 Zhiyang Liu
Zhiyang Liu Mei Meng
Mei Meng ShiJian Ding
ShiJian Ding XiaoChao Zhou
XiaoChao Zhou KaiYan Feng
KaiYan Feng Tao Huang
Tao Huang Yu-Dong Cai
Yu-Dong Cai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 September 2022
Sec. Systems Microbiology
Volume 13 - 2022 | https://doi.org/10.3389/fmicb.2022.1007295
This article is part of the Research Topic Computational and Systems Biology Methods for Elucidating Associations Between Cancer and Microbes View all 19 articles
Patients infected with SARS-CoV-2 at various severities have different clinical manifestations and treatments. Mild or moderate patients usually recover with conventional medical treatment, but severe patients require prompt professional treatment. Thus, stratifying infected patients for targeted treatment is meaningful. A computational workflow was designed in this study to identify key blood methylation features and rules that can distinguish the severity of SARS-CoV-2 infection. First, the methylation features in the expression profile were deeply analyzed by a Monte Carlo feature selection method. A feature list was generated. Next, this ranked feature list was fed into the incremental feature selection method to determine the optimal features for different classification algorithms, thereby further building optimal classifiers. These selected key features were analyzed by functional enrichment to detect their biofunctional information. Furthermore, a set of rules were set up by a white-box algorithm, decision tree, to uncover different methylation patterns on various severity of SARS-CoV-2 infection. Some genes (PARP9, MX1, IRF7), corresponding to essential methylation sites, and rules were validated by published academic literature. Overall, this study contributes to revealing potential expression features and provides a reference for patient stratification. The physicians can prioritize and allocate health and medical resources for COVID-19 patients based on their predicted severe clinical outcomes.
Since its outbreak in late 2019, COVID-19, which is caused by SARS-CoV-2, has resulted in more than 5 million deaths. SARS-CoV-2 binds to the spike (S) protein primarily through its functional receptor ACE2, an 805-amino acid type I transmembrane protein, allowing the virus to attach to the host cell membrane. This process results in alteration of the extracellular domain of ACE2 and internalization of the transmembrane domain, leading to further fusion of the viral particle with the host cell (Schulte-Schrepping et al., 2020). The SARS-CoV-2 infection progresses to different severities, including discharge from the emergency department, hospitalization, transfer to the ICU, and death, due to a variety of factors, such as age, gender, and other underlying diseases (Konigsberg et al., 2021). Therefore, rapidly determining the severity of the patient and taking corresponding treatment measures for timely and effective diagnosis and treatment is crucial.
Viruses can escape the immune clearance of the body through a variety of ways, among which epigenetic modification is an important way for respiratory viruses to resist the immune response of the body. DNA methylation, mainly of CpG islands, is a crucial reversible epigenetic regulation process (Fan et al., 2017; Benhamida et al., 2020). The regulation of the activity of a variety of DNA/RNA viruses, including HIV, HBV, and HPV, is related to changes in DNA methylation (Castro De Moura et al., 2021). Studies have shown that MERS-CoV and H5N1 influenza virus infection leads to methylation of antigen-presenting gene promoters in infected cells, which eliminates the expression of related genes, thereby antagonizing antigen presentation, resulting in impaired T-lymphocyte function during acute infection and aggravating the degree of virus infection in the body (Hatta et al., 2010; Menachery et al., 2018). Similarly, as a respiratory virus, SARS-CoV infection also results in DNA methylation in host cells (Menachery et al., 2018). Among them, the hypermethylation of the IFN pathway and inflammation-related genes is an important feature of severe COVID-19 (Corley et al., 2021). The study of ACE2 revealed that the DNA in the CpG island of the ACE2 promoter in lung epithelial cells is hypomethylated, indicating its high expression in the lung. Moreover, its methylation status was significantly correlated with age and gender, explaining the effect of age and gender on the severity of COVID-19 (Kianmehr et al., 2021). In addition, ACE2 mRNA is highly expressed in various diseases, especially cancer, which may be an important reason for the severe COVID-19 caused by the underlying disease in SARS-CoV infection (Sen et al., 2021). RNA modification, namely N 6-methylation of adenosine (m6A), also plays an important role in evading the innate immune recognition of exogenous RNA of the host, affecting virus structure and replication (Eberle et al., 2021). The study of human metapneumovirus showed that m6A-binding protein can label viral RNA as the RNA of the host after binding to m6A, thereby evading the antiviral response of the host (Durbin et al., 2016; Chen et al., 2019). In addition, some studies have found that the N region of the SARS-CoV-2 virus genome is rich in m6A modification and is regulated by the host cell methyltransferase METTL3. The reduced expression level of METTL3 will lead to a decrease in the level of SARS-CoV-2 m6A and correspondingly increased expression of inflammatory genes (Li S. et al., 2021). This process is more pronounced in severely infected patients than that in moderately infected patients. These findings suggest the possibility of using methylation to characterize disease states, and numerous studies have demonstrated the feasibility of this approach.
This study conducted a computational investigation on the blood methylation profile on severity of SARS-CoV-2 infection. Several advanced machine learning methods were adopted. First, the profile was analyzed by the Monte Carlo feature selection (MCFS) method (Dramiński et al., 2007) to analyze the importance of methylation features. One feature list was produced, which was further analyzed by incremental feature selection (IFS) (Liu and Setiono, 1998) method. Four classification algorithms were adopted in the IFS method to discover their optimal features, and build the optimal classifiers and classification rules. For the essential methylation features, their corresponding genes were picked up for gene ontology (GO) and KEGG enrichment analysis. Some results, including essential methylation sites, classification rules, and enrichment analysis results, were extensively discussed and can be validated by existing literature. The results reported in this study are helpful for the stratification of clinical patients and provide an effective reference for clinical diagnosis and treatment.
The blood DNA methylation dataset investigated in this study was obtained from the Gene Expression Omnibus (GEO) database with the accession ID of GSE167202 (Konigsberg et al., 2021). This dataset comprised 164 SARS-CoV-2-positive samples, 296 SARS-CoV-2-negative infection samples, and 65 other infection samples. In addition, the positive samples were classified into four categories based on severity score. The severity score is determined primarily by discharge from emergency, admission to inpatient care, progression to the ICU, and death. The above four categories, negative infection, and other infections were termed as six classes in this study. The methylation dataset was deeply analyzed by modeling a classification problem on the dataset. The sample size of each class is listed in Table 1. Each sample was represented by 655,010 methylation features. This dataset would be analyzed in the following steps.

Table 1. Sample size of each class for the methylation profile.
A large number of methylation features were used to represent each sample. However, only a few of them were highly related to the severity of SARS-CoV-2 infection. It was necessary to reveal essential methylation features with advanced computer techniques. Here, MCFS method was employed (Dramiński et al., 2007).
MCFS is a tree-based feature selection method that is widely used in methylation profiling analysis as it is deemed to be good at dealing with datasets containing small number of samples and huge number of features. It randomly constructs several decision trees (DTs) from the original training dataset and uses these DTs to evaluate the importance of features. More specifically, s subsets with m features are randomly selected from the original training dataset. t trees for each subset are then constructed based on samples randomly sampled from the original dataset. The performance of each tree is evaluated on test samples that are not selected as training samples. Overall, DTs are built in this process. The overall position of a feature on the tree node partition is used to estimate a measurement, called relative importance (RI). A high RI score of a feature indicates the importance of a feature. The RI score is defined as follows:
where indicates the information gain of tree node , and represent the number of samples in node and tree , respectively, and indicates the weighted accuracy of the DT . In addition, u and v are the two parameters for RI calculation.
The MCFS program developed by Dramiński et al. was applied in this study, which can be downloaded at https://home.ipipan.waw.pl/m.draminski/mcfs.html, to rank the methylation features. Default parameters were used, where u and v were set to 1. By applying the MCFS program on the methylation dataset, a ranked feature list was obtained.
Based on the MCFS method, the methylation features were ranked in a list. However, the threshold was difficult to determine, that is, which features were selected for further analysis. In view of this, we further employed the IFS method (Liu and Setiono, 1998).
The IFS method is always used to determine the optimal number of features in a ranked feature list combined with one supervised classification algorithm, such as random forest (RF). More specifically, IFS first generates a series of feature subsets based on a step size. For example, the first and second subsets, respectively, comprise the top 5 and 10 features when the step size is five. Next, on each feature subset, the samples represented by features in such subset are learned by the given classification algorithm, thereby building a classifier. Its performance is evaluated by the 10-fold cross-validation (Kohavi, 1995). After the evaluation metrics of all classifiers are obtained, the classifier with the highest performance is easy to find. Such classifier is called the optimal classifier. The corresponding feature subset is picked up and features in this subset are termed as the optimal features for the used classification algorithm.
As shown in Table 1, the sample sizes under six classes were quite different. The largest class contained samples about 17 times as many as those in the smallest class. This may lead to biased performance of the established classifiers. Therefore, the synthetic minority oversampling technique (SMOTE) algorithm (Chawla et al., 2002; Ding et al., 2022; Zhou et al., 2022), an oversampling method, was applied to solve the problem. The core idea of SMOTE is to generate new samples to each minor class for enlarging its size. For each minor class, SMOTE randomly selects one sample, say x, from this class and finds its k-nearest neighbor samples in the same class. One sample, say y, is randomly selected from these k-nearest neighbor samples. One new sample is synthesized by the linear combination of x and y. As such new sample is highly related to x and y, it belongs to the same class with a high probability. Thus, it is put into the minor class. Such procedures execute several times until the size of the minor class is equal to that of the major class.
In this study, the SMOTE program from the imblearn package1 was used to process the methylation data for solving the imbalanced problem when constructing classifiers in the IFS method.
As the execution of IFS method needs one classification algorithm, four classic classification algorithms were attempted in this study to fully assess each constructed feature subset. They were k-nearest neighbor (kNN; Cover and Hart, 1967), RF (Breiman, 2001), support vector machine (SVM; Cortes and Vapnik, 1995), and DT (Safavian and Landgrebe, 1991). Their brief descriptions were as follows.
k-Nearest neighbor is one of the most classic classification algorithms. It determines the class of a sample based on measuring the distance between samples. Given a training dataset, for a new test sample, the k neighbors closest to such sample are found in the training dataset. By counting the classes of its k neighbors, the class of the test sample can be determined. Generally, the class that occurs most for its k neighbors is assigned to the test sample.
RF is an ensemble algorithm that contains several DTs. Each DT is constructed by randomly selecting samples from the original dataset and features from all features. RF provides the final prediction result using the voting strategy on predictions yielded by DTs. RF is generally much more powerful than its component DT, and few parameters are involved in this algorithm.
SVM is an excellent classification algorithm in machine learning. The original SVM can only tackle binary classification. It separates samples into two classes by constructing a hyperplane, which can separate samples into two classes with the maximum interval. However, such hyperplane does not always exist or is not easy to find out. SVM maps samples into a high-dimensional space using one kernel function. In the new space, the hyperplane can be easily constructed. For a test sample, it is also mapped into the high-dimensional space. Its class is determined by the side it lies. The “one-versus-rest” or “one-versus-one” can be adopted to generalize the original SVM so that it can tackle multi-class classification problems.
Different from the above algorithms, which are deemed as black-box algorithms, DT can make the classification procedures interpretable. By learning the distributions of samples under each feature, a tree-like structure is built by DT. In this structure, each internal node indicates a decision on an attribute, outputting a judgment result, and each leaf node denotes a classification outcome. Besides, DT can also be represented by a set of rules. Each rule is obtained by a path from the root node to one leaf node in the tree. In terms of these rules, the class of a test sample can be determined. This operation also makes the classification procedures completely open, giving more chances for us to understand the procedures. In this case, more meaningful and hidden information in the dataset can be mined.
Above classification algorithms have wide applications in many fields. They are always important candidates for building classifiers in tackling various biological and medical problems (Zhou et al., 2020a,b, 2022; Chen et al., 2021, 2022; Onesime et al., 2021; Zhang Y. et al., 2021; Ding et al., 2022; Li et al., 2022; Ran et al., 2022; Tang and Chen, 2022; Wang and Chen, 2022; Wu and Chen, 2022; Yang and Chen, 2022). These algorithms were implemented in this study through the scikit-learn (Pedregosa et al., 2011) program in Python and run with default parameters.
The prediction performance of each classifier was mainly evaluated with the weighted F1. Its calculation is based on the F1 score on each class. The F1 score for one class can be computed by
where TP, FP, and FN represent true-positive, false-positive, and false-negative for the class, respectively. The weighted F1 is defined as the weighed mean of F1 scores on all classes. The direct mean of F1 scores on all classes was also provided, which was called macro F1.
To fully evaluate the performance of classifiers in the IFS method, we also adopted overall accuracy (ACC) and Matthews correlation coefficients (MCC; Matthews, 1975; Gorodkin, 2004). ACC is defined as the ratio of correctly predicted samples and all samples, which is the most accepted measurement. However, it is not perfect when the class sizes are of great differences. In view of this, MCC was proposed, which is deemed as a balanced measurement. For computing MCC, two binary matrices X and Y should be constructed first, where X stands for the true class of each sample and Y represents the predicted class of each sample. Then, MCC can be computed by
According to the IFS results, the essential methylation features for severity of SARS-CoV-2 infection can be obtained. Their corresponding genes can be picked up for further analysis. GO and KEGG enrichment analysis is a common method for uncovering biological meanings behind a set of genes. Here, it was applied to discover the biofunctional information of the genes corresponding to essential methylation features. Such analysis was performed by using the R package clusterProfiler 4.0 (Wu et al., 2021) with a threshold of 0.05.
This study conducted a deep computational investigation on the blood methylation profile with six severity types from the GEO database. The entire procedures are illustrated in Figure 1. The MCFS method was first used to rank methylation features based on their importance, and a ranked feature list was generated. This list was then fed into the IFS method with different classification algorithms to determine the optimal features for each classification algorithm and construct optimal classifiers. Classification rules generated by the optimal DT classifier were used to analyze the expression pattern of key methylation features.
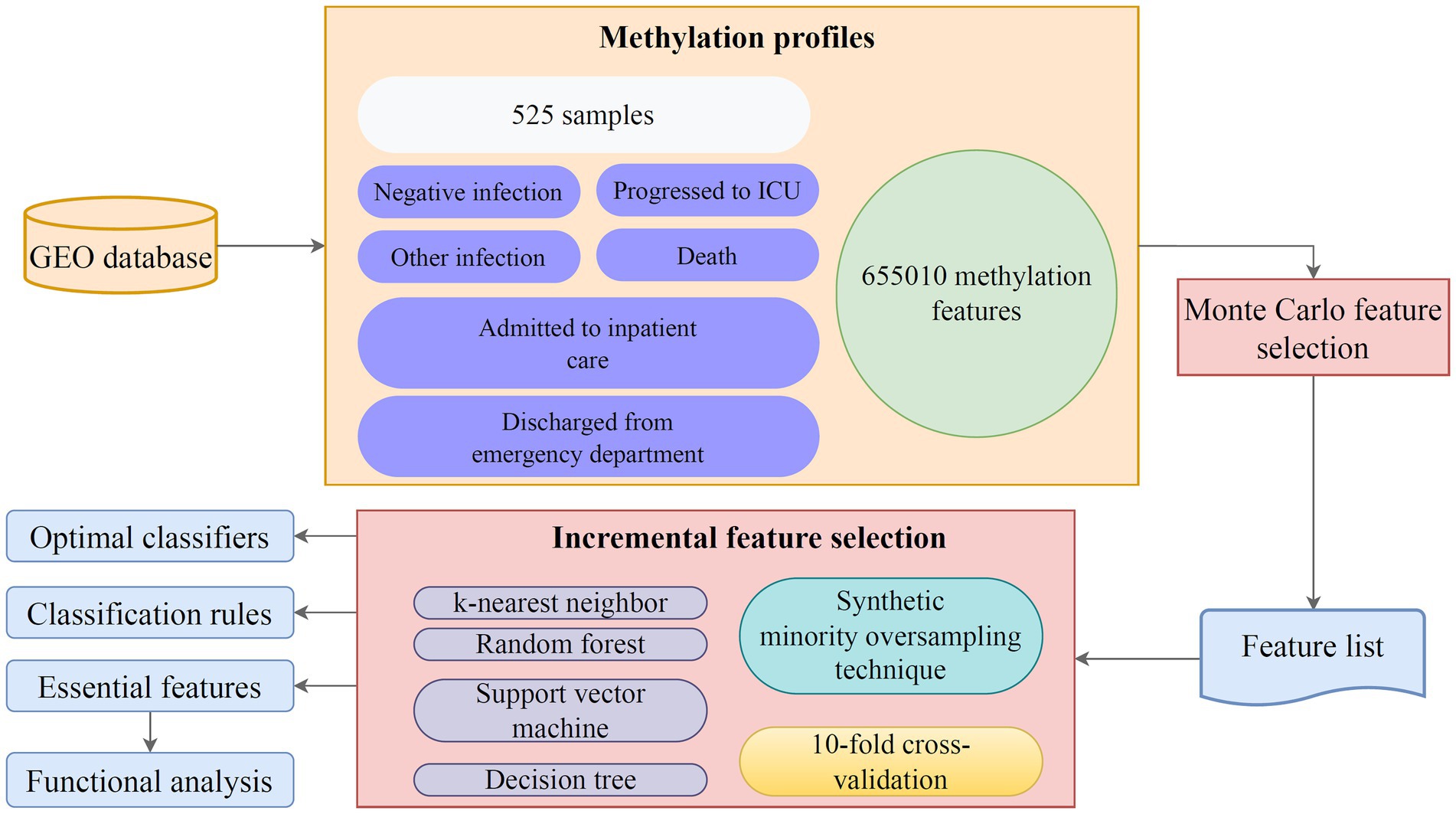
Figure 1. Workflow of this study. First, the Monte Carlo feature selection (MCFS) method was used to rank methylation signatures based on their importance, and a ranked feature list was generated. This list was then fed into the incremental feature selection (IFS) method with different classification algorithms to determine the optimal features for each classification algorithm. Optimal classifiers were set up. Classification rules generated by the optimal decision tree (DT) classifier were used to analyze the methylation expression pattern. The genes corresponding to essential methylation sites were subjected to functional enrichment analysis.
Initially, the MCFS method was used to rank 655,010 methylation features contained in blood the methylation profile. Each feature was assigned a RI score. A ranked feature list in descending order based on RI scores was generated. As some features were assigned RI scores of 0, they were removed. Thus, the final list contained 654,081 features with RI scores larger than 0, which is provided in Supplementary Table S1. The top 10 features alone with their RI score are plotted in Figure 2.
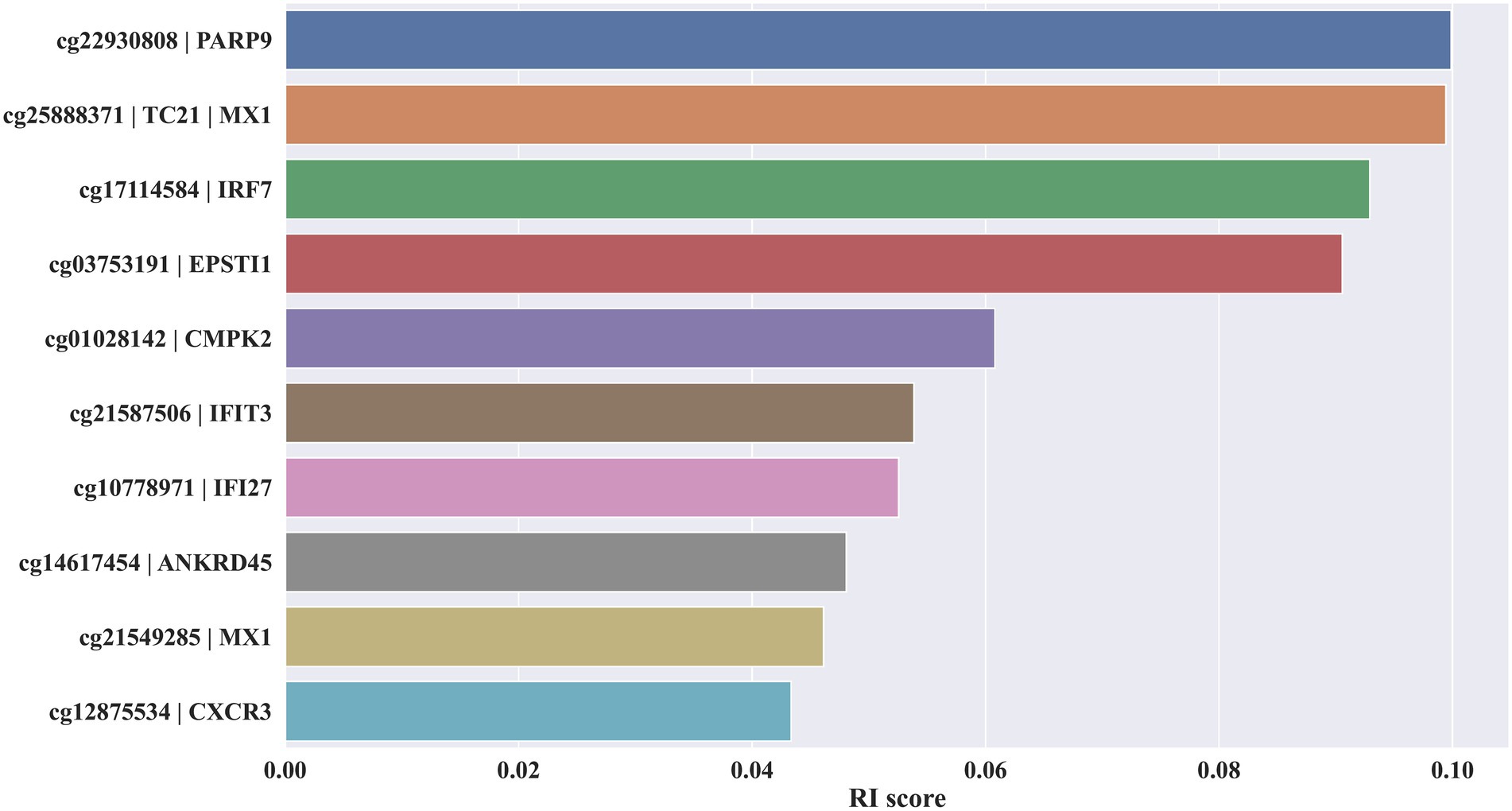
Figure 2. Bar chart to show top 10 key methylation features and their relative importance scores.
The IFS method was applied to determine the optimal features in the ranked feature list for each classification algorithm. To save time, we only considered top 2000 features in the list because of the huge number of features. The step size is set to five in the IFS method, thereby generating 400 feature subsets. The sample dataset comprising these feature subsets was learned by each of four classification algorithms, namely DT, kNN, RF, and SVM. Lots of classifiers were built, which were evaluated by 10-fold cross-validation. The evaluation metrics for each classifier are provided in Supplementary Table S2. To clearly display the performance of classifiers under different feature subsets, an IFS curve is plotted for each classification algorithm, which is provided in Figure 3. For SVM, the highest weighted F1 was 0.921 when top 1,025 features were adopted. These features constituted the optimal features for SVM and an optimal SVM classifier was built based on these features. As for kNN and RF, their highest weighted F1 values were 0.790 and 0.895, respectively. Their optimal features were top 10 and 35 features in the list. Furthermore, the optimal kNN and RF classifiers were set up with their optimal features, respectively. For DT, its highest weighted F1 was 0.780, which was obtained by using top 590 features. Such features comprised the optimal features for DT and the optimal DT classifier was built using these optimal features. According to the weighted F1 values of above optimal classifiers, the optimal SVM classifier was best, followed by the optimal RF and kNN classifiers, whereas the optimal DT classifier provided the lowest performance. Table 2 further lists other overall measurements for four optimal classifiers. It can be observed that on each measurement, the optimal SVM classifier always provided the highest performance, and the optimal RF classifier yielded slightly lower performance than the optimal SVM classifier. The performance of the other two optimal classifiers was much lower. The optimal DT classifier was a little inferior to the optimal kNN classifier. As for the performance of the above four optimal classifiers on six classes, it is illustrated in Figure 4. Clearly, the optimal SVM classifier generated the highest performance on all classes. On most classes, the optimal RF classifier occupied the second places. The optimal kNN and DT classifiers gave an almost equal performance. These results conformed to the overall performance of four optimal classifiers mentioned above.
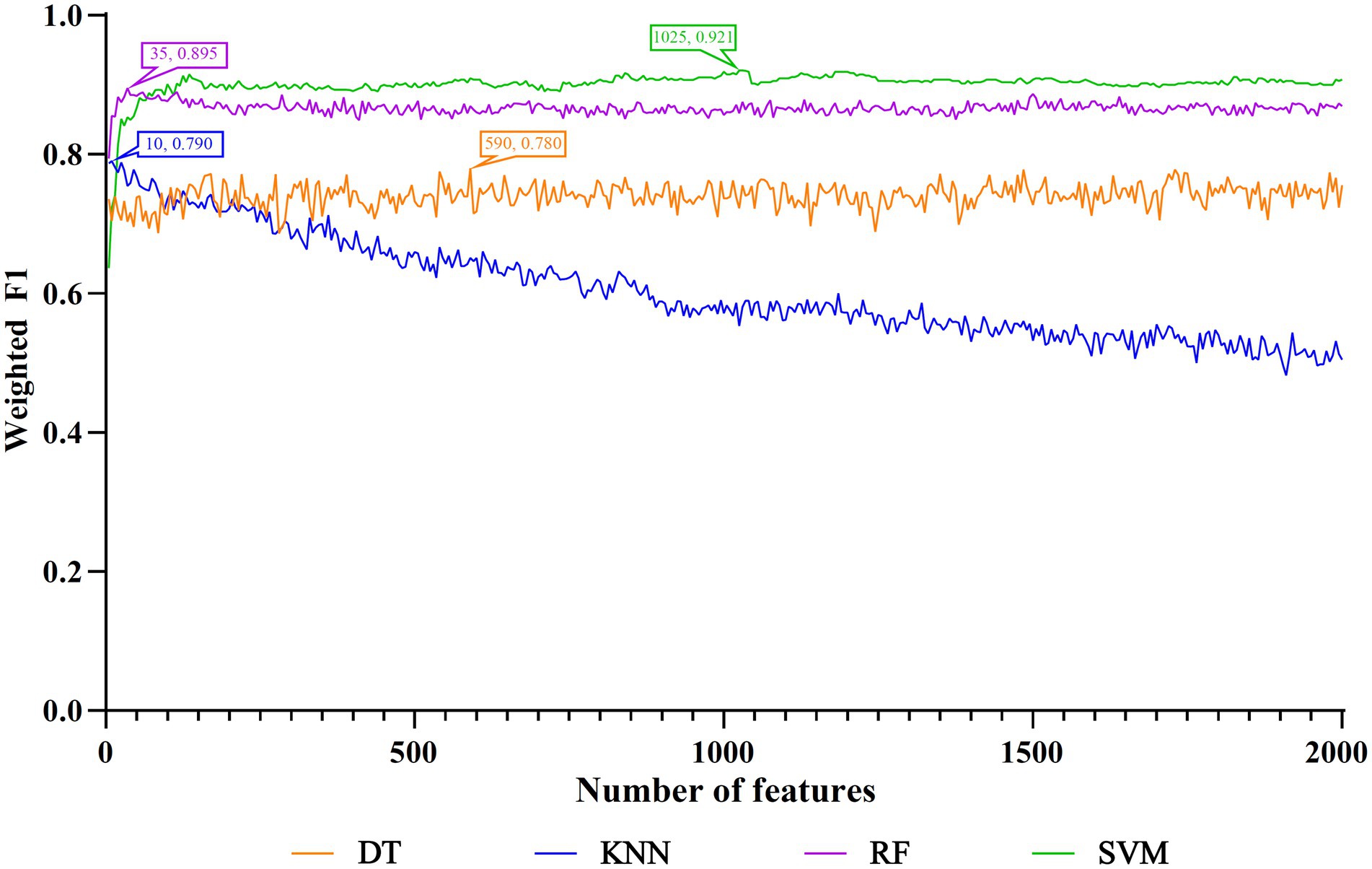
Figure 3. IFS curves to show the performance of different classification algorithms under different feature subsets. The highest weighted F1 for each classification algorithm was marked on the corresponding IFS curve. The SVM yielded the highest weighted F1 of 0.921 when top 1,025 features were used.
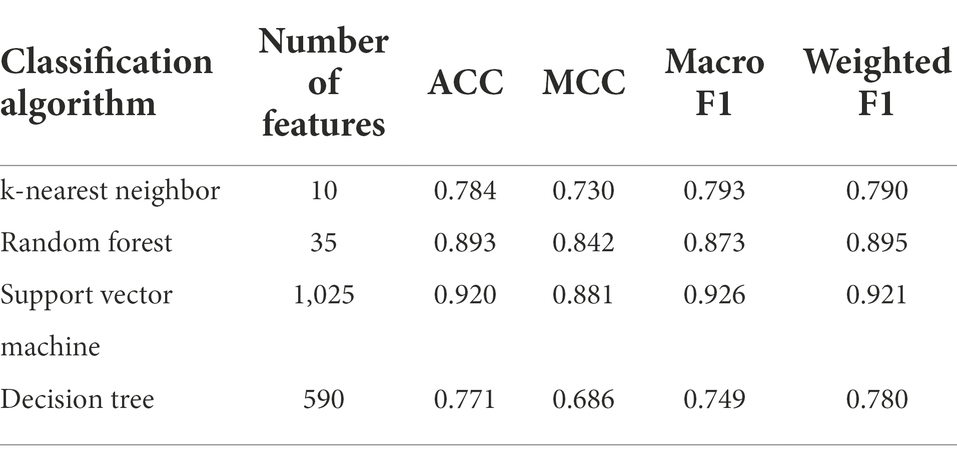
Table 2. Overall performance of the optimal classifiers.
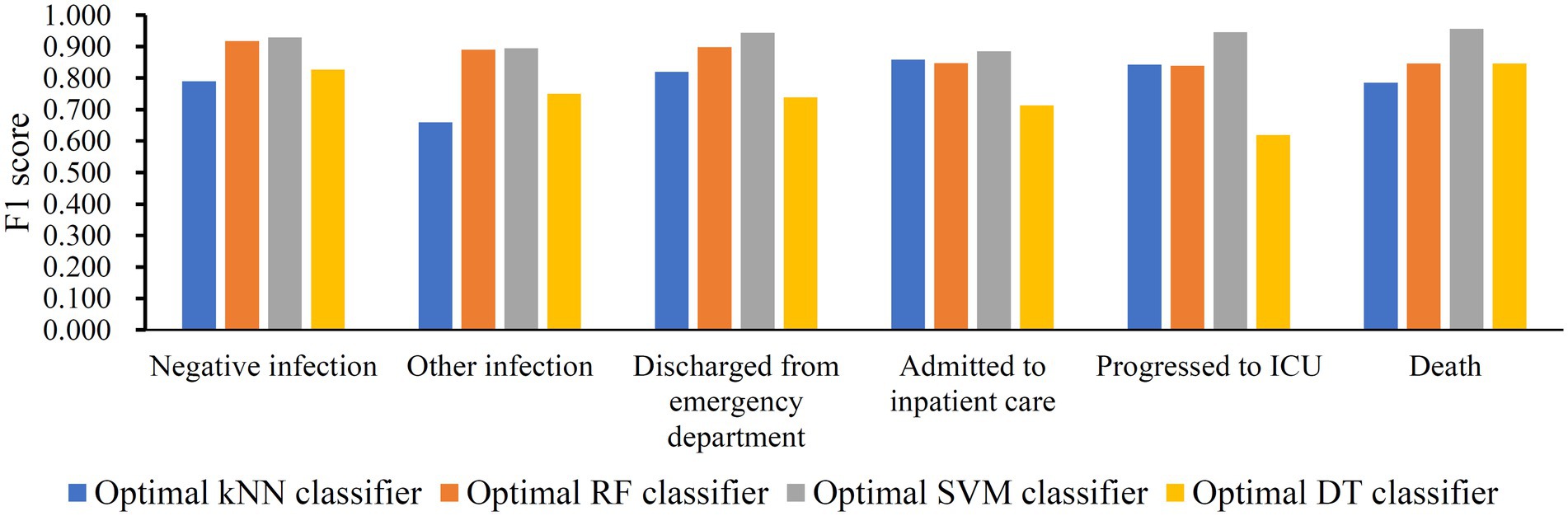
Figure 4. Performance of four optimal classifiers on six classes. The optimal SVM classifier produced best performance on all classes.
With the above arguments, the optimal SVM classifier was best. It can be an efficient tool to determine the severity of SARS-CoV-2 infection. The optimal RF classifier was inferior to the optimal SVM classifier. However, its efficiency was much higher than that of the optimal SVM classifier as much less features were used. This classifier can be used to conduct large-scale tests.
Although the optimal DT classifier provided lower performance than the other three optimal classifiers, it can provide much more explicable information than other classifiers. As the optimal DT classifier adopted top 590 features in the list, a DT classifier trained with all samples comprising these features was built. Classification rules were extracted from the tree, resulting in 77 rules. These rules are provided in Supplementary Table S3. The number of rules for each class is displayed in Figure 5. The rules for “negative infection” were most, whereas those for “Death” were least. In section “Analysis of rules for different classes”, some rules would be discussed.
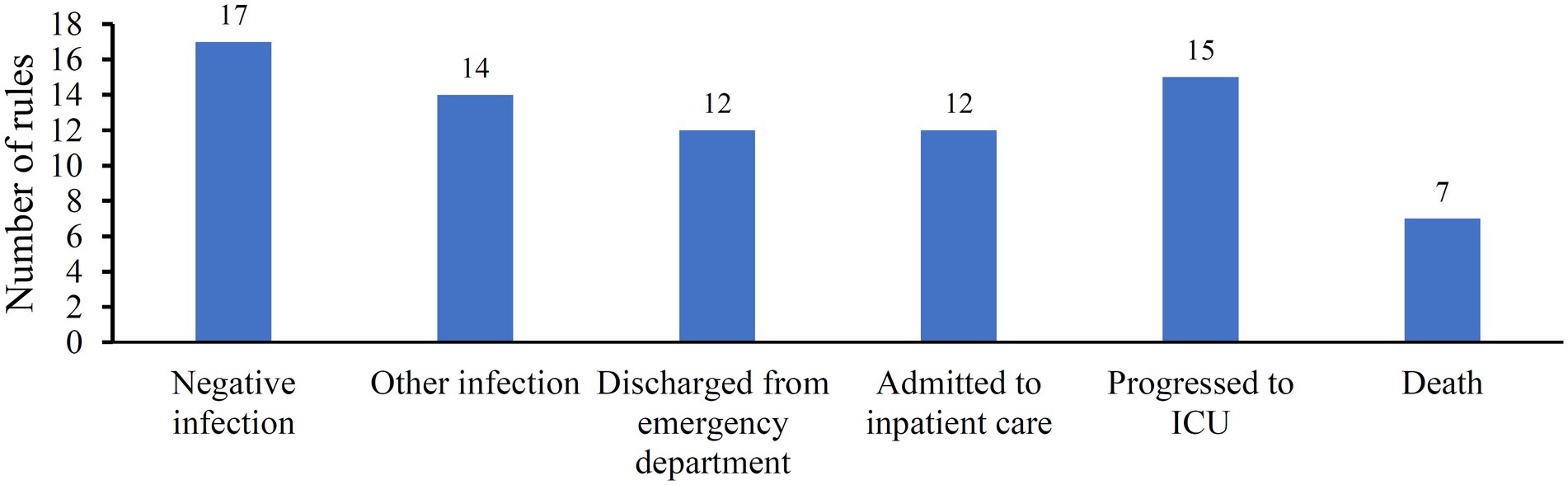
Figure 5. Distribution of classification rules on six classes.
As the optimal SVM classifier gave the best performance. This meant that features used in this classifier, that is the optimal features for SVM, were essential for determining the severity of SARS-CoV-2 infection. The corresponding genes of these features were picked up and the GO and KEGG enrichment analyses were performed on these genes, uncovering the biological meaning behind these genes. The detailed results are listed in Supplementary Table S4. Figures 6, 7 reveal that these genes were mainly enriched in biological processes, such as T-cell activation, regulation of neurotransmitter levels, and type I interferon signaling and KEGG pathways (e.g., Rap1 signaling pathway, Yersinia infection, and T-cell receptor signaling pathway). The role of these biological functions in SARS-CoV-2 infection will be verified in the section “Functional analysis based on GO and KEGG pathway”.
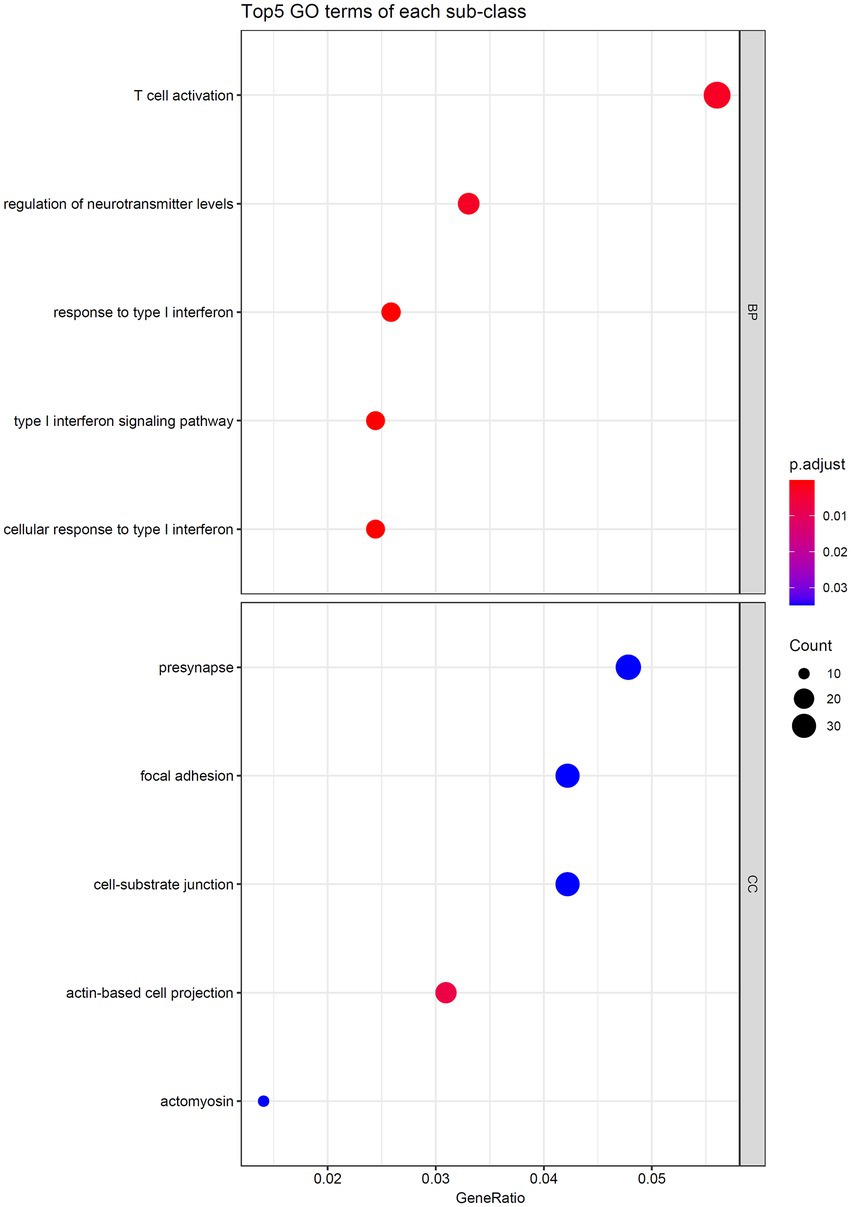
Figure 6. Top five GO terms enriched by the genes converted by the top 1,025 methylation features.
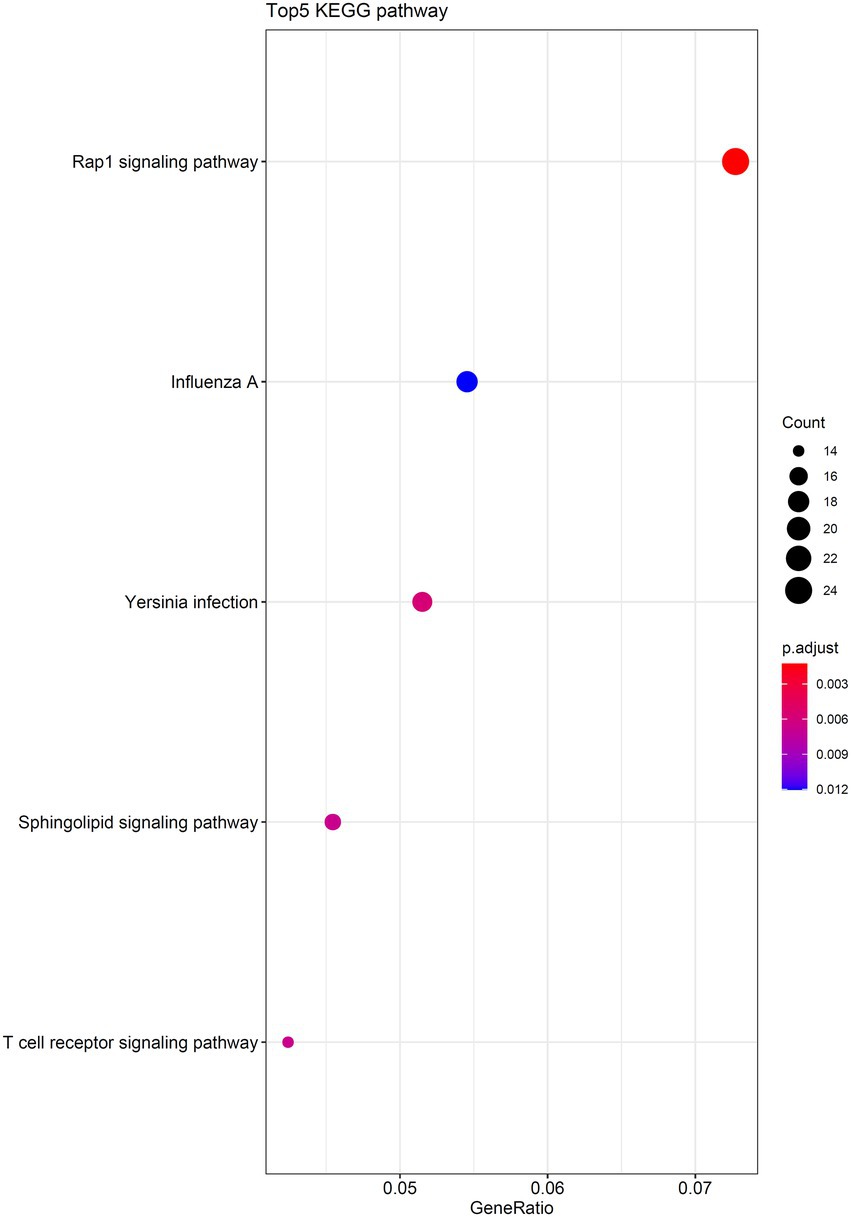
Figure 7. Top five KEGG pathways enriched by the genes converted by the top 1,025 methylation features.
Most studies only distinguish COVID-19-positive and negative samples. In this study, based on blood methylation biomarkers, we can not only classify COVID-19 from negative controls and other infections, but also accurately predict the clinical outcome of COVID-positive patients in detail. In practice, the physicians can prioritize and allocate health and medical resources for COVID-19 patients based on their predicted severe clinical outcomes. For the least severe patient, they can be discharged from hospital and avoid medical resource overstretch. For the second least severe patient, they can be hospitalized but without intensive health care. For the severe patient, intensive health care should be prepared. For the most severe patient who may die from COVID-19, life support system should be prepared.
A variety of machine learning methods were used to investigate the methylation profile on severity of SARS-CoV-2 infection. Some essential methylation features that can characterize the severity of SARS-CoV-2 infection were identified. Furthermore, a set of rules were also set up, which can not only classify SARS-CoV-2 infection samples, but also depict the methylation patterns for different severity of SARS-CoV-2 infection. These methylation features and rules would then be discussed below.
Key methylation signatures that can be used to distinguish severity of SARS-CoV-2 infection were obtained by using a set of machine learning methods. The genes corresponding to the top-ranked methylation signatures, listed in Table 3, were analyzed to demonstrate the reliability of the results.

Table 3. Essential methylation sites and their corresponding genes for distinguishing severity of SARS-CoV-2 infection.
As a type I IFN regulatory gene, high expressions of polyadenosine diphosphate ribose polymerase 9 (PARP9, cg22930808) accompanied by hypomethylation at relevant sites can enhance IFN signaling (Zhu et al., 2019), thereby playing a role in solid tumors, macrophage regulation, and antiviral immunity (Xing et al., 2021). PARP9 mediates the production of type I interferon after binding to viral RNA by activating the PI3K/AKT3 signaling pathway, thereby protecting against viral infection (Zhang et al., 2015). In addition, PARP9 is involved in the activation of anti-inflammatory M2 macrophages. This condition showed that the SARS-CoV-2 Nsp3 protein is similar to PARP9 and can inhibit PARP9 through molecular mimicry, depleting M2 macrophages, and weakening interferon signaling, which then weakens the ability of the host to resist viral infection (Da Silva et al., 2020; Fehr et al., 2020). The reduction of PARP9 combined with the reduction of NK and CD8+ cells leads to a weak viral response of the host, which may be an important reason for the life-threatening severe infections in patients.
Similar to PARP9, as an important host interferon-stimulated gene in antiviral infection (Anderson et al., 2021), MX1 (cg25888371) is hypomethylated in CpG after viral infection (Luo et al., 2021) and then participates in regulating the defense response of the host to infection. The study found that the expression of MX1 was significantly increased in COVID-19 patients compared with non-COVID-19 patients and increased with the viral load (Bizzotto et al., 2020). In addition, the methylation of CpG in MX1 is associated with the severity of HIV patients using cocaine in HIV infection studies (Shu et al., 2020), suggesting that MX1 methylation levels may be a reliable predictor of COVID-19 severity.
IRF7 (cg17114584), a member of the interferon regulatory factor (IRF) family, can regulate the response of type I IFN to viral infection. Phosphorylation of IRF7 upon pathogen stimulation followed by nuclear translocation induces the expression of IFN-α (Puthia et al., 2016). The methylation level of its promoter region affects the clinical manifestations of diseases (Konigsberg et al., 2021). Studies have shown that the expression level of IRF7 is increased in patients with mild/moderate COVID-19 (Li N. et al., 2021), while those with reduced IRF7 expression due to hypermethylation of the IRF7 promoter gene are likely to develop severe infection after SARS-CoV-2 infection (Liu and Hill, 2020).
Overall, the obtained genes showed differential expression of methylation in different infection groups, suggesting that the methylation status of different genes may be an important feature to distinguish different SARS-CoV2 infection severities.
The decision rules (Supplementary Table S3) revealed the importance of IRF7 (cg17114584) in predicting the clinical outcome of SARS-CoV-2 infection. IRF7 is markedly hypermethylated in patients with poor clinical response (progressed to ICU or death) compared with patients with mild clinical response (discharged from the emergency department or admitted to inpatient care). This finding is consistent with a previous result, in which IRF7 can regulate the response of type I IFN to viral infection and the expression level is negatively correlated with clinical manifestations. Recent studies show that methylation levels of IRF7 correlate with COVID-19 severity (Barturen et al., 2021), which is also consistent with the conclusions in the data source literature (Konigsberg et al., 2021).
The decision rule for distinguishing between other infections and non-COVID-19/COVID-19 infections indicated that FHL1 (cg00012680) was highly methylated in patients with other infections. As a member of the FHL protein family, FHL1 is mainly expressed in the heart and skeletal muscles (Shathasivam et al., 2010). As a tumor suppressor gene, FHL1 is downregulated in a variety of tumors (Wang et al., 2017). Studies have also shown that FHL1 is associated with viral infections (e.g., acting as a host factor to promote chikungunya virus infection; Meertens et al., 2019). Conversely, patients in the “death” cohort had low levels of FHL1 methylation. A study has shown that in COVID-19 patients, FHL1 is associated with the JAK–STAT pathway, which can indirectly activate STATs and induce various inflammatory responses (Bass et al., 2021). Another key criterion in distinguishing patients from other infections is the methylation level of TGFB3 (cg06958766), which is hypomethylated in COVID-19 patients (especially ICU and death patients). Existing studies have demonstrated that TGFB3 is a gene related to immune dysregulation in cardiovascular disease, and its expression is also dysregulated in COVID-19 (Lee et al., 2021). The association of the methylation level of TGFB3 with the clinical outcome of COVID-19 infection has not been revealed, and such level is speculated to be possibly associated with poor clinical response to COVID-19.
The result also indicated that the methylation level of the interferon type I pathway-related gene RSAD2 (cg10549986) for COVID-19 patients was negatively correlated to the severity of COVID-19, and the expression of RSAD2 is reported to have reached the highest level in the early stage compared with the late stage of COVID-19 (Zhang C. et al., 2021). This finding may be related to the decrease in IFN activity in patients with severe infections. In COVID-19 patients, RSAD2 can enhance antiviral and immunomodulatory functions after viral infection, and patients discharged from the emergency department in the current results had lower levels of RSAD2 methylation, possibly related to high RSAD2 expression levels and enhanced antiviral immunity (Zhu et al., 2020).
T-cell activation is the most significantly enriched pathway. Studies have shown that RNA m6A methylation is crucial for controlling the activation and differentiation of T lymphocytes (Qiu et al., 2021). m6A with T-cell activation function mainly mediates the activation and proliferation of T cells by increasing TGF-β and PI3K-AKT signaling necessary for T-cell differentiation and plays an anti-COVID-19 role (Li et al., 2017). Increased m6A regulator expression in COVID-19 patients results in the high expression of activated CD4 memory T cells (Yao et al., 2021). As a crucial immune cell in SARS-CoV-2 infection, T cells have dual roles in patients with COVID-19. The expression level of T cells is increased in patients with mild infection; among which, CD8+ T cells highly express cytotoxic molecules, such as granzyme A, which play an antiviral immune effect (Liao et al., 2020). Meanwhile, the expression levels of cytotoxic molecules and Tregs in severe patients are reduced (De Biasi et al., 2020; Toor et al., 2021). Studies have shown the presence of a complete memory T-cell response in asymptomatic or mildly infected COVID-19 patients (Sekine et al., 2020) and detected SARS-CoV-2-related T-cell responses in healthy blood samples, which may be due to seasonal coronavirus-induced T-cell responses and may further prevent serious infections (Braun et al., 2020; Mateus et al., 2020).
The current study also observed enrichment of pathways that regulate the level of neurotransmitters, suggesting the role of methylation of neurotransmitter-related genes in immunity to virus infection. Studies have shown that in addition to macrophages, viral infection also activates mast cells to release histamine, arachidonic acid, and other neurotransmitters, and histamine can strongly raise the level of IL-1, which, in turn, increases lung inflammation in SARS-CoV-2 infection (Conti et al., 2020). Furthermore, SARS-CoV2 infection will reduce the synthesis of dopamine and acetylcholine, resulting in the weakened immune function of the body (Blum et al., 2020; Alexandris et al., 2021).
Type I interferon plays a crucial role in antiviral immunity, and studies have shown that hypermethylation of IFN-related genes is a unique methylation signature of severe COVID-19. Moreover, three of the significant enrichment pathways are related to type I interferon response, further confirming the important role of IFN-related methylation in determining the severity of COVID-19 and the reliability of the current study. In vivo, IFN can bind to IFN receptors in an autocrine and paracrine manner to activate the JAK/STAT signaling pathway, thus demonstrating antiviral effects (Liu et al., 2012). IFN activity was also lower in patients with severe infection than mild infection patients, and impaired IFN-α production is an important sign of severe infection (Hadjadj et al., 2020), which may be related to the hypermethylation of IFN-related genes and the inhibition of the expression of related genes. In addition, studies have shown that IFN expression is delayed in SARS-CoV-2 infection (Kim et al., 2016). Such a delay leads to high levels of interferon expression in severely infected patients but does not reduce viral load; meanwhile, IFN pretreatment can significantly reduce viral infection levels, suggesting that drugs that can boost IFN production may be an effective option for early treatment of SARS-CoV-2 (Park and Iwasaki, 2020).
The enrichment of cellular components of differentially methylated genes mainly focused on the virus infection process of cells, including cell junction and migration. Synapses mainly mediate information transmission between neurons; they can also transmit large particles and mediate virus particles into the central nervous system, thus reflecting the neuroinvasiveness of coronaviruses (Li et al., 2020). As the main site of cell adhesion, focal adhesions help viral particles enter cells, and its functional integrity is critical to the infection and spread of SARS-CoV-2 (Sulzmaier et al., 2014).
This study also found enrichment of differentially methylated genes in the RAP1 pathway. RAP1 pathway plays an important role in processes, such as cell adhesion, junction, and polarity, and promotes tumor cell invasion and migration (Looi et al., 2020). Pulmonary vascular barrier integrity defection is a fatal factor in severe COVID-19 patients (Yamamoto et al., 2021). Meanwhile, studies have found that RAP1 can enhance endothelial cell–cell junctions mediated by VE-cadherin and regulate vascular permeability (Rho et al., 2017), suggesting that the RAP1 signaling pathway may serve as a potential therapeutic target for COVID-19.
The analysis of key features and related decision rules verified the effectiveness of methylation status in distinguishing different states of SARS-CoV-2 infection, which will provide a reference for studying the stratification of patients and help develop new treatment strategies.
A computational workflow containing several machine learning methods was designed to identify the blood methylation features and their expression rules, which can distinguish the severity of SARS-CoV-2 infection. First, the methylation features in the expression profile were analyzed by the MCFS algorithm, producing a ranked feature list. Next, this list was introduced into the IFS method to generate a series of feature subsets. Different classification algorithms were used to train samples comprising these feature subsets to build classifiers. After evaluating their performance, the optimal features were determined. The classification rules were extracted by the optimal DT classifier. The essential features were analyzed by functional enrichment to detect their biofunctional information. Some key features and rules are justified by recently published academic literature, which provides a reference for further related research.
Publicly available datasets were analyzed in this study. These data can be found at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167202.
TH and Y-DC designed the study. SD and KF performed the experiments. ZL, MM, and XZ analyzed the results. ZL, MM, and SD wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDA26040304 and XDB38050200), National Key R&D Program of China (2018YFC0910403), and the Fund of the Key Laboratory of Tissue Microenvironment and Tumor of Chinese Academy of Sciences (202002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1007295/full#supplementary-material
Alexandris, N., Lagoumintzis, G., Chasapis, C. T., Leonidas, D. D., Papadopoulos, G. E., Tzartos, S. J., et al. (2021). Nicotinic cholinergic system and COVID-19: in silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicol. Rep. 8, 73–83. doi: 10.1016/j.toxrep.2020.12.013
Anderson, D., Neri, J., Souza, C. R. M., Valverde, J. G., De Araújo, J. M. G., Nascimento, M., et al. (2021). Zika virus changes methylation of genes involved in immune response and neural development in Brazilian babies born with congenital microcephaly. J. Infect. Dis. 223, 435–440. doi: 10.1093/infdis/jiaa383
Barturen, G., Carnero-Montoro, E., Martínez-Bueno, M., Rojo-Rello, S., Sobrino, B., Alcántara-Domínguez, C., et al. (2021). Whole-blood DNA methylation analysis reveals respiratory environmental traits involved in COVID-19 severity following SARS-CoV-2 infection. medRXiv.
Bass, A., Liu, Y., and Dakshanamurthy, S. (2021). Single-cell and bulk RNASeq profiling of COVID-19 patients reveal immune and inflammatory mechanisms of infection-induced organ damage. Viruses 13:2418. doi: 10.3390/v13122418
Benhamida, J. K., Hechtman, J. F., Nafa, K., Villafania, L., Sadowska, J., Wang, J., et al. (2020). Reliable clinical MLH1 promoter Hypermethylation assessment using a high-throughput genome-wide methylation Array platform. J. Mol. Diagn. 22, 368–375. doi: 10.1016/j.jmoldx.2019.11.005
Bizzotto, J., Sanchis, P., Abbate, M., Lage-Vickers, S., Lavignolle, R., Toro, A., et al. (2020). SARS-CoV-2 infection boosts MX1 antiviral effector in COVID-19 patients. iScience 23:101585. doi: 10.1016/j.isci.2020.101585
Blum, K., Cadet, J. L., Baron, D., Badgaiyan, R. D., Brewer, R., Modestino, E. J., et al. (2020). Putative COVID-19 induction of reward deficiency syndrome (RDS) and associated behavioral addictions with potential concomitant dopamine depletion: is COVID-19 social distancing a double edged sword? Subst. Use Misuse 55, 2438–2442. doi: 10.1080/10826084.2020.1817086
Braun, J., Loyal, L., Frentsch, M., Wendisch, D., Georg, P., Kurth, F., et al. (2020). SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 587, 270–274. doi: 10.1038/s41586-020-2598-9
Castro De Moura, M., Davalos, V., Planas-Serra, L., Alvarez-Errico, D., Arribas, C., Ruiz, M., et al. (2021). Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66:103339. doi: 10.1016/j.ebiom.2021.103339
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357. doi: 10.1613/jair.953
Chen, Y. G., Chen, R., Ahmad, S., Verma, R., Kasturi, S. P., Amaya, L., et al. (2019). N6-Methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109.e9. doi: 10.1016/j.molcel.2019.07.016
Chen, W., Chen, L., and Dai, Q. (2021). iMPT-FDNPL: identification of membrane protein types with functional domains and a natural language processing approach. Comput. Math. Methods Med. 2021, 1–10. doi: 10.1155/2021/7681497
Chen, L., Li, Z., Zhang, S., Zhang, Y.-H., Huang, T., and Cai, Y.-D. (2022). Predicting RNA 5-methylcytosine sites by using essential sequence features and distributions. Biomed. Res. Int. 2022, 1–11. doi: 10.1155/2022/4035462
Conti, P., Caraffa, A., Tetè, G., Gallenga, C. E., Ross, R., Kritas, S. K., et al. (2020). Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J. Biol. Regul. Homeost. Agents 34, 1629–1632. doi: 10.23812/20-2EDIT
Corley, M. J., Pang, A. P. S., Dody, K., Mudd, P. A., Patterson, B. K., Seethamraju, H., et al. (2021). Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 110, 21–26. doi: 10.1002/JLB.5HI0720-466R
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Cover, T., and Hart, P. (1967). Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 13, 21–27. doi: 10.1109/TIT.1967.1053964
Da Silva, S. J. R., Alves Da Silva, C. T., Mendes, R. P. G., and Pena, L. (2020). Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J. Med. Virol. 92, 1427–1429. doi: 10.1002/jmv.25858
De Biasi, S., Meschiari, M., Gibellini, L., Bellinazzi, C., Borella, R., Fidanza, L., et al. (2020). Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 11:3434. doi: 10.1038/s41467-020-17292-4
Ding, S., Wang, D., Zhou, X., Chen, L., Feng, K., Xu, X., et al. (2022). Predicting heart cell types by using Transcriptome profiles and a machine learning method. Life 12:228. doi: 10.3390/life12020228
Dramiński, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., and Komorowski, J. (2007). Monte Carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Durbin, A. F., Wang, C., Marcotrigiano, J., and Gehrke, L. (2016). RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 7, e00833–e00816. doi: 10.1128/mBio.00833-16
Eberle, C., James-Todd, T., and Stichling, S. (2021). SARS-CoV-2 in diabetic pregnancies: a systematic scoping review. BMC Pregnancy Childbirth 21:573. doi: 10.1186/s12884-021-03975-3
Fan, R., Mao, S. Q., Gu, T. L., Zhong, F. D., Gong, M. L., Hao, L. M., et al. (2017). Preliminary analysis of the association between methylation of the ACE2 promoter and essential hypertension. Mol. Med. Rep. 15, 3905–3911. doi: 10.3892/mmr.2017.6460
Fehr, A. R., Singh, S. A., Kerr, C. M., Mukai, S., Higashi, H., and Aikawa, M. (2020). The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 34, 341–359. doi: 10.1101/gad.334425.119
Gorodkin, J. (2004). Comparing two K-category assignments by a K-category correlation coefficient. Comput. Biol. Chem. 28, 367–374. doi: 10.1016/j.compbiolchem.2004.09.006
Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., et al. (2020). Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724. doi: 10.1126/science.abc6027
Hatta, Y., Hershberger, K., Shinya, K., Proll, S. C., Dubielzig, R. R., Hatta, M., et al. (2010). Viral replication rate regulates clinical outcome and CD8 T cell responses during highly pathogenic H5N1 influenza virus infection in mice. PLoS Pathog. 6:e1001139. doi: 10.1371/journal.ppat.1001139
Kianmehr, A., Faraoni, I., Kucuk, O., and Mahrooz, A. (2021). Epigenetic alterations and genetic variations of angiotensin-converting enzyme 2 (ACE2) as a functional receptor for SARS-CoV-2: potential clinical implications. Eur. J. Clin. Microbiol. Infect. Dis. 40, 1587–1598. doi: 10.1007/s10096-021-04264-9
Kim, E. S., Choe, P. G., Park, W. B., Oh, H. S., Kim, E. J., Nam, E. Y., et al. (2016). Clinical progression and cytokine profiles of Middle East respiratory syndrome coronavirus infection. J. Korean Med. Sci. 31, 1717–1725. doi: 10.3346/jkms.2016.31.11.1717
Kohavi, R. (1995). “A study of cross-validation and bootstrap for accuracy estimation and model selection,” in Proceedings of the 14th International Joint Conference on Artificial Intelligence, Vol. 2 (Montreal, QC: Morgan Kaufmann Publishers Inc.).
Konigsberg, I. R., Barnes, B., Campbell, M., Davidson, E., Zhen, Y., Pallisard, O., et al. (2021). Host methylation predicts SARS-CoV-2 infection and clinical outcome. Commun Med (London) 1:42. doi: 10.1038/s43856-021-00042-y
Lee, A. C., Castaneda, G., Li, W. T., Chen, C., Shende, N., Chakladar, J., et al. (2021). COVID-19 severity potentially modulated by cardiovascular-disease-associated immune dysregulation. Viruses 13:1018. doi: 10.3390/v13061018
Li, Y. C., Bai, W. Z., and Hashikawa, T. (2020). The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 92, 552–555. doi: 10.1002/jmv.25728
Li, S., Duan, X., Li, Y., Li, M., Gao, Y., Li, T., et al. (2021). Differentially expressed immune response genes in COVID-19 patients based on disease severity. Aging (Albany NY) 13, 9265–9276. doi: 10.18632/aging.202877
Li, N., Hui, H., Bray, B., Gonzalez, G. M., Zeller, M., Anderson, K. G., et al. (2021). METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 35:109091. doi: 10.1016/j.celrep.2021.109091
Li, X., Lu, L., and Chen, L. (2022). Identification of protein functions in mouse with a label space partition method. Math. Biosci. Eng. 19, 3820–3842. doi: 10.3934/mbe.2022176
Li, H. B., Tong, J., Zhu, S., Batista, P. J., Duffy, E. E., Zhao, J., et al. (2017). M(6)a mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. doi: 10.1038/nature23450
Liao, M., Liu, Y., Yuan, J., Wen, Y., Xu, G., Zhao, J., et al. (2020). Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844. doi: 10.1038/s41591-020-0901-9
Liu, B. M., and Hill, H. R. (2020). Role of host immune and inflammatory responses in COVID-19 cases with underlying primary immunodeficiency: a review. J. Interf. Cytokine Res. 40, 549–554. doi: 10.1089/jir.2020.0210
Liu, S. Y., Sanchez, D. J., Aliyari, R., Lu, S., and Cheng, G. (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U. S. A. 109, 4239–4244. doi: 10.1073/pnas.1114981109
Liu, H., and Setiono, R. (1998). Incremental feature selection. Appl. Intell. 9, 217–230. doi: 10.1023/A:1008363719778
Looi, C. K., Hii, L. W., Ngai, S. C., Leong, C. O., and Mai, C. W. (2020). The role of Ras-associated protein 1 (Rap1) in cancer: bad actor or good player? Biomedicine 8:334. doi: 10.3390/biomedicines8090334
Luo, X., Peng, Y., Chen, Y. Y., Wang, A. Q., Deng, C. W., Peng, L. Y., et al. (2021). Genome-wide DNA methylation patterns in monocytes derived from patients with primary Sjogren syndrome. Chin. Med. J. 134, 1310–1316. doi: 10.1097/CM9.0000000000001451
Mateus, J., Grifoni, A., Tarke, A., Sidney, J., Ramirez, S. I., Dan, J. M., et al. (2020). Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370, 89–94. doi: 10.1126/science.abd3871
Matthews, B. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA)-protein. Structure 405, 442–451. doi: 10.1016/0005-2795(75)90109-9
Meertens, L., Hafirassou, M. L., Couderc, T., Bonnet-Madin, L., Kril, V., Kümmerer, B. M., et al. (2019). FHL1 is a major host factor for chikungunya virus infection. Nature 574, 259–263. doi: 10.1038/s41586-019-1578-4
Menachery, V. D., Schäfer, A., Burnum-Johnson, K. E., Mitchell, H. D., Eisfeld, A. J., Walters, K. B., et al. (2018). MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc. Natl. Acad. Sci. U. S. A. 115, E1012–e1021. doi: 10.1073/pnas.1706928115
Onesime, M., Yang, Z., and Dai, Q. (2021). Genomic Island prediction via Chi-Square test and random Forest algorithm. Comput. Math. Methods Med. 2021, 1–9. doi: 10.1155/2021/9969751
Park, A., and Iwasaki, A. (2020). Type I and type III Interferons – induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27, 870–878. doi: 10.1016/j.chom.2020.05.008
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830.
Puthia, M., Ambite, I., Cafaro, C., Butler, D., Huang, Y., Lutay, N., et al. (2016). IRF7 inhibition prevents destructive innate immunity-a target for nonantibiotic therapy of bacterial infections. Sci. Transl. Med. 8:336ra359. doi: 10.1126/scitranslmed.aaf1156
Qiu, X., Hua, X., Li, Q., Zhou, Q., and Chen, J. (2021). M(6)a regulator-mediated methylation modification patterns and characteristics of immunity in blood leukocytes of COVID-19 patients. Front. Immunol. 12:774776. doi: 10.3389/fimmu.2021.774776
Ran, B., Chen, L., Li, M., Han, Y., and Dai, Q. (2022). Drug-drug interactions prediction using fingerprint only. Comput. Math. Methods Med. 2022, 1–14. doi: 10.1155/2022/7818480
Rho, S. S., Ando, K., and Fukuhara, S. (2017). Dynamic regulation of vascular permeability by vascular endothelial cadherin-mediated endothelial cell-cell junctions. J. Nippon Med. Sch. 84, 148–159. doi: 10.1272/jnms.84.148
Safavian, S. R., and Landgrebe, D. (1991). A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 21, 660–674. doi: 10.1109/21.97458
Schulte-Schrepping, J., Reusch, N., Paclik, D., Baßler, K., Schlickeiser, S., Zhang, B., et al. (2020). Severe COVID-19 is marked by a Dysregulated myeloid cell compartment. Cells 182, 1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
Sekine, T., Perez-Potti, A., Rivera-Ballesteros, O., Strålin, K., Gorin, J. B., Olsson, A., et al. (2020). Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cells 183, 158–168.e14. doi: 10.1016/j.cell.2020.08.017
Sen, R., Garbati, M., Bryant, K., and Lu, Y. (2021). Epigenetic mechanisms influencing COVID-19. Genome 64, 372–385. doi: 10.1139/gen-2020-0135
Shathasivam, T., Kislinger, T., and Gramolini, A. O. (2010). Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J. Cell. Mol. Med. 14, 2702–2720. doi: 10.1111/j.1582-4934.2010.01176.x
Shu, C., Justice, A. C., Zhang, X., Wang, Z., Hancock, D. B., Johnson, E. O., et al. (2020). DNA methylation mediates the effect of cocaine use on HIV severity. Clin. Epigenetics 12:140. doi: 10.1186/s13148-020-00934-1
Sulzmaier, F. J., Jean, C., and Schlaepfer, D. D. (2014). FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610. doi: 10.1038/nrc3792
Tang, S., and Chen, L. (2022). iATC-NFMLP: identifying classes of anatomical therapeutic chemicals based on drug networks, fingerprints and multilayer perceptron. Curr. Bioinforma. 17. doi: 10.2174/1574893617666220318093000 [Epub ahead of print].
Toor, S. M., Saleh, R., Sasidharan Nair, V., Taha, R. Z., and Elkord, E. (2021). T-cell responses and therapies against SARS-CoV-2 infection. Immunology 162, 30–43. doi: 10.1111/imm.13262
Wang, R., and Chen, L. (2022). Identification of human protein subcellular location with multiple networks. Current. Proteomics 19, 344–356. doi: 10.2174/1570164619666220531113704
Wang, J., Huang, F., Huang, J., Kong, J., Liu, S., and Jin, J. (2017). Epigenetic analysis of FHL1 tumor suppressor gene in human liver cancer. Oncol. Lett. 14, 6109–6116. doi: 10.3892/ol.2017.6950
Wu, Z., and Chen, L. (2022). Similarity-based method with multiple-feature sampling for predicting drug side effects. Comput. Math. Methods Med. 2022, 1–13. doi: 10.1155/2022/9547317
Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovations 2:100141. doi: 10.1016/j.xinn.2021.100141
Xing, J., Zhang, A., Du, Y., Fang, M., Minze, L. J., Liu, Y. J., et al. (2021). Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat. Commun. 12:2681. doi: 10.1038/s41467-021-23003-4
Yamamoto, K., Takagi, Y., Ando, K., and Fukuhara, S. (2021). Rap1 small GTPase regulates vascular endothelial-cadherin-mediated endothelial cell-cell junctions and vascular permeability. Biol. Pharm. Bull. 44, 1371–1379. doi: 10.1248/bpb.b21-00504
Yang, Y., and Chen, L. (2022). Identification of drug–disease associations by using multiple drug and disease networks. Curr. Bioinforma. 17, 48–59. doi: 10.2174/1574893616666210825115406
Yao, Y., Yang, Y., Guo, W., Xu, L., You, M., Zhang, Y. C., et al. (2021). METTL3-dependent m(6)a modification programs T follicular helper cell differentiation. Nat. Commun. 12:1333. doi: 10.1038/s41467-021-21594-6
Zhang, C., Feng, Y. G., Tam, C., Wang, N., and Feng, Y. (2021). Transcriptional profiling and machine learning unveil a concordant biosignature of type I interferon-inducible host response across nasal swab and pulmonary tissue for COVID-19 diagnosis. Front. Immunol. 12:733171. doi: 10.3389/fimmu.2021.733171
Zhang, Y., Mao, D., Roswit, W. T., Jin, X., Patel, A. C., Patel, D. A., et al. (2015). PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 16, 1215–1227. doi: 10.1038/ni.3279
Zhang, Y.-H., Zeng, T., Chen, L., Huang, T., and Cai, Y.-D. (2021). Determining protein–protein functional associations by functional rules based on gene ontology and KEGG pathway. Biochim. Biophys. Acta Proteins Proteom. 1869:140621. doi: 10.1016/j.bbapap.2021.140621
Zhou, J.-P., Chen, L., and Guo, Z.-H. (2020a). iATC-NRAKEL: an efficient multi-label classifier for recognizing anatomical therapeutic chemical classes of drugs. Bioinformatics 36, 1391–1396. doi: 10.1093/bioinformatics/btz757
Zhou, J.-P., Chen, L., Wang, T., and Liu, M. (2020b). iATC-FRAKEL: a simple multi-label web-server for recognizing anatomical therapeutic chemical classes of drugs with their fingerprints only. Bioinformatics 36, 3568–3569. doi: 10.1093/bioinformatics/btaa166
Zhou, X., Ding, S., Wang, D., Chen, L., Feng, K., Huang, T., et al. (2022). Identification of cell markers and their expression patterns in skin based on single-cell RNA-sequencing profiles. Life 12:550. doi: 10.3390/life12040550
Zhu, H., Wu, L. F., Mo, X. B., Lu, X., Tang, H., Zhu, X. W., et al. (2019). Rheumatoid arthritis-associated DNA methylation sites in peripheral blood mononuclear cells. Ann. Rheum. Dis. 78, 36–42. doi: 10.1136/annrheumdis-2018-213970
Keywords: SARS-CoV-2, severity, methylation, machine learning, classification rule
Citation: Liu Z, Meng M, Ding S, Zhou X, Feng K, Huang T and Cai Y-D (2022) Identification of methylation signatures and rules for predicting the severity of SARS-CoV-2 infection with machine learning methods. Front. Microbiol. 13:1007295. doi: 10.3389/fmicb.2022.1007295
Edited by:
Fei Ma, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Martina Barchitta, University of Catania, ItalyCopyright © 2022 Liu, Meng, Ding, Zhou, Feng, Huang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Huang, dG9odWFuZ3Rhb0AxMjYuY29t; Yu-Dong Cai, Y2FpX3l1ZEAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.