C., Oliveira, S. C., Miyoshi, A., and Azevedo, V. (2006). Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 37, 201–218. doi: 10.1051/vetres:2005056
Dover, L. G., Thompson, A. R., Sutcliffe, I. C., and Sangal, V. (2021). Phylogenomic reappraisal of fatty acid biosynthesis, mycolic acid biosynthesis and clinical relevance among members of the genus Corynebacterium. Front. Microbiol. 12:802532. doi: 10.3389/fmicb.2021.802532
Dragomirescu, C. C., Lixandru, B. E., Coldea, I. L., Corneli, O. N., Pana, M., Palade,, et al. (2020). Antimicrobial susceptibility testing for Corynebacterium species isolated from clinical samples in Romania. Antibiotics 9:31. doi: 10.3390/antibiotics9010031
Drazek, E. S., Hammack, C. A., and Schmitt, M. P. (2000). Corynebacterium diphtheriae genes required for acquisition of iron from haemin and haemoglobin are homologous to ABC haemin transporters. Mol. Microbiol. 36, 68–84. doi: 10.1046/j.1365-2958.2000.01818.x
Echelman, D. J., Alegre-Cebollada, J., Badilla, C. L., Chang, C., Ton-That, H., and Fernández, J. M. (2016). CnaA domains in bacterial pili are efficient dissipaters of large mechanical shocks. Proc. Natl. Acad. Sci. U. S. A. 113, 2490–2495. doi: 10.1073/pnas.1522946113
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Funke, G., Lawson, P. A., Bernard, K. A., and Collins, M. D. (1996). Most Corynebacterium xerosis strains identified in the routine clinical laboratory correspond to Corynebacterium amycolatum. J. Clin. Microbiol. 34, 1124–1128. doi: 10.1128/jcm.34.5.1124-1128.1996
Gaspar, A. H., and Ton-That, H. (2006). Assembly of distinct pilus structures on the surface of Corynebacterium diphtheriae. J. Bacteriol. 188, 1526–1533. doi: 10.1128/JB.188.4.1526-1533.2006
Gavalda, S., Léger, M., van der Rest, B., Stella, A., Bardou, F., Montrozier, H., et al. (2009). The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis*. J. Biol. Chem. 284, 19255–19264. doi: 10.1074/jbc.M109.006940
Gladysheva, I. V., Cherkasov, S. V., Khlopko, Y. A., and Plotnikov, A. O. (2022). Genome characterization and probiotic potential of Corynebacterium amycolatum human vaginal isolates. Microorganisms 10:249. doi: 10.3390/microorganisms10020249
Haas, W., Shepard, B. D., and Gilmore, M. S. (2002). Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 415, 84–87. doi: 10.1038/415084a
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Hudson, C. M., Lau, B. Y., and Williams, K. P. (2015). Islander: a database of precisely mapped genomic islands in tRNA and tmRNA genes. Nucleic Acids Res. 43, D48–D53. doi: 10.1093/nar/gku1072
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Ibraim, I. C., Parise, M. T. D., Parise, D., Sfeir, M. Z. T., de Paula Castro, T. L., Wattam, A. R., et al. (2019). Transcriptome profile of Corynebacterium pseudotuberculosis in response to iron limitation. BMC Genomics 20:663. doi: 10.1186/s12864-019-6018-1
Ioneda, T. (1993). Chromatographic and mass spectrometric characterization of 3-O-benzoyl methyl ester derivatives of mycolic acid fractions from Corynebacterium pseudotuberculosis, C. diphtheriae and Rhodococcus rhodochrous. Chem. Phys. Lipids 65, 93–101. doi: 10.1016/0009-3084(93)90042-2
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kim, Y., Gu, C., Kim, H. U., and Lee, S. Y. (2020). Current status of pan-genome analysis for pathogenic bacteria. Curr. Opin. Biotechnol. 63, 54–62. doi: 10.1016/j.copbio.2019.12.001
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Korf, J., Stoltz, A., Verschoor, J., De Baetselier, P., and Grooten, J. (2005). The Mycobacterium tuberculosis cell wall component mycolic acid elicits pathogen-associated host innate immune responses. Eur. J. Immunol. 35, 890–900. doi: 10.1002/eji.200425332
Kunkle, C. A., and Schmitt, M. P. (2003). Analysis of the Corynebacterium diphtheriae DtxR regulon: identification of a putative siderophore synthesis and transport system that is similar to the yersinia high-pathogenicity Island-encoded yersiniabactin synthesis and uptake system. J. Bacteriol. 185, 6826–6840. doi: 10.1128/JB.185.23.6826-6840.2003
Kunkle, C. A., and Schmitt, M. P. (2005). Analysis of a DtxR-regulated iron transport and siderophore biosynthesis gene cluster in Corynebacterium diphtheriae. J. Bacteriol. 187, 422–433. doi: 10.1128/JB.187.2.422-433.2005
Langille, M. G., Hsiao, W. W., and Brinkman, F. S. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformat. 9:329. doi: 10.1186/1471-2105-9-329
Lea-Smith, D. J., Pyke, J. S., Tull, D., McConville, M. J., Coppel, R. L., and Crellin, P. K. (2007). The reductase that catalyzes mycolic motif synthesis is required for efficient attachment of mycolic acids to arabinogalactan*. J. Biol. Chem. 282, 11000–11008. doi: 10.1074/jbc.M608686200
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Moradigaravand, D., Palm, M., Farewell, A., Mustonen, V., Warringer, J., and Parts, L. (2018). Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 14:e1006258. doi: 10.1371/journal.pcbi.1006258
Mori, M., Stelitano, G., Gelain, A., Pini, E., Chiarelli, L. R., Sammartino,, et al. (2020). Shedding X-ray light on the role of magnesium in the activity of mycobacterium tuberculosis salicylate synthase (MbtI) for drug design. J. Med. Chem. 63, 7066–7080. doi: 10.1021/acs.jmedchem.0c00373
Ojha, A., and Hatfull, G. F. (2007). The role of iron in Mycobacterium smegmatis biofilm formation: the exochelin siderophore is essential in limiting iron conditions for biofilm formation but not for planktonic growth. Mol. Microbiol. 66, 468–483. doi: 10.1111/j.1365-2958.2007.05935.x
Oliveira, A., Oliveira, L. C., Aburjaile, F., Benevides, L., Tiwari, S., Jamal, S. B., et al. (2017). Insight of genus Corynebacterium: ascertaining the role of pathogenic and non-pathogenic species. Front. Microbiol. 8:1937. doi: 10.3389/fmicb.2017.01937
Parveen, S., Bishai, W. R., and Murphy, J. R. (2019). Corynebacterium diphtheriae: diphtheria toxin, the tox operon, and its regulation by Fe2+ activation of apo-DtxR. Microbiol. Spectr. 7:7.4.4. doi: 10.1128/microbiolspec.GPP3-0063-2019
Pearce, M. J., Mintseris, J., Ferreyra, J., Gygi, S. P., and Darwin, K. H. (2008). Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322, 1104–1107. doi: 10.1126/science.1163885
Portevin, D., de Sousa-D’Auria, C., Montrozier, H., Houssin, C., Stella, A., Lanéelle, M.-A., et al. (2005). The acyl-AMP ligase FadD32 and AccD4-containing Acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the Acyl-CoA carboxylase components*. J. Biol. Chem. 280, 8862–8874. doi: 10.1074/jbc.M408578200
Qian, Y., Lee, J. H., and Holmes, R. K. (2002). Identification of a DtxR-regulated operon that is essential for siderophore-dependent iron uptake in Corynebacterium diphtheriae. J. Bacteriol. 184, 4846–4856. doi: 10.1128/JB.184.17.4846-4856.2002
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rocha, D., Azevedo, V., Brenig, B., Silva, A., Blom, J., Ramos, R. T., et al. (2020). Whole-genome sequencing reveals misidentification of a multidrug-resistant urine clinical isolate as Corynebacterium urealyticum. J. Glob. Antimicrob. Resist. 23, 16–19. doi: 10.1016/j.jgar.2020.07.020
Sánchez Hernández, J., Mora Peris, B., Yagüe Guirao, G., Gutiérrez Zufiaurre, N., Muñoz Bellido, J. L., Segovia Hernández, M., et al. (2003). In vitro activity of newer antibiotics against Corynebacterium jeikeium, Corynebacterium amycolatum and Corynebacterium urealyticum. Int. J. Antimicrob. Agents 22, 492–496. doi: 10.1016/s0924-8579(03)00121-3
Santos, A. S., Ramos, R. T., Silva, A., Hirata, R. Jr., Mattos-Guaraldi, A. L., Meyer, R., et al. (2018). Searching whole genome sequences for biochemical identification features of emerging and reemerging pathogenic Corynebacterium species. Funct. Integr. Genomics 18, 593–610. doi: 10.1007/s10142-018-0610-3
Santos, C. S., Ramos, J. N., Vieira, V. V., Pinheiro, C. S., Meyer, R., Alcantara-Neves, N. M., et al. (2017). Efficient differentiation of Corynebacterium striatum, Corynebacterium amycolatum and Corynebacterium xerosis clinical isolates by multiplex PCR using novel species-specific primers. J. Microbiol. Methods 142, 33–35. doi: 10.1016/j.mimet.2017.09.002
Sengupta, M., Naina, P., Balaji, V., and Anandan, S. (2015). Corynebacterium amycolatum: an unexpected pathogen in the ear. J. Clin. Diagn. Res. 9:DD01–DD3. doi: 10.7860/JCDR/2015/15134.7002
Soltan Mohammadi, N., Mafakheri, S., Abdali, N., Bárcena-Uribarri, I., Tauch, A., and Benz, R. (2013). Identification and characterization of the channel-forming protein in the cell wall of Corynebacterium amycolatum. Biochim. Biophys. Acta 1828, 2574–2582. doi: 10.1016/j.bbamem.2013.06.024
Sreevatsan, S., Pan, X., Stockbauer, K. E., Williams, D. L., Kreiswirth, B. N., and Musser, J. M. (1996). Characterization of rpsL and rrs mutations in streptomycin-resistant Mycobacterium tuberculosis isolates from diverse geographic localities. Antimicrob. Agents Chemother. 40, 1024–1026. doi: 10.1128/AAC.40.4.1024
Swaminathan, A., Mandlik, A., Swierczynski, A., Gaspar, A., Das, A., and Ton-That, H. (2007). Housekeeping sortase facilitates the cell wall anchoring of pilus polymers in Corynebacterium diphtheriae. Mol. Microbiol. 66, 961–974. doi: 10.1111/j.1365-2958.2007.05968.x
Swierczynski, A., and Ton-That, H. (2006). Type III pilus of corynebacteria: pilus length is determined by the level of its major pilin subunit. J. Bacteriol. 188, 6318–6325. doi: 10.1128/JB.00606-06
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tauch, A., and Burkovski, A. (2015). Molecular armory or niche factors: virulence determinants of Corynebacterium species. FEMS Microbiol. Lett. 362:fnv185. doi: 10.1093/femsle/fnv185
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U. S. A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
van der Graaf-van Bloois, L., Wagenaar, J. A., and Zomer, A. L. (2021). RFPlasmid: predicting plasmid sequences from short-read assembly data using machine learning. Microb. genomics 7:000683. doi: 10.1099/mgen.0.000683
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformat. 7:142. doi: 10.1186/1471-2105-7-142
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Wauters, G., Van Bosterhaut, B., Janssens, M., and Verhaegen, J. (1998). Identification of Corynebacterium amycolatum and other nonlipophilic fermentative corynebacteria of human origin. J. Clin. Microbiol. 36, 1430–1432. doi: 10.1128/JCM.36.5.1430-1432.1998
Wyckoff, E. E., and Payne, S. M. (2011). The vibrio cholerae VctPDGC system transports catechol siderophores and a siderophore-free iron ligand. Mol. Microbiol. 81, 1446–1458. doi: 10.1111/j.1365-2958.2011.07775.x
Keywords: Corynebacterium amycolatum, pan-genome, multidrug resistance, emerging pathogen, virulence factor
Citation: Jesus HNR, Rocha DJPG, Ramos RTJ, Silva A, Brenig B, Góes-Neto A, Costa MM, Soares SC, Azevedo V, Aguiar ERGR, Martínez-Martínez L, Ocampo A, Alibi S, Dorta A, Pacheco LGC and Navas J (2022) Pan-genomic analysis of Corynebacterium amycolatum gives insights into molecular mechanisms underpinning the transition to a pathogenic phenotype. Front. Microbiol. 13:1011578. doi: 10.3389/fmicb.2022.1011578
Edited by:
Iain Sutcliffe, Northumbria University, United KingdomReviewed by:
Irina Gladysheva, Institute of Cellular and Intracellular Symbiosis (RAS), RussiaAndreas Burkovski, University of Erlangen Nuremberg, Germany
Michael Goodfellow, Newcastle University, United Kingdom
Copyright © 2022 Jesus, Rocha, Ramos, Silva, Brenig, Góes-Neto, Costa, Soares, Azevedo, Aguiar, Martínez-Martínez, Ocampo, Alibi, Dorta, Pacheco and Navas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis G. C. Pacheco, bGdjcGFjaGVjb0BnbWFpbC5jb20=; Jesus Navas, bmF2YXNqQHVuaWNhbi5lcw==